Disrupting VEGF–VEGFR1 Interaction: De Novo Designed Linear Helical Peptides to Mimic the VEGF13-25 Fragment


Abstract
1. Introduction
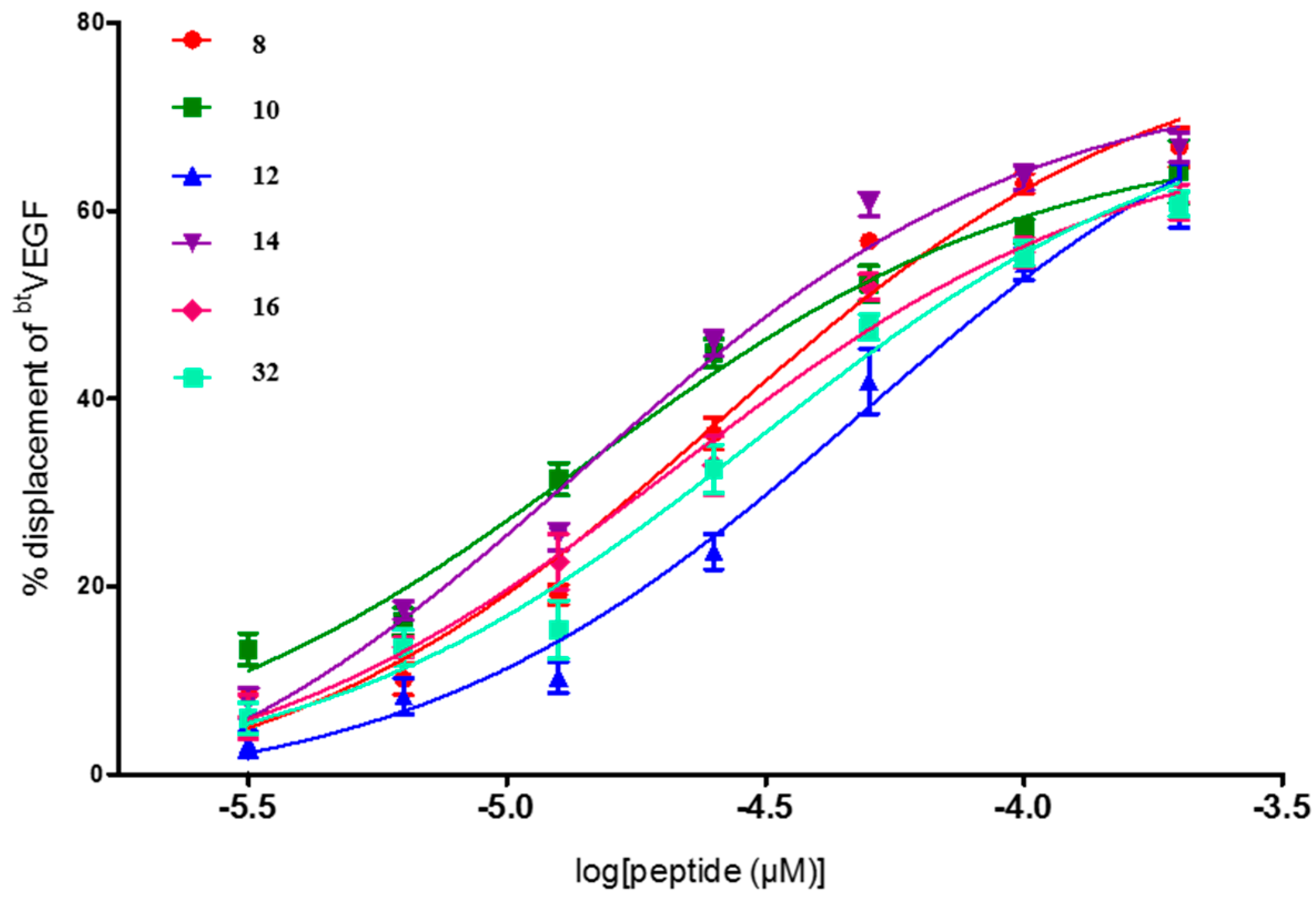
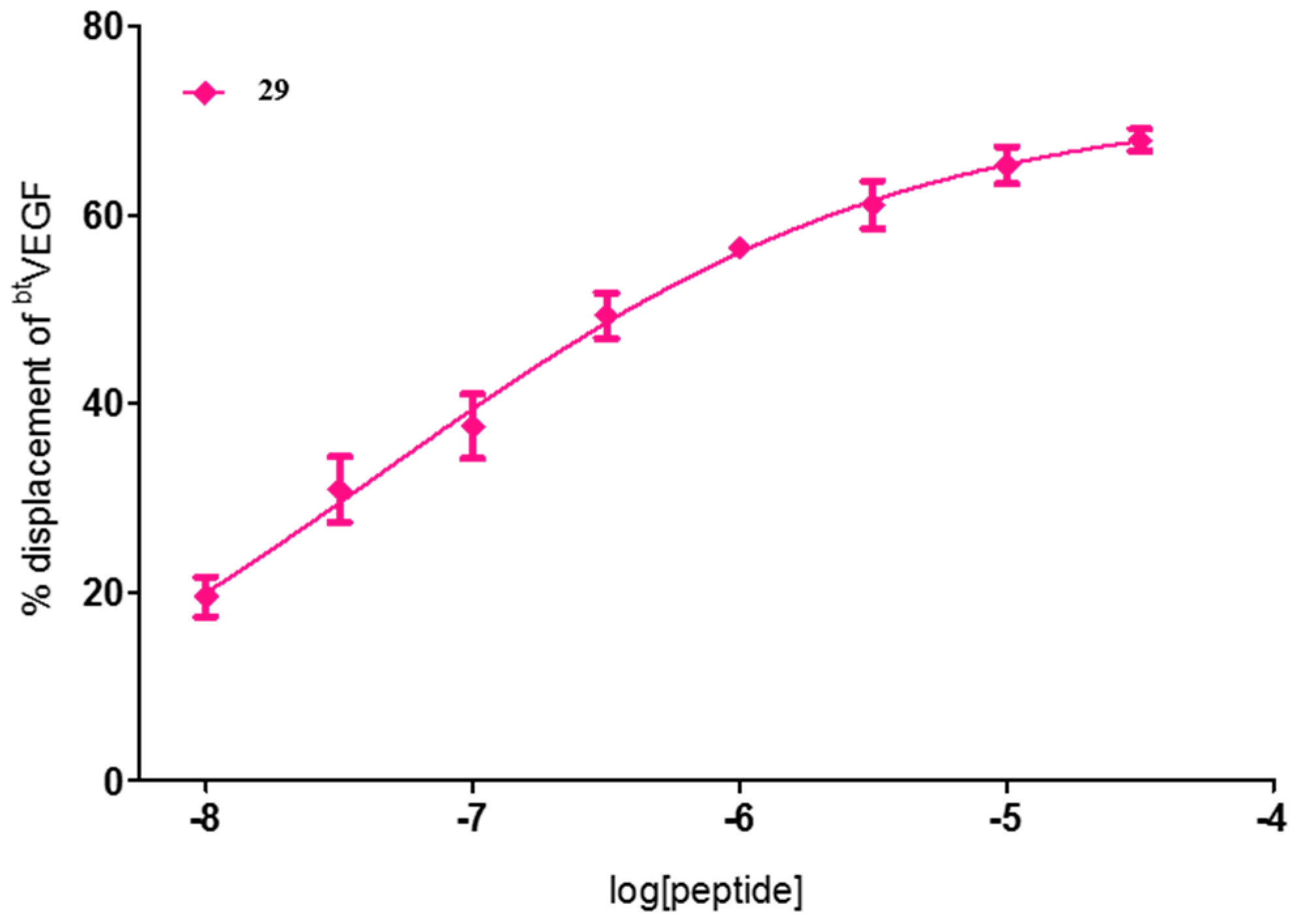
2. Results and Discussion
3. Experimental Section
3.1. Synthesis
3.2. Chemiluminescent Competition Assays
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ottmann, C. New Compound Classes: Protein–protein Interactions. Handb. Exp. Pharmacol. 2015, 232, 125–138. [Google Scholar] [CrossRef]
- Gurung, A.B.; Bhattacharjee, A.; Ajmal Ali, M.; Al-Hemaid, F.; Lee, J. Binding of small molecules at interface of protein–protein complex—A newer approach to rational drug design. Saudi J. Biol. Sci. 2017, 24, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Cardote, T.A.F.; Ciulli, A. Cyclic and macrocyclic peptides as chemical tools to recognise protein surfaces and probe protein–protein interactions. ChemMedChem 2016, 11, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Petta, I.; Lievens, S.; Libert, C.; Tavernier, J.; De Bosscher, K. Modulation of protein–protein interactions for the development of novel therapeutics. Mol. Ther. 2016, 24, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Zarzycka, B.; Kuenemann, M.A.; Miteva, M.A.; Nicolaes, G.A.F.; Vriend, G.; Sperandio, O. Stabilization of protein–protein interaction complexes through small molecules. Drug Discov. Today 2016, 21, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Aguilar, A.; Bernard, D.; Wang, S. Small-molecule inhibitors of the MDM2–p53 protein–protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment. J. Med. Chem. 2015, 58, 1038–1052. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.-H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef] [PubMed]
- Aileron Therapeutics. Available online: https://www.aileronrx.com/clinical-trials (accessed on 27 October 2017).
- Perez de Vega, M.J.; Martin-Martinez, M.; Gonzalez-Muniz, R. Modulation of protein–protein interactions by stabilizing/mimicking protein secondary structure elements. Curr. Top. Med. Chem. 2007, 7, 33–62. [Google Scholar] [CrossRef]
- Sawyer, N.; Watkins, A.M.; Arora, P.S. Protein domain mimics as modulators of protein–protein interactions. Acc. Chem. Res. 2017, 50, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Aiello, C.; Grieco, P.; Marasco, D. Targeting “undruggable” proteins: Design of synthetic cyclopeptides. Curr. Med. Chem. 2016, 23, 748–762. [Google Scholar] [CrossRef] [PubMed]
- Nevola, L.; Giralt, E. Modulating protein–protein interactions: The potential of peptides. Chem. Commun. 2015, 51, 3302–3315. [Google Scholar] [CrossRef] [PubMed]
- Scognamiglio, P.L.; Di Natale, C.; Perretta, G.; Marasco, D. From peptides to small molecules: An intriguing but intricated way to new drugs. Curr. Med. Chem. 2013, 20, 3803–3817. [Google Scholar] [CrossRef] [PubMed]
- Bonache, M.Á.; Balsera, B.; López-Méndez, B.; Millet, O.; Brancaccio, D.; Gómez-Monterrey, I.; Carotenuto, A.; Pavone, L.M.; Reille-Seroussi, M.; Gagey-Eilstein, N.; et al. De novo designed library of linear helical peptides: An exploratory tool in the discovery of protein–protein interaction modulators. ACS Comb. Sci. 2014, 16, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Horta, B.A.C.; Sodero, A.C.R.; de Alencastro, R.B. Investigating the differential activation of vascular endothelial growth factor (VEGF) receptors. J. Mol. Graph. Model. 2009, 28, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Muller, Y.A.; Christinger, H.W.; Keyt, B.A.; de Vos, A.M. The crystal structure of vascular endothelial growth factor (VEGF) refined to 1.93 A resolution: Multiple copy flexibility and receptor binding. Structure 1997, 5, 1325–1338. [Google Scholar] [CrossRef]
- Garcia-Aranda, M.I.; Gonzalez-Lopez, S.; Santiveri, C.M.; Gagey-Eilstein, N.; Reille-Seroussi, M.; Martin-Martinez, M.; Inguimbert, N.; Vidal, M.; Garcia-Lopez, M.T.; Jimenez, M.A.; et al. Helical peptides from VEGF and Vammin hotspots for modulating the VEGF-VEGFR interaction. Org. Biomol. Chem. 2013, 11, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, L.D.; Iaccarino, G.; Fattorusso, R.; Sorriento, D.; Carannante, C.; Capasso, D.; Trimarco, B.; Pedone, C. Targeting angiogenesis: Structural characterization and biological properties of a de novo engineered VEGF mimicking peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 14215–14220. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, V.; Gautier, B.; Garbay, C.; Vidal, M.; Inguimbert, N. Development of a chemiluminescent screening assay for detection of vascular endothelial growth factor receptor 1 ligands. Anal. Biochem. 2007, 366, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Ziaco, B.; Diana, D.; Capasso, D.; Palumbo, R.; Celentano, V.; Di Stasi, R.; Fattorusso, R.; D’Andrea, L.D. C-terminal truncation of Vascular Endothelial Growth Factor mimetic helical peptide preserves structural and receptor binding properties. Biochem. Biophys. Res. Commun. 2012, 424, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Basile, A.; Del Gatto, A.; Diana, D.; Di Stasi, R.; Falco, A.; Festa, M.; Rosati, A.; Barbieri, A.; Franco, R.; Arra, C.; et al. Characterization of a designed vascular endothelial growth factor receptor antagonist helical peptide with antiangiogenic activity in vivo. J. Med. Chem. 2011, 54, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, L.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Broussy, S.; Zhang, T.; Ji, L.; Vidal, M.; Liu, W.-Q. Identification of peptidic antagonists of vascular endothelial growth factor receptor 1 by scanning the binding epitopes of its ligands. J. Med. Chem. 2017, 60, 6598–6606. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Hensgens, C.M.; van der Laan, J.M.; Sutherland, J.D.; Hart, D.J.; Dijkstra, B.W. An approach to prevent aggregation during the purification and crystallization of wild type acyl coenzyme A: Isopenicillin N acyltransferase from Penicillium chrysogenum. Protein Expr. Purif. 2005, 41, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, J.-F.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Broussy, S.; Coric, P.; Seijo, B.; Lascombe, M.-B.; Gautier, B.; Liu, W.-Q.; Huguenot, F.; et al. Biophysical studies of the induced dimerization of human VEGF receptor 1 binding domain by divalent metals competing with VEGF-A. PLoS ONE 2016, 11, e0167755. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not Available. |



{kind=link}
{kind=link}
| Compd. No. | Compd. b X5X9X12 | % of Displacement c (100 μM) | IC50 (µM) d |
|---|---|---|---|
| 1 | FYY | 29 ± 3 | ND f |
| 2 | FYW | 48 ± 6 | 29 ± 5 |
| 3 | FWY | 50 ± 5 | 23 ± 4 |
| 4 | WYY | 45 ± 4 | >100 |
| 5 | FFY | 39 ± 6 | >100 |
| 6 | YYY | 33 ± 4 | ND |
| 7 | FYI | n.a. e | ND |
| QK g | - | 69 ± 3 | 32 ± 8 g |
| Compd. No. | Compd. X5X9X12 | % of Displacement b | ||
|---|---|---|---|---|
| 100 μM | 30 μM | 30 μM + EDTA | ||
| 8 | YWW | 63 ± 2 | 63 ± 1 | 40 ± 7 |
| 9 | FFF | 37 ± 3 | 47 ± 2 | - d |
| 10 | WFF | 55 ± 1 | 64 ± 3 | 55 ± 4 |
| 11 | YFF | 7 ± 4 | 5 ± 3 | - |
| 12 | FWF | 55 ± 1 | 66 ± 1 | 53 ± 4 |
| 13 | WWF | 58 ± 5 | 50 ± 4 | 34 ± 3 |
| 14 | YWF | 58 ± 1 | 62 ± 4 | 56 ± 6 |
| 15 | FYF | 27 ± 5 | 5 ± 4 | - |
| 16 | WYF | 62 ± 1 | 71 ± 2 | 44 ± 5 |
| 17 | YYF | 6 ± 1 | n.a c | - |
| 18 | WFW | 65 ± 2 | 61 ± 1 | 31 ± 2 |
| 19 | YFW | 54 ± 3 | 37 ± 1 | - |
| 20 | FWW | 43 ± 3 | 41 ± 3 | - |
| 21 | WWW | 78 ± 2 | 62 ± 6 | 21 ± 4 |
| 22 | FFW | 82 ± 1 | 63 ± 2 | 19 ± 3 |
| 23 | WYW | 50 ± 1 | 28 ± 2 | - |
| 24 | YYW | 70 ± 2 | 41 ± 2 | n.a |
| 25 | WFY | 43 ± 5 | 22 ± 3 | - |
| 26 | YFY | 38 ± 4 | 12 ± 5 | - |
| 27 | WWY | 64 ± 5 | 57 ± 3 | 32 ± 4 |
| 28 | YWY | 44 ± 4 | 44 ± 2 | - |
| 29 | FWI | 72 ± 1 | 60 ± 3 | 71 ± 2 |
| 30 | FIW | 47 ± 6 | 29 ± 7 | - |
| 31 | FYL | n.a | n.a | - |
| 32 | FIY | 60 ± 1 | 59 ± 4 | 55 ± 1 |
| 33 | FLY | 19 ± 7 | 6 ± 4 | - |
| Entry | Compd. X5X9X12 | IC50 a (μM) (95% Confident Interval) |
|---|---|---|
| 8 | YWW | 25.6 [18.6–35.1] |
| 10 | WFF | 14.5 [9.4–22.2] |
| 12 | FWF | 48.6 [31.8–74.3] |
| 14 | YWF | 14.0 [10.0–19.6] |
| 16 | WYF | 21.3 [13.9–32.7] |
| 29 | FWI | 4.6 × 10−2 [1.4 × 10−3–1.5] |
| 32 | FIY | 30.4 [19.4–47.5] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balsera, B.; Bonache, M.Á.; Reille-Seroussi, M.; Gagey-Eilstein, N.; Vidal, M.; González-Muñiz, R.; Pérez de Vega, M.J. Disrupting VEGF–VEGFR1 Interaction: De Novo Designed Linear Helical Peptides to Mimic the VEGF13-25 Fragment. Molecules 2017, 22, 1846. https://doi.org/10.3390/molecules22111846
Balsera B, Bonache MÁ, Reille-Seroussi M, Gagey-Eilstein N, Vidal M, González-Muñiz R, Pérez de Vega MJ. Disrupting VEGF–VEGFR1 Interaction: De Novo Designed Linear Helical Peptides to Mimic the VEGF13-25 Fragment. Molecules. 2017; 22(11):1846. https://doi.org/10.3390/molecules22111846
Chicago/Turabian StyleBalsera, Beatriz, M. Ángeles Bonache, Marie Reille-Seroussi, Nathalie Gagey-Eilstein, Michel Vidal, Rosario González-Muñiz, and María Jesús Pérez de Vega. 2017. "Disrupting VEGF–VEGFR1 Interaction: De Novo Designed Linear Helical Peptides to Mimic the VEGF13-25 Fragment" Molecules 22, no. 11: 1846. https://doi.org/10.3390/molecules22111846
APA StyleBalsera, B., Bonache, M. Á., Reille-Seroussi, M., Gagey-Eilstein, N., Vidal, M., González-Muñiz, R., & Pérez de Vega, M. J. (2017). Disrupting VEGF–VEGFR1 Interaction: De Novo Designed Linear Helical Peptides to Mimic the VEGF13-25 Fragment. Molecules, 22(11), 1846. https://doi.org/10.3390/molecules22111846