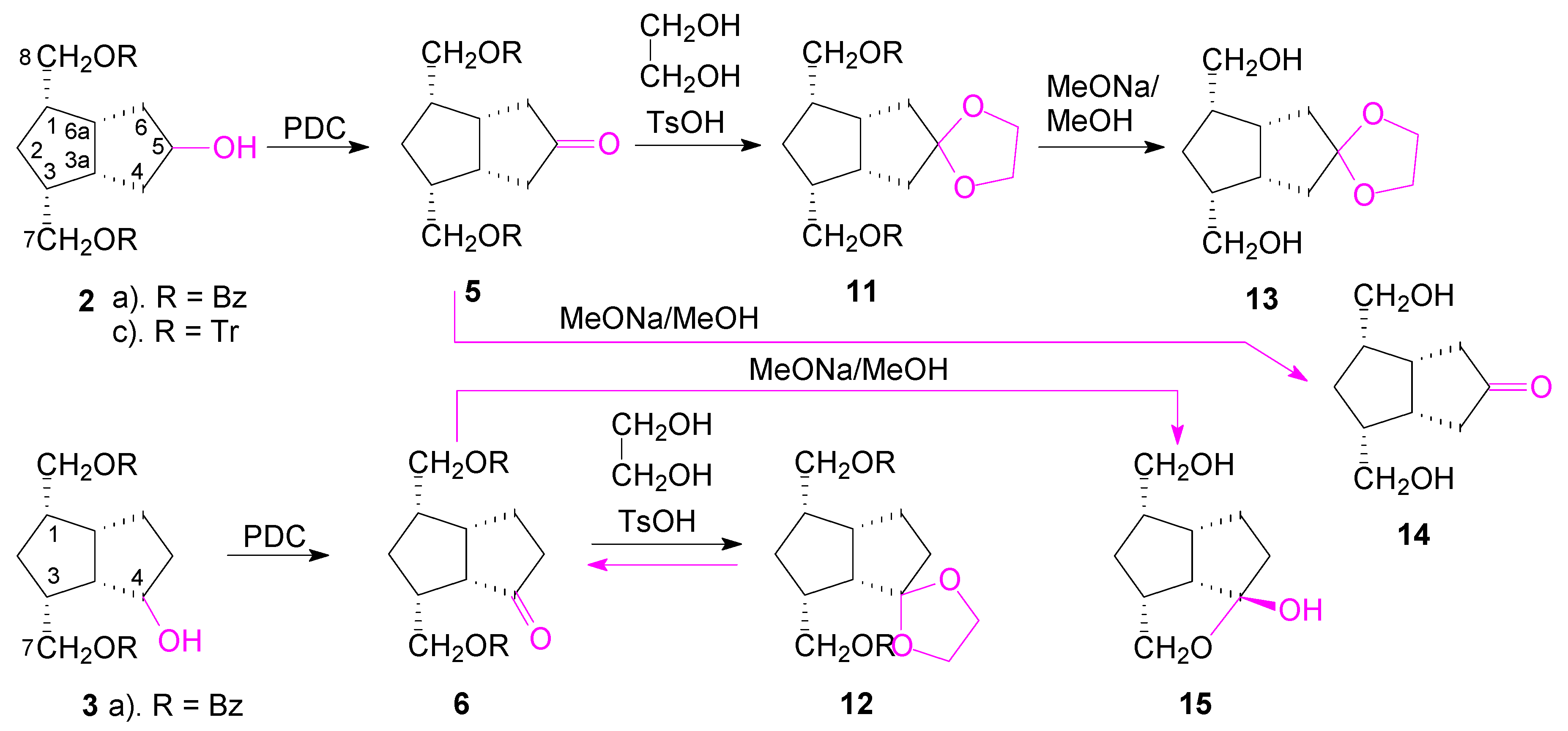
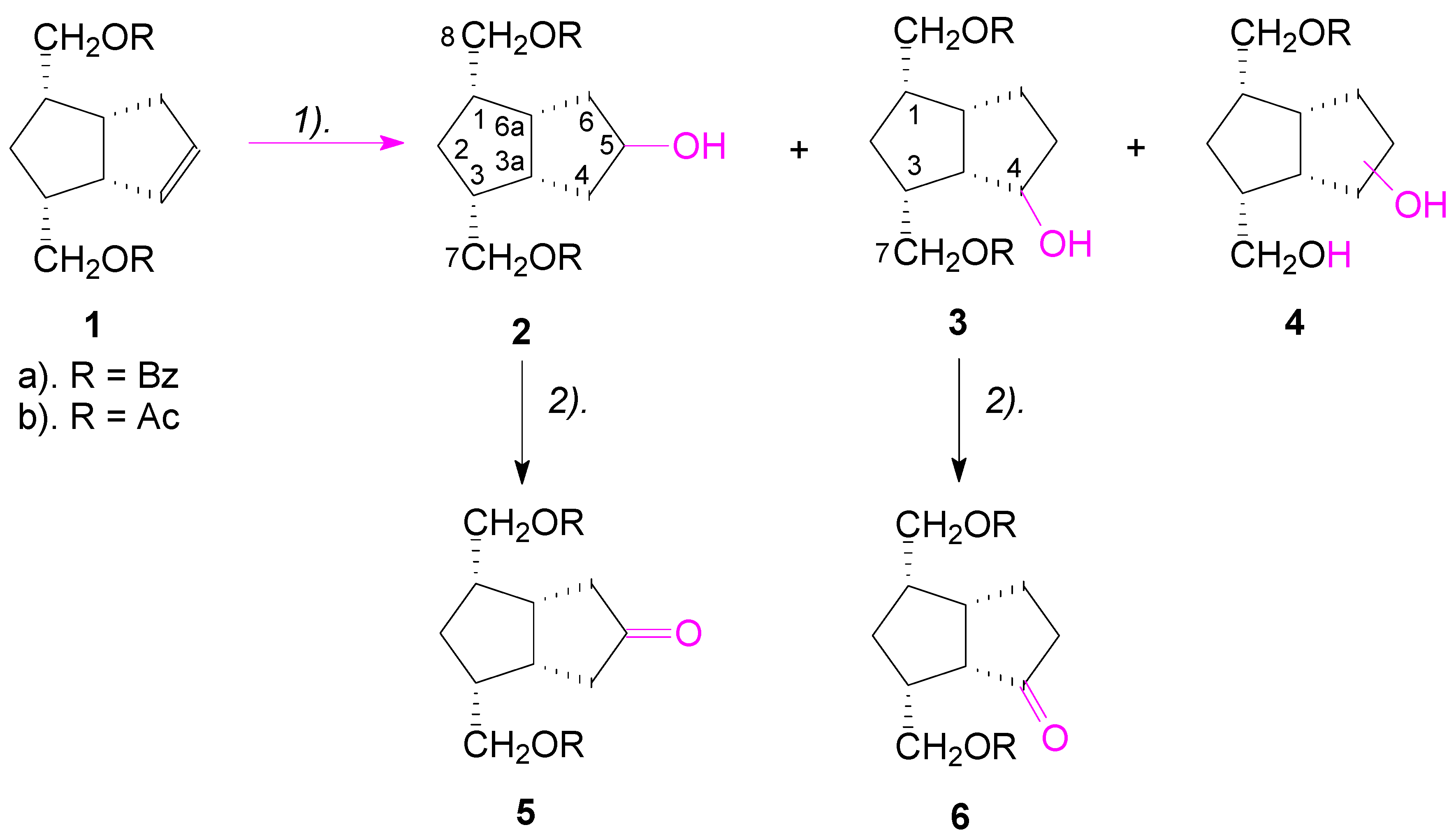
2. Results and Discussion
Retrosynthetic analysis of the key intermediates
5 and
6 (
Scheme 1), indicates that these compounds could be obtained by a sequence of two reactions: hydration of the double bond of alkene
1 to an alcohol, followed by the oxidation of the alcohol to the corresponding ketone. The starting compounds
1 were previously synthesized [
25] and have already been used for the synthesis of “pseudocarbacyclin” type compounds [
26,
27], of the corresponding diols by hydroxylation with KMnO
4 [
28], of the corresponding α and β-epoxides, by epoxidation of the double bond with MPBA [
29], and of pentalenofuranic compounds by regioselective reactions [
30]. Following our retrosynthetic analysis, we decided to hydrate the double bond of alkene
1 by hydroboration-oxidation with sodium acetoxyborohydride and with borane.
Hydroboration-oxidation of alkene
1a with sodium acetoxyborohydride [
31] resulted in the formation, in a non-regioselective manner, of both alcohols
2a (34%) and
3a (25%), slightly in favor of the symmetrical alcohol
2. In the reaction, the monobenzoate-triol
4a was also formed in a great quantity (38%) (
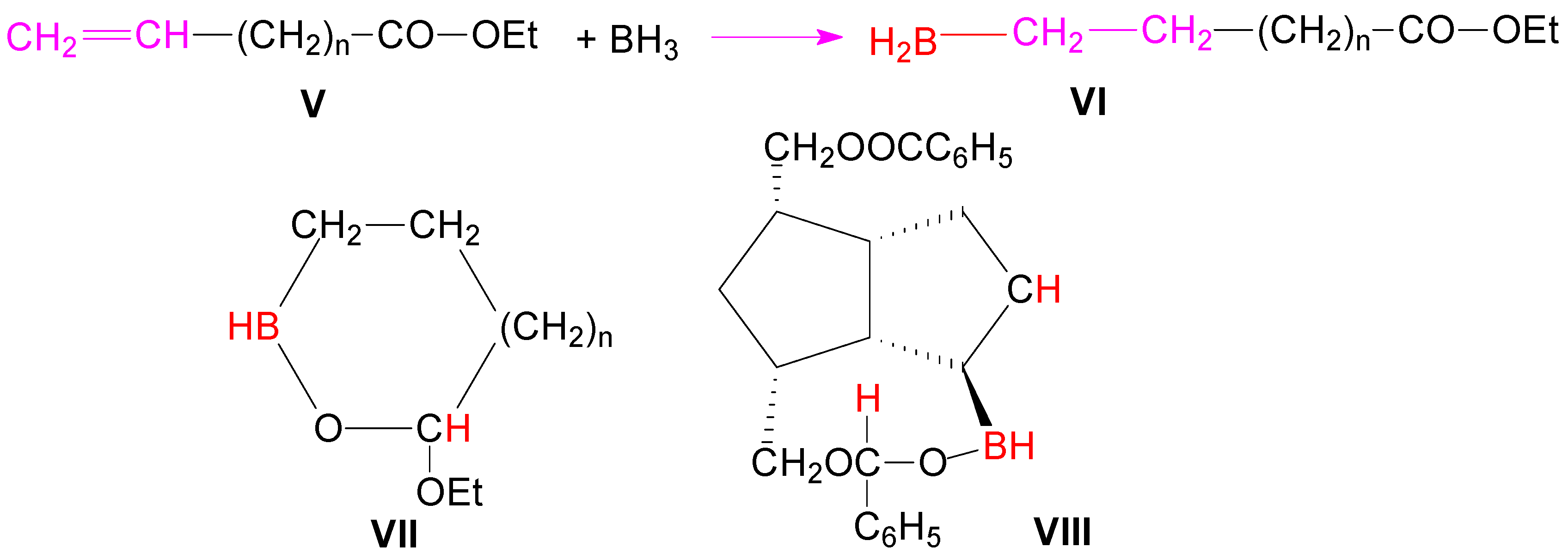
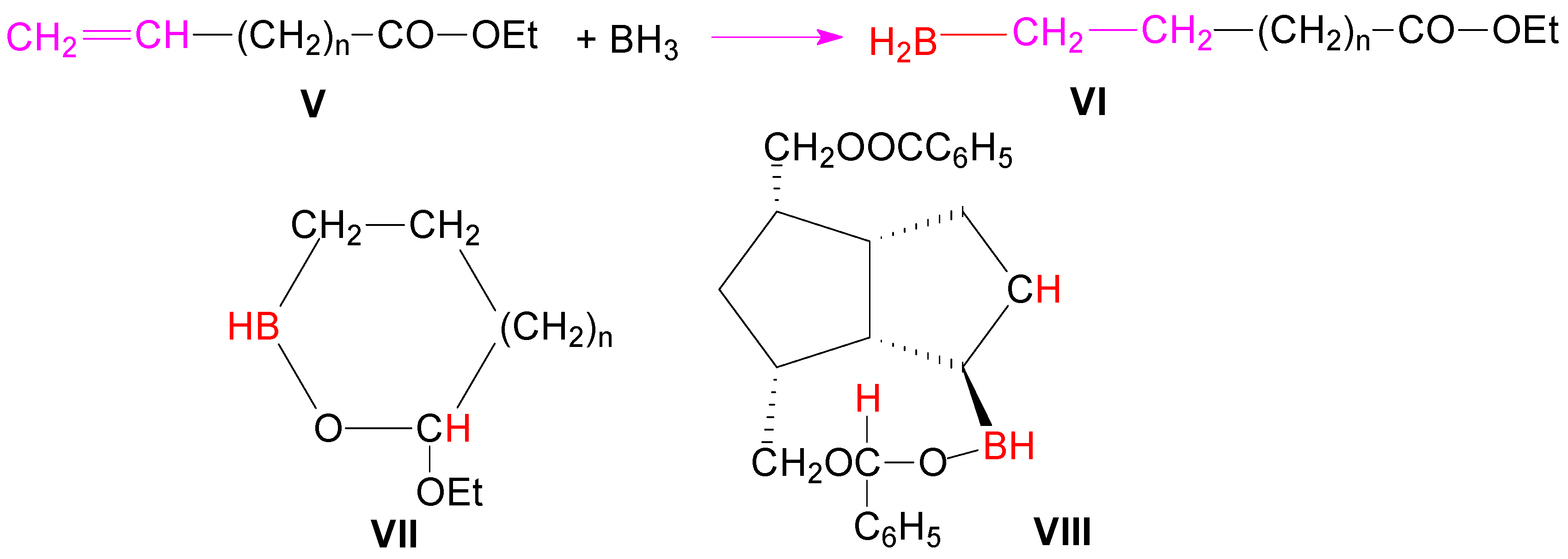
Scheme 1). Browsing the literature, we found that in the case of the unsaturated esters
V, by forming a cyclic intermediate with 5 or 6 atoms
VII, hydroboration of the double bond proceeded with concomitant reduction of the ester group (
Scheme 2) [
32].
Probably it is the same formation of a similar cyclic ester VIII in the hydroboration of diester compound 1, which favored the reduction of the closer ester group to double bond, this reduction being responsible for the formation of the monoacylated triols 4a and 4b.
In the hydroboration-oxidation of diacetate
1b, the monoacetate-triol
4b was formed also in 38% yield, and this is consistent with the mechanism presented in
Figure 2 for the formation of monoesters
4; the corresponding alcohols
2b and
3b were formed in about 1:1 ratio, but their isolation in pure form by low-pressure chromatography (LPC) was more difficult than in the case of the benzoate compounds
2a and
2b.
The hydroboration of 1 with BH3·THF, obtained in situ from NaBH4 and dimethyl sulfate, followed by H2O2 oxidation gave alcohols 2a, 3a and 4a in 31.5%, 28.8% and 34.5% yield; hence, there is no significant difference in the yields and the ratio of the alcohols between the hydroboration with NaBH3OOCCH3 and BH3. It is worth mentioning that the isolation of the alcohols by PC is easier for the benzoate esters than the acetate esters. The formation of the unwanted secondary by-products 4a and 4b could represent also an advantage, because it is easy to selectively protect the primary hydroxyl group, with a group different from benzoate, like an ether, trityl, tert-butyldimethylsilyl or other bulky silyl-protecting group, by the methods known in the art, and thus to obtain different protection of the hydroxymethyl groups, useful for the next steps.
The fact that by-product 4 has the hydroxyl group linked at the C4 atom (see below) means that the hydroboration-oxidation of benzoate 1a gave the C4-alcohol in a total yield of 62–63%.
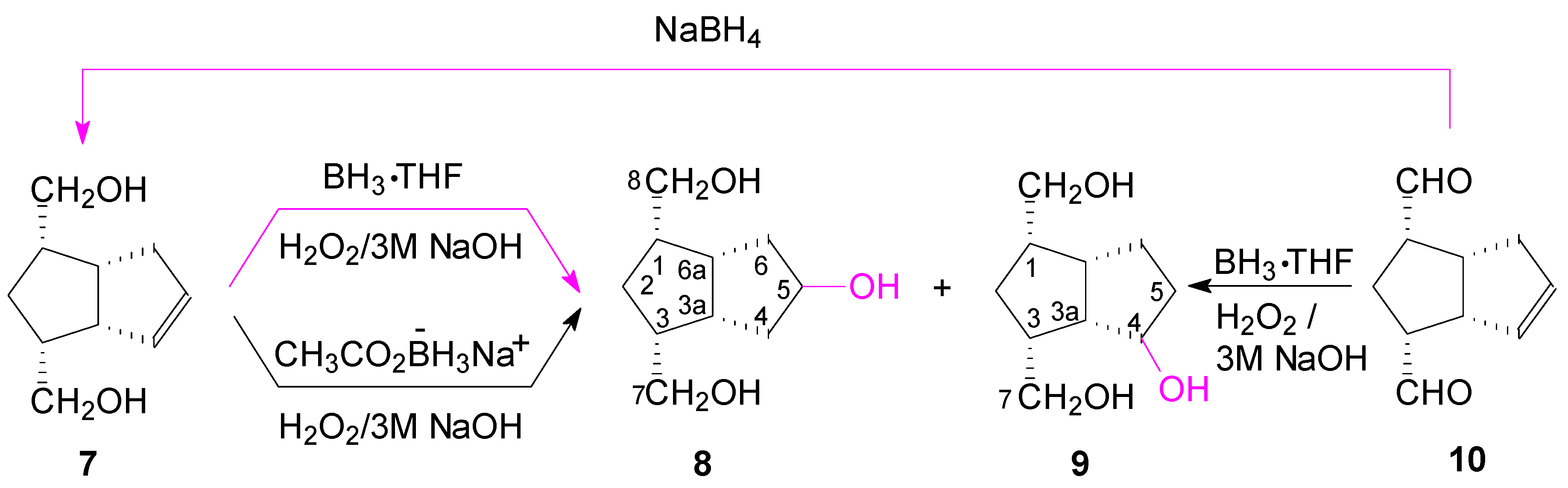
We then used the hydroboration-oxidation on the diol
7 (
Scheme 3). We observed that hydroboration with borane is still a slow reaction and a certain amount of alkene
7 remains unreacted. We used different molar ratios of BH
3·THF/
7, from 1.2:1 to 4:1, and the results are presented in
Table 1.
At a ratio of 2:1 BH
3·THF/
7, the borane reacted with the hydroxymethyl groups forming two alcoxyborane groups. The alkoxyborane group, closer to the double bond, hydroborated at the nearest carbon atom (C-4) with the formation in excess of the un-symmetrical alcohol
9, through an intermediate of type
VIII (
Scheme 2); the yield of alcohols
8 and
9 was still low (40.8%). By increasing the ratio of BH
3·THF/
7 to 3:1 and 4:1, there remained free borane which increased the yield of alcohols to 64.4%, and respectively to 73.3%, but there was no selectivity against
9 and the ratio of alcohols was nearly 1:1 (
8/
9).
Finally, we performed the hydroboration-oxidation of the double bond, concomitant with the reduction of the aldehyde groups of dialdehyde
10 (from which we previously [
25] obtained the alkene-diol
7 by NaBH
4 reduction of the aldehyde groups) with 2.2 molar equivalents of BH
3·THF (20 h), and a mixture of alkene
7, alcohols
9 and
8 was obtained in a ratio of 1.0:1.1:1.2. When the hydroboration was done with a greater molar ratio BH
3·THF:
7 of 3:1 (preparative scale on 0.359 M alkene
7) and time was increased to 72 h, the ratio of alcohols (
8/
9) was 1.0:1.2. Then another reaction was performed for 24 h at r.t. and for 2 h at 45–50 °C and the ratio of the alcohols changed to ~1.0:2.2 (
8/
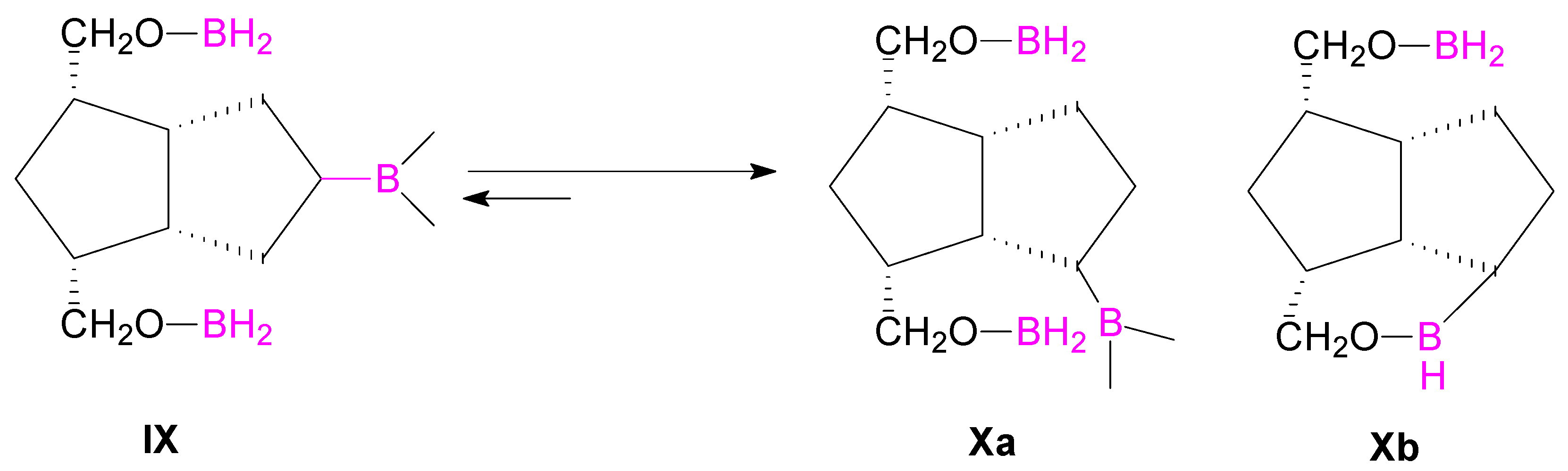
9). These suggest that an isomerization of the alkyl-boranes took place from type
IX to
Xa or
Xb (
Scheme 4), more thermodinamically stable in the reaction conditions.
Such isomerizations between alkyl-boranes are known in the literature [
33,
34,
35,
36,
37]. The α- or β-configuration of the hydroxyl groups introduced is not important, because the OH is oxidized in the next step to a ketone. Nonetheless, we believe that the secondary hydroxyl group is mainly introduced in a β orientation, because the access of the hydroboration reagent to the double bond ocurrs from the
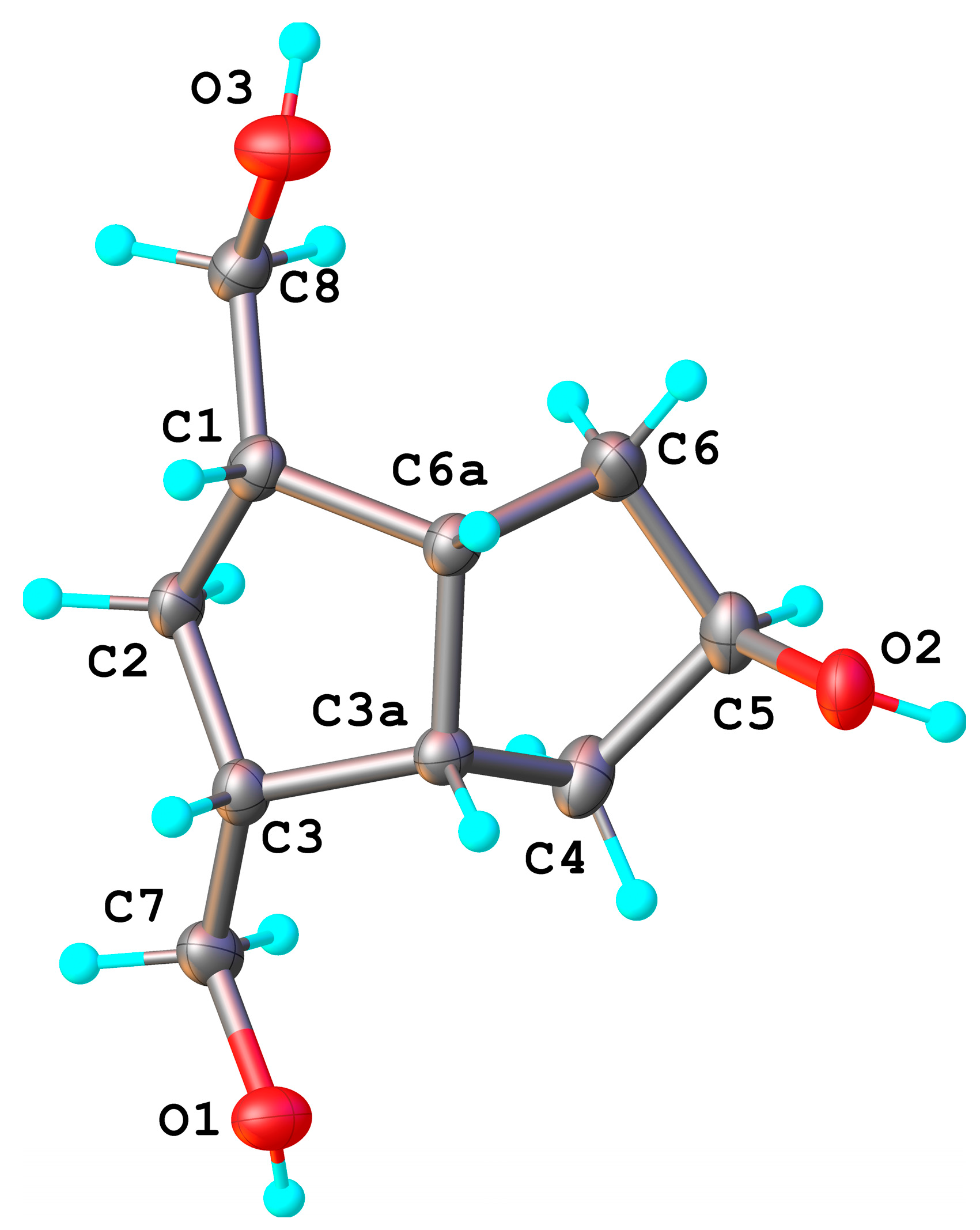
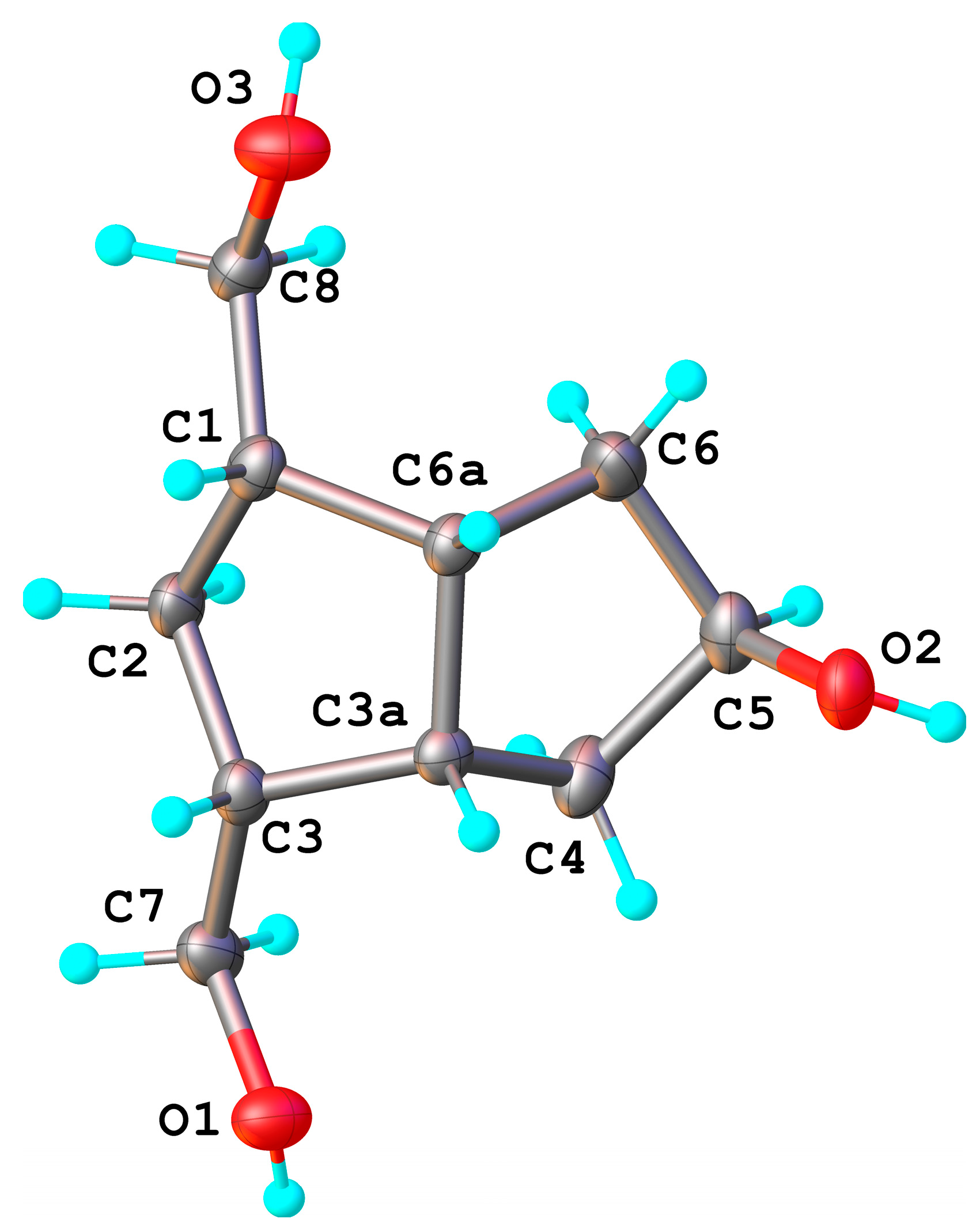
exo-side (the bulky crystallized compounds were only analyzed; the compound(s) remaining in the mother liquors were not analyzed for α/β isomers). The H
5 appears in
1H-NMR as a broad singlet and is not a clear evidence for the α-configuration, but the X-ray diffraction analysis (
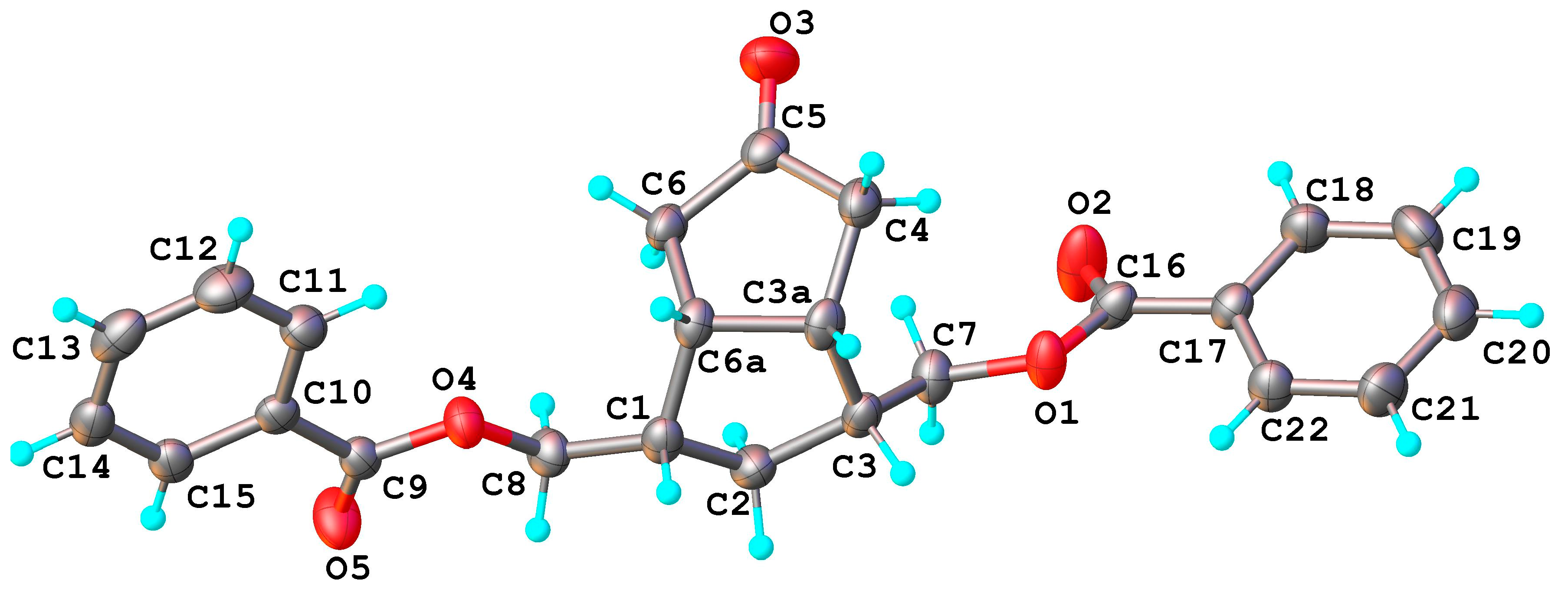
Figure 2) of the crystallized triol
8 confirmed the
exo-configuration of the secondary alcohol linked to the C
5 position, 5β-OH. Bond distances and angles for compound
8 are listed in
Table S1 and crystallographic data, details of data collection and structure refinement parameters in
Table S2 (also for compounds
3,
5 and
14,
Supplementary Material).
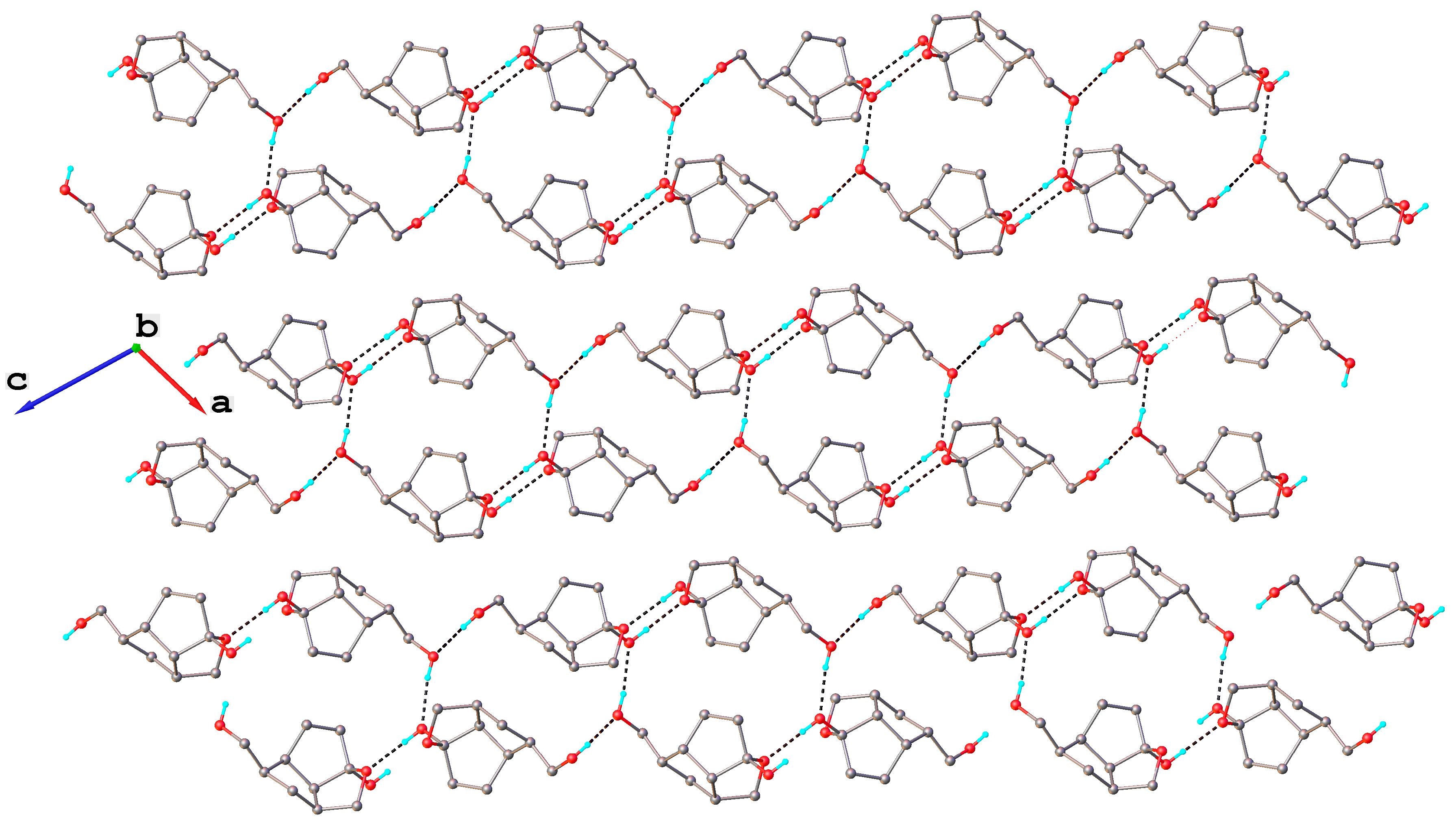
The compound
8 exhibits a molecular crystal structure where the neutral molecules interact through O-H···O hydrogen bonding to form a three-dimensional supramolecular network, as shown in
Figure 3. H-bonding parameters are listed in
Table 2.
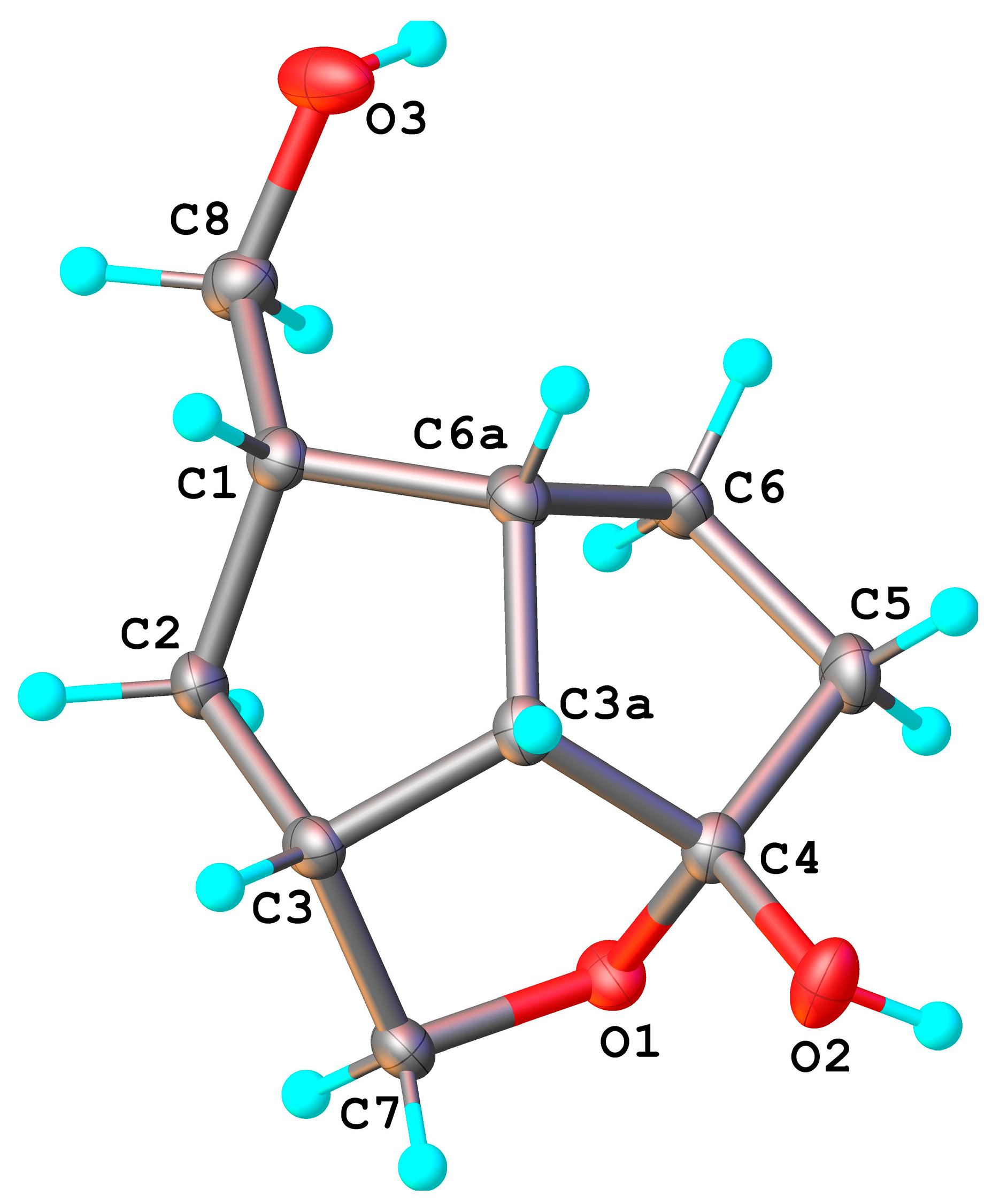
The configuration of the 4-OH in
9 was not studied, since the compound was obtained as an oil. For confirming the structure of the unsymmetrical alcohols (OH linked to C
4), we synthesized the tri-benzoates of
3 and
4 (and also of
2) to obtain suitable crystals for X-ray analysis, and their preparation is presented in the paper; their characterization by NMR was also done. At least for the fractions used for the analysis of the trisbenzoate obtained from
3, X-ray crystallography confirmed that the secondary 4-OH group are linked
exo (β) to the carbon atom, as in the case of
8. The result of single crystal X-ray diffraction study for this compound is shown in
Figure 4, while bond distances and angles are summarized in
Table S1.
Thus, the hydroboration-oxidation of alcohol 7 or dialdehyde 10 gives the symmetrical alcohol 8 in about 38% yield, but by increasing the reaction time and using hydroboration at elevated temperatures, the unsymmetrical alcohol 9 is formed in excess due to isomerization of the intermediary alkylboranes.
The regioisomers
8 (crystallized, m.p. 98–99 °C) and
9 (oil) were separated by LPC; their use in the next steps requires the selective protection of the primary hydroxyl groups, as exemplified for the obtaining of
2c (R = Tr) by treating
8 with trityl chloride; the following sequence is similar to the one of
2a (
Scheme 5).
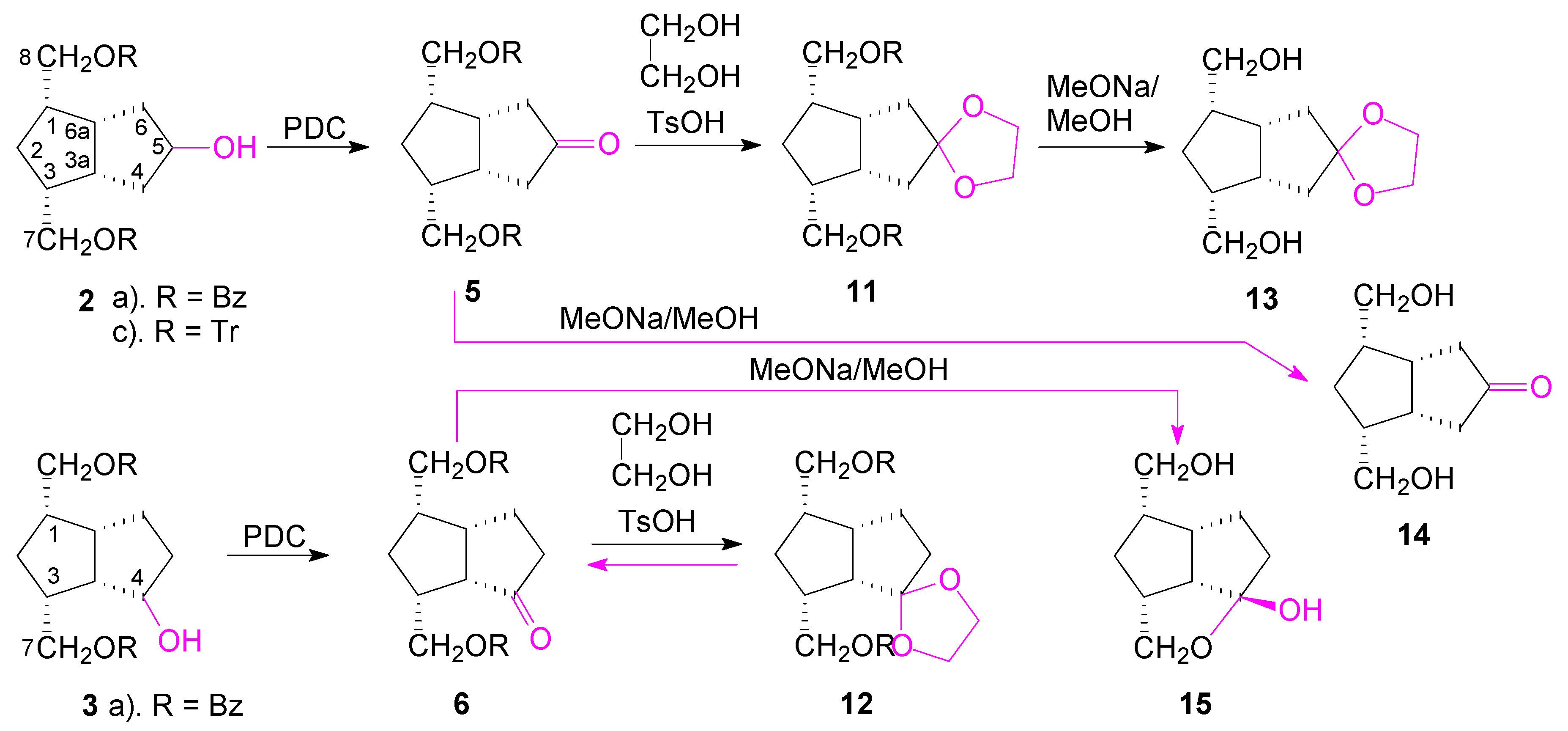
The secondary alcohols of
2 and
3 were oxidized with PDC to the corresponding ketones
5 and
6 and then the benzoate protecting groups were cleanly removed by transesterification with MeONa in MeOH (
Scheme 5). The structure of ketone
5 is easily established by NMR, just like the structure of the symmetrical alcohols
2 and
8, and that of the following compounds obtained from it: the ketone
14 and the ethylene ketal compounds
11 and
13. The X-ray diffraction investigation has confirmed that compound
5 crystalizes in
P2
1/n space group of monoclinic system and its molecular crystal consists of isolated neutral molecules, as illustrated in
Figure 5. Bond distances and angles are summarized in
Table S1.
The structure of ketone
6 was also established by NMR spectroscopy. By transesterification (MeONa/MeOH), the benzoate groups were removed and the symmetrical ketone bis-hydroxymethyl compound
14 was obtained from ketone
5. In the case of the unsymmetrical ketone
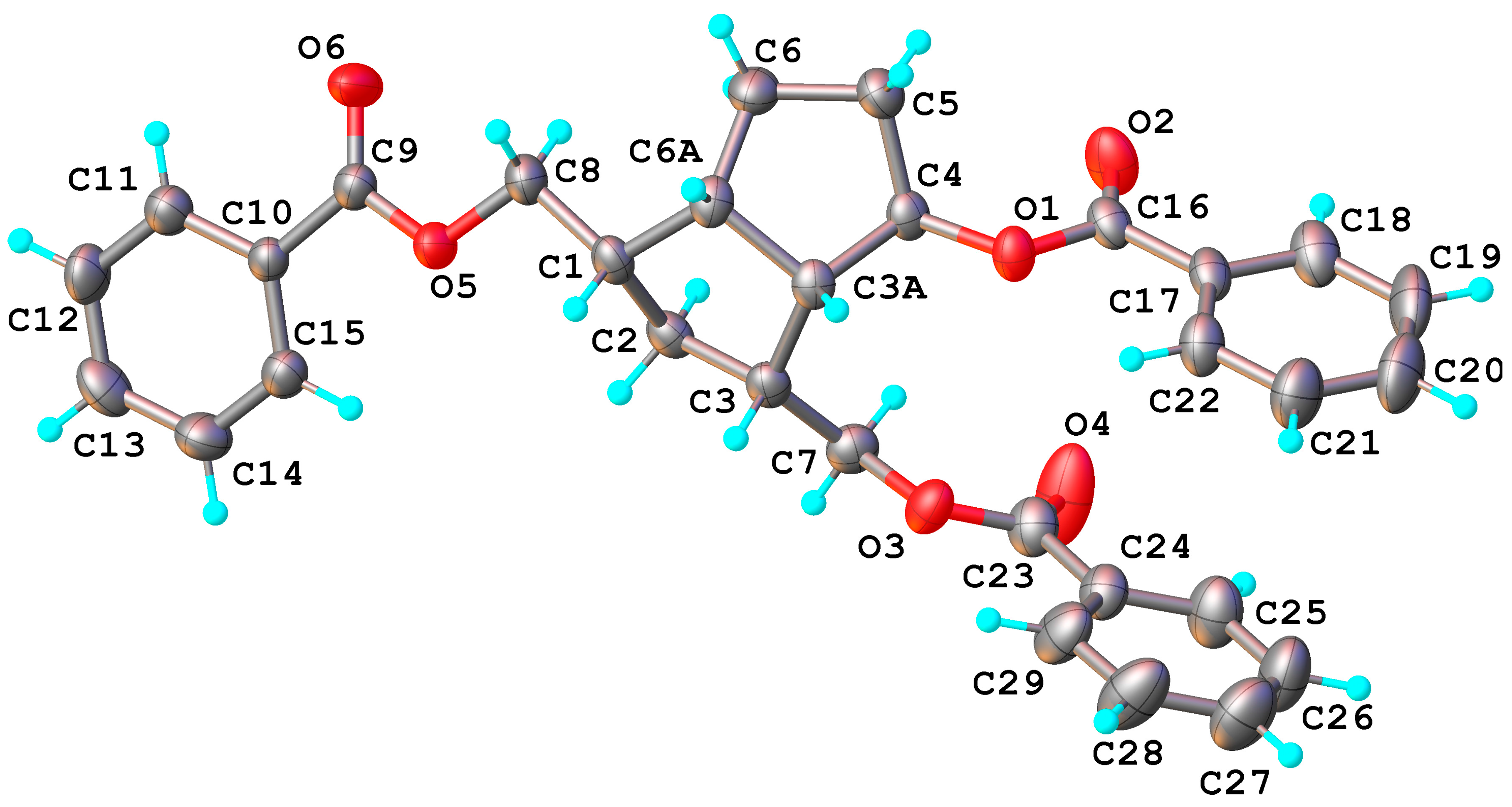
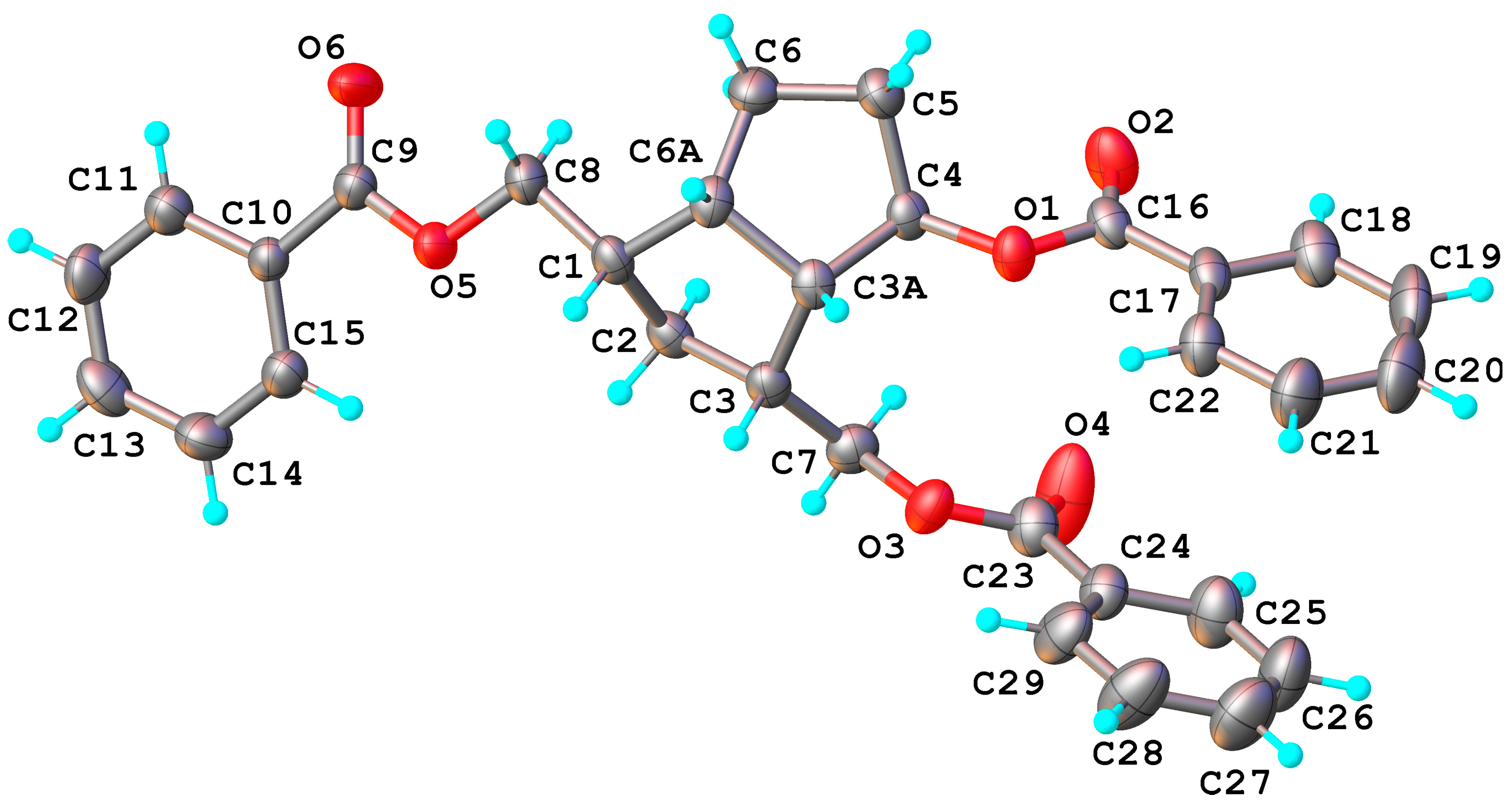
6, during the transesterification of the benzoate groups, the closer hydroxymethyl reacted with the ketone C
4=O and gave a cyclic hemiketal
15. Its molecular structure was also confirmed by X-ray crystallography (
Figure 6), showing the formation of the tetrahydrofuran ring with
exo-linked 4-hydroxyl of the hemiketal. According to X-ray crystallography, compound
15 crystallizes in
P2
1/n space group of monoclinic system with two crystallographically independent but chemically identical units, denoted as molecules
A and
B. In
Figure 6 only the structure of molecule
B is shown.
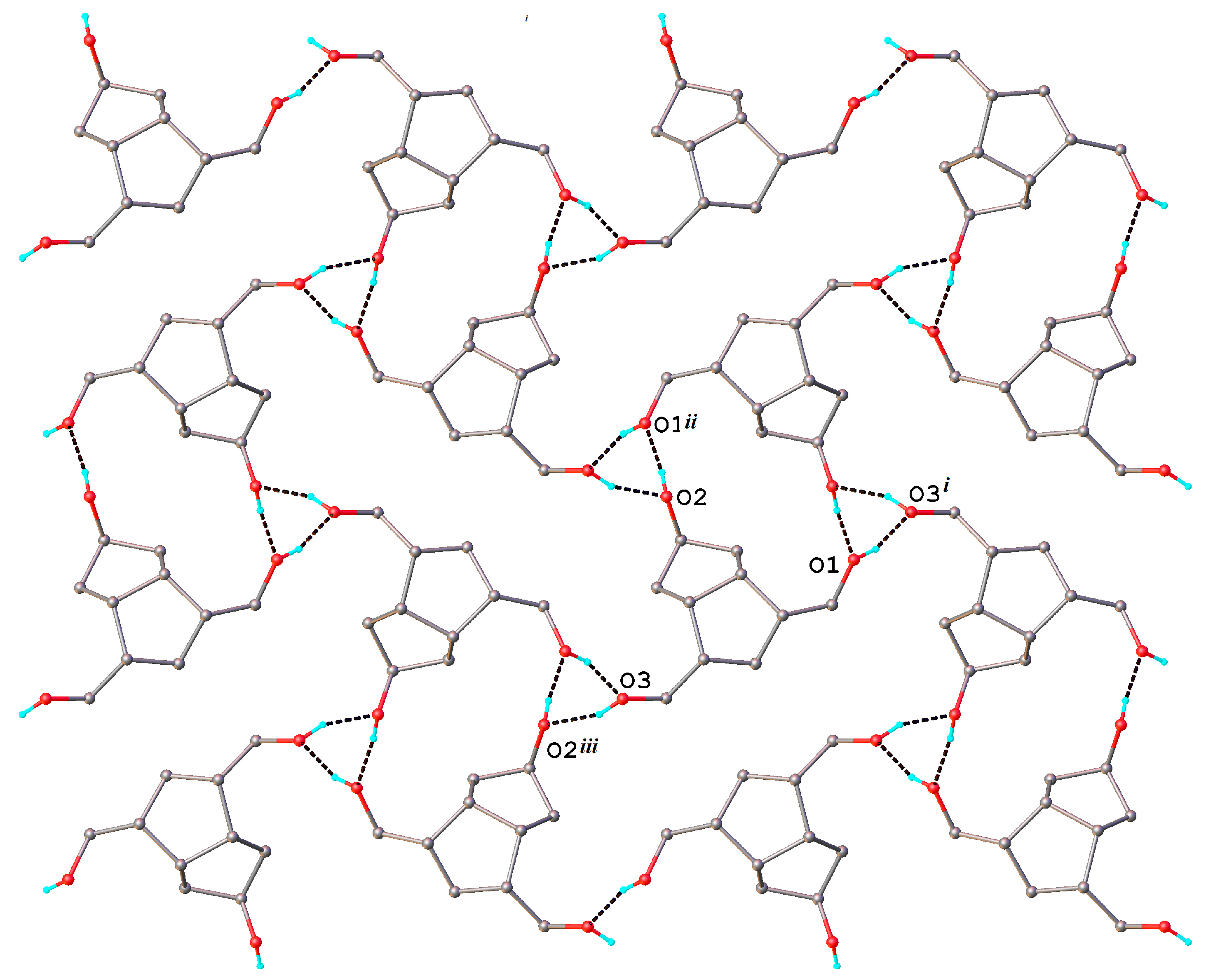
The crystal packing shows a parallel arrangement of two-dimensional supramolecular layers extended in the 101 plane. A partial view of the crystal structure is shown in
Figure 7. Each layer involves both independent molecules
A and
B connected by O-H···O hydrogen bonds, the formation of which is completely realized in the crystal. The corresponding hydrogen bond parameters are listed in
Table 3.
Both ketone groups of compounds 5 and 6 were transformed into the corresponding ethylene ketals by standard treatment with ethylene glycol (in C6H6 at reflux, TsOH catalyst). In the first case, we obtained compound 11 which by a similar transesterification reaction, gave compound 13, with the ketone protected as an ethylene ketal, a group useful for the next reactions for discrimination between the two hydroxymethyl groups. In the case of compound 12, though the reaction proceeded until all 6 reacted (TLC), during work-up or column chromatography purification, only the starting compound 6 was isolated, indicating that slightly acid conditions favored the deprotection of the ethylene ketal group. The applications of the symmetric-ketone compounds in the synthesis of new carbacyclin analogues are in progress.
3. Materials and Methods
3.1. General Information
Melting points (uncorrected) were determined in open capillaries on an OptiMelt apparatus (MPA 100, Stanford Research System, Inc., Sunnyvale, CA, USA). The progress of the reactions was monitored by TLC on silica gel 60F254 plates (Merck, Darmstadt, Germany) in solvent systems: I (benzene–ethyl acetate–hexane, 5:3:2), II (cyclohexane–ethyl acetate, 5:1), III (ethyl acetate–methanol–acetic acid, 90:13:1), IV (acetone–hexanes, 2:1), V (hexane–ethyl acetate–acetic acid, 5:3:0.1), VI (hexane–ethyl acetate–acetic acid, 5:3:0.1), VII (ethyl acetate–hexane–acetic acid, 5:1:0.1). Spots were visualized in UV or with 15% H2SO4 in MeOH (heating at 110 °C, 10 min) and 2,4-dinitrophenylhydrazine reagent for ketones. The compounds were purified by low pressure chromatography (<2 atm) (LPC), on a glass column, in the solvent systems presented at experimental. IR spectra were recorded on an FT-IR spectrometer 100 (Perkin Elmer, Shelton, CT, USA) and frequencies are expressed in cm−1. MS were recorded on a 1200 L/MS/MS triple-quadrupole instrument (Varian, Inc., Walnut Creek, CA, USA) equipped with an ESI interface, fragments obtained by collision with Ar and relative abundances (%) are given in parenthesis. 1H-NMR and 13C-NMR spectra were recorded on a Varian Gemini 300 BB spectrometer (300 MHz for 1H and 75 MHz for 13C, Varian, Inc., Palo Alto, CA, USA). Chemical shifts are given in ppm relative to TMS as an internal standard. Complementary spectra: 2D-NMR and decoupling were done for correct assignment of NMR signals. The numbering of the carbon atoms in the compounds is presented in the Schemes.
Crystallographic measurements were carried out with an XCALIBUR E CCD diffractometer (Oxford-Diffraction, Ltd., Abingdon, Oxfordshire, UK) equipped with graphite-monochromated Mo-Kα radiation. Single crystals were positioned at 40 mm from the detector and 481, 258, 251, and 774 frames were measured each for 20, 10, 5, and 20 s over 1° scan width for
8,
3,
5 and
15, respectively. The unit cell determination and data integration were carried out using the CrysAlis package of Oxford Diffraction [
38]. The structures were solved by direct methods using the Olex2 [
39] software (OlexSys Ltd., Durham University, UK) with the SHELXS structure solution program and refined by full-matrix least-squares method on
F2 with SHELXL-97 [
40]. The atomic displacements for the non-hydrogen atoms were refined using an anisotropic model. Hydrogen atoms were placed in fixed, idealized positions and refined as rigidly bonded to the corresponding atoms. Positional parameters of the H attached to the O atoms were obtained from difference Fourier syntheses and verified by the geometric parameters of the corresponding hydrogen bonds. In the absence of significant anomalous scattering, the absolute configuration for
8 could not be reliably determined. Friedel pairs were merged and any references to the Flack parameter were removed. The molecular plots were obtained using the Olex2 program. CCDC: 1566141 (for
8), 1566142 (for
3), 1566143 (for
5) and 1566145 (for
15). These data can be obtained free of charge via
www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or
deposit@ccdc.ca.ac.uk).
3.2. Hydroboration of 2α,4α-Dimethanol-1β,5β-bicyclo[3.3.0]oct-6-en dibenzoate with Sodium Acetoxy-Borohydride
To a suspension of 98% NaBH4 (772 mg, 20 mmol) in anhydrous THF (60 mL), cooled on an ice-water bath, 1.14 mL (1.2 g, 20 mmol) 99.9% acetic acid were added in Ar atmosphere, under stirring for 10 min. and stirring was continued for 20 min. A solution of 1a (7.53 g, 20 mmol) in THF (15 mL) was added during 20 min, stirring was continued overnight, monitoring the reaction by TLC (I, Rf1b = 0.91, Rf3a = 0.47, Rf2a = 0.35, Rf4a = 0.05). Water (5 mL) was added, the reaction mixture was cooled on an ice-water bath, then the alkylboranes were oxidized with 30% H2O2 (6 mL) and 3 M sodium acetate (10 mL) (instead of 3 M NaOH to prevent the hydrolysis of the benzoate protecting groups) both added at the same time during ~15 min, and stirred for 20 min. Ethyl ether (50 mL) was added, phases were separated, aqueous phase was extracted with ether (3 × 50 mL), organic phases were washed with sat. soln. NaHCO3 (50 mL), dried (MgSO4) and concentrated, obtaining 7.77 of crude product, which was purified by low-pressure chromatography (LPC) (eluent: hexanes, then solvent system: benzene–hexane–ethyl acetate, 5:3:2). The following fractions were eluted:
- (a)
1.6 g (21.2%) of unreacted 1a, (the yields below are based on the reacted 1a).
- (b)
1.54 g (24.8%, based on the reacted alkene) of pure unsymmetrical alcohol (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,5,6,6a-octahydro-4-hydroxy-1,3-diyl)bis(methylene) dibenzoate (3a) as an oil, IR (2% CHCl3): 3550–3400, 3350, 3035, 2910–2885, 2850, 1695, 1595, 1580, 1440, 1350, 1270–1200, 1090, 1055, 1040, 945 cm−1, 1H-NMR (DMSO-d6, δ ppm, J Hz): 7.98 (dd, 2H, 8.0, 1.2, H-o), 7.95 (dd, 2H, 8.0, 1.2, H-o), 7.67–7.61 (m, 2H, 8.0, 1.2, H-p), 7.51 (apparent t, 2H, 8.0, H-m), 7.50 (t, 2H, 8.0, H-m), 4.60 (d, 1H, OH, exchangeable with D2O, 4.8), 4.51 (dd,1H, 11.1, 5.0, H-7), 4.18–4.45 (m, 3H, H-7, H-8), 3.82 (m, 1H, H-4), 2.70 (m, 1H, H-3a), 2.20–2.40 (m, 3H, H-6a, H-1, H-3), 1.05–1.90 (m, 6H, H-5, H-6, H-2), 13C-NMR (DMSO-d6, δ ppm): 165.75, 165.58 (COO), 133.19, 133.08 (C-p), 130.04, 129.81 (C-q), 129.12, 129.03 (C-o), 128.71, 128.61 (C-m); 72.98 (C-4), 65.60 (C-7 or C-8), 65.21 (C-8 or C-7), 51.58 (C-3a), 42.61 (C-1), 40.42 (C-3 or C-6a), 40. 40 (C-6a or C-3), 35.85 (C-5), 31.02 (C-2), 23.01 (C-6), elem. anal. calcd for C24H26O5 (%), C: 73.07, H: 6.64, found: C: 72.80, H: 6.42.
- (c)
2.11 g (34%) of pure symmetrical more polar alcohol, (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,5,6,6a-5-hydroxyoctahydropentalene-1,3-diyl)bis(methylene) dibenzoate (2a) which was crystallized (1.69 g) by ethyl acetate-gas extraction, m.p. 115–117 °C. A fraction recrystallized twice had m.p. 119.5–120.5 °C, 1H-NMR (CDCl3, δ ppm, J Hz): 8.03 (dd, 4H, 7.1, 1.4, H-o), 7.56 (tt, 2H, 7.1, 1.4, H-p), 7.44 (t, 4H, 7.1, H-m), 4.41 (dd, 2H, 11.0, 7.1, H-7, H-8), 4.31 (dd, 2H, 11.0, 8.2, H-7, H-8), 4.11 (tt, 1H, 5.9, 10.4, H-5), 2.69–2.64 (m, 2H, H-3a, H-6a), 2.48–2.38 (m, 2H, 8.2, 7.1, H-1, H-3), 2.05–1.97 (m, 2H, 10.4, 5.9, H-4, H-6), 1.89 (dt, 1H, 11.1, 5.4, H-2), 1.34 (q, 1H, 11.1, H-2), 1.36–1.26 (m, 2H, 10.4, 5.9, H-4, H-6), 13C-NMR (CDCl3, δ ppm): 166.45 (COO), 132.88 (2C-p), 130.24 (2C-q), 129.48 (4C-o), 128.32 (4C-m), 73.33 (C5), 65.58 (C-7, C-8), 41.05 (C-1, C-3 or C-3a, C-6a), 40.85 (C-3a, C-6a or C-1, C-3), 35.98 (C-4, C-6), 30.01 (C-2), elem. anal. Calcd. for C24H26O5, C:d 73.07, H: 6.64, found: C: 72.88, H: 6.52.
- (d)
1.72 g (37.6%) pure (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,5,6,6a-4-hydroxy-3-(hydroxymethyl)-octahydropentalen-1-yl)methyl benzoate (4a), which was crystallized by ethyl acetate-gas extraction to give 1.12 g of product, m.p. 74–77 °C, 1H-NMR (CDCl3, δ ppm, J Hz): 8.02 (dd, 2H, 1.4, 7.4, H-o), 7.53 (t, 1H, 1.4, 7.4, H-p), 7.44 (t, 2H, 7.4, 2H-m), 4.36 (dd, 1H, 7.1, 11.0, H-8), 4.27 (dd, 1H, 8.0, 11.0, H-8), 4.02 (ddd, 1H, 5.5, 8.2, 10.4, H-4), 3.85 (dd, 1H, 5.8, 11.0, H-7), 3.76 (t, 1H, 11.0, H-7), 3.14–3.00 (br d, 2H, OH), 2.86 (qv, 1H, 9.9, H-6a), 2.51 (dt, 1H, 8.2, 9.9, H-3a), 2.44–2.28 (m, 2H, H-1, H-3), 1.98 (dt, 1H, 5.5, 11.0, H-5), 1.75 (m, 1H, H-6), 1.67 (dt, 1H, 5.5, 12.6, H-2), 1.47 (dd, 1H, 5.8, 11.0, H-5), 1.27 (m, 1H, H-6), 0.99 (q, 1H, 12.6, H-2), 13C-NMR (CDCl3, δ ppm): 166.68 (COO), 133.06 (C-p), 130.45 (C-q), 129.65 (C-o), 128.51 (C-m), 73.99 (C-4), 65.56 (C-8), 63.50 (C-7), 52.09 (C-3a), 43.68 (C-1), 42.98 (C-6a), 41.11 (C-3), 35.10 (C-5), 30.43 (C-2), 23.76 (C-6).
3.3. Hydroboration of 2α,4α-Dimethanol-1β,5β-bicyclo[3.3.0]oct-6-en diacetate with Sodium Acetoxy-Borohydride
Hydroboration of diacetate 1b (20 mmol) was realized as for 1a, affording 5.23 g of crude product which was similarly purified. The following fractions were collected: 0.86 g (3.41 mmol) of starting alkene 1b, (16.9%); 0.13 g slightly impure unsymmetrical alcohol 3b, as an oil; 1.62 g of a mixture of alcohols 2b and 3b, as an oil; 0.26 g of slightly impure symmetric alcohol 2b, as an oil and 1.74 g (7.6 mmol, 38%) of (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,5,6,6a-4-hydroxy-3-(hydroxymethyl)octahydro-pentalen-1-yl)methyl acetate (4b) which gave 1.32 g (28.8%) as of the title product as needles, m.p. 58–59.5 °C, upon crystallization from ethyl acetate-hexane; 1H-NMR (DMSO-d6, δ ppm, J Hz): 3.97 (d, 2H, 7.7, H-8), 3.77 (dd + TFA, 1H, 3.3, 6.3, H-4), 3.62 (dd, 1H, 7.6, 10.8, H-7), 3.54–3.40 (br, 2H, OH), 3.45 (dd, 1H, 7.4, 10.8, H-7), 2.57 (dt + TFA, 1H, 6.2, 9.2, H-6a), 2.20 (dt, 1H, 6.3, 8.8, H-3a), 2.08–1.98 (m, 2H, H-1, H-3), 1.98 (s, 3H, CH3), 1.68 (m, 1H, H-5), 1.61 (dt, 1H, 6.5, 12.8, H-2), 1.49 (m, 1H, H-6), 1.28 (m, 1H, H-5), 1.13 (m, 1H, H-6), 0.85 (q, 1H, 12.8, H-2), 13C-NMR (CDCl3, δ ppm): 170.40 (COO), 72.72 (C-4), 64.69 (C-8), 61.97 (C-7), 51.83 (C-3a), 43.83 (C-1), 42.53 (C-6a), 40.50 (C-3), 35.40 (C-5), 31.03 (C-2), 23.73 (C-6), 20.73 (CH3), elem. anal. calcd. for C12H20O4 (%), C: 63.12, H: 8.83, found: C: 63.20, H: 8.65.
3.4. Hydroboration of (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,6a-Hexahydro-1,3-pentalenodimethanol Dibenzoate with Borane (BH3·THF)
To a suspension of 98% NaBH4 (0.3 g, 7.8 mmol) in anhydrous THF (25 mL), dimethyl sulfate (DMS, 0.76 mL, 1.014 g, 7.8 mmol) was added dropwise under mechanical stirring under an Ar atmosphere. The reaction mixture was stirred for 2 h at room temperature (r.t.) and for 30 min at 50–60 °C, cooled under −10 °C, and a solution of alkene dibenzoate 1a (7.58 g, 20 mmol; alkene/borane molar ratio of 2.56:1, for forming the trialkylborane) in THF (15 mL) was added dropwise and stirred for 20 h at r.t., following the reaction by TLC. An equal quantity of borane was prepared and added to the reaction mixture at 10–13 °C, then stirred overnight at r.t. Though all the alkene was not consumed, the alkylboranes formed were oxidized, after cooling on an ice-water bath, with 30% H2O2 (4.4 mL) and 1 N NaOH (8.1 mL), added dropwise at the same time. The stirring was continued then for 20 min at r.t., 40 mL water were added and the mixture was extracted with dichloromethane (3 × 100 mL). Organic phases were washed with brine (2 × 50 mL), dried (MgSO4), concentrated and the crude product (7.2 g) was purified as in ex. 3.2, resulting 1.25 g of unreacted alkene (16.6%), 1.90 g (28.8%) 3a, 2.07 g (31.5%) 2a and 1.67 g (34.5%) 4a.
3.5. Hydroboration of Alkene-Diol 7 with Sodium Acetoxyborohydride
2α,4α-Dimethanol-1β,5β-bicyclo[3.3.0]octene-6
7 (6.72 g, 40 mmol) dissolved in anhydrous THF (50 mL), was hydroborated with acetoxyborohydride (as described in
Section 3.2), obtained from NaBH
4 (4.63 g, 120 mmol) and 99.9% acetic acid (7.2 mL, 120 mmol) in THF (100 mL) for 3 h at 0–5 °C, and over a weekend at r.t. TLC (III, R
f7 = 0.84, R
f9 = 0.62, R
f8 = 0.41). The alkylborane was oxidized with 30% H
2O
2 (33 mL) and 3 N NaOH (60 mL) added dropwise in the same time. The solvent was removed under reduced pressure, the concentrate was co-evaporated with anhydrous ethanol (3 × 50 mL), extracted with ethanol, resulting in an 8.9 g of a mixture of triols
8 and
9. The pure products were obtained by LPC (silica gel, eluent: acetone–hexanes, 1:2, then 1:1), resulting in 2.57 g (34.5%) of the symmetric alcohol
8 as an oil and 3.58 g (48%) of pure unsymmetric alcohol
9 as a slightly yellow oil (the isolated alcohols were obtained in a 1.0:1.4 ratio of
8/9).
- (a)
(±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,5,6,6a-5-Hydroxyoctahydropentalene-1,3-diyl)dimethanol (8). The symmetric alcohol was crystallized from acetone-hexanes giving 1.35 g 8 as needle crystals, m.p. 98–99 °C. IR (KBr): 3380–3180, 2945, 2980, 2850, 1480, 1460, 1440, 1350, 1200, 1080, 1020, 960, 870 cm−1, 1H-NMR (DMSO-d6, δ ppm, J Hz): 4.26 (t, 2H, 5.0, HO-C7,8, exchangeable with D2O), 4.16 (d, 1H, 3.0, HO-C5, exchangeable with D2O), 4.10 (br s, 1H, H-5), 3.34 (dd, 4H, 7.3, 5.0, H-7, H-8), 2.67–2.60 (m, 2H, H-3a, H-6a), 1.99–1.89 (m, 2H, 7.3, 5.5, H-1, H-3), 1.56 (dt, 1H, 12.1, 5.5, H-2), 1.52–1.44 (m, 2H, H-4, H-6), 1.30–1.20 (m, 2H, H-4, H-6), 0.71 (q, 1H, 12.1, H-2), 13C-NMR (DMSO-d6, δ ppm): 72.92 (C-5), 61.97 (C-7, C-8), 44.31 (C-1, C-3 or C-3a, C-6a), 41.98 (C-3a, C-6a or C-1, C-3), 35.30 (C-4, C-6), 30.62 (C-2), MS, m/z: 168 (PM), 150, 137 (68%), 109 (73%), 95, 93, 91, 79 (PB).
- (b)
(±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,5,6,6a-4-Hydroxyoctahydropentalene-1,3-diyl)dimethanol (9). IR: practically identical to that of 8, 1H-NMR (DMSO-d6, δ ppm, J Hz): 4.65 (dd, 1H, 3.8, 0.5, HO-C4, exchangeable with D2O), 4.43 (t, 1H, 5.6, HO-C7, exchangeable with D2O), 4.37 (t, 1H, 4.7, HO-C8, exchangeable with D2O), 3.76 (dt, 1H, 6.7, 7.3, H-4), 3.62 (dd, 1H, 10.7, 7.7, H-7), 3.46 (dd, 1H, 10.7, 7.5, H-7), 3.37 (d, 2H, 6.0, H-8), 2.56 (d, 1H, 9.0, H-6a), 2.20 (q, 1H, 8.8, H-3a), 2.00 (m, 1H, H-3), 1.90 (m, 1H, H-1), 1.69–1.44 (m, 3H, H-2, H-6, H-5), 1.28 (m, 1H, H-5), 1.17 (m, 1H, H-6), 0.74 (q, 1H, 12.3, H-2), 13C-NMR (DMSO-d6, δ ppm): 72.61 (C-4), 61.88 (C-7), 61.37 (C-8), 51.71 (C-3a), 44.30 (C-1), 43.62 (C-3), 42.45 (C-6a), 35.24 (C-5), 31.01 (C-2), 22.83 (C-6), MS, m/z: 168 (PM), 150, 138, 119, 106, 93, 91, 83 (PB).
3.6. Hydroboration of Alkene-Diol 7 with Borane
Alkene
7 (3.26 mmol) was hydroborated with borane in the conditions mentioned in
Section 3.4 and using the ratios of borane/
7 mentioned in
Table 1. The purification of the crude mixtures was performed as in
Section 3.5 and the yields are presented in
Table 1.
3.7. Hydroboration of Alkene-Dialdehyde 10 with Borane (at a 0.359 M Scale)
To a solution of BH3·THF, prepared from 98% NaBH4 (41 g, 1.056 mol) and 97% dimethyl sulfate (105 mL, 1.056 mol) in THF (500 mL), cooled to <−10 °C, a solution of 2α,4α-diformyl-1β,5β-bicyclo[3.3.0]octene-6 (0.359 mol, previously co-evaporated with benzene) in THF (150 mL) was added dropwise in 90 min, maintaining the temperature under −5 °C. The temperature was left to reach the r.t. and the reaction mixture was further stirred over weekend. TLC (IV, Rf7 = 0.43, Rf9 = 0.23, Rf8 = 0.09). The crude product (75 g) was purified (3×) by LPC (acetone–hexanes, 1:1, then 2:1), resulting 7.55 g (12.5%) alkene 7, 29.77 g (44.5%) 9 as an oil and 25.1 g (37.5%) 8, crystallized in mass, m.p. 102–104 °C (twice recrystallized from acetone). The ratio of 9/8 was 1.185/1.0.
3.8. Benzoylation of Symmetrical Alcohol 2a
The dibenzoate alcohol 2a (160 mg, 0.4 mmol) in pyridine (5 mL) was benzoylated with benzoyl chloride (0.24 mL) in usual conditions. TLC (V, Kieselgel plastifollien, Merck, Rf2a = 0.57, Rf2a-triBz = 0.93). The crude product was purified by LPC, resulting 190 mg (95.2%) of symmetrical tribenzoate 2a, m.p. 85–87 °C (crystallized from hexane–ethyl acetate), 1H-NMR (CDCl3, δ ppm, J Hz): 8.05–7.99 (m, 6H, H-o), 7.58–7.53 (m, 3H, H-p), 7.46–7.40 (m, 6H, H-m), 5.57 (t, 1H, 3.3, H-5), 4.43 (dd, 2H, 7.1, 11.0, H-7, H-8), 4.33 (dd, 2H, 8.0, 11.0, H-7, H-8), 3.12–3.03 (m, 2H, H-3a, H-6a), 2.60–2.51 (m, 2H, H-1, H-3), 2.11–2.04 (m, 2H, H-4, H-6), 1.95 (dt, 1H, 5.5, 12.4, H-2), 1.73–1.62 (m, 2H, H-4, H-6), 1.26 (q, 1H, 12.4, H-2), 13C-NMR (CDCl3, δ ppm): 166.63 (COO), 133.09, 132.97 (3C-p), 130.24 (3C-q), 129.71, 129.67 (6C-o), 128.53, 128.46 (6C-m), 79.06 (C-5), 65.58 (C-7, C-8), 42.97 (C-1, C-3 or C-3a, C-6a), 40.99 (C-3a, C-6a or C-1, C-3), 33.52 (C-4, C-6), 30.59 (C-2).
3.9. Benzoylation of Unsymmetrical Alcohol 3a
The dibenzoate alcohol 3a (160 mg, 0.4 mmol) was benzoylated as above; 150 mg (73%) of un-symmetrical tribenzoate 3a, m.p. 82–83 °C (hexane-ethyl acetate) were obtained, 1H-NMR (CDCl3, δ ppm, J Hz): 8.05 (dd, 2H, 1.4, 7.8, H-o), 7.94 (dd, 2H, 1.4, 7.8, H-o), 7.87 (dd, 2H, 1.4, 7.4, H-o), 7.57 (tt, 1H, 1.4, 7.8, H-p-in 4-O-Bz), 7.50–7.23 (m, 8H, 2H-p, 6H-m), 5.33 (dt, 1H, 5.5, 8.2, H-4), 4.51 (dd, 1H, 6.9, 11.3, H-7 or H-8), 4.51 (dd, 1H, 8.0, 11.3, H-7 or H-8), 4.43 (dd, 1H, 6.9, 11.0, H-8 or H-7), 4.37 (dd, 1H, 8.0, 11.0, H-8 or H-7), 3.00–2.89 (m, 2H, H-3a, H-6a), 2.59–2.42 (m, 2H, H-1, H-3), 2.23 (ddt, 1H, 3.6, 5.8, 12.1, H-5), 1.95 (dt, 1H, 5.5, 12.6, H-2), 1.87 (m, 1H, H-6), 1.68 (m, 1H, H-5), 1.51 (m, 1H, H-6), 1.26 (q, 1H, 12.6, H-2), 13C-NMR (CDCl3, δ ppm): 166.63, 166.45, 166.19 (3COO), 133.11 (C-p), 132.85 (2C-p), 130.45 (2-C-q), 130.24 (C-q), 129.65 (6C-o), 128.55, 128.31, 128.27 (6C-m), 76.58 [C-4, with Cr(acac)3], 65.49 (C-7 or C-8), 64.94 (C-8 or C-7), 49.67 (C-3a), 43.38 (C-1), 41.09, 40.98 (C-3, C-6a), 33.26 (C-5), 31.43 (C-2), 24.23 (C-6).
3.10. Benzoylation of Monobenzoate Alcohol 4a
The monobenzoate alcohol 4a (145 mg, 0.5 mmol) was benzoylated as above; 230 mg (73%) of tribenzoate 4a, m.p. 83–85 °C (hexane–ethyl acetate); 1H-NMR (CDCl3, δ ppm, J Hz): 8.05 (dd, 2H, 1.4, 7.4, H-o), 7.94 (dd, 2H, 1.4, 7.4, H-o), 7.87 (dd, 2H, 1.4, 7.4, H-o), 7.57 (tt, 1H, 1.4, 7.8, H-p-in 4-O-Bz), 7.50–7.42 (m, 2H-p), 7.35–7.20 (m, 6H-m), 5.33 (dt, 1H, 5.5, 8.2, H-4), 4.51 (dd, 1H, 6.9, 11.3, H-7 or 8), 4.46 (dd, 1H, 8.0, 11.3, H-7 or H-8), 4.43 (dd, 1H, 6.9, 11.0, H-8 or H-7), 4.37 (dd, 1H, 8.0, 11.0, H-8 or H-7), 3.00–2.88 (m, 2H, H-3a, H-6a), 2.60–2.39 (m, 2H, H-1, H-3), 2.23 (ddt, 1H, 3.5, 5.8, 12.1, H-5), 1.94 (dt, 1H, 5.5, 12.6, H-2), 1.87 (m, 1H, H-6), 1.68 (m, 1H, H-5), 1.51 (m, 1H, H-6), 1.26 (q, 1H, 12.6, H-2), 13C-NMR (CDCl3, δ ppm): 166.62, 166.43, 166.18 (3COO), 133.10, 132.85, 132.81 (3C-p), 130.445 (2-C-q), 130.25 (C-q), 129.70, 129.65 (6C-o), 128.55, 128.31, 128.27 (6C-m), 76.76, 65.49 (C-7 or C-8), 64.94 (C-8 or C-7), 49.70 (C-3a), 43.38 (C-1), 41.09, 40.99 (C-3, C-6a), 33.27 (C-5), 31.43 (C-2), 24.23 (C-6); mixt. m.p. tri-Bz-2a + triBz-3a = 69–72 °C; mixt. m.p. tri-Bz-2a + triBz-4a = 72–74 °C; mixt. m.p. tri-Bz-3a + triBz-4a = 83–85 °C.
3.11. Tritylation of Symmetric Triol 8
The symmetric triol 7 (3.17 g, 17.02 mmol) was dissolved in pyridine (40 mL) and 97% trityl chloride (10.2 g, 35.6 mmol) was added in portions over 30 min at r.t. under stirring. Stirring was continued over weekend (3 days) and TLC (IV, Rf7 = 0.11, Rf2c = 0.53, Rf2c-tris-Tr = 0.80) still showed the presence of a minor quantity of the starting triol. Another 1 g of trityl chloride was added and the solution was stirred for 3 h at 45–50 °C. The cooled reaction mixture was poured over crushed ice, stirred for 1 h, the product was extracted with dichloromethane (3 × 100 mL), the unified organic solutions were washed with sat. soln. NaHCO3 (2 × 100 mL), brine (100 mL), dried (MgSO4), filtered, concentrated and co-evaporated with toluene. The crude product was crystallized from methanol and recrystallized from methanol-ethyl acetate, resulting 3.84 g of pure compound 2c, m.p. 98–101 °C [By LPC purification of the concentrated mother liquors (eluent, hexanes–ethyl acetate, 3:1), 6.65 g (total yield: 92%) of pure compound 2c and 700 mg of almost pure tristrityl derivative (4.5%) were obtained], IR (KBr): 3600–3300, 3040–3020, 2920–2850, 1585, 1480, 1440, 1150, 1055, 740, 695 cm−1, 1H-NMR (CDCl3, δ ppm, J Hz): 7.43 (dd, 12H, 1.7, 8.5, H-o), 7.28 (t, 12H, 8.5, H-m), 7.21 (td, 6H, 1.7, 8.5, H-p), 4.21 (br s, 1H, H-5), 3.14 (dd, 2H, 6.5, 8.6, H-7, H-8), 2.96–2.85 (m, 2H, H-3a, H-6a), 2.80 (t, 2H, 8.6, H-7, H-8), 2.42–2.34 (m, 2H, 6.5, H-1, H-3), 1.71 (dt, 1H, 12.2, 5.1, H-2), 1.55–1.44 (m, 2H, 6.5, 13.1, H-4, H-6), 1.03–0.95 (m, 2H, H-4, H-6), 0,66 (q, 1H, 12.2, H-2), 13C-NMR (CDCl3, δ ppm): 144.42 (6C-q), 128.70 (12C, C-o), 127.65 (12C, C-m), 126.79 (6C, C-p), 86.22 (2C-q), 75.10 (C-5), 64.26 (2C, C-7, C-8), 42.34 (2C, C-3a, C-6a), 42.14 (2C, C-1, C-3), 35.91 (2C, C-4, C-6), 30.87 (C-2), elem. anal. calcd for C48H46O3: th. C: 85.93, H: 6.91, found: C: 85.10 (−0.83, due to traces of solvent, not determined), H: 6.79.
3.12. PDC Oxidation of Alcoholdibenzoate 2a
To alcohol 2a (1.2 g, 2.84 mmol) dissolved in dichloromethane (60 mL), PDC (1.65 g, 4.26 mM) and molecular sieve (2 g) were added and stirred overnight, monitoring the reaction by TLC (I, Rf2a = 0.40, Rf5a = 0.66). The reaction mixture was diluted with ethyl ether, filtered of a anh. MgSO4 bed, the bed was washed with ether, the filtrate was concentrated and the crude product was purified by LPC (hexanes–ethyl acetate, 3:1 or benzene–ethyl acetate–hexane 5:3:2) and the pure ketone, (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,6a-5-oxooctahydropentalene-1,3-diyl)bis(methylene) dibenzoate (5) was obtained in more than 90% yield as an oil, which crystallized in time, m.p. 79.5–81.5 °C (ethyl ether–hexane), IR (1% in CHCl3): 3030, 2910–1880, 1700–1690, 1595, 1580, 1450, 1250–1200, 1100, 950 cm−1, 1H-NMR (CDCl3, δ ppm, J Hz): 8.00 (d, 4H, 7.1, H-o), 7.57 (t, 2H, 7.1, H-p), 7.44 (d, 4H, 7.1, H-m), 4.39 (dd, 2H, 11.4, 6.3, H-7, H-8), 4.30 (dd, 2H, 11.4, 7.6, H-7, H-8), 3.10–3.03 (m, 2H, H-1, H-3), 2.38 (dd, 2H, 18.6, 6.2, H-4, H-6), 2.30 (dd, 2H, 18.6, 6.5, H-4, H-6), 2. 08 (dt, 1H, 12.3, 6.5, H-2), 1.46 (q, 1H, 12.3, H-2), 13C-NMR (CDCl3, δ ppm): 218.50 (C-5), 166.26 (COO), 133.01 (C-p), 129.87 (C-q), 129.42 (C-o), 128.36 (C-m), 65.25 (2C, C-7, C-8), 41.09 (2C, C-3a, C-6a), 40.29 (2C, C-1, C-3), 38.71 (2C, C-4, C-6), 30.47 (C-2), elem. anal. calcd. for C24H24O5, C: 73.45, H: 6.16, found: C: 73.49, H: 6.22.
3.13. PDC Oxidation of Alcoholdibenzoate 3a
Dibenzoate alcohol 3a (0.81 g, 2.84 mmol) in dichloromethane (50 mL), was oxidized as before with PDC (1.20 g, 3.19 mmol) and molecular sieve (1.35 g), overnight. TLC (I, Rf3a = 0.50, Rf6a = 0.68) showed still the presence of 3a and more PDC (0.35 g) were added and stirring was continued for 3 days. After work-up and purification as above, 0.67 g (83.3%) of pure ketone, (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,6a-4-oxooctahydropentalene-1,3-diyl)bis(methylene) dibenzoate (6) were obtained as an oil, which crystallized overnight in the refrigerator, m.p. 61–63 °C, IR (1% CHCl3), practically the same as those of ketone 5: 3030, 2910–2880, 1700–1690, 1595, 1580, 1450, 1250–1200, 1100, 1060, 950 cm−1, 1H-NMR (CDCl3, δ ppm, J Hz): 8.05 (dd, 2H, 8.3, 1.4, H-o), 7.98 (dd, 2H, 8.3, 1.4, H-o), 7.58 (tt, 1H, 8.3, 1.4, H-p), 7.55 (tt, 1H, 8.3, 1.4, H-p), 7.45 (t, 2H, 8.3, H-m), 7.43 (t, 2H, 8.3, H-m), 4.55 (dd, 1H, 11.4, 3.6, H-7 or H-8), 4.48 (dd, 1H, 6.9, 11.1, H-8 or H-7), 4.44 (dd, 1H, 11.1, 6.9, H-7 or H-8), 4.43 (dd, 1H, 11.4, 7.9, H-8 or H-7), 3.01 (m, 1H, H-1), 2.83 (t, 1H, 10.8, H-3a), 2.78 (m, 1H, H-3), 2.65 (m, 1H, H-6a), 2.20–2.28 (m, 2H, H-5), 2.00–2.15 (m, 2H, H-2, H-6), 1.80 (dq, 1H, 13.0, 10.2, H-6), 1.62 (q, 1H, 12.5, H-2), 13C-NMR (CDCl3, δ ppm): 219.83 (C-6), 166.30, 166.05 (COO), 132.95, 132.84 (C-p), 129.98, 129.93 (C-q), 129.40, 129.22 (C-o), 128.31, 128.28 (C-m), 64.81 (C-8 or C-7), 63.73 (C-7 or C-8), 51.90 (C-3a), 43.83 (C-1), 41.29, 41.20 (2C, C-3, C-6a), 39.66 (C-5), 30.76 (C-2), 22.16 (C-6), elem. anal. calcd for C24H24O5, C: 73.45, H: 6.16, found: C: 73.90, H: 6.24.
3.14. PDC Oxidation of Alcohol 2c
The compound 2c (3.42 g, 5.1 mmol) was oxidized (as in 3.12) [CH2Cl2 (100 mL), PDC (3 g), molecular sieves (3.8 g), r.t., 2 days]; TLC (II, Rf2c = 0.32, Rf5c = 0.57); 3.31 g (97%) of pure 5c were obtained. By recrystallization from ethyl ether, 2.35 g of crystallized (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,6a-1,3-bis((trityloxy)methyl)hexahydropentalen-5-one (5) were obtained, m.p. 189–191 °C, IR (KBr): 3020, 2910, 2860, 1730, 1590, 1480, 1445, 1150, 1060, 750, 700 cm−1, 1H-NMR (CDCl3, δ ppm, J Hz): 7.40 (dd, 12H, 7.9, 1.2, H-o), 7.28 (t, 12H, 7.9, H-m), 7.23 (tt, 6H, 7.9, 1.2, H-p), 3.13 (dd, 2H, 8.8, 5.8, H-7, H-8), 3.04–2.95 (m, 2H, H-3a, H-6a), 2.80 (t, 2H, 8.8, H-7, H-8), 2.57–2.45 (m, 2H, H-1, H-3), 2.06 (dd, 2H, 19.1, 9.5, H-4, H-6), 1.87 (dd, 2H, 19.1, 5.9, H-4, H-6), 1.82 (dt, 1H, 12.4, 6.2, H-2), 1.00 (q, 1H, 12.4, H-2), 13C-NMR (CDCl3, δ ppm): 220.42 (C-5), 144.05 (6 Cq, Tr), 128.53 (12 C-m), 127.72 (12 C-o), 126.93 (6 C-p), 86.40 (2Cq, Tr), 64.30 (C-7, C-8), 41.75 (2C, C-3a, C-6a or C-1, C-3), 40.88 (2C, C-1, C-3 or C-3a, C-6a), 38.76 (2C, C-4, C-6), 30.89 (C-2).
3.15. Synthesis of (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,6,6a-Hexahydro-1H-spiro[pentalene-4,4′-[1,3]dioxolane]-1,3-diyl)bis(methylene)dibenzoate (11) from Ketone 5a
The symmetric ketone 5a (0.93g, 2.37 mmol) was dissolved in benzene (25 mL), ethylene glycol (0.4 mL) and TsOH (10 mg) were added, and refluxed for 5 h (Dean-Stark trap for removing the water) monitoring the reaction by TLC (VI, Rf5a = 0.69, Rf11 = 0.77). The reaction mixture was cooled, washed with sat. sol. NaHCO3 (2 × 20 mL), brine (20 mL), concentrated, and ketone 11 was obtained in quantitative yield by crystallization from benzene-hexane; the product has m.p. 99–100 °C, IR (KBr): 3045, 2945, 2870, 1690, 1590, 1580, 1445, 1260, 1160, 1090, 1060, 1015, 960, 940, 870, 695 cm−1, 1H-NMR (CDCl3, δ ppm, J Hz): 8.04 (dd, 4H, 8.3, 1.2, H-o), 7.75 (tt, 2H, 7.2, 1.2, H-p), 7.44 (td, 4H, 8.3, 7.2, H-m), 4.40 (dd, 2H, 11.1, 6.8, H-7, H-8), 4.27 (dd, 2H, 11.1, 8.4, H-7, H-8), 3.92 (apparent t, 4H, O-CH2CH2-O), 2.90–2.83 (m, 2H, H-3a, H-6a), 2.55–2.46 (m, 2H, 6.8, 8.4, H-1, H-3), 1.91 (dt, 1H, 12.5, 5.3, H-2), 1.82 (dd, 2H, 12.7, 8.6, H4, H-6), 1.68 (dd, 2H, 12.7, 9.6, H-4, H-6), 1.32 (q, 1H, 12.5, H-2), 13C-NMR (CDCl3, δ ppm): 166.46 (COO), 132.93 (C-p), 130.54 (C-q), 129.60 (C-o), 128.40 (C-m), 117.86 (C-5), 65.54 (C-7, C-8), 64.91, 64.26 (OCH2CH2O), 41.14 (C-3a, C-6a or C-1, C-3), 41.03 (C-1, C-3 or C-3a, C-6a), 35.46 (C-4, C-6), 30.29 (C-2).
3.16. Synthesis of the Ethylene Ketal of the Unsymmetric Ketone 6a
The same reaction conditions: 6a (0.93 g, 2.37 mmol); TLC (VI, Rf5a = 0.69, Rf11 = 0.83). The crude product (0.73 g) was purified similarly, resulting 605 mg (81.2%) of pure starting ketone 6a (IR and NMR as for compound 6a).
3.17. Synthesis of Symmetrical Ketone 14 by Deprotection of the Benzoate Groups of 5a
To a soln. of bisbenzoate ketone 5a (0.4 g, 1.02 mmol) in methanol (30 mL), a solution of MeONa (0.065 g/mL) (1 mL) was added and stirred at r.t. for 3 h, monitoring the reaction by TLC (VII, Rf14 = 0.18). The base was neutralized with methanol-washed Dowex 50 × 4, the filtrate was concentrated and the crude product was purified by LPC (eluent, ethyl acetate–hexanes, 5:1), resulting a pure fraction of 165 mg (86.8%) of (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,6,6a-1,3-bis(hydroxymethyl)-hexahydropentalen-5-one (14) as an oil, 1H-NMR (DMSO-d6, δ ppm, J Hz): 3.55 (dd, 2H, 10.7, 5.7, H-7, H-8), 3.27 (dd, 2H, 10.7, 7.4, H-7, H-8), 2.81–2.70 (m, 2H, H-3a, H-6a), 2.22–2.12 (m, 2H, H-1, H-3), 2.15 (dd, 2H, 18.5, 5.9, H-4, H-6), 2.07 (dd, 2H, 18.5, 9.5, H-4, H-6), 1.66 (dt, 1H, 12.5, 5.0, H-2), 1.11 (q, 1H, 12.5, H-2), 13C-NMR (DMSO-d6, δ ppm): 220.02 (C=O), 61.90 (2C, C-7, C-8), 42.92 (2C, C-1, C-3), 41.02 (2C, C-3a, C-6a), 38.36 (2C, C-4, C-6), 30.07 (C-2).
3.18. Synthesis of (±)-(1α,3α,3aβ,6aβ)-1,2,3,3a,4,6,6a-Hexahydro-1H-spiro[pentalene-4,4'-[1,3]dioxolane]-1,3-diyl)dimethanol (13)
Ethylene ketal 11 (0.49 g, 1.12 mmol) was hydrolyzed as described above for compound 5a: TLC (VII, Rf11 = 0.81, Rf13 = 0.20); LPC (ethyl acetate-hexane, 5:1); 235 mg (91.2%) of pure compound 13 were obtained as an oil, 1-H-NMR (CDCl3, δ ppm, J Hz): 3.91 (m, 4H, OCH2CH2O), 3.59 (d, 4H, 7.4, H-7, H-8), 3.17 (br s, 2H, exchangeable with D2O, OH), 2.75–2.68 (m, 2H, H-3a, H-6a), 2.20–2.10 (m, 2H, H-1, H-3), 1.79 (dt, 1H, 12.4, 5.4, H-2), 1.72 (dd, 2H, 14.6, 8.5, H-4, H-6), 1.55 (dd, 2H, 14.6, 7.7, H-4, H-6), 1.05 (q, 1H, 12.4, H-2), 13C-NMR (CDCl3, δ ppm): 118.02 (C-5), 64.63, 63.95 (OCH2CH2O), 63.30 (2C, C-7, C-8), 44.32 (C-1, C-3), 40.73 (C-3a, 6a), 34.84 (2C, C-4, C-6), 30.02 (C-2).
3.19. Deprotection of Benzoate Groups of Compound 6a by Transesterification
The benzoate groups of the compound
6a (0.41 g, 1.04 mmol) were hydrolyzed by transesterification, as described above (TLC, VII, R
f12 = 0.24, less active to dinitrophenylhydrazine); to give 185 mg (quantitative yield) of (±)-(2
aα,2
a1α,4α,4
aα,6
aα)-4-(hydroxymethyl)octahydro-6
aH-pentaleno[1,6-
bc]furan-6
a-ol (
15) as a cyclic hemiketal. The compound was crystallized from ethyl acetate, m.p. 106–108 °C, IR: 3442, 3353, 3250, 2952, 2913, 2878, 2858, 1462, 1428, 1315, 1078, 1056, 1024, 981, 898, 790 cm
−1,
1H-NMR (DMSO-
d6,
δ ppm,
J Hz): 5.75 (s, 1H, OH-4, exchangeable with D
2O), 4.29 (t, 1H, 4.7, OH-8, exchangeable with D
2O,), 3.82 (dd, 1H, 8.5, 4.8, H-7), 3.53 (d, 1H, 8.5, H-7), 3.28 (d, 2H, 7.3, H-8), 2.61 (t, 1H, 8.6, H-3a), 2.45–2.60 (m, 2H, H-6a. H-3), 1.96 (m, 1H, 6.6, H-1), 1.86 (dd, 1H, 12.4, 6.5, H-5), 1.80 (m, 1H, 12.6, H-2), 1.54 (dt, 1H, 13.3, 6.5, H-6), 1.43 (dt, 1H, 13.3, 6.9, H-5), 1.25 (m, 1H, 12.4, 10.2, 6.0, H-6), 1.01 (td, 1H, 12.6, 8.9, H-2),
13C-NMR (DMSO-
d6,
δ ppm): 115.00 (C-4), 71.74 (C-7 ), 61.38 (C-8), 58.90 (C-3a), 47.16 (C-1), 44.14 (C-6a or C-3), 43.72 (C-3 or C-6a), 38.44 (C-5), 33.37 (C-2), 24.02 (C-6).
1H-NMR (CDCl
3,
δ ppm,
J ppm): 4.05 (dd, 1H, 8.8, 5.1, H-7), 3.75 (d, 1H, 8.8, H-7), 3.58 (d, 2H, 7.3, H-8), 2.86 (t, 1H, 9.0, H-3a), 2.77–2.66 (m, 2H, H-3, H-6a), 2.15 (m, 1H, H-1), 2.05 (dd, 1H, 6.3, 12.4, H-6), 1.93 (ddd, 1H, 12.6, 8.4, 6.1, H-2), 1.78 (td, 1H, 12.5, 6.8, H-5), 1.59 (dt, 1H, 12.5, 7.4, H-5), 1.43 (ddt, 1H, 11.7, 12.4, H-6), 1.20 (dt, 1H, 12.6, 9.6, H-2),
13C-NMR (CDCl
3,
δ ppm): 115.00 (C-5), 71.44 (C-7), 61.38 (C-8), 58.90 (C-3a), 47.16 (C-1), 44.14 (C-6a or C-3), 43.72 (C-3 or C-6a), 38.44 (C-5), 33.72 (C-2), 24.02 (C-6). The molecular structure was confirmed by X-ray crystallography (
Figure 7).

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}