Biophysical Properties and Antiviral Activities of Measles Fusion Protein Derived Peptide Conjugated with 25-Hydroxycholesterol



Abstract
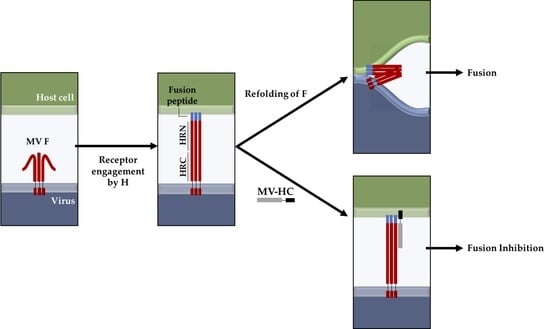
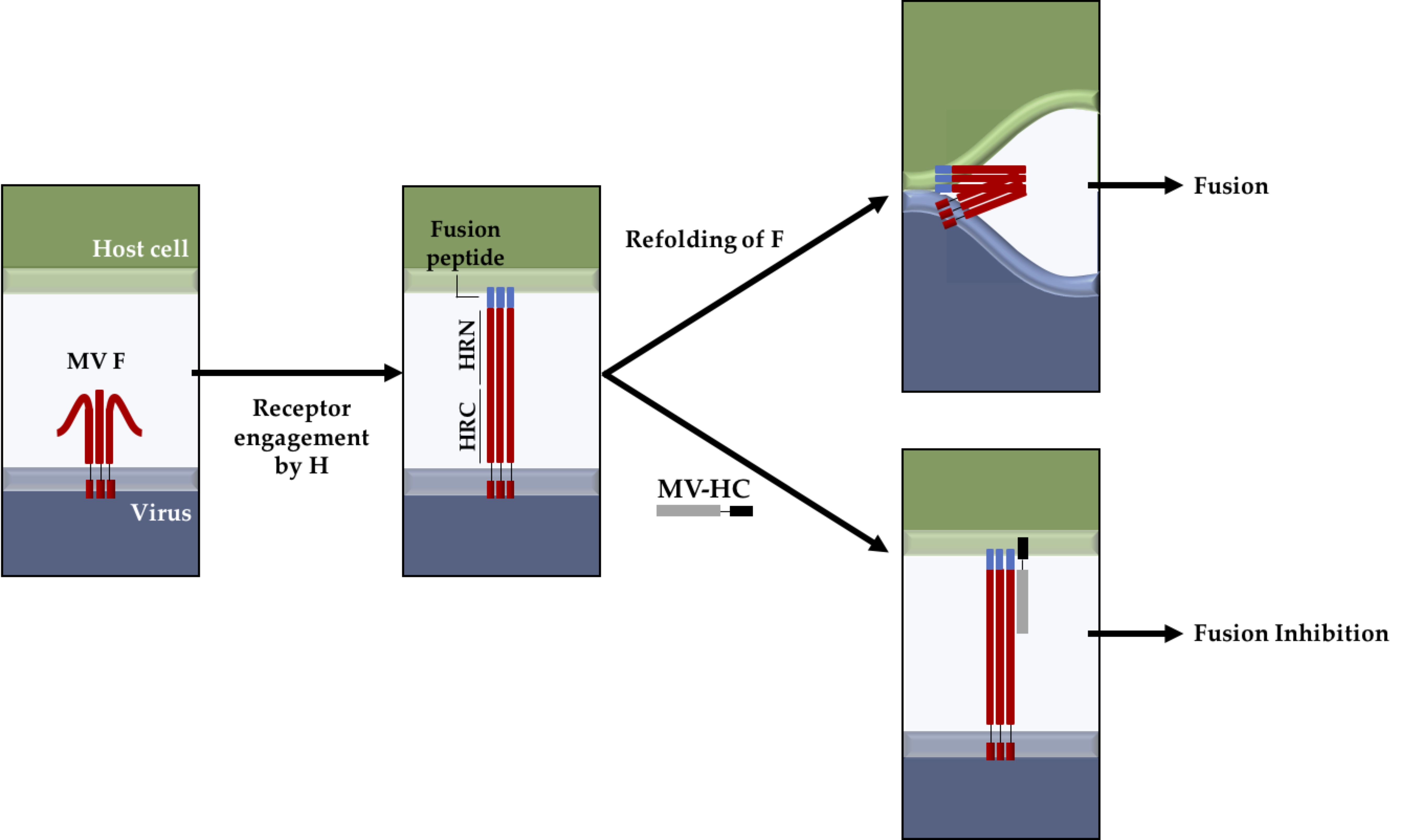
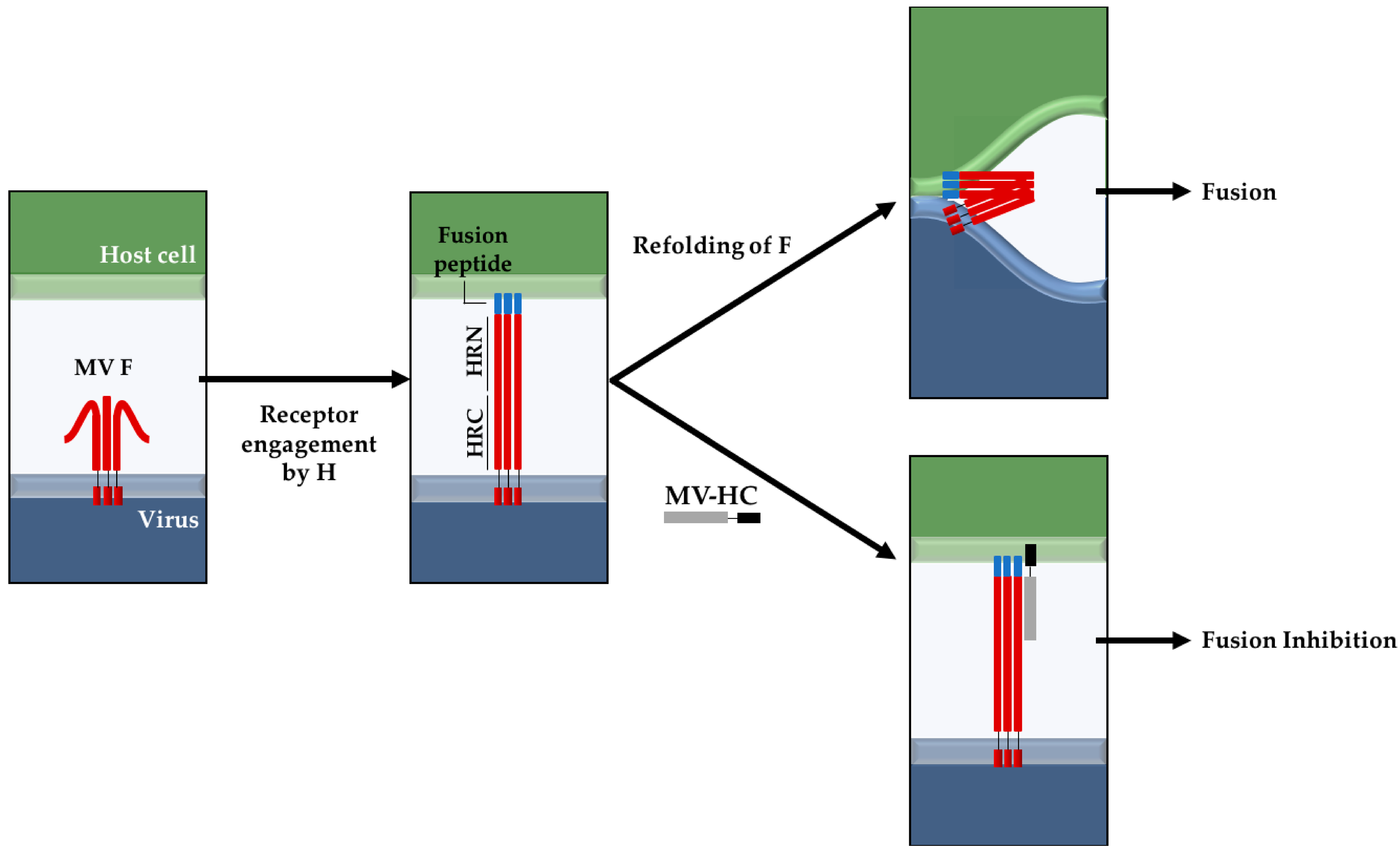
1. Introduction
2. Results
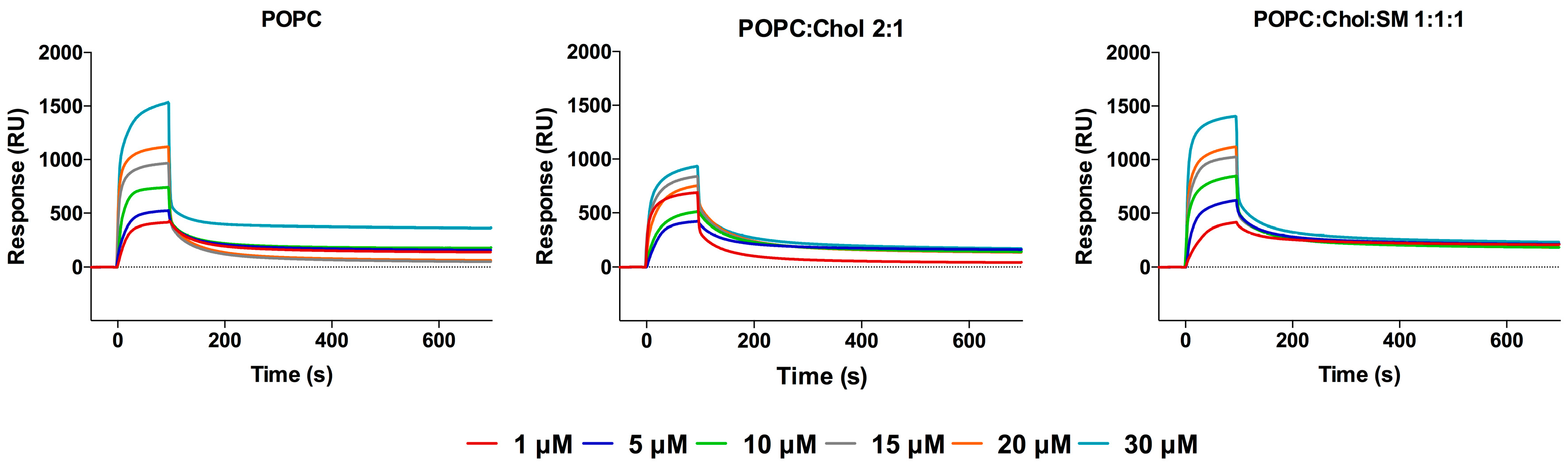
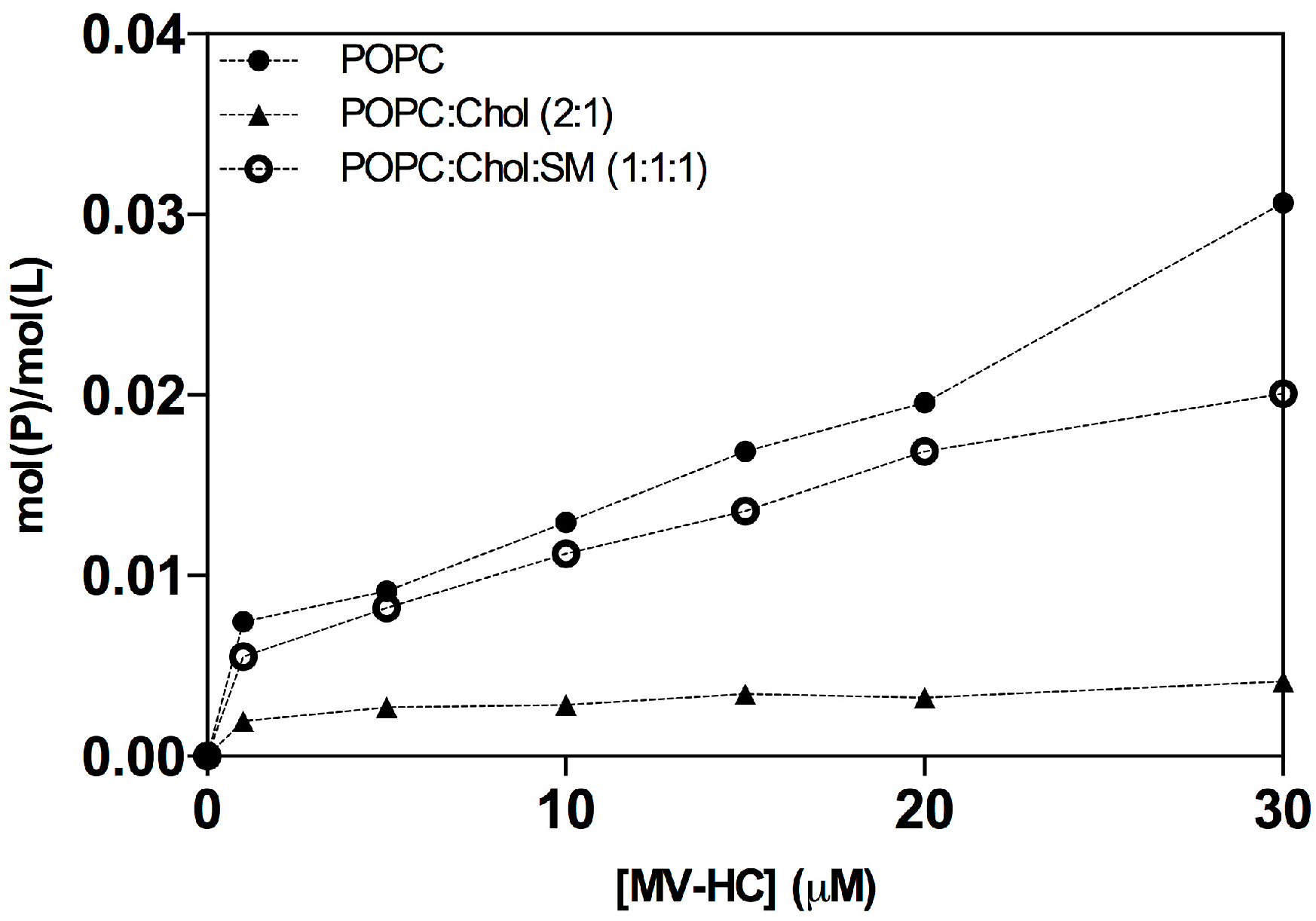
2.1. Interaction with Small Unilamellar Vesicles
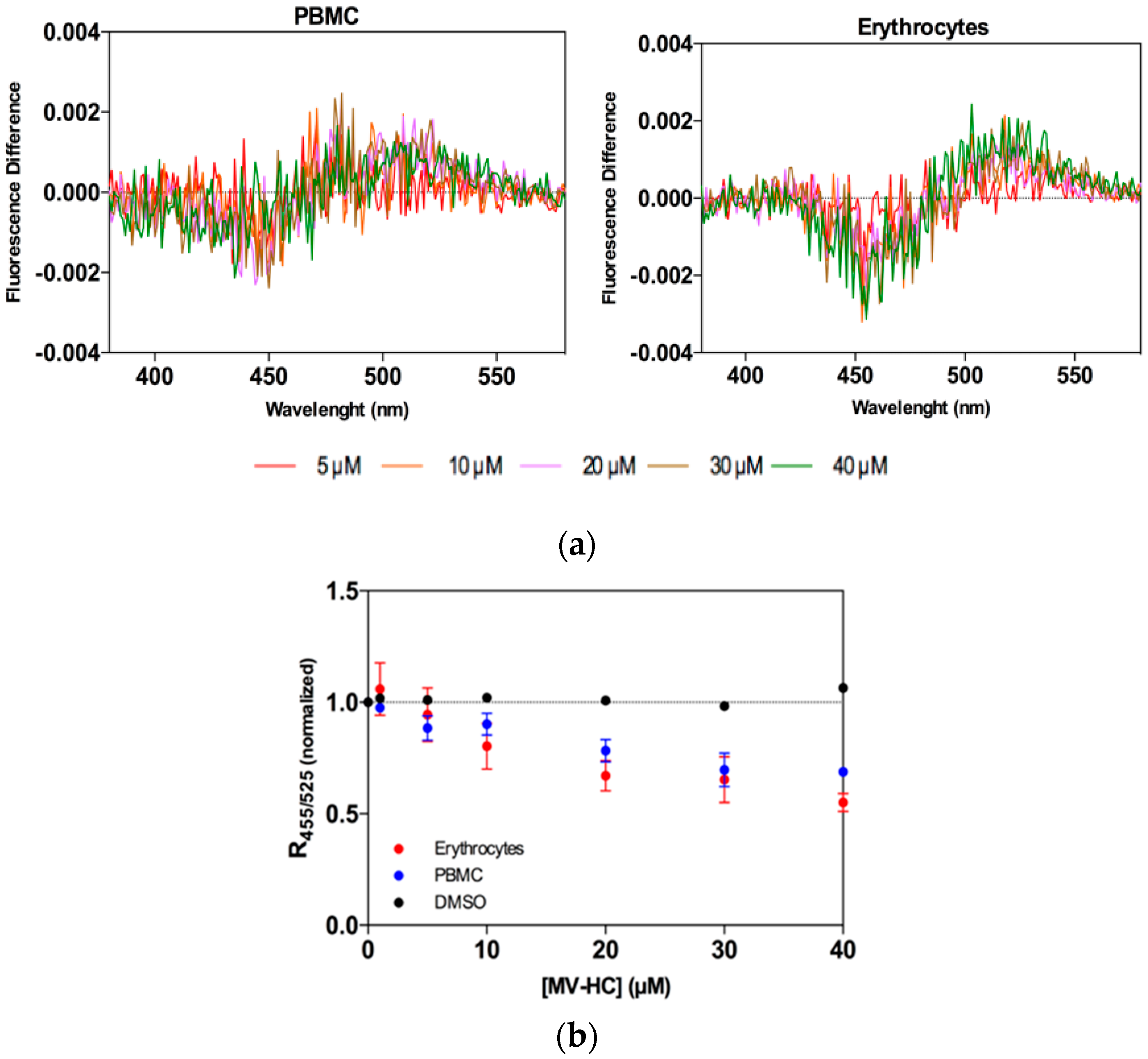
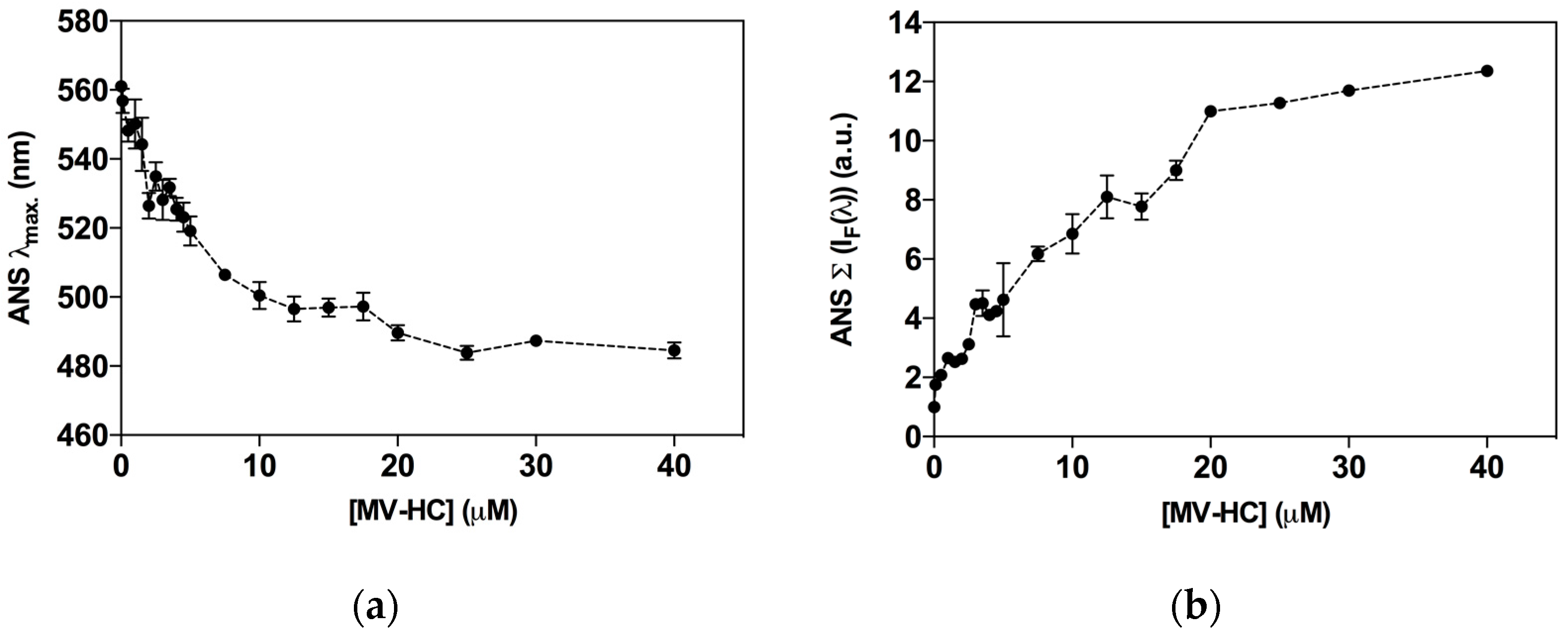
2.2. Interaction with Blood Cells
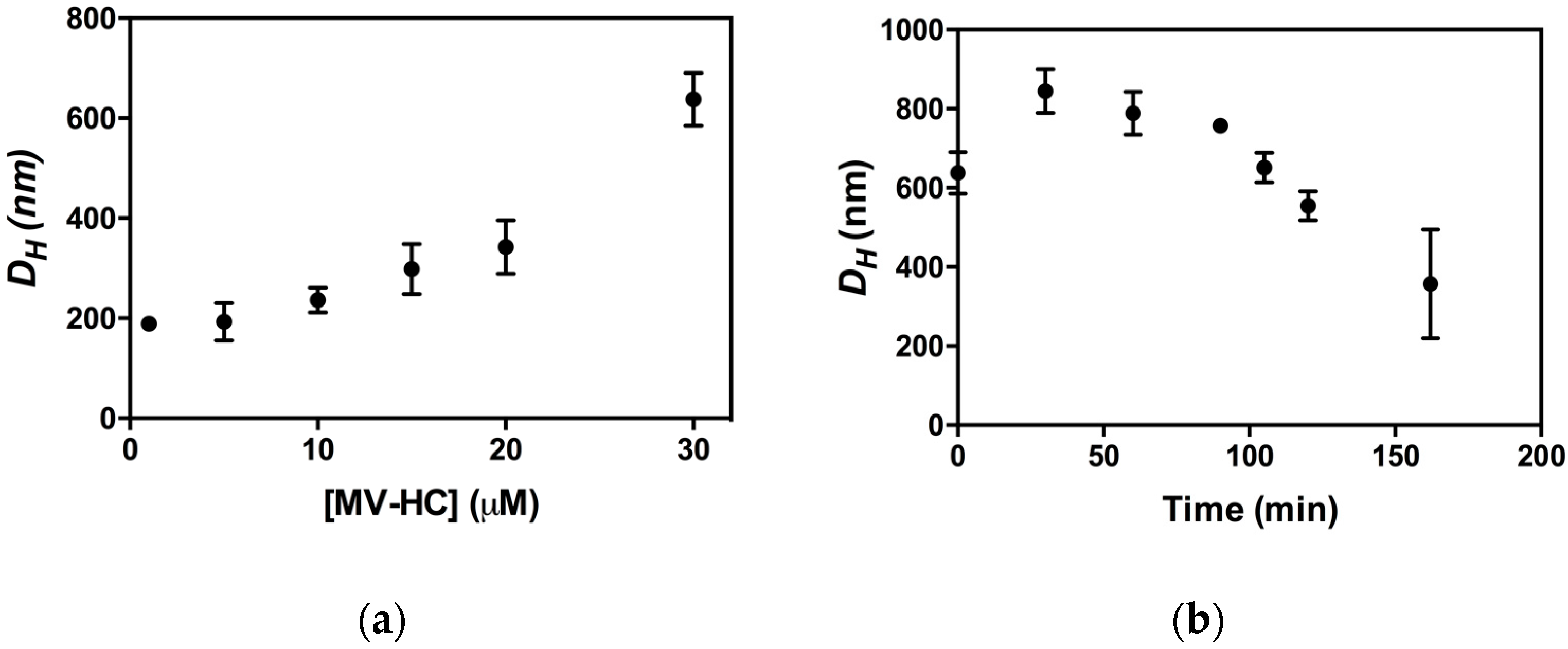
2.3. Aggregation Studies
2.4. Lipid-Conjugated Inhibitory Peptides Inhibit Cell-to-Cell Fusion
3. Discussion
4. Material and Methods
4.1. Reagents
4.2. Sample Preparation
4.3. Surface Plasmon Resonance
4.4. Membrane Dipole Potential Assessment with Di-8-ANEPPS
4.5. ANS Fluorescence Studies
4.6. Dynamic Light Scattering
4.7. Cells
4.8. Transient Expression of H and F Genes
4.9. β-Galactosidase Complementation-Based Fusion Assay
4.10. Data Analysis
Acknowledgments
Author contributions
Conflicts of Interest
References
- De Vries, R.D.; Duprex, W.P.; De Swart, R.L. Morbillivirus infections: An introduction. Viruses 2015, 7, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Progress Toward Regional Measles Elimination—Worldwide. 2000–2015. Available online: https://www.cdc.gov/mmwr/volumes/65/wr/mm6544a6.htm (accessed on 10 July 2017).
- Holzmann, H.; Hengel, H.; Tenbusch, M.; Doerr, H.W. Eradication of measles: Remaining challenges. Med. Microbiol. Immunol. 2016, 205, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Measles Outbreaks Across Europe Threaten Progress Towards Elimination. Available online: http://www.euro.who.int/en/media-centre/sections/press-releases/2017/measles-outbreaks-across-europe-threaten-progress-towards-elimination (accessed on 10 July 2017).
- De Serres, G.; Boulianne, N.; Defay, F.; Brousseau, N.; Benoit, M.; Lacoursiere, S.; Guillemette, F.; Soto, J.; Ouakki, M.; Ward, B.J.; et al. Higher risk of measles when the first dose of a 2-dose schedule of measles vaccine is given at 12–14 months versus 15 months of age. Clin. Infect. Dis. 2012, 55, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Haralambieva, I.H.; Ovsyannikova, I.G.; O’Byrne, M.; Pankratz, V.S.; Jacobson, R.M.; Poland, G.A. A large observational study to concurrently assess persistence of measles specific B-cell and T-cell immunity in individuals following two doses of MMR vaccine. Vaccine 2011, 29, 4485–4491. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Lee, P.I.; Hsieh, Y.C.; Chen, P.Y.; Ho, Y.H.; Chang, C.J.; Liu, D.P.; Chang, F.Y.; Chiu, C.H.; Huang, Y.C.; et al. Waning population immunity to measles in Taiwan. Vaccine 2012, 30, 6721–6727. [Google Scholar] [CrossRef] [PubMed]
- Wendorf, K.A.; Winter, K.; Zipprich, J.; Schechter, R.; Hacker, J.K.; Preas, C.; Cherry, J.D.; Glaser, C.; Harriman, K. Subacute sclerosing panencephalitis: The devastating measles complication that might be more common than previously estimated. Clin. Infect. Dis. 2017, 65, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.; Hung, T.M.; Wertheim, H.; Hoa le, N.M.; Vincent, A.; Lang, B.; Waters, P.; Ha, N.H.; Trung, N.V.; Farrar, J.; et al. Acute measles encephalitis in partially vaccinated adults. PLoS ONE 2013, 8, e71671. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.R.; Albertyn, C.; Heckmann, J.M.; Smuts, H.E. Molecular characterisation of virus in the brains of patients with measles inclusion body encephalitis (MIBE). Virol. J. 2013, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Mahil, S.K.; Fleming, J.; Robson, A.; Sarkany, R. Measles in a previously vaccinated human immunodeficiency virus-positive adult. Clin. Exp. Dermatol. 2014, 39, 117–118. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, L.A.; Rall, G.F. Blue moon neurovirology: The merits of studying rare CNS diseases of viral origin. J. Neuroimmune Pharmacol. 2010, 5, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Young, V.A.; Rall, G.F. Making it to the synapse: Measles virus spread in and among neurons. Curr. Top. Microbiol. Immunol. 2009, 330, 3–30. [Google Scholar] [PubMed]
- Makhortova, N.R.; Askovich, P.; Patterson, C.E.; Gechman, L.A.; Gerard, N.P.; Rall, G.F. Neurokinin-1 enables measles virus trans-synaptic spread in neurons. Virology 2007, 362, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Reuter, D.; Schneider-Schaulies, J. Measles virus infection of the CNS: Human disease, animal models, and approaches to therapy. Med. Microbiol. Immunol. 2010, 199, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.H.; Meatherall, B.; Nikolic, A.; Cannon, K.; Fonseca, K.; Joseph, J.T.; MacDonald, J.; Pabbaraju, K.; Tellier, R.; Wong, S.; et al. Subacute sclerosing panencephalitis in pregnancy. Lancet Infect. Dis. 2016, 16, 366–375. [Google Scholar] [CrossRef]
- De Vries, R.D.; Mesman, A.W.; Geijtenbeek, T.B.; Duprex, W.P.; De Swart, R.L. The pathogenesis of measles. Curr. Opin. Virol. 2012, 2, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, C.S.; Frenzke, M.; Leonard, V.H.; Welstead, G.G.; Richardson, C.D.; Cattaneo, R. Measles virus infection of alveolar macrophages and dendritic cells precedes spread to lymphatic organs in transgenic mice expressing human signaling lymphocytic activation molecule (SLAM, CD150). J. Virol. 2010, 84, 3033–3042. [Google Scholar] [CrossRef] [PubMed]
- Avota, E.; Koethe, S.; Schneider-Schaulies, S. Membrane dynamics and interactions in measles virus dendritic cell infections. Cell Microbiol. 2013, 15, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Koethe, S.; Avota, E.; Schneider-Schaulies, S. Measles virus transmission from dendritic cells to T cells: Formation of synapse-like interfaces concentrating viral and cellular components. J. Virol. 2012, 86, 9773–9781. [Google Scholar] [CrossRef] [PubMed]
- Lemon, K.; de Vries, R.D.; Mesman, A.W.; McQuaid, S.; van Amerongen, G.; Yuksel, S.; Ludlow, M.; Rennick, L.J.; Kuiken, T.; Rima, B.K.; et al. Early target cells of measles virus after aerosol infection of non-human primates. PLoS Pathog. 2011, 7, e1001263. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y.; Ono, N.; Tatsuo, H.; Hashimoto, K.; Minagawa, H. Measles virus receptor SLAM (CD150). Virology 2002, 299, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y.; Takeda, M.; Ohno, S.; Hashiguchi, T. Measles virus receptors. Curr. Top. Microbiol. Immunol. 2009, 329, 13–30. [Google Scholar] [PubMed]
- Hashiguchi, T.; Maenaka, K.; Yanagi, Y. Measles virus hemagglutinin: Structural insights into cell entry and measles vaccine. Front. Microbiol. 2011, 2, 247. [Google Scholar] [CrossRef] [PubMed]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature 2000, 406, 893–897. [Google Scholar] [PubMed]
- Muhlebach, M.D.; Mateo, M.; Sinn, P.L.; Prufer, S.; Uhlig, K.M.; Leonard, V.H.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 2011, 480, 530–553. [Google Scholar] [CrossRef] [PubMed]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.T.; Sisson, G.; Tsao, M.S.; Richardson, C.D. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.K.; Li, N.; Mark, A.C.; Mateo, M.; Cattaneo, R.; Sinn, P.L. Cell-to-cell contact and nectin-4 govern spread of measles virus from primary human myeloid cells to primary human airway epithelial cells. J. Virol. 2016, 90, 6808–6817. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.T.; Richardson, C.D. The host cell receptors for measles virus and their interaction with the viral hemagglutinin (H) protein. Viruses 2016, 8, 250. [Google Scholar] [CrossRef] [PubMed]
- Laksono, B.M.; De Vries, R.D.; McQuaid, S.; Duprex, W.P.; De Swart, R.L. Measles virus host invasion and pathogenesis. Viruses 2016, 8, E210. [Google Scholar] [CrossRef] [PubMed]
- Delpeut, S.; Sawatsky, B.; Wong, X.X.; Frenzke, M.; Cattaneo, R.; Von Messling, V. Nectin-4 interactions govern measles virus virulence in a new model of pathogenesis, the squirrel monkey (Saimiri sciureus). J. Virol. 2017, 91, e02490-16. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Paterson, R.G.; Wen, X.; Lamb, R.A.; Jardetzky, T.S. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9288–9293. [Google Scholar] [CrossRef] [PubMed]
- Lamb, R.A.; Paterson, R.G.; Jardetzky, T.S. Paramyxovirus membrane fusion: Lessons from the F and HN atomic structures. Virology 2006, 344, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Dutch, R.E. Paramyxovirus fusion and entry: Multiple paths to a common end. Viruses 2012, 4, 613–636. [Google Scholar] [CrossRef] [PubMed]
- Plemper, R.K.; Brindley, M.A.; Iorio, R.M. Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 2011, 7, e1002058. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins: Multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Sapir, A.; Avinoam, O.; Podbilewicz, B.; Chernomordik, L.V. Viral and developmental cell fusion mechanisms: Conservation and divergence. Dev. Cell. 2008, 14, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Pessi, A.; Langella, A.; Capito, E.; Ghezzi, S.; Vicenzi, E.; Poli, G.; Ketas, T.; Mathieu, C.; Cortese, R.; Horvat, B.; et al. A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS ONE 2012, 7, e36833. [Google Scholar] [CrossRef] [PubMed]
- Vigant, F.; Lee, B. Hendra and nipah infection: Pathology, models and potential therapies. Infect. Disord. Drug Targets 2011, 11, 315–336. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.G.; Yang, P.L.; Harrison, S.C. Peptide inhibitors of flavivirus entry derived from the E protein stem. J. Virol. 2010, 84, 12549–12554. [Google Scholar] [CrossRef] [PubMed]
- Steffen, D.L.; Xu, K.; Nikolov, D.B.; Broder, C.C. Henipavirus mediated membrane fusion, virus entry and targeted therapeutics. Viruses 2012, 4, 280–309. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.J.; Ma, X.T.; Liu, C.; Zhang, X.Y.; Wang, C.X. The current status and challenges in the development of fusion inhibitors as therapeutics for HIV-1 infection. Curr. Pharm. Des. 2012, 19, 1810–1817. [Google Scholar] [CrossRef]
- Miyamoto, F.; Kodama, E.N. Novel HIV-1 fusion inhibition peptides: Designing the next generation of drugs. Antivir. Chem. Chemother. 2012, 22, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Eckert, D.M.; Kim, P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001, 70, 777–810. [Google Scholar] [CrossRef] [PubMed]
- Eckert, D.M.; Kim, P.S. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc. Natl. Acad. Sci. USA 2001, 98, 11187–11192. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Pessi, A.; Gui, L.; Santoprete, A.; Talekar, A.; Moscona, A.; Porotto, M. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011, 286, 42141–42149. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Carta, P.; Deng, Y.; Kellogg, G.E.; Whitt, M.; Lu, M.; Mungall, B.A.; Moscona, A. Molecular determinants of antiviral potency of paramyxovirus entry inhibitors. J. Virol. 2007, 81, 10567–10574. [Google Scholar] [CrossRef] [PubMed]
- Lambert, D.M.; Barney, S.; Lambert, A.L.; Guthrie, K.; Medinas, R.; Davis, D.E.; Bucy, T.; Erickson, J.; Merutka, G.; Petteway, S.R., Jr. Peptides from conserved regions of paramyxovirus fusion (F) proteins are potent inhibitors of viral fusion. Proc. Natl. Acad. Sci. USA 1996, 93, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Yokoyama, C.C.; Orefice, G.; Kim, H.S.; Aljofan, M.; Mungall, B.A.; Moscona, A. Kinetic dependence of paramyxovirus entry inhibition. J. Virol. 2009, 83, 6947–6951. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Yokoyama, C.C.; Palermo, L.M.; Mungall, B.; Aljofan, M.; Cortese, R.; Pessi, A.; Moscona, A. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010, 84, 6760–6768. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Rockx, B.; Yokoyama, C.C.; Talekar, A.; Devito, I.; Palermo, L.M.; Liu, J.; Cortese, R.; Lu, M.; Feldmann, H.; et al. Inhibition of Nipah virus infection in vivo: Targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 2010, 6, e1001168. [Google Scholar] [CrossRef] [PubMed]
- Welsch, J.C.; Talekar, A.; Mathieu, C.; Pessi, A.; Moscona, A.; Horvat, B.; Porotto, M. Fatal measles virus infection prevented by brain-penetrant fusion inhibitors. J. Virol. 2013, 87, 13785–13794. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.G.; DeVito, I.; Jenkins, S.G.; Niewiesk, S.; Porotto, M.; Moscona, A. Circulating clinical strains of human parainfluenza virus reveal viral entry requirements for in vivo infection. J. Virol. 2014, 88, 13495–13502. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Augusto, M.T.; Niewiesk, S.; Horvat, B.; Palermo, L.M.; Sanna, G.; Madeddu, S.; Huey, D.; Castanho, M.A.; Porotto, M.; et al. Broad spectrum antiviral activity for paramyxoviruses is modulated by biophysical properties of fusion inhibitory peptides. Sci. Rep. 2017, 7, 43610. [Google Scholar] [PubMed]
- Figueira, T.N.; Palermo, L.M.; Veiga, A.S.; Huey, D.; Alabi, C.A.; Santos, N.C.; Welsch, J.C.; Mathieu, C.; Horvat, B.; Niewiesk, S.; et al. In vivo efficacy of measles virus fusion protein-derived peptides is modulated by properties of self-assembly and membrane residence. J. Virol. 2016, 91, e01554-16. [Google Scholar] [PubMed]
- Chen, Y.; Wang, S.; Yi, Z.; Tian, H.; Aliyari, R.; Li, Y.; Chen, G.; Liu, P.; Zhong, J.; Chen, X.; et al. Interferon-inducible cholesterol-25-hydroxylase inhibits hepatitis C virus replication via distinct mechanisms. Sci. Rep. 2014, 4, 7242. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Aliyari, R.; Chikere, K.; Li, G.M.; Marsden, M.D.; Smith, J.K.; Pernet, O.; Guo, H.T.; Nusbaum, R.; Zack, J.A.; et al. Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 2013, 38, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Bauman, D.R.; Bitmansour, A.D.; McDonald, J.G.; Thompson, B.M.; Liang, G.S.; Russell, D.W. 25-Hydroxycholesterol secreted by macrophages in response to toll-like receptor activation suppresses immunoglobulin A production. Proc. Natl. Acad. Sci. USA 2009, 106, 16764–16769. [Google Scholar] [CrossRef] [PubMed]
- Cyster, J.G.; Dang, E.V.; Reboldi, A.; Yi, T. 25-Hydroxycholesterols in innate and adaptive immunity. Nat. Rev. Immunol. 2014, 14, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Besenicar, M.; Macek, P.; Lakey, J.H.; Anderluh, G. Surface plasmon resonance in protein-membrane interactions. Chem. Phys. Lipids 2006, 141, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Manie, S.N.; de Breyne, S.; Vincent, S.; Gerlier, D. Measles virus structural components are enriched into lipid raft microdomains: A potential cellular location for virus assembly. J. Virol. 2000, 74, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.; Gerlier, D.; Manie, S.N. Measles virus assembly within membrane rafts. J. Virol. 2000, 74, 9911–9915. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S. Real-Time Analysis of Biomolecular Interactions—Principles of BIACORE; Nagata, K., Handa, H., Eds.; Springer: Tokyo, Japan, 2000; pp. 23–30. [Google Scholar]
- Matos, P.M.; Castanho, M.A.; Santos, N.C. HIV-1 fusion inhibitor peptides enfuvirtide and T-1249 interact with erythrocyte and lymphocyte membranes. PLoS ONE 2010, 5, e9830. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.M.; Franquelim, H.G.; Castanho, M.A.; Santos, N.C. Quantitative assessment of peptide-lipid interactions. Ubiquitous fluorescence methodologies. Biochim. Biophys. Acta. 2010, 1798, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Augusto, M.T.; Hollmann, A.; Castanho, M.A.; Porotto, M.; Pessi, A.; Santos, N.C. Improvement of HIV fusion inhibitor C34 efficacy by membrane anchoring and enhanced exposure. J. Antimicrob. Chemother. 2014, 69, 1286–1297. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Matos, P.M.; Augusto, M.T.; Castanho, M.A.; Santos, N.C. Conjugation of cholesterol to HIV-1 fusion inhibitor C34 increases peptide-membrane interactions potentiating its action. PLoS ONE 2013, 8, e60302. [Google Scholar] [CrossRef] [PubMed]
- Slavik, J. Anilinonaphthalene sulfonate as a probe of membrane composition and function. Biochim. Biophys. Acta 1982, 694, 1–25. [Google Scholar] [CrossRef]
- Stetefeld, J.; McKenna, S.A.; Patel, T.R. Dynamic light scattering: A practical guide and applications in biomedical sciences. Biophys. Rev. 2016, 8, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, E.M.; Mathieu, C.; Palermo, L.M.; Hardie, D.; Horvat, B.; Moscona, A.; Porotto, M. Measles fusion machinery is dysregulated in neuropathogenic variants. MBio 2015, 6, e02528-14. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Huey, D.; Jurgens, E.; Welsch, J.C.; DeVito, I.; Talekar, A.; Horvat, B.; Niewiesk, S.; Moscona, A.; Porotto, M. Prevention of measles virus infection by intranasal delivery of fusion inhibitor peptides. J. Virol. 2015, 89, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Moosmann, P.; Rusconi, S. Alpha complementation of LacZ in mammalian cells. Nucleic Acids Res. 1996, 24, 1171–1172. [Google Scholar] [CrossRef] [PubMed]
- Hawe, A.; Sutter, M.; Jiskoot, W. Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 2008, 25, 1487–1499. [Google Scholar] [CrossRef] [PubMed]
- Oldendorf, W.H. Lipid solubility and drug penetration of the blood brain barrier. Proc. Soc. Exp. Biol. Med. 1974, 147, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, J.A.; Gomes, B.; Coelho, M.A.; do Carmo Pereira, M.; Rocha, S. Targeting nanoparticles across the blood-brain barrier with monoclonal antibodies. Nanomedicine 2014, 9, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Testa, B.; Fahr, A. Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Brossi, A.; Pei, X.F.; Ingram, D.K.; Soncrant, T.T. Designing Drugs for Optimal Nervous System Activity. In New Concepts of a Blood-Brain Barrier; Greenwood, J., Begley, D.J., Segal, M.B., Eds.; Springer: Boston, MA, USA, 1995; pp. 251–264. [Google Scholar]
- Vemuri, S.; Rhodes, C.T. Preparation and characterization of liposomes as therapeutic delivery systems: A review. Pharm. Acta Helv. 1995, 70, 95–111. [Google Scholar] [CrossRef]
- Craik, D.J.; Henriques, S.T.; Mylne, J.S.; Wang, C.K. Cyclotide isolation and characterization. Methods Enzymol. 2012, 516, 37–62. [Google Scholar] [PubMed]
- Gross, E.; Bedlack, R.S.; Loew, L.M. Dual-wavelength ratiometric fluorescence measurement of the membrane dipole potential. Biophys. J. 1994, 67, 208–216. [Google Scholar] [CrossRef]
- Clarke, R.J.; Kane, D.J. Optical detection of membrane dipole potential: Avoidance of fluidity and dye-induced effects. Biochim. Biophys. Acta-Biomembr. 1997, 1323, 223–239. [Google Scholar] [CrossRef]
- Provencher, S. CONTIN: A general purpose constrained regularization program for inverting noisy linear algebraic and integral equations. Comput. Phys. Commun. 1982, 27, 13. [Google Scholar] [CrossRef]
- Berne, B.J.; Pecora, R. Dynamic Light Scattering: With Applications to Chemistry, Biology, and Physics; Dover Publications: Mineola, NY, USA, 1990. [Google Scholar]
- Porotto, M.; Fornabaio, M.; Kellogg, G.E.; Moscona, A. A second receptor binding site on human parainfluenza virus type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism. J. Virol. 2007, 81, 3216–3228. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequences and Modifications |
|---|---|
| MV HRC1 a | Ac–PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR-GSGSG–C–(CH2CONH2) |
| MV HRC2 a | Ac–PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR–GSGSG–C–(PEG4–Chol) |
| MV–HC | Ac–PPISLERLDVGTNLGNAIAKLEDAKELLESSDQILR–GSGSG–C–(25HC) |
| Peptide | Fusion Inhibition | |
|---|---|---|
| IC50 (μM) | IC90 (μM) | |
| MV HRC1 a | >10 | >10 |
| MV HRC2 a | 0.05 ± 0.01 | ~10 |
| MV–HC | 0.07 ± 0.005 | ~0.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomes, B.; Santos, N.C.; Porotto, M. Biophysical Properties and Antiviral Activities of Measles Fusion Protein Derived Peptide Conjugated with 25-Hydroxycholesterol. Molecules 2017, 22, 1869. https://doi.org/10.3390/molecules22111869
Gomes B, Santos NC, Porotto M. Biophysical Properties and Antiviral Activities of Measles Fusion Protein Derived Peptide Conjugated with 25-Hydroxycholesterol. Molecules. 2017; 22(11):1869. https://doi.org/10.3390/molecules22111869
Chicago/Turabian StyleGomes, Bárbara, Nuno C. Santos, and Matteo Porotto. 2017. "Biophysical Properties and Antiviral Activities of Measles Fusion Protein Derived Peptide Conjugated with 25-Hydroxycholesterol" Molecules 22, no. 11: 1869. https://doi.org/10.3390/molecules22111869
APA StyleGomes, B., Santos, N. C., & Porotto, M. (2017). Biophysical Properties and Antiviral Activities of Measles Fusion Protein Derived Peptide Conjugated with 25-Hydroxycholesterol. Molecules, 22(11), 1869. https://doi.org/10.3390/molecules22111869