
How Does Thymine DNA Survive Ultrafast Dimerization Damage?


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
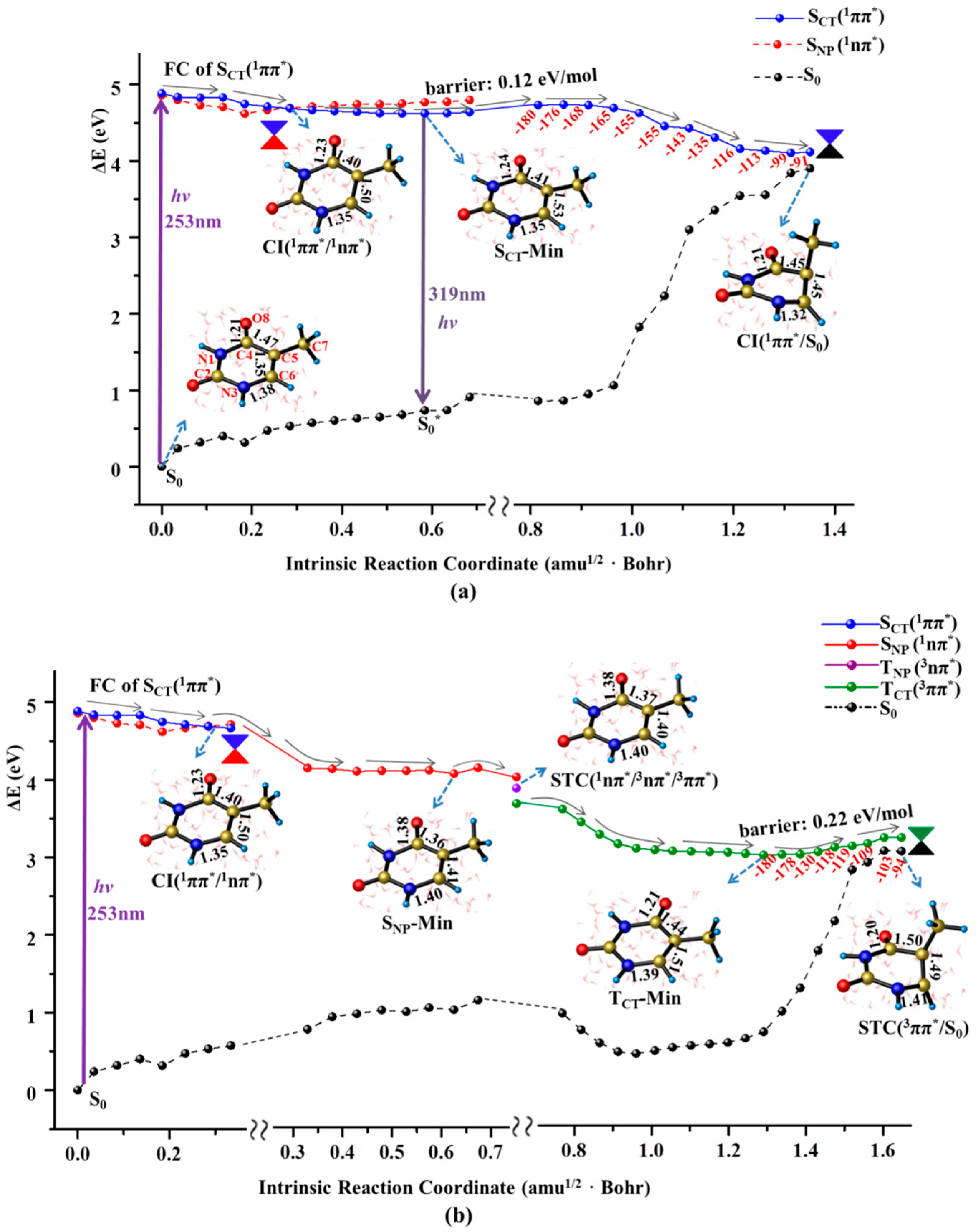
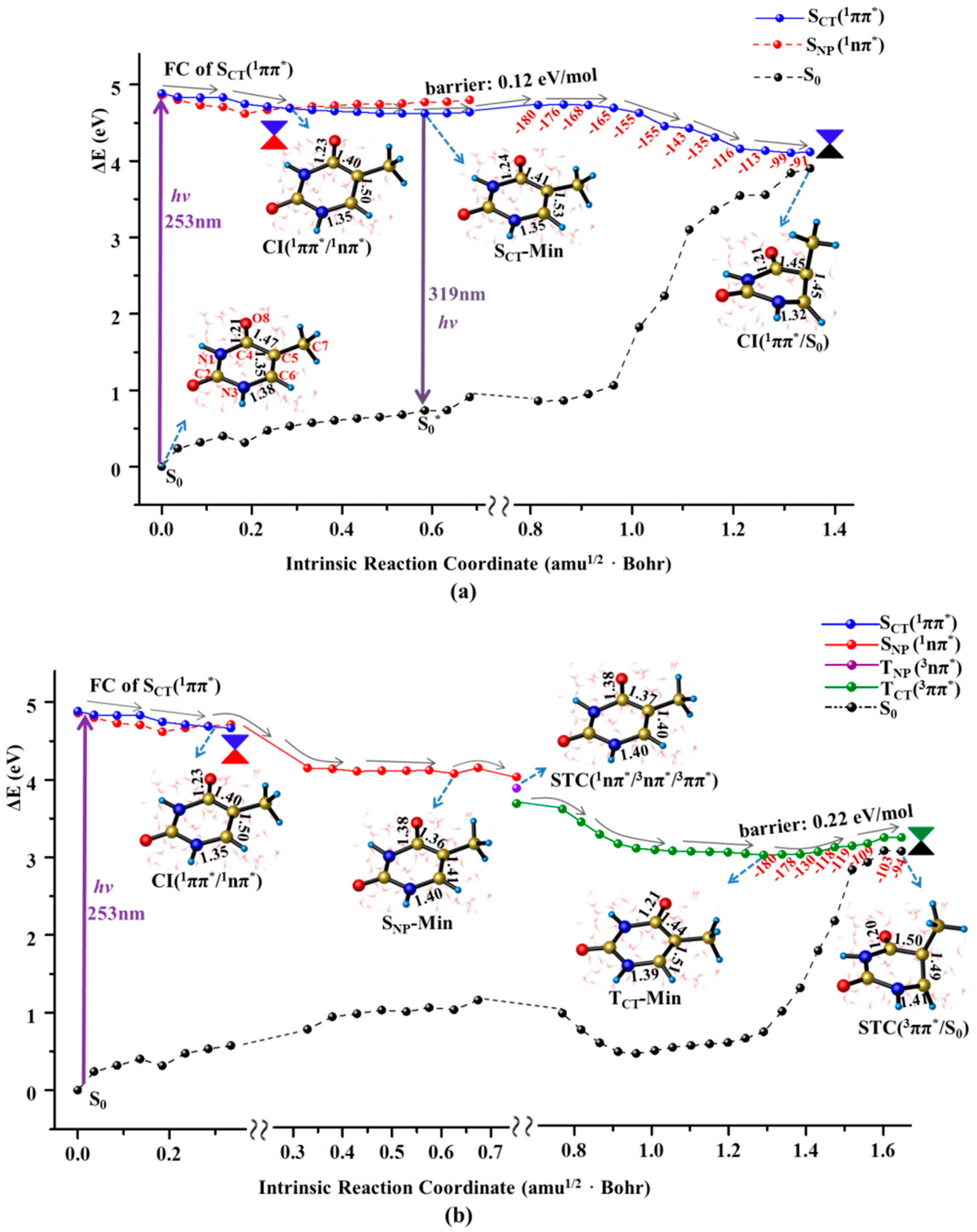
2.1. Ultrafast Decay of Thymine Monomer in Bright SCT(1ππ*) and TCT(3ππ*) States
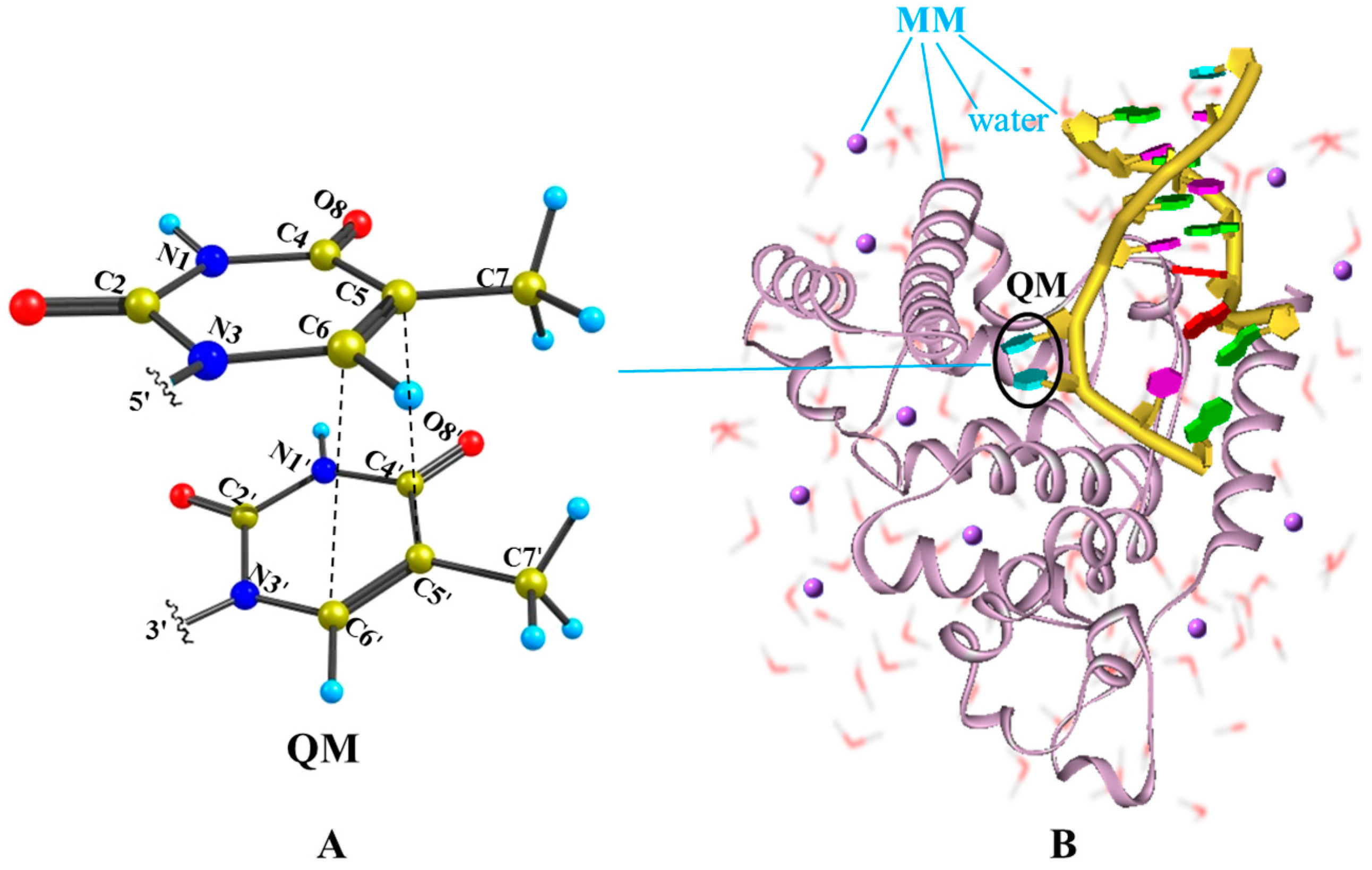
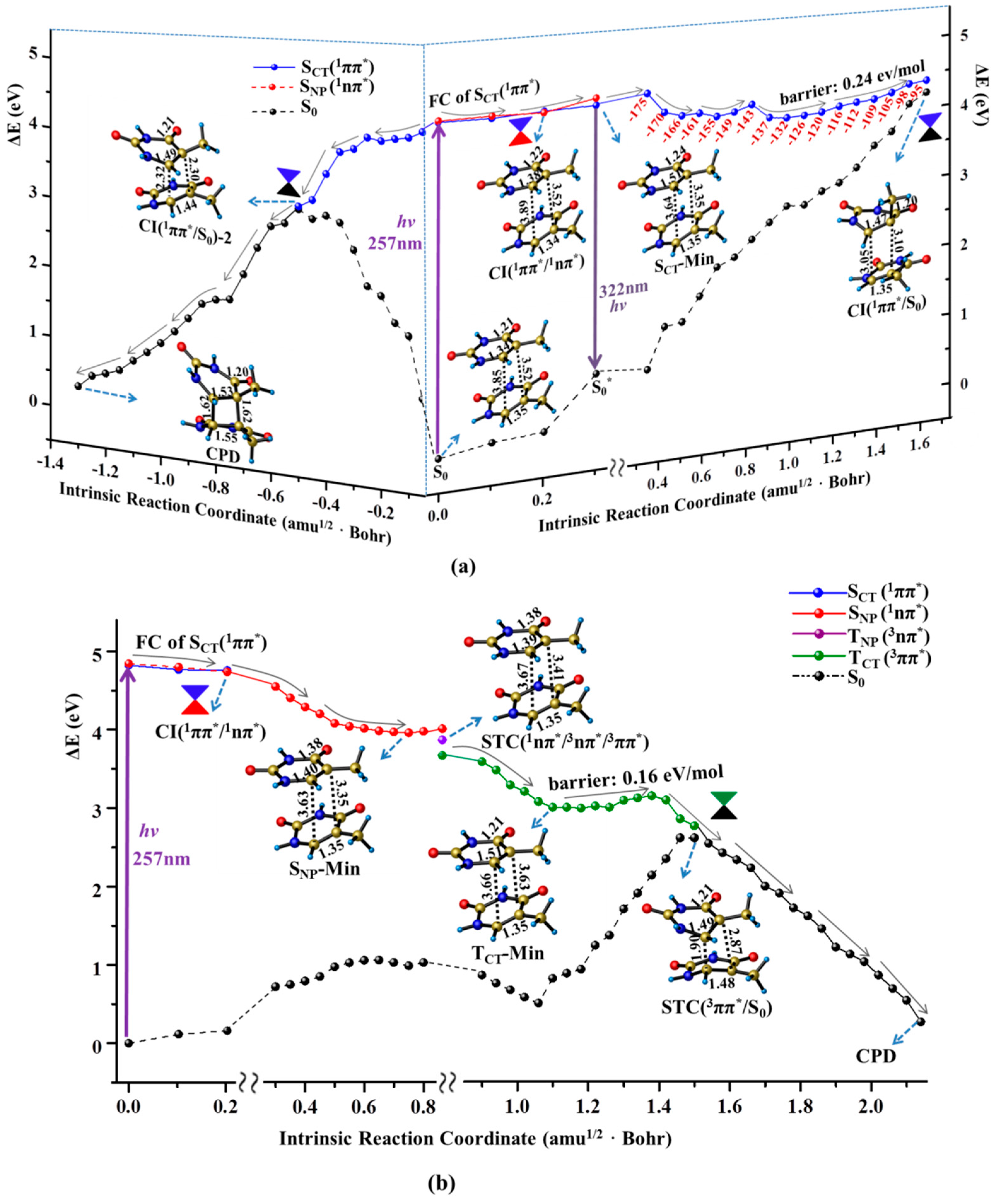
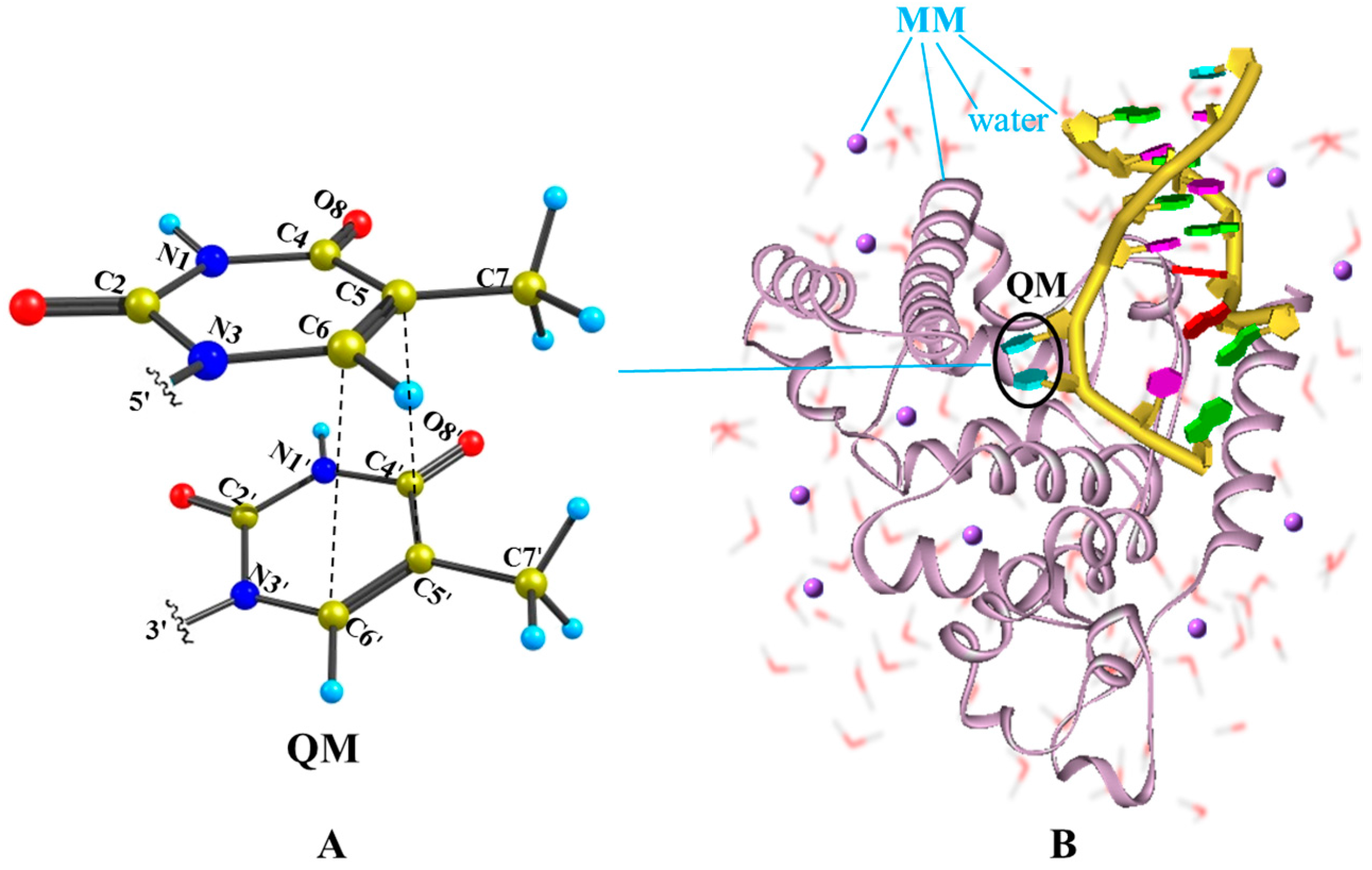
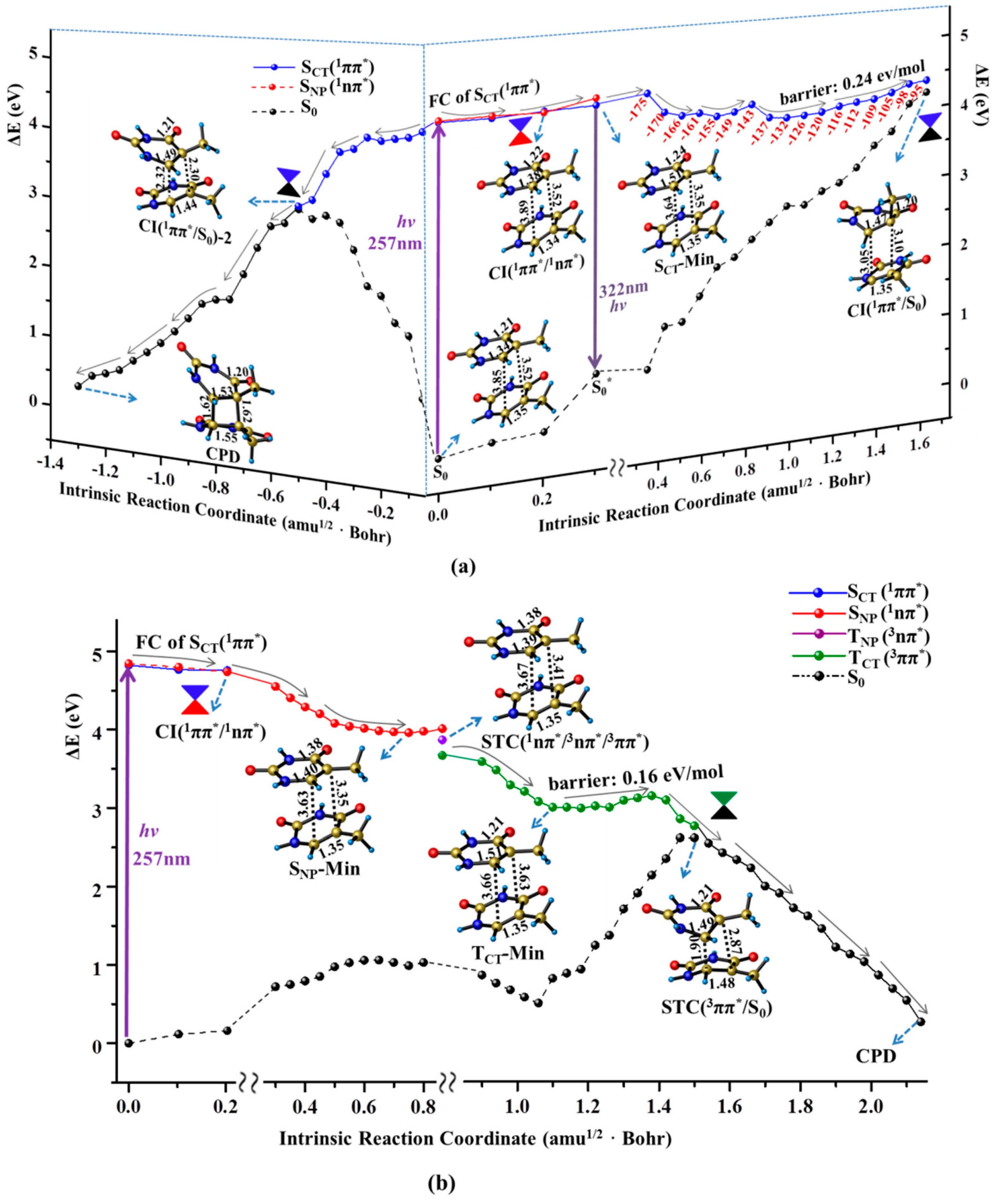
2.2. Nonradiative Decay and Dimerization of Thymine Oligomer in Bright SCT(1ππ*) and TCT(3ππ*) States
3. Computational Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cadet, J.; Vigny, P. The Photochemistry of Nucleic Acids. In Bioorganic Photochemistry; Morrison, H., Ed.; John Wiley & Sons: New York, NY, USA, 1990; Volume 1, pp. 1–272. [Google Scholar]
- Taylor, J.S. Unraveling the Molecular Pathway from Sunlight to Skin Cancer. Acc. Chem. Res. 1994, 27, 76–82. [Google Scholar] [CrossRef]
- Vink, A.A.; Roza, L. Biological Consequences of Cyclobutane Pyrimidine Dimmers. J. Photochem. Photobiol. B 2001, 65, 101–104. [Google Scholar] [CrossRef]
- Melnikova, V.O.; Ananthaswamy, H.N. Cellular and Molecular Events Leading to the Development of Skin Cancer. Mutat. Res. 2005, 571, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Salet, C.; Bensasson, R.; Becker, R.S. Triplet Excited States of Pyrimidine Nucleosides and Nucleotides. Photochem. Photobiol. 1979, 30, 325–329. [Google Scholar] [CrossRef]
- Lamola, A.A.; Mittal, J.P. Solution Photochemistry of Thymine and Uracil. Science 1966, 154, 1560–1561. [Google Scholar] [CrossRef] [PubMed]
- Moysan, A.; Viari, A.; Vigny, P.; Voituriez, L.; Cadet, J.; Moustacchi, E.; Sage, E. Formation of Cyclobutane Thymine Dimers Photosensitized by Pyridopsoralens: Quantitative and Qualitative Distribution within DNA. Biochemistry 1991, 30, 7080–7088. [Google Scholar] [CrossRef] [PubMed]
- Pecourt, J.L.; Peon, J.; Kohler, B. Ultrafast Internal Conversion of Electronically Excited RNA and DNA Nucleosides in Water. J. Am. Chem. Soc. 2000, 122, 9348–9349. [Google Scholar] [CrossRef]
- Pecourt, J.L.; Peon, J.; Kohler, B. DNA Excited-State Dynamics: Ultrafast Internal Conversion and Vibrational Cooling in a Series of Nucleosides. J. Am. Chem. Soc. 2001, 123, 10370–10378. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernández, C.E.; Cohen, B.; Hare, P.M.; Kohler, B. Ultrafast Excited-State Dynamics in Nucleic Acids. Chem. Rev. 2004, 104, 1977–2019. [Google Scholar] [CrossRef] [PubMed]
- Cuquerella, M.C.; Lhiaubet-Vallet, V.; Bosca, F.; Miranda, M.A. Photosensitised Pyrimidine Dimerisation in DNA. Chem. Sci. 2011, 2, 1219–1232. [Google Scholar] [CrossRef]
- Asturiol, D.; Lasorne, B.; Robb, M.A.; Blancafort, L. Photophysics of the π,π* and n,π* States of Thymine: MS-CASPT2 Minimum-Energy Paths and CASSCF on-the-fly Dynamics. J. Phys. Chem. A 2009, 113, 10211–10218. [Google Scholar] [CrossRef] [PubMed]
- Kwok, W.M.; Ma, C.; Phillips, D.L. A Doorway State Leads to Photostability or Triplet Photodamage in Thymine DNA. J. Am. Chem. Soc. 2008, 130, 5131–5139. [Google Scholar] [CrossRef] [PubMed]
- Kohler, B. Nonradiative Decay Mechanisms in DNA Model Systems. J. Phys. Chem. Lett. 2010, 1, 2047–2053. [Google Scholar] [CrossRef]
- Etinski, M.; Fleig, T.; Marian, C.A. Intersystem Crossing and Characterization of Dark States in the Pyrimidine Nucleobases Uracil, Thymine, and 1-Methylthymine. J. Phys. Chem. A 2009, 113, 11809–11816. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pérez, J.J.; González-Luque, R.; Merchán, M.; Serrano-Andrés, L. On the Intrinsic Population of the Lowest Triplet State of Thymine. J. Phys. Chem. B 2007, 111, 11880–11883. [Google Scholar] [CrossRef] [PubMed]
- Marguet, S.; Markovitsi, D. Time-Resolved Study of Thymine Dimer Formation. J. Am. Chem. Soc. 2005, 127, 5780–5781. [Google Scholar] [CrossRef] [PubMed]
- Schreier, W.J.; Schrader, T.E.; Koller, F.O.; Gilch, P.; Crespo-Hernández, C.E.; Swaminathan, V.N.; Carell, T.; Zinth, W.; Kohler, B. Thymine Dimerization in DNA is an Ultrafast Photoreaction. Science 2007, 315, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Schreier, W.J.; Kubon, J.; Regner, N.; Haiser, K.; Schrader, T.E.; Zinth, W.; Clivio, P.; Gilch, P. Thymine Dimerization in DNA Model Systems: Cyclobutane Photolesion is Predominantly Formed via the Singlet Channel. J. Am. Chem. Soc. 2009, 131, 5038–5039. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Douki, T.; Improta, R.; Gustavsson, T.; Onidas, D.; Vaya, I.; Perron, M.; Markovitsi, D. Electronic Excited States Responsible for Dimer Formation upon UV Absorption Directly by Thymine Strands: Joint Experimental and Theoretical Study. J. Am. Chem. Soc. 2012, 134, 14834–14845. [Google Scholar] [CrossRef] [PubMed]
- Nikogosyan, D.N. Two-quantum UV Photochemistry of Nucleic Acids: Comparison with Conventional Low-intensity UV Photochemistry and Radiation Chemistry. Int. J. Radiat. Biol. 1990, 57, 233–299. [Google Scholar] [CrossRef] [PubMed]
- McCullagh, M.; Hariharan, M.; Lewis, F.; Markovitsi, D.; Douki, T.; Schatz, G.C. Conformational Control of TT Dimerization in DNA Conjugates. A Molecular Dynamics Study. J. Phys. Chem. B 2010, 114, 5215–5221. [Google Scholar] [CrossRef] [PubMed]
- Beukers, R.; Eker, A.P.M.; Lohman, P.H.M. 50 Years Thymine Dimer. DNA Repair 2008, 7, 530–543. [Google Scholar] [CrossRef] [PubMed]
- Banyasz, A.; Vayá, I.; Changenet-Barret, P.; Gustavsson, T.; Douki, T.; Markovitsi, D. Base Pairing Enhances Fluorescence and Favors Cyclobutane Dimer Formation Induced upon Absorption of UVA Radiation by DNA. J. Am. Chem. Soc. 2011, 133, 5163–5165. [Google Scholar] [CrossRef] [PubMed]
- Görner, H. Transients of Uracil and Thymine Derivatives and the Quantum Yields of Electron and Intersystem Crossing upon 20 ns Photolysis at 248 nm. Photochem. Photobiol. 1990, 52, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Court, M.; Sauvaigo, S.; Odin, F.; Cadet, J. Formation of the Main UV-induced Thymine Dimeric Lesions within Isolated and Cellular DNA as Measured by High Performance Liquid Chromatography-Tandem Mass Spectrometry. J. Biol. Chem. 2000, 275, 11678–11685. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Angelov, D.; Cadet, J. UV Laser Photolysis of DNA: Effect of Duplex Stability on Charge-Transfer Efficiency. J. Am. Chem. Soc. 2001, 123, 11360–11366. [Google Scholar] [CrossRef] [PubMed]
- Douki, T.; Reynaud-Angelin, A.; Cadet, J.; Sage, E. Bipyrimidine Photoproducts Rather than Oxidative Lesions Are the Main Type of DNA Damage Involved in the Genotoxic Effect of Solar UVA Radiation. Biochemistry 2003, 42, 9221–9226. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernández, C.E.; Cohen, B.; Kohler, B. Base stacking controls excited-state dynamics in A-T DNA. Nature 2005, 436, 1141–1144. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Marquetand, P.; González-Vázquez, J.; Sola, I.; González, L. Femtosecond Intersystem Crossing in the DNA Nucleobase Cytosine. J. Phys. Chem. Lett. 2012, 3, 3090–3095. [Google Scholar] [CrossRef] [PubMed]
- Markovitsi, D.; Talbot, F.; Gustavsson, T.; Onidas, D.; Lazzarotto, E.; Marguet, S. Complexity of excited-state dynamics in DNA. Nature 2006, 441, E7. [Google Scholar] [CrossRef] [PubMed]
- Markovitsi, D.; Onidas, D.; Gustavsson, T.; Talbot, F.; Lazzarotto, E. Collective Behavior of Franck-Condon Excited States and Energy Transfer in DNA Double Helices. J. Am. Chem. Soc. 2005, 127, 17130–17131. [Google Scholar] [CrossRef] [PubMed]
- Kwok, W.-M.; Ma, C.; Phillips, D.L. Femtosecond Time- and Wavelength-Resolved Fluorescence and Absorption Spectroscopic Study of the Excited States of Adenosine and an Adenine Oligomer. J. Am. Chem. Soc. 2006, 62, 11894–11905. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Fang, W.H.; Wang, H.B. Slow deactivation channels in UV-photoexcited adenine DNA. Phys. Chem. Chem. Phys. 2014, 9, 4210–4219. [Google Scholar] [CrossRef] [PubMed]
- Blancafort, L.; Migani, A. Modeling Thymine Photodimerizations in DNA: Mechanism and Correlation Diagrams. J. Am. Chem. Soc. 2007, 129, 14540–14541. [Google Scholar] [CrossRef] [PubMed]
- Boggio-Pasqua, M.; Groenhof, G.; Schäfer, L.V.; Grubmüller, H.; Robb, M.A. Ultrafast Deactivation Channel for Thymine Dimerization. J. Am. Chem. Soc. 2007, 129, 10996–10997. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pérez, J.J.; González-Ramírez, I.; Coto, P.B.; Merchán, M.; Serrano-Andrés, L. Theoretical Insight into the Intrinsic Ultrafast Formation of Cyclobutane Pyrimidine Dimers in UV-Irradiated DNA: Thymine versus Cytosine. J. Phys. Chem. B 2008, 112, 14096–14098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.B.; Eriksson, L.A. A Triplet Mechanism for the Formation of Cyclobutane Pyrimidine Dimers in UV-Irradiated DNA. J. Phys. Chem. B 2006, 110, 7556–7562. [Google Scholar] [CrossRef] [PubMed]
- Climent, T.; González-Ramírez, I.; González-Luque, R.; Merchán, M.; Serrano-Andrés, L. Cyclobutane Pyrimidine Photodimerization of DNA/RNA Nucleobases in the Triplet State. J. Phys. Chem. Lett. 2010, 1, 2072–2076. [Google Scholar] [CrossRef]
- Law, Y.K.; Azadi, J.; Crespo-Hernandez, C.E.; Olmon, E.; Kohler, B. Predicting Thymine Dimerization Yields from Molecular Dynamics Simulations. Biophys. J. 2008, 94, 3590–3600. [Google Scholar] [CrossRef] [PubMed]
- Tramer, Z.; Wierzcho, K.; Shugar, D. Influence of polynucleotide secondary structure on thymine photodimerization. Acta Biochim. Pol. 1969, 16, 83–107. [Google Scholar] [PubMed]
- Görner, H. Photochemistry of DNA and related biomolecules: Quantum yields and consequences of photoionization. J. Photochem. Photobiol. B Biol. 1994, 26, 117–139. [Google Scholar] [CrossRef]
- Patrick, M.H. Studies on thymine-derived UV photoproducts in DNA-I. Formation and biological role of pyrimidine adducts in DNA. Photochem. Photobiol. 1977, 25, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Games, F.; Davila, C.A. Alterations in DNA irradiated with ultraviolet radiation-I. The formation process of cyclobutylpyrimidine dimers: Cross sections, action spectra and quantum yields. Photochem. Photobiol. 1982, 35, 9–16. [Google Scholar]
- Desnous, C.; Babu, B.R.; McIriou, C.; Mayo, J.U.O.; Favre, A.; Wengel, J.; Clivio, P. The sugar conformation governs (6–4) photoproduct formation at the dinucleotide level. J. Am. Chem. Soc. 2008, 130, 30–31. [Google Scholar] [CrossRef] [PubMed]
- Merchán, M.; González-Luque, R.; Climent, T.; Serrano-Andrés, L. Unified model for the ultrafast decay of pyrimidine nucleobases. J. Phys. Chem. B 2006, 110, 26471–26476. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.B.; Fang, W.H. Insights into photodissociation dynamics of benzamide and formanilide from ab initio calculations. J. Am. Chem. Soc. 2004, 126, 8976–8980. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.H. Ab initio determination of dark structures in radiationless transitions for aromatic carbonyl compounds. Acc. Chem. Res. 2008, 41, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Buchner, F.; Nakayama, A.; Yamazaki, S.; Ritze, H.-H.; Lübcke, A. Excited-State Relaxation of Hydrated Thymine and Thymidine Measured by Liquid-Jet Photoelectron Spectroscopy: Experiment and Simulation. J. Am. Chem. Soc. 2015, 137, 2931–2938. [Google Scholar] [CrossRef] [PubMed]
- Hare, P.M.; Crespo-Hernandez, C.E.; Kohler, B. Internal Conversion to the Electronic Ground State Occurs via Two Distinct Pathways for Pyrimidine Bases in Aqueous Solution. Proc. Natl. Acad. Sci. USA 2007, 104, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Conti, I.; Altoe, P.; Stenta, M.; Garavelli, M.; Orlandi, G. Adenine Deactivation in DNA Resolved at the CASPT2//CASSCF/AMBER Level. Phys. Chem. Chem. Phys. 2010, 12, 5016–5023. [Google Scholar] [CrossRef] [PubMed]
- Segarra-Martí, J.; Garavelli, M.; Aquilante, F. Multiconfigurational Second-Order Perturbation Theory with Frozen Natural Orbitals Extended to the Treatment of Photochemical Problems. J. Chem. Theory Comput. 2015, 11, 3772–3784. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, T.; Banyasz, A.; Lazzarotto, E.; Markovitsi, D.; Scalamani, G.; Frisch, M.J.; Barone, V.; Improta, R. Singlet Excited-State Behavior of Uracil and Thymine in Aqueous Solution: A Combined Experimental and Computational Study of 11 Uracil Derivatives. J. Am. Chem. Soc. 2006, 128, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Giussani, A.; Segarra-Marti, J.; Roca-Sanjuán, D.; Merchán, M. Excitation of nucleobases from a computational perspective I: Reaction paths. Top. Curr. Chem. 2015, 355, 57–97. [Google Scholar] [PubMed]
- Francés-Monerris, A.; Segarra-Martí, J.; Merchán, M.; Roca-Sanjuán, D. Theoretical study on the excited-state π-stacking versus intermolecular hydrogen-transfer processes in the guanine–cytosine/cytosine trimer. Theor. Chem. Acc. 2016, 135, 31. [Google Scholar] [CrossRef]
- Nachtigallova, D.; Zeleny, T.; Ruckenbauer, M.; Muller, T.; Barbatti, M.; Hobza, P.; Lischka, H. Does Stacking Restrain the Photodynamics of Individual Nucleobases? J. Am. Chem. Soc. 2010, 132, 8261–8263. [Google Scholar] [CrossRef] [PubMed]
- Mendieta-Moreno, J.I.; Trabada, D.G.; Mendieta, J.; Lewis, J.P.; Gómez-Puertas, P.; Ortega, J. Quantum Mechanics/Molecular Mechanics Free Energy Maps and Nonadiabatic Simulations for a Photochemical Reaction in DNA: Cyclobutane Thymine Dimer. J. Phys. Chem. Lett. 2016, 7, 4391–4397. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.J.; Chen, X.B.; Fang, W.H. Excited-state proton coupled electron transfer between photolyase and the damaged DNA through water wire: A photo-repair mechanism. Phys. Chem. Chem. Phys. 2014, 16, 25432–25441. [Google Scholar] [CrossRef] [PubMed]
- Rauer, C.; Nogueira, J.J.; Marquetand, P.; Gonzalez, L. Cyclobutane Thymine Photodimerization Mechanism Revealed by Nonadiabatic Molecular Dynamics. J. Am. Chem. Soc. 2016, 138, 15911–15916. [Google Scholar] [CrossRef] [PubMed]
- Mees, A.; Klar, T.; Gnau, P.; Hennecke, U.; Eker, A.P.M.; Carell, T.; Essen, L.O. Crystal structure of a photolyase bound to a CPD-like DNA lesion after in situ repair. Science 2004, 306, 1789–1793. [Google Scholar] [CrossRef] [PubMed]
- Roca-Sanjuán, D.; Olaso-González, G.; González-Ramírez, I.; Serrano-Andrés, L.; Merchán, M. Molecular basis of DNA photodimerization: Intrinsic production of cyclobutane cytosine dimers. J. Am. Chem. Soc. 2008, 130, 10768–10779. [Google Scholar] [CrossRef] [PubMed]
- Giussani, A.; Serrano-Andres, L.; Merchan, M.; Roca-Sanjuan, D.; Garavelli, M. Photoinduced Formation Mechanism of the Thymine–Thymine (6–4) adduct. J. Phys. Chem. B 2013, 117, 1999–2004. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O.; Sadlej, A.J.; Wolinski, K. Second-Order Perturbation Theory with a CASSCF Reference Function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.A.; Roos, B.O. Second-Order Perturbation Theory with a Complete Active Space Self-Consistent Field Reference Function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Merchán, M.; Serrano-Andrés, L. Ab Initio Methods for Excited States. In Theoretical and Computational Chemistry; Olivucci, M., Ed.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 16, pp. 35–91. [Google Scholar]
- Roos, B.O.; Andersson, K.; Fulscher, M.P.; Malmqvist, P.A.; Serrano-Andrés, L.; Pierloot, K.; Merchán, M. Multiconfigurational Perturbation Theory: Applications in Electronic Spectroscopy. In Advances in Chemical Physics: New Methods in Computational Quantum Mechanics; Prigogine, I., Rice, S.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; Volume 93, pp. 219–331. [Google Scholar]
- Roca-Sanjuán, D.; Aquilante, F.; Lindh, R. Multiconfiguration Second-Order Perturbation Theory Approach to Strong Electron Correlation in Chemistry and Photochemistry. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 585–603. [Google Scholar] [CrossRef]
- Olaso-González, G.; Roca-Sanjuán, D.; Serrano-Andrés, L.; Merchán, M. Toward the understanding of DNA fluorescence: The singlet excimer of cytosine. J. Chem. Phys. 2006, 125, 231102. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian03; revision D.02; Gaussian, Inc.: Pittsburgh, PA, USA, 2004. [Google Scholar]
- Aquilante, F.; De Vico, L.; Ferré, N.; Lindh, R.; Ghigo, G.; Malmqvist, P.A.; Neogrády, P.; Pedersen, T.B.; Pitonák, M.; Reiher, M.; et al. Software news and update MOLCAS 7: The next generation. J. Comput. Chem. 2010, 31, 224–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Cieplak, P.; Kollman, P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000, 21, 1049–1074. [Google Scholar] [CrossRef]
- Ponder, J.W.; Richards, F.M. An efficient newton-like method for molecular mechanics energy minimization of large molecules. J. Comput. Chem. 1987, 8, 1016–1024. [Google Scholar] [CrossRef]
- Ferré, N.; Cembran, A.; Garavelli, M.; Olivucci, M. Complete-active-space self-consistent-field/Amber parameterization of the Lys296-retinal-Glu113 rhodopsin chromophore-counterion system. Theor. Chem. Acc. 2004, 112, 335–341. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Chen, X. How Does Thymine DNA Survive Ultrafast Dimerization Damage? Molecules 2017, 22, 60. https://doi.org/10.3390/molecules22010060
Wang H, Chen X. How Does Thymine DNA Survive Ultrafast Dimerization Damage? Molecules. 2017; 22(1):60. https://doi.org/10.3390/molecules22010060
Chicago/Turabian StyleWang, Hongjuan, and Xuebo Chen. 2017. "How Does Thymine DNA Survive Ultrafast Dimerization Damage?" Molecules 22, no. 1: 60. https://doi.org/10.3390/molecules22010060
APA StyleWang, H., & Chen, X. (2017). How Does Thymine DNA Survive Ultrafast Dimerization Damage? Molecules, 22(1), 60. https://doi.org/10.3390/molecules22010060