Studies for Improving a Rat Model of Alzheimer’s Disease: Icv Administration of Well-Characterized β-Amyloid 1-42 Oligomers Induce Dysfunction in Spatial Memory


 ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Pilot Experiment with Icv Administration of AMCA-Labeled Aβ1-42 Oligomers
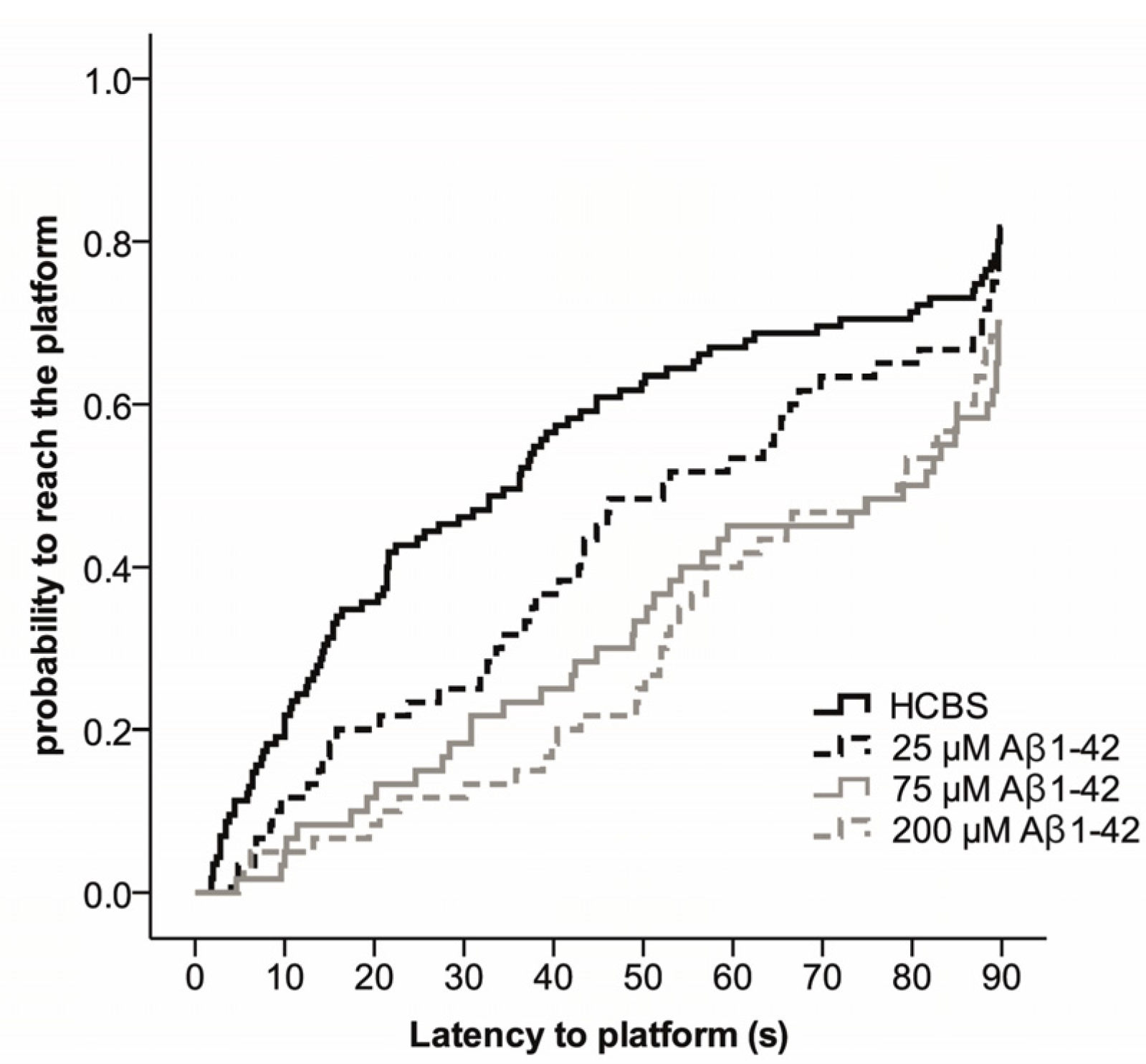
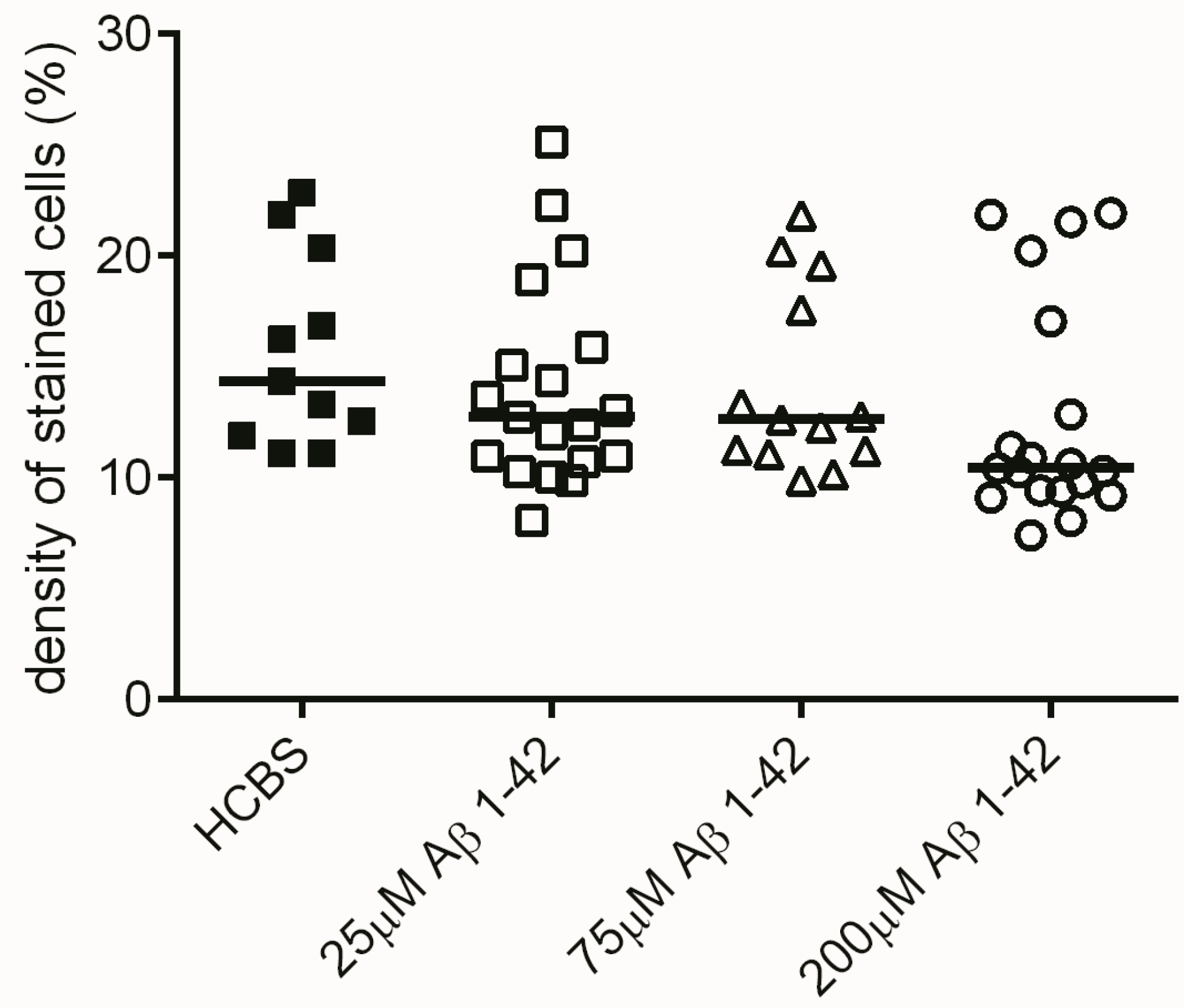
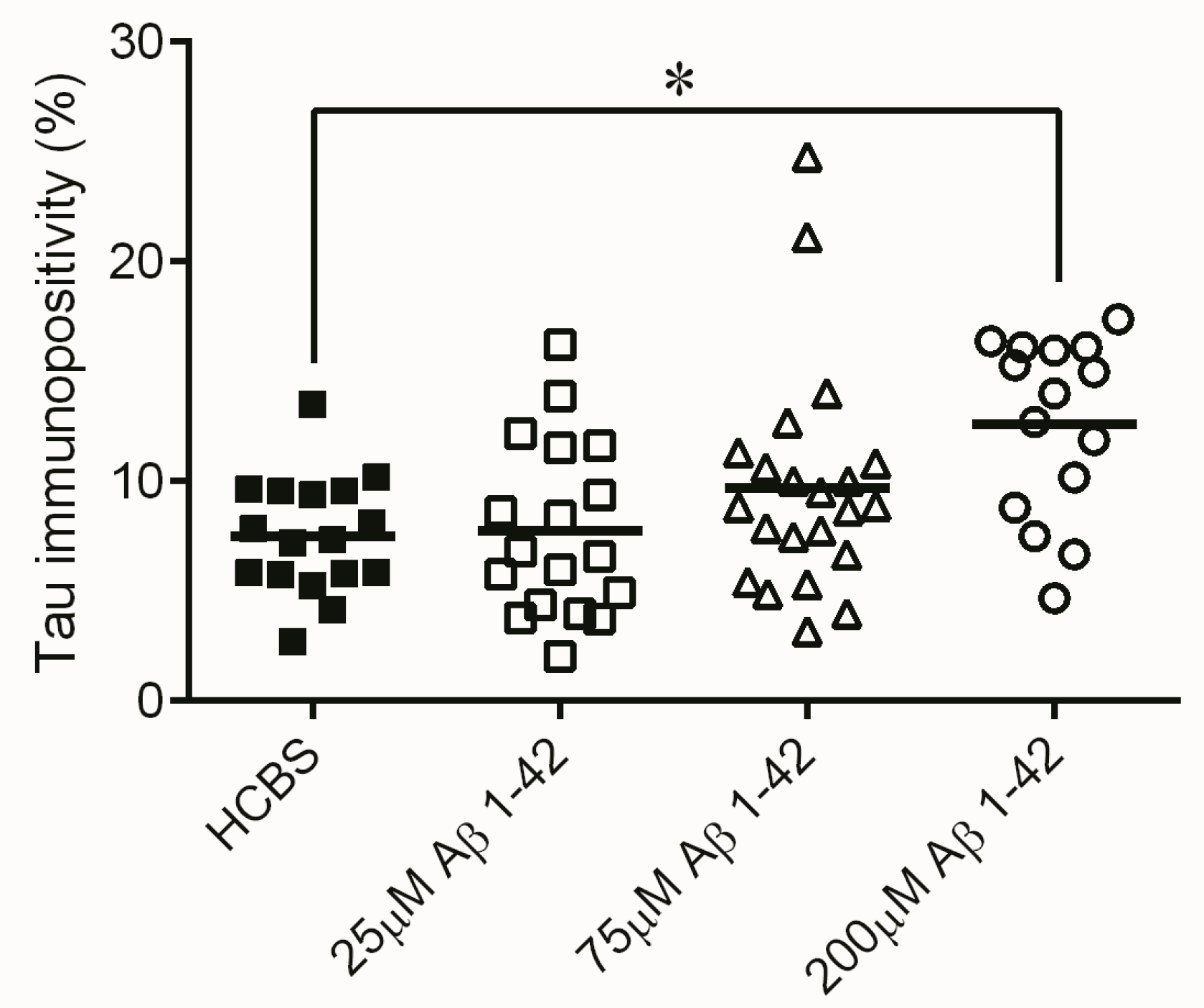
2.2. Studies on the Neurotoxic Effect of Two Different, Icv Administered Aβ1-42 Aggregates into Rat Brains
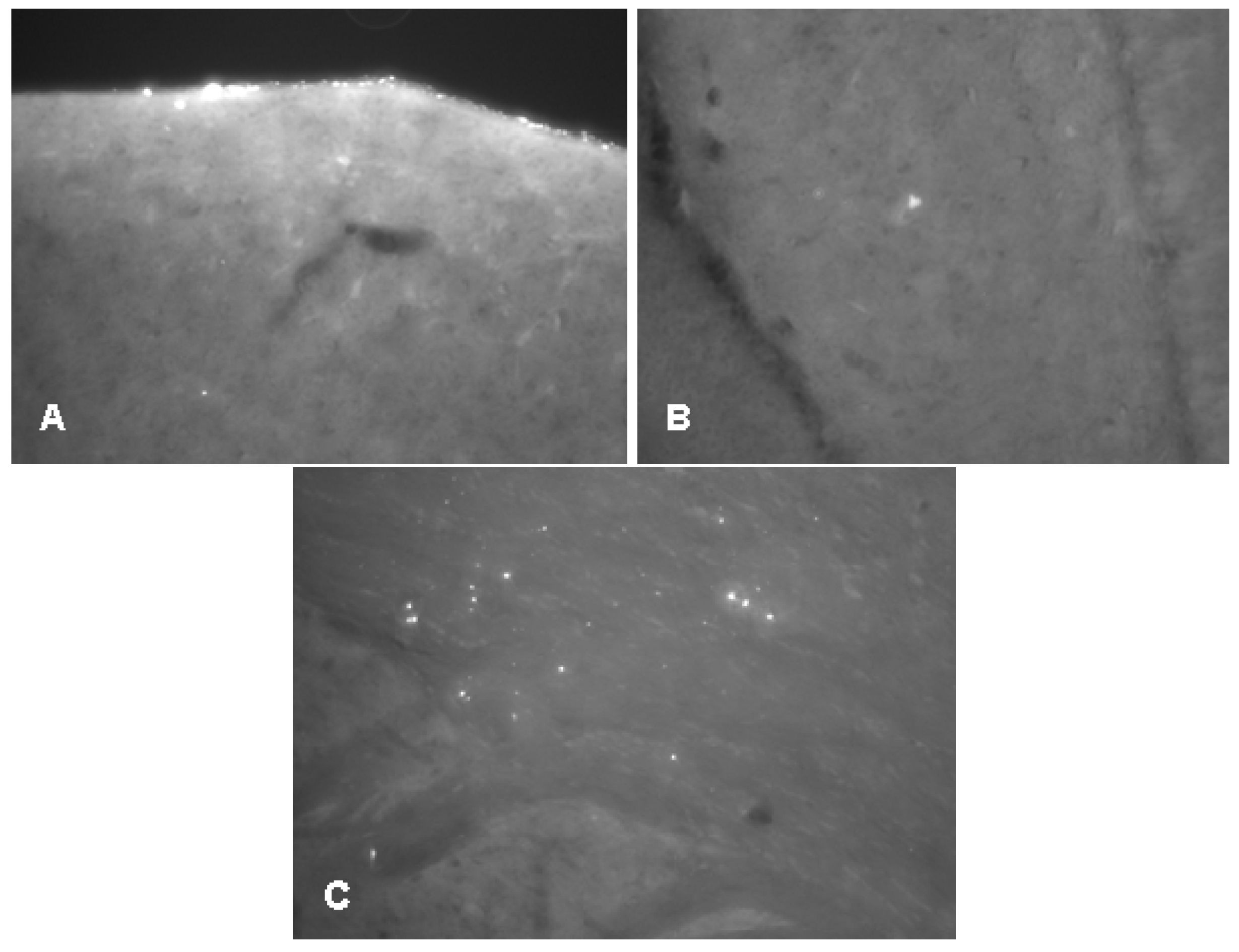
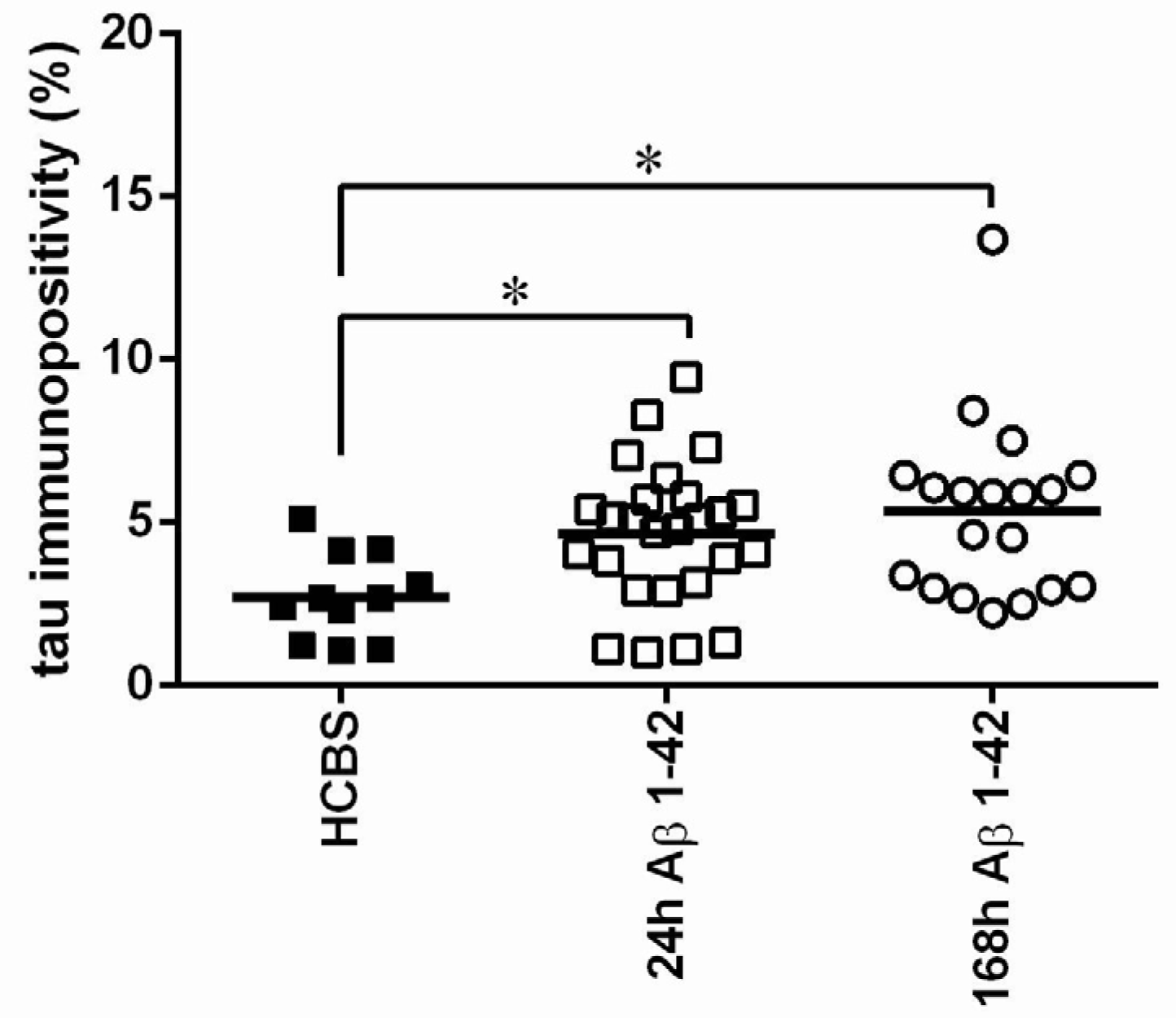
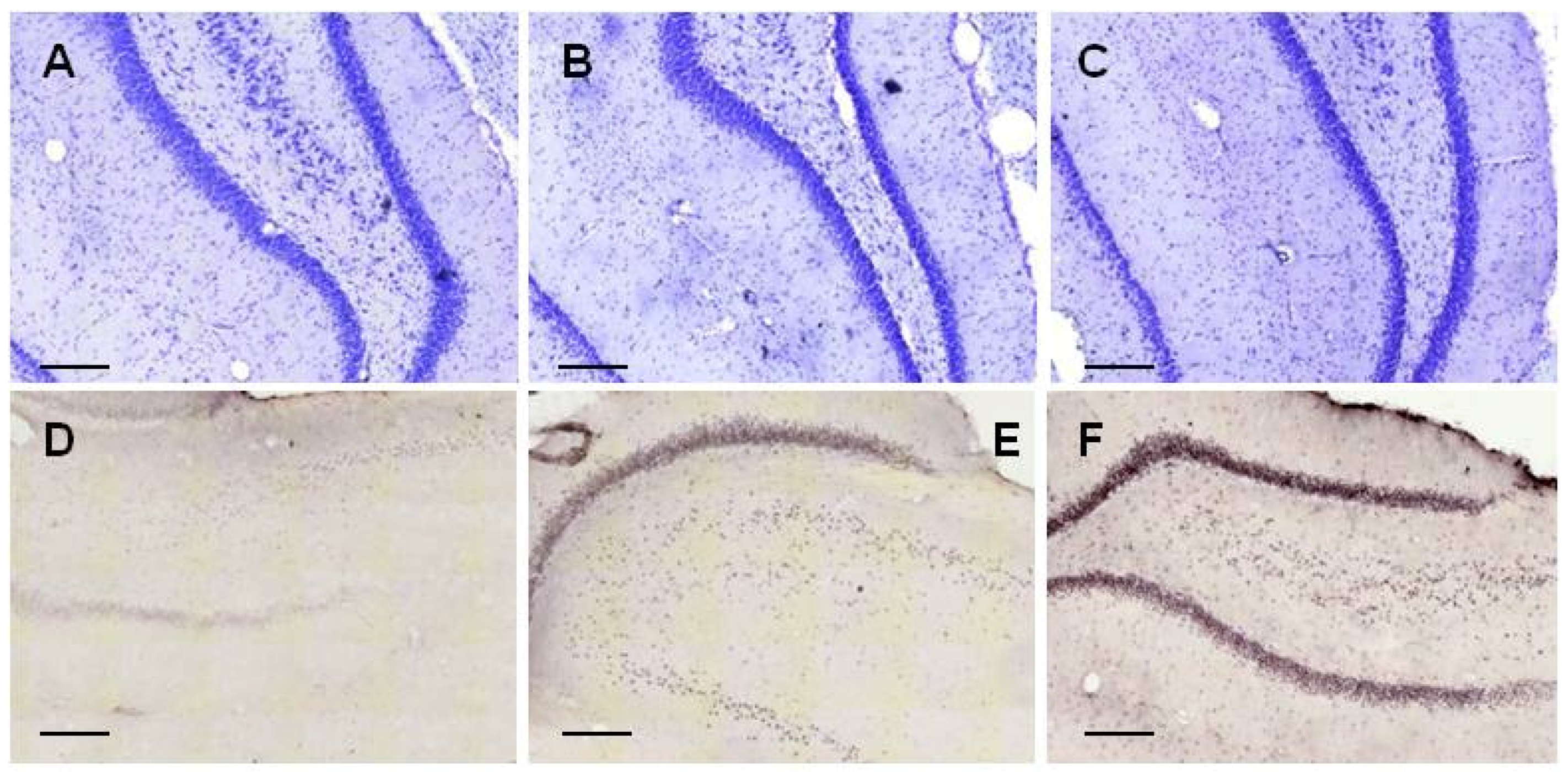
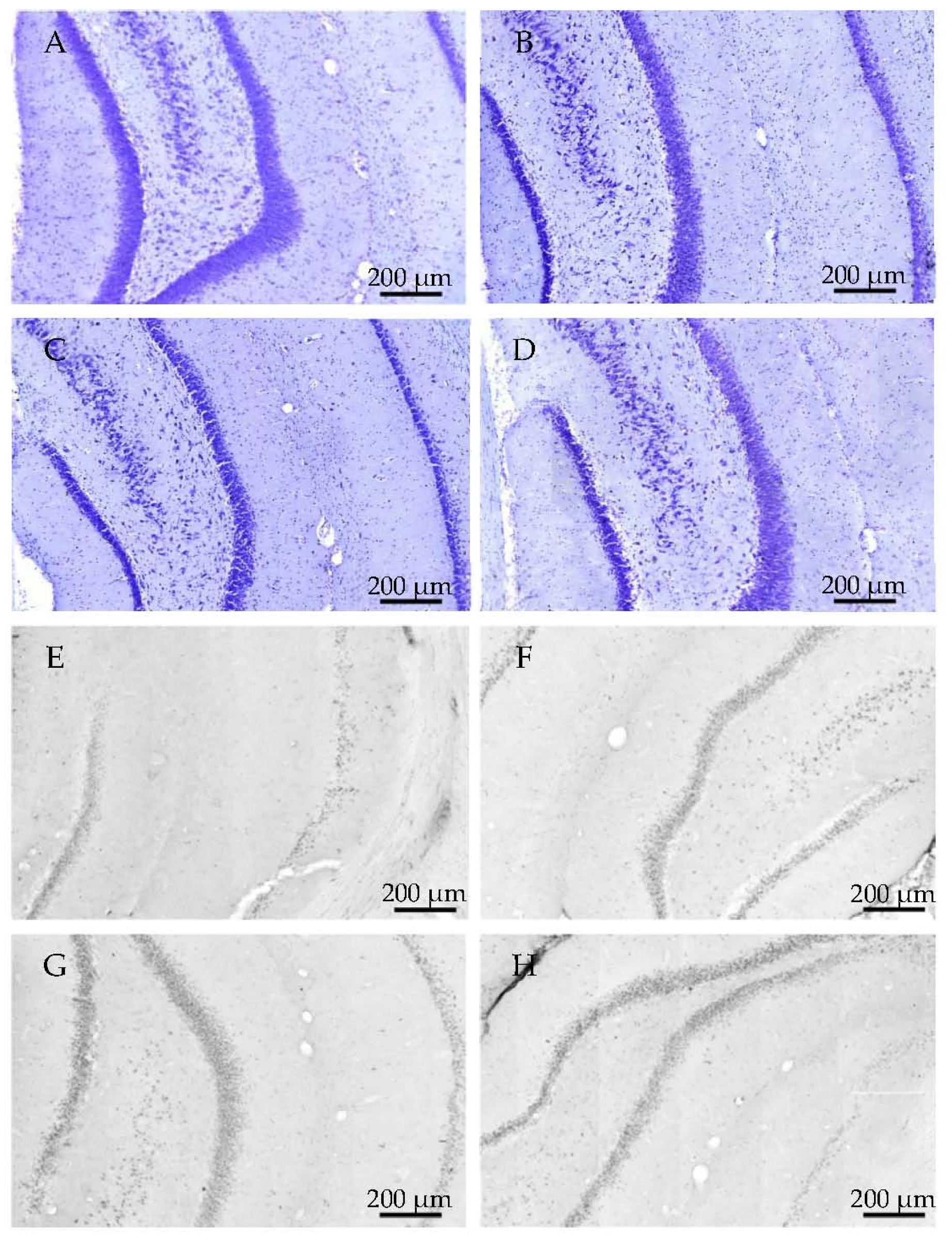
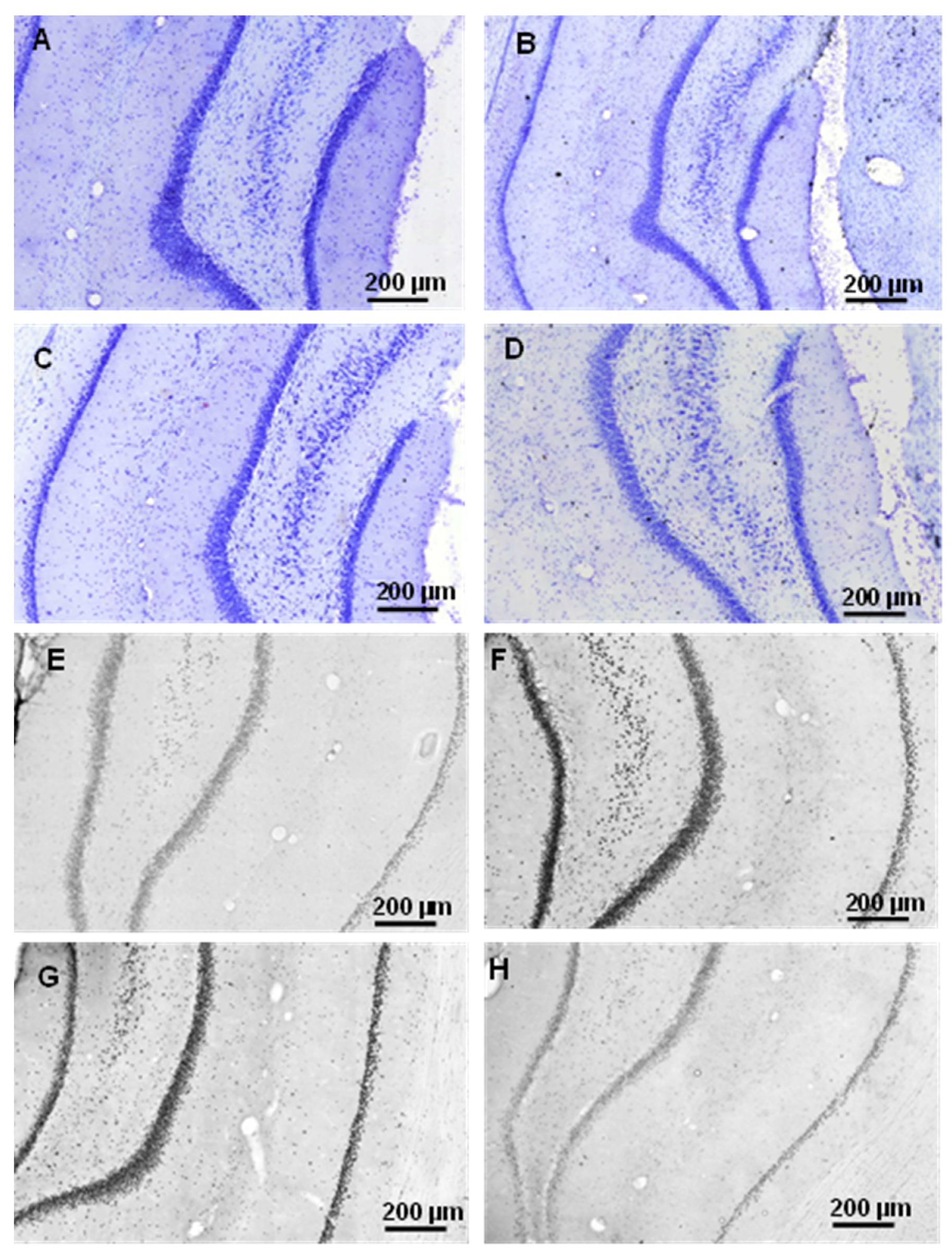
2.2.1. Histology
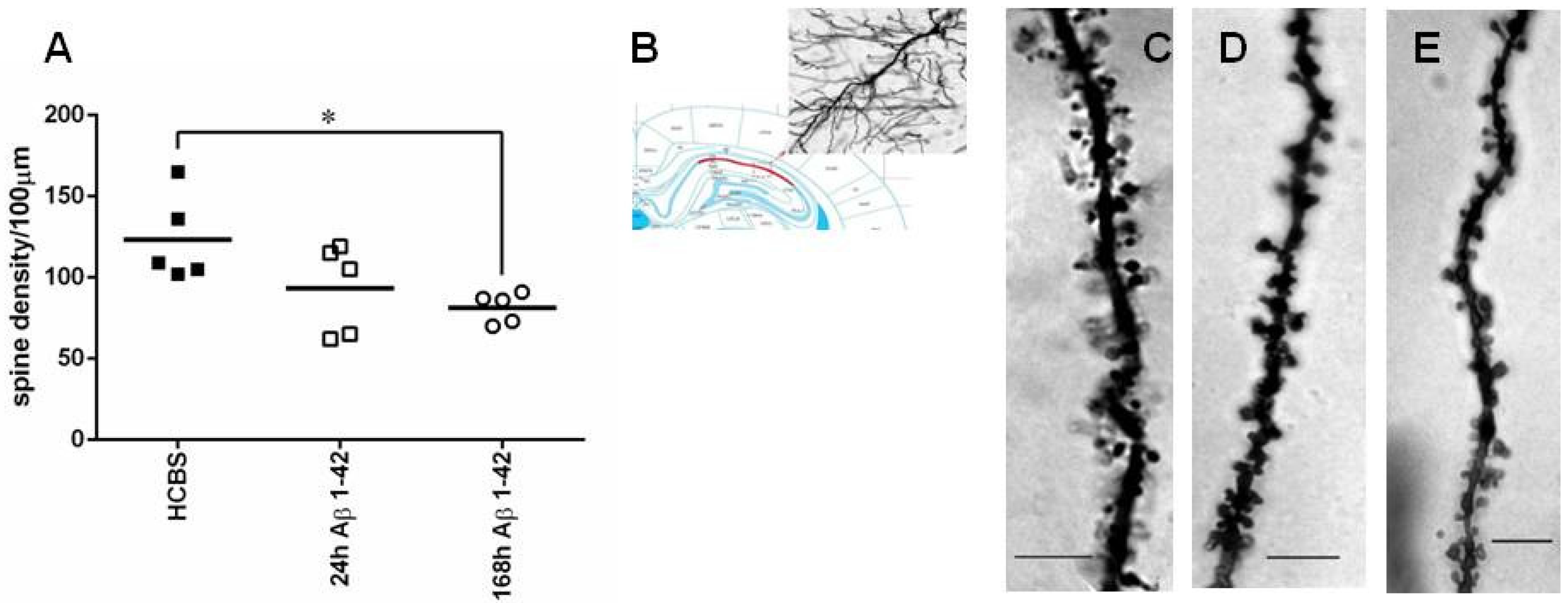
2.2.2. Studying the Change of Dendritic Spine Density Using Golgi-Cox Impregnation
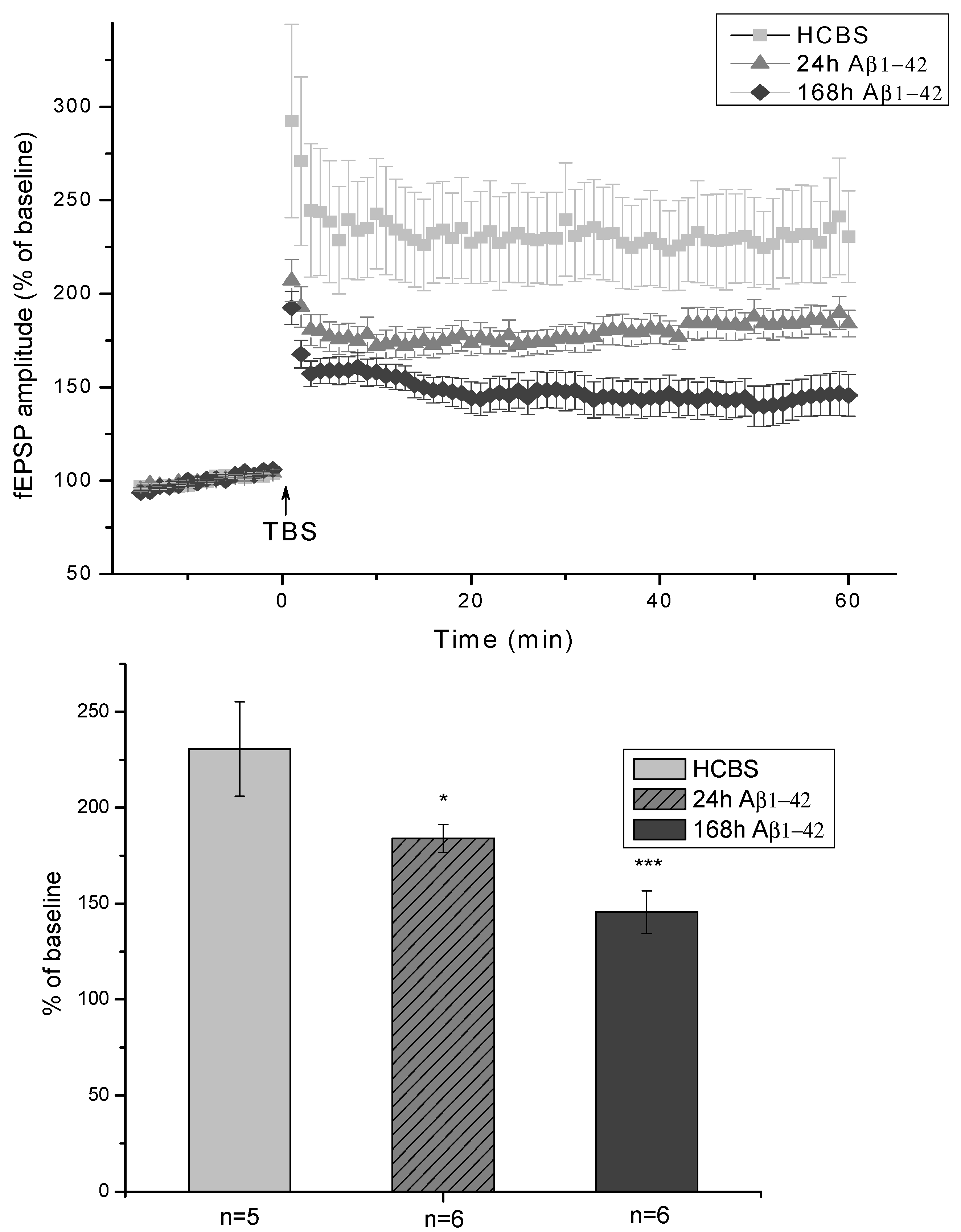
2.2.3. Electrophysiological Studies
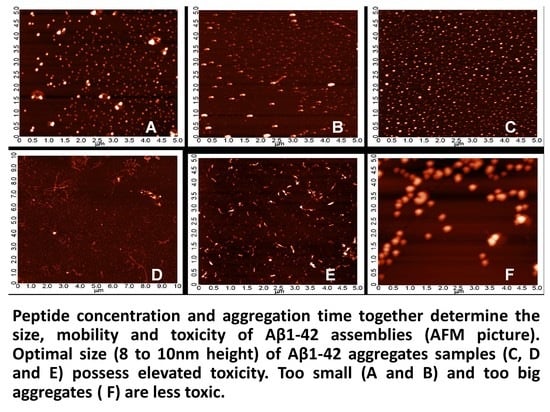
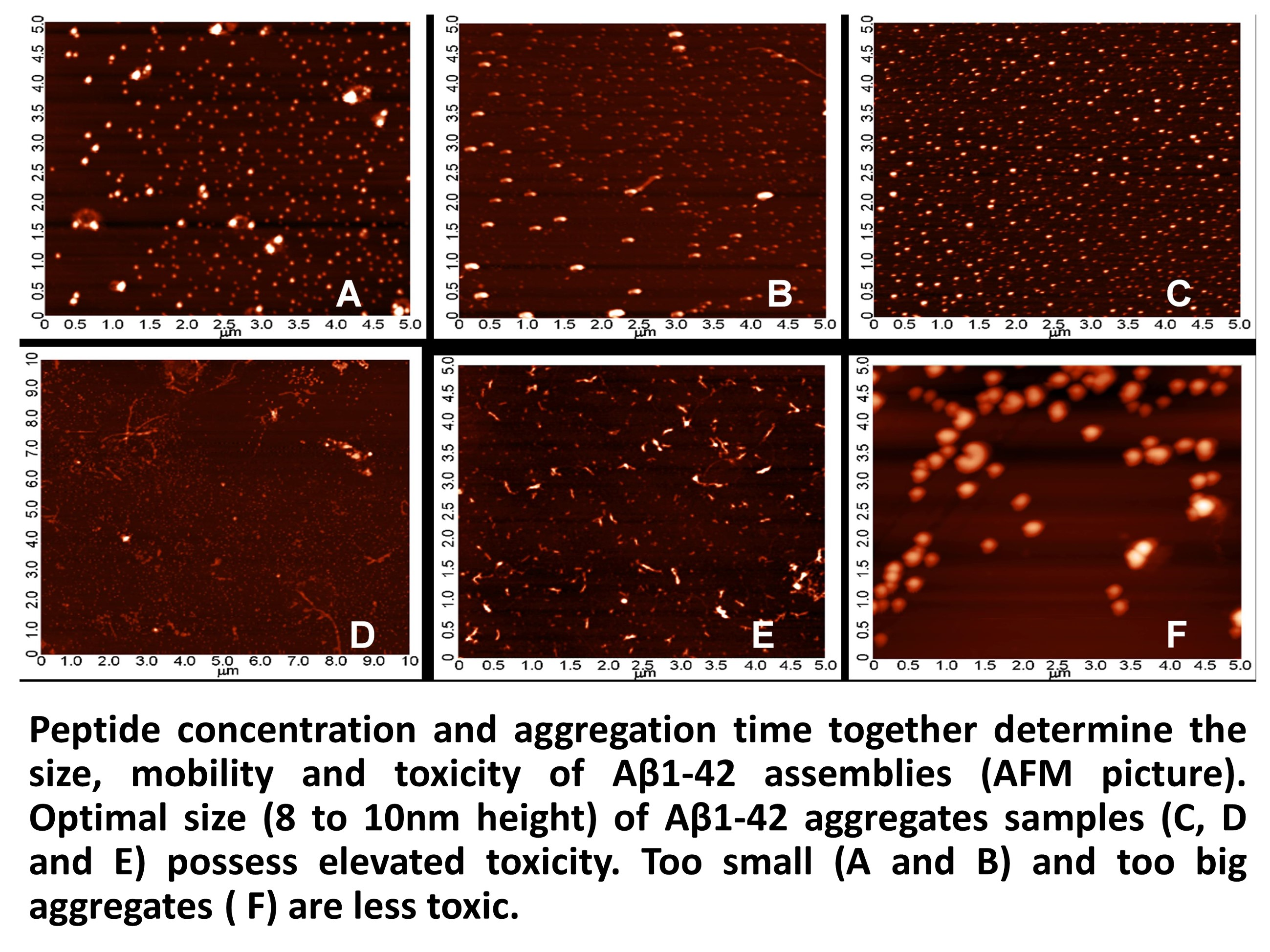
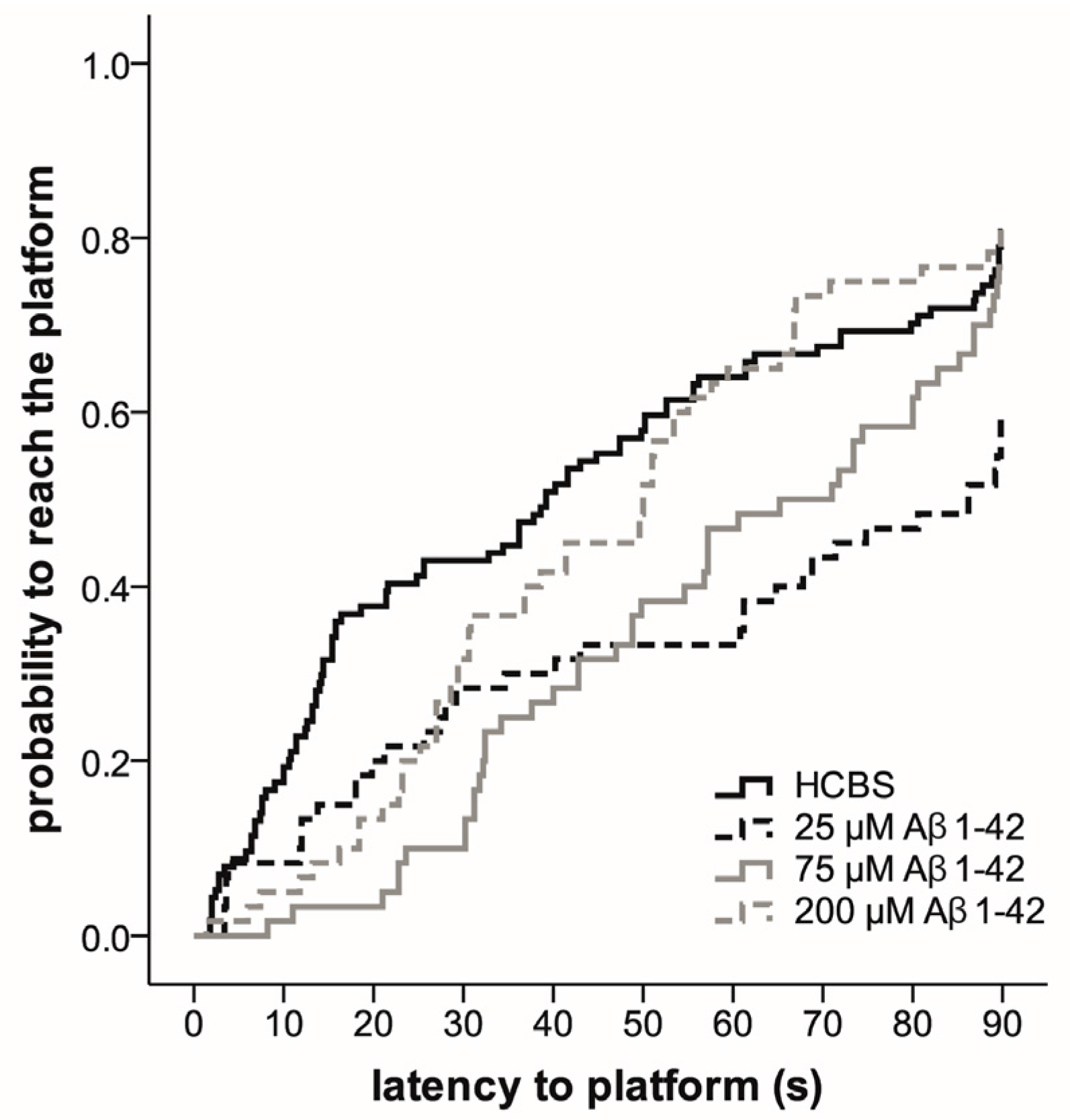
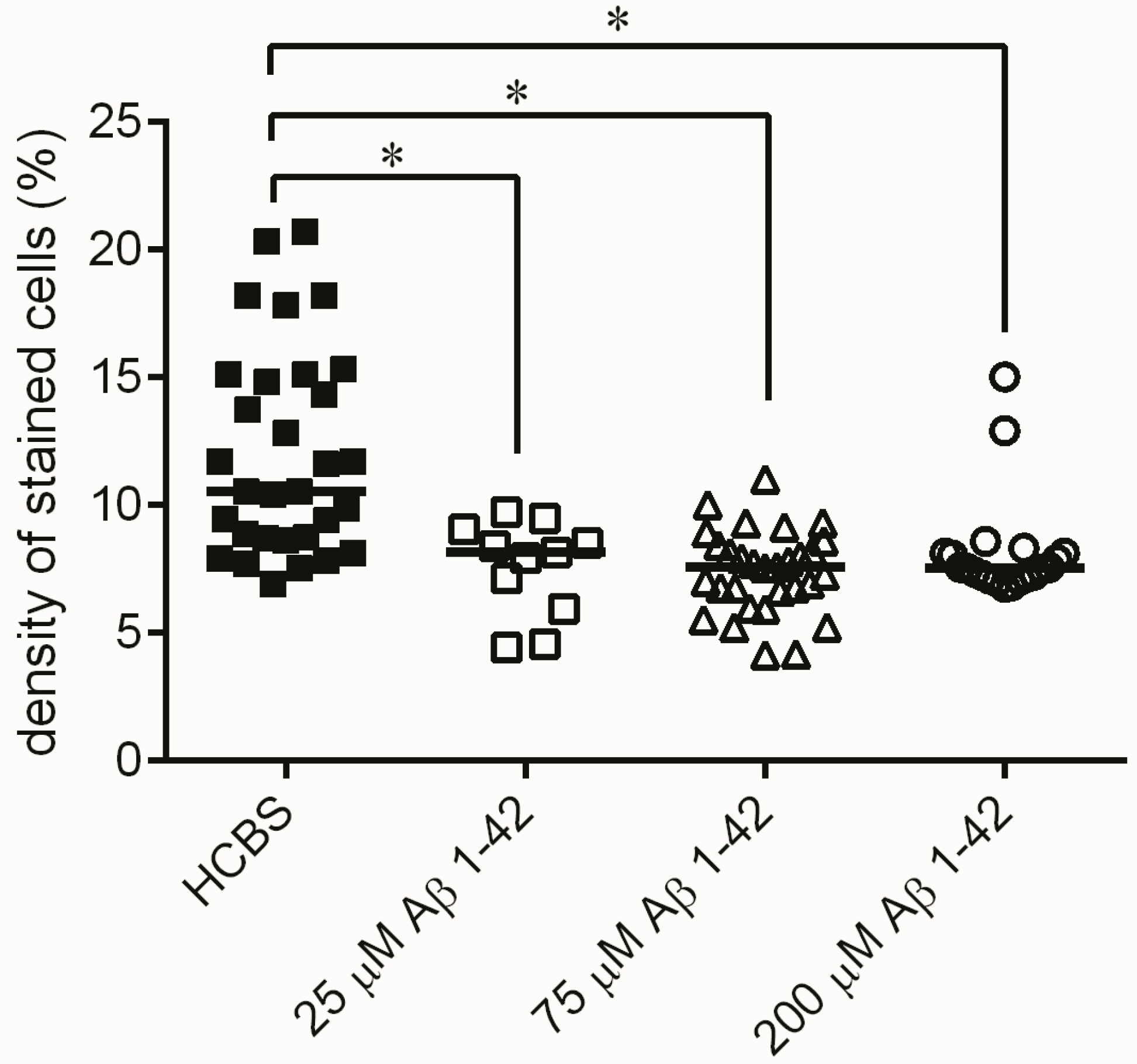
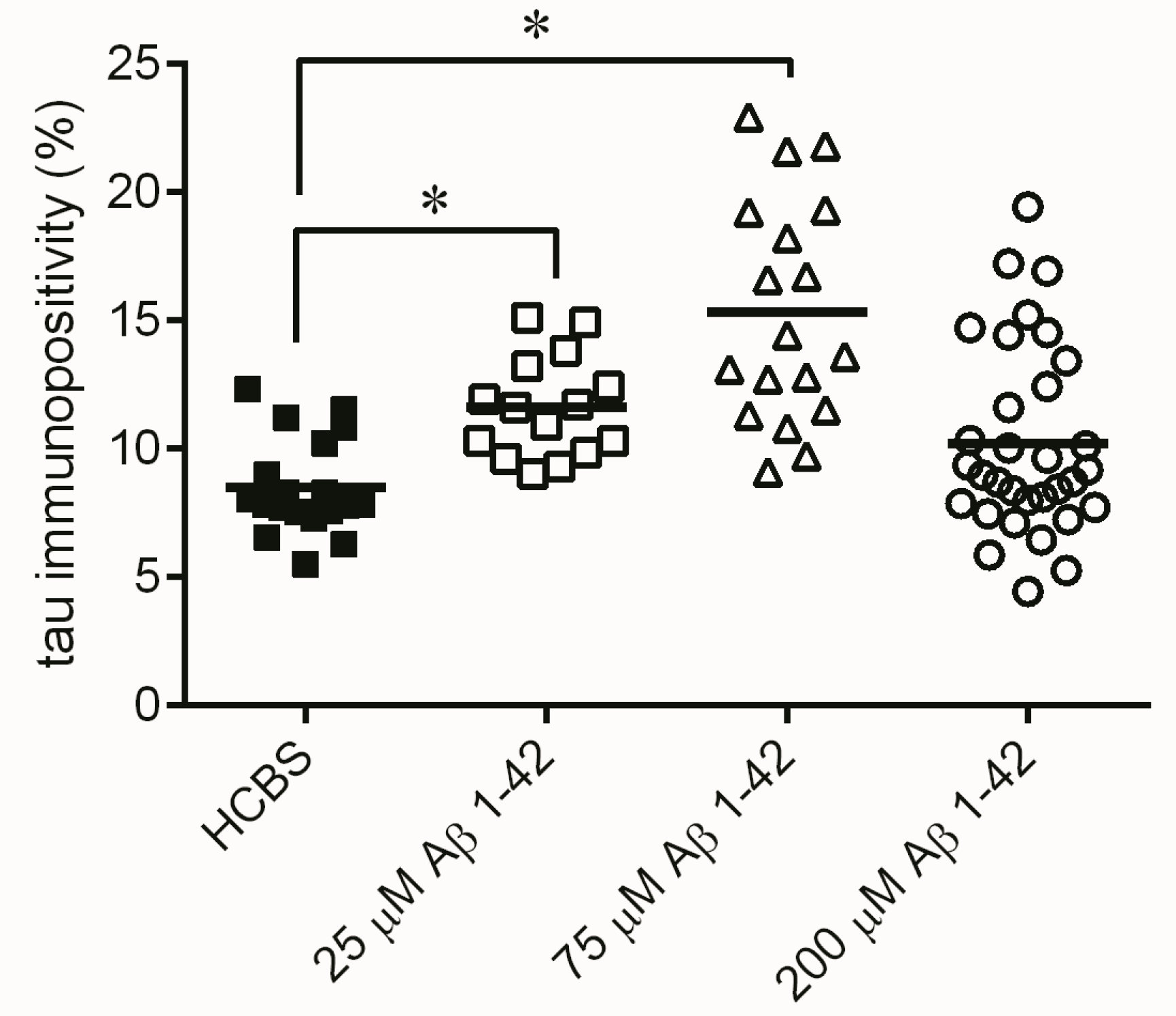
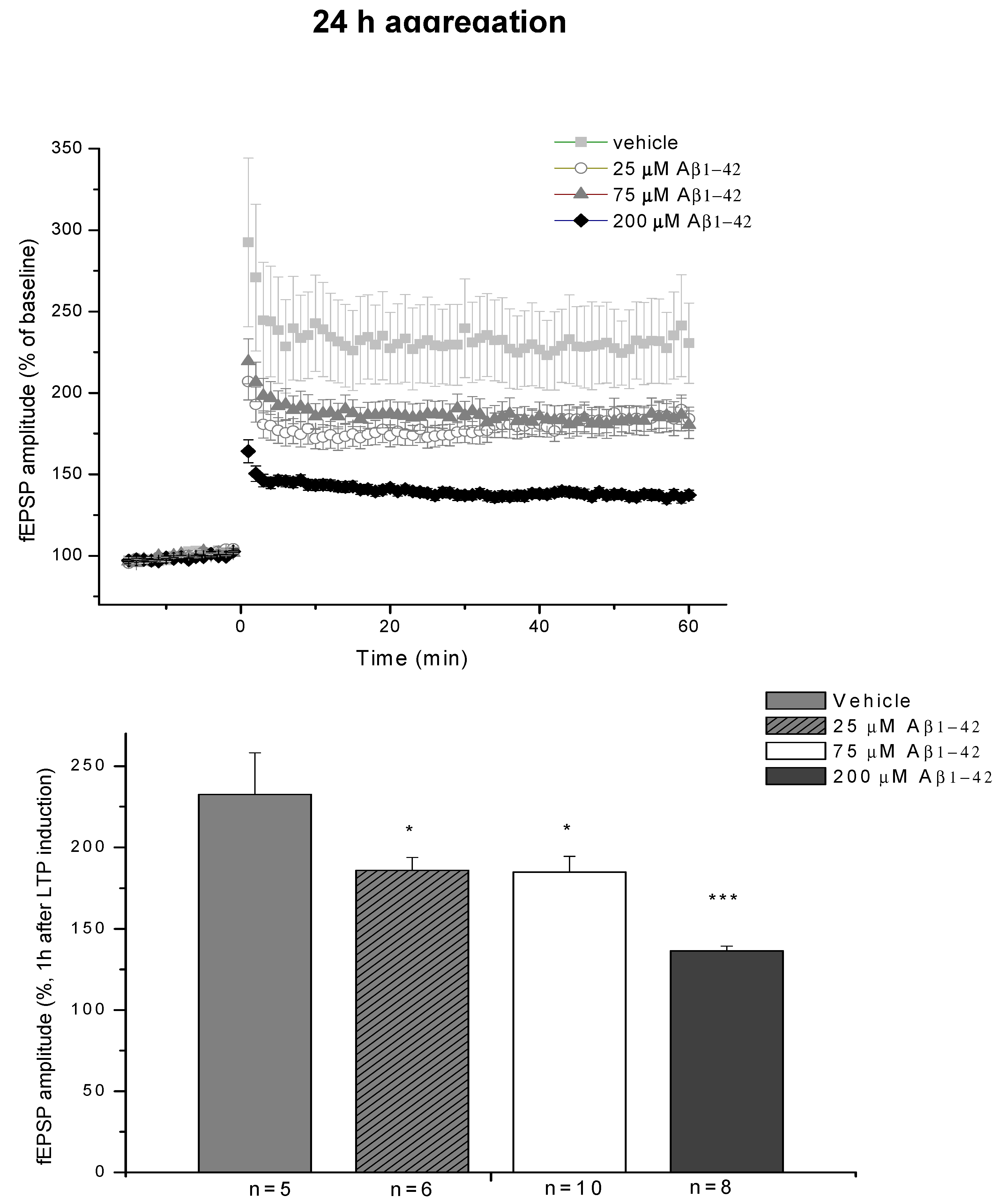
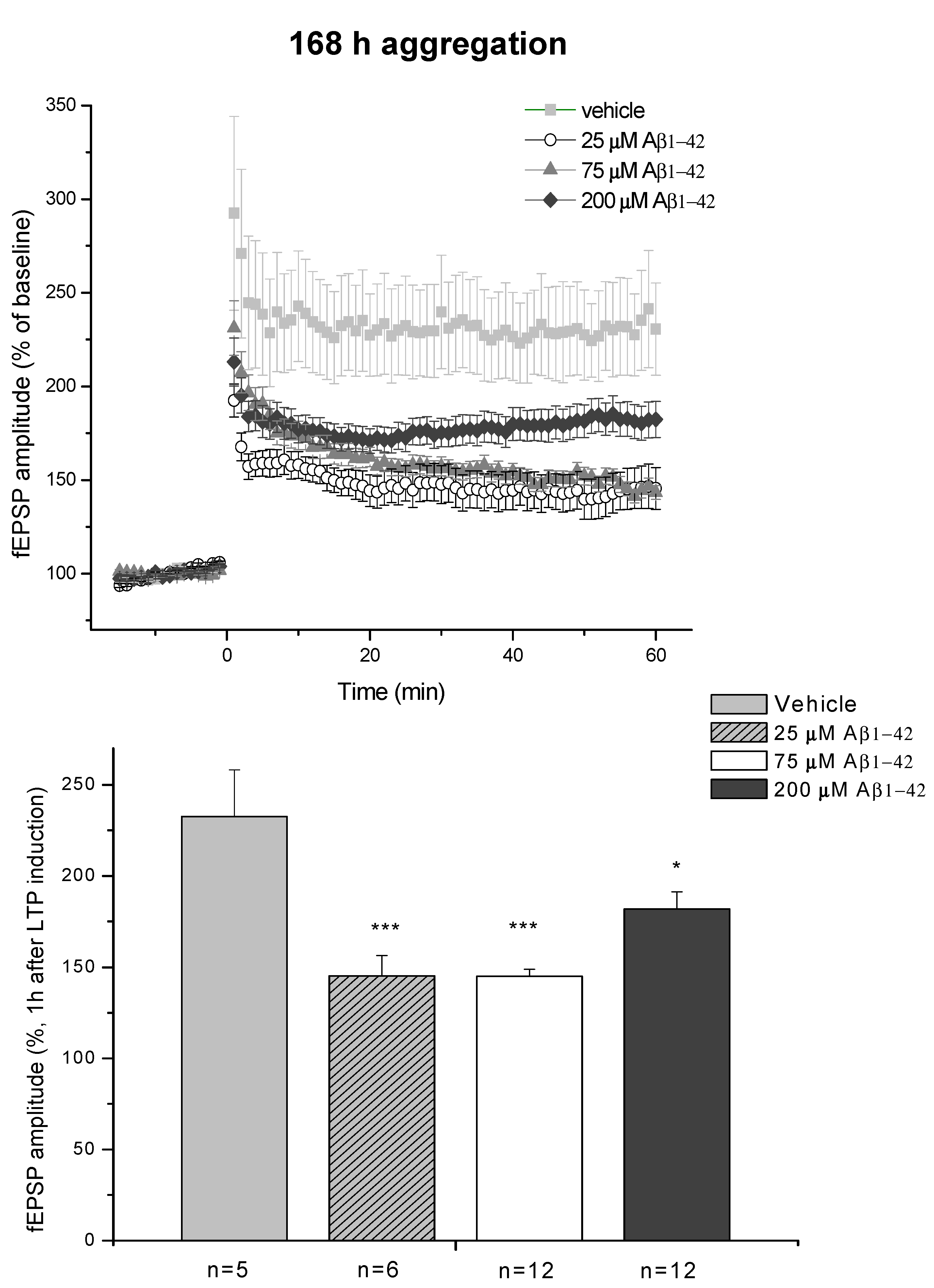
2.3. Systematic Studies for Finding the Most Toxic form of the Aβ1-42 Oligomers
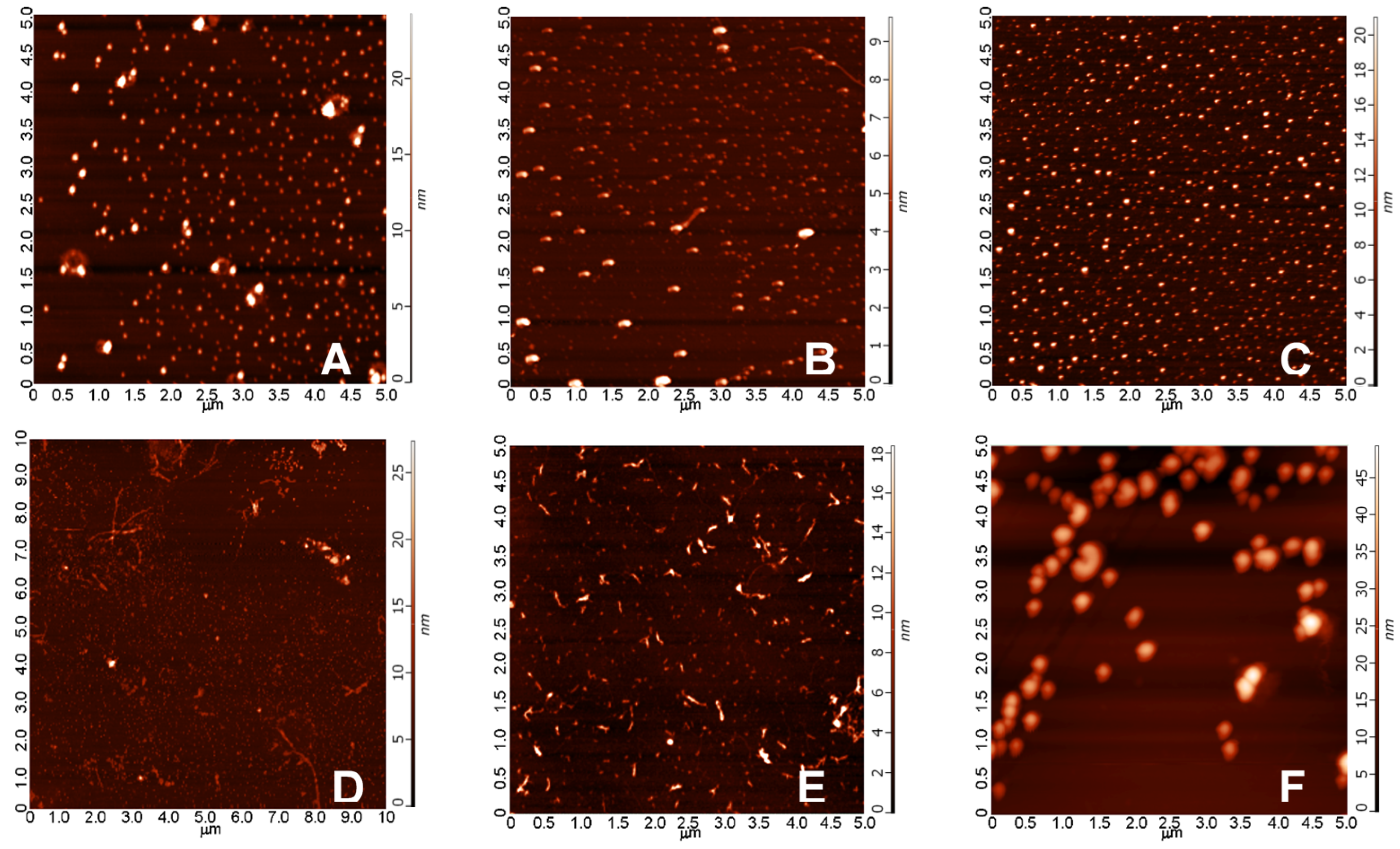
2.3.1. AFM Studies of the Effect of the Concentration and the Aggregation Time on the Size of Aβ1-42 Oligomers
2.3.2. Spatial Navigation in Morris Water Maze
2.3.3. Histology
2.3.4. Ex Vivo Electrophysiological Recordings with Multi-Electrode Array (MEA)
3. Discussion
- (1)
- Preparation of toxic Aβ1-42 oligomers from a precursor peptide (iso-Aβ1-42) and standardization of the method of the synthesis and characterization of oAβ1-42 assemblies.
- (2)
- Measurement of the effect of different oAβ1-42 samples on neuron viability, NFT-formation, dendritic spine density, synaptic plasticity and spatial behavior in nontransgenic rats.
- (3)
- Development of a novel rat model of AD using icv administration of well-characterized oAβ1-42 samples.
- (1)
- Mice are typically more variable in their behavior than rats; thus, we were able to use fewer animals for getting significant results in rats than in mice in MWM and other behavior experiments.
- (2)
- Rats are physiologically, genetically and morphologically closer to humans than mice [46]. Their larger body and brain size facilitate neurosurgical procedures, neuroimaging and in vivo electrophysiology.
- (3)
- Our research group also successfully used intranasal delivery of human Aβ 1-42 for rat brain targeting, but intranasal administration is complicated and very time-consuming.
- (4)
- The recent method is a short-term model: it is possible to get pathology and behavior changes within two weeks after the icv administration into rats.
- (5)
- Several laboratories have used short human Aβ fragments (e.g., Aβ25–35) in rat experiments; however, these kinds of short Aβ peptides are not natural metabolites of Aβ degradation, only aggregation-prone synthetic products.
- (6)
- Other laboratories (e.g., [49]) used a rat model of AD injecting fibrillary Aβ1-42 into the rat brain. In this model, the injected fibrillar Aβ aggregates form a deposit in the brain and a long time is necessary for destabilization and disintegration of the assemblies to diffusible toxic Aβ-oligomers. The use of icv administration of toxic oAβ1-42 samples is advantageous, and the optimal size of the Aβ1-42 assemblies is 8 to 10 nm height.
4. Materials and Methods
4.1. Preparation of Aβ1-42 Peptide Samples and Different Oligomeric Assemblies
4.1.1. Preparation of Different Aβ1-42 Oligomeric Assemblies
4.1.2. Atomic Force Microscopy Studies (AFM) of Aβ1-42 Assemblies
4.1.3. Fluorescent Microscopy
4.2. Treatment Groups
4.2.1. Studies on Two Different Aβ1-42 Oligomers (24 h and 168 h Aggregation Time, Concentration 25 µM)
4.2.2. Systematic Studies for Finding the Most Toxic Form among Six Different Aβ1-42 Oligomers
- A/ Aβ1-42, 24 h aggregation time, c = 25 µM; mean particle diameter 6.5 nm,
- B/ Aβ1-42, 24 h aggregation time, c = 75 µM; mean particle diameter 6.5 nm,
- C/ Aβ1-42, 24 h aggregation time, c = 200 µM; mean particle diameter 10.3 nm,
- D/ Aβ1-42, 168 h aggregation time, c = 25 µM; mean particle diameter 8.2 nm,
- E/ Aβ1-42, 168 h aggregation time, c = 75 µM; mean particle diameter 8.0 nm,
- F/ Aβ1-42, 168 h aggregation time, c = 200 µM; mean particle diameter 21.5 nm.
4.2.3. Surgery and Icv Administration of oAβ1-42
4.3. Spatial Navigation of Rats in a Morris Water Maze (MWM)
4.3.1. Experimental Animals and Housing
4.3.2. Morris-Water Maze Experiments
4.4. Histology
4.4.1. Cresyl Violet (Nissl) Staining
4.4.2. Tau-Immunohistology
4.5. Quantification of Dendritic Spine Density Using Golgi Impregnation
4.6. Ex Vivo Electrophysiological Studies
4.6.1. Stimulation Protocols
4.6.2. Multi-Electrode Array (MEA) Recordings
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACSF | Artificial cerebrospinal fluid |
| AD | Alzheimer’s disease |
| AFM | Atomic force microscopy |
| AMCA | 7 Amino-4-methylcoumarin-3-acetic acid |
| ANOVA | Analysis of variance |
| ApoE | Apolipoprotein E |
| APP | Amyloid precursor protein |
| Aβ | Beta-amyloid |
| CA | Cornu Ammonis |
| EOAD | Early-onset AD |
| ER | Endoplasmic reticulum |
| fAβ | Fibrillar form of Aβ |
| fEPSP | Field excitatory postsynaptic potential |
| HC | Hippocampus |
| HCBS | Hydrocarbonate buffered saline |
| icv | Intracerebroventricular |
| LOAD | Late-onset AD |
| LTP | Long term potentiation |
| MEA | Multi-electrode array |
| MWM | Morris water maze |
| NFTs | Neurofibrillary tangles |
| oAβ | Oligomeric form of Aβ |
| PBS | Phosphate-buffered saline solution |
| PCA-AD | Posterial cortical atrophy variant of AD |
| PSEN1, 2 | Presenilin-1 and 2 |
| r-AD | Rapidy progressive form of AD |
| t-AD | Typical prolonged-duration form of AD |
| TBS | Theta-burst stimulation |
References
- Halliday, M.; Mallucci, G.R. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology 2014, 7, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; Rao, R.V.; Mehlen, P. Cell death in the nervous system. Nature 2006, 443, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Lam, B.; Masellis, M.; Freedman, M.; Stuss, D.T.; Black, S.E. Clinical, imaging, and pathological heterogeneity of the Alzheimer’s disease syndrome. Alzheimers Res. Ther. 2013, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. (Elite Ed.) 2013, 5, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Bettens, K.; Sleegers, K.; Van Broeckhoven, C. Genetic insights in Alzheimer’s disease. Lancet Neurol. 2013, 12, 92–104. [Google Scholar] [CrossRef]
- Giri, M.; Zhang, M.; Lu, Y. Genes associated with Alzheimer’s disease: An overview and current status. Clin. Interv. Aging 2016, 11, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J. Neurochem. 2009, 110, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the etiology of Alzheimers-Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Cavallucci, V.; D’Amelio, M.; Cecconi, F. Abeta toxicity in Alzheimer’s disease. Mol. Neurobiol. 2012, 45, 366–378. [Google Scholar] [CrossRef] [PubMed]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of A-beta oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Upadhaya, A.R.; Capetillo-Zarate, E.; Kosterin, I.; Abramowski, D.; Kumar, S.; Yamaguchi, H.; Walter, J.; Fandrich, M.; Staufenbiel, M.; Thal, D.R. Dispersible amyloid beta-protein oligomers, protofibrils, and fibrils represent diffusible but not soluble aggregates: Their role in neurodegeneration in amyloid precursor protein (APP) transgenic mice. Neurobiol. Aging 2012, 33, 2641–2660. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.Y.; Teplow, D.B. Amyloid beta-protein oligomers and Alzheimer’s disease. Alzheimers Res. Ther. 2013, 5, 60–70. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Munoz, D.P. Opportunities and challenges in developing relevant animal models for Alzheimer’s disease. Ageing Res. Rev. 2016, 26, 112–114. [Google Scholar] [CrossRef] [PubMed]
- Casadesus, G. Ebook: Handbook of Animal Models in Alzheimer’s Disease, 1st ed.; IOS Press: Amsterdam, The Netherlands, 2011; pp. 1–352. ISBN 978-1-60750-733-8. [Google Scholar]
- Lecanu, L.; Papadopoulos, V. Modeling Alzheimer’s disease with non-transgenic rat models. Alzheimers Res. Ther. 2013, 5. [Google Scholar] [CrossRef]
- Sasaguri, H.; Nilsson, P.; Hashimoto, S.; Nagata, K.; Saito, T.; De Strooper, B.; Hardy, J.; Vassar, R.; Winblad, B.; Saido, T.C. App mouse models for Alzheimer’s disease preclinical studies. EMBO J. 2017, 36, 2473–2487. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic caenorhabditis-elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef] [PubMed]
- Link, C.D. C-elegans models of age-associated neurodegenerative diseases: Lessons from transgenic worm models of Alzheimer’s disease. Exp. Gerontol. 2006, 41, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Bouleau, S.; Tricoire, H. Drosophila models of Alzheimer’s disease: Advances, limits, and perspectives. J. Alzheimers Dis. 2015, 45, 1015–1038. [Google Scholar] [PubMed]
- Crowther, D.C.; Kinghorn, K.J.; Miranda, E.; Page, R.; Curry, J.A.; Duthie, F.A.I.; Gubb, D.C.; Lomas, D.A. Intraneuronal a beta, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer’s disease. Neuroscience 2005, 132, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Dodart, J.C.; May, P. Overview on rodent models of Alzheimer’s disease. Curr. Protoc. Neurosci. 2005. [Google Scholar] [CrossRef]
- Gotz, J.; Schonrock, N.; Vissel, B.; Ittner, L.M. Alzheimer’s disease selective vulnerability and modeling in transgenic mice. J. Alzheimers Dis. 2009, 18, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Puzzo, D.; Lee, L.; Palmeri, A.; Calabrese, G.; Arancio, O. Behavioral assays with mouse models of Alzheimer’s disease: Practical considerations and guidelines. Biochem. Pharmacol. 2014, 88, 450–467. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Bachstetter, A.D.; Nelson, P.T.; Schmitt, F.A.; Van Eldik, L.J. Using mice to model Alzheimer’s dementia: An overview of the clinical disease and the preclinical behavioral changes in 10 mouse models. Front. Genet. 2014, 5, 88. [Google Scholar] [CrossRef] [PubMed]
- Klohs, J.; Rudin, M.; Shimshek, D.R.; Beckmann, N. Imaging of cerebrovascular pathology in animal models of Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, J.S.O.; Castren, E. Mice with altered BDNF signaling as models for mood disorders and antidepressant effects. Front. Behav. Neurosci. 2014, 8, 143. [Google Scholar] [CrossRef] [PubMed]
- Onos, K.D.; Rizzo, S.J.S.; Howell, G.R.; Sasnera, M. Toward more predictive genetic mouse models of Alzheimer’s disease. Brain Res. Bull. 2016, 122, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Orta-Salazar, E.; Vargas-Rodríguez, I.; Castro-Chavira, S.A.; Feria-Velasco, A.I.; Díaz-Cintra, S. Alzheimer’s disease: From animal models to the human syndrome. In Update on Dementia; Moretti, D., Ed.; Intech: Rijeka, Croatia, 2016; ISBN 978-953-51-2654-6. [Google Scholar]
- Esquerda-Canals, G.; Montoliu-Gaya, L.; Guell-Bosch, J.; Villegas, S. Mouse models of Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Cavanaugh, S.E.; Pippin, J.J.; Barnard, N.D. Animal models of Alzheimer disease: Historical pitfalls and a path forward. ALTEX 2014, 31, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Filipcik, P.; Zilka, N.; Bugos, O.; Kucerak, J.; Koson, P.; Novak, P.; Novak, M. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol. Aging 2012, 33, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.M.; Rezai-Zadeh, K.; Weitz, T.M.; Rentsendorj, A.; Gate, D.; Spivak, I.; Bholat, Y.; Vasilevko, V.; Glabe, C.G.; Breunig, J.J.; et al. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J. Neurosci. 2013, 33, 6245–6256. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 2008, 192, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Walsh, D.M. Alzheimer’s disease: Synaptic dysfunction and A-beta. Mol. Neurodegener. 2009, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Beeg, M.; Stravalaci, M.; Bastone, A.; Sclip, A.; Biasini, E.; Tapella, L.; Colombo, L.; Manzoni, C.; Borsello, T.; et al. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Sato, N.; Niisato, K.; Takeuchi, D.; Kurinami, H.; Shinohara, M.; Rakugi, H.; Kano, M.; Morishita, R. Validation of a beta 1–40 administration into mouse cerebroventricles as an animal model for Alzheimer disease. Brain Res. 2009, 1280, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Cleary, J.P.; Mehta, T.; Hofmeister, J.; Lesne, S.; O’Hare, E.; Walsh, D.M.; Selkoe, D.J. Orally available compound prevents deficits in memory caused by the Alzheimer amyloid-beta oligomers. Ann. Neurol. 2006, 60, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Zussy, C.; Brureau, A.; Delair, B.; Marchal, S.; Keller, E.; Ixart, G.; Naert, G.; Meunier, J.; Chevallier, N.; Maurice, T.; et al. Time-course and regional analyses of the physiopathological changes induced after cerebral injection of an amyloid beta fragment in rats. Am. J. Pathol. 2011, 179, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, D.K.; Chung, B.R.; Kim, H.V.; Kim, Y. Intracerebroventricular injection of amyloid-beta peptides in normal mice to acutely induce Alzheimer-like cognitive deficits. J. Vis. Exp. 2016. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Jacob, H.J.; Kwitek, A.E. Rat genetics: Attaching physiology and pharmacology to the genome. Nat. Rev. Genet. 2002, 3, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Joghataei, M.T.; Mohseni, S.; Roghani, M. Genistein ameliorates learning and memory deficits in amyloid beta((1–40)) rat model of Alzheimer’s disease. Neurobiol. Learn. Mem. 2011, 95, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.Z.; Bayer, T.A.; Wirths, O. Formic acid is essential for immunohistochemical detection of aggregated intraneuronal a beta peptides in mouse models of Alzheimer’s disease. Brain Res. 2009, 1301, 116–125. [Google Scholar] [CrossRef] [PubMed]
- He, F.Q.; Qiu, B.Y.; Zhang, X.H.; Li, T.K.; Xie, Q.; Cui, D.J.; Huang, X.L.; Gan, H.T. Tetrandrine attenuates spatial memory impairment and hippocampal neuroinflammation via inhibiting NF-kappaB activation in a rat model of Alzheimer’s disease induced by amyloid-beta(1–42). Brain Res. 2011, 1384, 89–96. [Google Scholar] [CrossRef] [PubMed]
- O’Hare, E.; Weldon, D.T.; Mantyh, P.W.; Ghilardi, J.R.; Finke, M.P.; Kuskowski, M.A.; Maggio, J.E.; Shephard, R.A.; Cleary, J. Delayed behavioral effects following intrahippocampal injection of aggregated a beta(1–42). Brain Res. 1999, 815, 1–10. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.M.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Yang, N.; Liu, Y.Y.; Zheng, J.; Ji, C.; Zuo, P.P. M2 macrophage transplantation ameliorates cognitive dysfunction in amyloid-beta-treated rats through regulation of microglial polarization. J. Alzheimers Dis. 2016, 52, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Sipos, E.; Kurunczi, A.; Andras, F.; Penke, Z.; Fulop, L.; Kasza, A.; Janos, H.; Sandor, H.; Veszelka, S.; Balogh, G.; et al. Intranasal delivery of human beta-amyloid peptide in rats: Effective brain targeting. Cell. Mol. Neurobiol. 2010, 30, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Sipos, E.; Kurunczi, A.; Kasza, A.; Horvath, J.; Felszeghy, K.; Laroche, S.; Toldi, J.; Parducz, A.; Penke, B.; Penke, Z. Beta-amyloid pathology in the entorhinal cortex of rats induces memory deficits: Implications for Alzheimer’s disease. Neuroscience 2007, 147, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Aggarwal, A.; Singh, A.; Naidu, P.S. Animal models in drug discovery of Alzheimer’s disease: A mini review. EC Pharmacol. Toxicol. 2016, 2, 60–79. [Google Scholar]
- Salari, S.; Bagheri, M. A review of animal models of Alzheimer’s disease: A brief insight into pharmacologic and genetic models. Physiol. Pharmacol. 2016, 20, 5–11. [Google Scholar]
- Ruggeri, F.S.; Habchi, J.; Cerreta, A.; Dietler, G. AFM-based single molecule techniques: Unraveling the amyloid pathogenic species. Curr. Pharm. Des. 2016, 22, 3950–3970. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.H.; Liao, Y.H.; Plogg, B.A.; Peng, W.G.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates csf flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Lopez, M.; Mendez, M.; Sampedro-Piquero, P.; Arias, J.L. Spatial learning-related changes in metabolic activity of limbic structures at different posttask delays. J. Neurosci. Res. 2013, 91, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Morris, R. Developments of a water-maze procedure for studying spatial-learning in the rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Morris, R.G.M.; Anderson, E.; Lynch, G.S.; Baudry, M. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-d-aspartate receptor antagonist, AP5. Nature 1986, 319, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.G.M.; Garrud, P.; Rawlins, J.N.P.; Okeefe, J. Place navigation impaired in rats with hippocampal-lesions. Nature 1982, 297, 681–683. [Google Scholar] [CrossRef] [PubMed]
- Vorhees, C.V.; Williams, M.T. Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat. Protoc. 2006, 1, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Yau, W.M.; Lu, J.X.; Collinge, J.; Tycko, R. Structural variation in amyloid-beta fibrils from Alzheimer’s disease clinical subtypes. Nature 2017, 541, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Nguyen, L.N.; Kessels, H.W.; Hagiwara, H.; Sisodia, S.; Malinow, R. Amyloid beta from axons and dendrites reduces local spine number and plasticity. Nat. Neurosci. 2010, 13, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Chang, L.; Fernandez, S.J.; Gong, Y.S.; Viola, K.L.; Lambert, M.P.; Velasco, P.T.; Bigio, E.H.; Finch, C.E.; et al. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J. Neurosci. 2004, 24, 10191–10200. [Google Scholar] [CrossRef] [PubMed]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Klein, W.L. A beta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci. 2007, 27, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Chacon, M.A.; Barria, M.I.; Soto, C.; Inestrosa, N.C. Beta-sheet breaker peptide prevents a beta-induced spatial memory impairments with partial reduction of amyloid deposits. Mol. Psychiatry 2004, 9, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Borbely, E.; Horvath, J.; Furdan, S.; Bozso, Z.; Penke, B.; Fulop, L. Simultaneous changes of spatial memory and spine density after intrahippocampal administration of fibrillar Aβ(1–42) to the rat brain. Biomed. Res. Int. 2014, 2014, 345305. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, A.; Luheshi, L.M.; Sollvander, S.; de Barros, T.P.; Macao, B.; Knowles, T.P.J.; Biverstal, H.; Lendel, C.; Ekholm-Petterson, F.; Dubnovitsky, A.; et al. Stabilization of neurotoxic Alzheimer amyloid-beta oligomers by protein engineering. Proc. Natl. Acad. Sci. USA 2010, 107, 15595–15600. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, A.C.; Oliveira, C.R.; Pereira, C.F.; Cardoso, S.M. Loss of proteostasis induced by amyloid beta peptide in brain endothelial cells. Biochim. Biophys. Acta 2014, 1843, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Kopniczky, Z.; Dochnal, R.; Macsai, M.; Pal, A.; Kiss, G.; Mihaly, A.; Szabo, G. Alterations of behavior and spatial learning after unilateral entorhinal ablation of rats. Life Sci. 2006, 78, 2683–2688. [Google Scholar] [CrossRef] [PubMed]
- Avraamides, M.N.; Kelly, J.W. Multiple systems of spatial memory and action. Cogn. Process. 2008, 9, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Bozso, Z.; Penke, B.; Simon, D.; Laczko, I.; Juhasz, G.; Szegedi, V.; Kasza, A.; Soos, K.; Hetenyi, A.; Weber, E.; et al. Controlled in situ preparation of a beta(1–42) oligomers from the isopeptide “iso-a beta(1–42)”, physicochemical and biological characterization. Peptides 2010, 31, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Lavenex, P.; Amaral, D.G. Hippocampal-neocortical interaction: A hierarchy of associativity. Hippocampus 2000, 10, 420–430. [Google Scholar] [CrossRef]
- Fulop, L.; Penke, B.; Zarandi, M. Synthesis and fluorescent labeling of beta-amyloid peptides. J. Pept. Sci. 2001, 7, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: London, UK; Sydney, Australia, 2006; pp. 1–456. ISBN 0080475159. [Google Scholar]
- Nagy, D.; Kocsis, K.; Fuzik, J.; Marosi, M.; Kis, Z.; Teichberg, V.I.; Toldi, J.; Farkas, T. Kainate postconditioning restores LTP in ischemic hippocampal ca1: Onset-dependent second pathophysiological stress. Neuropharmacology 2011, 61, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds iso-Aβ(1-42) and Aβ(1-42) are available from the authors. |



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups of Aβ-Treated Animals | Aggregation Time (Hour) | Concentration (μM) of Aβ during Aggregation |
|---|---|---|
| A (n = 11), 24 h/25 μM | 24 | 25 |
| B (n = 11), 24 h/75 μM | 24 | 75 |
| C (n = 11), 24 h/200 μM | 24 | 200 |
| D (n = 12), 168 h/25 μM | 168 | 25 |
| E (n = 12), 168 h/75 μM | 168 | 75 |
| F (n = 12), 168 h/200 μM | 168 | 200 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasza, Á.; Penke, B.; Frank, Z.; Bozsó, Z.; Szegedi, V.; Hunya, Á.; Németh, K.; Kozma, G.; Fülöp, L. Studies for Improving a Rat Model of Alzheimer’s Disease: Icv Administration of Well-Characterized β-Amyloid 1-42 Oligomers Induce Dysfunction in Spatial Memory. Molecules 2017, 22, 2007. https://doi.org/10.3390/molecules22112007
Kasza Á, Penke B, Frank Z, Bozsó Z, Szegedi V, Hunya Á, Németh K, Kozma G, Fülöp L. Studies for Improving a Rat Model of Alzheimer’s Disease: Icv Administration of Well-Characterized β-Amyloid 1-42 Oligomers Induce Dysfunction in Spatial Memory. Molecules. 2017; 22(11):2007. https://doi.org/10.3390/molecules22112007
Chicago/Turabian StyleKasza, Ágnes, Botond Penke, Zsuzsanna Frank, Zsolt Bozsó, Viktor Szegedi, Ákos Hunya, Klaudia Németh, Gábor Kozma, and Lívia Fülöp. 2017. "Studies for Improving a Rat Model of Alzheimer’s Disease: Icv Administration of Well-Characterized β-Amyloid 1-42 Oligomers Induce Dysfunction in Spatial Memory" Molecules 22, no. 11: 2007. https://doi.org/10.3390/molecules22112007
APA StyleKasza, Á., Penke, B., Frank, Z., Bozsó, Z., Szegedi, V., Hunya, Á., Németh, K., Kozma, G., & Fülöp, L. (2017). Studies for Improving a Rat Model of Alzheimer’s Disease: Icv Administration of Well-Characterized β-Amyloid 1-42 Oligomers Induce Dysfunction in Spatial Memory. Molecules, 22(11), 2007. https://doi.org/10.3390/molecules22112007