Antimicrobial Abietane-Type Diterpenoids from Plectranthus punctatus


Abstract
:
1. Introduction
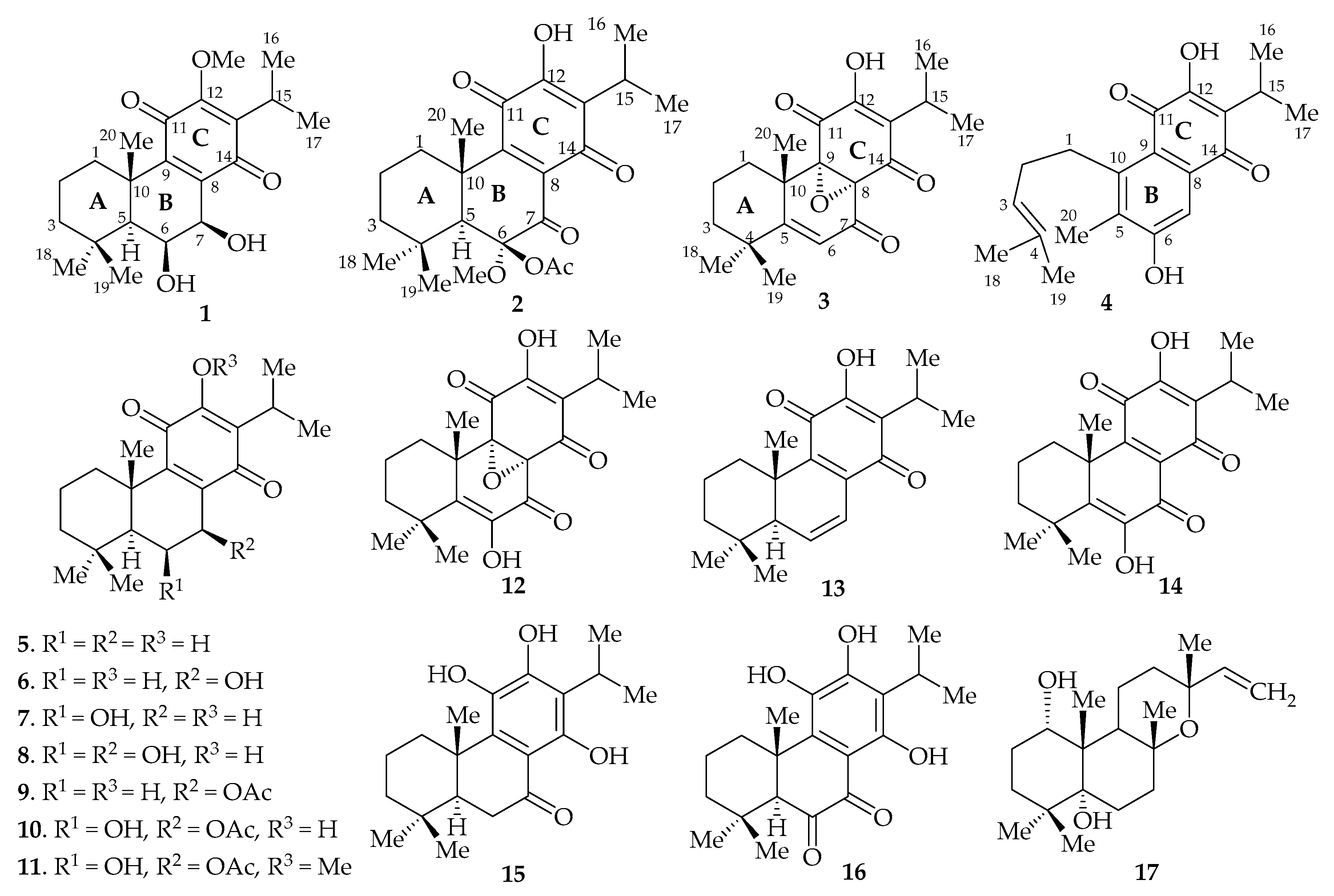
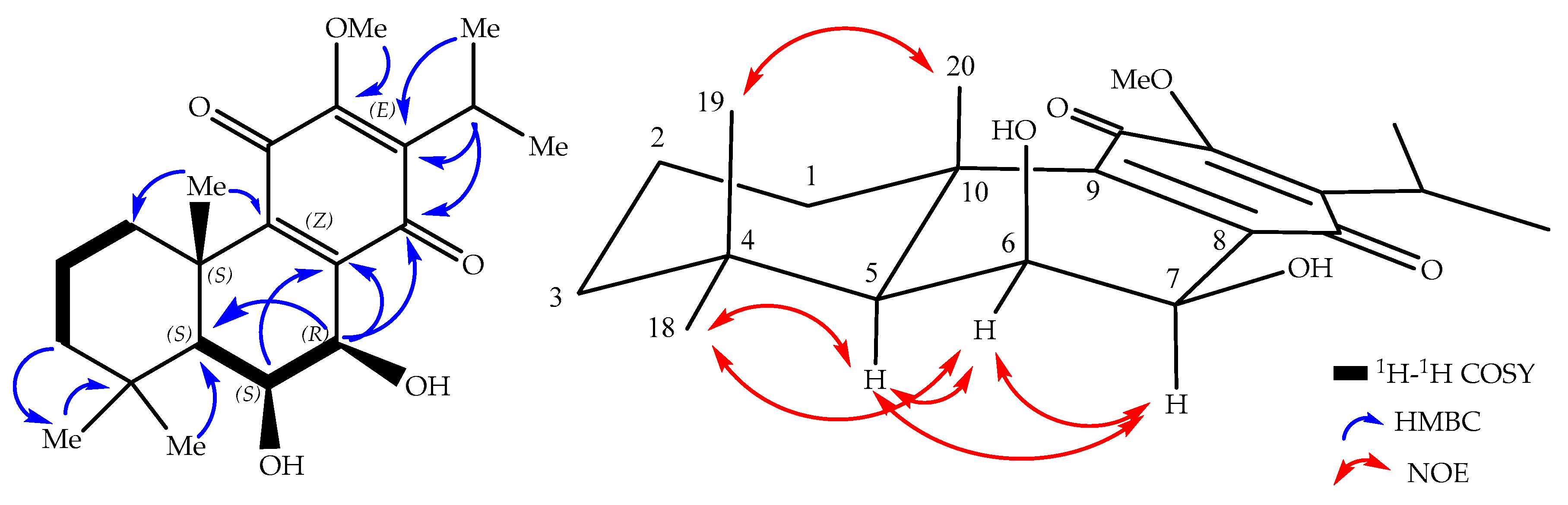
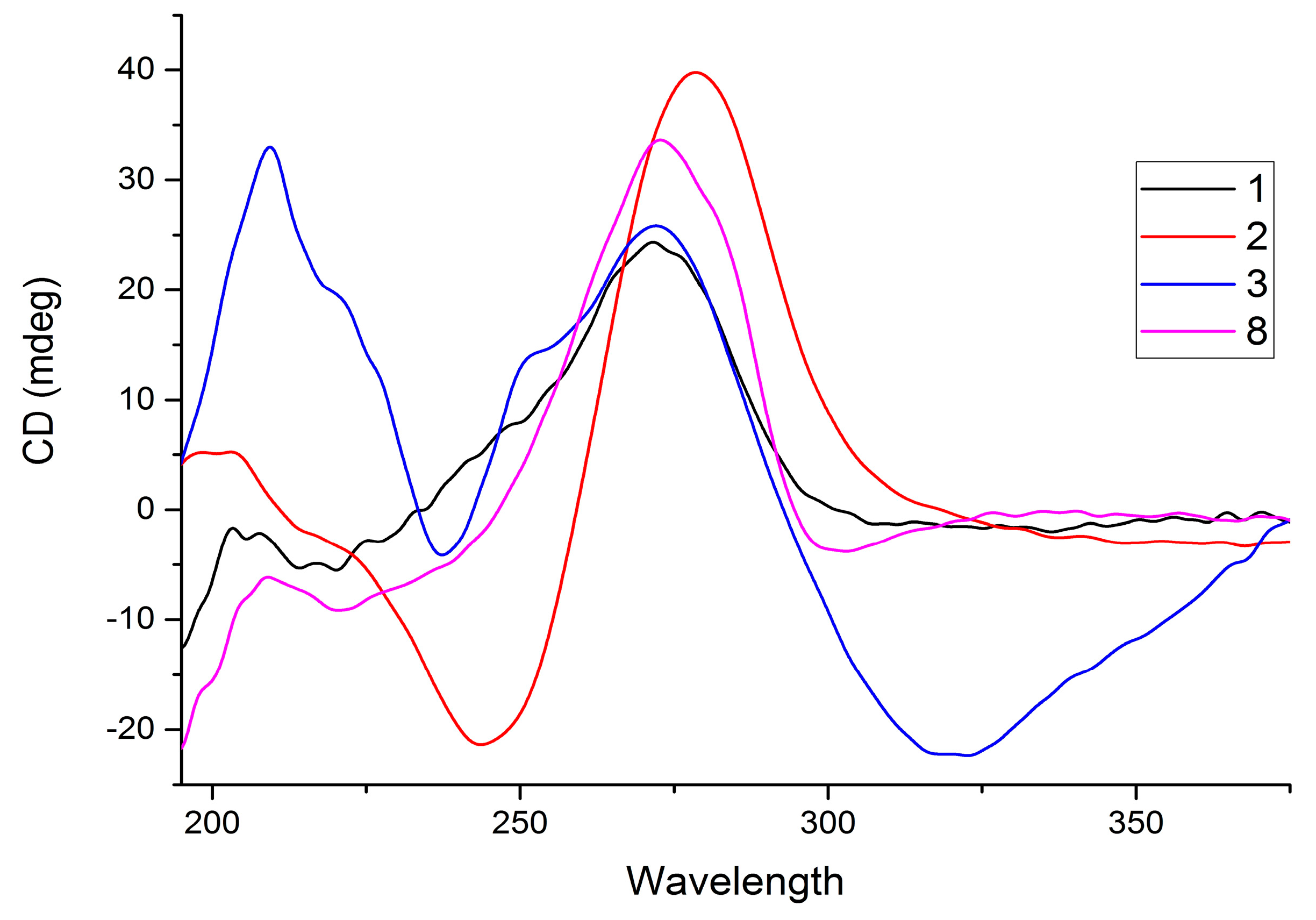
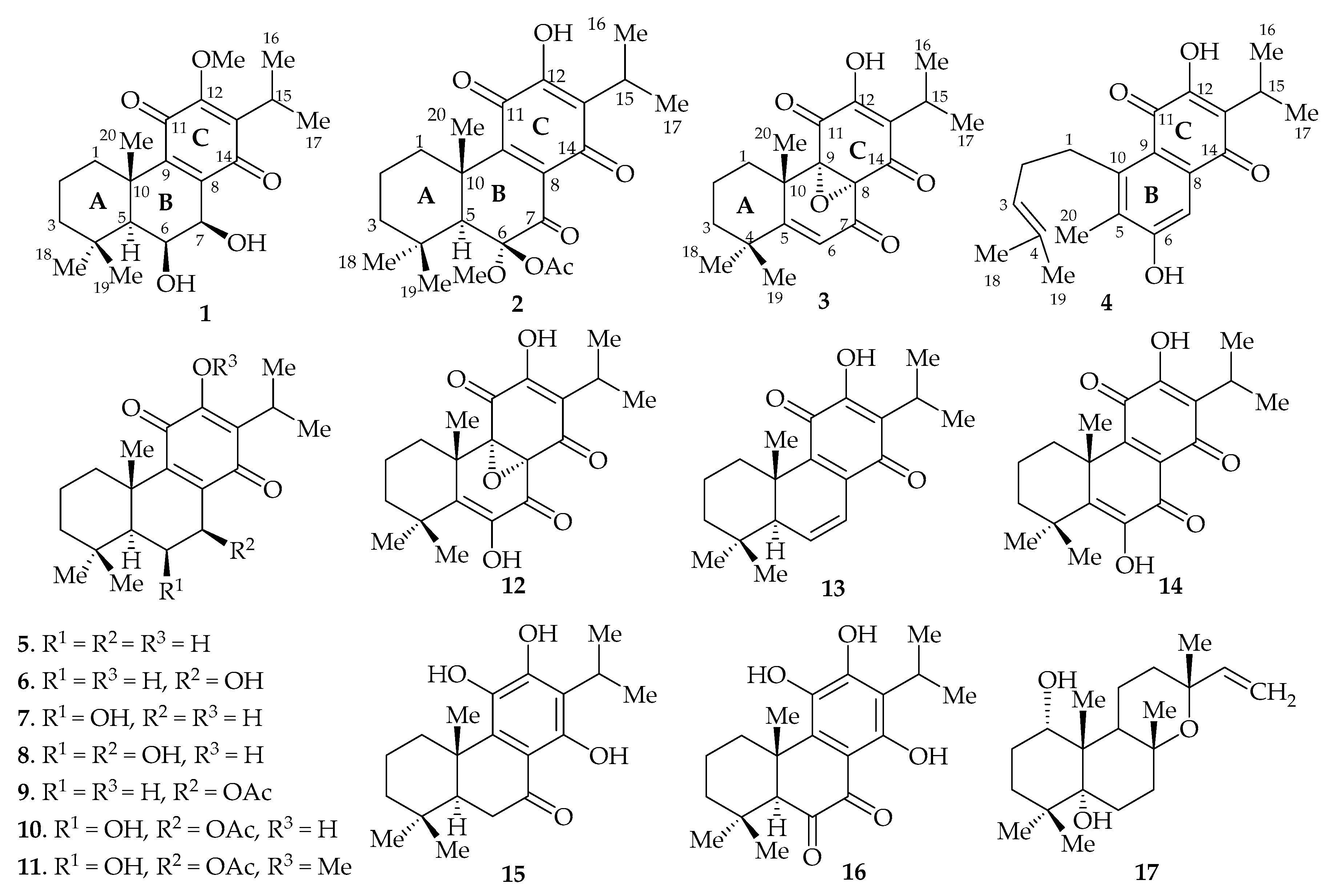
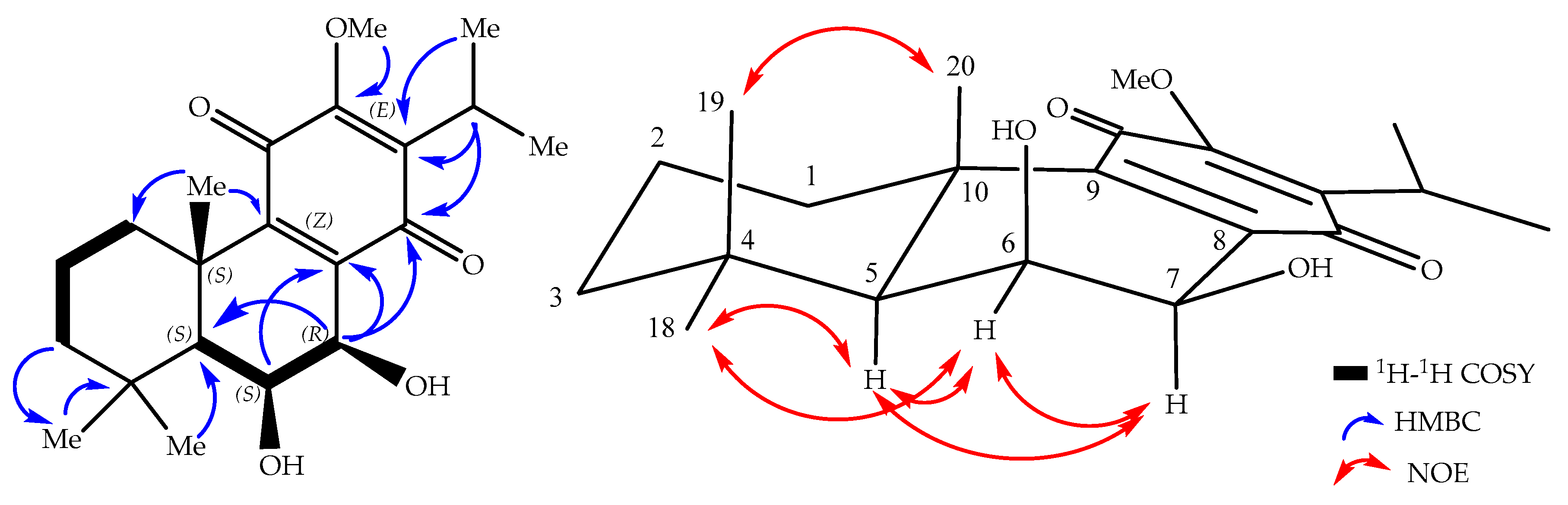
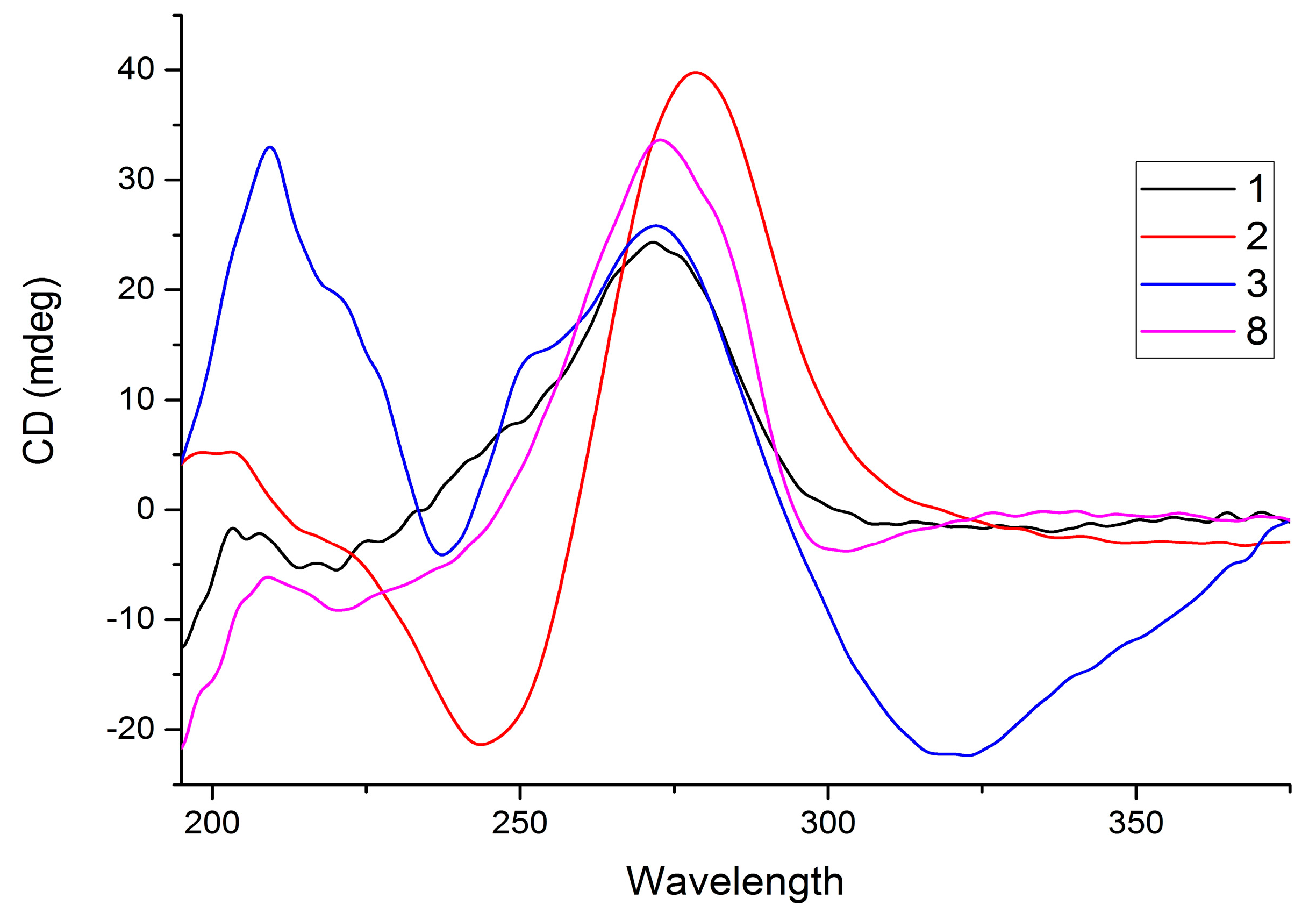
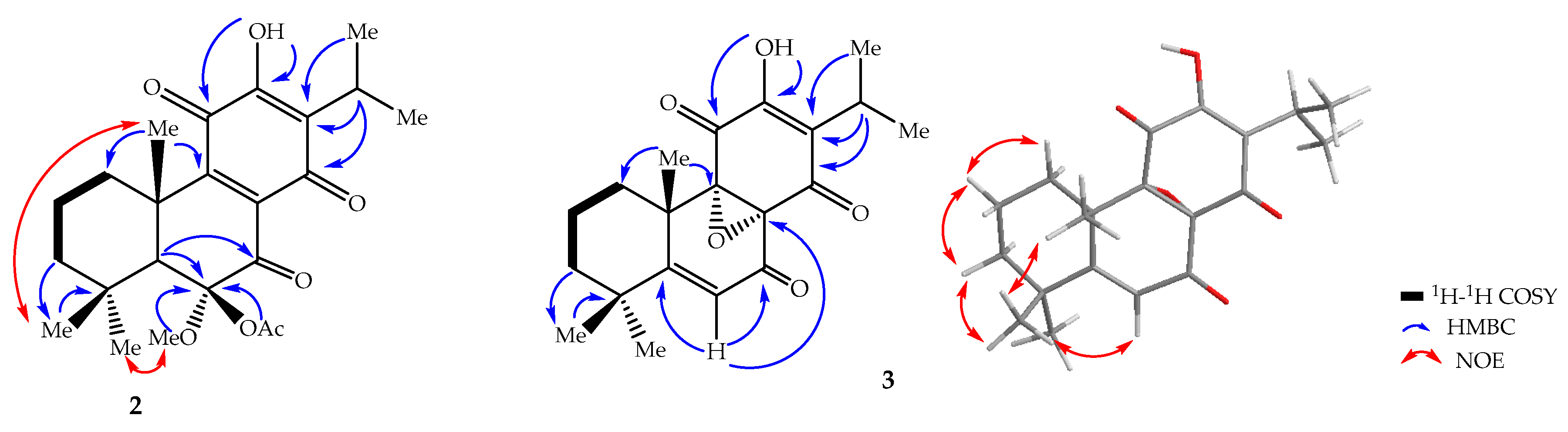
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Plant Materials
3.3. Extraction and Isolation
3.4. Antimicrobial Assay Using Agar Diffusion Test
3.5. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Abdel Khalik, K.N. A Systematic Revision of the Genus Plectranthus L. (Lamiaceae) in Saudi Arabia Based on Morphological, Palynological, and Micromorphological Characters of Trichomes. Am. J. Plant Sci. 2016, 7, 1429–1444. [Google Scholar] [CrossRef]
- Van Jaarsveld, E.J. The Plectranthus Handbook; National Botanic Gardens of South Africa, CTP Book Printers: Cape Town, South Africa, 1988; p. 23. [Google Scholar]
- Lukhoba, C.W.; Simmonds, M.S.J.; Paton, A.J. Plectranthus: A review of ethnobotanical uses. J. Ethnopharmacol. 2006, 103, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Al-Saleem, M.S.M.; Alkhathlan, H.Z. A detailed study on chemical characterization of essential oil components of two Plectranthus species grown in Saudi Arabia. J. Saudi Chem. Soc. 2016, 20, 711–721. [Google Scholar] [CrossRef]
- Stavri, M.; Paton, A.J.; Skelton, B.W.; Gibbons, S. Antibacterial diterpenes from Plectranthus ernstii. J. Nat. Prod. 2009, 72, 1191–1194. [Google Scholar] [CrossRef] [PubMed]
- Gaspar-Marques, C.; Rijo, P.; Simões, M.F.; Duarte, M.A.; Rodriguez, B. Abietanes from Plectranthus grandidentatus and Plectranthus hereroensis against methicillin and vancomycin-resistant bacteria. Phytomedicine 2006, 13, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl, R.L.; Khan, F.; Edwards, T.J.; Drewes, S.E. Antiplasmodial activities of some abietane diterpenes from the leaves of five Plectranthus species. S. Afr. J. Sci. 2008, 104, 62–64. [Google Scholar]
- Simões, M.F.; Rijo, P.; Duarte, A.; Barbosa, D.; Matias, D.; Delgado, J.; Cirilo, N.; Rodriguez, B. Two new diterpenoids from Plectranthus species. Phytochem. Lett. 2010, 3, 221–225. [Google Scholar] [CrossRef]
- Grayer, R.J.; Eckert, M.R.; Lever, A.; Veitch, N.C.; Kite, G.C.; Paton, A.J. Distribution of exudate flavonoids in the genus Plectranthus. Biochem. Sys. Ecol. 2010, 38, 335–341. [Google Scholar] [CrossRef]
- Fichtl, R.; Adi, A. Honeybee Flora of Ethiopia; Margraf: Weikersheim, Germany, 1994; pp. 118–121. [Google Scholar]
- Allemann, J.; Laurie, S.M.; Thiart, S.; Vorster, H.J.; Bornman, C.H. Sustainable production of root and tuber crops (potato, sweet potato, indigenous potato, cassava) in southern Africa. S. Afr. J. Bot. 2004, 70, 60–66. [Google Scholar] [CrossRef]
- Tadesse, D.; Eguale, T.; Giday, M.; Mussa, A. Ovicidal and larvicidal activity of crude extracts of Maesa lanceolata and Plectranthus punctatus against Haemonchus contortus. J. Ethnopharmacol. 2009, 122, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Edwards, O.E.; Feniak, G.; Los, M. Diterpenoid quinones of Inula royleana. Can. J. Chem. 1962, 40, 1540–1546. [Google Scholar]
- Hensch, M.; Rüedi, P.; Eugster, C.H. Horminon, Taxochinon und weitere Royleanone aus 2 abessinischen Plectranthus-Spezies (Labiatae). Helv. Chim. Acta 1975, 58, 1921–1934. [Google Scholar] [CrossRef]
- Mehrotra, R.; Vishwakarma, R.A.; Thakur, R.S. Abietane diterpenoids from Coleus zeylanicus. Phytochemistry 1989, 28, 3135–3137. [Google Scholar] [CrossRef]
- Alder, A.C.; Rüedi, P.; Eugster, C.H. Drüsenfarbstoffe aus Labiaten: Die polaren Diterpenoide aus Plectranthus argentatus S.T. BLAKE. Helv. Chim. Acta 1984, 67, 1523–1530. [Google Scholar] [CrossRef]
- Miyase, T.; Rüedi, P.; Eugster, C.H. Diterpenoide Drüsenfarbstoffe aus Labiaten: Coleone U, V, W und 14-O-Formyl-coleon-V sowie 2 Royleanone aus Plectranthus myrianthus BRIQ.; cis- und trans-A/B-6, 7-Dioxoroyleanon. Helv. Chim. Acta 1977, 60, 2770–2779. [Google Scholar] [CrossRef]
- Chang, C.I.; Tseng, M.H.; Kuo, Y.H. Five new diterpenoids from the bark of Taiwania cryptomerioides. Chem. Pharm. Bull. 2005, 53, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Labbé, C.; Castillo, M.; Fainia, F.; Coll, J.; Connolly, J.D. Rearranged isopimarenes and other diterpenoids from Satureja gilliesii. Phytochemistry 1994, 36, 735–738. [Google Scholar] [CrossRef]
- Inabuy, F.S.; Fischedick, J.T.; Lange, I.; Hartmann, M.; Srividya, N.; Parrish, A.N.; Xu, M.; Peters, R.J.; Lange, B.M. Biosynthesis of diterpenoids in Tripterygium adventitious root cultures. Plant Physiol. 2017, 175, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Lu, Y.; Zheng, Q.T.; Xiao-Li, X.L.; Zhao, Q.S. 8α,9α-Epoxy-7-oxoroyleanon. Acta Cryst. E 2006, 62, 3269–3270. [Google Scholar] [CrossRef]
- Topcu, G.; Eriş, C.; Ulubelen, A. Rearranged abietane diterpenes from Salvia limbata. Phytochemistry 1996, 41, 1143–1147. [Google Scholar] [CrossRef]
- Mehta, J.P.; Davariya, V.S.; Parmar, P.H. An antimicrobial activity of anthraquinones from Cassia occidentalis. Int. J. Chem. Sci. 2012, 10, 413–419. [Google Scholar]
- Rasheed, M.U.; Thajuddin, N.; Ahamed, P.; Teklemariam, Z.; Jamil, K. Antimicrobial drug resistance in strains of Escherichia coli isolated from food sources. Rev. Inst. Med. Trop. Sao Paulo 2014, 56, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Sammet, B.; Bogner, T.; Nahrwold, M.; Weiss, C.; Sewald, N. Approaches for the synthesis of functionalized cryptophycins. J. Org. Chem. 2010, 75, 6953–6960. [Google Scholar] [CrossRef] [PubMed]
- Baur, A.W.; Kirby, W.M.; Sherris, J.C.; Truck, M. Antibiotic susceptibility testing by a standardized single disk method. Am. J. Clin. Pathol. 1966, 45, 493. [Google Scholar]
Sample Availability: Samples of the compounds 7, 8, 9, 10 and 13 are available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|---|
| δH (m, J in Hz) | δC | δH (m, J in Hz) | δC | δH (m, J in Hz) | δC | δH (m, J in Hz) | δC | |
| 1 | 1.18 (m), 2.51 (m) | 39.2 | 1.36 (m), 2.83 (m) | 36.7 | 1.73 (m), 2.98 (m) | 33.9 | 3.23 (m) | 30.7 |
| 2 | 1.12 (m), 1.44 (m) | 24.4 | 1.34 (m), 1.64 (m) | 18.9 | 1.23 (m), 1.70 (m) | 17.4 | 2.19 (m) | 28.0 |
| 3 | 1.47 (m), 1.50 (m) | 43.3 | 1.42 (m), 1.27 (m) | 41.6 | 1.58 (m), 1.44 (m) | 39.1 | 5.31 (br t, 3.4) | 123.8 |
| 4 | 34.5 | 32.7 | 37.7 | 132.7 | ||||
| 5 | 1.54 (d, 3.7) | 49.6 | 3.00 (s) | 58.7 | 169.5 | 129.1 | ||
| 6 | 4.35 (dd, 3.7, 2.2) | 69.9 | 98.3 | 6.04 (s) | 120.9 | 159.6 | ||
| 7 | 4.56 (d, 2.2) | 67.9 | 199.7 | 189.0 | 7.69 (s) | 112.8 | ||
| 8 | 140.5 | 138.1 | 60.0 | 135.2 | ||||
| 9 | 150.3 | 147.6 | 67.7 | 119.9 | ||||
| 10 | 39.9 | 45.2 | 41.6 | 148.1 | ||||
| 11 | 184.9 | 183.8 | 187.0 | 181.4 | ||||
| 12 | 157.8 | 150.5 | 151.2 | 154.2 | ||||
| 13 | 136.2 | 125.2 | 128.4 | 125.4 | ||||
| 14 | 189.3 | 183.5 | 185.5 | 185.4 | ||||
| 15 | 3.18 (qq, 7.1, 7.1) | 25.3 | 3.15 (qq, 7.3, 7.3) | 24.4 | 3.19 (qq, 7.2, 7.2) | 24.9 | 3.35 (qq, 7.1, 7.1) | 24.6 |
| 16 | 1.73 (d, 7.1) | 20.7 | 1.19 (d, 7.3) | 19.9 | 1.23 (d, 7.2) | 19.2 | 1.30 (d, 7.1) | 20.0 |
| 17 | 1.21 (d, 7.1) | 19.9 | 1.20 (d, 7.3) | 19.8 | 1.21 (d, 7.2) | 19.4 | 1.30 (d, 7.1) | 20.0 |
| 18 | 1.00 (s) | 34.2 | 1.00 (s) | 32.8 | 1.26 (s) | 30.7 | 1. 64 (s) | 17.8 |
| 19 | 1.27 (s) | 22.3 | 1.37 (s) | 22.0 | 1.18 (s) | 32.2 | 1.75 (s) | 25.9 |
| 20 | 1.66 (s) | 21.0 | 1.36 (s) | 18.6 | 1.50 (s) | 24.0 | 2.31 (s) | 11.5 |
| 12-OMe/OH | 3.91 (s) | 61.1 | 7.03 (s) | 7.04 (s) | 8.05 (s) | |||
| 6-OMe | 3.42 (s) | 52.7 | ||||||
| 6-OAc | 2.13 (s) | 20.9, 169.3 | ||||||
| Compound | E. coli DSMZ1058 | S. warneri DSMZ20036 | B. subtlis DSMZ704 | M. luteus DSMZ1605 | P. agarici DSMZ11810 |
|---|---|---|---|---|---|
| 1 | n.a. | 22 | 20 | 23 | 21 |
| 2 | n.a. | 9 | 10 | 14 | 9 |
| 3 | n.a. | n.a. | 7 | n.a. | 14 |
| 4 | n.a. | n.a. | 7 | n.a. | 8 |
| 6 | 8 | 26 | 20 | 18 | 23 |
| 7 | 8 | 20 | 19 | 21 | 19 |
| 8 | 7 | 28 | 15 | 8 | 20 |
| 9 | 7 | 9 | 12 | 9 | 9 |
| 10 | 7 | 27 | 25 | 25 | 21 |
| 11 | 8 | 26 | 25 | 24 | 21 |
| 13 | 7 | 7 | 9 | 9 | n.a. |
| 14 | 8 | 19 | 13 | 24 | 14 |
| 15 | n.a. | 18 | 13 | 23 | 16 |
| 16 | 8 | 19 | 14 | 23 | 16 |
| Gentamycin | 23 | 21 | 26 | 23 | 24 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdissa, N.; Frese, M.; Sewald, N. Antimicrobial Abietane-Type Diterpenoids from Plectranthus punctatus. Molecules 2017, 22, 1919. https://doi.org/10.3390/molecules22111919
Abdissa N, Frese M, Sewald N. Antimicrobial Abietane-Type Diterpenoids from Plectranthus punctatus. Molecules. 2017; 22(11):1919. https://doi.org/10.3390/molecules22111919
Chicago/Turabian StyleAbdissa, Negera, Marcel Frese, and Norbert Sewald. 2017. "Antimicrobial Abietane-Type Diterpenoids from Plectranthus punctatus" Molecules 22, no. 11: 1919. https://doi.org/10.3390/molecules22111919
APA StyleAbdissa, N., Frese, M., & Sewald, N. (2017). Antimicrobial Abietane-Type Diterpenoids from Plectranthus punctatus. Molecules, 22(11), 1919. https://doi.org/10.3390/molecules22111919