How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair

 ,
, 
Abstract
:1. Introduction
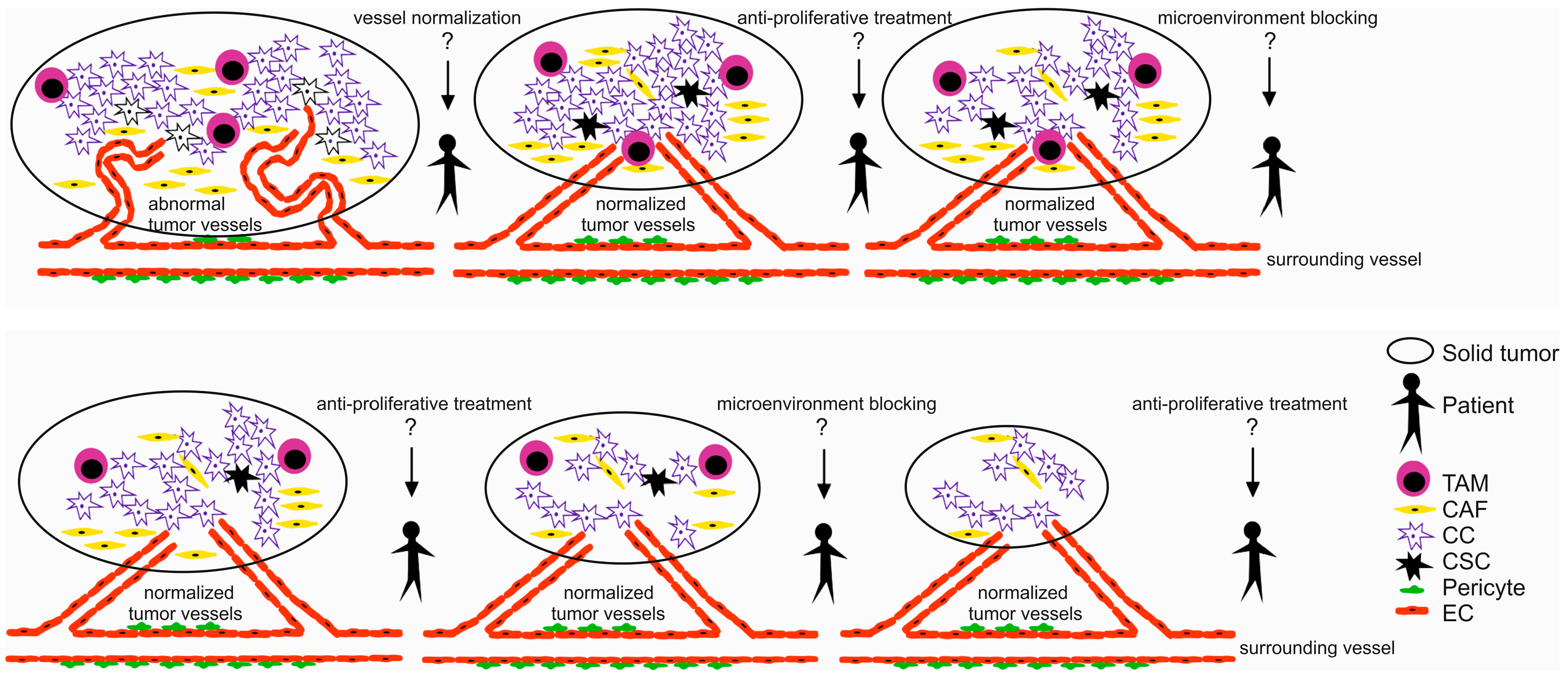
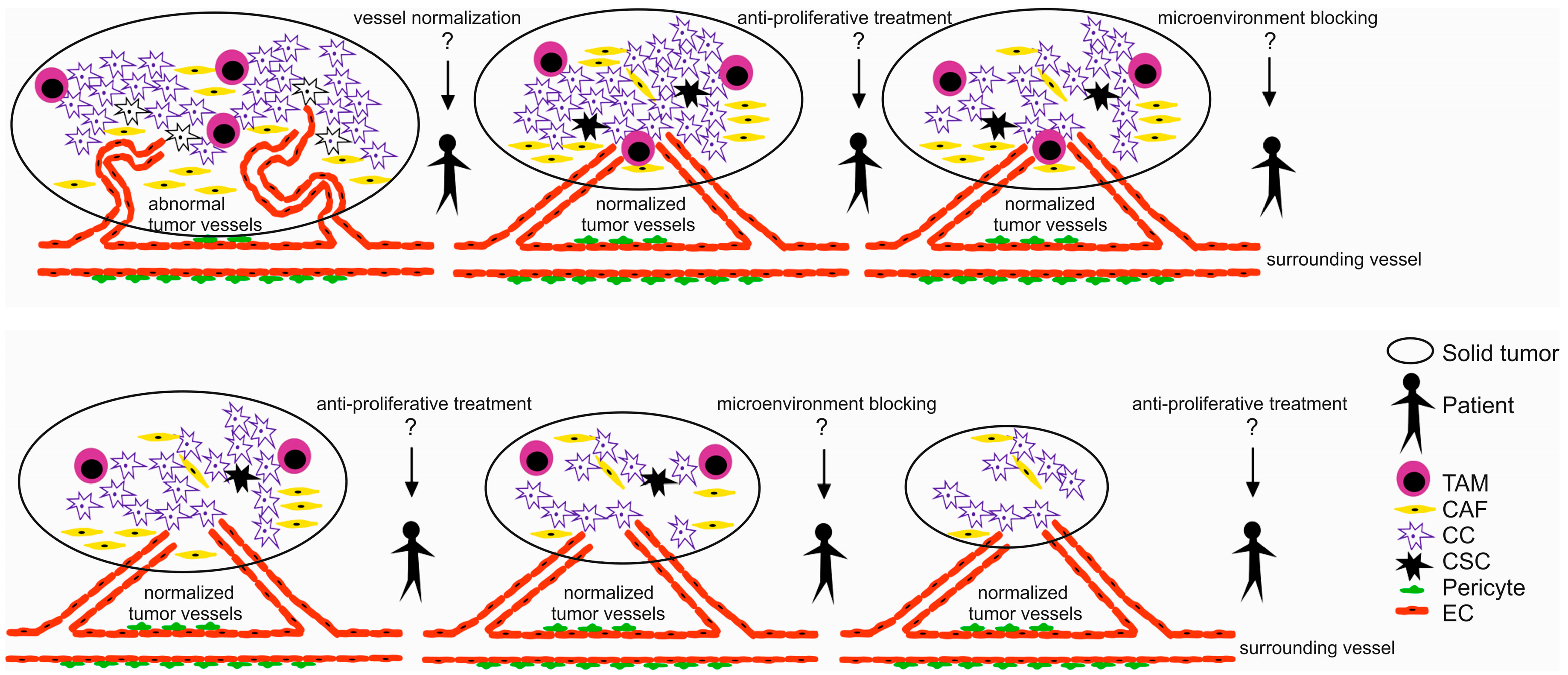
2. Tumor Microenvironment
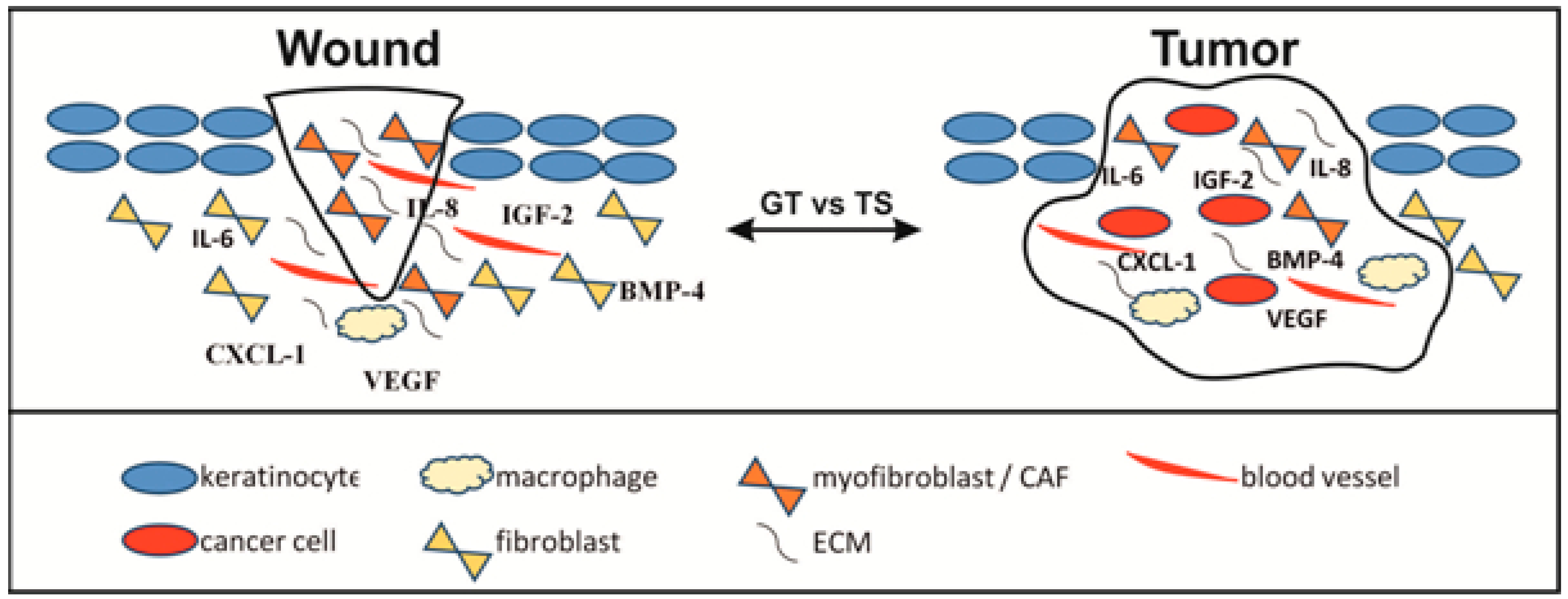
3. Wound/Keloid Scar Microenvironment and Its Similarity to TME
4. Roles of Cytokines/Chemokines and the Immune System in the Tumor/Wound Microenvironment Formation
5. Roles of Growth Factors in the Tumor/Wound Microenvironment Formation
6. Roles of Galectins in the Tumor/Wound Microenvironment Formation
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Korkaya, H.; Liu, S.; Wicha, M.S. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J. Clin. Investig. 2011, 121, 3804–3809. [Google Scholar] [CrossRef] [PubMed]
- Scatena, R.; Bottoni, P.; Pontoglio, A.; Giardina, B. Cancer stem cells: The development of new cancer therapeutics. Expert. Opin. Biol. Ther. 2011, 11, 875–892. [Google Scholar] [CrossRef] [PubMed]
- Gandalovicova, A.; Rosel, D.; Fernandes, M.; Vesely, P.; Heneberg, P.; Cermak, V.; Petruzelka, L.; Kumar, S.; Sanz-Moreno, V.; Brabek, J. Migrastatics-anti-metastatic and anti-invasion drugs: Promises and challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [PubMed]
- Dvorankova, B.; Szabo, P.; Lacina, L.; Gal, P.; Uhrova, J.; Zima, T.; Kaltner, H.; Andre, S.; Gabius, H.J.; Sykova, E.; et al. Human galectins induce conversion of dermal fibroblasts into myofibroblasts and production of extracellular matrix: Potential application in tissue engineering and wound repair. Cells Tissues Organs 2011, 194, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolar, M.; Szabo, P.; Dvorankova, B.; Lacina, L.; Gabius, H.J.; Strnad, H.; Sachova, J.; Vlcek, C.; Plzak, J.; Chovanec, M.; et al. Upregulation of il-6, il-8 and cxcl-1 production in dermal fibroblasts by normal/malignant epithelial cells in vitro: Immunohistochemical and transcriptomic analyses. Biol. Cell 2012, 104, 738–751. [Google Scholar] [CrossRef] [PubMed]
- Lacina, L.; Plzak, J.; Kodet, O.; Szabo, P.; Chovanec, M.; Dvorankova, B.; Smetana, K., Jr. Cancer microenvironment: What can we learn from the stem cell niche. Int. J. Mol. Sci. 2015, 16, 24094–24110. [Google Scholar] [CrossRef] [PubMed]
- Braund, R.; Hook, S.; Medlicott, N.J. The role of topical growth factors in chronic wounds. Curr. Drug Deliv. 2007, 4, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Plzak, J.; Lacina, L.; Chovanec, M.; Dvorankova, B.; Szabo, P.; Cada, Z.; Smetana, K., Jr. Epithelial-stromal interaction in squamous cell epithelium-derived tumors: An important new player in the control of tumor biological properties. Anticancer Res. 2010, 30, 455–462. [Google Scholar] [PubMed]
- Strnad, H.; Lacina, L.; Kolar, M.; Cada, Z.; Vlcek, C.; Dvorankova, B.; Betka, J.; Plzak, J.; Chovanec, M.; Sachova, J.; et al. Head and neck squamous cancer stromal fibroblasts produce growth factors influencing phenotype of normal human keratinocytes. Histochem. Cell Biol. 2010, 133, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Valach, J.; Fik, Z.; Strnad, H.; Chovanec, M.; Plzak, J.; Cada, Z.; Szabo, P.; Sachova, J.; Hroudova, M.; Urbanova, M.; et al. Smooth muscle actin-expressing stromal fibroblasts in head and neck squamous cell carcinoma: Increased expression of galectin-1 and induction of poor prognosis factors. Int. J. Cancer 2012, 131, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Formation and function of the myofibroblast during tissue repair. J. Investig. Dermatol. 2007, 127, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Smyth, M.J. Targeting cancer-related inflammation in the era of immunotherapy. Immunol. Cell Biol. 2017, 95, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Krejčí, E.; Dvořánková, B.; Szabo, P.; Naňka, O.; Strnad, H.; Kodet, O.; Lacina, L.; Kolář, M.; Smetana, K.J. Fibroblasts as drivers of healing and cancer progression: From in vitro experiments to clinics. In Molecular Mechanisms of Skin Aging and Age-Related Diseases; Quan, T., Ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 121–138. [Google Scholar]
- Majidinia, M.; Yousefi, B. Breast tumor stroma: A driving force in the development of resistance to therapies. Chem. Biol. Drug Des. 2017, 89, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Tlsty, T.D.; Coussens, L.M. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 2006, 1, 119–150. [Google Scholar] [CrossRef] [PubMed]
- Appleby, T.C.; Greenstein, A.E.; Hung, M.; Liclican, A.; Velasquez, M.; Villasenor, A.G.; Wang, R.; Wong, M.H.; Liu, X.; Papalia, G.A.; et al. Biochemical characterization and structure determination of a potent, selective antibody inhibitor of human MMP9. J. Biol. Chem. 2017, 292, 6810–6820. [Google Scholar] [CrossRef] [PubMed]
- Patra, D.; Sandell, L.J. Antiangiogenic and anticancer molecules in cartilage. Expert Rev. Mol. Med. 2012, 14, e10. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Robertson, A.A.; Cooper, M.A. Natural product and natural product derived drugs in clinical trials. Nat. Prod. Rep. 2014, 31, 1612–1661. [Google Scholar] [CrossRef] [PubMed]
- Hofheinz, R.D.; al-Batran, S.E.; Hartmann, F.; Hartung, G.; Jager, D.; Renner, C.; Tanswell, P.; Kunz, U.; Amelsberg, A.; Kuthan, H.; et al. Stromal antigen targeting by a humanised monoclonal antibody: An early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 2003, 26, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.; Chaitanya, K.; Wuest, T.; Wadle, A.; Scott, A.M.; van den Broek, M.; Schibli, R.; Bauer, S.; Renner, C. Radioimmunotherapy of fibroblast activation protein positive tumors by rapidly internalizing antibodies. Clin. Cancer Res. 2012, 18, 6208–6218. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Wang, C.T.; Ma, T.T.; Li, Z.Y.; Zhou, L.N.; Mu, B.; Leng, F.; Shi, H.S.; Li, Y.O.; Wei, Y.Q. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010, 101, 2325–2332. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Saga, Y.; Sato, N.; Nakamura, T.; Takikawa, O.; Mizukami, H.; Matsubara, S.; Fujiwara, H. The hepatocyte growth factor antagonist NK4 inhibits indoleamine-2,3-dioxygenase expression via the c-Met-phosphatidylinositol 3-kinase-AKT signaling pathway. Int. J. Oncol. 2016, 48, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hong, S.H.; Kim, J.Y.; Kim, I.C.; Park, Y.W.; Lee, S.J.; Song, S.W.; Kim, J.J.; Park, G.; Kim, T.M.; et al. Preclinical development of a humanized neutralizing antibody targeting hgf. Exp. Mol. Med. 2017, 49, e309. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Longoni, N.; Costales, P.; Dallavalle, C.; Garcia Inclan, C.; Albino, D.; Nunez, L.E.; Moris, F.; Carbone, G.M.; Catapano, C.V. EC-70124, a novel glycosylated indolocarbazole multikinase inhibitor, reverts tumorigenic and stem cell properties in prostate cancer by inhibiting STAT3 and NF-κB. Mol. Cancer Ther. 2016, 15, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, M.; Steurer, M.; Caligaris-Cappio, F.; Montillo, M.; Janssens, A.; Trentin, L.; Dummler, T.; Zollner, S.; Zeitler, S.; Riecke, K.; et al. Anti-CXCL12/SDF-1 spiegelmer (r) NOX-A12 alone and in combination with bendamustine and rituximab in patients with relapsed chronic lymphocytic leukemia (CLL): Results from a phase IIa study. Blood 2013, 122, 1635. [Google Scholar]
- Abraham, M.; Klein, S.; Bulvik, B.; Wald, H.; Weiss, I.D.; Olam, D.; Weiss, L.; Beider, K.; Eizenberg, O.; Wald, O.; et al. The CXCR4 inhibitor BL-8040 induces the apoptosis of aml blasts by downregulating ERK, BCL-2, MCL-1 and cyclin-C1 via altered miR-15a/16-1 expression. Leukemia 2017. [Google Scholar] [CrossRef] [PubMed]
- Cooper, T.M.; Sison, E.A.R.; Baker, S.D.; Li, L.; Ahmed, A.; Trippett, T.; Gore, L.; Macy, M.E.; Narendran, A.; August, K.; et al. A phase 1 study of the CXCR4 antagonist plerixafor in combination with high-dose cytarabine and etoposide in children with relapsed or refractory acute leukemias or myelodysplastic syndrome: A Pediatric Oncology Experimental Therapeutics Investigators' consortium study (POE 10-03). Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Onoyama, M.; Kitadai, Y.; Tanaka, Y.; Yuge, R.; Shinagawa, K.; Tanaka, S.; Yasui, W.; Chayama, K. Combining molecular targeted drugs to inhibit both cancer cells and activated stromal cells in gastric cancer. Neoplasia 2013, 15, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Kindler, H.; Gelderblom, H.; Schoffski, P.; Bauer, S.; Hohenberger, P.; Kopp, H.G.; Lopez-Martin, J.A.; Peeters, M.; Reichardt, P.; et al. A phase II study of a human anti-PDGFRα monoclonal antibody (olaratumab, IMC-3G3) in previously treated patients with metastatic gastrointestinal stromal tumors. Ann. Oncol. 2017, 28, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Appiah-Kubi, K.; Wang, Y.; Qian, H.; Wu, M.; Yao, X.; Wu, Y.; Chen, Y. Platelet-derived growth factor receptor/platelet-derived growth factor (PDGFR/PDGF) system is a prognostic and treatment response biomarker with multifarious therapeutic targets in cancers. Tumour. Biol. 2016, 37, 10053–10066. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFβ) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.J.; Santoro, A.; Gane, E.; Kelley, R.K.; Hourmand, I.I.; Assenat, E.; Gueorguieva, I.; Cleverly, A.; Desaiah, D.; Lahn, M.M.F.; et al. A phase 2 study of galunisertib, a novel transforming growth factor-beta (TGFβ) receptor I kinase inhibitor in patients with advanced hepatocellular carcinoma (HCC) and low serum alpha fetoprotein (AFP). J. Clin. Oncol. 2016, 34, 4070. [Google Scholar]
- Shepard, H.M. Breaching the castle walls: Hyaluronan depletion as a therapeutic approach to cancer therapy. Front. Oncol. 2015, 5, 192. [Google Scholar] [CrossRef] [PubMed]
- Roy, K.; Kanwar, R.K.; Kanwar, J.R. Targeted inhibition of tumour vascularisation using anti-PDGF/VEGF aptamers. Austin J. Nanomed. Nanotechnol. 2014, 2, 1027. [Google Scholar]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Tabernero, J.; Elez, E.; Cohen, S.J.; Bahleda, R.; van Laethem, J.L.; Ottensmeier, C.; Lopez-Martin, J.A.; Clive, S.; Joly, F.; et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 2192–2204. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Future options of anti-angiogenic cancer therapy. Chin. J. Cancer 2016, 35, 21. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.S.; Quinn, D.I.; Dorff, T.B. Clinical use of cabozantinib in the treatment of advanced kidney cancer: Efficacy, safety, and patient selection. OncoTargets Ther. 2016, 9, 5825–5837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, F.; Hsieh, T.C.; Wu, J.M.; Wu, E. Thalidomide-a notorious sedative to a wonder anticancer drug. Curr. Med. Chem. 2013, 20, 4102–4108. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhou, J.; Wu, J.; Shen, Y.; Li, X. Anti-angiogenic therapy: Strategies to develop potent VEGFR-2 tyrosine kinase inhibitors and future prospect. Curr. Med. Chem. 2016, 23, 1000–1040. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.C.; van Zandwijk, N.; Reid, G. Cilengitide inhibits attachment and invasion of malignant pleural mesothelioma cells through antagonism of integrins αvβ3 and αvβ5. PLoS ONE 2014, 9, e90374. [Google Scholar] [CrossRef] [PubMed]
- Millard, M.; Odde, S.; Neamati, N. Integrin targeted therapeutics. Theranostics 2011, 1, 154–188. [Google Scholar] [CrossRef] [PubMed]
- Colon-Otero, G.; Weroha, S.J.; Foster, N.R.; Haluska, P.; Hou, X.; Wahner-Hendrickson, A.E.; Jatoi, A.; Block, M.S.; Dinh, T.A.; Robertson, M.W.; et al. Phase 2 trial of everolimus and letrozole in relapsed estrogen receptor-positive high-grade ovarian cancers. Gynecol. Oncol. 2017, 146, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Walia, A.; Yang, J.F.; Huang, Y.H.; Rosenblatt, M.I.; Chang, J.H.; Azar, D.T. Endostatin’s emerging roles in angiogenesis, lymphangiogenesis, disease, and clinical applications. Biochim. Biophys. Acta 2015, 1850, 2422–2438. [Google Scholar] [CrossRef] [PubMed]
- Jeanne, A.; Schneider, C.; Martiny, L.; Dedieu, S. Original insights on thrombospondin-1-related antireceptor strategies in cancer. Front. Pharmacol. 2015, 6, 252. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Giobbie-Hurder, A.; Liao, X.; Connelly, C.; Connolly, E.M.; Li, J.; Manos, M.P.; Lawrence, D.; McDermott, D.; Severgnini, M.; et al. Angiopoietin-2 as a biomarker and target for immune checkpoint therapy. Cancer Immunol. Res. 2017, 5, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti- CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Reilley, M.J.; Bailey, A.; Subbiah, V.; Janku, F.; Naing, A.; Falchook, G.; Karp, D.; Piha-Paul, S.; Tsimberidou, A.; Fu, S.; et al. Phase i clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J. Immunother. Cancer 2017, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Linch, S.; Kasiewicz, M.J.; McNamara, M.; Hilgart, I.; Farhad, M.; Redmond, W. Galectin-3 inhibition using novel inhibitor GR-MD-02 improves survival and immune function while reducing tumor vasculature. J. Immunother. Cancer 2015, 3, 306. [Google Scholar] [CrossRef]
- Feng, X.; Clark, R.A.; Galanakis, D.; Tonnesen, M.G. Fibrin and collagen differentially regulate human dermal microvascular endothelial cell integrins: Stabilization of αv/β3 mRNA by fibrin1. J. Investig. Dermatol. 1999, 113, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Perzelova, V.; Varinska, L.; Dvorankova, B.; Szabo, P.; Spurny, P.; Valach, J.; Mojzis, J.; Andre, S.; Gabius, H.J.; Smetana, K., Jr.; et al. Extracellular matrix of galectin-1-exposed dermal and tumor-associated fibroblasts favors growth of human umbilical vein endothelial cells in vitro: A short report. Anticancer Res. 2014, 34, 3991–3996. [Google Scholar] [PubMed]
- Rhodes, J.M.; Simons, M. The extracellular matrix and blood vessel formation: Not just a scaffold. J. Cell Mol. Med. 2007, 11, 176–205. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.A.; Ghosh, K.; Tonnesen, M.G. Tissue engineering for cutaneous wounds. J. Investig. Dermatol. 2007, 127, 1018–1029. [Google Scholar] [CrossRef] [PubMed]
- Deonarine, K.; Panelli, M.C.; Stashower, M.E.; Jin, P.; Smith, K.; Slade, H.B.; Norwood, C.; Wang, E.; Marincola, F.M.; Stroncek, D.F. Gene expression profiling of cutaneous wound healing. J. Transl. Med. 2007, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Smetana, K., Jr.; Szabo, P.; Gal, P.; Andre, S.; Gabius, H.J.; Kodet, O.; Dvorankova, B. Emerging role of tissue lectins as microenvironmental effectors in tumors and wounds. Histol. Histopathol. 2015, 30, 293–309. [Google Scholar] [PubMed]
- Werner, S.; Krieg, T.; Smola, H. Keratinocyte-fibroblast interactions in wound healing. J. Investig. Dermatol. 2007, 127, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Motlik, J.; Klima, J.; Dvorankova, B.; Smetana, K., Jr. Porcine epidermal stem cells as a biomedical model for wound healing and normal/malignant epithelial cell propagation. Theriogenology 2007, 67, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumor stroma, tumor blood vessels, and antiangiogenesis therapy. Cancer J. 2015, 21, 237–243. [Google Scholar] [CrossRef] [PubMed]
- van den Broek, L.J.; Limandjaja, G.C.; Niessen, F.B.; Gibbs, S. Human hypertrophic and keloid scar models: Principles, limitations and future challenges from a tissue engineering perspective. Exp. Dermatol. 2014, 23, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Gauglitz, G.G.; Korting, H.C.; Pavicic, T.; Ruzicka, T.; Jeschke, M.G. Hypertrophic scarring and keloids: Pathomechanisms and current and emerging treatment strategies. Mol. Med. 2011, 17, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Jumper, N.; Paus, R.; Bayat, A. Functional histopathology of keloid disease. Histol. Histopathol. 2015, 30, 1033–1057. [Google Scholar] [PubMed]
- Rees, P.A.; Greaves, N.S.; Baguneid, M.; Bayat, A. Chemokines in wound healing and as potential therapeutic targets for reducing cutaneous scarring. Adv. Wound Care (New Rochelle) 2015, 4, 687–703. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.; Budd, D.C.; Shih, B.; Seifert, O.; Beaton, A.; Wright, T.; Dempsey, M.; Kelly, F.; Egerton, J.; Marshall, R.P.; et al. Transforming growth factor beta gene signatures are spatially enriched in keloid tissue biopsies and ex vivo-cultured keloid fibroblasts. Acta Derm. Venereol. 2017, 97, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Dienus, K.; Bayat, A.; Gilmore, B.F.; Seifert, O. Increased expression of fibroblast activation protein-alpha in keloid fibroblasts: Implications for development of a novel treatment option. Arch. Dermatol. Res. 2010, 302, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Bremnes, R.M.; Donnem, T.; Al-Saad, S.; Al-Shibli, K.; Andersen, S.; Sirera, R.; Camps, C.; Marinez, I.; Busund, L.T. The role of tumor stroma in cancer progression and prognosis: Emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Augsten, M. Cancer-associated fibroblasts and tumor growth-bystanders turning into key players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Erez, N.; Glanz, S.; Raz, Y.; Avivi, C.; Barshack, I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem. Biophys. Res. Commun. 2013, 437, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chen, B. Siltuximab (CNTO 328): A promising option for human malignancies. Drug Des. Devel. Ther. 2015, 9, 3455–3458. [Google Scholar] [CrossRef] [PubMed]
- Flechsig, P.; Dadrich, M.; Bickelhaupt, S.; Jenne, J.; Hauser, K.; Timke, C.; Peschke, P.; Hahn, E.W.; Grone, H.J.; Yingling, J.; et al. LY2109761 attenuates radiation-induced pulmonary murine fibrosis via reversal of TGF-β and BMP-associated proinflammatory and proangiogenic signals. Clin. Cancer Res. 2012, 18, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef] [PubMed]
- McFarland-Mancini, M.M.; Funk, H.M.; Paluch, A.M.; Zhou, M.; Giridhar, P.V.; Mercer, C.A.; Kozma, S.C.; Drew, A.F. Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J. Immunol. 2010, 184, 7219–7228. [Google Scholar] [CrossRef] [PubMed]
- Luckett-Chastain, L.R.; Gallucci, R.M. Interleukin (IL)-6 modulates transforming growth factor-β expression in skin and dermal fibroblasts from IL-6-deficient mice. Br. J. Dermatol. 2009, 161, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Jobe, N.P.; Rosel, D.; Dvorankova, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brabek, J. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit caf-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Jayatilaka, H.; Tyle, P.; Chen, J.J.; Kwak, M.; Ju, J.; Kim, H.J.; Lee, J.S.H.; Wu, P.H.; Gilkes, D.M.; Fan, R.; et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 2017, 8, 15584. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Takubo, K.; Shimizu, T.; Ohno, H.; Kishi, K.; Shibuya, M.; Saya, H.; Suda, T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J. Exp. Med. 2009, 206, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Ashcroft, G.S.; Mills, S.J.; Lei, K.; Gibbons, L.; Jeong, M.J.; Taniguchi, M.; Burow, M.; Horan, M.A.; Wahl, S.M.; Nakayama, T. Estrogen modulates cutaneous wound healing by downregulating macrophage migration inhibitory factor. J. Clin. Investig. 2003, 111, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Gilliver, S.C.; Emmerson, E.; Bernhagen, J.; Hardman, M.J. MIF: A key player in cutaneous biology and wound healing. Exp. Dermatol. 2011, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Izzo, J.G.; Correa, A.M.; Wu, T.T.; Malhotra, U.; Chao, C.K.; Luthra, R.; Ensor, J.; Dekovich, A.; Liao, Z.; Hittelman, W.N.; et al. Pretherapy nuclear factor-κB status, chemoradiation resistance, and metastatic progression in esophageal carcinoma. Mol. Cancer Ther. 2006, 5, 2844–2850. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Masood, M.; Rasul, A.; Wei, W.; Wang, Y.; Ali, M.; Mustaqeem, M.; Li, J.; Li, X. Altholactone inhibits NF-κB and STAT3 activation and induces reactive oxygen species-mediated apoptosis in prostate cancer DU145 cells. Molecules 2017, 22, 240. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Goktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.T.; Nooh, N.; Gillen, T.; Davey, M.; Patel, S.; Cottrell, D.; Amar, S. Il-1 plays a critical role in oral, but not dermal, wound healing. J. Immunol. 2001, 167, 5316–5320. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers (Basel) 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Spaeth, E.L.; Dembinski, J.L.; Sasser, A.K.; Watson, K.; Klopp, A.; Hall, B.; Andreeff, M.; Marini, F. Mesenchymal stem cell transition to tumor-associated fibroblasts contributes to fibrovascular network expansion and tumor progression. PLoS ONE 2009, 4, e4992. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, H.; Mundel, T.M.; Kieran, M.W.; Kalluri, R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol. Ther. 2006, 5, 1640–1646. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, K.S.; Tham, S.T.; Mohamed, Z.; Woo, Y.L.; Mat Adenan, N.A.; Chung, I. Cancer-associated fibroblasts promote proliferation of endometrial cancer cells. PLoS ONE 2013, 8, e68923. [Google Scholar] [CrossRef] [PubMed]
- Owens, P.; Polikowsky, H.; Pickup, M.W.; Gorska, A.E.; Jovanovic, B.; Shaw, A.K.; Novitskiy, S.V.; Hong, C.C.; Moses, H.L. Bone morphogenetic proteins stimulate mammary fibroblasts to promote mammary carcinoma cell invasion. PLoS ONE 2013, 8, e67533. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 2016, 21, 965. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Lan, L.; Jiang, Y.G.; Zhao, J.H.; Li, M.C.; Wei, N.B.; Lin, Y.H. Epithelial-mesenchymal transition and migration of prostate cancer stem cells is driven by cancer-associated fibroblasts in an HIF-1α/β-catenin-dependent pathway. Mol. Cells 2013, 36, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Croce, K.; Steensma, D.P.; McDermott, D.F.; Ben-Yehuda, O.; Moslehi, J. Vascular and metabolic implications of novel targeted cancer therapies: Focus on kinase inhibitors. J. Am. Coll. Cardiol. 2015, 66, 1160–1178. [Google Scholar] [CrossRef] [PubMed]
- Losi, P.; Briganti, E.; Errico, C.; Lisella, A.; Sanguinetti, E.; Chiellini, F.; Soldani, G. Fibrin-based scaffold incorporating VEGF- and BFGF-loaded nanoparticles stimulates wound healing in diabetic mice. Acta Biomater. 2013, 9, 7814–7821. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.A.; Eaglstein, W.H. Recombinant human platelet-derived growth factor gel speeds healing of acute full-thickness punch biopsy wounds. J. Am. Acad. Dermatol. 2001, 45, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Steed, D.L. Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity diabetic ulcers. Diabetic ulcer study group. J. Vasc. Surg. 1995, 21, 71–81. [Google Scholar] [CrossRef]
- Fernandez-Montequin, J.I.; Betancourt, B.Y.; Leyva-Gonzalez, G.; Mola, E.L.; Galan-Naranjo, K.; Ramirez-Navas, M.; Bermudez-Rojas, S.; Rosales, F.; Garcia-Iglesias, E.; Berlanga-Acosta, J.; et al. Intralesional administration of epidermal growth factor-based formulation (Heberprot-P) in chronic diabetic foot ulcer: Treatment up to complete wound closure. Int. Wound J. 2009, 6, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.R.; Wang, Y. Controlled delivery of heparin-binding EGF-like growth factor yields fast and comprehensive wound healing. J. Control. Release 2013, 166, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Mast, B.A.; Schultz, G.S. Interactions of cytokines, growth factors, and proteases in acute and chronic wounds. Wound Repair Regen. 1996, 4, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.R.; Mooney, D.J. Polymeric growth factor delivery strategies for tissue engineering. Pharm. Res. 2003, 20, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Solis, D.; Bovin, N.V.; Davis, A.P.; Jimenez-Barbero, J.; Romero, A.; Roy, R.; Smetana, K., Jr.; Gabius, H.J. A guide into glycosciences: How chemistry, biochemistry and biology cooperate to crack the sugar code. Biochim. Biophys. Acta 2015, 1850, 186–235. [Google Scholar] [CrossRef] [PubMed]
- Gal, P.; Vasilenko, T.; Kostelnikova, M.; Jakubco, J.; Kovac, I.; Sabol, F.; Andre, S.; Kaltner, H.; Gabius, H.J.; Smetana, K., Jr. Open wound healing in vivo: Monitoring binding and presence of adhesion/growth-regulatory galectins in rat skin during the course of complete re-epithelialization. Acta Histochem. Cytochem. 2011, 44, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Klima, J.; Lacina, L.; Dvorankova, B.; Herrmann, D.; Carnwath, J.W.; Niemann, H.; Kaltner, H.; Andre, S.; Motlik, J.; Gabius, H.J.; et al. Differential regulation of galectin expression/reactivity during wound healing in porcine skin and in cultures of epidermal cells with functional impact on migration. Physiol. Res. 2009, 58, 873–884. [Google Scholar] [PubMed]
- Thijssen, V.L.; Griffioen, A.W. Galectin-1 and -9 in angiogenesis: A sweet couple. Glycobiology 2014, 24, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, V.L.; Heusschen, R.; Caers, J.; Griffioen, A.W. Galectin expression in cancer diagnosis and prognosis: A systematic review. Biochim. Biophys. Acta 2015, 1855, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Stannard, K.; Gabutero, E.; Clark, A.M.; Neo, S.Y.; Onturk, S.; Blanchard, H.; Ralph, S.J. Galectin-1 as a potent target for cancer therapy: Role in the tumor microenvironment. Cancer Metastasis Rev. 2012, 31, 763–778. [Google Scholar] [CrossRef] [PubMed]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat. Rev. 2014, 40, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Rabien, A.; Sanchez-Ruderisch, H.; Schulz, P.; Otto, N.; Wimmel, A.; Wiedenmann, B.; Detjen, K.M. Tumor suppressor p16INK4a controls oncogenic K-Ras function in human pancreatic cancer cells. Cancer Sci. 2012, 103, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ruderisch, H.; Detjen, K.M.; Welzel, M.; Andre, S.; Fischer, C.; Gabius, H.J.; Rosewicz, S. Galectin-1 sensitizes carcinoma cells to anoikis via the fibronectin receptor α5β1-integrin. Cell Death Differ. 2011, 18, 806–816. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Ji, B.; Ramachandran, V.; Wang, H.; Hafley, M.; Logsdon, C.; Bresalier, R.S. Overexpressed galectin-3 in pancreatic cancer induces cell proliferation and invasion by binding Ras and activating Ras signaling. PLoS ONE 2012, 7, e42699. [Google Scholar] [CrossRef] [PubMed]
- Griffioen, A.W.; Thijssen, V.L. Galectins in tumor angiogenesis. Ann. Transl. Med. 2014, 2, 90. [Google Scholar] [PubMed]
- Cedeno-Laurent, F.; Dimitroff, C.J. Galectins and their ligands: Negative regulators of anti-tumor immunity. Glycoconj. J. 2012, 29, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Perillo, N.L.; Pace, K.E.; Seilhamer, J.J.; Baum, L.G. Apoptosis of t cells mediated by galectin-1. Nature 1995, 378, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.A.; Bianco, G.A.; Ilarregui, J.M.; Croci, D.O.; Correale, J.; Hernandez, J.D.; Zwirner, N.W.; Poirier, F.; Riley, E.M.; Baum, L.G.; et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 2007, 8, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Dalotto-Moreno, T.; Croci, D.O.; Cerliani, J.P.; Martinez-Allo, V.C.; Dergan-Dylon, S.; Mendez-Huergo, S.P.; Stupirski, J.C.; Mazal, D.; Osinaga, E.; Toscano, M.A.; et al. Targeting galectin-1 overcomes breast cancer-associated immunosuppression and prevents metastatic disease. Cancer Res. 2013, 73, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Juszczynski, P.; Ouyang, J.; Monti, S.; Rodig, S.J.; Takeyama, K.; Abramson, J.; Chen, W.; Kutok, J.L.; Rabinovich, G.A.; Shipp, M.A. The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classical Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2007, 104, 13134–13139. [Google Scholar] [CrossRef] [PubMed]
- Grigorian, A.; Torossian, S.; Demetriou, M. T-cell growth, cell surface organization, and the galectin-glycoprotein lattice. Immunol. Rev. 2009, 230, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Said, N.; Amin, S.; Wu, H.K.; Bruce, A.; Garate, M.; Hsu, D.K.; Kuwabara, I.; Liu, F.T.; Panjwani, N. Galectins-3 and -7, but not galectin-1, play a role in re-epithelialization of wounds. J. Biol. Chem. 2002, 277, 42299–42305. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Chen, J.S.; Wu, M.H.; Hsieh, I.S.; Liang, C.H.; Hsu, C.L.; Hong, T.M.; Chen, Y.L. Galectin-1 accelerates wound healing by regulating the neuropilin-1/SMAD3/NOX4 pathway and ROS production in myofibroblasts. J. Investig. Dermatol. 2015, 135, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, P.P. Galectin 3 as a guardian of the tumor microenvironment. Biochim. Biophys. Acta 2016, 1863, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Galectin Inhibitor (GR-MD-02) and Ipilimumab in Patients with Metastatic Melanoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02117362 (accessed on 19 October 2017).
- Walker, J.T.; Elliott, C.G.; Forbes, T.L.; Hamilton, D.W. Genetic deletion of galectin-3 does not impair full-thickness excisional skin healing. J. Investig. Dermatol. 2016, 136, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Seki, M.; Oomizu, S.; Sakata, K.M.; Sakata, A.; Arikawa, T.; Watanabe, K.; Ito, K.; Takeshita, K.; Niki, T.; Saita, N.; et al. Galectin-9 suppresses the generation of TH17, promotes the induction of regulatory t cells, and regulates experimental autoimmune arthritis. Clin. Immunol. 2008, 127, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Sindrewicz, P.; Lian, L.Y.; Yu, L.G. Interaction of the oncofetal Thomsen-Friedenreich antigen with galectins in cancer progression and metastasis. Front. Oncol. 2016, 6, 79. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.H.; Ying, N.W.; Wu, M.H.; Chiang, W.F.; Hsu, C.L.; Wong, T.Y.; Jin, Y.T.; Hong, T.M.; Chen, Y.L. Galectin-1, a novel ligand of neuropilin-1, activates VEGFR-2 signaling and modulates the migration of vascular endothelial cells. Oncogene 2008, 27, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Ying, N.W.; Hong, T.M.; Chiang, W.F.; Lin, Y.T.; Chen, Y.L. Galectin-1 induces vascular permeability through the neuropilin-1/vascular endothelial growth factor receptor-1 complex. Angiogenesis 2014, 17, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Gao, J.; Wang, S.; Ye, N.; Chong, Y.; Huang, Y.; Wang, J.; Li, B.; Yin, W.; Wang, D. Cancer-associated fibroblasts promote angiogenesis in gastric cancer through galectin-1 expression. Tumour. Biol. 2016, 37, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.I.; Jefferies, K.C.; Panjwani, N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J. Biol. Chem. 2011, 286, 29913–29921. [Google Scholar] [CrossRef] [PubMed]
- Van der Veldt, A.A.; Lammertsma, A.A.; Smit, E.F. Scheduling of anticancer drugs: Timing may be everything. Cell Cycle 2012, 11, 4339–4343. [Google Scholar] [CrossRef] [PubMed]
- Van der Veldt, A.A.; Lubberink, M.; Bahce, I.; Walraven, M.; de Boer, M.P.; Greuter, H.N.; Hendrikse, N.H.; Eriksson, J.; Windhorst, A.D.; Postmus, P.E.; et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: Implications for scheduling of anti-angiogenic drugs. Cancer Cell 2012, 21, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.J.; Gheordunescu, E.; Kakarala, P.; Newman, B.; Korkaya, H.; Heath, A.N.; Clouthier, S.G.; Wicha, M.S. Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, 2784–2789. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O. Cancer: Limitations of therapies exposed. Nature 2012, 484, 44–46. [Google Scholar] [CrossRef] [PubMed]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Jo, M.; Kim, Y.R.; Lee, C.K.; Hong, J.T. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Kwee, J.K. A paradoxical chemoresistance and tumor suppressive role of antioxidant in solid cancer cells: A strange case of Dr. Jekyll and Mr. Hyde. Biomed. Res. Int. 2014, 2014, 209845. [Google Scholar] [CrossRef] [PubMed]
- Koria, P. Delivery of growth factors for tissue regeneration and wound healing. Biodrugs 2012, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hwang, S.R.; Yoon, I.S. Advanced growth factor delivery systems in wound management and skin regeneration. Molecules 2017, 22, 1259. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Event | Wound | Squamous/Basal Cell Cancer |
|---|---|---|
| Infiltration by leukocytes | Wound bed, GT/Transitory | Stroma and between cancer cells/Continuous |
| Accumulation of fibroblasts | GT/Transitory | Stroma/Continuous |
| Production of ECM | ||
| New capillaries formation | ||
| Myofibroblast formation | ||
| Secretion of proteases from fibroblasts/myofibroblasts | ECM of GT remodelation/Transitory | Stroma remodelation/Continuous |
| Proliferation of epithelial cells | Reepithelisation/Transitory | Tumor growth/Continuous |
| EMT | Reepithelisation/Transitory | Locally aggressive growth and metastazing/Continuous |
| TME Targets | Strategy | Target + Drugs (Examples) | References | |
|---|---|---|---|---|
| ECM | Inhibition of ECM degradation | MMP inhibitors | Andecaliximab (GS-5745) (anti-MMP-9 monoclonal antibody) | [18] |
| Neovastat (shark cartilage extract AE-941) | [19] | |||
| Growth factors and signalling pathways | Inhibiotion of kinases and kinase receptor activity | Inhibition of kinases and kinase receptor activity | Genistein (protein-tyrosine kinase inhibitor, antioxidant) | [20] |
| Plitidepsin 171 (VEGF and VEGFR1 inhibitor, marine invertebrate compound) | ||||
| CAFs | Direct targeting of CAFs | FAP-α antibodies | Sibrotuzumab | [21] |
| Lu-labeled ESC11; Lu-labeled ESC14 | [22] | |||
| Vaccines targeting FAPα | [23] | |||
| CAF-epithelial interaction | HGF-Met signalling | NK4 (HGF antagonist) | [24] | |
| YYB-101 (monoclonal anti-HGF antibody) | [25] | |||
| NF-κB and STAT3 signaling pathway | EC-70124 (multikinase inhibitor) | [26] | ||
| CXCL12/SDF-1 | NOX-A12 (L-stereoisomer RNA aptamer (Spiegelmer)) | [27] | ||
| CXCR4 | BL-8040 (CXCR4 inhibitor) | [28] | ||
| Plerixafor (CXCR4 antagonist) | [29] | |||
| PDGF-R | Nilotinib (PDGF-R tyrosine kinase inhibitor) | [30] | ||
| Olaratumab (IMC-3G3) (anti-PDGFR-α monoclonal antibody | [31] | |||
| Crenolanib (inhibitor of receptor tyrosine kinases PDGFRα, -β; FLT3) | [32] | |||
| TGF-β ligand inhibitors | Fresolimumab (GC1008) (human anti-TGF-β monoclonal antibody) | [33] | ||
| TGF-β receptor inhibitor | Galunisertib TGF-βRI (TGF-beta receptor I kinase inhibitor) | [34] | ||
| CAF-ECM interaction | Hyaluronan | rHuPH20 (recombinant human hyaluronidase enzyme) | [35] | |
| CAF-endothelial interaction | PDGF-B | E10030 (Fovista) RNA-based anti-PDGFR aptamer | [36] | |
| CAF—inflammatory immune cell interactions | IL-6 | Siltuximab (anti-IL-6 monoclonal antibody) | [37] | |
| TNF | Inflinximab and Etanercept (TNF inhibitors) | [38] | ||
| Angiogenesis | Growth factors | Bevacizumab (VEGF-A antibody); Aflibercept (chimeric soluble receptor); VEGF-trap; Thalidomide; Lenalidomide; IMC-18F1 (VEGFR-1 signaling); Ramucirumab (VEGFR-2 signaling) | [39,40,41] | |
| Small molecules tyrosine kinase inhibitors | Sunitinib; Sorafenib; Pazopanib; Axitinib; Vandetanib; Regorafenib; Cabozantinib; Motesanib; Cediranib; Tivozanib | [42] | ||
| Intergrin inhibitors | MEDI-522 (Vitaxin); Cilengitide (EMD 121974); Volociximab (chimeric monoclonal antibody) | [43,44] | ||
| mTOR | Everolimus | [45] | ||
| Human antiangiogenic factors | Endostatin | [46] | ||
| Thrombospondin-1 | [47] | |||
| Angiopoietin | Trebananib AGM 386 (angiopoietin-1/-2-neutralizing peptibody) | [48] | ||
| Immune system | CSF-1 | RG7155 (monoclonal antibody against CSF-1 receptor activation) | [49] | |
| CTLA-4 | Ipilimumab (CTLA-4 monoclonal antibody) | [50] | ||
| Galectins | Galectin-3 | GR-MD-02 | [51] | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gál, P.; Varinská, L.; Fáber, L.; Novák, Š.; Szabo, P.; Mitrengová, P.; Mirossay, A.; Mučaji, P.; Smetana, K. How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair. Molecules 2017, 22, 1818. https://doi.org/10.3390/molecules22111818
Gál P, Varinská L, Fáber L, Novák Š, Szabo P, Mitrengová P, Mirossay A, Mučaji P, Smetana K. How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair. Molecules. 2017; 22(11):1818. https://doi.org/10.3390/molecules22111818
Chicago/Turabian StyleGál, Peter, Lenka Varinská, Lenka Fáber, Štepán Novák, Pavol Szabo, Petra Mitrengová, Andrej Mirossay, Pavel Mučaji, and Karel Smetana. 2017. "How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair" Molecules 22, no. 11: 1818. https://doi.org/10.3390/molecules22111818
APA StyleGál, P., Varinská, L., Fáber, L., Novák, Š., Szabo, P., Mitrengová, P., Mirossay, A., Mučaji, P., & Smetana, K. (2017). How Signaling Molecules Regulate Tumor Microenvironment: Parallels to Wound Repair. Molecules, 22(11), 1818. https://doi.org/10.3390/molecules22111818