Valorization of Lignin by Partial Wet Oxidation Using Sustainable Heteropoly Acid Catalysts





Abstract
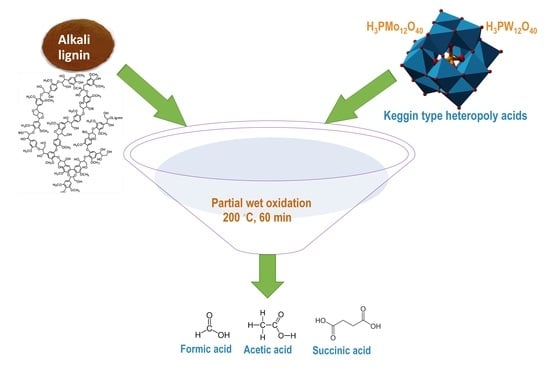
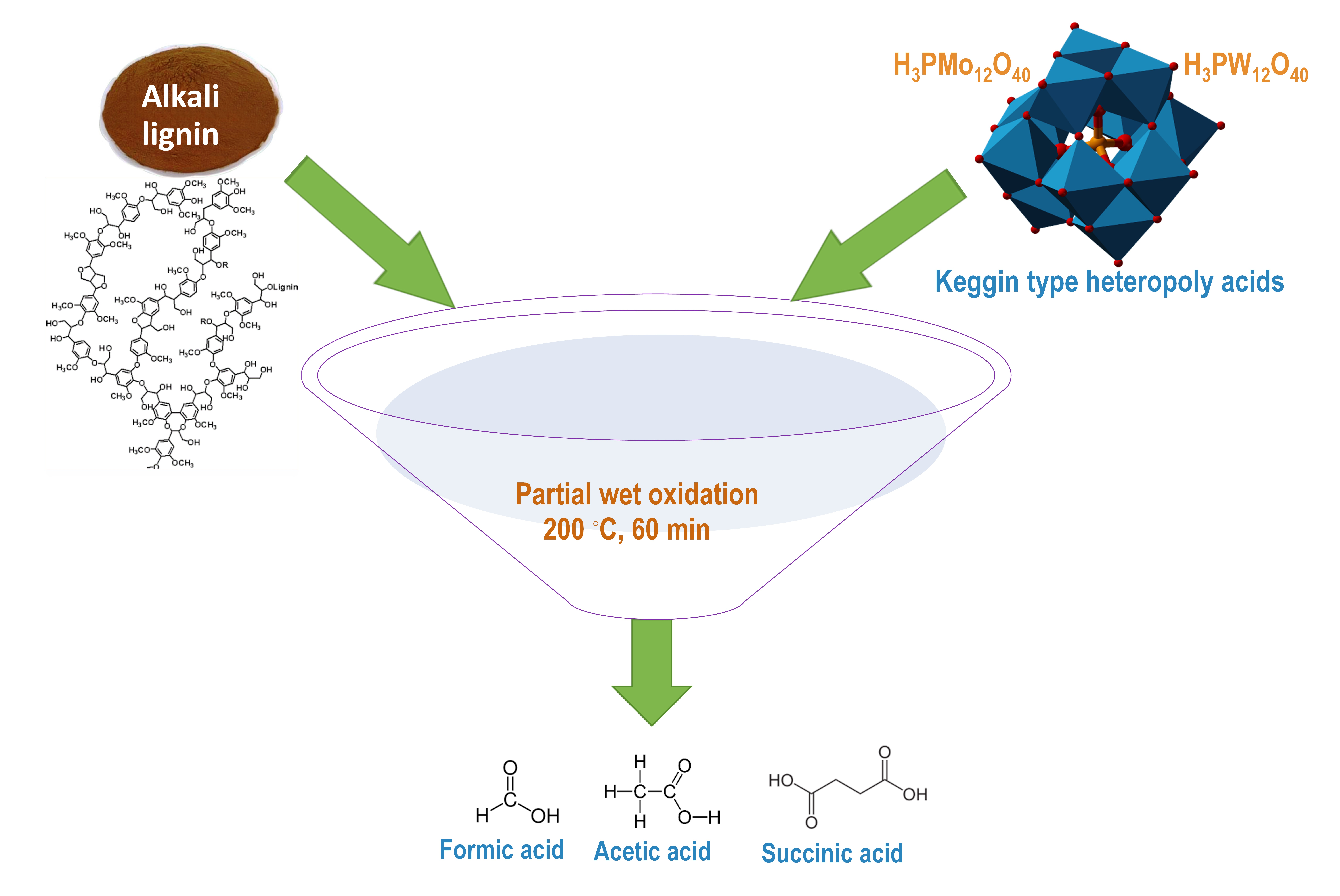
:
1. Introduction
2. Results and Discussion
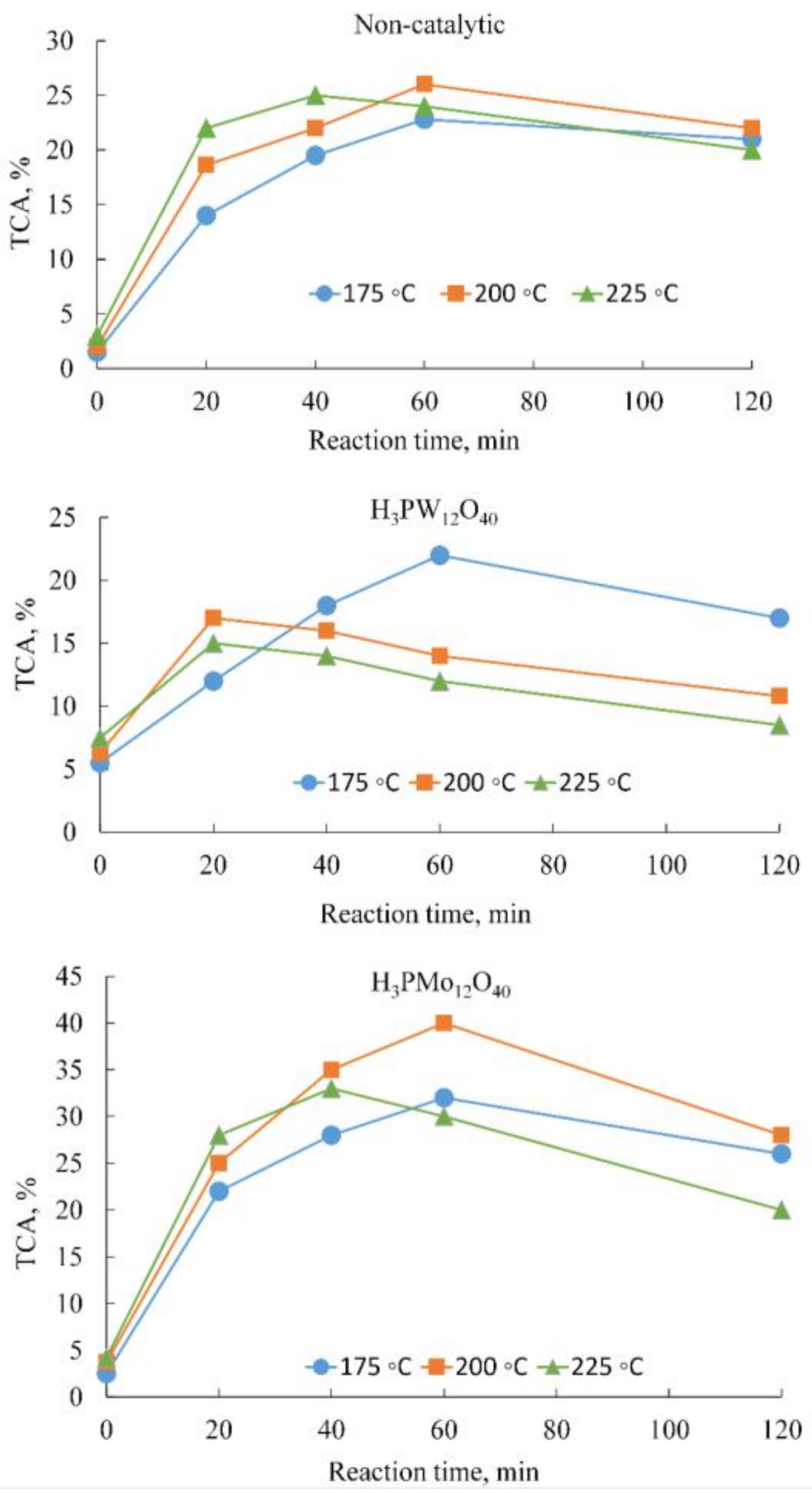
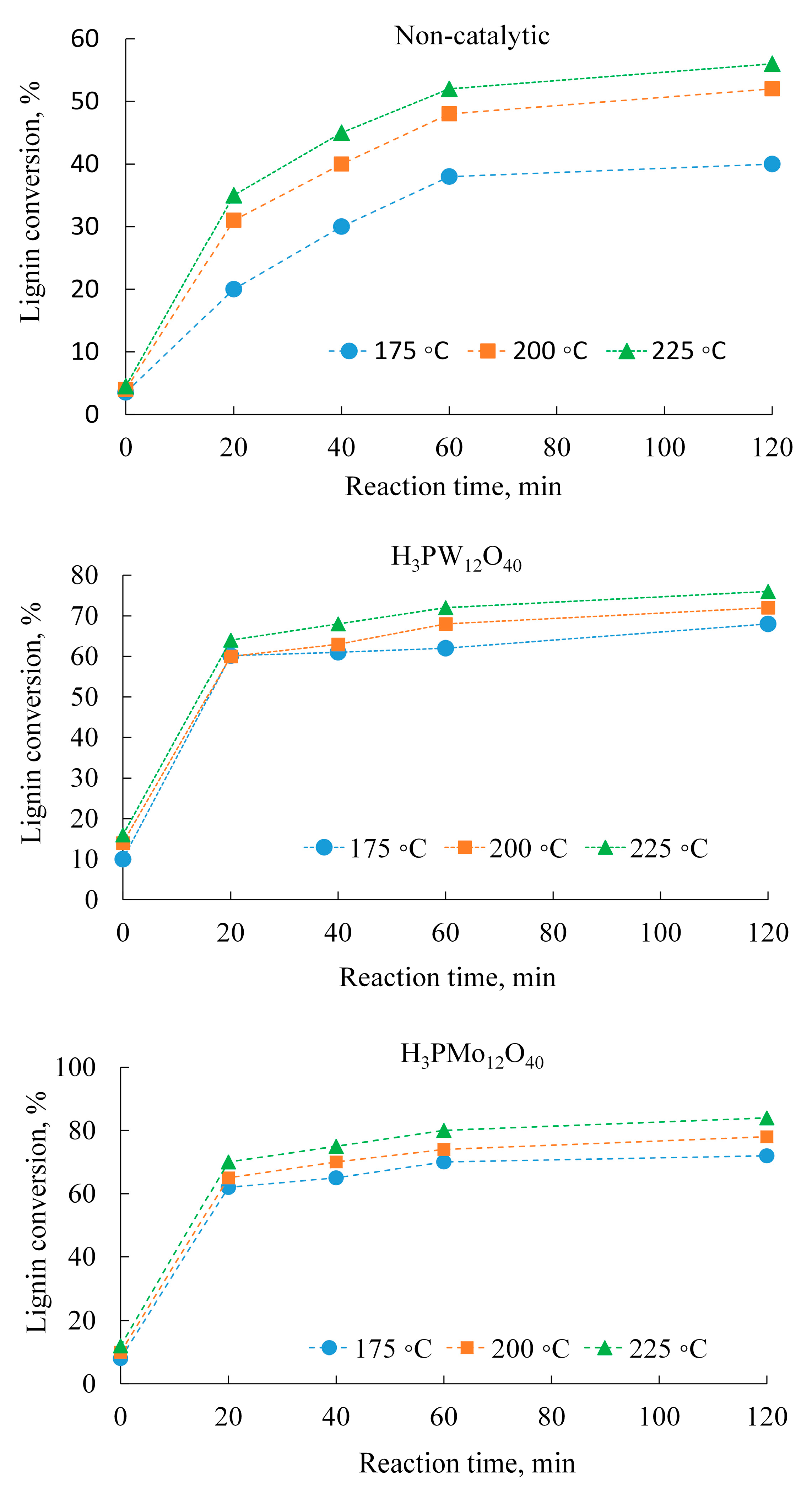
2.1. Influence of Temperature and Reaction Time
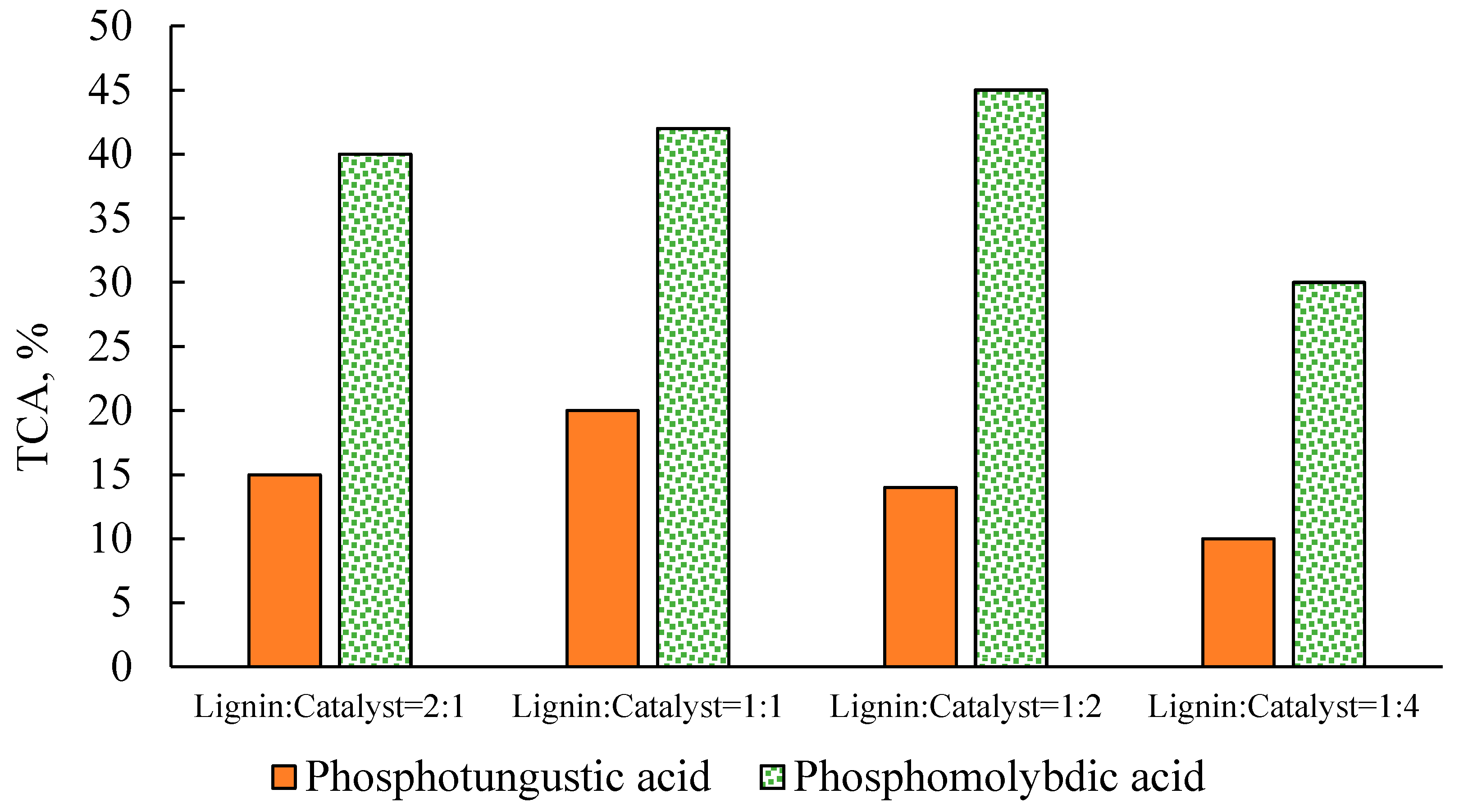
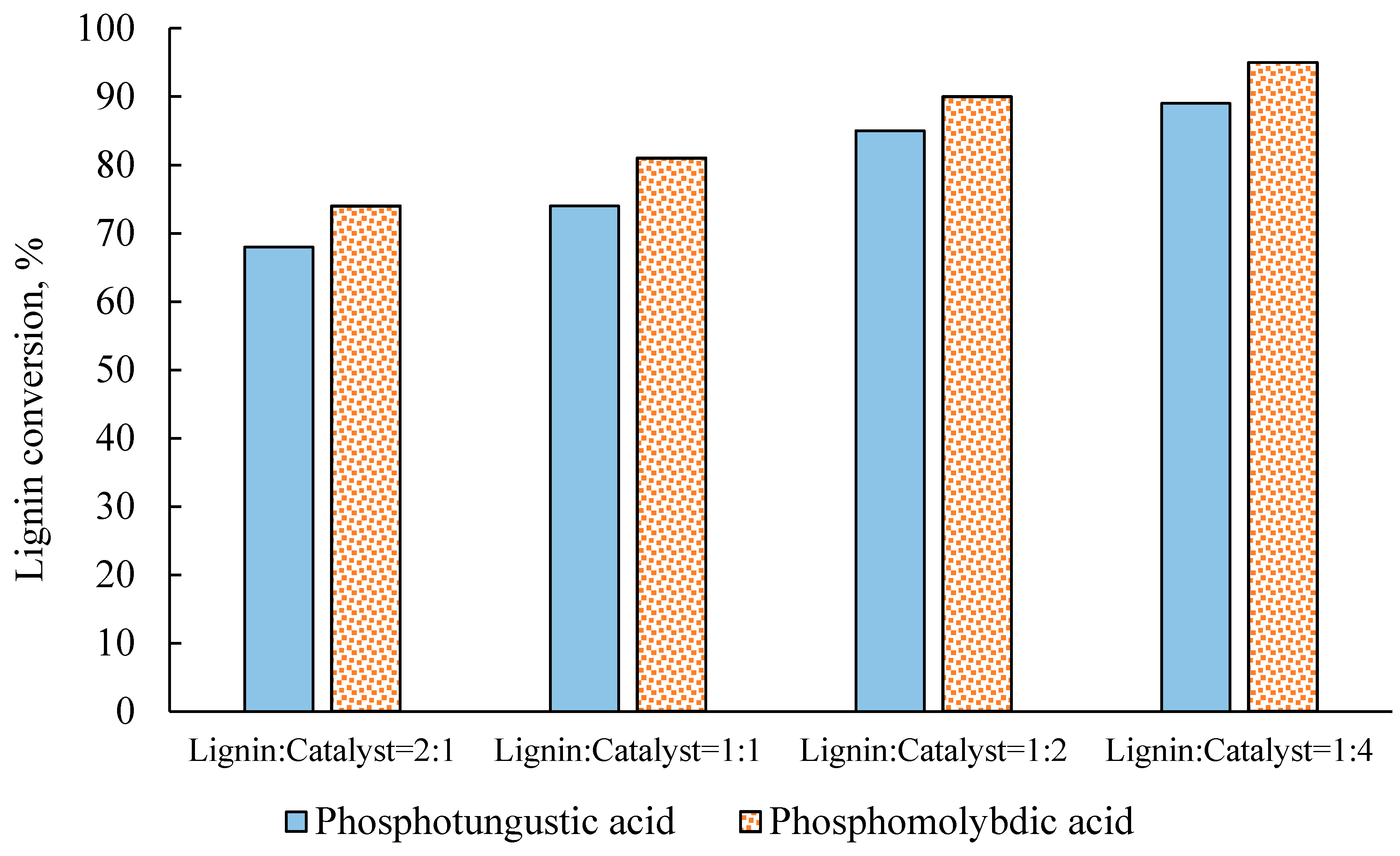
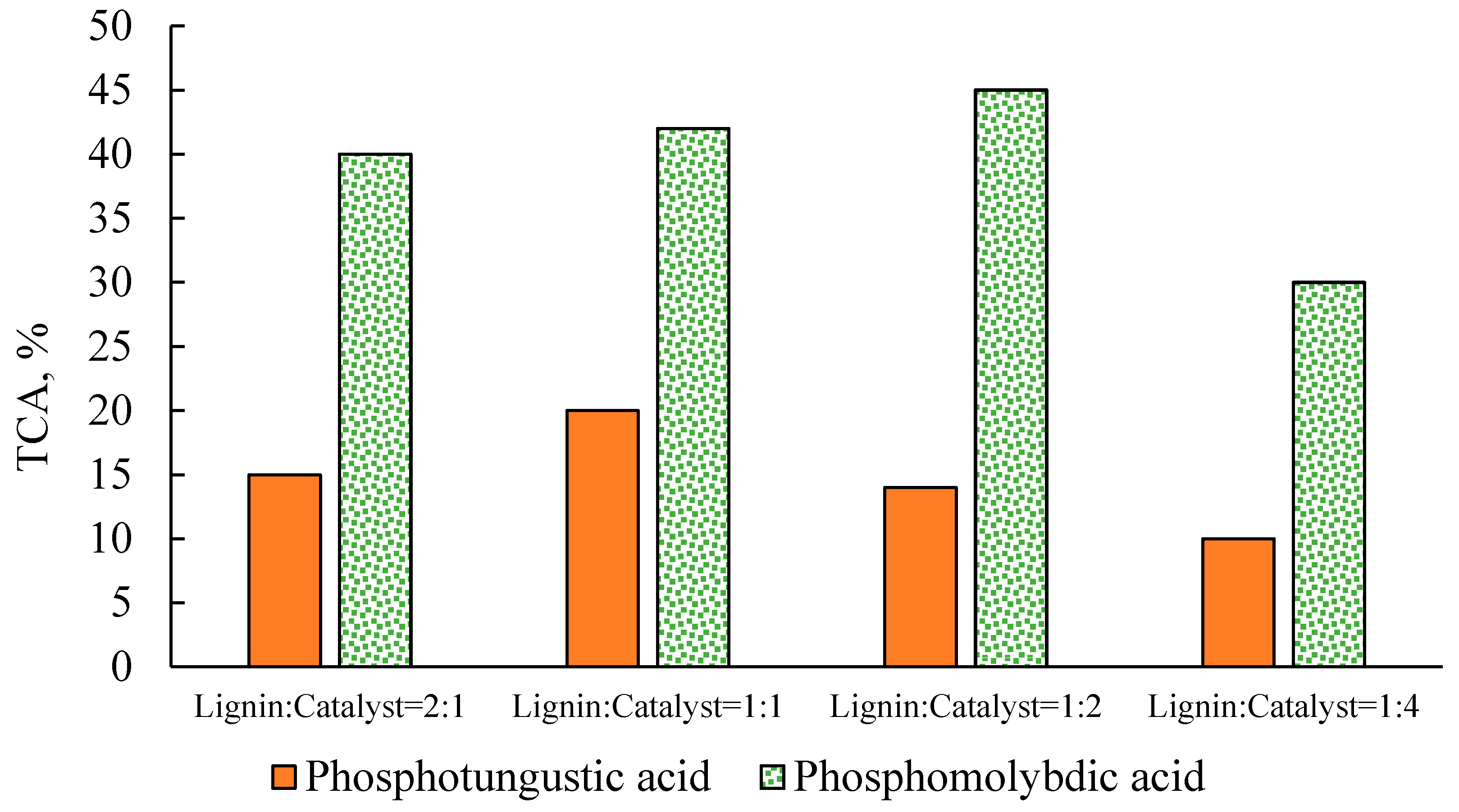
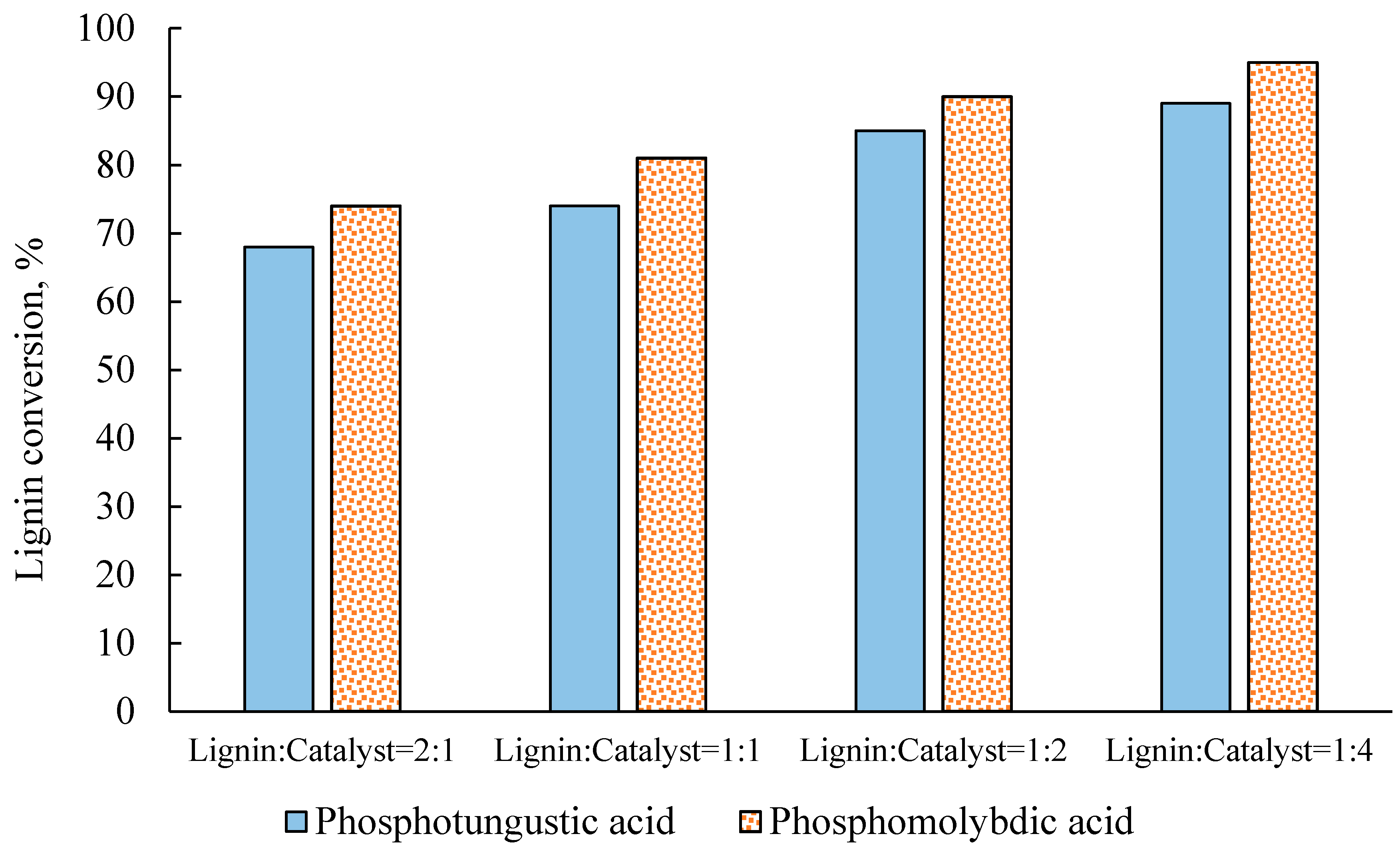
2.2. Effect of Catalyst Concentration
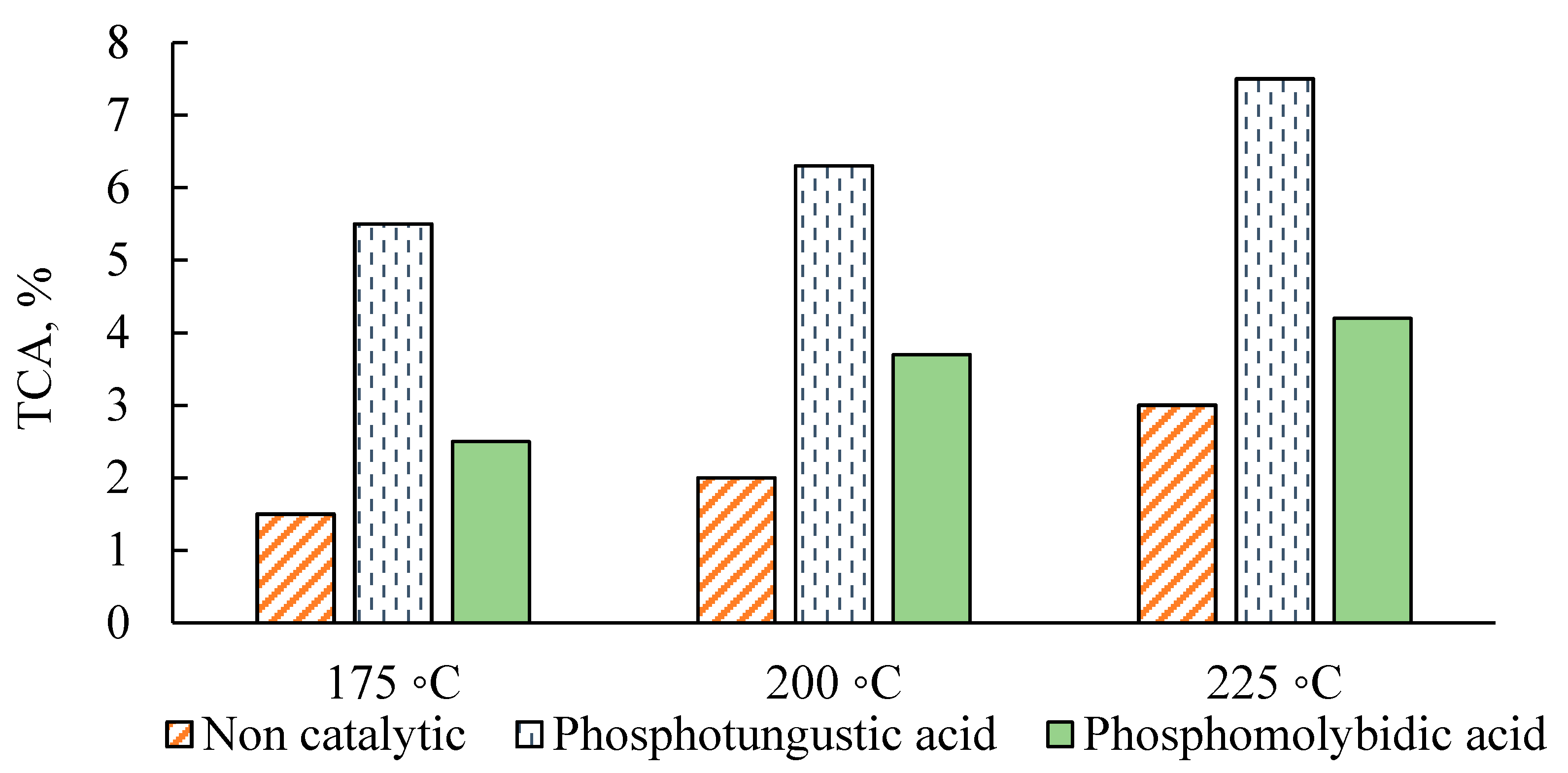
2.3. Recoverability of the Catalysts
3. Materials and Methods
3.1. Materials
3.2. Experimental Procedure and Method
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Serrano-Ruiz, J.C.; Luque, R.; Sepulveda-Escribano, A. Transformations of biomass-derived platform molecules: From high added-value chemicals to fuels via aqueous-phase processing. Chem. Soc. Rev. 2011, 40, 5266–5281. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Beltramini, J.N.; Fan, Y.X.; Lu, G.Q. Chemoselective catalytic conversion of glycerol as a biorenewable source to valuable commodity chemicals. Chem. Soc. Rev. 2008, 37, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Huber, G.W.; Beltramini, J.N.; Fan, Y.X.; Lu, G.Q. Synthesis of transportation fuels from biomass: Chemistry, catalysts, and engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Xia, X.; Lin, C.; Tong, D.; Beltramini, J. Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels. Chem. Soc. Rev. 2011, 40, 5588–5617. [Google Scholar] [CrossRef] [PubMed]
- Ragauskas, A.J.; Williams, C.K.; Davison, B.H.; Britovsek, G.; Cairney, J.; Eckert, C.A.; Frederick, W.J., Jr.; Hallett, J.P.; Leak, D.J.; Liotta, C.L.; et al. The path forward for biofuels and biomaterials. Science 2006, 311, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cheng, J. Hydrolysis of lignocellulosic materials for ethanol production: A review. Bioresour. Technol. 2002, 83, 1–11. [Google Scholar] [CrossRef]
- Isikgor, F.H.; Becer, C.R. Lignocellulosic biomass: A sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. [Google Scholar] [CrossRef]
- Huber, G.W. Breaking the Chemical and Engineering Barriers to Lignocellulosic Biofuels: Next Generation Hydrocarbon Biorefineries; University of Massachusetts Amherst: Amherst, MA, USA; National Science Foundation: Arlington, VA, USA, 2008.
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.P.; Kim, C.S. Lignin depolymerization and conversion: A review of thermochemical methods. Chem. Eng. Technol. 2010, 34, 29–41. [Google Scholar] [CrossRef]
- Demesa, A.G.; Laari, A.; Turunen, I.; Sillanpää, M. Alkaline partial wet oxidation of lignin for the production of carboxylic acids. Chem. Eng. Technol. 2015, 38, 2270–2278. [Google Scholar] [CrossRef]
- Varanasi, P.; Singh, P.; Auer, M.; Adams, P.D.; Simmons, B.A.; Singh, S. Survey of renewable chemicals produced from lignocellulosic biomass during ionic liquid pretreatment. Biotechnol. Biofuels 2013, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, S.K.; Tardio, J.; Prasad, J.; Föger, K.; Akolekar, D.B.; Grocott, S.C. Wet oxidation and catalytic wet oxidation. Ind. Eng. Chem. Res. 2006, 45, 1221–1258. [Google Scholar] [CrossRef]
- Sales, F.G.; Maranhão, L.C.A.; Filho, N.M.L.; Abreu, C.A.M. Kinetic Evaluation and Modeling of Lignin Catalytic Wet Oxidation to Selective Production of Aromatic Aldehydes. Ind. Eng. Chem. Res. 2006, 45, 6627–6631. [Google Scholar] [CrossRef]
- Sales, F.G.; Abreu, C.A.M.; Pereira, J.A.F.R. Catalytic wet-air oxidation of lignin in a three-phase reactor with aromatic aldehyde. Braz. J. Chem. Eng. 2004, 21, 211–218. [Google Scholar] [CrossRef]
- Sales, F.G.; Maranhão, L.C.A.; Filho, N.M.L.; Abreu, C.A.M. Experimental evaluation and continuous catalytic process for fine aldehyde production from lignin. Chem. Eng. Sci. 2007, 62, 5386–5391. [Google Scholar] [CrossRef]
- Mathias, A.L.; Rodrigues, A.E. Production of vanillin by oxidation of pine kraft lignins with oxygen. Holzforschung 1994, 49, 273. [Google Scholar] [CrossRef]
- Wu, G.; Heitz, M.; Chornet, E. Improved alkaline oxidation process for the production of aldehydes (vanillin and syringaldehyde) from steam-explosion hardwood lignin. Ind. Eng. Chem. Res. 1994, 33, 718. [Google Scholar] [CrossRef]
- Wu, G.; Heitz, M. Catalytic mechanism of Cu2+ and Fe3+ in alkaline O2 oxidation of lignin. J. Wood Chem. Technol. 1995, 15, 189–202. [Google Scholar] [CrossRef]
- Tarabanko, V.E.; Petukhov, D.V.; Selyutin, G.E. New mechanism for the catalytic oxidation of lignin to vanillin. Kinet. Catal. 2004, 45, 569–577. [Google Scholar] [CrossRef]
- Villar, J.C.; Caperos, A.; Garcìa-Ochoa, F. Oxidation of hardwood kraft-lignin to phenolic derivatives with oxygen as oxidant. Wood Sci. Technol. 2001, 35, 245–255. [Google Scholar] [CrossRef]
- Deng, H.; Lin, L.; Sun, Y.; Pang, C.; Zhuang, J.; Ouyang, P.; Li, J.; Liu, S. Perovskite-type oxide LaMnO3: An efficient and recyclable heterogeneous catalyst for the wet aerobic oxidation of lignin to aromatic aldehydes. Catal. Lett. 2008, 126, 106. [Google Scholar] [CrossRef]
- Yabushita, M.; Kobayashi, H.; Fukuoka, A. Catalytic transformation of cellulose into platform chemicals. Appl. Catal. B Environ. 2014, 145, 1–9. [Google Scholar] [CrossRef]
- Kruger, J.S.; Cleveland, N.S.; Zhang, S.; Katahira, R.; Black, B.A.; Chupka, G.M.; Lammens, T.; Hamilton, P.G.; Biddy, M.J.; Beckham, G.T. Lignin depolymerization with nitrate-intercalated hydrotalcite catalysts. ACS Catal. 2016, 6, 1316–1328. [Google Scholar] [CrossRef]
- Yan, k.; Liu, Y.; Lu, Y.; Chai, J.; Sun, L. Catalytic application of layered double hydroxidederived catalysts for the conversion of biomass-derived molecules. Catal. Sci. Technol. 2017, 7, 1622–1645. [Google Scholar] [CrossRef]
- Zhang, J. Biomass Conversion over Heteropoly Acid Catalysts. Ph.D. Thesis, King Abdullah University of Science and Technology, Thuwal, Saudi Arabia, 2015. [Google Scholar]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef]
- Lana, E.J.L.; Rocha, K.A.D.S.; Kozhevnikov, I.V.; Gusevskaya, E.V. One-pot synthesis of diisobornyl ether from camphene using heteropoly acid catalysts. J. Mol. Catal. A Chem. 2006, 243, 258–263. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Sun, M.; Ma, X.; Han, Y. Direct conversion of Cellulose to glycolic acid with a phosphomolybdic acid catalyst in a water medium. ACS Catal. 2012, 2, 1698–1702. [Google Scholar] [CrossRef]
- Deng, W.; Zhang, Q.; Wang, Y. Catalytic transformations of cellulose and cellulose-derivedcarbohydrates into organic acids. Catal. Today 2014, 234, 31–41. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Heterogeneous acid catalysis by heteropoly acids: Approaches to catalyst deactivation. J. Mol. Catal. A Chem. 2009, 305, 104–111. [Google Scholar] [CrossRef]
- Geboers, J.; Van de Vyver, S.; Carpentier, K.; Jacobs, P.; Sels, B. Hydrolytic hydrogenation of cellulose with hydrotreated caesium salts of heteropoly acids and Ru/C. Green Chem. 2011, 13, 2167–2174. [Google Scholar] [CrossRef]
- Sun, N.; Jiang, X.; Maxim, M.L.; Metlen, A.; Rogers, R.D. Use of polyoxometalate catalysts in ionic liquids to enhance the dissolution and delignification of woody biomass. ChemSusChem 2011, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Cheng, M.X.; Li, J.Z.; Tian, J.A.; Wang, X.H. One pot production of 5-hydroxymethylfurfural with high yield from cellulose by a Brønsted-Lewis-surfactant-combined heteropolyacid catalyst. Chem. Commun. 2011, 47, 2176–2178. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Furukawa, H.; Kobayashi, N.; Itaya, Y.; Satsuma, A. Effects of Brønsted and Lewis acidities on activity and selectivity of heteropolyacid-based catalysts for hydrolysis of cellobiose and cellulose. Green Chem. 2009, 11, 1627–1632. [Google Scholar] [CrossRef]
- Wolfel, R.; Taccardi, N.; Bosmann, A.; Wasserscheid, P. Selective catalytic conversion of biobased carbohydrates to formic acid using molecular oxygen. Green Chem. 2011, 13, 2759–2763. [Google Scholar] [CrossRef]
- Demesa, A.; Laari, A.; Sillanpää, M. Catalytic wet oxidation of lignin for production of carboxylic acids. In Proceedings of the 23rd European Biomass Conference and Exhibition, Vienna, Austria, 1–4 June 2015. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Advances in catalysis by heteropolyacids. Russ. Chem. Rev. 1987, 56, 811–825. [Google Scholar] [CrossRef]
- Tsigdinos, G.A. Heteropoly compounds of molybdenum and tungsten. Top. Curr. Chem. 1978, 76, 1–64. [Google Scholar]
- Jürgensen, A.; Moffat, J.B. The stability of 12-molybdosilicic, 12-tungstosilicic, 12-molybdophosphoric and 12-tungstophosphoric acids in aqueous solution at various pH. Catal. Lett. 1995, 34, 237–244. [Google Scholar] [CrossRef]
- Deng, H.; Lin, L.; Sun, Y.; Pang, C.; Zhuang, J.; Ouyang, P.; Li, J.; Liu, S. Activity and stability of Perovskite-type oxide LaCoO3 catalyst in lignin catalytic wet oxidation to aromatic aldehydes process. Energy Fuels 2009, 23, 19–24. [Google Scholar] [CrossRef]
- Toledano, A.; LSerrano, L.; Labidi, J. Organosolv lignin depolymerization with different base catalysts. J. Chem. Technol. Biotechnol. 2012, 87, 1593–1599. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Witte, P.T.; Blank, D.H.A.; Alsters, P.L.; ten Elshof, J.E. Recovery of homogeneous polyoxometallate catalysts from aqueous and organic media by a mesoporous ceramic membrane without loss of catalytic activity. Chem. Eur. J. 2006, 12, 3061–3066. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, J.; Zhao, S.; Jiang, C.; Zhang, X.; Wang, X. Hydrolysis of cellulose by the heteropoly acid H3PW12O40. Cellulose 2010, 17, 587–594. [Google Scholar] [CrossRef]
- Zhao, Y.; Xu, Q.; Pan, T.; Zuo, Y.; Fu, Y.; Guo, Q. Depolymerization of lignin by catalytic oxidation with aqueous polyoxometalates. Appl. Catal. A Gen. 2013, 467, 504–508. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Temperature (°C) | Conversion (%) | TCA (%) | Formic Acid | Acetic Acid | Succinic Acid | |||
|---|---|---|---|---|---|---|---|---|---|
| Yield (%) | Selectivity (%) | Yield (%) | Selectivity (%) | Yield (%) | Selectivity (%) | ||||
| 175 | 38 | 22.5 | 8.6 | 22.5 | 6.1 | 16.0 | 0.9 | 2.4 | |
| Non-catalytic | 200 | 48 | 26.0 | 16.0 | 33.3 | 6.5 | 13.5 | 1.2 | 2.5 |
| 225 | 52 | 24.0 | 12.0 | 23.1 | 4.8 | 9.2 | 0.8 | 1.5 | |
| 175 | 62 | 22.0 | 15.4 | 24.8 | 0.4 | 0.7 | 1.8 | 1.8 | |
| H3PW12O4 | 200 | 60 | 14.0 | 9.8 | 16.3 | 0.3 | 0.5 | 1.1 | 2.8 |
| 225 | 72 | 12.0 | 8.4 | 11.7 | 0.2 | 0.3 | 1.0 | 3.8 | |
| 175 | 70 | 33.0 | 21.5 | 30.6 | 8.9 | 12.7 | 1.0 | 1.4 | |
| H3PMo12O40 | 200 | 74 | 40.0 | 26.0 | 35.1 | 10.8 | 14.6 | 1.2 | 1.6 |
| 225 | 80 | 30.0 | 19.5 | 24.4 | 8.1 | 10.1 | 0.9 | 1.1 | |
| Acid | pK1 | pK2 | pK3 |
|---|---|---|---|
| H3PW12O40 | 1.6 | 3 | 4 |
| H3PMo12O40 | 2 | 3.6 | 5.3 |
| H2SO4 | 6.6 | ||
| HCl | 4.3 | ||
| HNO3 | 9.4 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demesa, A.G.; Laari, A.; Sillanpää, M.; Koiranen, T. Valorization of Lignin by Partial Wet Oxidation Using Sustainable Heteropoly Acid Catalysts. Molecules 2017, 22, 1625. https://doi.org/10.3390/molecules22101625
Demesa AG, Laari A, Sillanpää M, Koiranen T. Valorization of Lignin by Partial Wet Oxidation Using Sustainable Heteropoly Acid Catalysts. Molecules. 2017; 22(10):1625. https://doi.org/10.3390/molecules22101625
Chicago/Turabian StyleDemesa, Abayneh Getachew, Arto Laari, Mika Sillanpää, and Tuomas Koiranen. 2017. "Valorization of Lignin by Partial Wet Oxidation Using Sustainable Heteropoly Acid Catalysts" Molecules 22, no. 10: 1625. https://doi.org/10.3390/molecules22101625
APA StyleDemesa, A. G., Laari, A., Sillanpää, M., & Koiranen, T. (2017). Valorization of Lignin by Partial Wet Oxidation Using Sustainable Heteropoly Acid Catalysts. Molecules, 22(10), 1625. https://doi.org/10.3390/molecules22101625