Recent Development of Non-Peptide GnRH Antagonists

 ,
,
Abstract
:1. Introduction
2. Small Molecule GnRH Receptor Antagonist
2.1. Thieno[2,3-b]pyridine-4-one and Thieno[2,3-b]pyrimidine-2,4-one Derivatives
2.2. Pyrrolo[1,2-a]Pyrimidin-7-one Derivatives
2.3. Imidazolo[1,2-a]pyrimidin-5-one Derivatives
2.4. Uracil Derivatives
2.5. Indole Derivatives
2.6. Quinolone Derivatives
2.7. Furamide Derivatives
2.8. Benzimidazole Derivatives
2.9. Piperazine-Benzimidazole Derivatives
2.10. Spiroindoline Derivatives
2.11. Other Compounds
3. Clinical Developments of Small Molecule GnRH Receptor Antagonist
4. Potential New Indications for Non-Peptide GnRH Antagonists
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Matsuo, H.; Baba, Y.; Nair, R.M.G.; Arimura, A.; Schally, A.V. Structure of the porcine LH- and FSH-releasing hormone. I. The proposed amino acid sequence. Biochem. Biophys. Res. Commun. 1971, 43, 1334–1339. [Google Scholar] [CrossRef]
- Millar, R.P.; Lu, Z.-L.; Pawson, A.J.; Flanagan, C.A.; Morgan, K.; Maudsley, S.R. Gonadotropin-Releasing Hormone Receptors. Endocr. Rev. 2004, 25, 235–275. [Google Scholar] [CrossRef] [PubMed]
- Filicori, M. Gonadotrophin-Releasing Hormone Agonists. Drugs 1994, 48, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Emons, G.; Schally, A.V. The use of luteinizing hormone releasing hormone agonists and antagonists in gynaecological cancers. Hum. Reprod. 1994, 9, 1364–1379. [Google Scholar] [CrossRef] [PubMed]
- Belchetz, P.; Plant, T.; Nakai, Y.; Keogh, E.; Knobil, E. Hypophysial responses to continuous and intermittent delivery of hypopthalamic gonadotropin-releasing hormone. Science 1978, 202, 631–633. [Google Scholar] [CrossRef] [PubMed]
- Conn, P.M.; Crowley, W.F. Gonadotropin-Releasing Hormone and Its Analogues. N. Engl. J. Med. 1991, 324, 93–103. [Google Scholar] [PubMed]
- Schultze-Mosgau, A.; Griesinger, G.; Altgassen, C.; von Otte, S.; Hornung, D.; Diedrich, K. New developments in the use of peptide gonadotropin-releasing hormone antagonists versus agonists. Expert Opin. Investig. Drugs 2005, 14, 1085–1097. [Google Scholar] [PubMed]
- Barlow, D.H. GnRH agonists and in vitro fertilization. J. Reprod. Med. 1998, 43 (Suppl. 3), 245–251. [Google Scholar] [PubMed]
- Schally, A.V.; Nagy, A. New approaches to treatment of various cancers based on cytotoxic analogs of LHRH, somatostatin and bombesin. Life Sci. 2003, 72, 2305–2320. [Google Scholar] [CrossRef]
- Lemay, A.; Maheux, R.; Faure, N.; Jean, C.; Fazekas, A.T. Reversible hypogonadism induced by a luteinizing hormone-releasing hormone (LH-RH) agonist (Buserelin) as a new therapeutic approach for endometriosis. Fertil. Steril. 1984, 41, 863–871. [Google Scholar] [PubMed]
- Cetel, N.S.; Rivier, J.; Vale, W.; Yen, S.S.C. The Dynamics of Gonadotropin Inhibition in Women Induced by an Antagonistic Analog of Gonadotropin-Releasing Hormone. J. Clin. Endocrinol. Metab. 1983, 57, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.B.; Schally, A.V. Drug Insight: Clinical use of agonists and antagonists of luteinizing-hormone-releasing hormone. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Manea, M. Gonadotropin-releasing hormone receptors as molecular therapeutic targets in prostate cancer: Current options and emerging strategies. Cancer Treat. Rev. 2013, 39, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Marelli, M.M.; Mai, S.; Motta, M.; Martini, L.; Moretti, R.M. GnRH Receptors in Cancer: From Cell Biology to Novel Targeted Therapeutic Strategies. Endocr. Rev. 2012, 33, 784–811. [Google Scholar] [CrossRef] [PubMed]
- De, B.; Plattner, J.J.; Bush, E.N.; Jae, H.S.; Diaz, G.; Johnson, E.S.; Perun, T.J. LH-RH antagonists: Design and synthesis of a novel series of peptidomimetics. J. Med. Chem. 1989, 32, 2036–2038. [Google Scholar] [CrossRef] [PubMed]
- Ezzati, M.; Carr, B.R. Elagolix, a novel, orally bioavailable GnRH antagonist under investigation for the treatment of endometriosis-related pain. Women’s Health 2015, 11, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Mező, G.; Manea, M. Luteinizing hormone-releasing hormone antagonists. Expert Opin. Ther. Pat. 2009, 19, 1771–1785. [Google Scholar] [CrossRef] [PubMed]
- Betz, S.F.; Zhu, Y.-F.; Chen, C.; Struthers, R.S. Non-Peptide Gonadotropin-Releasing Hormone Receptor Antagonists. J. Med. Chem. 2008, 51, 3331–3348. [Google Scholar] [CrossRef] [PubMed]
- Heitman, L.H.; Ijzerman, A.P. G protein-coupled receptors of the hypothalamic–pituitary–gonadal axis: A case for gnrh, LH, FSH, and GPR54 receptor ligands. Med. Res. Rev. 2008, 28, 975–1011. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-F.; Chen, C.; Scott Struthers, R. Nonpeptide Gonadotropin Releasing Hormone Antagonists. In Annual Reports in Medicinal Chemistry; Academic Press: Cambridge, MA, USA, 2004; Volume 39, pp. 99–110. [Google Scholar]
- Zhu, Y.-F.; Chen, C. Recent advances in small molecule gonadotrophin-releasing hormone receptor antagonists. Expert Opin. Ther. Pat. 2004, 14, 187–199. [Google Scholar] [CrossRef]
- Cho, N.; Harada, M.; Imaeda, T.; Imada, T.; Matsumoto, H.; Hayase, Y.; Sasaki, S.; Furuya, S.; Suzuki, N.; Okubo, S.; et al. Discovery of a Novel, Potent, and Orally Active Nonpeptide Antagonist of the Human Luteinizing Hormone-Releasing Hormone (LHRH) Receptor. J. Med. Chem. 1998, 41, 4190–4195. [Google Scholar] [CrossRef] [PubMed]
- Imada, T.; Cho, N.; Imaeda, T.; Hayase, Y.; Sasaki, S.; Kasai, S.; Harada, M.; Matsumoto, H.; Endo, S.; Suzuki, N.; et al. Design, Synthesis, and Structure−Activity Relationships of Thieno[2,3-b]pyridin-4-one Derivatives as a Novel Class of Potent, Orally Active, Non-Peptide Luteinizing Hormone-Releasing Hormone Receptor Antagonists. J. Med. Chem. 2006, 49, 3809–3825. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Cho, N.; Nara, Y.; Harada, M.; Endo, S.; Suzuki, N.; Furuya, S.; Fujino, M. Discovery of a Thieno[2,3-d]pyrimidine-2,4-dione Bearing a p-Methoxyureidophenyl Moiety at the 6-Position: A Highly Potent and Orally Bioavailable Non-Peptide Antagonist for the Human Luteinizing Hormone-Releasing Hormone Receptor. J. Med. Chem. 2003, 46, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Miwa, K.; Hitaka, T.; Imada, T.; Sasaki, S.; Yoshimatsu, M.; Kusaka, M.; Tanaka, A.; Nakata, D.; Furuya, S.; Endo, S.; et al. Discovery of 1-{4-[1-(2,6-Difluorobenzyl)-5-[(dimethylamino)methyl]-3-(6-methoxypyridazin-3-yl)-2,4-dioxo-1,2,3,4-tetrahydrothieno[2,3-d]pyrimidin-6-yl]phenyl}-3-methoxyurea (TAK-385) as a Potent, Orally Active, Non-Peptide Antagonist of the Human Gonadotropin-Releasing Hormone Receptor. J. Med. Chem. 2011, 54, 4998–5012. [Google Scholar] [PubMed]
- Cho, N.; Imada, T.; Hitaka, T.; Miwa, K.; Kusaka, M.; Suzuki, N. Preparation of Thienopyrimidine Derivatives as Gonadotropin-Releasing Hormone Antagonists. J.P. Patent WO2004067535A1, 8 December 2004. [Google Scholar]
- Furuya, S.; Kusaka, M. Premature Ovulation Preventive Agent Containing Gonadotropin-Releasing Hormone Antagonists. J.P. Patent WO2007011072A1, 25 January 2007. [Google Scholar]
- Kamikawa, K.; Okabe, T.; Nakamura, M. Method for Improving Absorbability of Preparation, and Preparation Having Improved Absorbability. J.P. Patent WO2010026993A1, 11 March 2010. [Google Scholar]
- Fukuoka, K.; Miwa, K.; Sasaki, T.; Komura, F. Production Method of a Thienopyrimidine Derivative. J.P. Patent WO2014051164A2, 3 April 2014. [Google Scholar]
- Yamane, I.; Nomura, Y.; Nishimoto, Y.; Hoshina, W. Solid Preparation with Improved Stability of Thienopyrimidine Derivative. J.P. Patent WO2016136849A1, 1 September 2016. [Google Scholar]
- Zhu, Y.-F.; Struthers, R.S.; Connors, J.P.J.; Gao, Y.; Gross, T.D.; Saunders, J.; Wilcoxen, K.; Reinhart, G.J.; Ling, N.; Chen, C. Initial Structure–Activity Relationship Studies of a Novel Series of Pyrrolo[1,2-a]pyrimid-7-ones as GnRH Receptor Antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 399–402. [Google Scholar] [CrossRef]
- Zhu, Y.-F.; Wilcoxen, K.; Saunders, J.; Guo, Z.; Gao, Y.; Connors, J.P.J.; Gross, T.D.; Tucci, F.C.; Scott Struthers, R.; Reinhart, G.J.; et al. A Novel Synthesis of 2-Arylpyrrolo[1,2-a]pyrimid-7-ones and Their Structure–Activity Relationships as Potent GnRH Receptor Antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 403–406. [Google Scholar] [CrossRef]
- Tucci, F.C.; Zhu, Y.-F.; Guo, Z.; Gross, T.D.; Connors, P.J., Jr.; Struthers, R.S.; Reinhart, G.J.; Wang, X.; Saunders, J.; Chen, C. A novel synthesis of 7-aryl-8-fluoro-pyrrolo[1,2-a]pyrimid-4-ones as potent, stable GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 3491–3495. [Google Scholar] [CrossRef]
- Zhu, Y.-F.; Wilcoxen, K.M.; Struthers, R.S.; Chen, C.; Connors, P.J., Jr.; Gao, Y.; Tucci, F.C. Preparation of Imidazo- and Pyrrolo[1,2-a]pyrimid-4-ones as Gonadotropin-Releasing Hormone Receptor Antagonists. U.S. Patent 6346534B1, 12 February 2002. [Google Scholar]
- Sasaki, S.; Imaeda, T.; Hayase, Y.; Shimizu, Y.; Kasai, S.; Cho, N.; Harada, M.; Suzuki, N.; Furuya, S.; Fujino, M. A new class of potent nonpeptide luteinizing hormone-releasing hormone (LHRH) antagonists: Design and synthesis of 2-phenylimidazo[1,2-a]pyrimidin-5-ones. Bioorg. Med. Chem. Lett. 2002, 12, 2073–2077. [Google Scholar] [CrossRef]
- Furuya, S.; Imaeda, T.; Sasaki, S. Nitrogen-Containing Heterocyclic Compounds, including Imidazopyrimidines, and Their Production and Use as GnRH Antagonists. J.P. Patent WO9933831A1, July July 1999. [Google Scholar]
- Zhu, Y.-F.; Gross, T.D.; Gao, Y.; Connors, P.J., Jr.; Guo, Z.; Chen, C. Preparation of Imidazolo[1,2-a]pyrimid-4-one Derivatives and Related Compounds as Gonadotropin-Releasing Hormone Receptor Antagonists. U.S. Patent WO2001029044A1, 26 April 2001. [Google Scholar]
- Wilcoxen, K.M.; Zhu, Y.-F.; Connors, P.J., Jr.; Saunders, J.; Gross, T.D.; Gao, Y.; Reinhart, G.J.; Struthers, R.S.; Chen, C. Synthesis and initial structure–Activity relationships of a novel series of imidazolo[1,2-a]pyrimid-5-ones as potent GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 2179–2183. [Google Scholar] [PubMed]
- Gross, T.D.; Zhu, Y.-F.; Saunders, J.; Wilcoxen, K.M.; Gao, Y.; Connors, P.J., Jr.; Guo, Z.; Struthers, R.S.; Reinhart, G.J.; Chen, C. Design, synthesis and structure–Activity relationships of novel imidazolo[1,2-a]pyrimid-5-ones as potent GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 2185–2187. [Google Scholar] [PubMed]
- Zhu, Y.-F.; Guo, Z.; Gross, T.D.; Gao, Y.; Connors, P.J.; Struthers, R.S.; Xie, Q.; Tucci, F.C.; Reinhart, G.J.; Wu, D.; et al. Design and Structure−Activity Relationships of 2-Alkyl-3-aminomethyl-6-(3-methoxyphenyl)-7-methyl-8-(2-fluorobenzyl)imidazolo[1,2-a]pyrimid-5-ones as Potent GnRH Receptor Antagonists. J. Med. Chem. 2003, 46, 1769–1772. [Google Scholar] [PubMed]
- Zhu, Y.-F.; Gross, T.D.; Guo, Z.; Connors, P.J.; Gao, Y.; Tucci, F.C.; Struthers, R.S.; Reinhart, G.J.; Saunders, J.; Chen, T.K.; et al. Identification of 1-Arylmethyl-3- (2-aminoethyl)-5-aryluracil as Novel Gonadotropin-Releasing Hormone Receptor Antagonists. J. Med. Chem. 2003, 46, 2023–2026. [Google Scholar] [PubMed]
- Guo, Z.; Zhu, Y.-F.; Tucci, F.C.; Gao, Y.; Struthers, R.S.; Saunders, J.; Gross, T.D.; Xie, Q.; Reinhart, G.J.; Chen, C. Synthesis and Structure–Activity relationships of 1-arylmethyl-3-(2-aminopropyl)-5-aryl-6-methyluracils as potent GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 2003, 13, 3311–3315. [Google Scholar] [CrossRef]
- Guo, Z.; Zhu, Y.-F.; Gross, T.D.; Tucci, F.C.; Gao, Y.; Moorjani, M.; Connors, P.J.; Rowbottom, M.W.; Chen, Y.; Struthers, R.S.; et al. Synthesis and Structure−Activity Relationships of 1-Arylmethyl-5-aryl-6-methyluracils as Potent Gonadotropin-Releasing Hormone Receptor Antagonists. J. Med. Chem. 2004, 47, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Tucci, F.C.; Zhu, Y.-F.; Struthers, R.S.; Guo, Z.; Gross, T.D.; Rowbottom, M.W.; Acevedo, O.; Gao, Y.; Saunders, J.; Xie, Q.; et al. 3-[(2R)-Amino-2-phenylethyl]-1-(2,6-difluorobenzyl)-5-(2-fluoro-3-methoxyphenyl)- 6-methylpyrimidin-2,4-dione (NBI 42902) as a Potent and Orally Active Antagonist of the Human Gonadotropin-Releasing Hormone Receptor. Design, Synthesis, and in Vitro and in Vivo Characterization. J. Med. Chem. 2005, 48, 1169–1178. [Google Scholar] [PubMed]
- Struthers, R.S.; Xie, Q.; Sullivan, S.K.; Reinhart, G.J.; Kohout, T.A.; Zhu, Y.-F.; Chen, C.; Liu, X.-J.; Ling, N.; Yang, W.; et al. Pharmacological Characterization of a Novel Nonpeptide Antagonist of the Human Gonadotropin-Releasing Hormone Receptor, NBI-42902. Endocrinology 2007, 148, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Chen, Y.; Huang, C.Q.; Gross, T.D.; Pontillo, J.; Rowbottom, M.W.; Saunders, J.; Struthers, S.; Tucci, F.C.; Xie, Q.; et al. Uracils as potent antagonists of the human gonadotropin-releasing hormone receptor without atropisomers. Bioorg. Med. Chem. Lett. 2005, 15, 2519–2522. [Google Scholar] [CrossRef] [PubMed]
- Rowbottom, M.W.; Tucci, F.C.; Connors, P.J., Jr.; Gross, T.D.; Zhu, Y.-F.; Guo, Z.; Moorjani, M.; Acevedo, O.; Carter, L.; Sullivan, S.K.; et al. Synthesis and structure–activity relationships of uracil derived human GnRH receptor antagonists: (R)-3-[2-(2-Amino)phenethyl]-1-(2,6-difluorobenzyl)-6-methyluracils containing a substituted thiophene or thiazole at C-5. Bioorg. Med. Chem. Lett. 2004, 14, 4967–4973. [Google Scholar] [CrossRef]
- Tucci, F.C.; Hu, T.; Mesleh, M.F.; Bokser, A.; Allsopp, E.; Gross, T.D.; Guo, Z.; Zhu, Y.-F.; Struthers, R.S.; Ling, N.; et al. Atropisomeric property of 1-(2,6-difluorobenzyl)-3-[(2R)-amino-2-phenethyl]-5-(2-fluoro-3-methoxyphenyl)-6-methyluracil. Chirality 2005, 17, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, Y.; Pontillo, J.; Guo, Z.; Huang, C.Q.; Wu, D.; Madan, A.; Chen, T.; Wen, J.; Xie, Q.; et al. Potent and orally bioavailable zwitterion GnRH antagonists with low CYP3A4 inhibitory activity. Bioorg. Med. Chem. Lett. 2008, 18, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, D.; Guo, Z.; Xie, Q.; Reinhart, G.J.; Madan, A.; Wen, J.; Chen, T.; Huang, C.Q.; Chen, M.; et al. Discovery of Sodium R-(+)-4-{2-[5-(2-Fluoro-3-methoxyphenyl)-3-(2-fluoro-6-[trifluoromethyl]benzyl)-4-methyl-2,6-dioxo-3,6-dihydro-2H-pyrimidin-1-yl]-1-phenylethylamino}butyrate (Elagolix), a Potent and Orally Available Nonpeptide Antagonist of the Human Gonadotropin-Releasing Hormone Receptor. J. Med. Chem. 2008, 51, 7478–7485. [Google Scholar] [PubMed]
- Zhu, Y.-F.; Chen, C.; Tucci, F.C.; Guo, Z.; Gross, T.D.; Rowbottom, M.; Struthers, R.S. Preparation of Uracil Derivatives as Gonadotropin-Releasing Hormone Receptor Antagonists. U.S. Patent WO2001055119A2, 2 October 2001. [Google Scholar]
- Guo, Z.; Chen, Y.; Wu, D.; Chen, C.; Wade, W.; Dwight, W.J.; Huang, C.Q.; Tucci, F.C. Preparation of Pyrimidine-2,4(1H,3H)-dione Derivatives as Gonadotropin-Releasing Hormone Receptor Antagonists. U.S. Patent WO2005007165A1, 27 January 2005. [Google Scholar]
- Chwalisz, K.; Williams, L.; Jain, R.; North, J.; Ng, W.-K.J. Compositions for Use in Treating Heavy Menstrual Bleeding and Uterine Fibroids Comprising Elagolix with or without Ad-back Therapy. U.S. Patent WO2014143669A1, 18 September 2014. [Google Scholar]
- Gallagher, D.J.; Treiber, L.R.; Hughes, R.M.; Campopiano, O.; Wang, P.; Zhao, Y.; Chou, S.K.; Ouellette, M.A.; Hettinger, D.N. Processes for the Preparation of Uracil Derivatives. U.S. Patent 8765948B2, 1 July 2014. [Google Scholar]
- Kim, S.-M.; Lee, M.; Lee, S.Y.; Park, E.; Lee, S.-M.; Kim, E.J.; Han, M.Y.; Yoo, T.; Ann, J.; Yoon, S.; et al. Discovery of an Orally Bioavailable Gonadotropin-Releasing Hormone Receptor Antagonist. J. Med. Chem. 2016, 59, 9150–9172. [Google Scholar] [CrossRef] [PubMed]
- Goulet, M.; Chu, L.; Ashton, W.T.; Fisher, M.H.; Wyvratt, M.J.; Smith, R.G.; Bugianesi, R.L.; Ponpipom, M.M.; Yang, Y.T.; Lin, P. Preparation of N-aralkyl-2-(substituted-aryl)indole-3-alkanamines and Analogs as Gonadotropin Releasing Hormone Antagonists. U.S. Patent 5756507A, 26 May 1998. [Google Scholar]
- Goulet, M.; Chu, L.; Walsh, T.F.; Fisher, M.H.; Girotra, N.N.; Wyvratt, M.J.; Lin, P.; Ashton, W.T. Preparation of Indole Derivatives as Gonadotropin Releasing Hormone Antagonists. U.S. Patent 5849764A, 15 December 1998. [Google Scholar]
- Goulet, M.; Ashton, W.T.; Chu, L.; Fisher, M.H.; Lin, P.; Ponpipom, M.M.; Wyvratt, M.J.; Girotra, N.N.; Young, J. Preparation of Heterocyclic Compounds as Antagonists of Gonadotropin Releasing Hormone. WO2000004013A1, 27 January 2000. [Google Scholar]
- Goulet, M.; Ashton, W.T.; Chu, L.; Fisher, M.H.; Lin, P.; Ponpipom, M.M.; Wyvratt, M.J.; Girotra, N.N.; Young, J. Preparation of Indoles as Antagonists of Gonadotropin Releasing Hormone. U.S. Patent 6200957B1, 13 March 2001. [Google Scholar]
- Chu, L.; Hutchins, J.E.; Weber, A.E.; Lo, J.-L.; Yang, Y.-T.; Cheng, K.; Smith, R.G.; Fisher, M.H.; Wyvratt, M.J.; Goulet, M.T. Initial structure–activity relationship of a novel class of nonpeptidyl GnRH receptor antagonists: 2-arylindoles. Bioorg. Med. Chem. Lett. 2001, 11, 509–513. [Google Scholar] [CrossRef]
- Lin, P.; Marino, D.; Lo, J.-L.; Yang, Y.T.; Cheng, K.; Smith, R.G.; Fisher, M.H.; Wyvratt, M.J.; Goulet, M.T. 2-(3,5-Dimethylphenyl)tryptamine Derivatives that Bind to the GnRH Receptor. Bioorg. Med. Chem. Lett. 2001, 11, 1073–1076. [Google Scholar] [CrossRef]
- Ashton, W.T.; Sisco, R.M.; Yang, Y.T.; Lo, J.-L.; Yudkovitz, J.B.; Cheng, K.; Goulet, M.T. Substituted Indole-5-carboxamides and -acetamides as Potent Nonpeptide GnRH Receptor Antagonists. Bioorg. Med. Chem. Lett. 2001, 11, 1723–1726. [Google Scholar] [CrossRef]
- Ashton, W.T.; Sisco, R.M.; Kieczykowski, G.R.; Yang, Y.T.; Yudkovitz, J.B.; Cui, J.; Mount, G.R.; Ren, R.N.; Wu, T.-J.; Shen, X.; et al. Orally bioavailable, indole-based nonpeptide GnRH receptor antagonists with high potency and functional activity. Bioorg. Med. Chem. Lett. 2001, 11, 2597–2602. [Google Scholar] [CrossRef]
- Simeone, J.P.; Bugianesi, R.L.; Ponpipom, M.M.; Yang, Y.T.; Lo, J.-L.; Yudkovitz, J.B.; Cui, J.; Mount, G.R.; Ren, R.N.; Creighton, M.; et al. Modification of the pyridine moiety of non-peptidyl indole GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 3329–3332. [Google Scholar] [CrossRef]
- Goulet, M.; Allen, E.E.; Jiang, J.; Smith, R.G.; Walsh, T.F.; Wyvratt, M.J., Jr.; Yang, Y.T.; Young, J.R.; Devita, R.J. Preparation of 1H-quinolin-2-one Derivatives as Antagonists of Gonadotropin Releasing Hormone. WO9744321A1, 1997. [Google Scholar]
- Goulet, M.; Allen, E.E.; Devita, R.J.; Jiang, J.; Walsh, T.F.; Young, J.R.; Wyvratt, M.J., Jr.; Toupence, R.B.; Ujjainwalla, F. Preparation of 4-aminoalkoxy-2-quinolones and Analogs as Gonadotropin Releasing Hormone Antagonists. WO9744339A1, 1997. [Google Scholar]
- Devita, R.J.; Goulet, M.T.; Parikh, M. Preparation of Quinolonecarboxylic Acid Heterocyclylamides as Antagonists of Gonadotropin Releasing Hormone. U.S. Patent WO2001070228A1, 27 September 2001. [Google Scholar]
- DeVita, R.J.; Hollings, D.D.; Goulet, M.T.; Wyvratt, M.J.; Fisher, M.H.; Lo, J.-L.; Yang, Y.T.; Cheng, K.; Smith, R.G. Identification and initial structure-activity relationships of a novel non-peptide quinolone GnRH receptor antagonist. Bioorg. Med. Chem. Lett. 1999, 9, 2615–2620. [Google Scholar] [CrossRef]
- DeVita, R.J.; Goulet, M.T.; Wyvratt, M.J.; Fisher, M.H.; Lo, J.-L.; Yang, Y.T.; Cheng, K.; Smith, R.G. Investigation of the 4-O-alkylamine substituent of non-peptide quinolone GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 1999, 9, 2621–2624. [Google Scholar] [CrossRef]
- Walsh, T.F.; Toupence, R.B.; Young, J.R.; Huang, S.X.; Ujjainwalla, F.; DeVita, R.J.; Goulet, M.T.; Wyvratt, M.J., Jr.; Fisher, M.H.; Lo, J.-L.; et al. Potent antagonists of gonadotropin releasing hormone receptors derived from quinolone-6-carboxamides. Bioorg. Med. Chem. Lett. 2000, 10, 443–447. [Google Scholar] [CrossRef]
- Young, J.R.; Huang, S.X.; Chen, I.; Walsh, T.F.; DeVita, R.J.; Wyvratt, M.J., Jr.; Goulet, M.T.; Ren, N.; Lo, J.; Yang, Y.T.; et al. Quinolones as gonadotropin releasing hormone (GnRH) antagonists: Simultaneous optimization of the C(3)-aryl and C(6)-substituents. Bioorg. Med. Chem. Lett. 2000, 10, 1723–1727. [Google Scholar] [CrossRef]
- DeVita, R.J.; Walsh, T.F.; Young, J.R.; Jiang, J.; Ujjainwalla, F.; Toupence, R.B.; Parikh, M.; Huang, S.X.; Fair, J.A.; Goulet, M.T.; et al. A Potent, Nonpeptidyl 1H-Quinolone Antagonist for the Gonadotropin-Releasing Hormone Receptor. J. Med. Chem. 2001, 44, 917–922. [Google Scholar] [CrossRef] [PubMed]
- DeVita, R.J.; Parikh, M.; Jiang, J.; Fair, J.A.; Young, J.R.; Walsh, T.F.; Goulet, M.T.; Lo, J.-L.; Ren, N.; Yudkovitz, J.B.; et al. Identification of neutral 4-O-alkyl quinolone nonpeptide GnRH receptor antagonists. Bioorg. Med. Chem. Lett. 2004, 14, 5599–5603. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.B.; Vazir, H.N.; Luthin, D.R.; Paderes, G.D.; Pathak, V.P.; Christie, L.C.; Hong, Y.; Tompkins, E.V.; Li, H.; Faust, J. Non-Peptide GnRH Agents, Methods and Intermediates for Their Preparation. U.S. Patent WO2000020358A2, 13 April 2000. [Google Scholar]
- Anderson, M.B.; Christie, L.C.; Feng, J.; Hong, Y.; Li, H.; Pathak, V.P.; Rajapakse, R.; Tompkins, E.; Vazir, H.N. Preparation of Non-Peptide Compounds Affecting the Action of Gonadotropin-Releasing Hormone (GNRH). U.S. Patent EP1334972A1, 13 August 2003. [Google Scholar]
- Luthin, D.R.; Hong, Y.; Pathak, V.P.; Paderes, G.; Nared-Hood, K.D.; Castro, M.A.; Vazir, H.; Li, H.; Tompkins, E.; Christie, L.; et al. The discovery of novel small molecule non-peptide gonadotropin releasing hormone (GnRH) receptor antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 3467–3470. [Google Scholar] [CrossRef]
- Luthin, D.R.; Hong, Y.; Tompkins, E.; Anderes, K.L.; Paderes, G.; Kraynov, E.A.; Castro, M.A.; Nared-Hood, K.D.; Castillo, R.; Gregory, M.; et al. Characterization of mono- and diaminopyrimidine derivatives as novel, nonpeptide gonadotropin releasing hormone (GnRH) receptor antagonists. Bioorg. Med. Chem. Lett. 2002, 12, 3635–3639. [Google Scholar] [CrossRef]
- Anderes, K.L.; Luthin, D.R.; Castillo, R.; Kraynov, E.A.; Castro, M.; Nared-Hood, K.; Gregory, M.L.; Pathak, V.P.; Christie, L.C.; Paderes, G.; et al. Biological Characterization of a Novel, Orally Active Small Molecule Gonadotropin-Releasing Hormone (GnRH) Antagonist Using Castrated and Intact Rats. J. Pharmacol. Exp. Ther. 2003, 305, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Anderes, K.L.; Kraynov, E.A.; Luthin, D.R.; Do, Q.-Q.; Hong, Y.; Tompkins, E.; Sun, E.T.; Rajapakse, R.; Pathak, V.P.; et al. Discovery of a Novel, Orally Active, Small Molecule Gonadotropin-Releasing Hormone (GnRH) Receptor Antagonist. J. Med. Chem. 2006, 49, 3362–3367. [Google Scholar] [PubMed]
- Olberg, D.E.; Andressen, K.W.; Levy, F.O.; Klaveness, J.; Haraldsen, I.; Sutcliffe, J.L. Synthesis and in vitro evaluation of small-molecule [18F] labeled gonadotropin-releasing hormone (GnRH) receptor antagonists as potential PET imaging agents for GnRH receptor expression. Bioorg. Med. Chem. Lett. 2014, 24, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Olberg, D.E.; Bauer, N.; Andressen, K.W.; Hjørnevik, T.; Cumming, P.; Levy, F.O.; Klaveness, J.; Haraldsen, I.; Sutcliffe, J.L. Brain penetrant small molecule 18F-GnRH receptor (GnRH-R) antagonists: Synthesis and preliminary positron emission tomography imaging in rats. Nucl. Med. Biol. 2016, 43, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Tatsuta, M.; Kataoka, M.; Yasoshima, K.; Shogase, Y.; Shimazaki, M.; Yura, T.; Li, Y.; Yamamoto, N.; Gupta, J.B.; et al. Benzimidazole derivatives as novel nonpeptide luteinizing hormone-releasing hormone (LHRH) antagonists. Part 1: Benzimidazole-5-sulfonamides. Bioorg. Med. Chem. Lett. 2005, 15, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Kataoka, M.; Tatsuta, M.; Yasoshima, K.; Yamamoto, M.; Yura, T.; Yamamoto, N.; Urbahns, K.; Gupta, J.B.; Li, Y. Benzimidazole-5-sulfonamides as Novel Nonpeptide Luteinizing Hormone Releasing Hormone (LHRH) Antagonists: Minimization of Mechanism-Based CYP3A4 Inhibition. Chem. Pharm. Bull. 2005, 53, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, M.; Kataoka, M.; Yasoshima, K.; Sakakibara, S.; Shogase, Y.; Shimazaki, M.; Yura, T.; Li, Y.; Yamamoto, N.; Gupta, J.; et al. Benzimidazoles as non-peptide luteinizing hormone-releasing hormone (LHRH) antagonists. Part 3: Discovery of 1-(1H-benzimidazol-5-yl)-3-tert-butylurea derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 2265–2269. [Google Scholar] [CrossRef] [PubMed]
- Garrick, L.M.; Hauze, D.B.; Kees, K.L.; Lundquist, J.T., IV; Mann, C.W.; Mehlmann, J.F.; Pelletier, J.C.; Rogers, J.F., Jr.; Wrobel, J.E. Preparation of Benzimidazole Derivatives as Gonadotropin Releasing Hormone Receptor Antagonists. U.S. Patent WO2006009734A1, 26 January 2006. [Google Scholar]
- Gontcharov, A.V.; Khafizova, G.; Potoski, J.R.; Huryn, D.M. Processes and Intermediates for Preparation of 4-(piperazin-1-yl)-1H-benzimidazole Derivatives Useful as Gonadotropin Releasing Hormone Receptor Antagonists. U.S. Patent 20050282820A1, 22 December 2005. [Google Scholar]
- Pelletier, J.C.; Chengalvala, M.; Cottom, J.; Feingold, I.; Garrick, L.; Green, D.; Hauze, D.; Huselton, C.; Jetter, J.; Kao, W.; et al. 2-Phenyl-4-piperazinylbenzimidazoles: Orally active inhibitors of the gonadotropin releasing hormone (GnRH) receptor. Bioorg. Med. Chem. 2008, 16, 6617–6640. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.C.; Chengalvala, M.V.; Cottom, J.E.; Feingold, I.B.; Green, D.M.; Hauze, D.B.; Huselton, C.A.; Jetter, J.W.; Kopf, G.S.; Lundquist, J.T.; et al. Discovery of 6-({4-[2-(4-tert-Butylphenyl)-1H-Benzimidazol-4-yl]piperazin-1-yl}methyl)quinoxaline (WAY-207024): An Orally Active Antagonist of the Gonadotropin Releasing Hormone Receptor (GnRH-R). J. Med. Chem. 2009, 52, 2148–2152. [Google Scholar] [CrossRef] [PubMed]
- Panknin, O.; Baeurle, S.; Ring, S.; Schwede, W.; Bone, W.; Nowak-Reppel, K.; Bender, E.; Nubbemeyer, R.; Gnoth, M.J. Preparation of Spiroindoline Compounds as Gonadotropin- Releasing Hormone Receptor Antagonists Useful in the Treatment of Sex Hormone-Related Diseases. D.E. Patent WO2013107743A1, 25 July 2013. [Google Scholar]
- Panknin, O.; Ring, S.; Baeurle, S.; Wagenfeld, A.; Nubbemeyer, R.; Nowak-Reppel, K.; Langer, G. Preparation of 2-Cycloalkyl-1-phenylsulfonyl-hexahydrospiro[indolethiopyran]-5-carboxamide Derivatives as Gonadotropin-Releasing Hormone Receptor Antagonists. D.E. Patent WO2014166958A1, 16 October 2014. [Google Scholar]
- Panknin, O.; Ring, S.; Nowak-Reppel, K.; Langer, G. Preparation of Spiroindoline Derivatives as GnRH Receptor Antagonist. D.E. Patent WO2015007606A1, 22 January 2015. [Google Scholar]
- Panknin, O.; Ring, S.; Nowak-Reppel, K.; Langer, G. Preparation of Spiroindoline-thiopyran-imine-oxide Derivatives as Gonadotropin-Releasing Hormone Receptor Antagonists. D.E. Patent WO2015082374A1, 11 June 2015. [Google Scholar]
- Panknin, O.; Baeurle, S.; Ring, S.; Schwede, W.; Schmees, N.; Nowak-Reppel, K.; Langer, G. Spiro[indolin-3,4′-piperidine] Derivatives as GnRH Receptor Antagonists and Their Preparation, Pharmaceutical Compositions and Use in the Treatment of Sex-hormone-related Diseases. D.E. Patent WO2015091315A1, 25 June 2015. [Google Scholar]
- Hirano, M.; Kawaminami, E.; Kinoyama, I.; Matsumoto, S.; Ohnuki, K.; Obitsu, K.; Kusayama, T. Preparation of Benzimidazole Derivatives as GnRH Receptor Antagonists. J.P. Patent WO2005118556A1, 15 December 2005. [Google Scholar]
- Kim, S.-M.; Kim, J.-S.; Lee, M.; Lee, S.-Y.; Lee, B.-Y.; Shin, Y.A.; Park, E.; Lee, J.A.; Han, M.-Y.; Ahn, J.; et al. Spiro[furopyrimidine-piperidine] and Spiro[cyclopentapyrimidine-piperidine] Derivatives as Gonadotropin Releasing Hormone Receptor Antagonists, Method for Their Preparation and Pharmaceutical Composition Comprising the Same. K.R. Patent WO2013129879A1, 6 September 2013. [Google Scholar]
- Fjellaksel, R.; Boomgaren, M.; Sundset, R.; Haraldsen, I.H.; Hansen, J.H.; Riss, P.J. Small molecule piperazinyl-benzimidazole antagonists of the gonadotropin-releasing hormone (GnRH) receptor. MedChemComm 2017, 8, 1965–1969. [Google Scholar] [CrossRef]
- Struthers, R.S.; Chen, T.; Campbell, B.; Jimenez, R.; Pan, H.; Yen, S.S.C.; Bozigian, H.P. Suppression of Serum Luteinizing Hormone in Postmenopausal Women by an Orally Administered Nonpeptide Antagonist of the Gonadotropin-Releasing Hormone Receptor (NBI-42902). J. Clin. Endocrinol. Metab. 2006, 91, 3903–3907. [Google Scholar] [CrossRef] [PubMed]
- Alessandro, P.; Luigi, N.; Felice, S.; Maria, P.A.; Benedetto, M.G.; Stefano, A. Research development of a new GnRH antagonist (Elagolix) for the treatment of endometriosis: A review of the literature. Arch. Gynecol. Obstet. 2017, 295, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Melis, G.B.; Neri, M.; Corda, V.; Malune, M.E.; Piras, B.; Pirarba, S.; Guerriero, S.; Orru, M.; D’Alterio, M.N.; Angioni, S.; et al. Overview of elagolix for the treatment of endometriosis. Expert Opin. Drug Metab. Toxicol. 2016, 12, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Chwalisz, K.; Carter, D.C.; Klein, C.E. Dose-Dependent Suppression of Gonadotropins and Ovarian Hormones by Elagolix in Healthy Premenopausal Women. J. Clin. Endocrinol. Metab. 2017, 102, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Yoo, T.; Lee, S.Y.; Kim, E.J.; Lee, S.M.; Lee, M.H.; Han, M.Y.; Jung, S.-H.; Choi, J.-H.; Ryu, K.H.; et al. Effect of SKI2670, a novel, orally active, non-peptide GnRH antagonist, on hypothalamic–pituitary–gonadal axis. Life Sci. 2015, 139, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Clark, E.; Johnston, A.; George, M.; Davies, J.; Hibberd, M. Effects of single and repeated oral doses of TAK-013, a new non-peptide gonadotropin-releasing hormone (GnRH) antagonist, in healthy post-menopausal women. Fertil. Steril. 2002, 78, S281–S282. [Google Scholar] [CrossRef]
- MacLean, D.B.; Shi, H.; Faessel, H.M.; Saad, F. Medical Castration Using the Investigational Oral GnRH Antagonist TAK-385 (Relugolix): Phase 1 Study in Healthy Males. J. Clin. Endocrinol. Metab. 2015, 100, 4579–4587. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.P.; Carr, B.; Dmowski, W.P.; Koltun, W.; O’Brien, C.; Jiang, P.; Burke, J.; Jimenez, R.; Garner, E.; Chwalisz, K. Elagolix treatment for endometriosis-associated pain: Results from a phase 2, randomized, double-blind, placebo-controlled study. Reprod. Sci. 2014, 21, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Carr, B.; Chwalisz, K.; Jimenez, R.; Burke, J.; Jiang, P.; O’Brien, C. A novel oral GnRH antagonist, elagolix, is effective for reducing endometriosis-associated pelvic pain: Results of a 24-week randomized study. Fertil. Steril. 2011, 96, S45. [Google Scholar] [CrossRef]
- Imani, R.; Thai-Cuarto, D.; Jimenez, R.; Burke, J.; Kroll, R.; O’Brien, C. Petal study: Safety, tolerability and effectiveness of elagolix, an oral GnRH antagonist for endometriosis. Fertil. Steril. 2009, 92, S111–S112. [Google Scholar] [CrossRef]
- Struthers, R.S.; Nicholls, A.J.; Grundy, J.; Chen, T.; Jimenez, R.; Yen, S.S.C.; Bozigian, H.P. Suppression of gonadotropins and estradiol in premenopausal women by oral administration of the nonpeptide gonadotropin-releasing hormone antagonist elagolix. J. Clin. Endocrinol. Metab. 2009, 94, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=ASP-1707&cntry1=&state1=&recrs= (accessed on 27 September 2017).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=relugolix&cntry1=&state1=&Search=Search#tableTop (accessed on 27 September 2017).
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=Elagolix&cntry1=&state1=&Search=Search (accessed on 27 September 2017).
- Kass, A.; Hollan, I.; Fagerland, M.W.; Gulseth, H.C.; Torjesen, P.A.; Førre, O.T. Rapid Anti-Inflammatory Effects of Gonadotropin-Releasing Hormone Antagonism in Rheumatoid Arthritis Patients with High Gonadotropin Levels in the AGRA Trial. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Clinicaltrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=ASP1707&cntry1=&state1=&Search=Search (accessed on 27 September 2017).
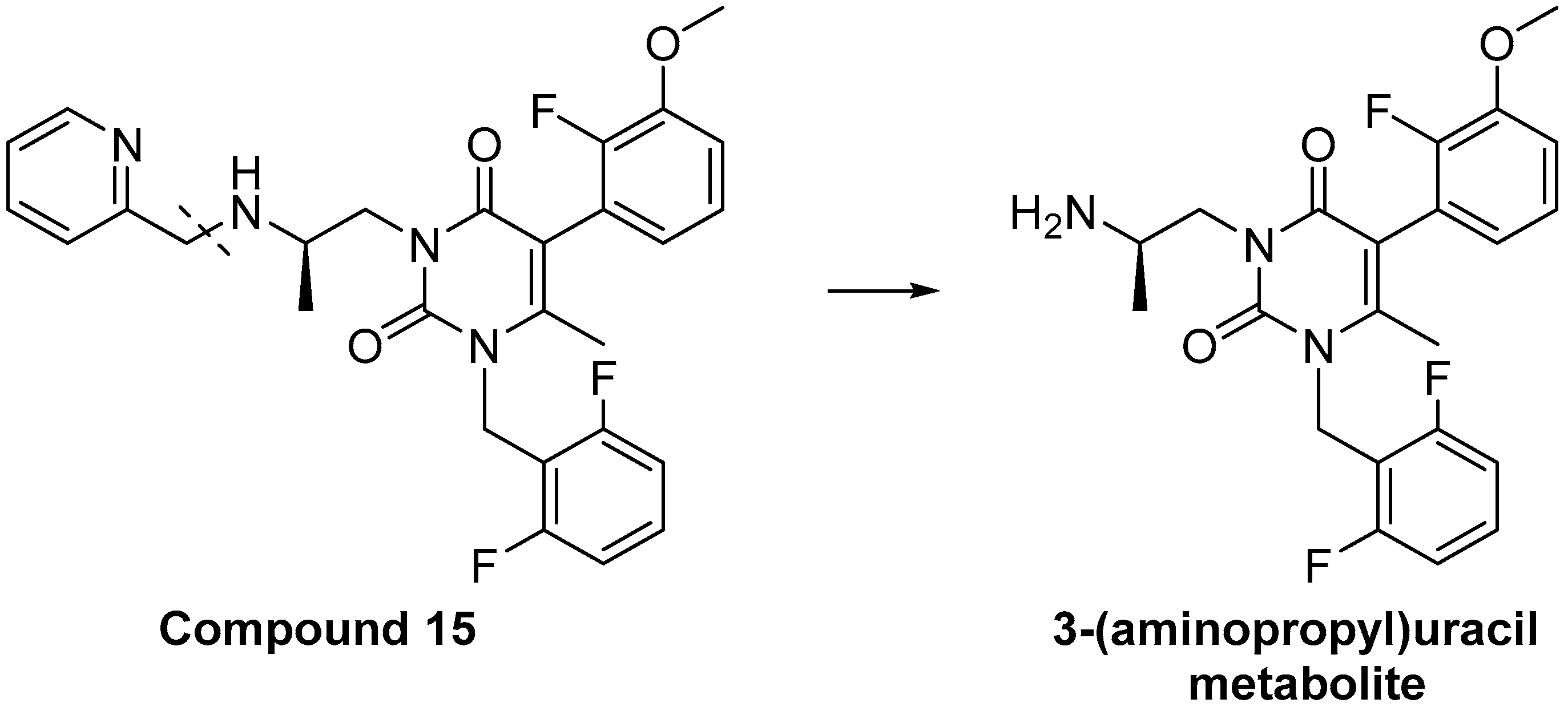

{kind=link}
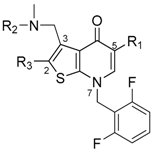
| Compound | R1 | R2 | R3 | IC50 (nM) |
|---|---|---|---|---|
| 1 |  | 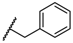 |  | 0.2 |
| 2 | 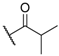 |  | 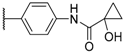 | 0.1 |
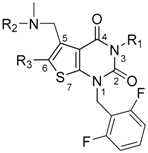
| Compound | R1 | R2 | R3 | IC50 (nM) |
|---|---|---|---|---|
| 3 TAK-013 Sufugolix | 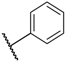 | 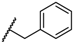 | 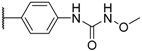 | 0.1 |
| 4 TAK-385 Relugolix |  | Me | 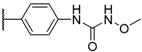 | 0.33 |
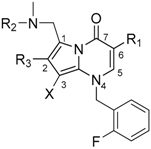
| Compound | R1 | R2 | R3 | X | Ki (nM) |
|---|---|---|---|---|---|
| 5 | 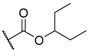 | 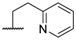 | 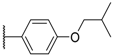 | CN | 25 |
| 6 |  | 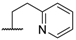 | 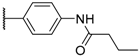 | H | 1.2 |
| 7 | 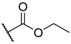 | 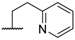 |  | F | 9 |

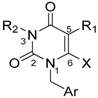
| Compound | R1 | R2 | R3 | Ar | X | Ki (nM) |
|---|---|---|---|---|---|---|
| 8 | 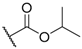 | 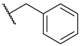 | 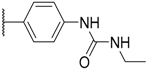 | 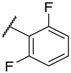 | H | 0.3 1 |
| 9 | 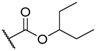 | 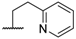 | 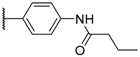 | 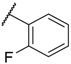 | H | 7.5 |
| 10 | 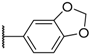 | 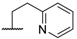 | 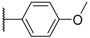 | 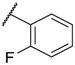 | Me | 11 |
| 11 | 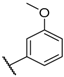 | 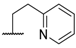 | 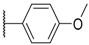 | 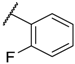 | Me | 4.6 |
| 12 | 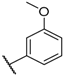 | 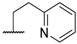 |  | 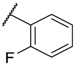 | Me | 5.2 ± 0.6 |
| Compound | R1 | R2 | Ar | X | Ki (nM) |
|---|---|---|---|---|---|
| 13 |  | 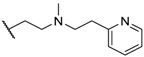 | 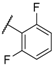 | Me | 34 |
| 14 | 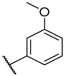 | 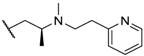 | 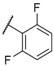 | Me | 5.2 |
| 15 | 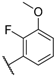 |  | 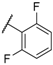 | Me | 1.1 |
| 16 | 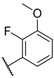 |  | 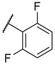 | Me | 8.1 |
| 17 NBI 42902 | 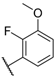 | 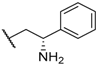 | 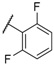 | Me | 0.56 |
| 18 | 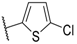 | 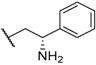 | 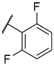 | Me | 2 |
| 19 | 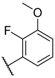 | 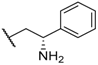 | 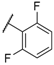 | H | 5.3 |
| 20 | 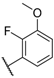 | 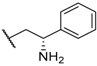 | 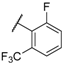 | H | 0.64 |
| 21 | 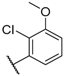 | 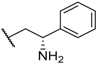 | 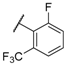 | H | 0.45 |
| 22 | 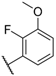 | 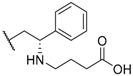 |  | H | 1.2 |
| 23 Elagolix | 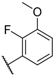 | 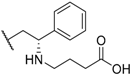 | 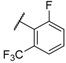 | Me | 0.9 |

| Compound | R1 | R2 | Ar | IC50 (nM) |
|---|---|---|---|---|
| 24 | 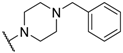 | 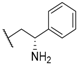 |  | 12 |
| 25 | 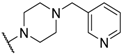 | 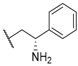 | 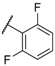 | 13.9 |
| 26 |  | 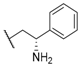 | 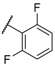 | 4.1 |
| 27 | 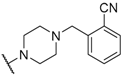 | 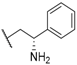 |  | 6.8 |
| 28 | 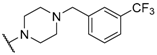 | 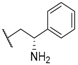 | 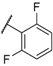 | 0.91 |
| 29 | 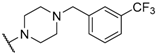 | 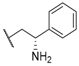 | 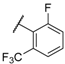 | 0.52 |
| 30 | 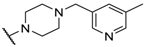 | 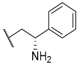 | 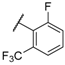 | 7.18 |
| 31 | 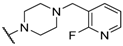 | 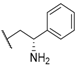 | 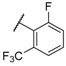 | 1.07 |
| 32 | 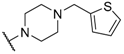 | 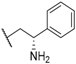 | 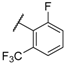 | 1.27 |
| 33 |  | 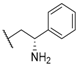 | 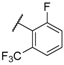 | 2.76 |
| 34 | 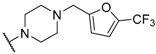 | 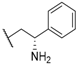 | 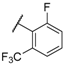 | 0.95 |
| 35 | 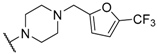 | 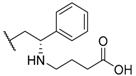 | 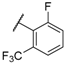 | 0.44 |
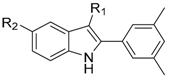
| Compound | R1 | R2 | IC50 (nM) |
|---|---|---|---|
| 36 | 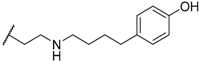 | H | 50 |
| 37 | 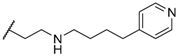 | H | 16 |
| 38 | 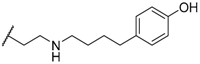 | 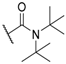 | 5.7 |
| 39 | 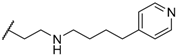 | 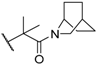 | 1.4 |
| 40 | 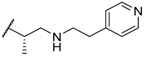 | 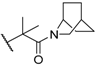 | 0.6 |
| 41 | 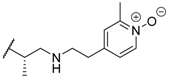 | 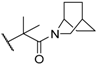 | 0.3 |

| Compound | R1 | R2 | Ar | IC50 (nM) |
|---|---|---|---|---|
| 42 | 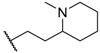 | NO2 | 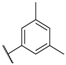 | 32 |
| 43 |  | NO2 | 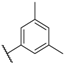 | 10 |
| 44 | 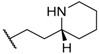 |  | 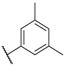 | 0.9 |
| 45 | 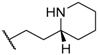 |  |  | 0.3 |
| 46 | 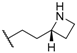 | 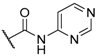 | 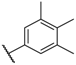 | 0.44 |
| 47 | 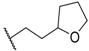 | 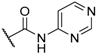 | 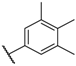 | 0.6 |
| 48 | 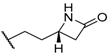 | 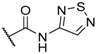 | 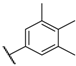 | 0.1 |
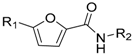
| Compound | R1 | R2 | Ki (nM) |
|---|---|---|---|
| 49 |  | 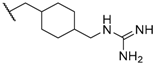 | 40 |
| 50 | | 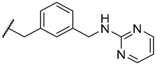 | 130 ± 10 |
| 51 | | 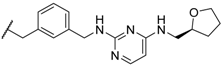 | 8 ± 0.8 |
| 52 CMPD1 | | 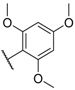 | 6.0 ± 0.8 |
| 53 |  | 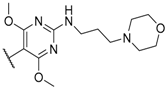 | 0.4 |
| 54 | 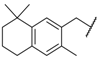 | 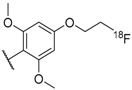 | 0.3 |
| 55 | 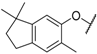 | 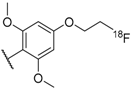 | 1.3 |
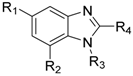
| Compound | R1 | R2 | R3 | R4 | IC50 (nM) |
|---|---|---|---|---|---|
| 56 | 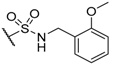 | H |  |  | 3400 |
| 57 | 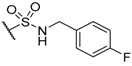 | H | 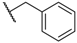 | 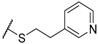 | 120 |
| 58 | 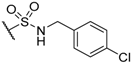 | H |  | 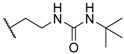 | 4.2 |
| 59 |  | H | 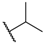 |  | 6500 |
| 60 | 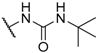 | H | 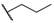 | 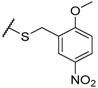 | 18 |
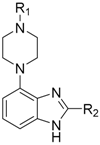
| Compound | R1 | R2 | IC50 (nM) |
|---|---|---|---|
| 61 | 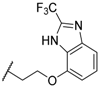 | 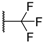 | 1540 |
| 62 |  | 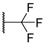 | 800 |
| 63 | 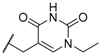 | 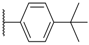 | 1.7 ± 0.65 |
| 64 WAY-207024 | 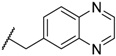 | 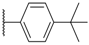 | 12 ± 4.2 |
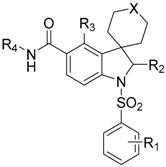
| Compound | R1 | R2 | R3 | R4 | X | IC50 (nM) |
|---|---|---|---|---|---|---|
| 65 | 4-F | 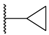 | H | 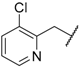 | SO2 | 104 |
| 66 | 4-F | 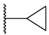 | H |  | SO2 | 21 |
| 67 | 3-CN |  | H |  | SO2 | 14 |
| 68 | 4-CN | 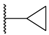 | H | 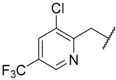 | SO2 | 10 |
| 69 | 4-F | 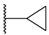 | Cl | 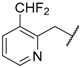 | SO2 | 3.5 |
| 70 | 4-F | 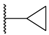 | H | 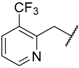 |  | 1.7 |
| 71 | 4-F | 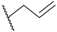 | H | 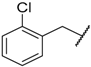 |  | 11.1 |
| Compound | Structure |
|---|---|
| 72 1 |  |
| 73 1 | 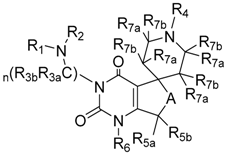 |
| 74 | 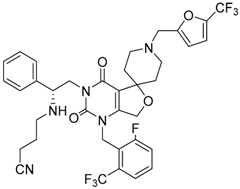 |
| 75 | 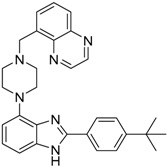 |
| GnRH Antagonists | Clinical Trial | Indications Evaluated in Clinical Trials | Industry | Published Human Data |
|---|---|---|---|---|
| TAK-013 (Sufugolix) Compound 3 | Phase II 1 | Endometriosis, uterine fibroids | Takeda | [102] |
| TAK-385 (Relugolix) Compound 4 | Phase III | Prostate Cancer | Takeda | - |
| Phase II | Endometriosis, uterine fibroids, prostate cancer | Takeda | [103] | |
| NBI-42902 Compound 17 | Phase II 1 | Hormone dependent diseases | Neurocrine Biosciences | [97] |
| Elagolix Compound 23 | Phase III | Endometriosis, uterine fibroids | Neurocrine Biosciences | [104,105,106,107] |
| ASP-1707 | Phase II | Endometriosis | Astellas Pharma | [108] |
| SKI2670 2 | Phase I | Endometriosis | SK Chemical | [101] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tukun, F.-L.; Olberg, D.E.; Riss, P.J.; Haraldsen, I.; Kaass, A.; Klaveness, J. Recent Development of Non-Peptide GnRH Antagonists. Molecules 2017, 22, 2188. https://doi.org/10.3390/molecules22122188
Tukun F-L, Olberg DE, Riss PJ, Haraldsen I, Kaass A, Klaveness J. Recent Development of Non-Peptide GnRH Antagonists. Molecules. 2017; 22(12):2188. https://doi.org/10.3390/molecules22122188
Chicago/Turabian StyleTukun, Feng-Ling, Dag Erlend Olberg, Patrick J. Riss, Ira Haraldsen, Anita Kaass, and Jo Klaveness. 2017. "Recent Development of Non-Peptide GnRH Antagonists" Molecules 22, no. 12: 2188. https://doi.org/10.3390/molecules22122188
APA StyleTukun, F.-L., Olberg, D. E., Riss, P. J., Haraldsen, I., Kaass, A., & Klaveness, J. (2017). Recent Development of Non-Peptide GnRH Antagonists. Molecules, 22(12), 2188. https://doi.org/10.3390/molecules22122188