Melatonin Treatment Reduces Oxidative Damage and Normalizes Plasma Pro-Inflammatory Cytokines in Patients Suffering from Charcot-Marie-Tooth Neuropathy: A Pilot Study in Three Children

 and
and 
Abstract
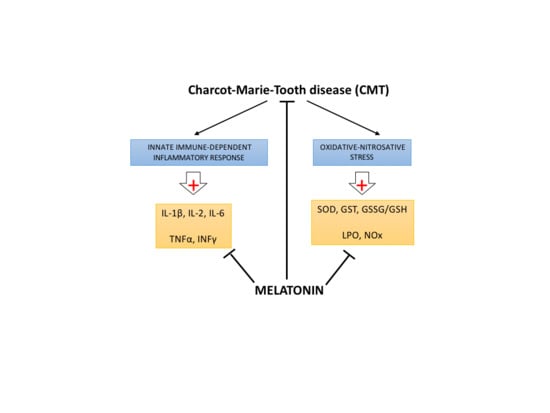
:
1. Introduction
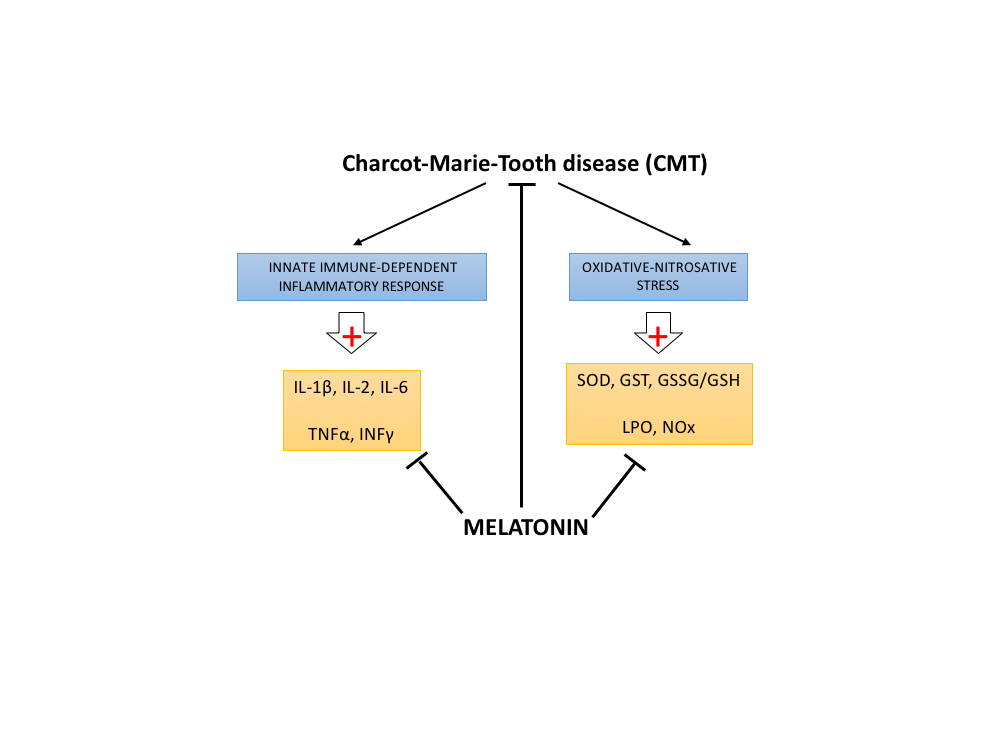
2. Results
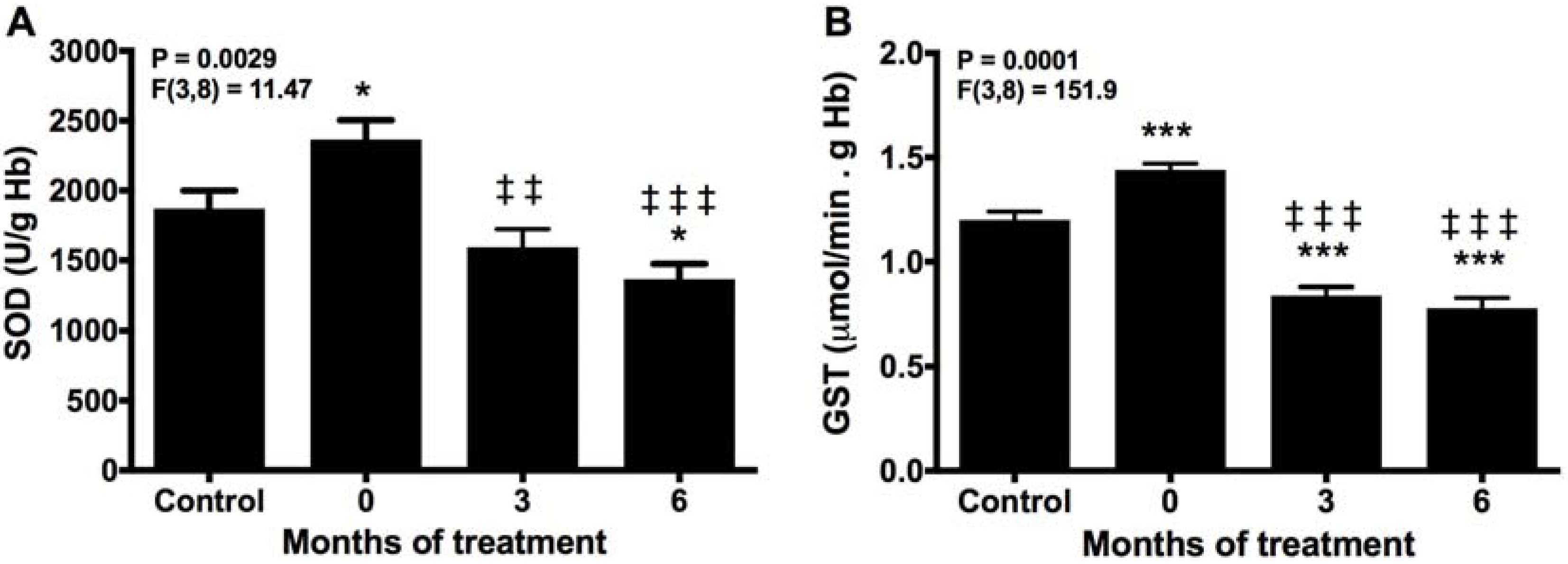
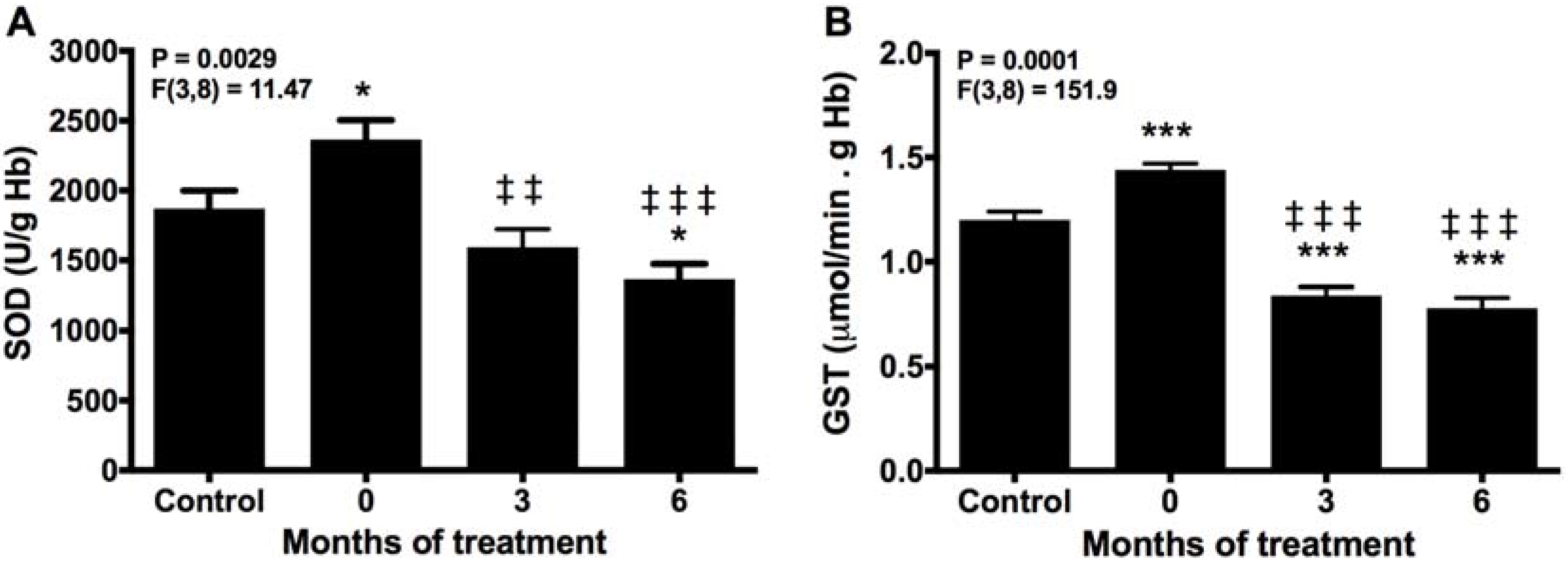
2.1. Melatonin Reduces SOD and GST Activities
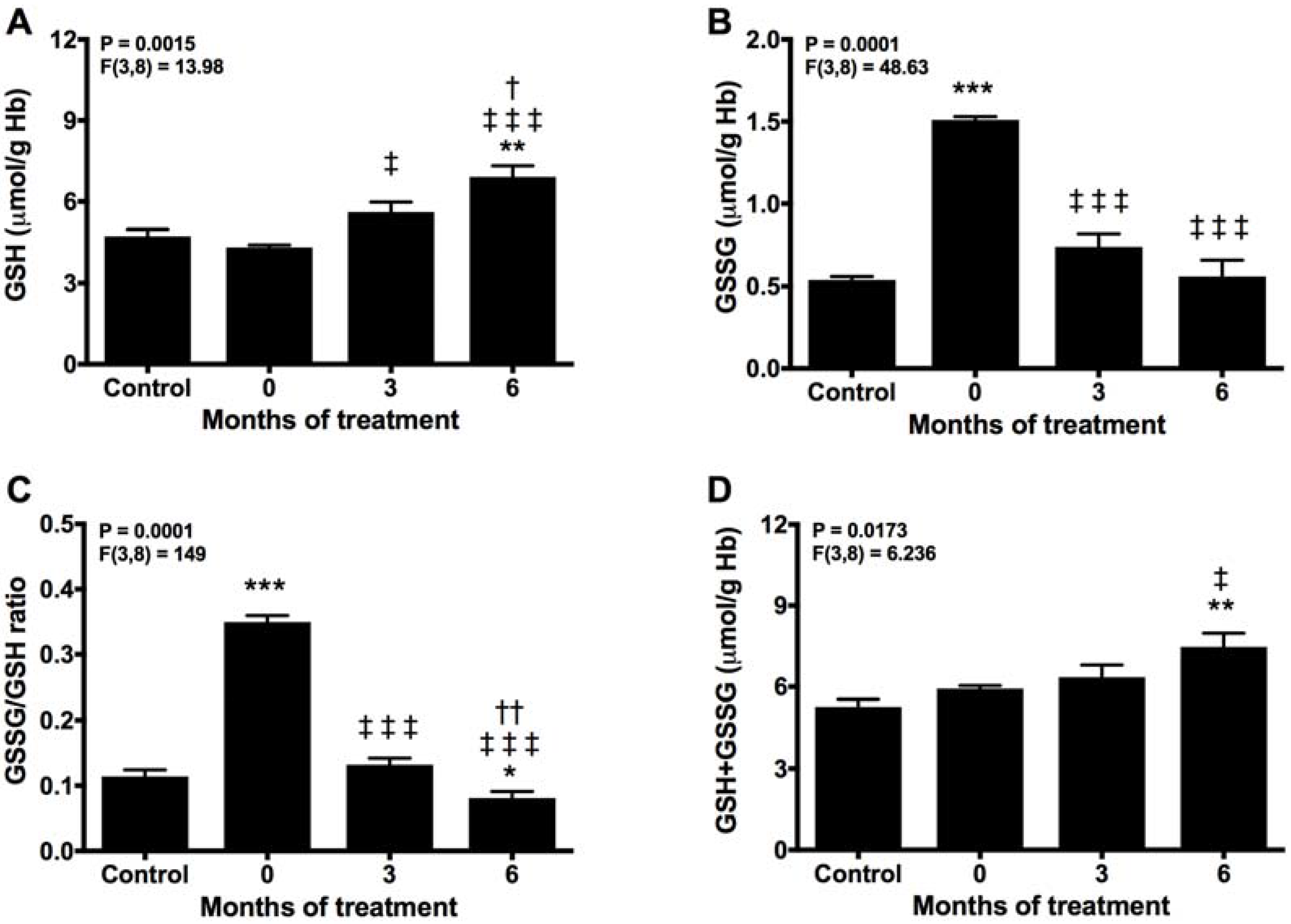
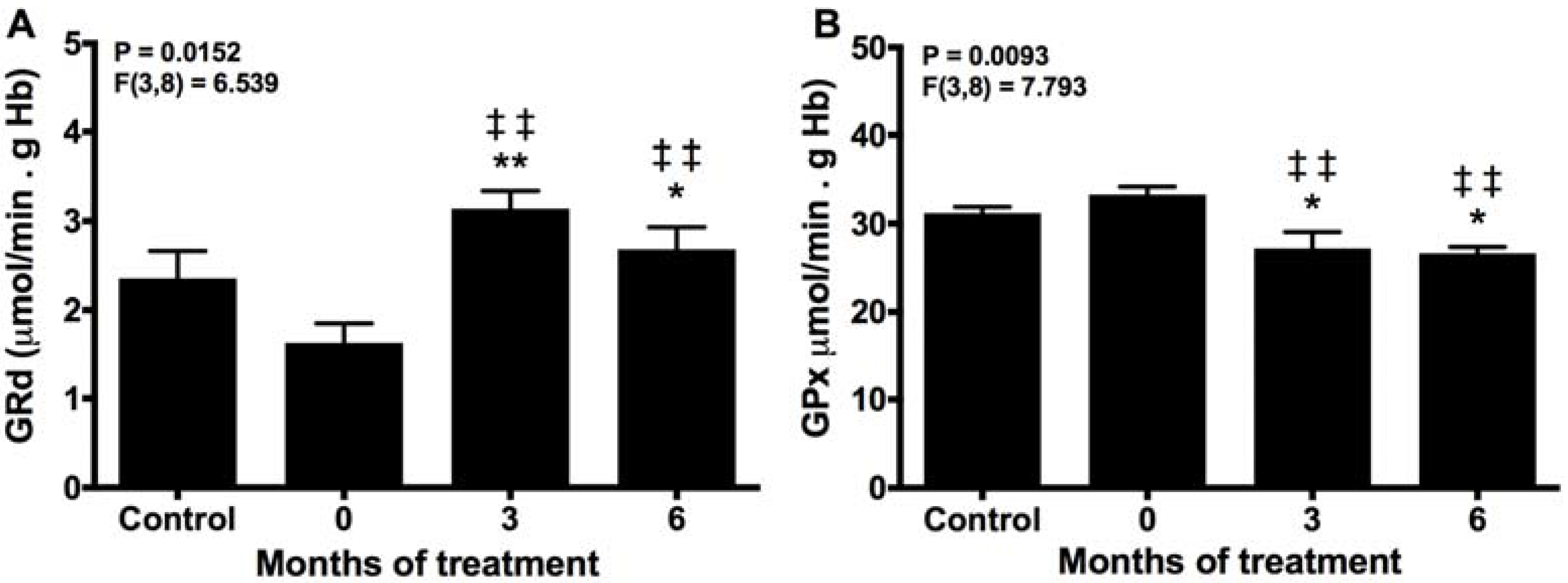
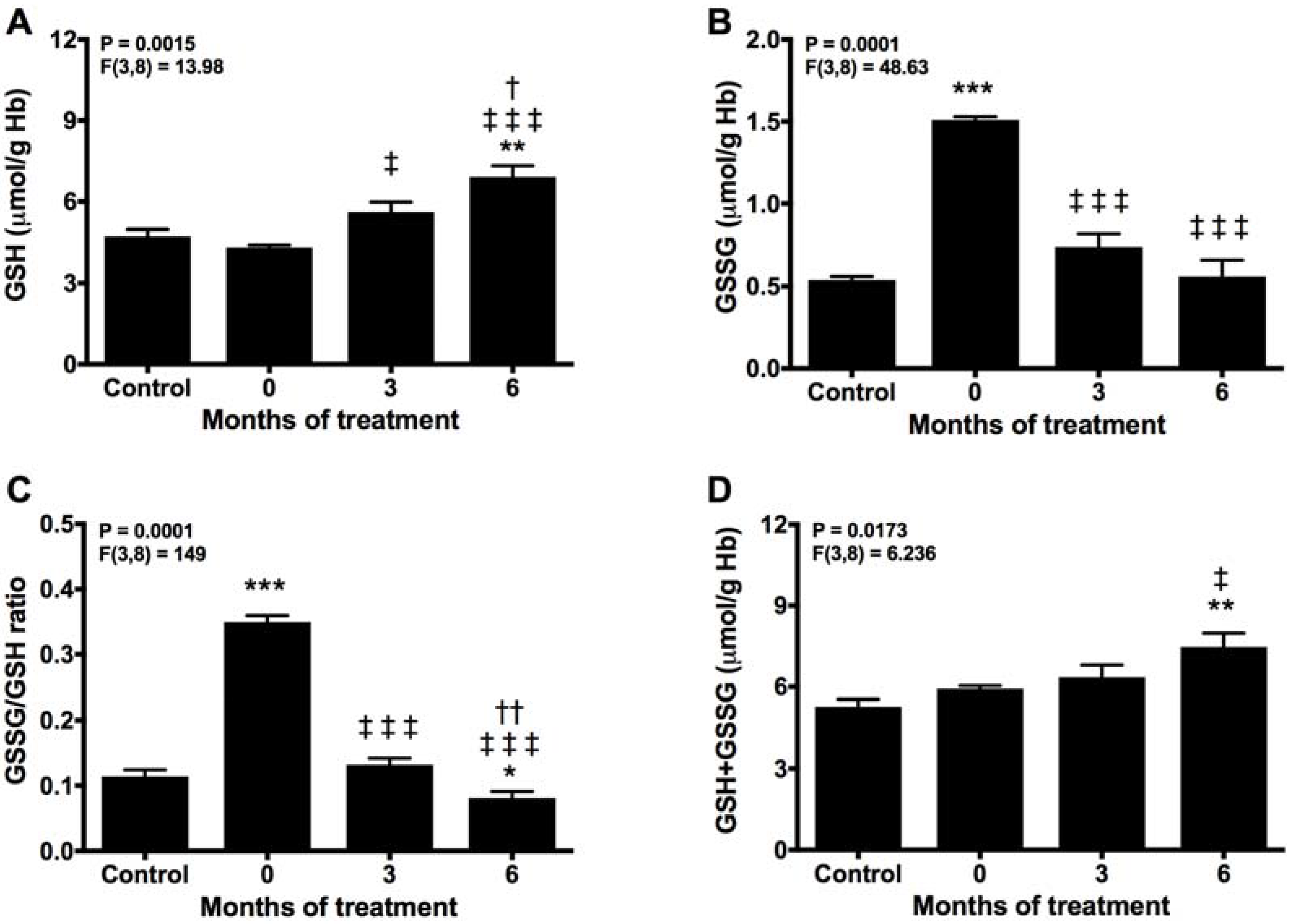
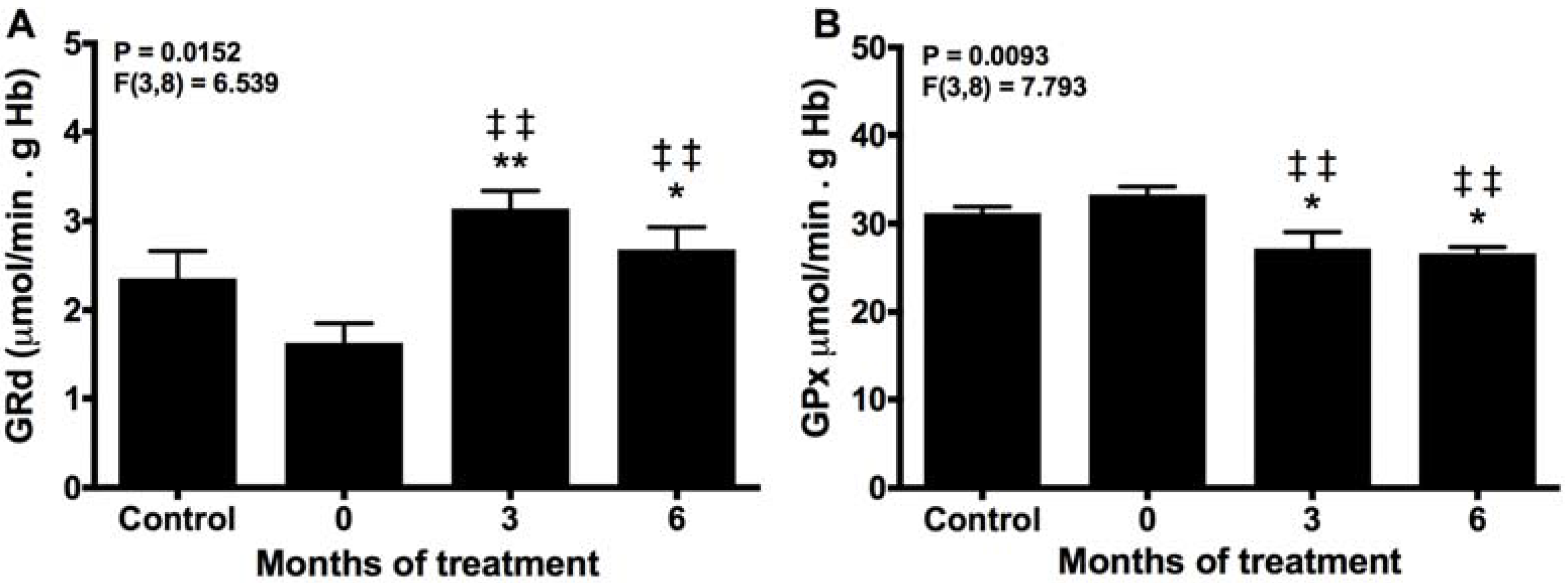
2.2. Melatonin Recovery of the Normal Turnover of the Glutathione Cycle
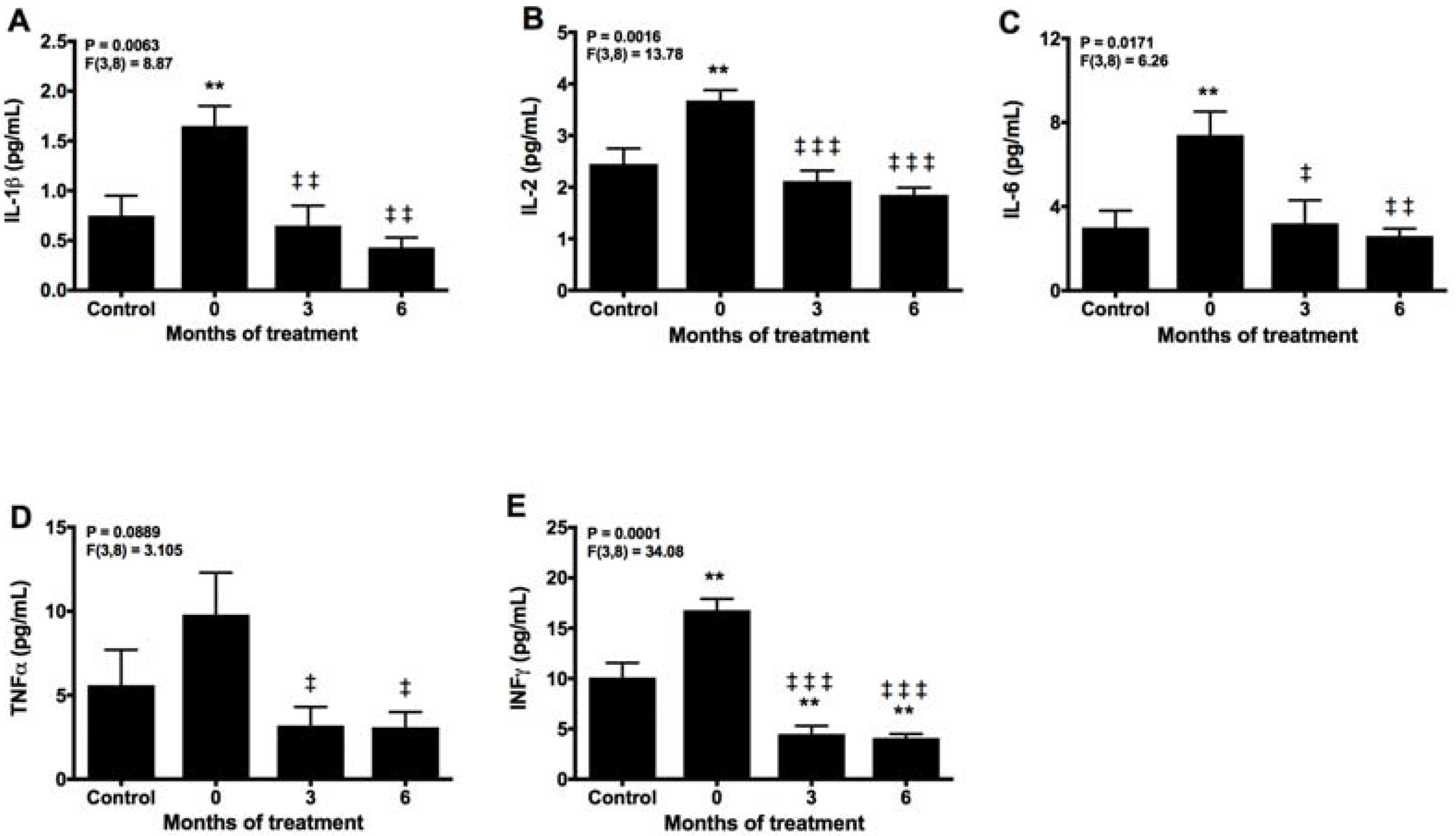
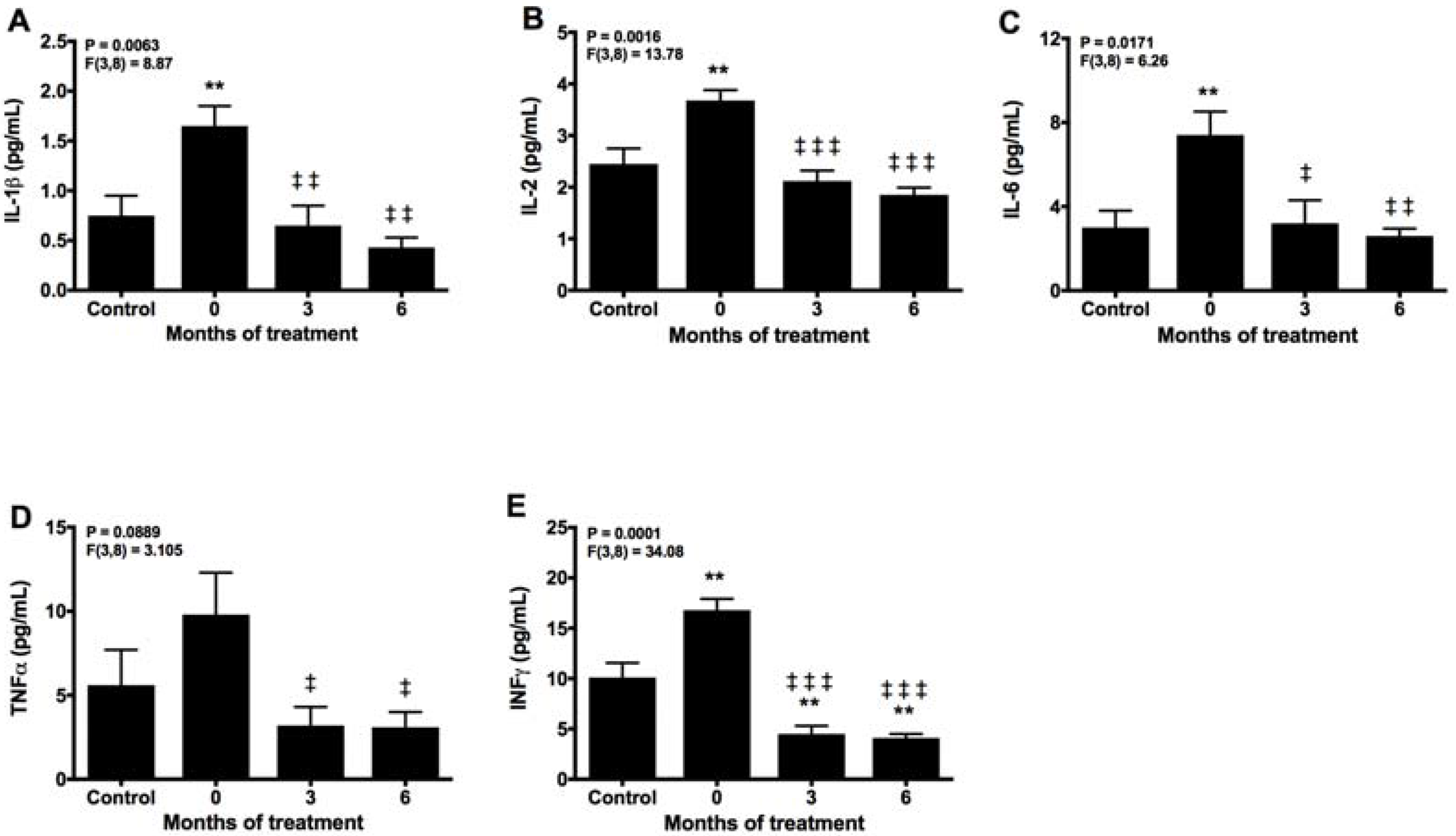
2.3. Melatonin Counteracts Proinflammatory Cytokines
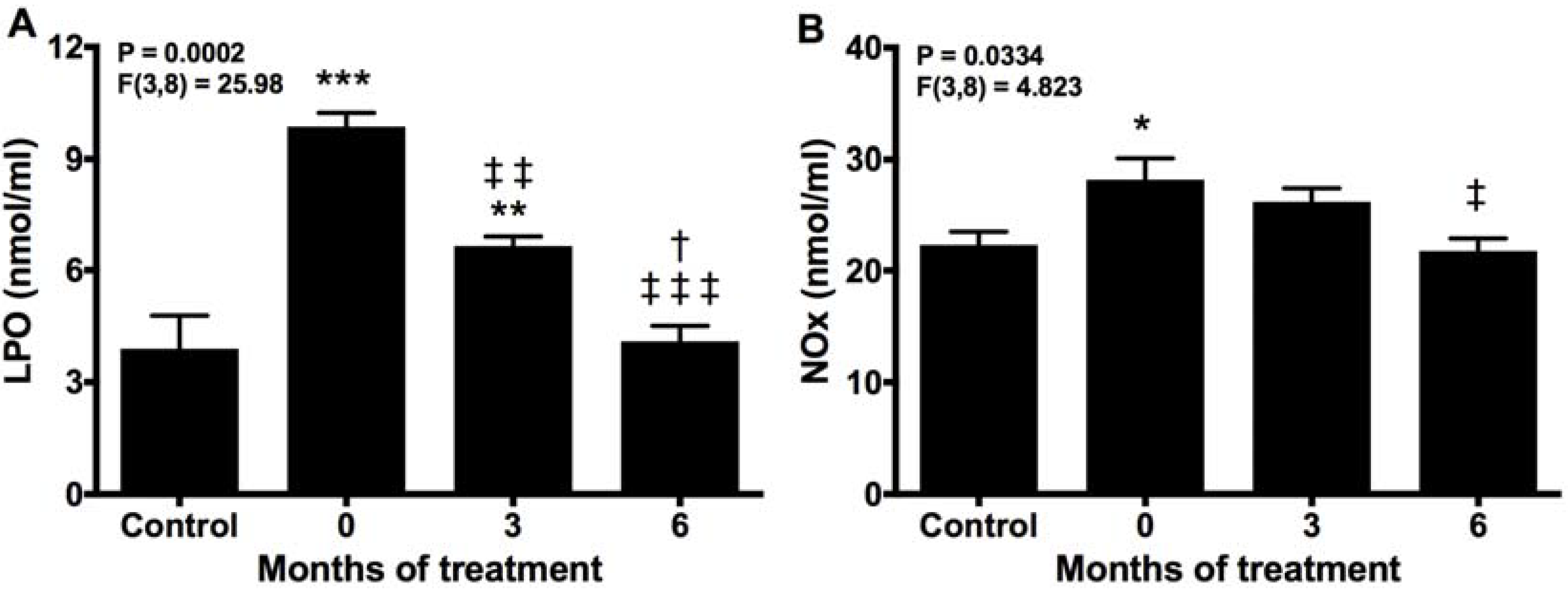
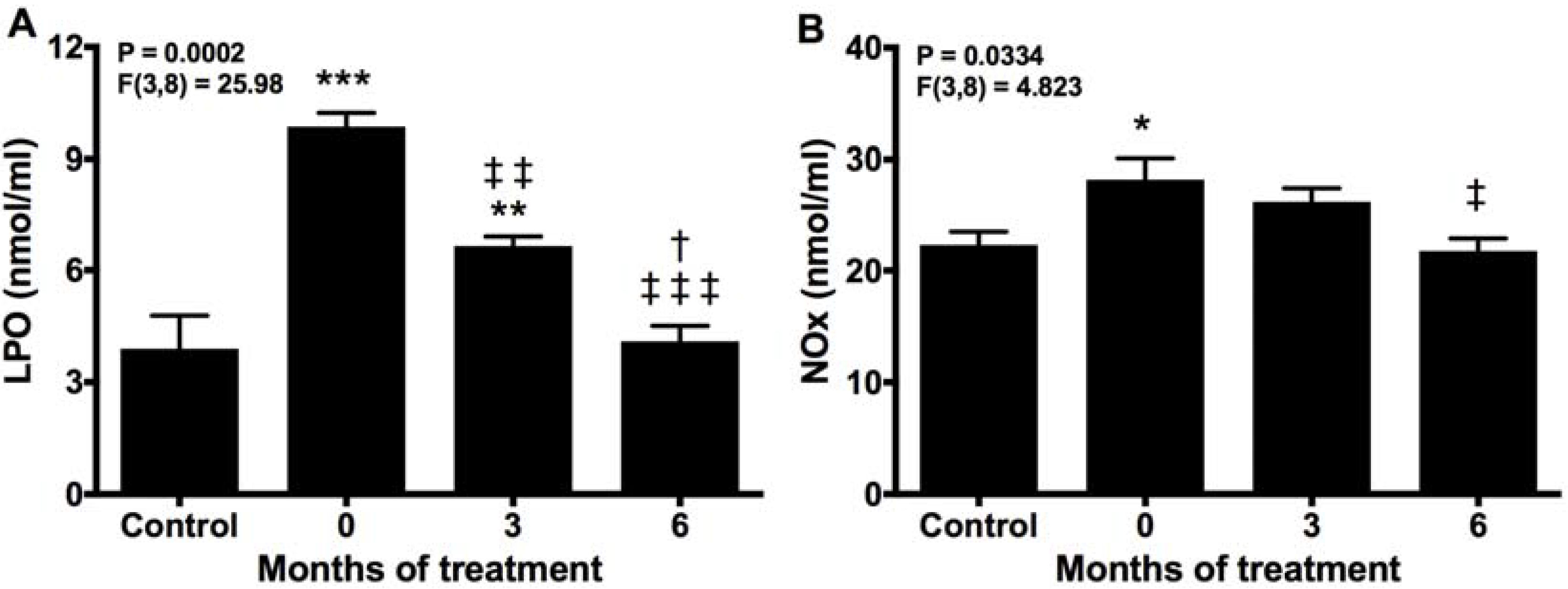
2.4. Melatonin Neutralizes Markers of Oxidative Damage and Inflammation
2.5. Effects of Melatonin on Biochemical Markers
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Samples
4.3. Superoxide Dismutase Assay
4.4. Glutathione S-Transferase Assay
4.5. GSH and GSSG Determinations
4.6. Glutathione Peroxidase and Reductase Assays
4.7. Plasma Cytokines Determination
4.8. Lipid Peroxidation Assay
4.9. Nitrite Plus Nitrate Determination
4.10. Biochemical Analysis
4.11. Statistical Analysis
4.12. Ethical Approval
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jani-Acsadi, A.; Ounpuu, S.; Pierz, K.; Acsadi, G. Pediatric Charcot-Marie-Tooth disease. Pediatr. Clin. N. Am. 2015, 62, 767–786. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, F. A Review of X-linked Charcot-Marie-Tooth Disease. J. Child Neurol. 2016, 31, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat. Rev. Neurol. 2013, 9, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Skre, H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974, 6, 98–118. [Google Scholar] [CrossRef] [PubMed]
- Dyck, P.J.; Lambert, E.H. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch. Neurol. 1968, 18, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Timmerman, V.; Strickland, A.V.; Züchner, S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes 2014, 5, 13–32. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; de Oca-Luna, R.M.; Slaugenhaupt, S.; Pentao, L.; Guzzetta, V.; Trask, B.J.; Saucedo-Cardenas, O.; Barker, D.F.; Killian, J.M.; Garcia, C.A.; et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 1991, 66, 219–232. [Google Scholar] [CrossRef]
- Cottenie, E.; Menezes, M.P.; Rossor, A.M.; Morrow, J.M.; Yousry, T.A.; Dick, D.J.; Anderson, J.R.; Jaunmuktane, Z.; Brandner, S.; Blake, J.C.; et al. Rapidly progressive asymmetrical weakness in Charcot-Marie-Tooth disease type 4J resembles chronic inflammatory demyelinating polyneuropathy. Neuromuscul. Disord. 2013, 23, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Desurkar, A.; Lin, J.P.; Mills, K.; Al-Sarraj, S.; Jan, W.; Jungbluth, H.; Wraige, E. Charcot-Marie-Tooth (CMT) disease 1A with superimposed inflammatory polyneuropathy in children. Neuropediatrics 2009, 40, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Kume, K.; Deguchi, K.; Ikeda, K.; Takata, T.; Kokudo, Y.; Kamada, M.; Tsukaguchi, M.; Touge, T.; Masaki, T. Usefulness of the modified F-ratio for assessments of proximal conduction in chronic inflammatory demyelinating polyneuropathy superimposed on Charcot Marie-Tooth disease type 1A. J. Neurol. Sci. 2014, 343, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Malandrini, A.; Villanova, M.; Dotti, M.T.; Federico, A. Acute inflammatory neuropathy in Charcot-Marie-Tooth disease. Neurology 1999, 52, 859–861. [Google Scholar] [CrossRef] [PubMed]
- Nakai, Y.; Okumura, A.; Takada, H.; Negoro, T.; Watanabe, K.; Hattori, N.; Sobue, G. Inflammatory pathological changes in a 2-year-old boy with Charcot-Marie-Tooth disease. Brain Dev. 2001, 23, 258–260. [Google Scholar] [CrossRef]
- Noack, R.; Frede, S.; Albrecht, P.; Henke, N.; Pfeiffer, A.; Knoll, K.; Dehmel, T.; Meyer Zu Hörste, G.; Stettner, M.; Kieseier, B.C.; et al. Charcot-Marie-Tooth disease CMT4A: GDAP1 increases cellular glutathione and the mitochondrial membrane potential. Hum. Mol. Genet. 2012, 21, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Watila, M.M.; Balarabe, S.A. Molecular and clinical features of inherited neuropathies due to PMP22 duplication. J. Neurol. Sci. 2015, 355, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.Q.; Li, A.W.; Li, J.; Li, D.G.; Li, Y.X.; Hao, F.; Gong, M.Z. Changes of lipid peroxidation in carbon disulfide-treated rat nerve tissues and serum. Chem. Biol. Interact. 2009, 179, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, H.; Zhao, X.; Brancho, D.; Liang, Y.; Zou, Y.; Bennett, C.; Chow, C.W. Dysregulated Inflammatory Signaling upon Charcot-Marie-Tooth Type 1C Mutation of SIMPLE Protein. Mol. Cell Biol. 2015, 35, 2464–2478. [Google Scholar] [CrossRef] [PubMed]
- Kobsar, I.; Hasenpusch-Theil, K.; Wessig, C.; Müller, H.W.; Martini, R. Evidence for macrophage-mediated myelin disruption in an animal model for Charcot-Marie-Tooth neuropathy type 1A. J. Neurosci. Res. 2005, 81, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J. The melatonin rhythm: Both a clock and a calendar. Experientia 1993, 49, 654–664. [Google Scholar] [CrossRef] [PubMed]
- García, J.A.; Volt, H.; Venegas, C.; Doerrier, C.; Escames, G.; López, L.C.; Acuña-Castroviejo, D. Disruption of the NF-κB/NLRP3 connection by melatonin requires retinoid-related orphan receptor-alpha and blocks the septic response in mice. FASEB J. 2015, 29, 3863–3875. [Google Scholar] [CrossRef] [PubMed]
- Leon, J.; Acuña-Castroviejo, D.; Sainz, R.M.; Mayo, J.C.; Tan, D.X.; Reiter, R.J. Melatonin and mitochondrial function. Life Sci. 2004, 75, 765–790. [Google Scholar] [CrossRef] [PubMed]
- Manchester, L.C.; Coto-Montes, A.; Boga, J.A.; Andersen, L.P.; Zhou, Z.; Galano, A.; Vriend, J.; Tan, D.X.; Reiter, R.J. Melatonin: An ancient molecule that makes oxygen metabolically tolerable. J. Pineal Res. 2015, 59, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Rahim, I.; Djerdjouri, B.; Sayed, R.K.; Fernández-Ortiz, M.; Fernández-Gil, B.; Hidalgo-Gutiérrez, A.; López, L.C.; Escames, G.; Reiter, R.J.; Acuña-Castroviejo, D. Melatonin administration to wild-type mice and non-treated NLRP3 mutant mice share similar inhibition of the inflammatory response during sepsis. J. Pineal Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Mayo, J.C.; Tan, D.X.; Sainz, R.M.; Alatorre-Jimenez, M.; Qin, L. Melatonin as an antioxidant: Under promises but over delivers. J. Pineal Res. 2016, 61, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Acuña Castroviejo, D.; López, L.C.; Escames, G.; López, A.; García, J.A.; Reiter, R.J. Melatonin-mitochondria Interplay in Health and Disease. Curr. Top. Med. Chem. 2011, 11, 221–240. [Google Scholar] [CrossRef] [PubMed]
- Martín, M.; Macías, M.; Escames, G.; León, J.; Acuña-Castroviejo, D. Melatonin but not vitamins C and E maintains glutathione homeostasis in t-butyl hydroperoxide-induced mitochondrial oxidative stress. FASEB J. 2000, 14, 2128. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; López, L.; Tapias, V.; Utrilla, P.; Reiter, R.; Hitos, A.; León, J.; Rodríguez, M.; Acuña-Castroviejo, D. Melatonin counteracts inducible mitochondrial nitric oxide synthase-dependent mitochondrial dysfunction in skeletal muscle of septic mice. J. Pineal Res. 2006, 40, 71–78. [Google Scholar] [CrossRef] [PubMed]
- López, L.C.; Escames, G.; Ortiz, F.; Ros, E.; Acuña-Castroviejo, D. Melatonin restores the mitochondrial production of ATP in septic mice. Neuroendocrinol. Lett. 2006, 27, 623–630. [Google Scholar] [PubMed]
- Rodríguez, M.I.; Carretero, M.; Escames, G.; López, L.C.; Maldonado, M.D.; Tan, D.X.; Reiter, R.J.; Acuña-Castroviejo, D. Chronic melatonin treatment prevents age-dependent cardiac mitochondrial dysfunction in senescence-accelerated mice. Free Radic. Res. 2007, 41, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.I.; Escames, G.; López, L.C.; López, A.; García, J.A.; Ortiz, F.; Sánchez, V.; Romeu, M.; Acuña-Castroviejo, D. Improved mitochondrial function and increased life span after chronic melatonin treatment in senescent prone mice. Exp. Gerontol. 2008, 43, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Chahbouni, M.; Escames, G.; López, L.C.; Sevilla, B.; Doerrier, C.; Muñoz-Hoyos, A.; Molina-Carballo, A.; Acuña-Castroviejo, D. Melatonin treatment counteracts the hyperoxidative status in erythrocytes of patients suffering from Duchenne muscular dystrophy. Clin. Biochem. 2011, 44, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Chahbouni, M.; Escames, G.; Venegas, C.; Sevilla, B.; Antonio García, J.; López, L.C.; Muñoz-Hoyos, A.; Molina-Carballo, A.; Acuña-Castroviejo, D. Melatonin treatment normalizes plasma pro-inflammatory cytokines and nitrosative/oxidative stress in patients suffering from Duchenne muscular dystrophy. J. Pineal Res. 2010, 48, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Escames, G.; Ozturk, G.; Baño-Otalora, B.; Pozo, M.J.; Madrid, J.A.; Reiter, R.J.; Serrano, E.; Concepción, M.; Acuña-Castroviejo, D. Exercise and melatonin in humans: Reciprocal benefits. J. Pineal Res. 2012, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Leonardo-Mendonça, R.C.; Ocaña-Wilhelmi, J.; de Haro, T.; de Teresa-Gálvan, C.; Guerra-Hernández, E.; Rusanova, I.; Fernández-Ortiz, M.; Sayed, R.K.; Escames, G.; Acuña-Castroviejo, D. The benefit of a supplement with the antioxidant melatonin on redox status and muscle damage in resistance trained athletes. Appl. Physiol. Nutr. Metab. 2017, 42, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Coto-Montes, A.; Boga, J.A.; Tan, D.X.; Reiter, R.J. Melatonin as a potential agent in the treatment of sarcopenia. Int. J. Mol. Sci. 2016, 17, 1771. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.; Igbaria, A.; D’Autreaux, B.; Planson, A.G.; Junot, C.; Godat, E.; Bachhawat, A.K.; Delaunay-Moisan, A.; Toledano, M.B. Glutathione revisited: A vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J. 2011, 30, 2044–2056. [Google Scholar] [CrossRef] [PubMed]
- Urata, Y.; Honman, S.; Todoroki, S.; Lida, T.; Cho, S.; Honma, K.; Kondo, T. Melatonin induces gamma-glutamylcysteine synthase mediated by activator proteín-1 in human vascular endothelial cells. Free Radic. Biol. Med. 1999, 27, 838–847. [Google Scholar] [CrossRef]
- Piperi, C.; Papapanagiotou, A.; Kalofoutis, C.; Zisaki, K.; Michalaki, V.; Tziraki, A.; Kalofoutis, A. Altered long chain fatty acids composition in Duchenne muscular dystrophy erythrocytes. In Vivo 2004, 18, 799–802. [Google Scholar] [PubMed]
- Seco-Cervera, M.; Ibañez-Cabellos, J.S.; Garcia-Gimenez, J.L.; Espinos, C.; Palau, F.; Pallardo, F.V. Biomarkers research in neuromuscular disease Charcot-Marie-Tooth. Free Radic. Biol. Med. 2014, 75, S48–S49. [Google Scholar] [CrossRef] [PubMed]
- Rajabally, Y.A.; Adams, D.; Latour, P.; Attarian, S. Hereditary and inflammatory neuropathies: A review of reported associations, mimics and misdiagnoses. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- García, A.; Combarros, O.; Calleja, J.; Berciano, J. Charcot-Marie-Tooth disease type 1A with 17p duplication in infancy and early childhood: A longitudinal clinical and electrophysiologic study. Neurology 1998, 50, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- López Del Amo, V.; Seco-Cervera, M.; García-Giménez, J.L.; Whitworth, A.J.; Pallardó, F.V.; Galindo, M.I. Mitochondrial defects and neuromuscular degeneration caused by altered expression of Drosophila Gdap1: Implications for the Charcot-Marie-Tooth neuropathy. Hum. Mol. Genet. 2015, 24, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Niemann, A.; Huber, N.; Wagner, K.M.; Somandin, C.; Horn, M.; Lebrun-Julien, F.; Angst, B.; Pereira, J.A.; Halfter, H.; Welzl, H.; et al. The Gdap1 knockout mouse mechanistically links redox control to Charcot-Marie-Tooth disease. Brain 2014, 137, 668–682. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Song, W.; Jang, Y.C.; Liu, Y.; Sabia, M.; Richardson, A.; van Remmen, H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1159–R1168. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, G.; Tangorra, A.; Curatola, G. Effects of intramembrane particle aggregation on erythrocyte membrane fluidity: An electron spin resonance study in normal and in dystrophic subjects. Exp. Cell Res. 1990, 191, 14–21. [Google Scholar] [CrossRef]
- Poderoso, J.J.; Boveris, A.; Cadenas, E. Mitochondrial oxidative stress: A self-propagating process with implications for signaling cascades. Biofactors 2000, 11, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Pickett, C.B.; Lu, A.Y. Glutathione S-transferases: Gene structure, regulation, and biological function. Annu. Rev. Biochem. 1989, 58, 743–764. [Google Scholar] [CrossRef] [PubMed]
- Jezek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Huber, N.; Bieniossek, C.; Wagner, K.M.; Elsasser, H.P.; Suter, U.; Berger, I.; Niemann, A. Glutathione-conjugating and membrane-remodeling activity of GDAP1 relies on amphipathic C-terminal domain. Sci. Rep. 2016, 6, 36930. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.; Patil, A.; Phatak, A.; Chandra, N. Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 2010, 4, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Cheeseman, K.H. Determination of aldehidic lipid peroxidation products: Malonaldehide and 4-hydroxynonenal. Methods Enzymol. 1990, 186, 407–421. [Google Scholar] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Singhal, S.S.; Singh, S.P.; Singhal, P.; Horne, D.; Singhal, J.; Awasthi, S. Antioxidant role of glutathione S-transferases: 4-Hydroxynonenal, a key molecule in stress-mediated signaling. Toxicol. Appl. Pharmacol. 2015, 289, 361–370. [Google Scholar] [CrossRef] [PubMed]
- García, J.J.; López-Pingarrón, L.; Almeida-Souza, P.; Tres, A.; Escudero, P.; García-Gil, F.A.; Tan, D.X.; Reiter, R.J.; Ramírez, J.M.; Bernal-Pérez, M. Protective effects of melatonin in reducing oxidative stress and in preserving the fluidity of biological membranes: A review. J. Pineal Res. 2014, 56, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Reiter, R.J.; Tan, D.X.; Galano, A. Melatonin: Exceeding expectations. Physiology 2014, 29, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Schwartz, R.J.; Waddell, I.D.; Holloway, B.R.; Reid, M.B. Skeletal muscle myocytes undergo protein loss and reactive oxygen-mediated NF-κB activation in response to tumor necrosis factor alpha. FASEB J. 1998, 12, 871–880. [Google Scholar] [PubMed]
- Ginsberg, L.; Malik, O.; Kenton, A.R.; Sharp, D.; Muddle, J.R.; Davis, M.B.; Winer, J.B.; Orrell, R.W.; King, R.H. Coexistent hereditary and inflammatory neuropathy. Brain 2004, 127, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Vital, A.; Vital, C.; Lagueny, A.; Ferrer, X.; Ribiére-Bachelier, C.; Latour, P.; Petry, K.G. Inflammatory demyelination in a patient with CMT1A. Muscle Nerve 2003, 28, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Crespo, E.; Macias, M.; Pozo, D.; Escames, G.; Martin, M.; Vives, F.; Guerrero, J.; Acuña-Castroviejo, D. Melatonin inhibits expression of the inducible NO synthase II in liver and lung and prevents endotoxemia in lipopolysaccharide-induced multiple organ dysfunction syndrome in rats. FASEB J. 1999, 13, 1537–1546. [Google Scholar] [PubMed]
- Volt, H.; García, J.A.; Doerrier, C.; Diaz-Casado, M.E.; Guerra-Librero, A.; López, L.C.; Escames, G.; Tresguerres, J.A.; Acuña-Castroviejo, D. Same molecule but different expression: Aging and sepsis trigger NLRP3 inflammasome activation, a target of melatonin. J. Pineal Res. 2016, 60, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Sajic, M. Mitochondrial dynamics in peripheral neuropathies. Antioxid. Redox Signal. 2014, 21, 601–620. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, F.; Ronchi, D.; Salani, S.; Nizzardo, M.; Fortunato, F.; Bordoni, A.; Stuppia, G.; Del Bo, R.; Piga, D.; Fato, R.; et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot-Marie-Tooth 2A motor neurons. Hum. Mol. Genet. 2016, 25, 4266–4281. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, L.; Shy, M. Update on Charcot-Marie-Tooth disease. Curr. Opin. Neurol. 2015, 28, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Sao Paulo Med. J. 2015, 133, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Oe, T.; Blair, I.A. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science 2001, 292, 2083–2086. [Google Scholar] [CrossRef] [PubMed]
- Posa, L.; De Gregorio, D.; Gobbi, G.; Comai, S. Targeting melatonin MT2 receptors: A novel pharmacological avenue for inflammatory and neuropathic pain. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- López-Canul, M.; Palazzo, E.; Dominguez-Lopez, S.; Luongo, L.; Lacoste, B.; Comai, S.; Angeloni, D.; Fraschini, F.; Boccella, S.; Spadoni, G.; et al. Selective melatonin MT2 receptor ligands relieve neuropathic pain through modulation of brainstem descending antinociceptive pathways. Pain 2015, 156, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Misra, H.P.; Fridovich, I. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J. Biol. Chem. 1972, 247, 3170–3175. [Google Scholar] [PubMed]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem. 1974, 249, 7130–7139. [Google Scholar] [PubMed]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Jaskot, R.H.; Charlet, E.G.; Grose, E.C.; Grady, M.A.; Roycroft, J.H. An automated analysis of glutathione peroxidase, S-transferase, and reductase activity in animal tissue. J. Anal. Toxicol. 1983, 7, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.L.; Taintor, R.R.; Boockvar, K.S.; Hibbs, J.B., Jr. Measurement of nitrate and nitrite in biological samples using nitrate reductase and Griess reaction. Methods Enzymol. 1996, 268, 142–151. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Control | Melatonin Treatment (Months) | ||
|---|---|---|---|---|
| 0 | 3 | 6 | ||
| CPK | 114.3 ± 45.2 | 146.3 ± 73.2 | 155.5 ± 70.5 | 83.7 ± 40.8 ***,### |
| LDH | 248.4 ± 44.6 | 407.2 ± 79.2 ** | 380.4 ± 46.2 * | 339.1 ± 17.5 *,# |
| AST | 23.4 ± 5.1 | 29.7 ± 5.3 * | 25.6 ± 5.3 | 23.8 ± 6.0 # |
| ALT | 24.4 ± 3.2 | 17.7 ± 0.6 * | 14.8 ± 3.0 ** | 12.2 ± 2.1 *** |
| γGT | 25.5 ± 2.5 | 11.9 ± 5.5 ** | 13.2 ± 2.5 ** | 16.3 ± 8.9 **,# |
| Aldolase | 6.7 ± 2.1 | 4.7 ± 1.6 | 3.5 ± 0.7 ** | 2.4 ± 0.2 ***,# |
| Alkaline phosphatase | <350 | 188.5 ± 101.2 | 174.2 ± 75.0 | 153.2 ± 75.8 |
| Myoglobin | 50.6 ± 10.4 | 23.7 ± 2.5 *** | 22.9 ± 3.1 *** | 24.7 ± 2.4 *** |
| Calcium | 9.7 ± 0.5 | 9.3 ± 0.1 | 9.5 ± 0.3 | 9.4 ± 0.3 |
| Phosphate | 3.8 ± 0.8 | 4.3 ± 0.1 | 4.7 ± 0.1 | 4.4 ± 0.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chahbouni, M.; López, M.D.S.; Molina-Carballo, A.; De Haro, T.; Muñoz-Hoyos, A.; Fernández-Ortiz, M.; Guerra-Librero, A.; Acuña-Castroviejo, D. Melatonin Treatment Reduces Oxidative Damage and Normalizes Plasma Pro-Inflammatory Cytokines in Patients Suffering from Charcot-Marie-Tooth Neuropathy: A Pilot Study in Three Children. Molecules 2017, 22, 1728. https://doi.org/10.3390/molecules22101728
Chahbouni M, López MDS, Molina-Carballo A, De Haro T, Muñoz-Hoyos A, Fernández-Ortiz M, Guerra-Librero A, Acuña-Castroviejo D. Melatonin Treatment Reduces Oxidative Damage and Normalizes Plasma Pro-Inflammatory Cytokines in Patients Suffering from Charcot-Marie-Tooth Neuropathy: A Pilot Study in Three Children. Molecules. 2017; 22(10):1728. https://doi.org/10.3390/molecules22101728
Chicago/Turabian StyleChahbouni, Mariam, María Del Señor López, Antonio Molina-Carballo, Tomás De Haro, Antonio Muñoz-Hoyos, Marisol Fernández-Ortiz, Ana Guerra-Librero, and Darío Acuña-Castroviejo. 2017. "Melatonin Treatment Reduces Oxidative Damage and Normalizes Plasma Pro-Inflammatory Cytokines in Patients Suffering from Charcot-Marie-Tooth Neuropathy: A Pilot Study in Three Children" Molecules 22, no. 10: 1728. https://doi.org/10.3390/molecules22101728
APA StyleChahbouni, M., López, M. D. S., Molina-Carballo, A., De Haro, T., Muñoz-Hoyos, A., Fernández-Ortiz, M., Guerra-Librero, A., & Acuña-Castroviejo, D. (2017). Melatonin Treatment Reduces Oxidative Damage and Normalizes Plasma Pro-Inflammatory Cytokines in Patients Suffering from Charcot-Marie-Tooth Neuropathy: A Pilot Study in Three Children. Molecules, 22(10), 1728. https://doi.org/10.3390/molecules22101728