
Pharmacomodulation of the Antimalarial Plasmodione: Synthesis of Biaryl- and N-Arylalkylamine Analogues, Antimalarial Activities and Physicochemical Properties

 ,
,
Abstract
: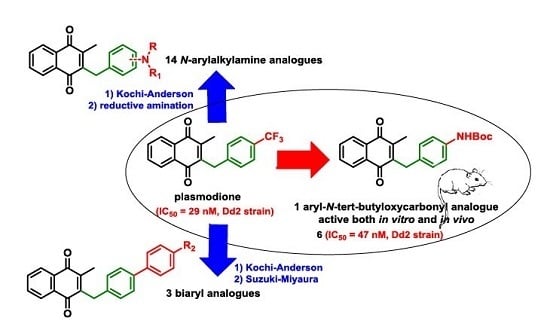
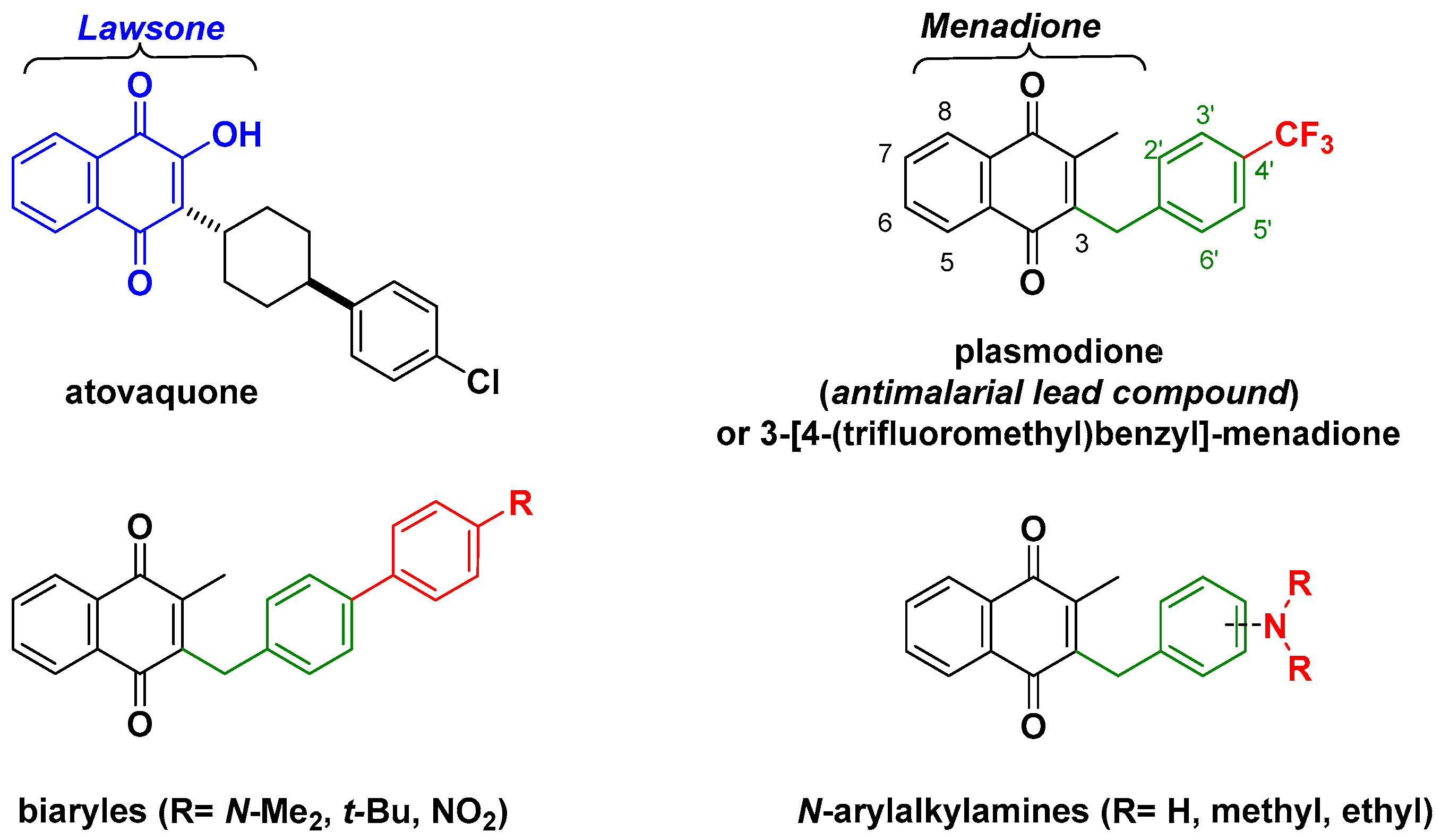
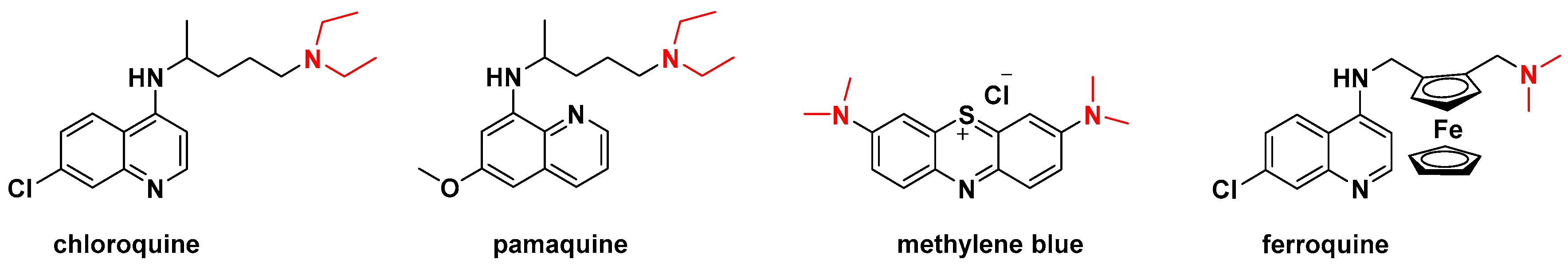
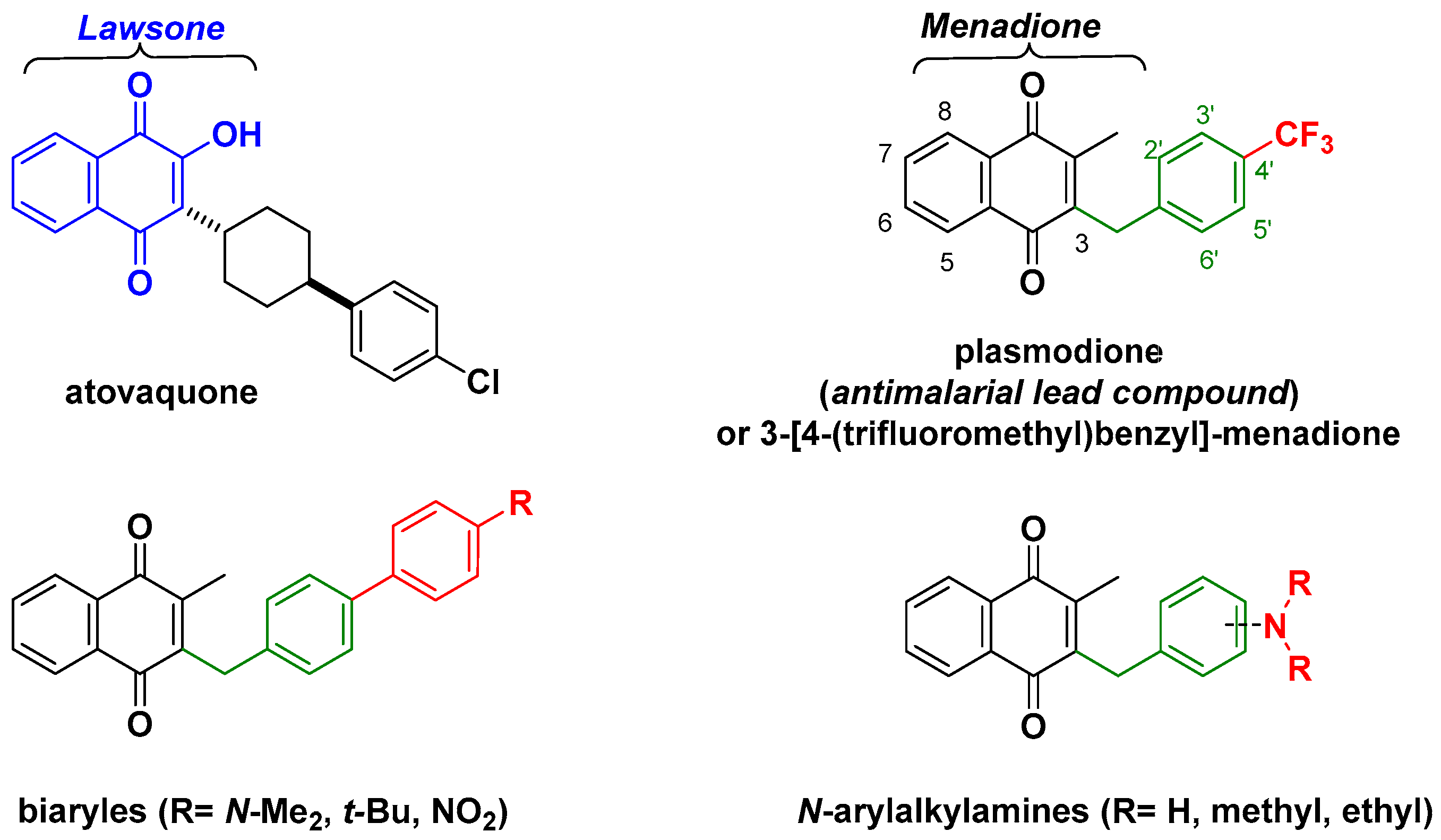
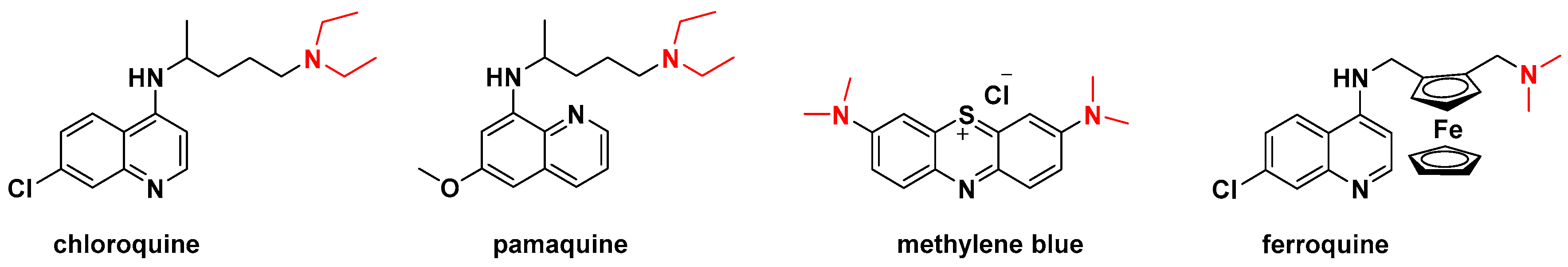
1. Introduction
2. Results
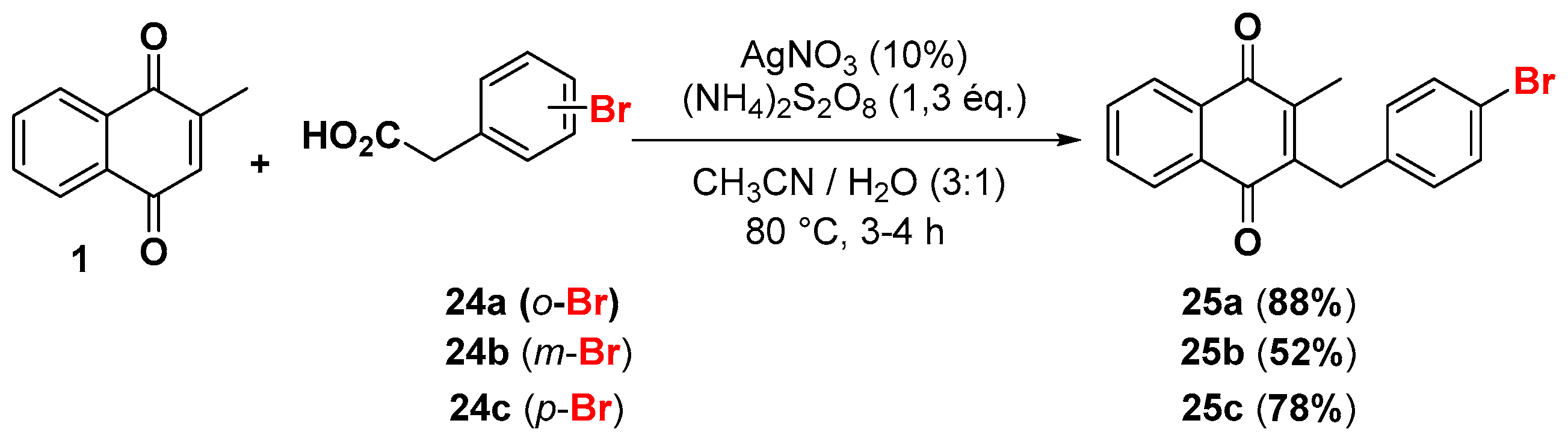
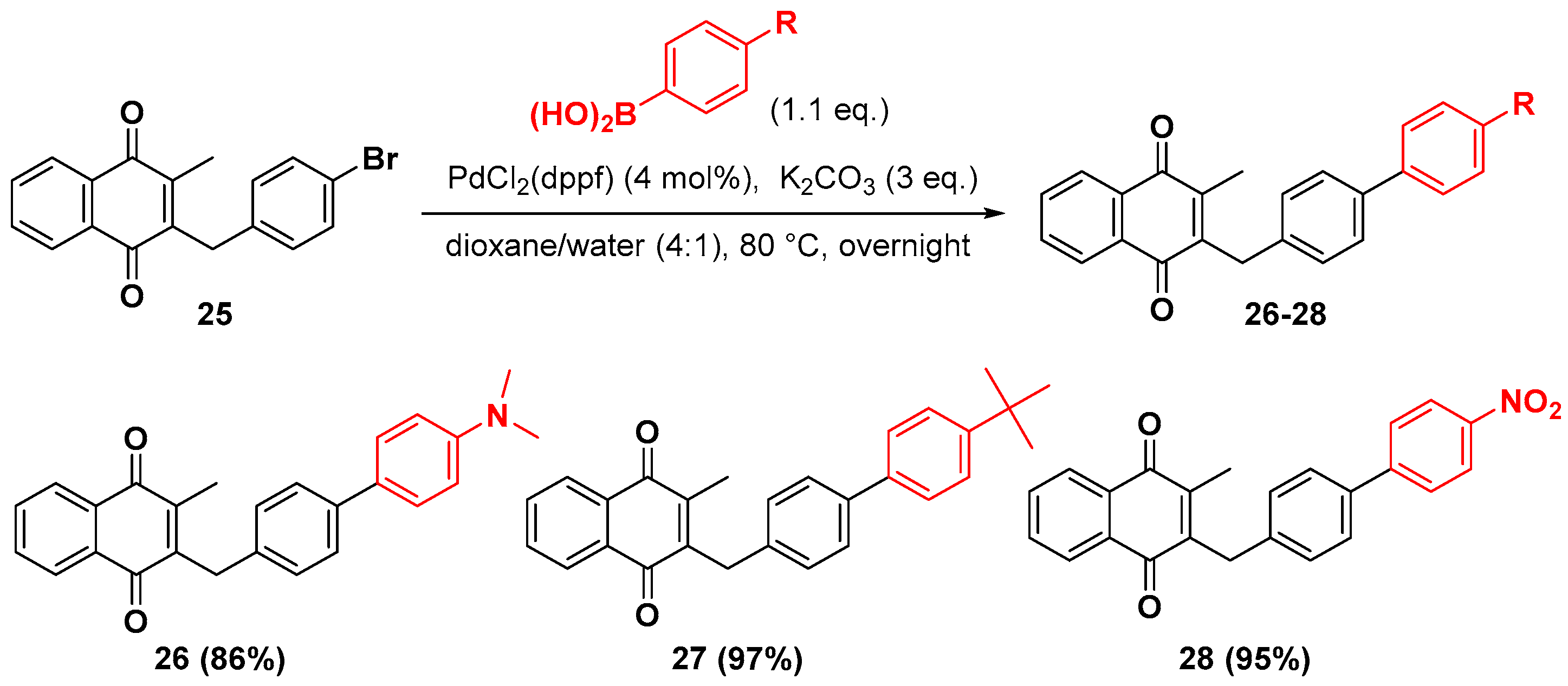
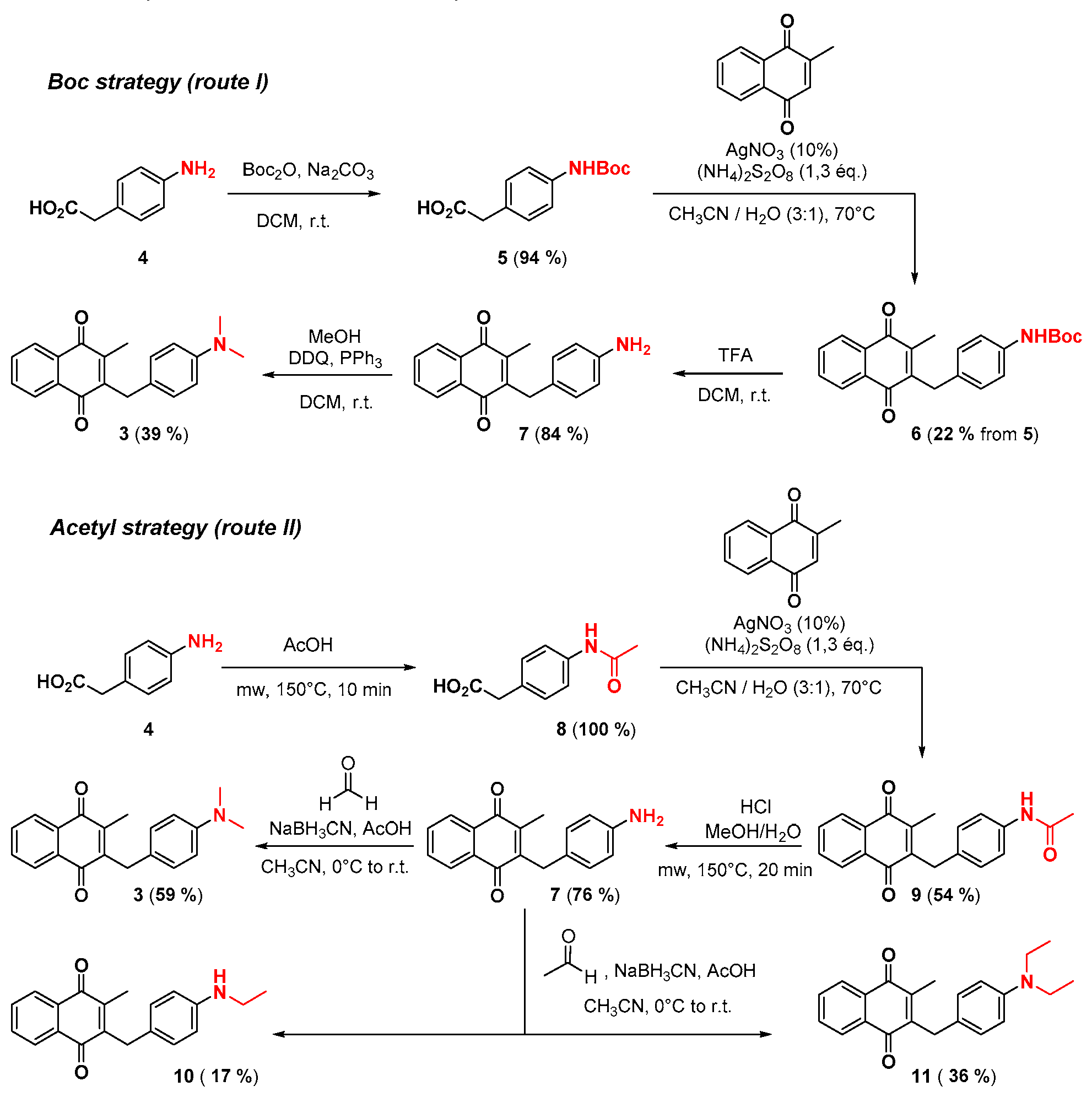
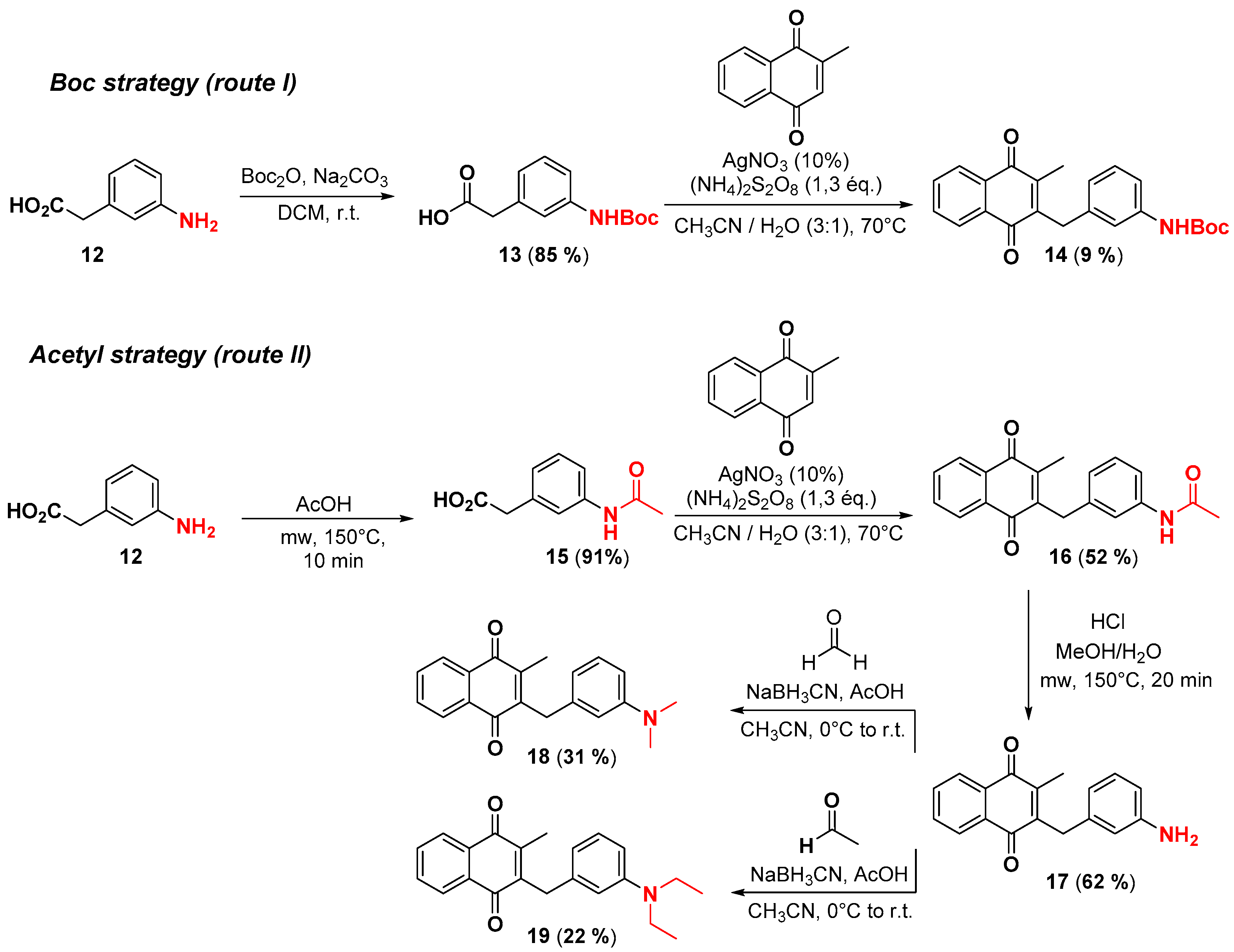
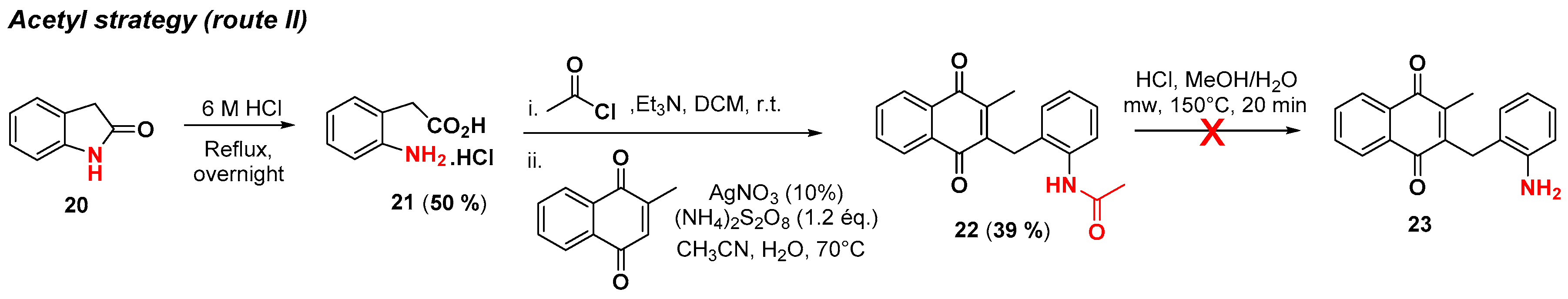
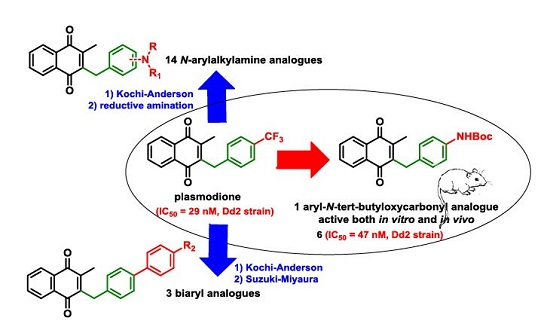
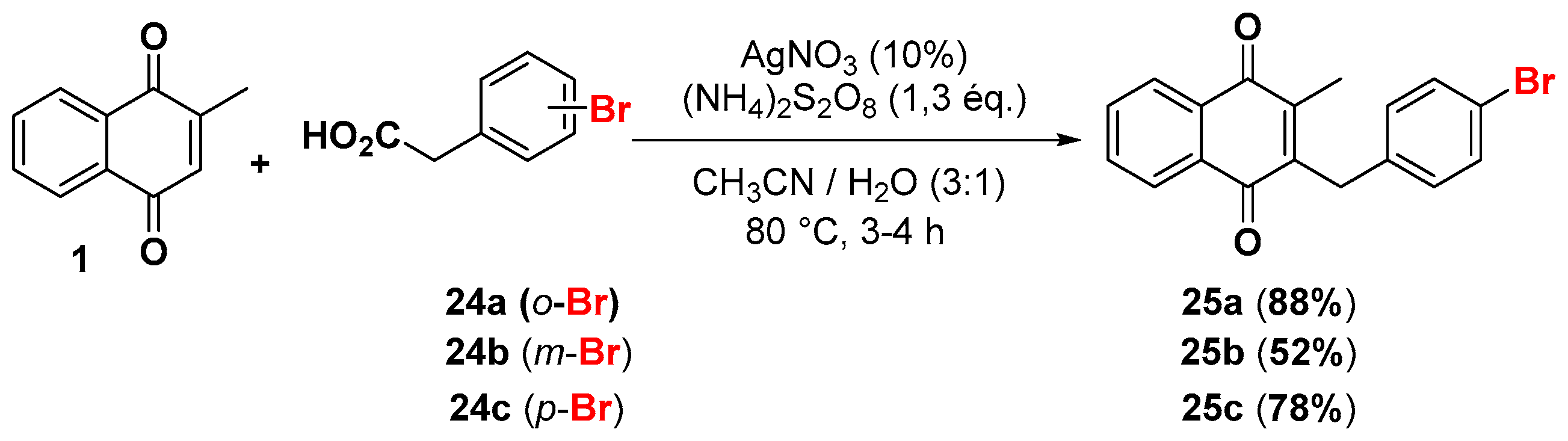
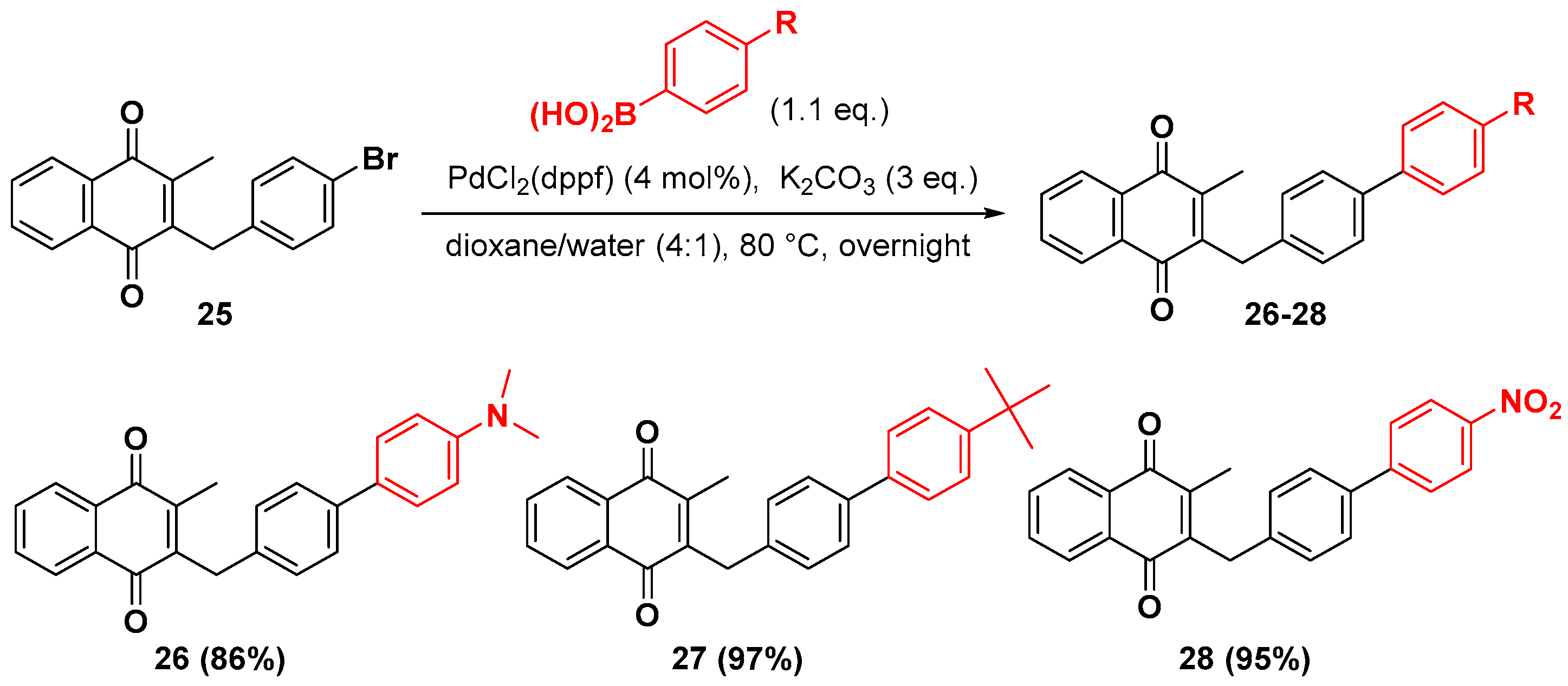
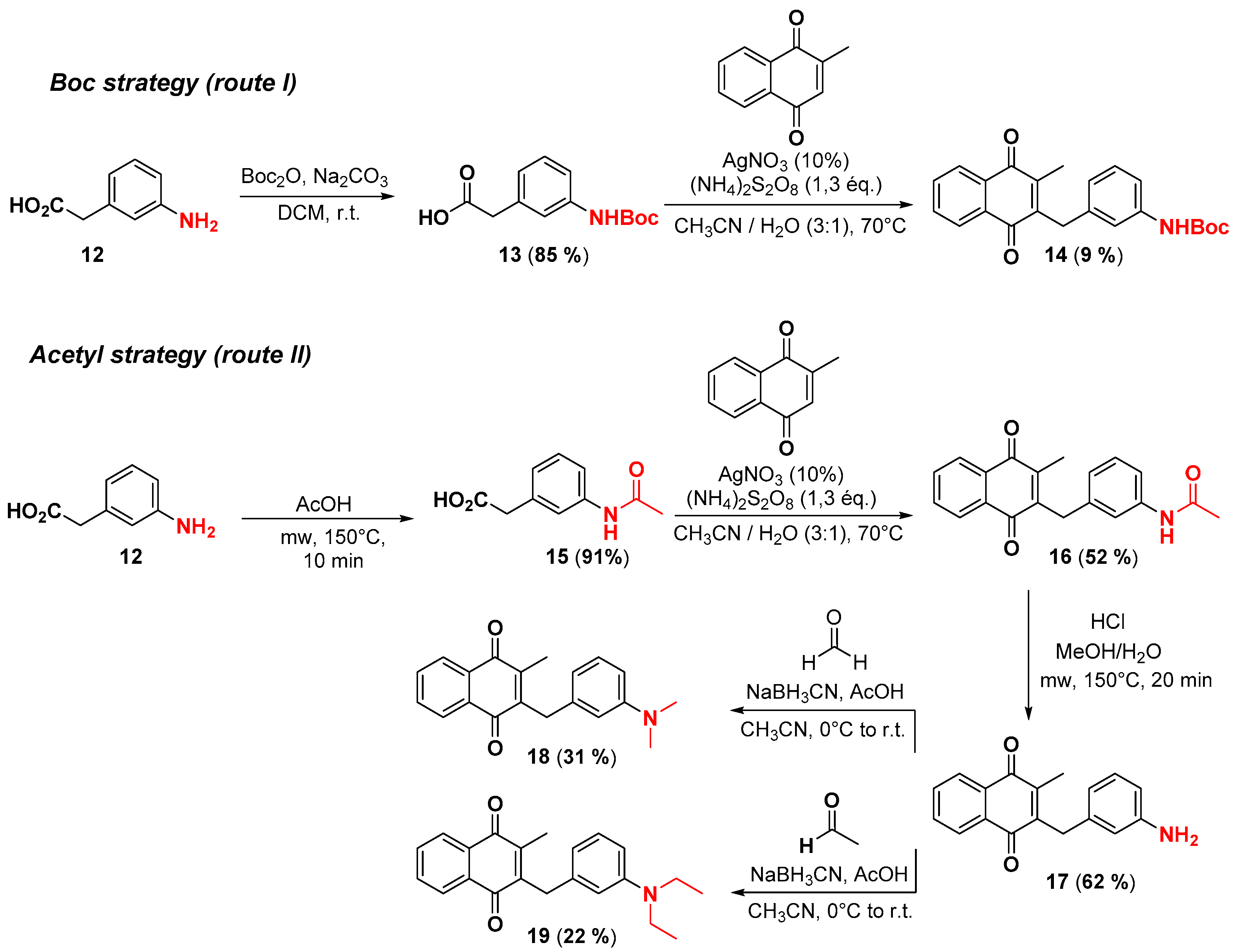
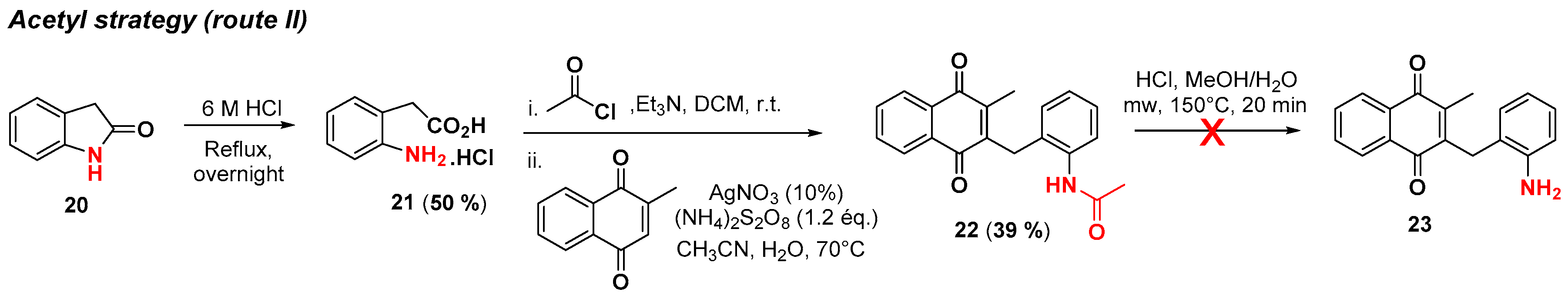
2.1. Chemistry
2.2. Biological Activities
2.2.1. In Vitro Antimalarial Activity
2.2.2. Cytotoxicity of Plasmodione Derivatives in Human Cell Assays
2.2.3. In Vivo Antimalarial Activity
2.3. Physicochemical Properties
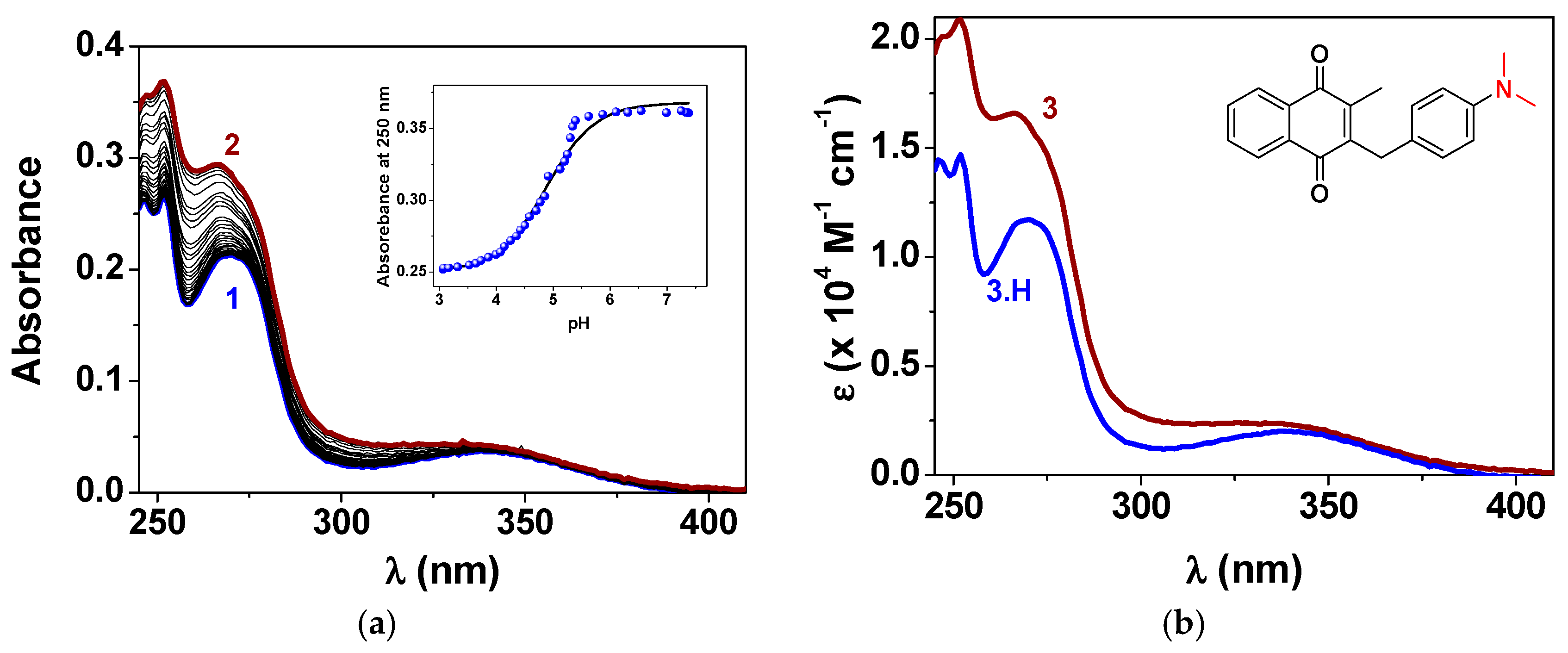
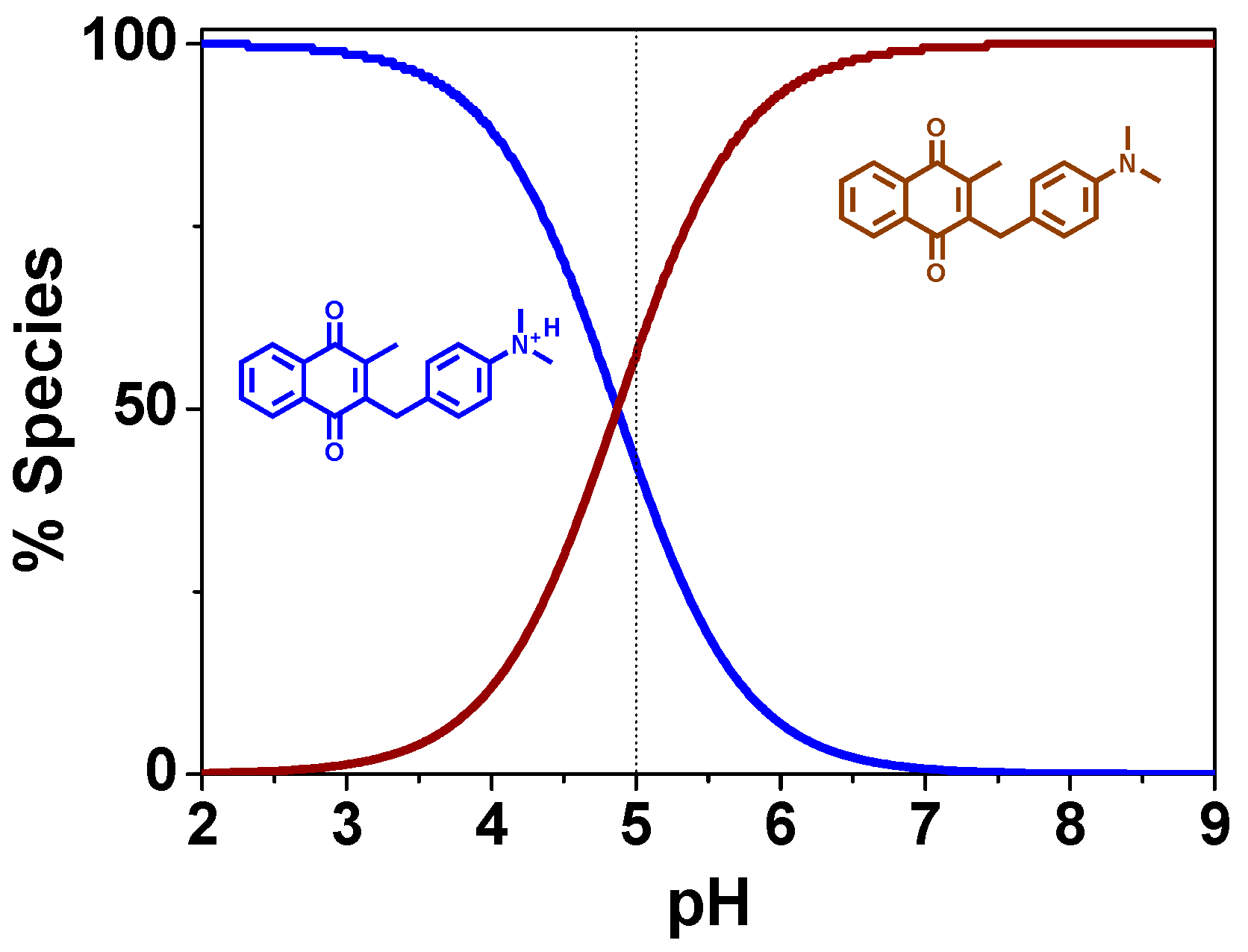
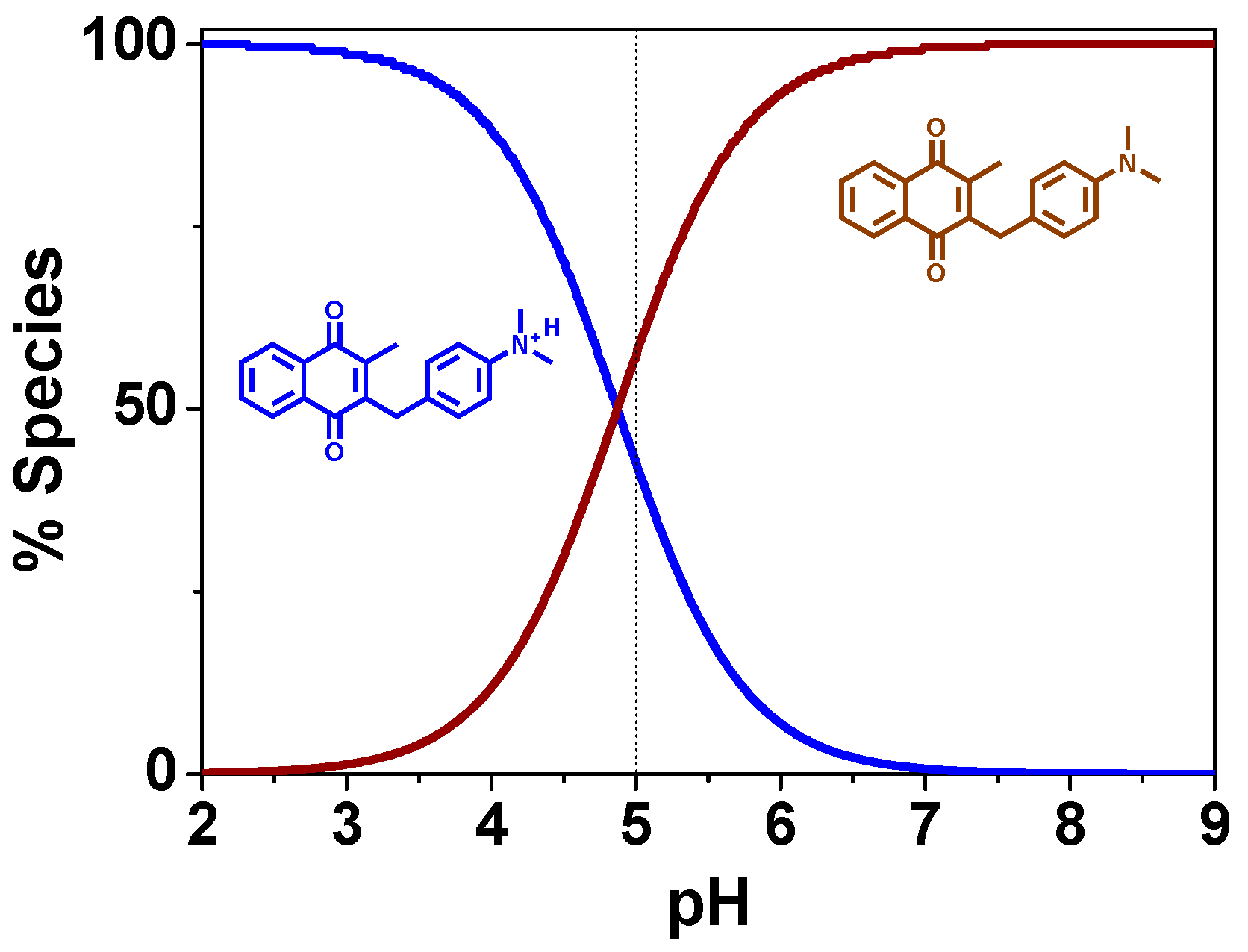
Acid-Base Properties
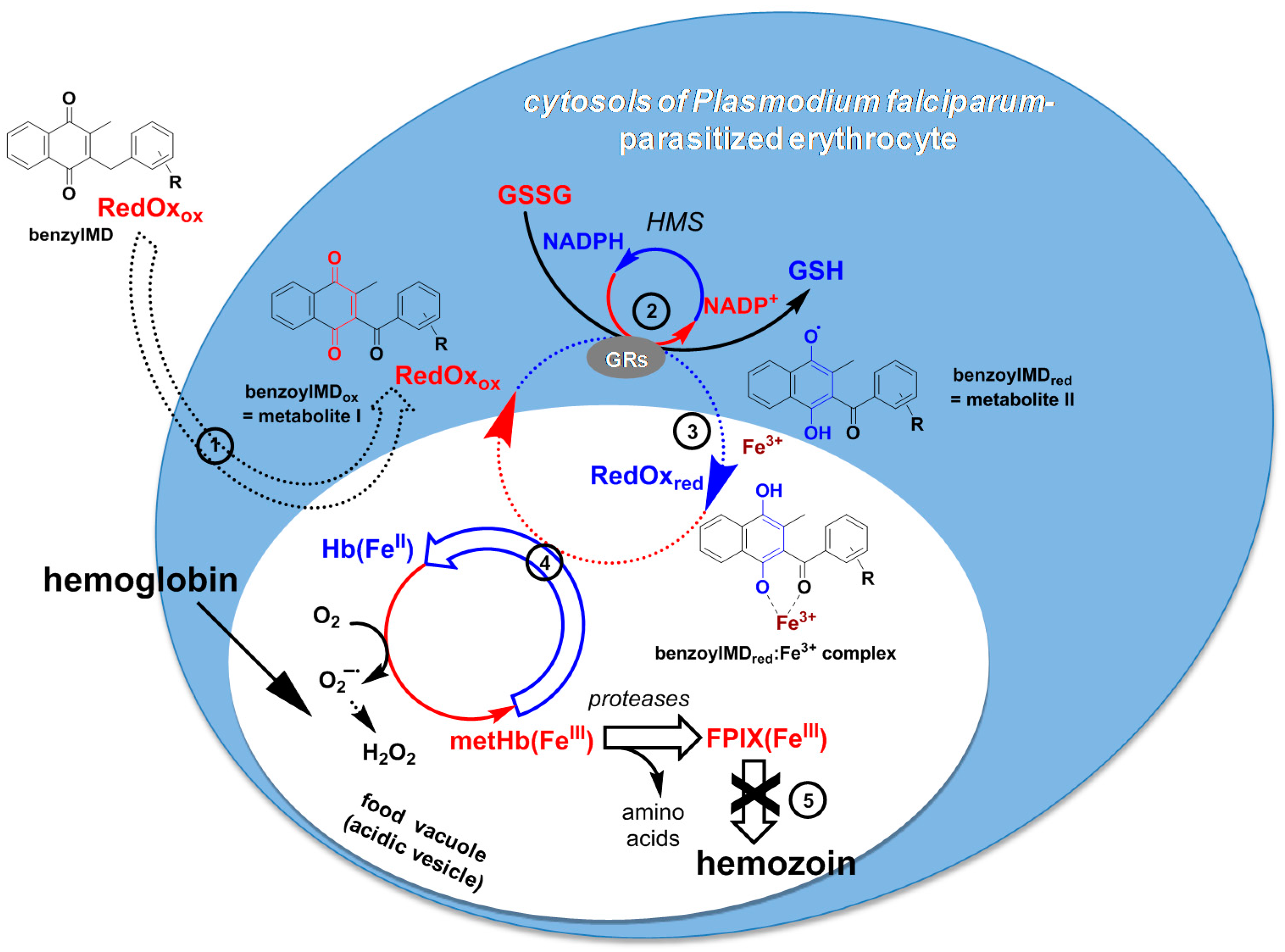
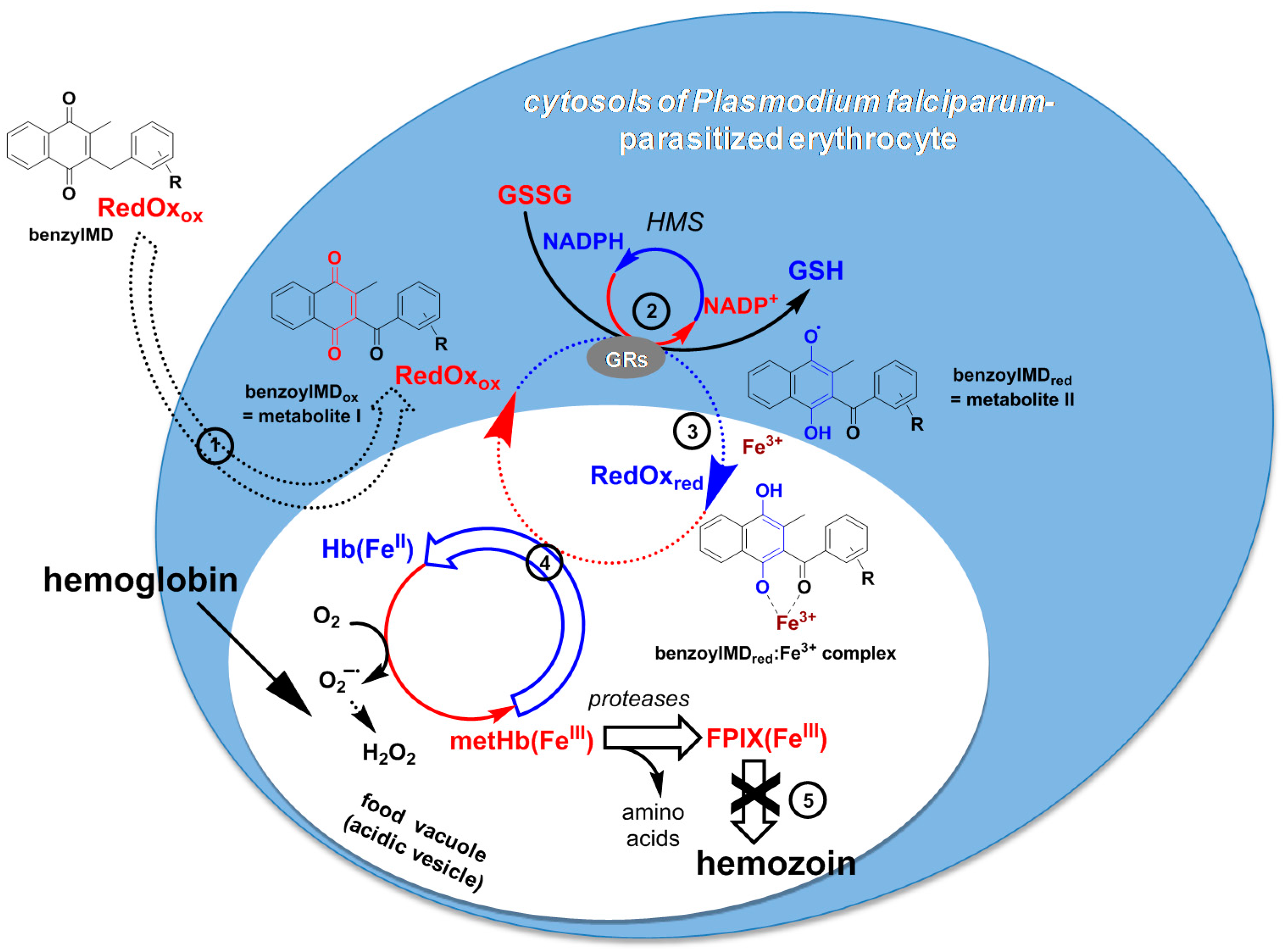
3. Discussion
4. Materials and Methods
4.1. General Information
4.1.1. Solvents and Reagents
4.1.2. Instruments
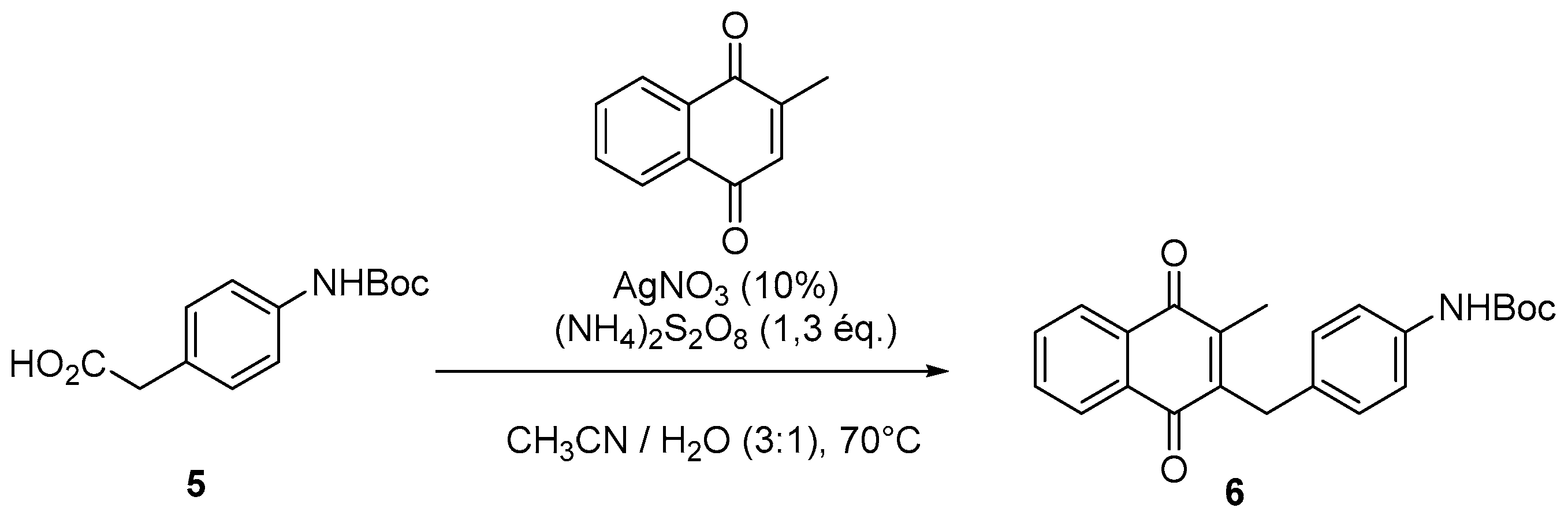
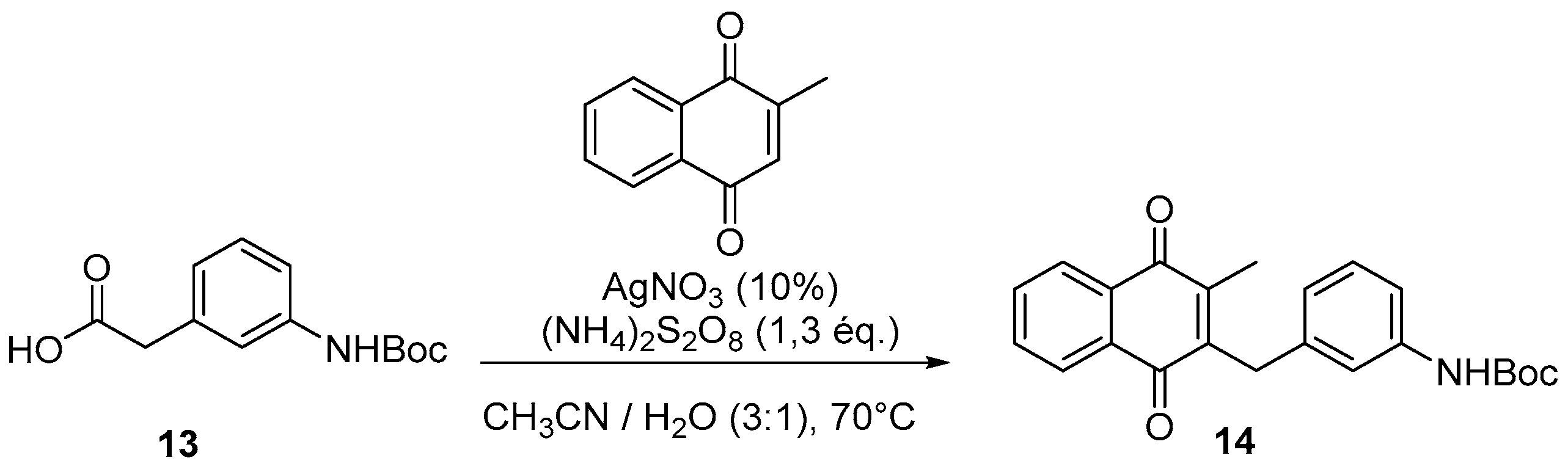
4.2. Protection of Aminophenylacetic acids with Boc Groups (Synthesis of 5 and 13)
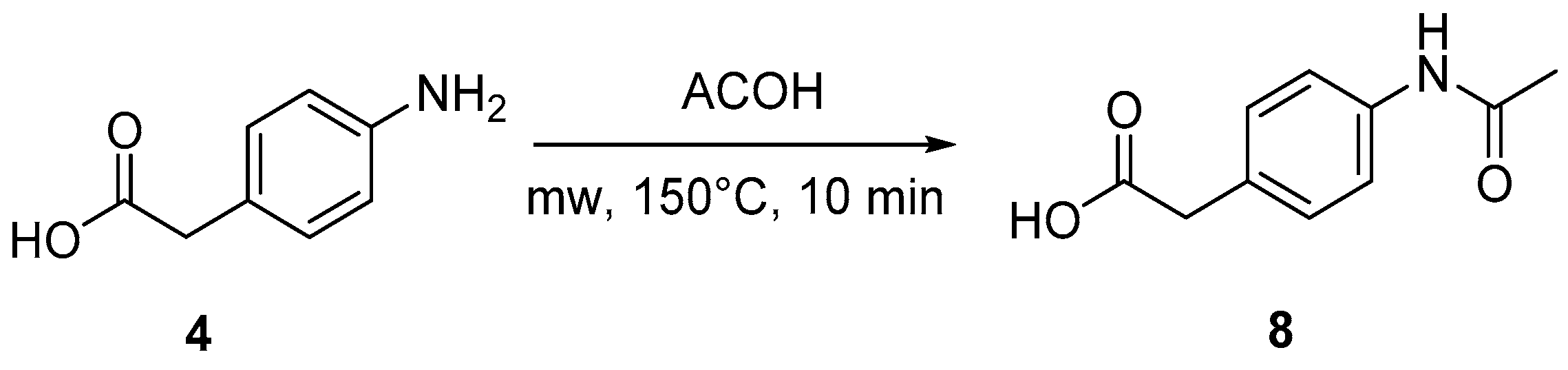
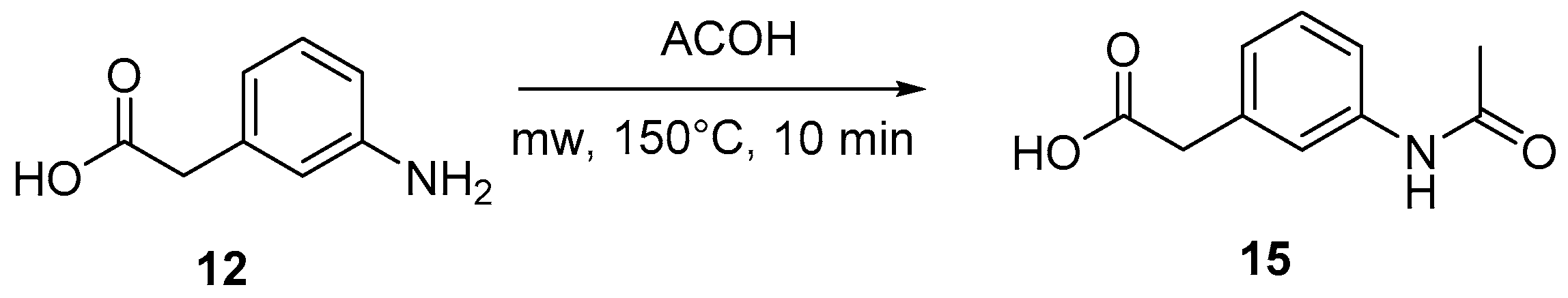
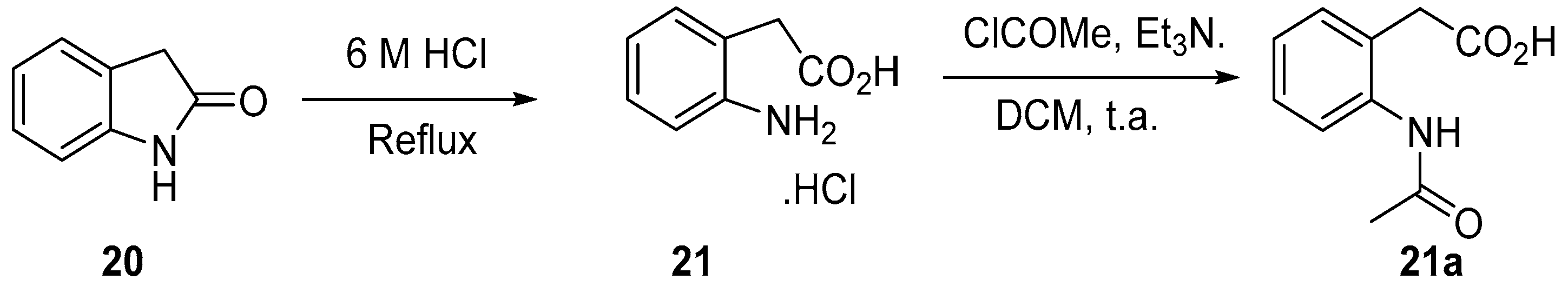
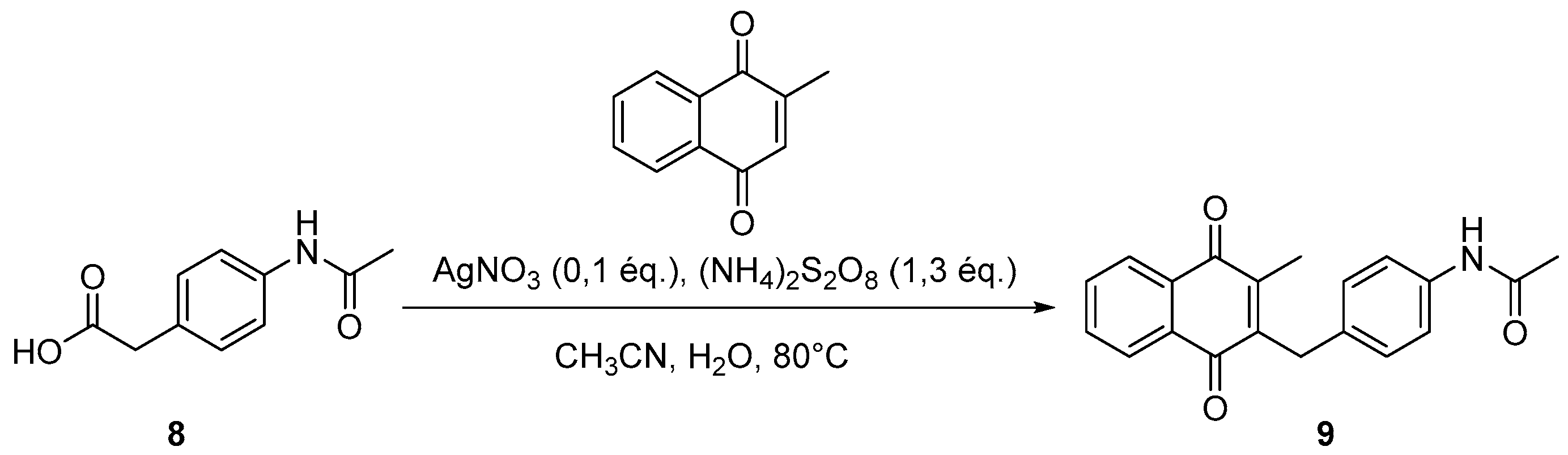
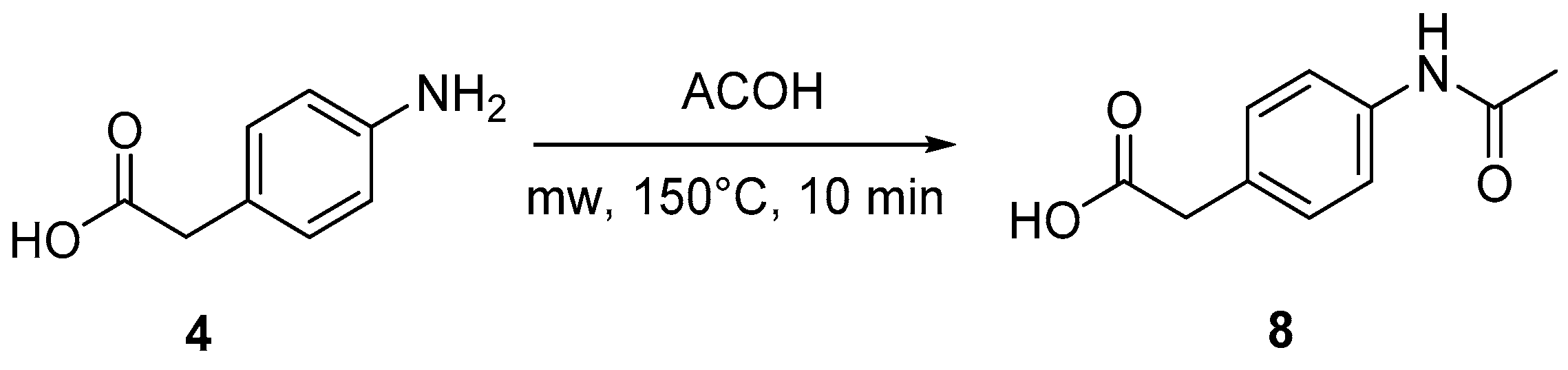
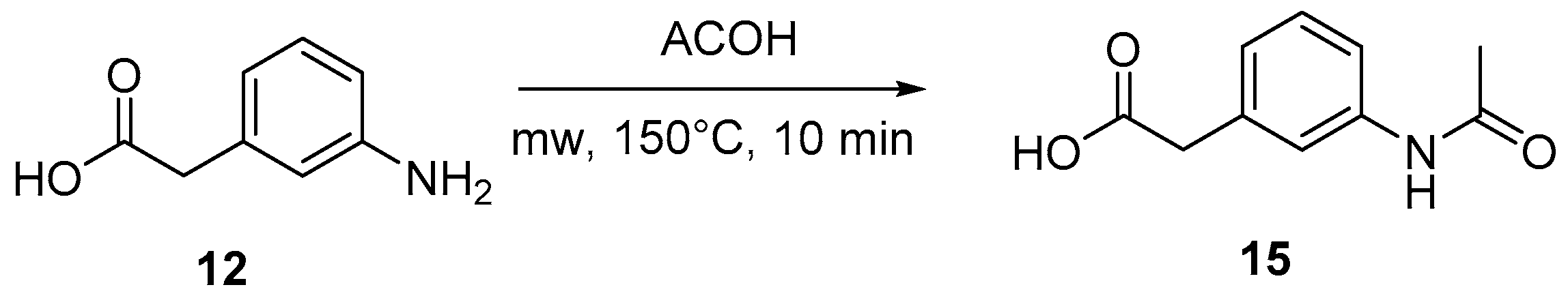
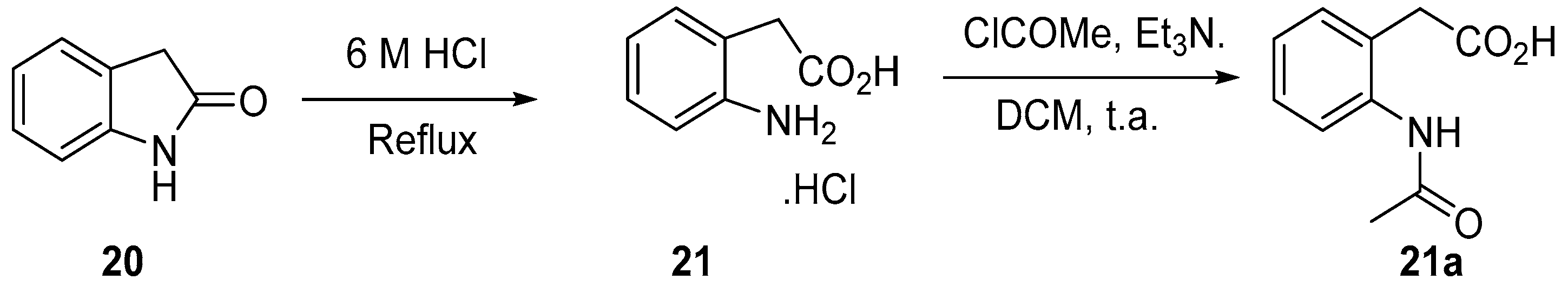
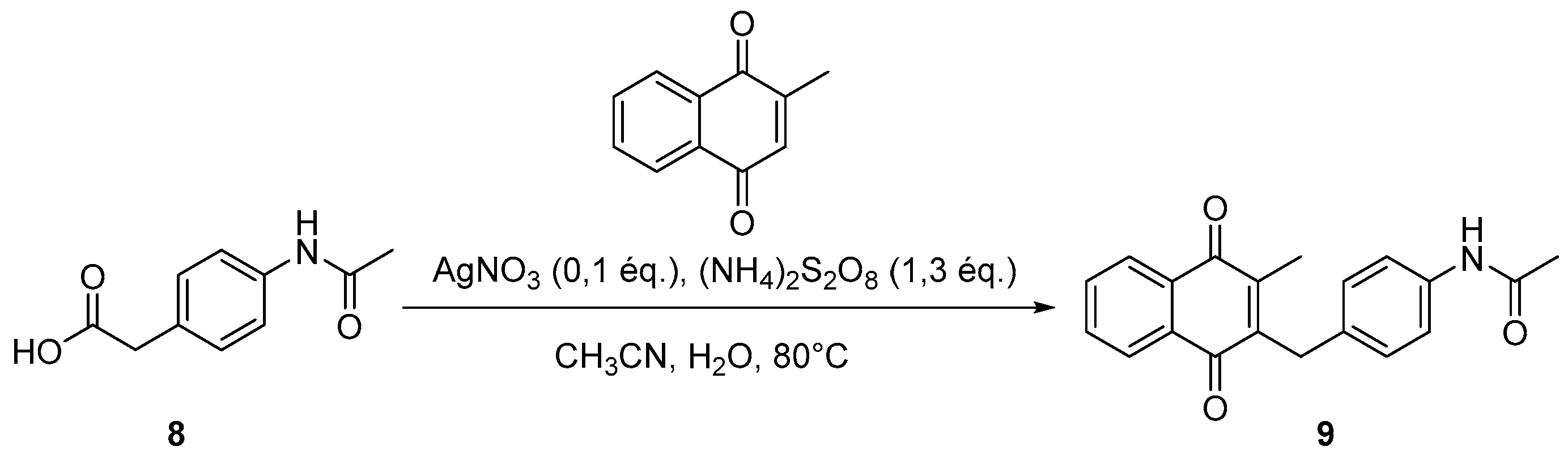
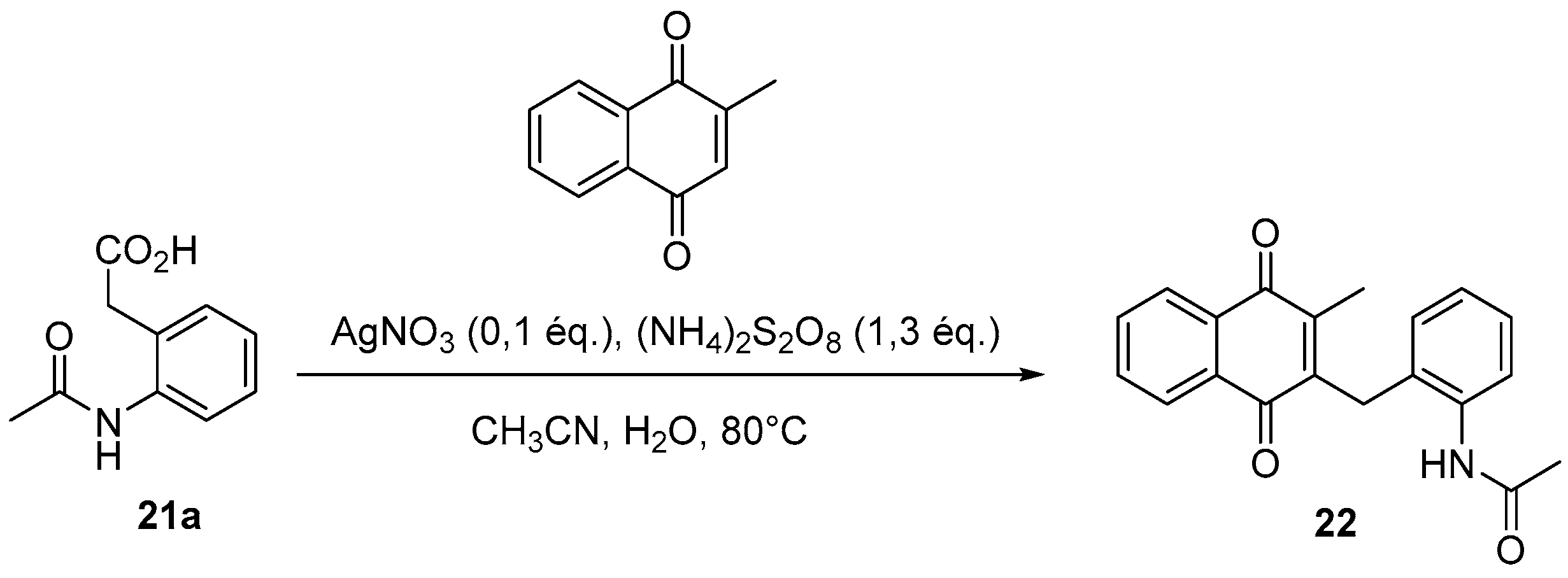
4.3. Protection of Aminophenylacetic Acids with Acetyl Groups (Synthesis of 8, 15 and 21a)
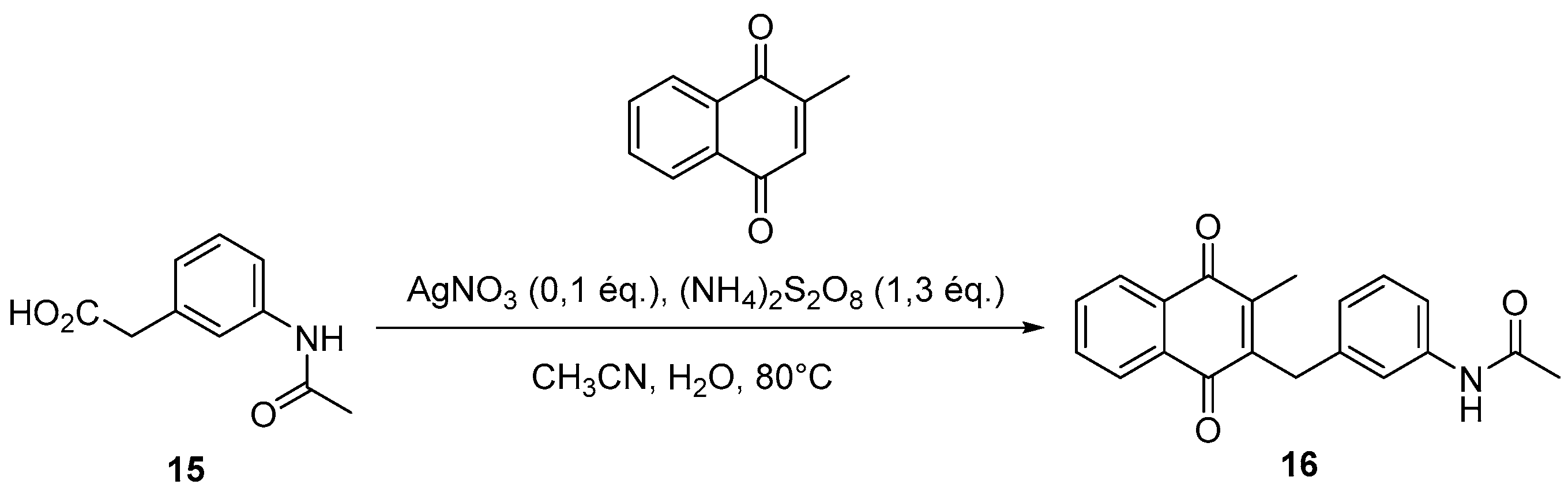
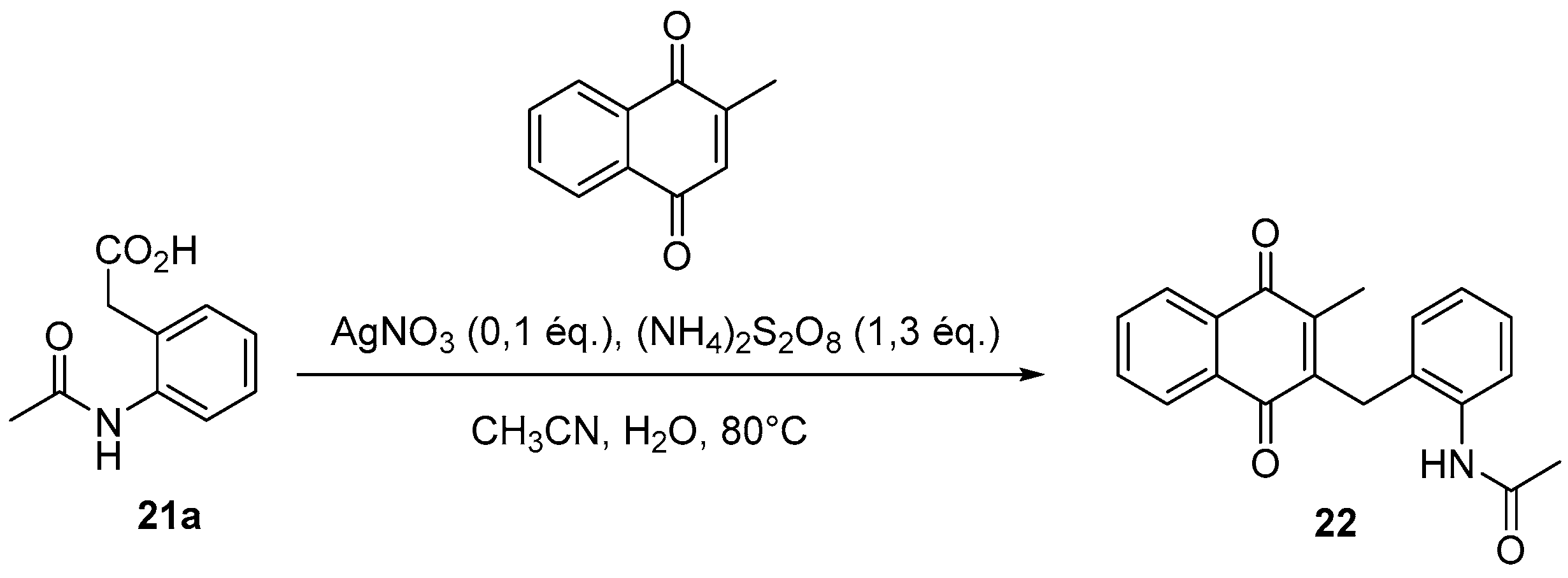
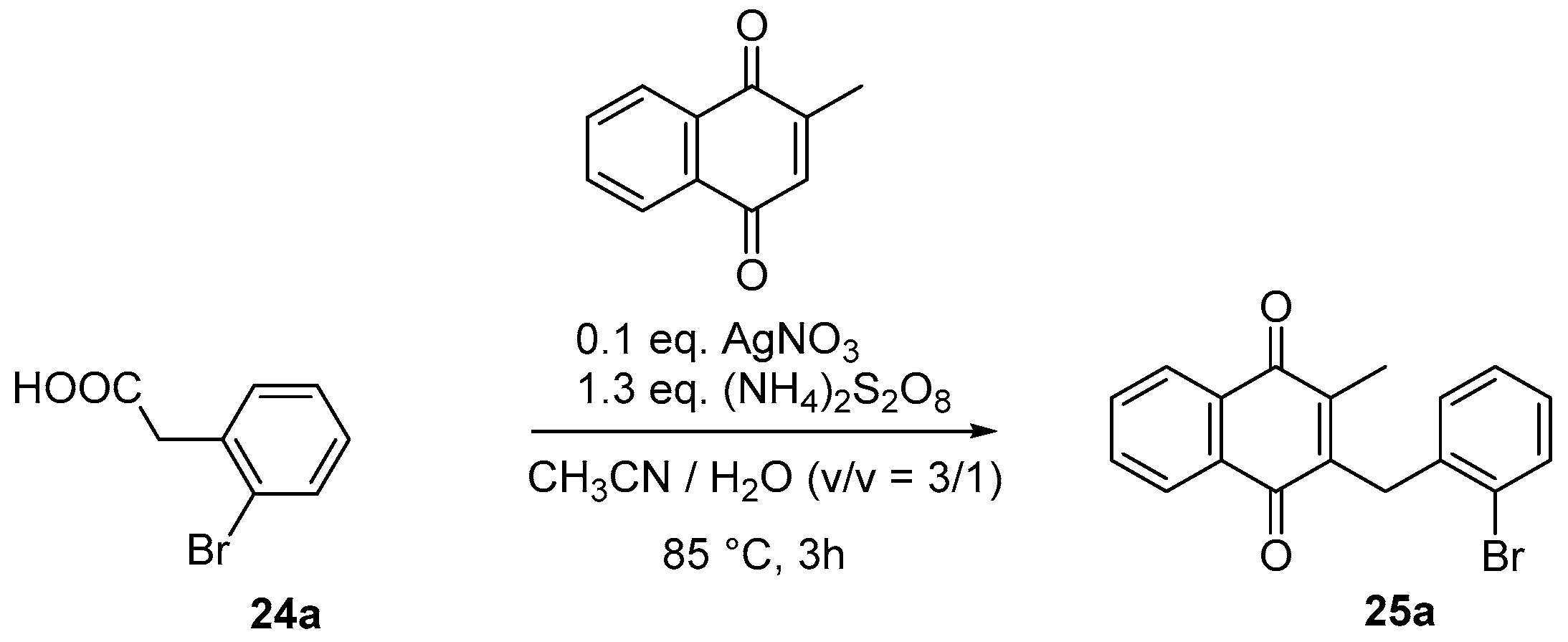
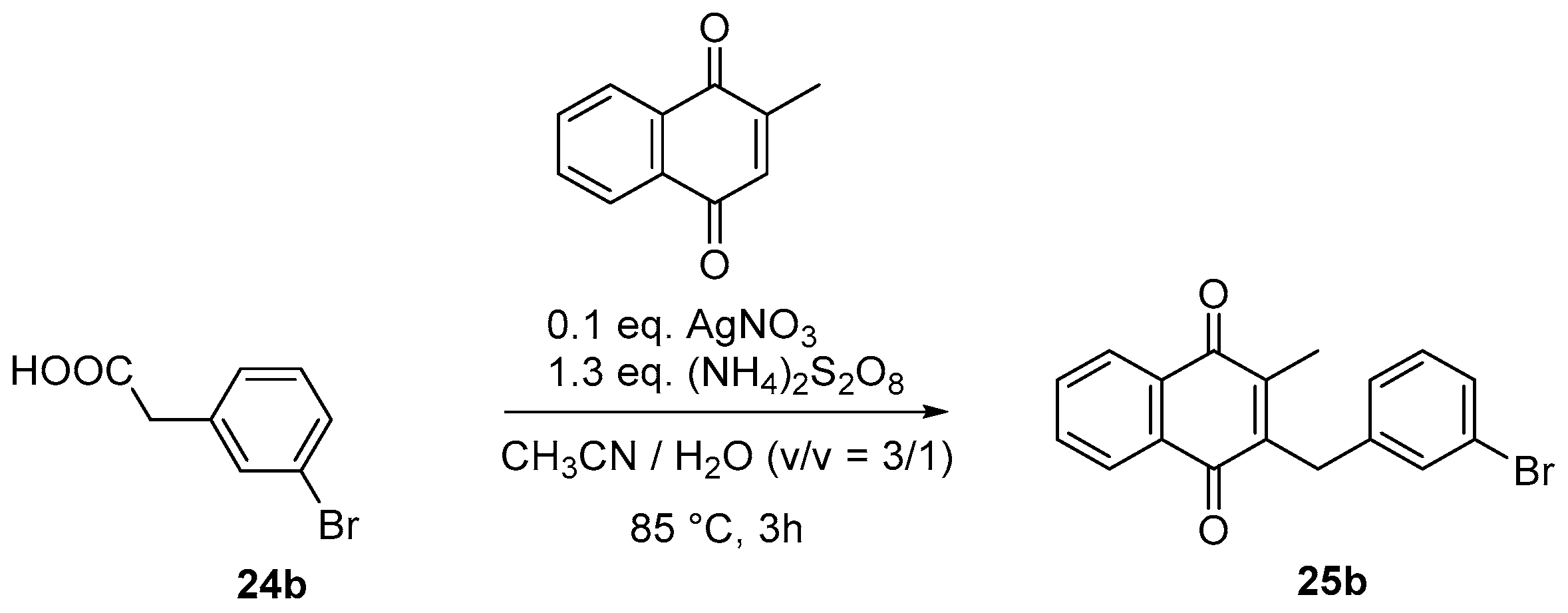
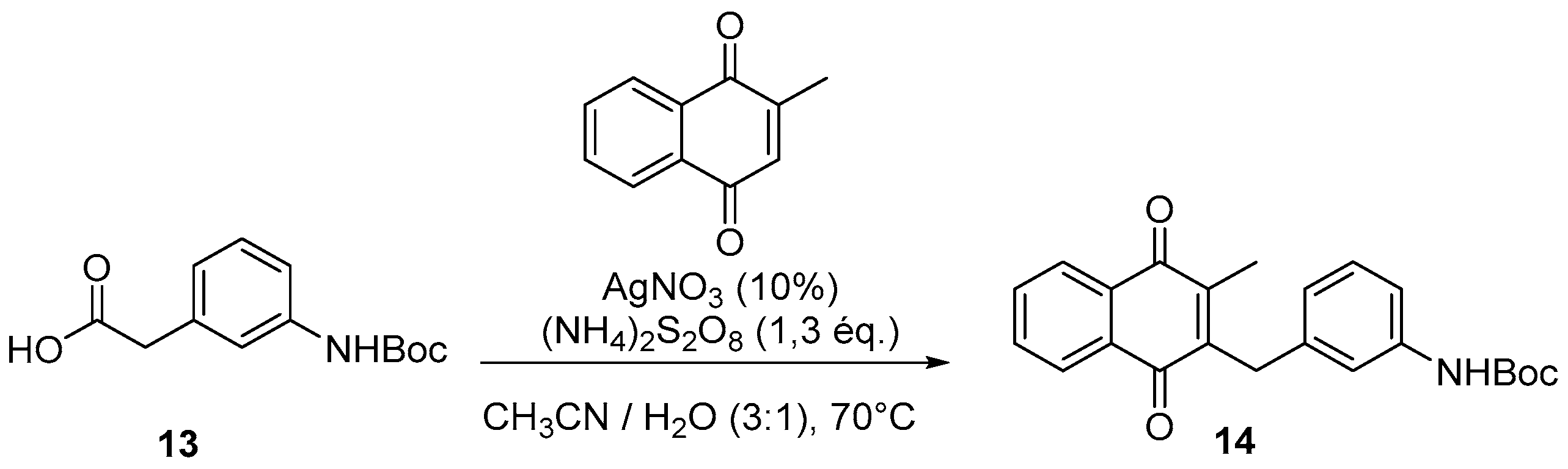
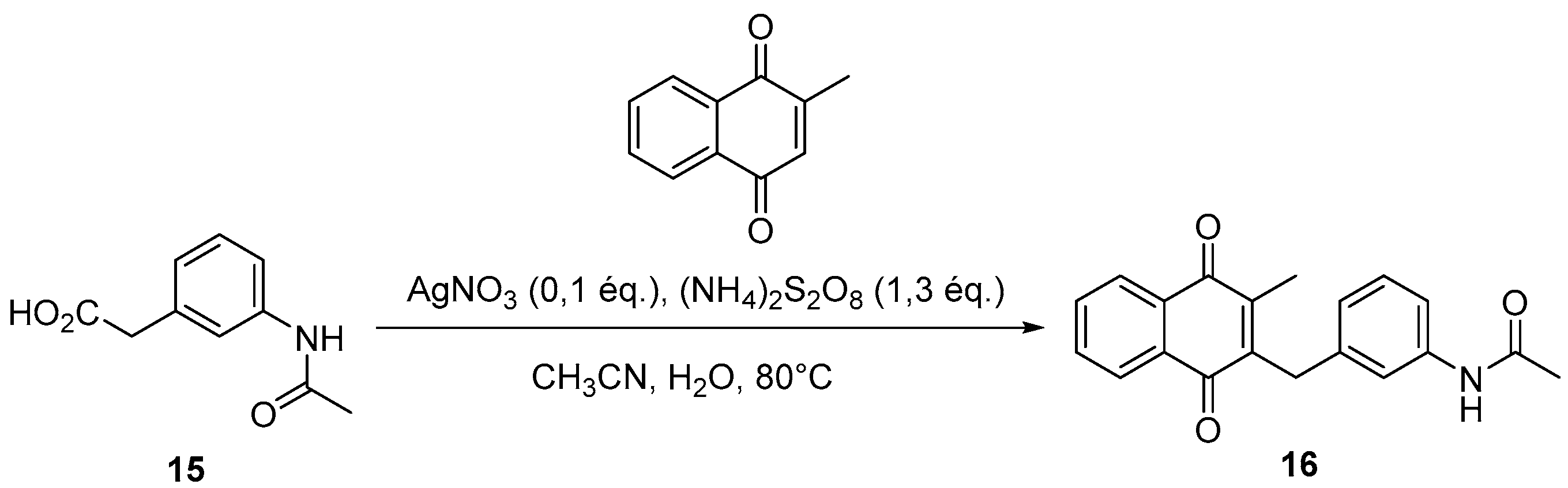
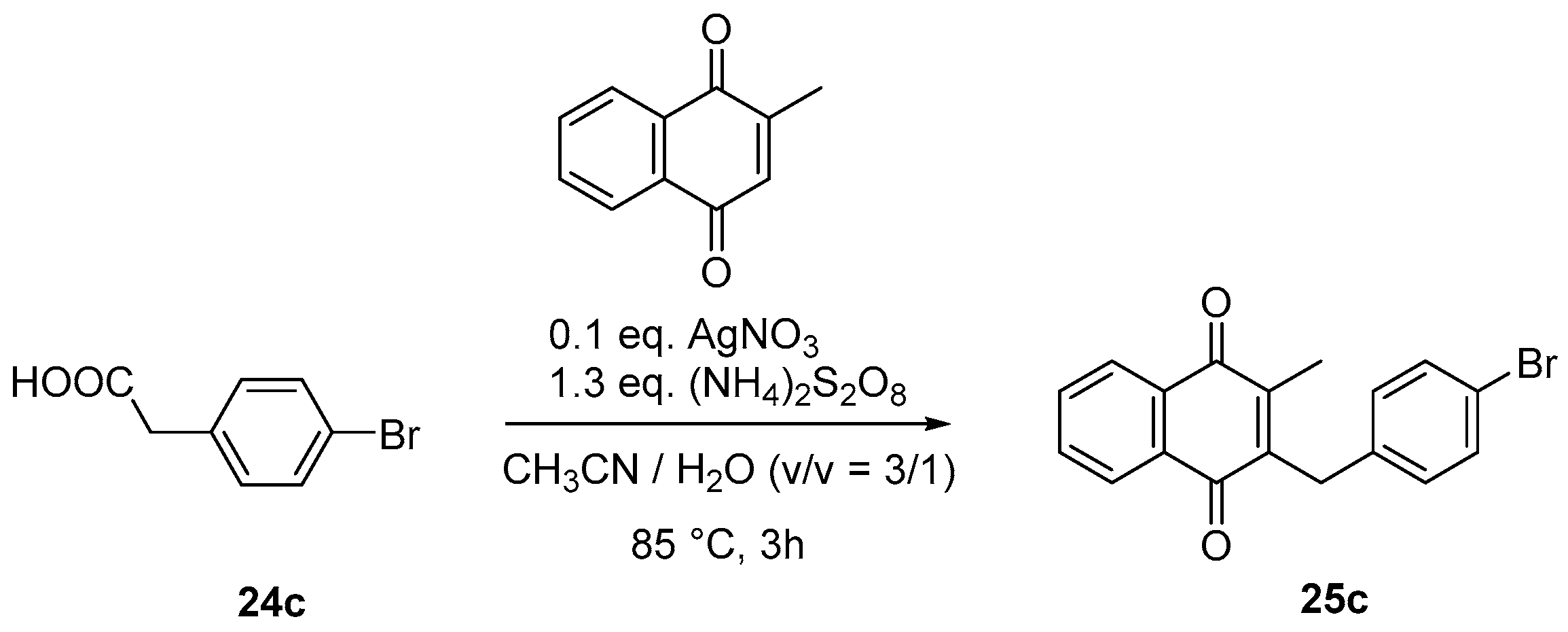
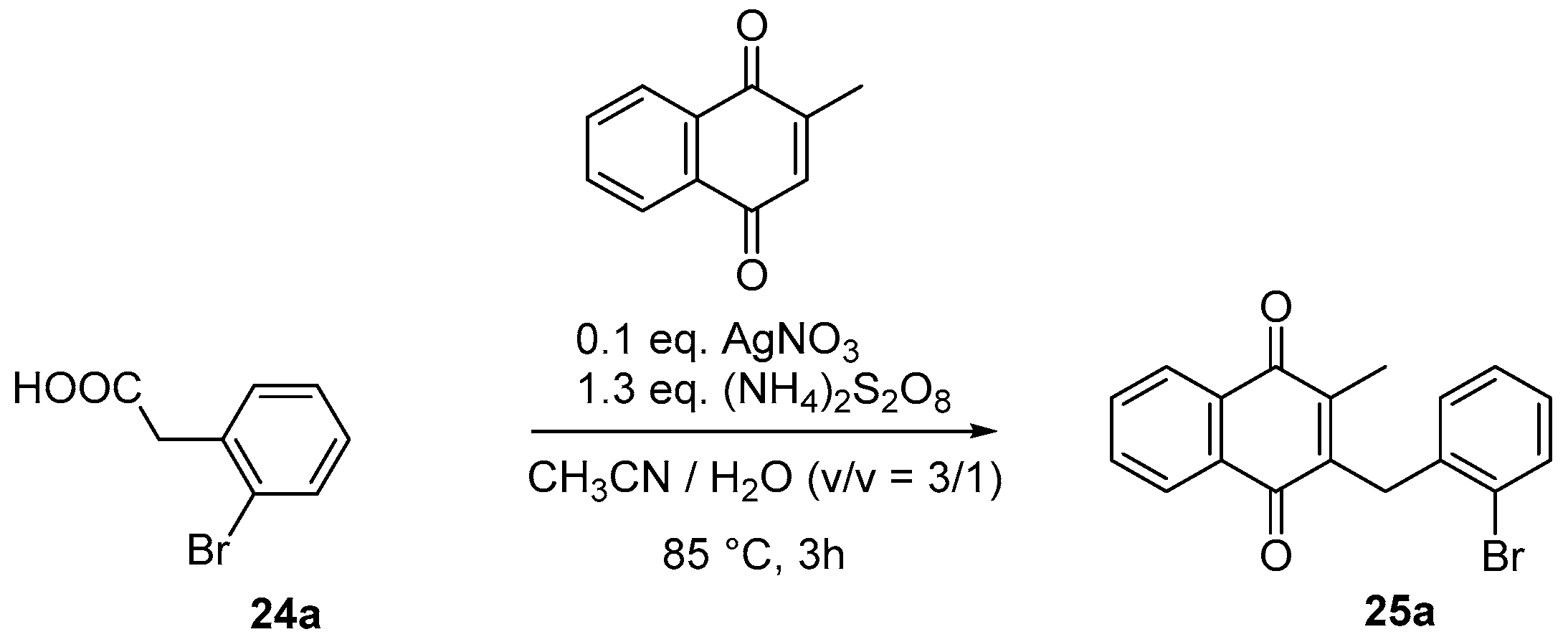
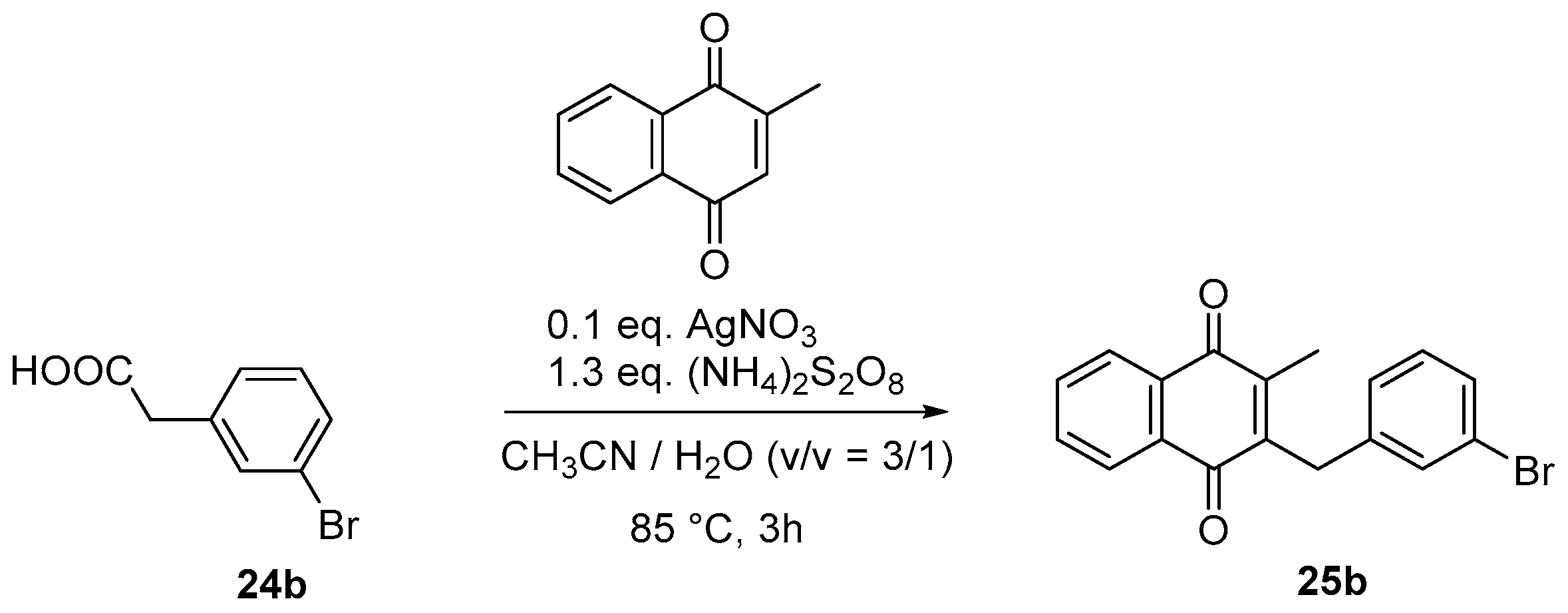
4.4. General Procedure for the Kochi-Anderson Reaction of Menadione with N-Protected Phenylacetic Acid Derivatives (Synthesis of 6, 14, 9, 16, 22 and 25)
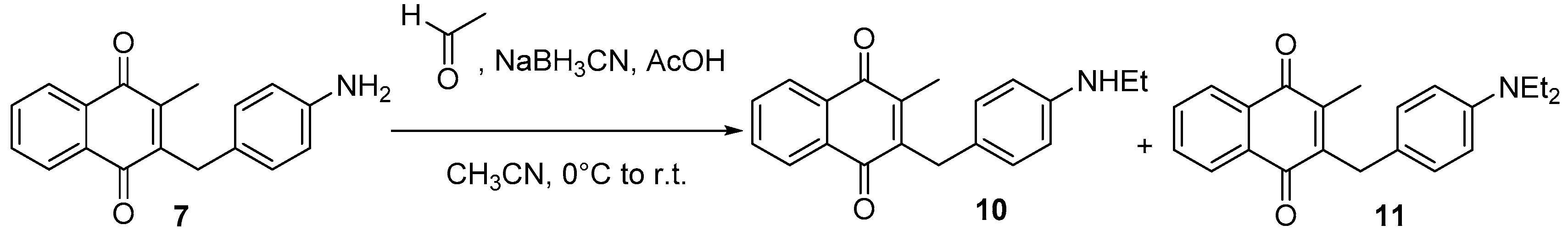
4.5. Deprotection of the Benzylamine Acteyl Group (Synthesis of 7 and 17)
4.6. Dialkylation of Benzylamine Menadione Derivatives (Synthesis of 3, 18, 11 and 19)
4.7. General Procedure for the Suzuki-Miyaura Coupling Reaction (Synthesis of 26–28)
4.8. In Vitro Antimalarial Activity
4.8.1. Hypoxanthine Method Determination of IC50 Values of Dd2 Growth Inhibition
4.8.2. SYBRgreen Method for Determination of IC50 Values of P. falciparum Dd2
4.9. Cytotoxicity against Human MRC-5 Cells
4.10. In Vivo Antimalarial Activity
4.10.1. Animals
4.10.2. Infection and Drug Treatment of P. berghei-Infected Mice
4.11. Physicochemistry
4.11.1. Starting Materials and Solvents
4.11.2. Spectrophotometric Titrations Versus pH
4.11.3. Analysis and Processing of the Spectroscopic Data
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACT | Artemisinin-based Combination Therapy |
| CQ | chloroquine |
| DHODH | dihydroorotate dehydrogenase |
| DMSO | dimethylsulfoxide |
| EA | Elemental analyses |
| equiv. | equivalent |
| ESI-MS | Electronspray-Mass spectrometry |
| HRMS | High Resolution Mass Spectra |
| ip | intraperitoneal |
| MB | methylene blue |
| mETC | mitochondrial electron transport |
| M.p. | Melting point |
| N-Boc | N-tert-butyloxycarbonyl |
| yDHODH | dihydroorotate dehydrogenase from yeast |
References
- World Malaria Report 2014. Available online: http://www.who.int/malaria/publications/world_malaria_report_2014/wmr-2014-summary-eng.pdf (accessed on 25 November 2016).
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735. [Google Scholar] [CrossRef]
- Müller, T.; Johann, L.; Jannack, B.; Bruckner, M.; Lanfranchi, D.A.; Bauer, H.; Sanchez, C.; Yardley, V.; Deregnaucourt, C.; Schrevel, J.; et al. Glutathione reductase-catalyzed cascade of redox reactions to bioactivate potent antimalarial 1,4-naphthoquinones—A new strategy to combat malarial parasites. J. Am. Chem. Soc. 2011, 133, 11557–11571. [Google Scholar] [CrossRef] [PubMed]
- Bielitza, M.; Belorgey, D.; Ehrhardt, K.; Johann, L.; Lanfranchi, D.A.; Gallo, V.; Schwarzer, E.; Mohring, F.; Jortzik, E.; Williams, D.L.; et al. Antimalarial NADPH-consuming redox-cyclers as superior glucose-6-phosphate dehydrogenase deficiency copycats. Antioxid. Redox Signal. 2015, 22, 1337–1351. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, K.; Davioud-Charvet, E.; Ke, H.; Vaidya, A.; Lanzer, M.; Deponte, M. The antimalarial activities of methylene blue and the 1,4-naphthoquinone 3-[4-(trifluoromethyl) benzyl]-menadione are not due to inhibition of the mitochondrial electron transport chain. Antimicrob. Agents Chemother. 2013, 57, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, K.; Deregnaucourt, C.; Goetz, A.-A.; Tzanova, T.; Pradines, B.; Adjalley, S.H.; Blandin, S.; Bagrel, D.; Lanzer, M.; Davioud-Charvet, E. The redox cycler plasmodione is a fast-acting antimalarial lead compound with pronounced activity against sexual and early asexual blood-stage parasites. Antimicrob. Agents Chemother. 2016, 60, 5146–5158. [Google Scholar] [CrossRef] [PubMed]
- Sidorov, P.; Desta, I.; Chessé, M.; Horvath, D.; Marcou, G.; Varnek, A.; Davioud-Charvet, E.; Elhabiri, M. Redox polypharmacology as an emerging strategy to combat malarial parasites. ChemMedChem 2016, 11, 1339–1351. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.L.; Moss, D.M.; Shone, A.E.; Lalloo, D.G.; Fisher, N.; O’Neill, P.M.; Ward, S.A.; Biagini, G.A. Antimalarial pharmacology and therapeutics of atovaquone. J. Antimicrob. Chemother. 2013, 68, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Morrisey, J.M.; Ganesan, S.M.; Painter, H.J.; Mather, M.W.; Vaidya, A.B. Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain function. Eukaryot. Cell 2011, 10, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Lanfranchi, D.A.; Belorgey, D.; Müller, T.; Vezin, H.; Lanzer, M.; Davioud-Charvet, E. Exploring the trifluoromenadione core as a template to design antimalarial redox-active agents interacting with glutathione reductase. Org. Biomol. Chem. 2012, 10, 4795–4806. [Google Scholar] [CrossRef] [PubMed]
- Painter, H.J.; Morrisey, J.M.; Vaidya, A.B. Mitochondrial electron transport inhibition and viability of intraerythrocytic Plasmodium falciparum. Antimicrob. Agents Chemother. 2010, 54, 5281–5287. [Google Scholar] [CrossRef] [PubMed]
- Elhabiri, M.; Sidorov, P.; Cesar-Rodo, E.; Marcou, G.; Lanfranchi, D.A.; Davioud-Charvet, E.; Horvath, D.; Varnek, A. Electrochemical properties of substituted 2-methyl-1,4-naphthoquinones: Redox behavior predictions. Chem. Eur. J. 2015, 21, 3415–3424. [Google Scholar] [CrossRef] [PubMed]
- Cesar Rodo, E.; Feng, L.; Jida, M.; Ehrhardt, K.; Bielitza, M.; Boilevin, J.; Lanzer, M.; Williams, D.L.; Lanfranchi, D.A.; Davioud-Charvet, E. A platform of regioselective methodologies to access polysubstituted 2-methyl-1,4-naphthoquinone derivatives: Scope and limitations. Eur. J. Org. Chem. 2016, 11, 1982–1993. [Google Scholar] [CrossRef]
- Wolf, C.; Lerebours, R. Use of highly active palladium-phosphinous acid catalysts in Stille, Heck, amination, and thiation reactions of chloroquinolines. J. Org. Chem. 2003, 68, 7077–7084. [Google Scholar] [CrossRef] [PubMed]
- Verhaeghe, P.; Azas, N.; Gasquet, M.; Hutter, S.; Ducros, C.; Laget, M.; Rault, S.; Rathelot, P.; Vanelle, P. Synthesis and antiplasmodial activity of new 4-aryl-2-trichloromethylquinazolines. Bioorg. Med. Chem. Lett. 2008, 18, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, T.; Imam, M.; Kaushik, N.K.; Chauhan, V.S.; Verma, A.K. Pyrano [4,3-b] quinolines library generation via iodocyclization and palladium-catalyzed coupling reactions. ACS Comb. Sci. 2011, 13, 530–536. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.; Chicharro, J.; Bueno, J.M.; Lorenzo, M. Isoxazole mediated synthesis of 4-(1H)pyridones: Improved preparation of antimalarial candidate GSK932121. Chem. Commun. 2016, 52, 10190–10192. [Google Scholar] [CrossRef] [PubMed]
- Leung, S.C.; Gibbons, P.; Amewu, R.; Nixon, G.L.; Pidathala, C.; Hong, W.D.; Pacorel, B.; Berry, N.G.; Sharma, R.; Stocks, P.A.; et al. Identification, Design and Biological Evaluation of Heterocyclic Quinolones Targeting Plasmodium falciparum Type II NADH:Quinone Oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55, 1844–1857. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, C.; Amewu, R.; Pacorel, B.; Nixon, G.L.; Gibbons, P.; Hong, W.D.; Leung, S.C.; Berry, N.G.; Sharma, R.; Stocks, P.A.; et al. Identification, Design and Biological Evaluation of Bisaryl Quinolones Targeting Plasmodium falciparum Type II NADH:Quinone Oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.; Mehlhorn, H. Seventy-five years of Resochin in the fight against malaria. Parasitol. Res. 2009, 105, 609–627. [Google Scholar] [CrossRef] [PubMed]
- Cheruku, S.R.; Maiti, S.; Dorn, A.; Scorneaux, B.; Bhattacharjee, A.K.; Ellis, W.Y.; Vennerstrom, J.L. Carbon isosteres of the 4-aminopyridine substructure of chloroquine: Effects on pKa, hematin binding, inhibition of hemozoin formation, and parasite growth. J. Med. Chem. 2003, 46, 3166–3169. [Google Scholar] [CrossRef] [PubMed]
- Biot, C.; Glorian, G.; Maciejewski, L.A.; Brocard, J.S. Synthesis and antimalarial activity in vitro and in vivo of a new ferrocene-chloroquine analogue. J. Med. Chem. 1997, 40, 3715–3718. [Google Scholar] [CrossRef] [PubMed]
- Domarle, O.; Blampain, G.; Agnanet, H.; Nzadiyabi, T.; Lebibi, J.; Brocard, J.S.; Maciejewski, L.A.; Biot, C.; Georges, A.J.; Millet, P. In vitro antimalarial activity of a new organometallic analog, ferrocene-chloroquine. Antimicrob. Agents Chemother. 1998, 42, 540–544. [Google Scholar] [PubMed]
- Roehl, W. Die Wirkung des Plasmochins auf die Vogelmalaria. Archiv für Schiffs-und. Tropenhygiene 1926, 30, 311–318. [Google Scholar] [CrossRef]
- Roehl, W. Die Wirkung des Plasmochins auf die Vogelmalaria. Naturwissenschaften 1926, 14, 1156–1159. [Google Scholar] [CrossRef]
- Greenwood, D. Conflicts of interest: The genesis of synthetic antimalarial agents in peace and war. J. Antimicrob. Chemother. 1995, 36, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Vennerstrom, J.L.; Makler, M.T.; Angerhofer, C.K.; Williams, J.A. Antimalarial dyes revisited: Xanthenes, azines, oxazines, and thiazines. Antimicrob. Agents Chemother. 1995, 39, 2671–2677. [Google Scholar] [CrossRef] [PubMed]
- Blank, O.; Davioud-Charvet, E.; Elhabiri, M. Interactions of the antimalarial drug methylene blue with methemoglobin and heme targets in Plasmodium falciparum: A physico-biochemical study. Antioxid. Redox Signal. 2012, 17, 544–554. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, B.; Zoungrana, A.; Mockenhaupt, F.P.; Schirmer, R.H.; Klose, C.; Mansmann, U.; Meissner, P.E.; Müller, O. Strong gametocytocidal effect of methylene blue-based combination therapy against falciparum malaria: A randomised controlled trial. PLoS ONE 2009, 4, e5318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohring, F.; Pretzel, J.; Jortzik, E.; Becker, K. The redox systems of Plasmodium falciparum and Plasmodium vivax: Comparison, in silico analyses and inhibitor studies. Curr. Med. Chem. 2014, 21, 1728–1756. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, K.; Schirmer, R.H.; Eubel, J.K.; Akoachere, M.B.; Dandekar, T.; Becker, K.; Gromer, S. Interactions of methylene blue with human disulfide reductases and their orthologues from Plasmodium falciparum. Antimicrob. Agents Chemother. 2008, 52, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Johann, L.; Lanfranchi, D.A.; Davioud-Charvet, E.; Elhabiri, M. A physico-biochemical study on potential redox-cyclers as antimalarial and anti-schistosomal drugs. Curr. Pharm. Des. 2012, 18, 3539–3566. [Google Scholar] [PubMed]
- Loria, P.; Miller, S.; Foley, M.; Tilley, L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 1999, 339, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Lanfranchi, D.A.; Cesar-Rodo, E.; Bertrand, B.; Huang, H.H.; Day, L.; Johann, L.; Elhabiri, M.; Becker, K.; Williams, D.L.; Davioud-Charvet, E. Synthesis and biological evaluation of 1,4-naphthoquinones and quinoline-5,8-diones as antimalarial and schistosomicidal agents. Org. Biomol. Chem. 2012, 10, 6375–6387. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Kochi, J.K. Silver(I)-catalyzed oxidative decarboxylation of acids by peroxydisulfate. Role of silver(II). J. Am. Chem. Soc. 1970, 92, 1651–1659. [Google Scholar] [CrossRef]
- Jacobsen, N.; Torssell, K. Radikalische Alkylierung von Chinonen: Erzeugung von Radikalen in Redoxreaktionen. Liebigs Ann. Chem. 1972, 763, 135–147. [Google Scholar] [CrossRef]
- Jacobsen, N.; Torssell, K. Synthesis of Naturally Occurring Quinones. Alkylation with the Silver Ion-Peroxydisulphate-Carboxylic Acid System. Acta Chem. Scand. 1973, 27, 3211–3216. [Google Scholar] [CrossRef]
- Salmon-Chemin, L.; Buisine, E.; Yardley, V.; Kohler, S.; Debreu, M.-A.; Landry, V.; Sergheraert, C.; Croft, S.L.; Krauth-Siegel, R.L.; Davioud-Charvet, E. 2- and 3-substituted 1,4-naphthoquinone derivatives as subversive substrates of trypanothione reductase and lipoamide dehydrogenase from Trypanosoma cruzi: Synthesis and correlation between redox cycling activities and in vitro cytotoxicity. J. Med. Chem. 2001, 44, 548–565. [Google Scholar] [CrossRef] [PubMed]
- Salmon-Chemin, L.; Lemaire, A.; De Freitas, S.; Deprez, B.; Sergheraert, C.; Davioud-Charvet, E. Parallel synthesis of a library of 1,4-naphthoquinones and automated screening of potential inhibitors of trypanothione reductase from Trypanosoma cruzi. Bioorg. Med. Chem. Lett. 2000, 10, 631–635. [Google Scholar] [CrossRef]
- Bauer, H.; Fritz-Wolf, K.; Winzer, A.; Kühner, S.; Little, S.; Yardley, V.; Vezin, H.; Palfey, B.; Schirmer, R.H.; Davioud-Charvet, E. A fluoro analogue of the menadione derivative 6-[2′-(3′-methyl)-1′,4′-naphthoquinolyl]hexanoic acid is a suicide substrate of glutathione reductase. Crystal structure of the alkylated human enzyme. J. Am. Chem. Soc. 2006, 128, 10784–10794. [Google Scholar] [CrossRef] [PubMed]
- Diederich, F.; Stang, P.J. Metal-Catalyzed Cross-Coupling Reactions; Wiley-VCH: Weinheim, Germany, 1998. [Google Scholar]
- Stille, J.K. Palladium-katalysierte Kupplungsreaktionen organischer Elektrophile mit Organozinn-Verbindungen (Palladium-catalyzed coupling reactions of organic electrophiles with organic tin compounds). Angew. Chem. 1986, 98, 504–519, The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles [New Synthetic Methods (58)]. Angew. Chem. Int. Ed. Engl. 1986, 25, 508–524.. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Lin, S.; Yang, Z.Q.; Kwok, B.H.; Koldobskiy, M.; Crews, C.M.; Danishefsky, S.J. Total Synthesis of TMC-95A and -B via a New Reaction Leading to Z-Enamides. Some Preliminary Findings as to SAR. J. Am. Chem. Soc. 2004, 126, 6347–6355. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, R.; Najera, C. The Sonogashira reaction: A booming methodology in synthetic organic chemistry. Chem. Rev. 2007, 107, 874–922. [Google Scholar] [CrossRef] [PubMed]
- Iranpoor, N.; Firouzabadi, H.; Nowrouzi, N.; Khalili, D. Selective mono- and di-N-alkylation of aromatic amines with alcohols and acylation of aromatic amines using Ph3P/DDQ. Tetrahedron 2009, 65, 3893–3899. [Google Scholar] [CrossRef]
- Wang, X.-J.; Yang, Q.; Liu, F.; You, Q.-D. Microwave-assisted synthesis of amide under solvent-free conditions. Synth. Commun. 2008, 38, 1028–1035. [Google Scholar] [CrossRef]
- Majetich, G.; Hicks, R. Applications of microwave accelerated organic chemistry. Res. Chem. Intermed. 1994, 20, 61–77. [Google Scholar] [CrossRef]
- Chiranjeevi, B.; Vinayak, B.; Parsharamulu, T.; PhaniBabu, V.S.; Jagadeesh, B.; Sridhar, B.; Chandrasekharam, M. Iron(III)-catalyzed C–H functionalization: Ortho-benzoyloxylation of N,N-dialkylanilines and its application to 1,4-benzoxazepines. Eur. J. Org. Chem. 2014, 35, 7839–7849. [Google Scholar] [CrossRef]
- Clift, M.D.; Silverman, R.B. Synthesis and Evaluation of Novel Aromatic Substrates and Competitive Inhibitors of GABA Aminotransferase. Bioorg. Med. Chem. Lett. 2008, 18, 3122–3125. [Google Scholar] [CrossRef] [PubMed]
- Woods, K.W.; Fischer, J.P.; Claiborne, A.; Li, T.; Thomas, S.A.; Zhu, G.D.; Diebold, R.B.; Liu, X.; Shi, Y.; Klinghofer, V.; et al. Synthesis and SAR of indazole-pyridine based protein kinase B/Akt inhibitors. Bioorg. Med. Chem. 2006, 14, 6832–6846. [Google Scholar] [CrossRef] [PubMed]
- Bunting, J.W.; Stefanidis, D. A systematic entropy relationship for the general-base catalysis of the deprotonation of a carbon acid. A quantitative probe of transition-state salvation. J. Am. Chem. Soc. 1990, 112, 779–786. [Google Scholar] [CrossRef]
- Crans, D.C.; Boukhobza, I. Vanadium(V) complexes of polydentate amino alcohols: Fine-tuning complex properties. J. Am. Chem. Soc. 1998, 120, 8069–8078. [Google Scholar] [CrossRef]
- Jida, M.; Sanchez, C.; Ehrhardt, K.; Urgin, K.; Mounien, S.; Geyer, A.; Elhabiri, M.; Lanzer, M.; Davioud-Charvet, E. A redox-active fluorescent pH indicator for detecting P. falciparum strains with reduced responsiveness to quinoline antimalarial drugs. ACS Inf. Dis. 2016. [Google Scholar] [CrossRef]
- Ishikawa, F.; Tsumuraya, T.; Fujii, I. A single antibody catalyzes multiple chemical transformations upon replacement of the functionalized small nonprotein components. J. Am. Chem. Soc. 2009, 131, 456–457. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, R.E.; Canfield, C.J.; Haynes, J.D.; Chulay, J.D. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 1979, 16, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Beez, D.; Sanchez, C.P.; Stein, W.D.; Lanzer, M. Genetic predisposition favors the acquisition of stable artemisinin resistance in malaria parasites. Antimicrob. Agents Chemother. 2011, 55, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48, 1803–1806. [Google Scholar] [CrossRef] [PubMed]
- Raymond, K.N. Chemical safety. Chem. Eng. News 1983, 61, 4–5. [Google Scholar]
- Gans, P.; O’Sullivan, B. GLEE, a new computer program for glass electrode calibration. Talanta 2000, 51, 33–37. [Google Scholar] [CrossRef]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data-I Mathematical considerations. Talanta 1985, 32, 95–101. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data—II: SPECFIT: Two user-friendly programs in basic and standard FORTRAN 77. Talanta 1985, 32, 257–264. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbühler, A.D. Calculation of equilibrium constants from multiwavelength spectroscopic data—III: Model-free analysis of spectrophotometric and ESR titrations. Talanta 1985, 32, 1133–1139. [Google Scholar] [CrossRef]
- Marquardt, D.W. An algorithm for least-squares estimation of nonlinear parameters. J. Soc. Indust. Appl. Math. 1963, 11, 431–441. [Google Scholar] [CrossRef]
- Maeder, M.; Zuberbühler, A.D. Nonlinear least-squares fitting of multivariate absorption data. Anal. Chem. 1990, 62, 2220–2224. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 3, 6, 7, 9–11, 16–19, 22, 25a, 25b, 25c, 26–28 are available from the authors.
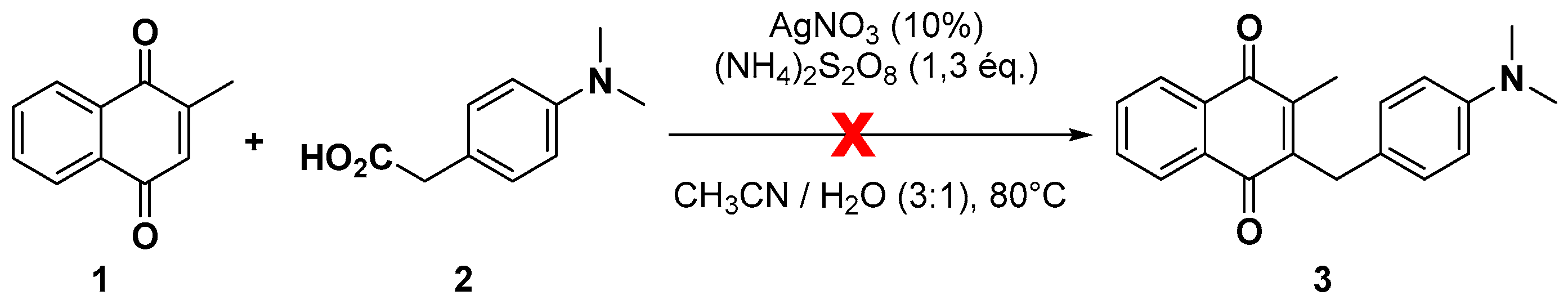
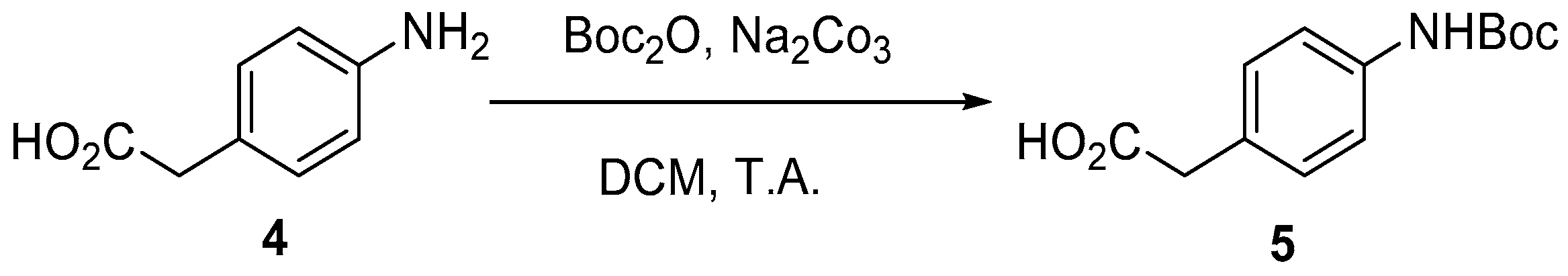
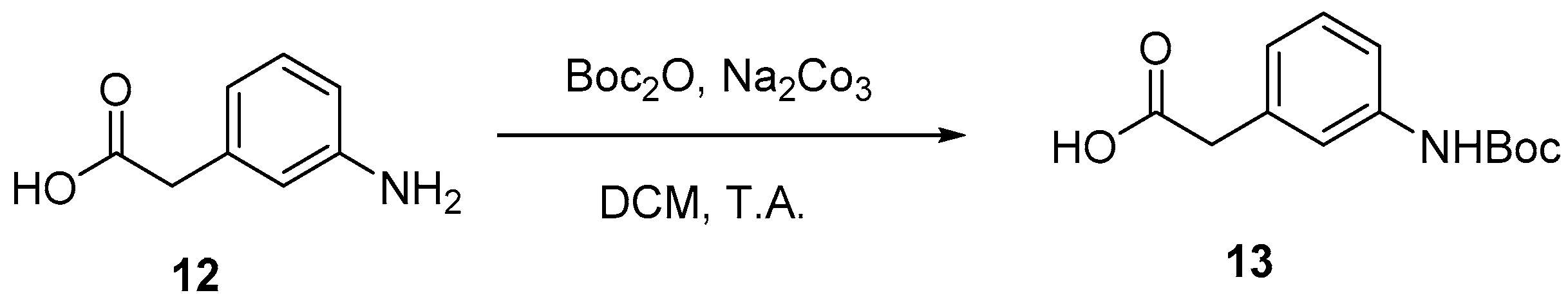

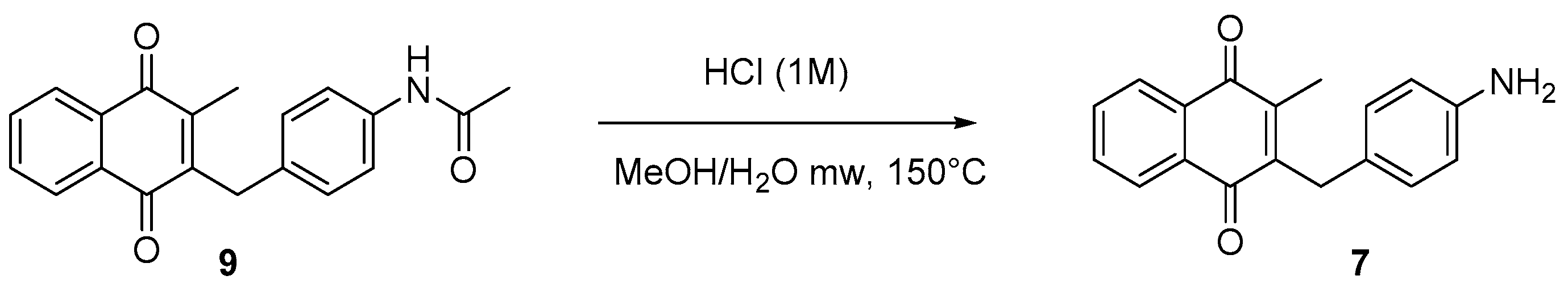
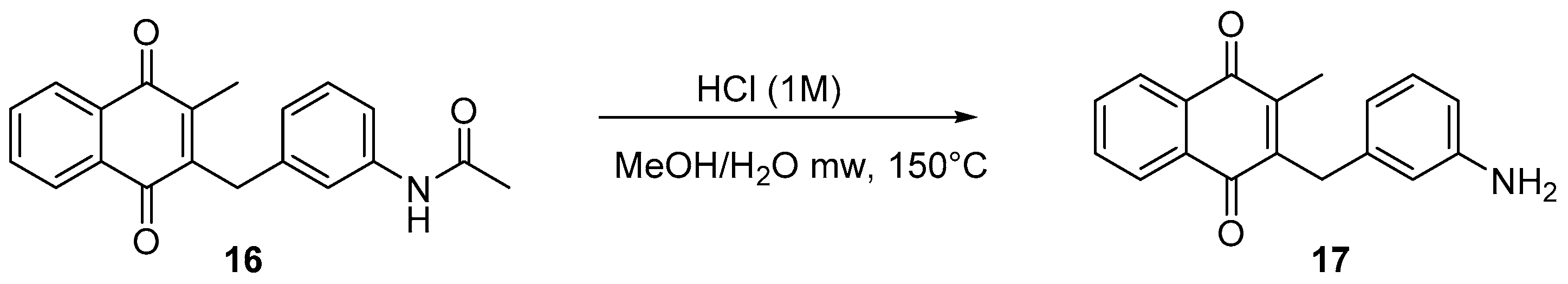
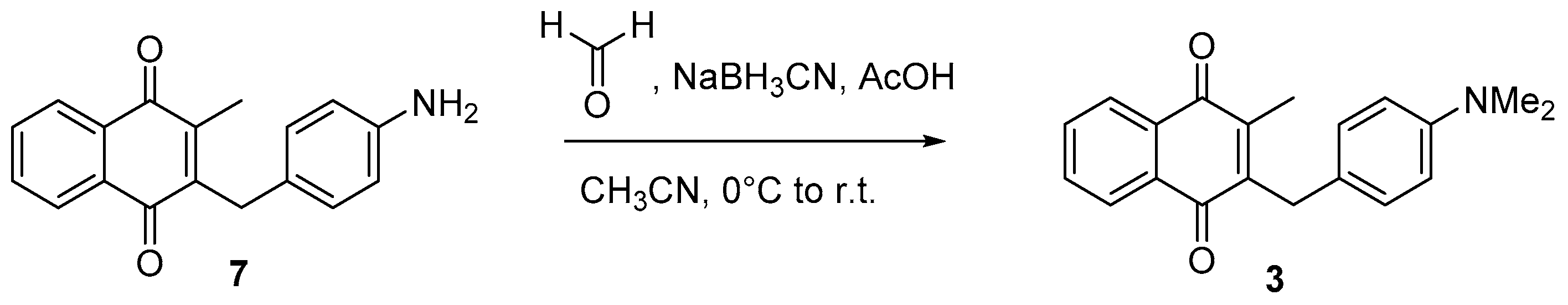
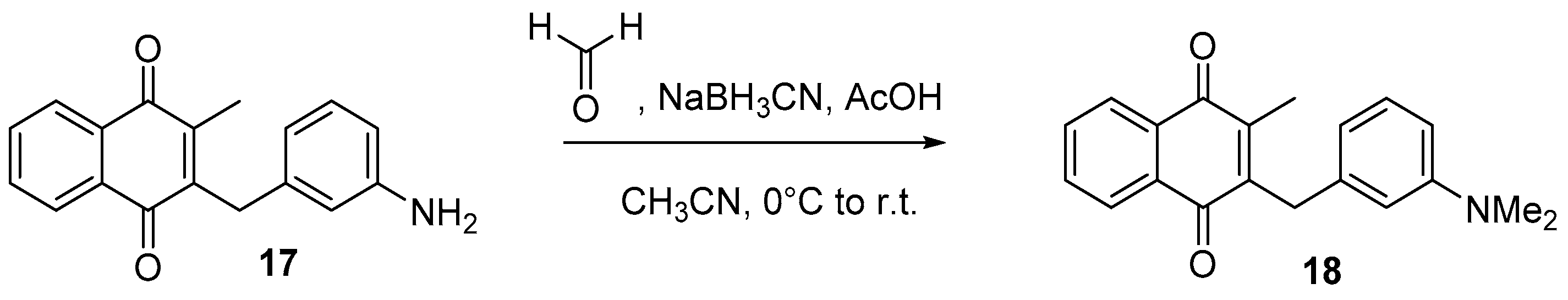
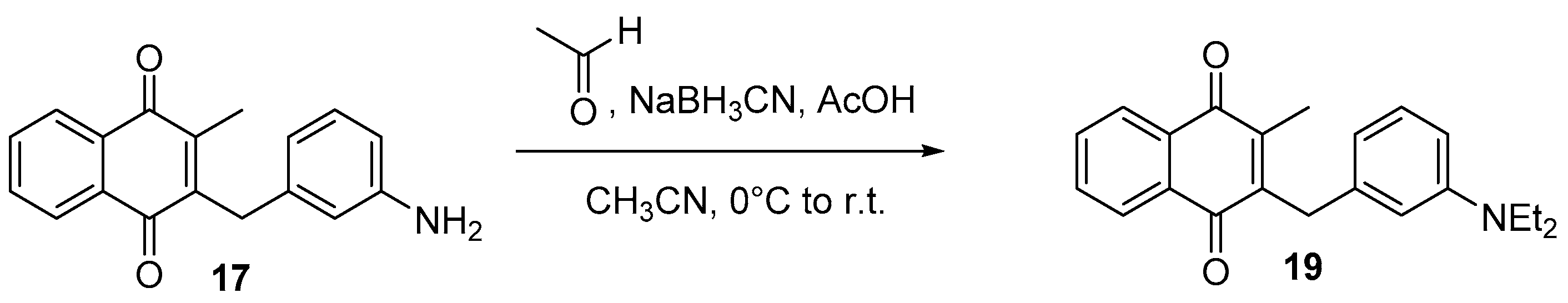
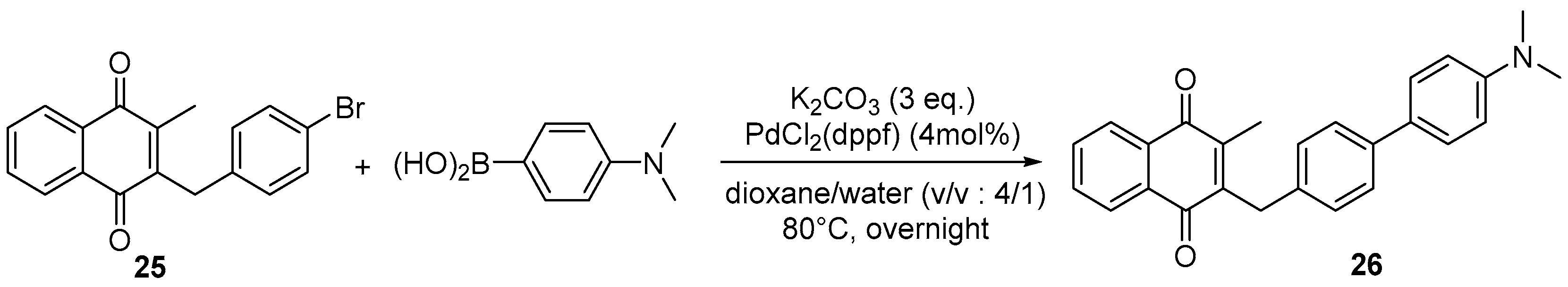
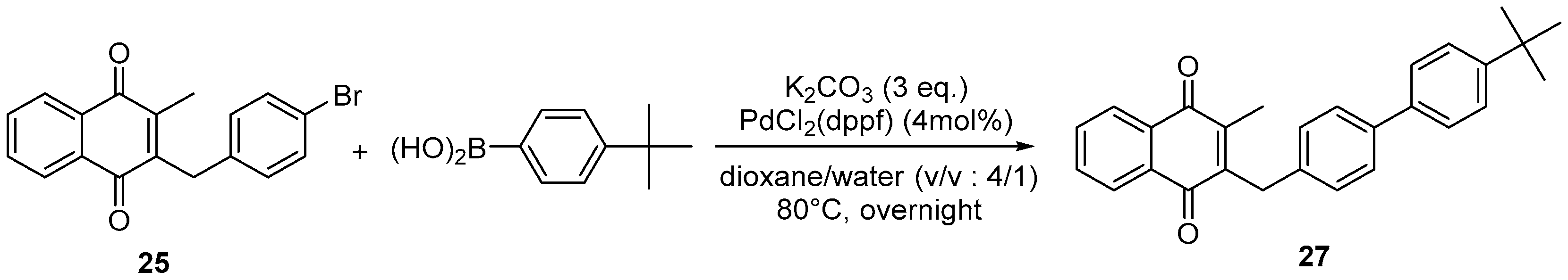
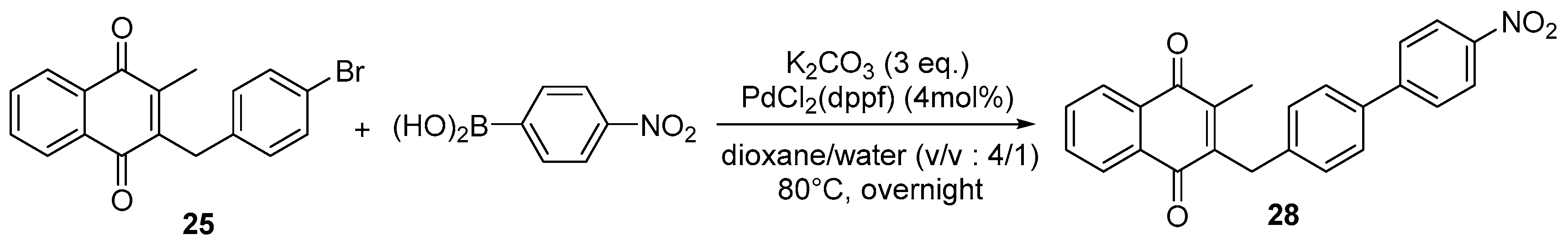

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
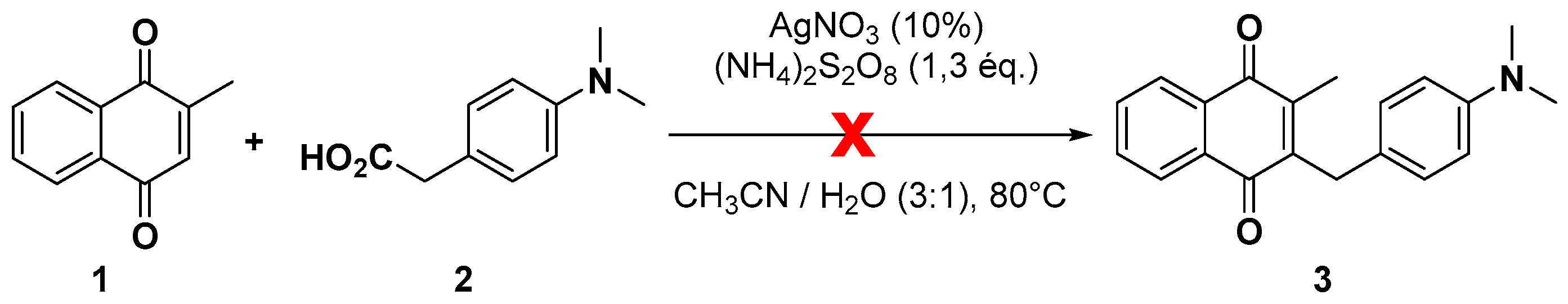{kind=link}
{kind=link}
{kind=link}
{kind=link}
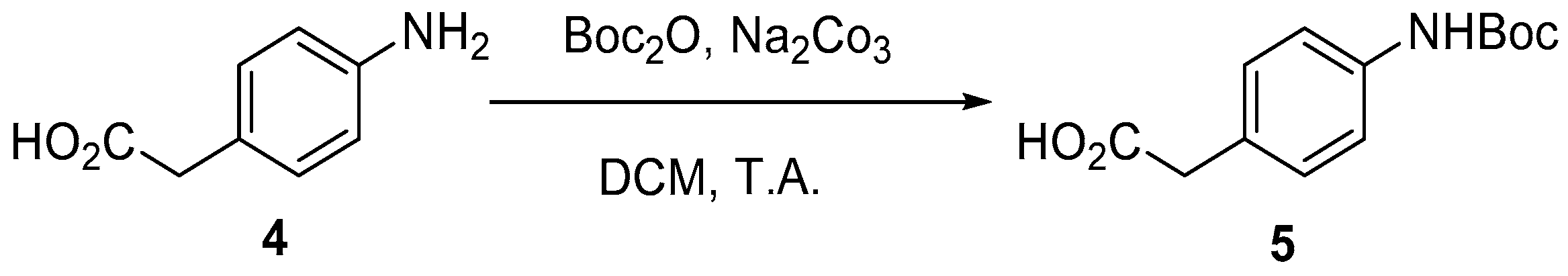{kind=link}
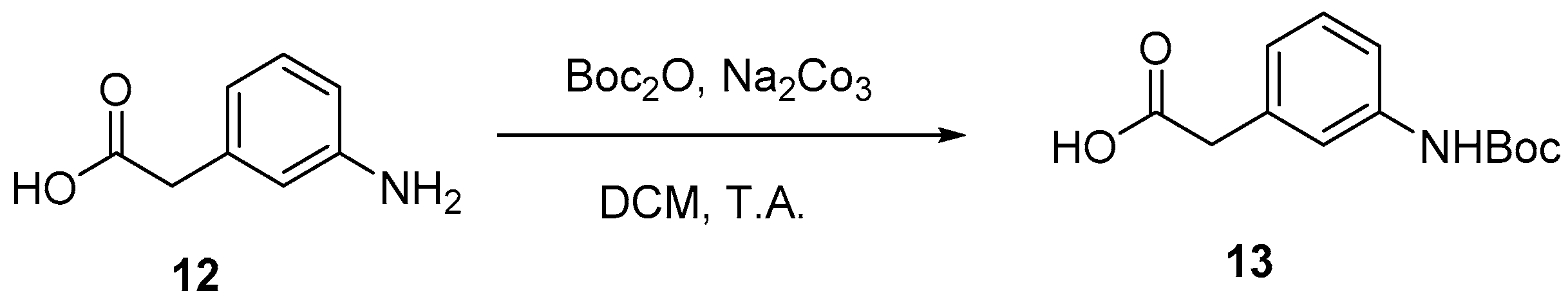{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
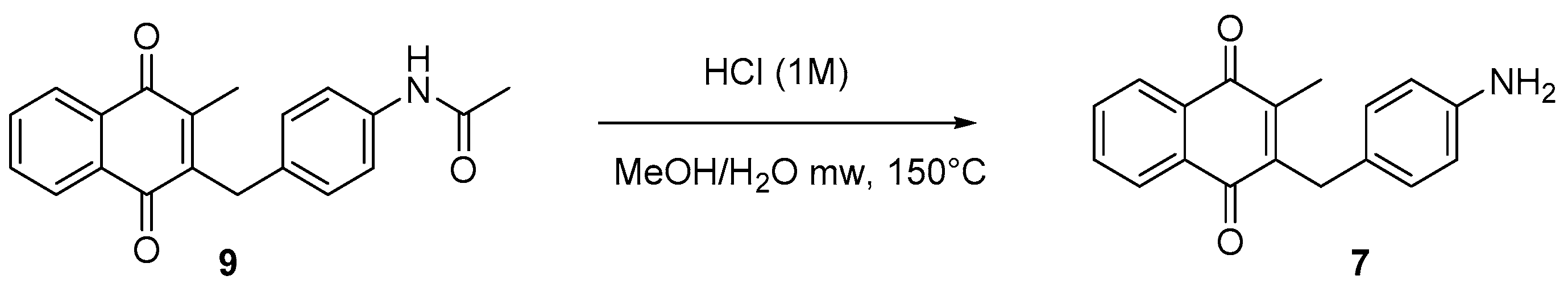{kind=link}
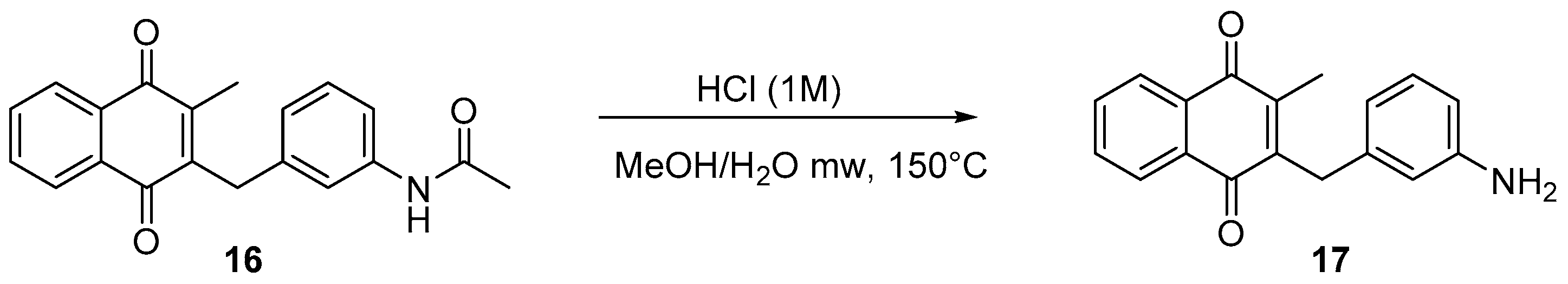{kind=link}
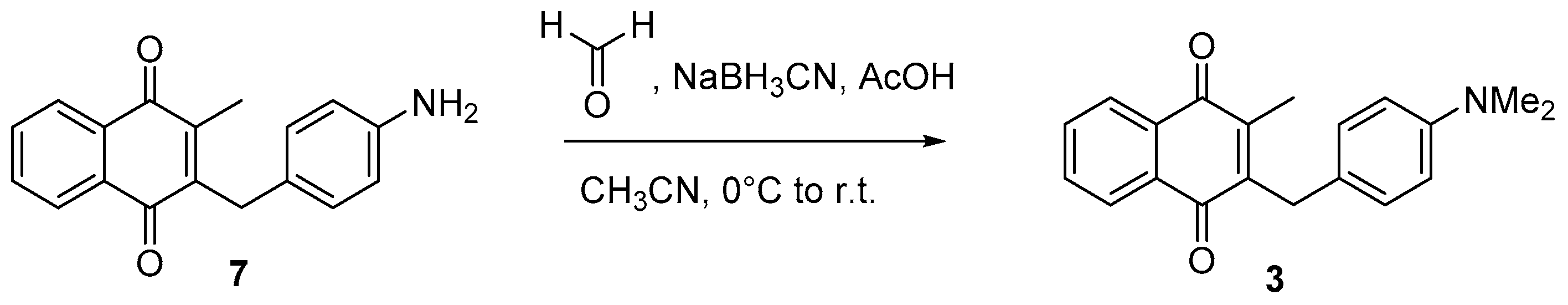{kind=link}
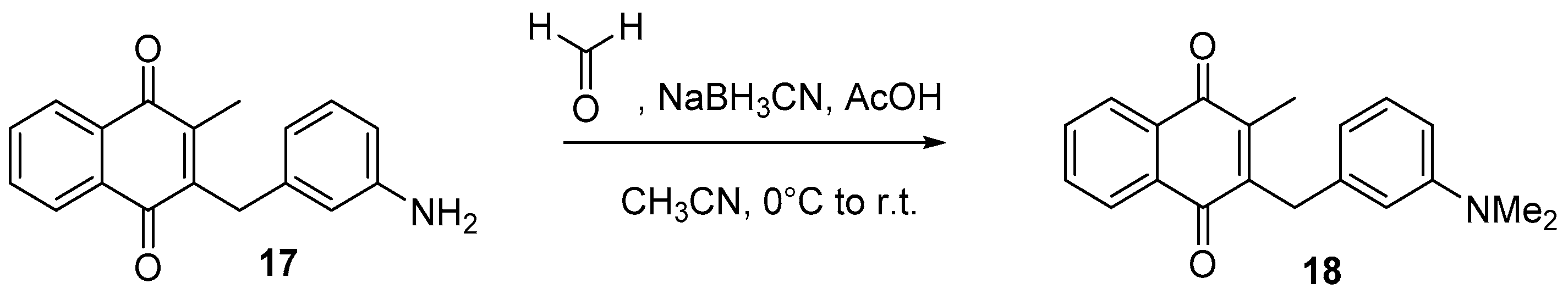{kind=link}
{kind=link}
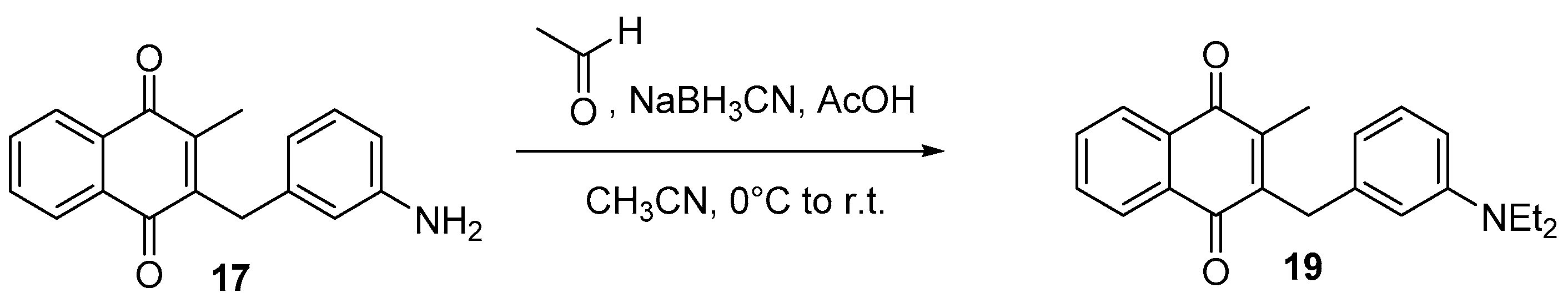{kind=link}
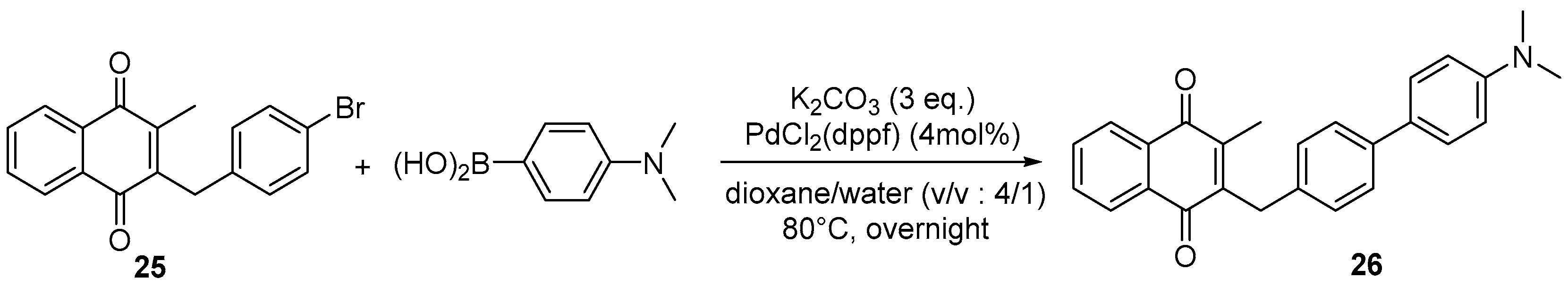{kind=link}
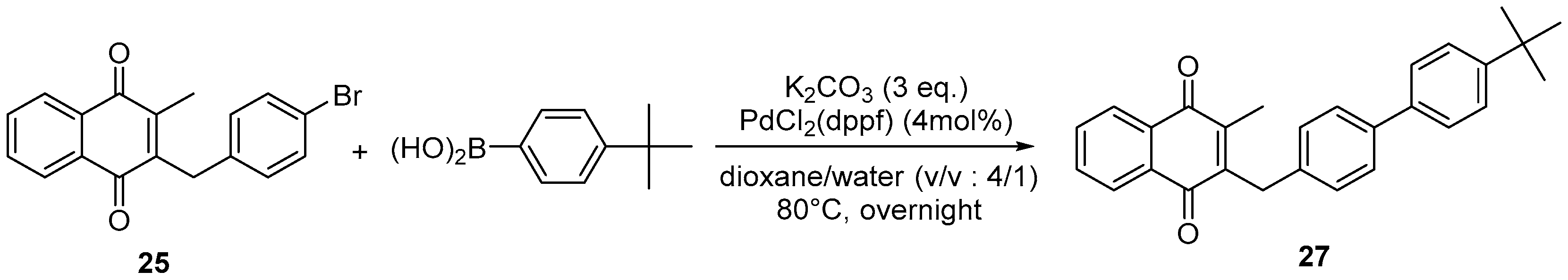{kind=link}
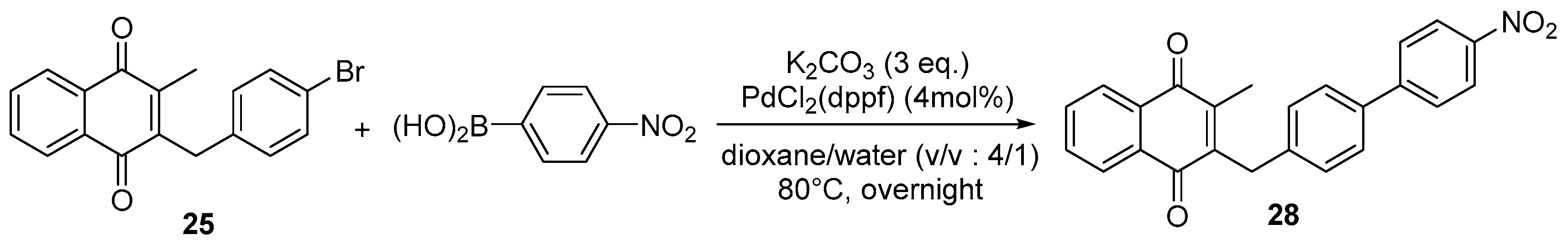{kind=link}
| Compound Code | Structure | IC50 (nM) | Tox/MRC-5 (µM) |
|---|---|---|---|
| Plasmodione b | 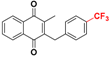 | 29 ± 2 (3) | >32.0 |
| 6 | 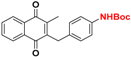 | 42.5 (2) | >64.0 |
| 25c b | 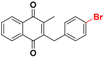 | 46 ± 4 (5) | >32.0 |
| 26 | 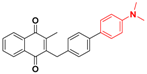 | 223 | >64.0 |
| 27 | 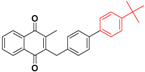 | 485 | >64.0 |
| 28 |  | 474 | >64.0 |
| Chloroquine b | 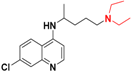 | 110 ± 20 (4) | 51.5 |
| Compound Code | Structure | IC50 (nM) ± SD (n) a | Tox/MRC-5 (µM) |
|---|---|---|---|
| 3 | 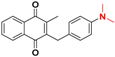 | 567 ± 217(3) | >64.0 |
| 6 | 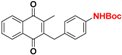 | 109 ± 46 (3) | >64.0 |
| 7 | 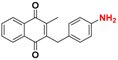 | 303 ± 138 (3) | >64.0 |
| 9 | 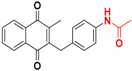 | 411 ± 220 (3) | >64.0 |
| 10 | 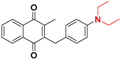 | 418 ± 166 (3) | >64.0 |
| 11 | 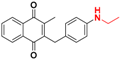 | 2056 ± 1073 (3) | 29.3 |
| 16 | 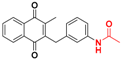 | 310 ± 134 (3) | 4.5 |
| 17 * | 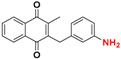 | 1198 ± 1651 (3) | >64.0 |
| 18 | 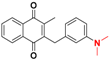 | 386 ± 151 (3) | >64.0 |
| 19 | 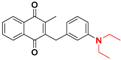 | 236 ± 97 (3) | >64.0 |
| 22 |  | 282 ± 86 (3) | 4.4 |
| 25a | 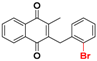 | 86.3 (1) | >64.0 |
| 25b | 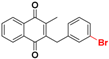 | 127.6 (1) | 20.8 |
| 25c | 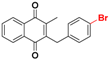 | 82 ± 2 (7) | >32.0 b |
| Plasmodione | 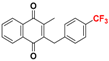 | 58 ± 11 (9) | >32.0 b |
| Chloroquine |  | 99 ± 19 (6) | 51.5 b |
| Compound | Treatment (ip for 5 Days) | Animals (n) | % Parasitaemia (dpi) | % Suppression (dpi) | Health Status | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 7 | 11 | 14 | 4 | 7 | 11 | 14 | ||||
| Untreated control | - | 5 | 38.3 | 42.2 | 67.7 | 61.9 | - | - | - | - | From day 4 post-infection (p.i.) onwards:poor appearance, tremor, body weight loss, rough hair, 4 animals died before day 7 p.i. |
| Chloroquine | 10 mg/kg | 5 | 5.2 | 9.8 | 32.1 | 56.2 | 86.3 | 76.8 | 52.7 | 9.2 | From day 4 p.i. onwards: poor appearance, tremor, body weight loss, rough hair, one animal died before day 14 p.i. |
| Compound 6 | 50 mg/kg | 4 | 9.4 | 7.7 | 41.9 | 69.3 | 75.5 | 81.8 | 38.1 | 12.0 | From day 4 p.i. onwards: poor appearance (intermittent), one animal died before day 14 p.i. |
| Compound Code | Structure | pKa ± σ [LH+]pH5/[LH+]pH7.4 |
|---|---|---|
| 3 | 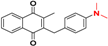 | 4.87 ± 0.03 144.7 |
| 7 b | 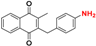 | 4.74 ± 0.05 162.5 |
| 10 | 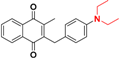 | e |
| 11 |  | 5.07 ± 0.06 112.0 |
| 17 *,b | 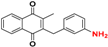 | 4.30 ± 0.02 209.6 |
| 18 | 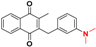 | 4.60 ± 0.04 179.9 |
| 19 | 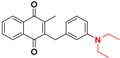 | e |
| Methylene blue c | 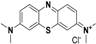 | pKa1 = 1.7 pKa2 = 4.5 pKa3 = 5.9 |
| Chloroquine d | 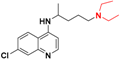 | pKa1 = 8.4 ± 0.2 pKa2 = 10.6 ± 0.2 1.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urgin, K.; Jida, M.; Ehrhardt, K.; Müller, T.; Lanzer, M.; Maes, L.; Elhabiri, M.; Davioud-Charvet, E. Pharmacomodulation of the Antimalarial Plasmodione: Synthesis of Biaryl- and N-Arylalkylamine Analogues, Antimalarial Activities and Physicochemical Properties. Molecules 2017, 22, 161. https://doi.org/10.3390/molecules22010161
Urgin K, Jida M, Ehrhardt K, Müller T, Lanzer M, Maes L, Elhabiri M, Davioud-Charvet E. Pharmacomodulation of the Antimalarial Plasmodione: Synthesis of Biaryl- and N-Arylalkylamine Analogues, Antimalarial Activities and Physicochemical Properties. Molecules. 2017; 22(1):161. https://doi.org/10.3390/molecules22010161
Chicago/Turabian StyleUrgin, Karène, Mouhamad Jida, Katharina Ehrhardt, Tobias Müller, Michael Lanzer, Louis Maes, Mourad Elhabiri, and Elisabeth Davioud-Charvet. 2017. "Pharmacomodulation of the Antimalarial Plasmodione: Synthesis of Biaryl- and N-Arylalkylamine Analogues, Antimalarial Activities and Physicochemical Properties" Molecules 22, no. 1: 161. https://doi.org/10.3390/molecules22010161
APA StyleUrgin, K., Jida, M., Ehrhardt, K., Müller, T., Lanzer, M., Maes, L., Elhabiri, M., & Davioud-Charvet, E. (2017). Pharmacomodulation of the Antimalarial Plasmodione: Synthesis of Biaryl- and N-Arylalkylamine Analogues, Antimalarial Activities and Physicochemical Properties. Molecules, 22(1), 161. https://doi.org/10.3390/molecules22010161