Differential Interaction of Antimicrobial Peptides with Lipid Structures Studied by Coarse-Grained Molecular Dynamics Simulations

Abstract
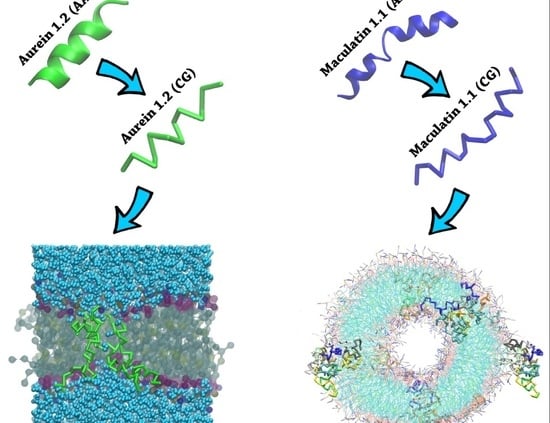
1. Introduction
2. Results and Discussion
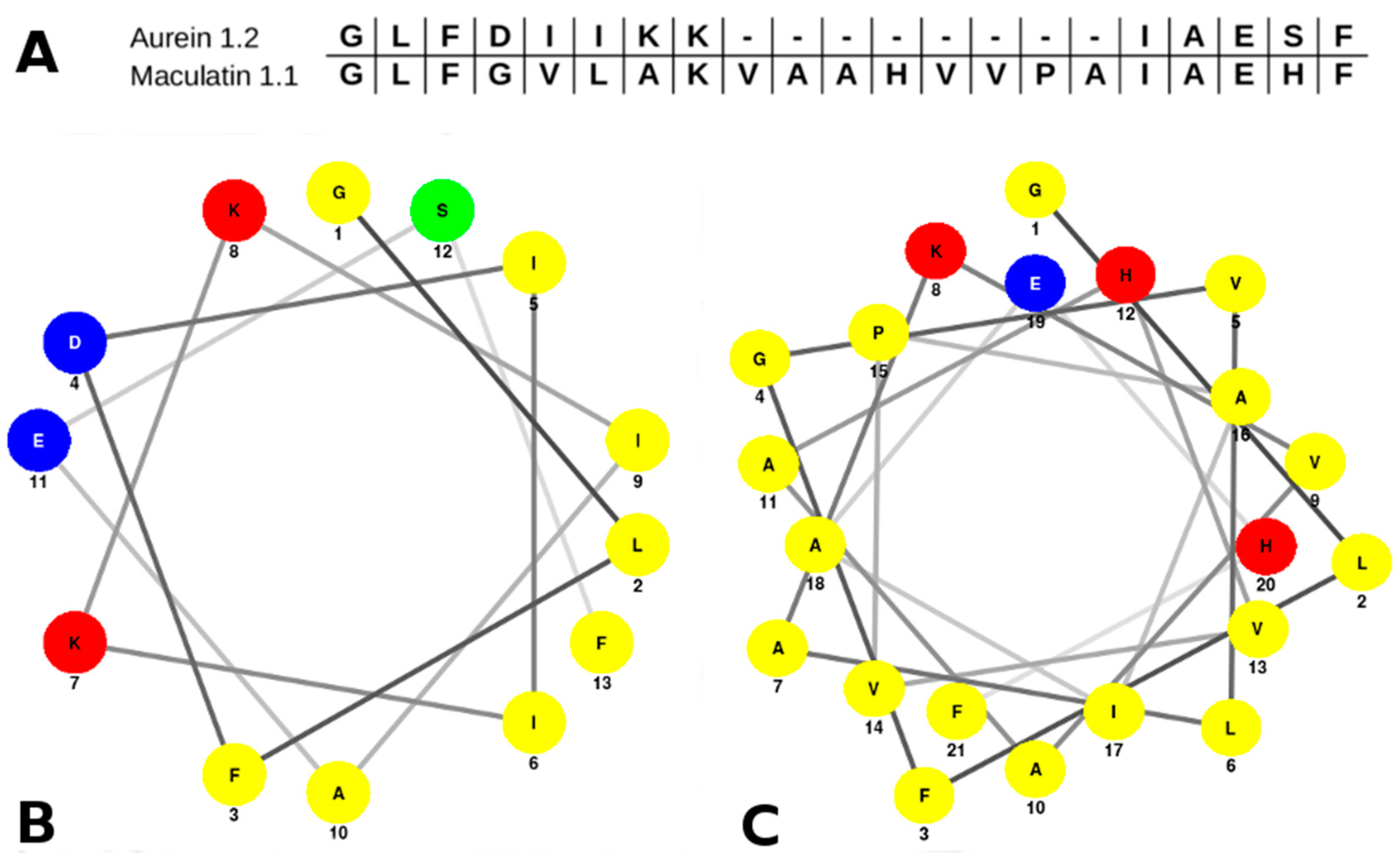
2.1. Aurein 1.2 and Maculatin 1.1 Structural Overview
2.2. MD Simulations
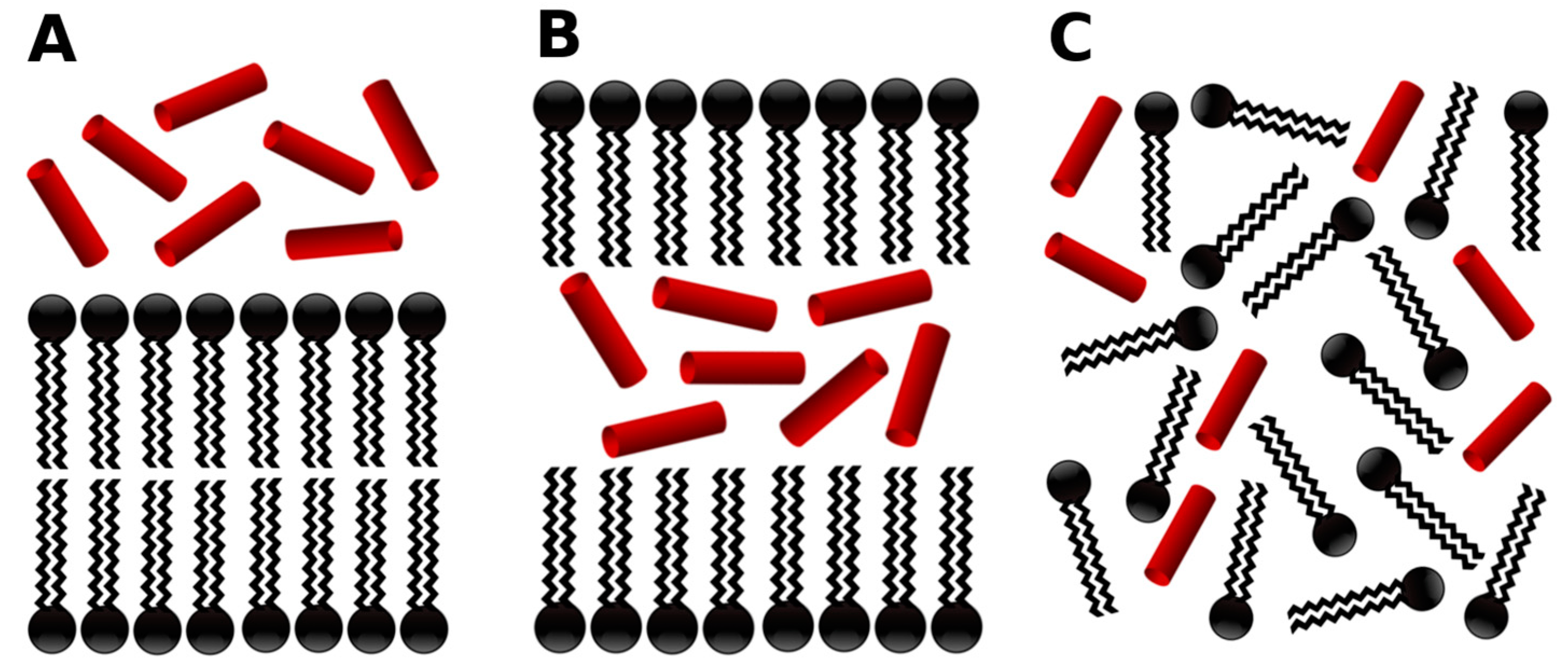
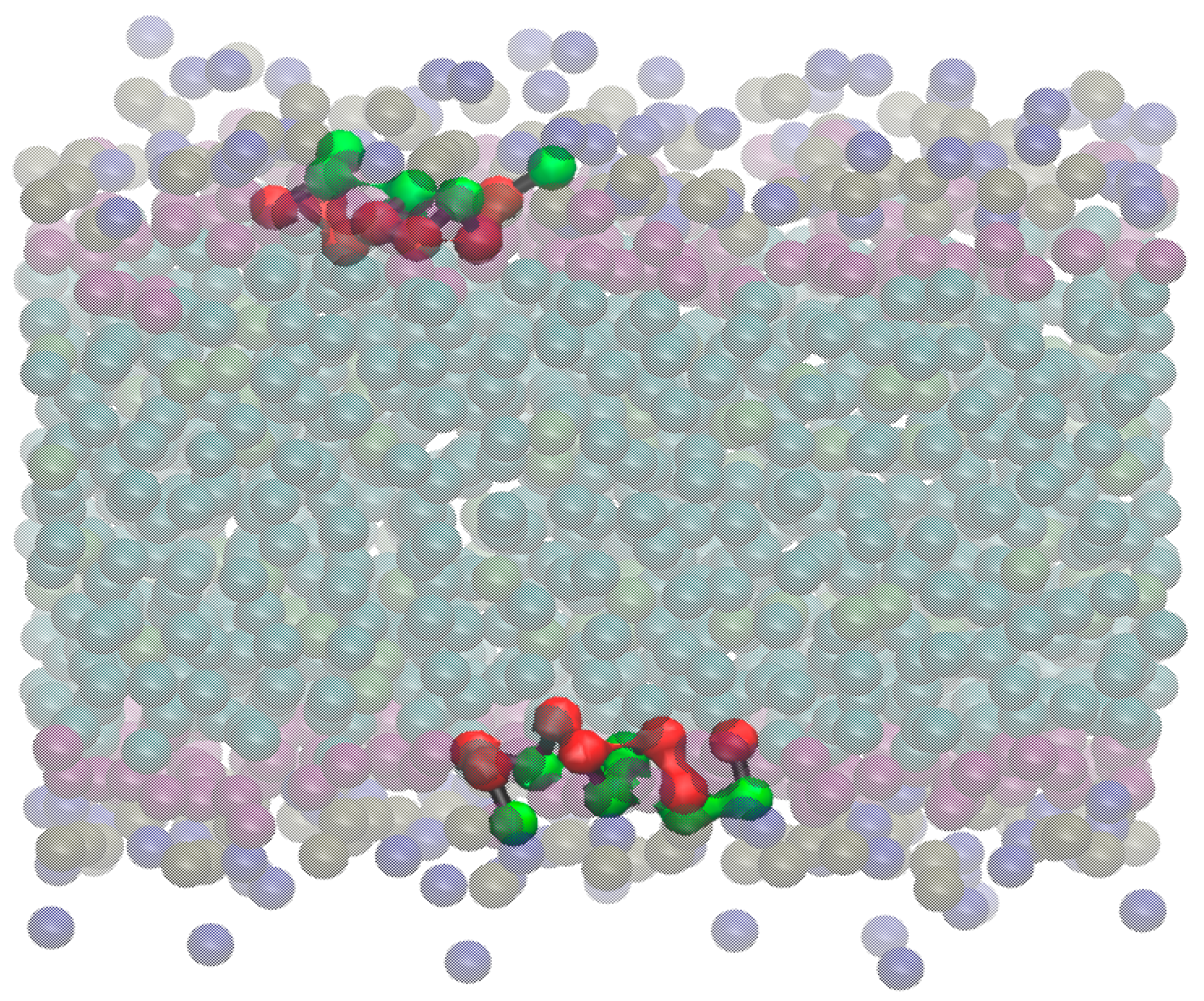
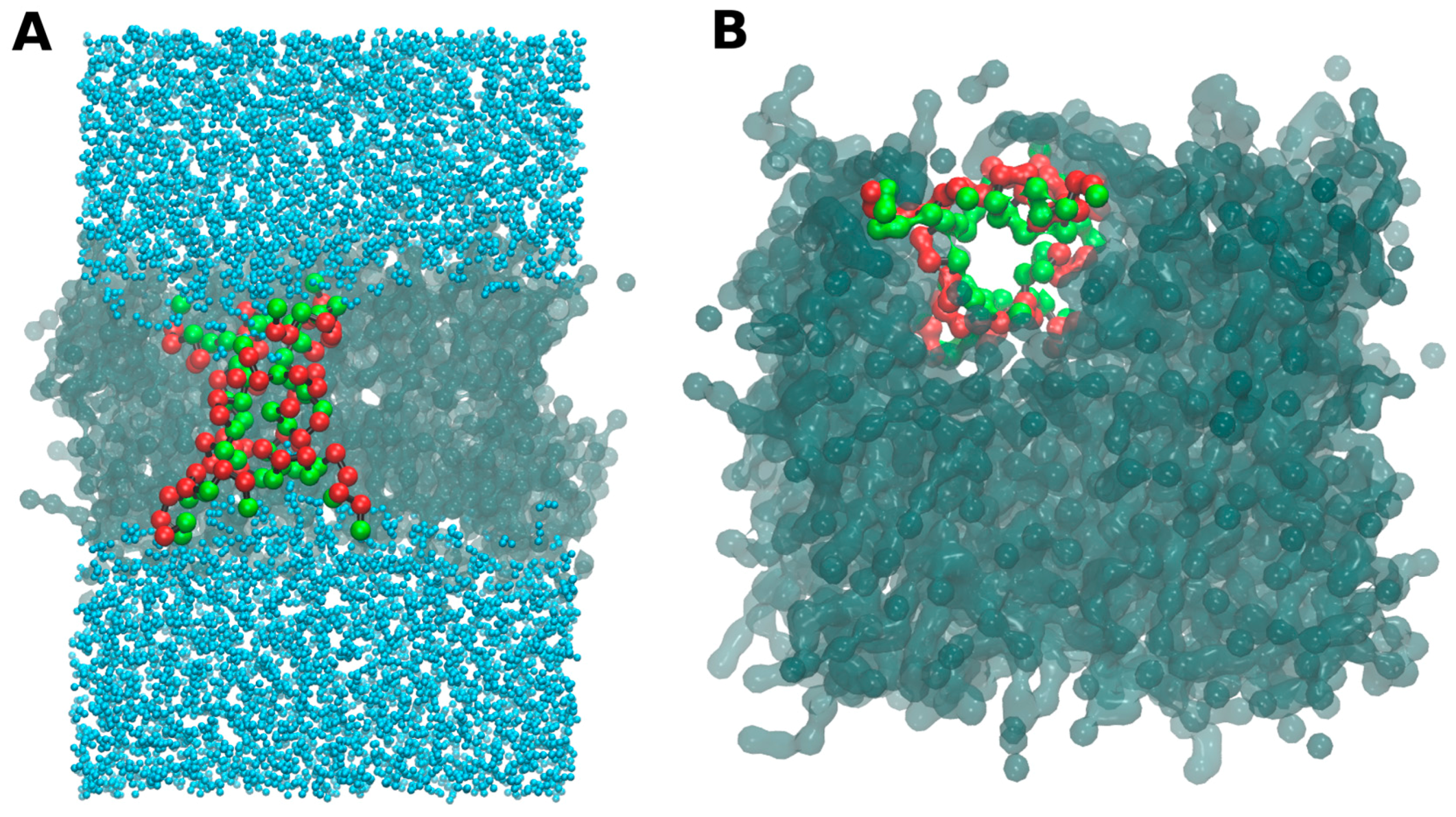
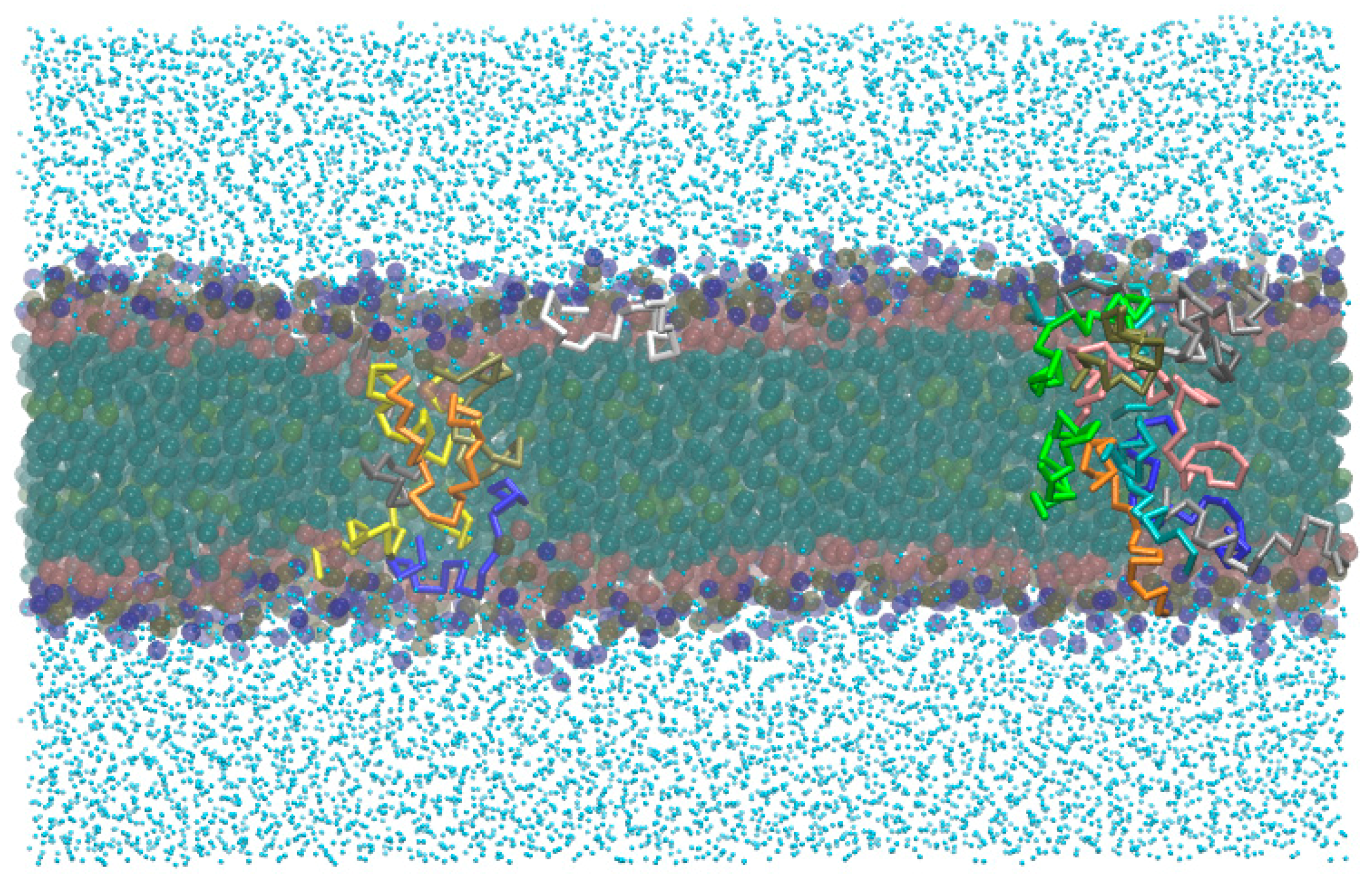
- -out systems: peptides were placed homogeneously distributed across the XY plane, 3–5 nm away from a pre-assembled lipid bilayer (Figure 2A).
- -in systems: peptides were placed inside the hydrophobic core of a pre-assembled lipid bilayer (Figure 2B).
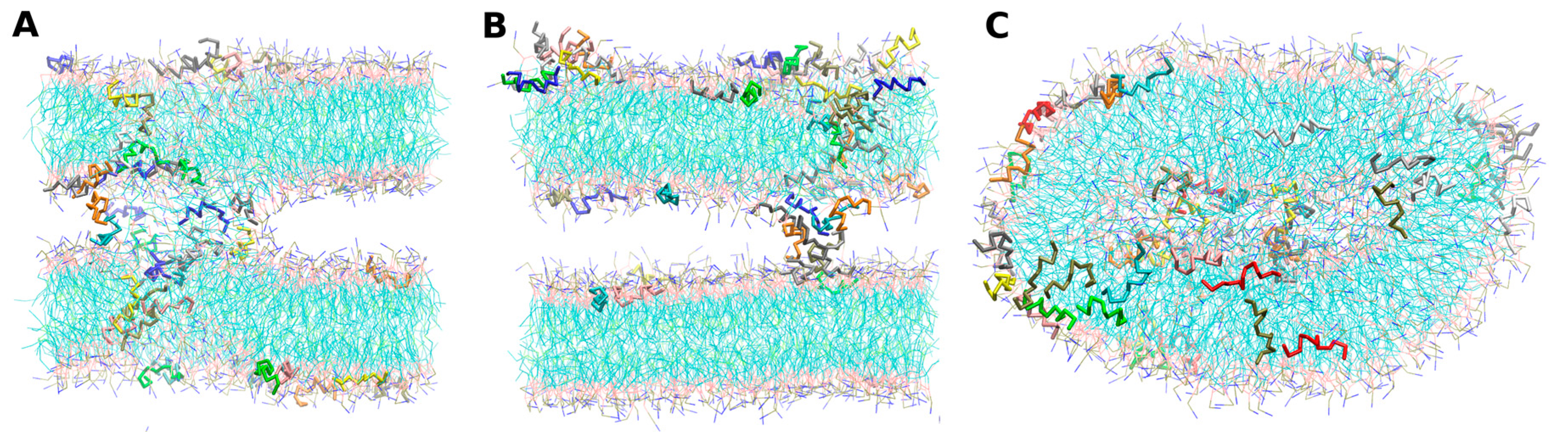
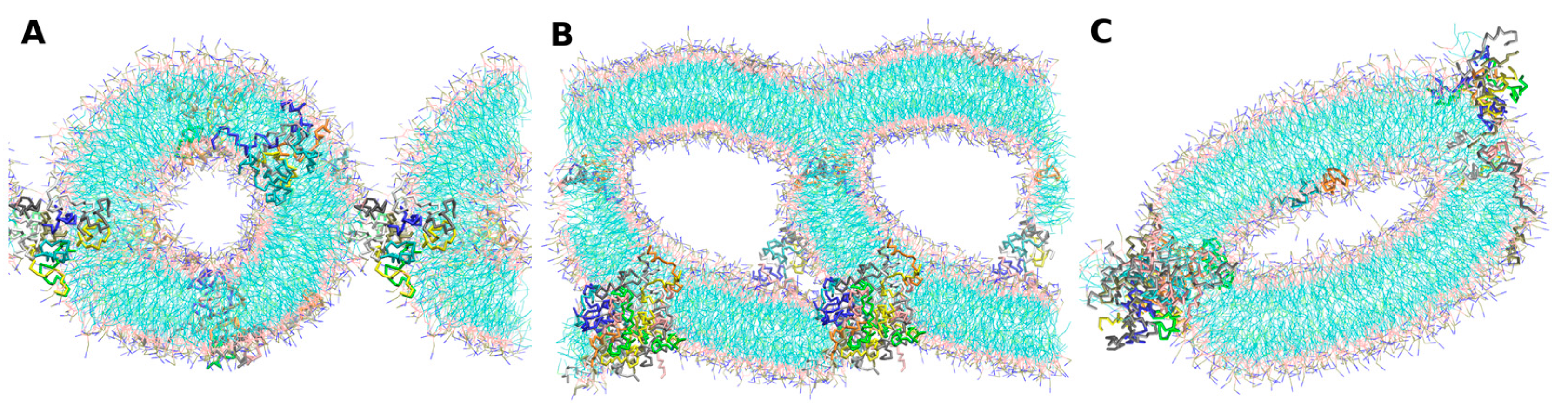
- -self systems: molecules were randomly distributed in the simulation box. We have followed three different randomization configurations in order to validate the strategy (as discussed below, and illustrated in Figure 2C).
- Self-A, a homogeneous distribution of molecules, where peptides and lipids were uniformly distributed with no preferential orientation or proximity.
- Self-B, with peptides closely located and, thus, prioritizing the peptide–peptide proximity.
- Self-C, where the lipid–lipid interaction was favored following the same as for self-B.
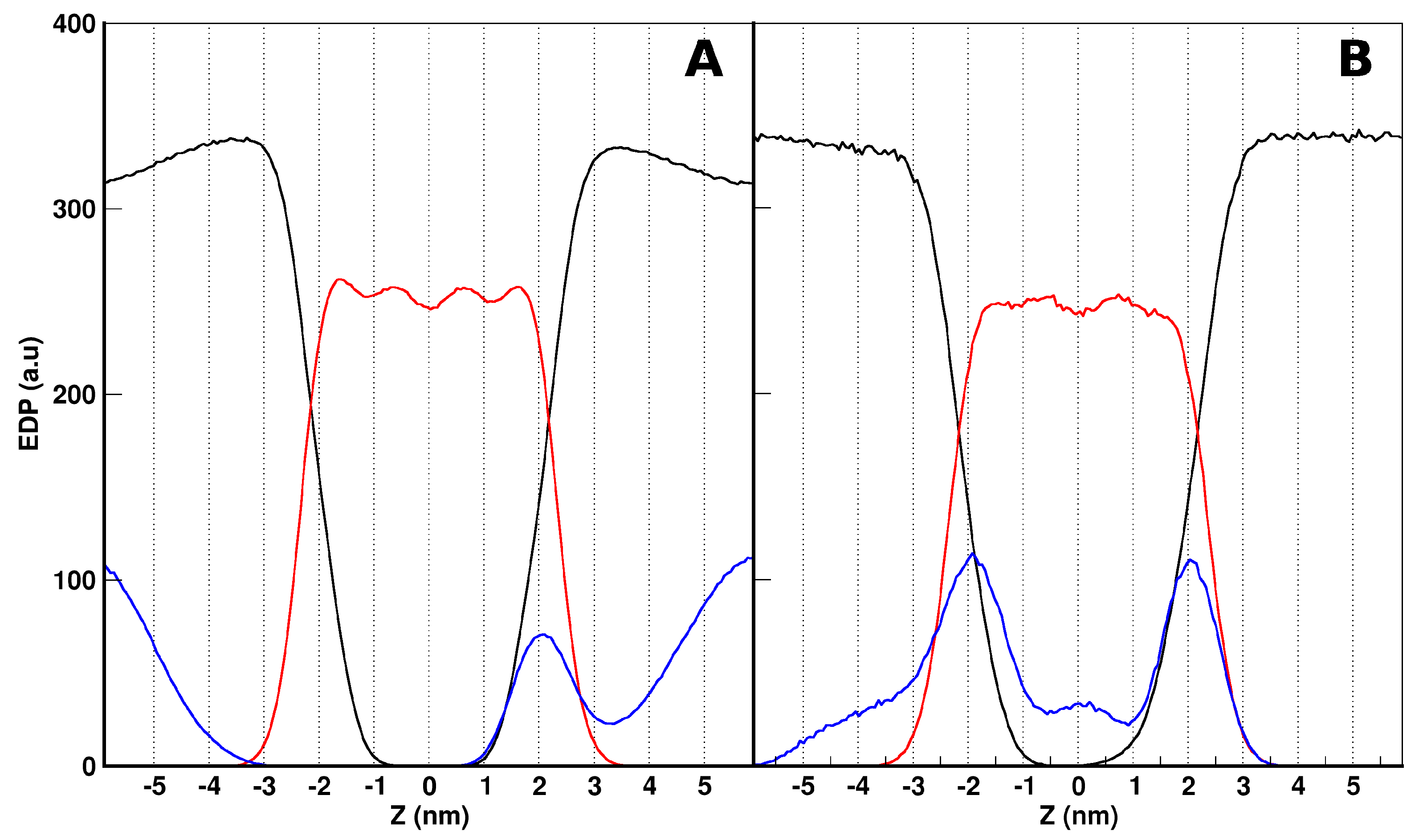
Aurein/POPC Simulations
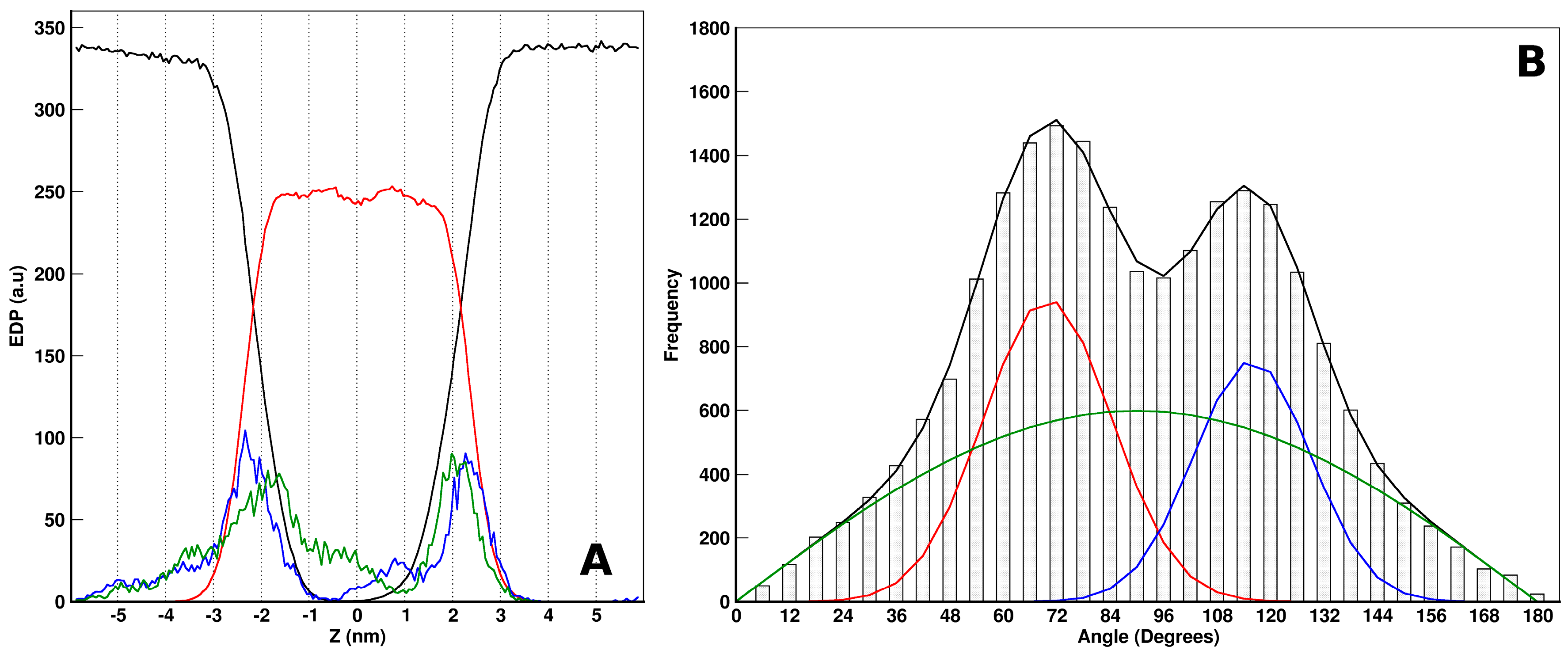
2.3. MD Simulations: Maculatin/POPC Systems
2.4. Peptides Differential Behavior
3. Methods
3.1. Coarse-Grain Model
3.2. Sequence and Structure Analysis Tools
3.3. MD Simulation Conditions
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski, L.S.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: The various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Wieprecht, T. Structural features of helical antimicrobial peptides: Their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1999, 1462, 71–87. [Google Scholar] [CrossRef]
- Reddy, K.V.R.; Yedery, R.D.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Kosikowska, P.; Lesner, A. Antimicrobial peptides (AMPs) as drug candidates: A patent review (2003–2015). Expert Opin. Ther. Pat. 2016, 26, 689–702. [Google Scholar] [CrossRef] [PubMed]
- Rozek, T.; Wegener, K.L.; Bowie, J.H.; Olver, I.N.; Carver, J.A.; Wallace, J.C.; Tyler, M.J. The antibiotic and anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria raniformis: The solution structure of aurein 1.2. Eur. J. Biochem. 2000, 267, 5330–5341. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar] [CrossRef] [PubMed]
- De la Fuente-Núñez, C.; Cardoso, M.H.; de Souza Cândido, E.; Franco, O.L.; Hancock, R.E.W. Synthetic antibiofilm peptides. Biochim. Biophys. Acta 2016, 1858, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E.W. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 2013, 9, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E.W. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2011. [Google Scholar] [CrossRef] [PubMed]
- Bahar, A.A.; Ren, D. Antimicrobial peptides. Pharmaceuticals 2013, 6, 1543–1575. [Google Scholar] [CrossRef] [PubMed]
- Tossi, A.; Sandri, L.; Giangaspero, A. Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides–Using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial Peptides in Action Antimicrobial Peptides in Action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef] [PubMed]
- King, M.J.; Bennett, A.L.; Almeida, P.F.; Lee, H.S. Coarse-grained simulations of hemolytic peptide δ-lysin interacting with a POPC bilayer. Biochim. Biophys. Acta 2016, 1858, 3182–3194. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sinha, S.K.; Patel, S. Reconciling structural and thermodynamic predictions using all-atom and coarse-grain force fields: The case of charged oligo-arginine translocation into dmpc bilayers. J. Phys. Chem. B 2014, 118, 11973–11992. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Forsman, J.; Woodward, C.E. Amphipathic membrane-active peptides recognize and stabilize ruptured membrane pores: Exploring cause and effect with coarse-grained simulations. Langmuir 2015, 31, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.J.; Parton, D.L.; Clark, J.F.; Sansom, M.S.P. Coarse-Grained Simulations of the Membrane-Active Antimicrobial Peptide Maculatin 1.1. Biophys. J. 2008, 95, 3802–3815. [Google Scholar] [CrossRef] [PubMed]
- Parton, D.L.; Akhmatskaya, E.V.; Sansom, M.S.P. Multiscale simulations of the antimicrobial peptide maculatin 1.1: Water permeation through disordered aggregates. J. Phys. Chem. B 2012, 116, 8485–8493. [Google Scholar] [CrossRef] [PubMed]
- Santo, K.P.; Irudayam, S.J.; Berkowitz, M.L. Melittin creates transient pores in a lipid bilayer: Results from computer simulations. J. Phys. Chem. B 2013, 117, 5031–5042. [Google Scholar] [CrossRef] [PubMed]
- Santo, K.P.; Berkowitz, M.L. Difference between magainin-2 and melittin assemblies in phosphatidylcholine bilayers: Results from coarse-grained simulations. J. Phys. Chem. B 2012, 116, 3021–3030. [Google Scholar] [CrossRef] [PubMed]
- Cruz, V.L.; Ramos, J.; Melo, M.N.; Martinez-Salazar, J. Bacteriocin AS-48 binding to model membranes and pore formation as revealed by coarse-grained simulations. Biochim. Biophys. Acta 2013, 1828, 2524–2531. [Google Scholar] [CrossRef] [PubMed]
- Chia, B.C.S.; Carver, J.A.; Mulhern, T.D.; Bowie, J.H. Maculatin 1.1, an anti-microbial peptide from the Australian tree frog, Litoria genimaculata. Solution structure and biological activity. Eur. J. Biochem. 2000, 267, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Ambroggio, E.E.; Separovic, F.; Bowie, J.H.; Fidelio, G.D.; Bagatolli, L.A. Direct Visualization of Membrane Leakage Induced by the Antibiotic Peptides: Maculatin, Citropin, and Aurein. Biophys. J. 2005, 89, 1874–1881. [Google Scholar] [CrossRef] [PubMed]
- Hopp, T.P.; Woods, K.R. Prediction of protein antigenic determinants from amino acid sequences. Immunology 1981, 78, 3824–3828. [Google Scholar] [CrossRef]
- Fernández-Vidal, M.; Jayasinghe, S.; Ladokhin, A.S.; White, S.H. Folding Amphipathic Helices Into Membranes: Amphiphilicity Trumps Hydrophobicity. J. Mol. Biol. 2007, 370, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bogaart, G.; Guzmán, J.V.; Mika, J.T.; Poolman, B. On the mechanism of pore formation by melittin. J. Biol. Chem. 2008, 283, 33854–33857. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.I.; Gehman, J.D.; Separovic, F. Membrane interactions of antimicrobial peptides from Australian frogs. Biochim. Biophys. Acta 2009, 1788, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Bond, P.J.; Holyoake, J.; Ivetac, A.; Khalid, S.; Sansom, M.S.P. Coarse-grained molecular dynamics simulations of membrane proteins and peptides. J. Struct. Biol. 2007, 157, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Shahmiri, M.; Enciso, M.; Mechler, A. Controls and constrains of the membrane disrupting action of Aurein 1.2. Sci. Rep. 2015, 5, 16378. [Google Scholar] [CrossRef] [PubMed]
- Bennett, W.F.D.; Tieleman, D.P. Water defect and pore formation in atomistic and coarse-grained lipid membranes: Pushing the limits of coarse graining. J. Chem. Theory Comput. 2011, 7, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, I.; Wegener, K.L.; Lam, Y.H.; Chia, B.C.S.; De Planque, M.R.R.; Bowie, J.H.; Auger, M.; Separovic, F. Interaction of antimicrobial peptides from Australian amphibians with lipid membranes. Chem. Phys. Lipids 2003, 122, 107–120. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Sugishita, K.I.; Ishibe, N.; Ueha, M.; Nakata, S.; Miyajima, K.; Epand, R.M. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry 1998, 37, 11856–11863. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Lai, G.H.; Schmidt, N.W.; Sun, V.Z.; Rodriguez, A.R.; Tong, R.; Tang, L.; Cheng, J.; Deming, T.J.; Kamei, D.T.; et al. Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. USA 2011, 108, 16883–16888. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.W.; Wong, G.C.L. Antimicrobial peptides and induced membrane curvature: Geometry, coordination chemistry, and molecular engineering. Curr. Opin. Solid State Mater. Sci. 2013, 17, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Marrink, S.J.; de Vries, A.H.; Mark, A.E. Coarse Grained Model for Semiquantitative Lipid Simulations. J. Phys. Chem. B 2004, 108, 750–760. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; De Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved parameters for the martini coarse-grained protein force field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Yesylevskyy, S.O.; Schäfer, L.V.; Sengupta, D.; Marrink, S.J. Polarizable water model for the coarse-grained MARTINI force field. PLoS Comput. Biol. 2010, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Uggerhøj, L.E.; Munk, J.K.; Hansen, P.R.; Güntert, P.; Wimmer, R. Structural features of peptoid-peptide hybrids in lipid-water interfaces. FEBS Lett. 2014, 588, 3291–3297. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Te Beek, T.A.H.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.W.; Schneider, R.; Sander, C.; Vriend, G. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- BACHEM Peptide Calculator. Available online: http://www.bachem.com/servicesupport/peptidecalculator/ (accessed on 19 October 2017).
- Snider, C.; Jayasinghe, S.; Hristova, K.; White, S.H. MPEx: A tool for exploring membrane proteins. Protein Sci. 2009, 18, 2624–2628. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Weiss, R.M.; Terwilliger, T.C. The hydrophobic moment detects periodicity in protein hydrophobicity. Proc. Natl. Acad. Sci. USA 1984, 81, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Mól, A.R.; Castro, M.S.; Fontes, W. NetWheels Tool. Available online: http://lbqp.unb.br/NetWheels/ (accessed on 19 October 2017).
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Description | System Configuration | Final Box Size | Time |
|---|---|---|---|---|
| Out | Outside bilayer, large | 1000 POPC/50 peptides/ 22,639 PW/50 NA/100 CL | 18.55 × 18.55 × 12.13 18.56 × 18.56 × 12.25 | 2 μs (aurein 1.2) 3 μs (maculatin 1.1) |
| Small-out | Outside bilayer, avoiding aggregation | 128 POPC/2 peptides/ PW/NA/CL | 6.50 × 6.50 × 12.43 6.50 × 6.50 × 12.46 | 2 μs |
| In-A | Inside bilayer, big | 1000 POPC/50 peptides/ 22,639 PW/50 NA/100 CL | 18.77 × 18.77 × 11.81 19.10 × 19.10 × 11.56 | 1 μs |
| In-B | Inside the bilayer large, low P:L ratio | 1000 POPC/20 peptides/ 22,639 PW/50 NA/70 CL | 18.58 × 18.58 × 11.80 (only maculatin 1.1) | 1 μs |
| Small-in | Inside the bilayer small | 128 POPC/8 Peptides/ 2976 PW/8 NA/16 CL | 6.84 × 7.29 × 10.82 6.86 × 7.32 × 10.89 | 4 μs |
| Self (A, B, and C) | Large self-assembly | 1000 POPC/50 Peptides/ 22,639 PW/50 NA/100 CL | 13,09 × 13,09 × 24.31 14.54 × 14.54 × 19.70 | 1 μs |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balatti, G.E.; Ambroggio, E.E.; Fidelio, G.D.; Martini, M.F.; Pickholz, M. Differential Interaction of Antimicrobial Peptides with Lipid Structures Studied by Coarse-Grained Molecular Dynamics Simulations. Molecules 2017, 22, 1775. https://doi.org/10.3390/molecules22101775
Balatti GE, Ambroggio EE, Fidelio GD, Martini MF, Pickholz M. Differential Interaction of Antimicrobial Peptides with Lipid Structures Studied by Coarse-Grained Molecular Dynamics Simulations. Molecules. 2017; 22(10):1775. https://doi.org/10.3390/molecules22101775
Chicago/Turabian StyleBalatti, Galo E., Ernesto E. Ambroggio, Gerardo D. Fidelio, M. Florencia Martini, and Mónica Pickholz. 2017. "Differential Interaction of Antimicrobial Peptides with Lipid Structures Studied by Coarse-Grained Molecular Dynamics Simulations" Molecules 22, no. 10: 1775. https://doi.org/10.3390/molecules22101775
APA StyleBalatti, G. E., Ambroggio, E. E., Fidelio, G. D., Martini, M. F., & Pickholz, M. (2017). Differential Interaction of Antimicrobial Peptides with Lipid Structures Studied by Coarse-Grained Molecular Dynamics Simulations. Molecules, 22(10), 1775. https://doi.org/10.3390/molecules22101775