
Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Treg-Cells
2.1. Transcription Factors and Surface Markers
2.2. Cytokines
3. Th17-Cells
3.1. Transcription Factors and Surface Markers
3.2. Cytokines
3.3. Pathogenic vs. Non-Pathogenic
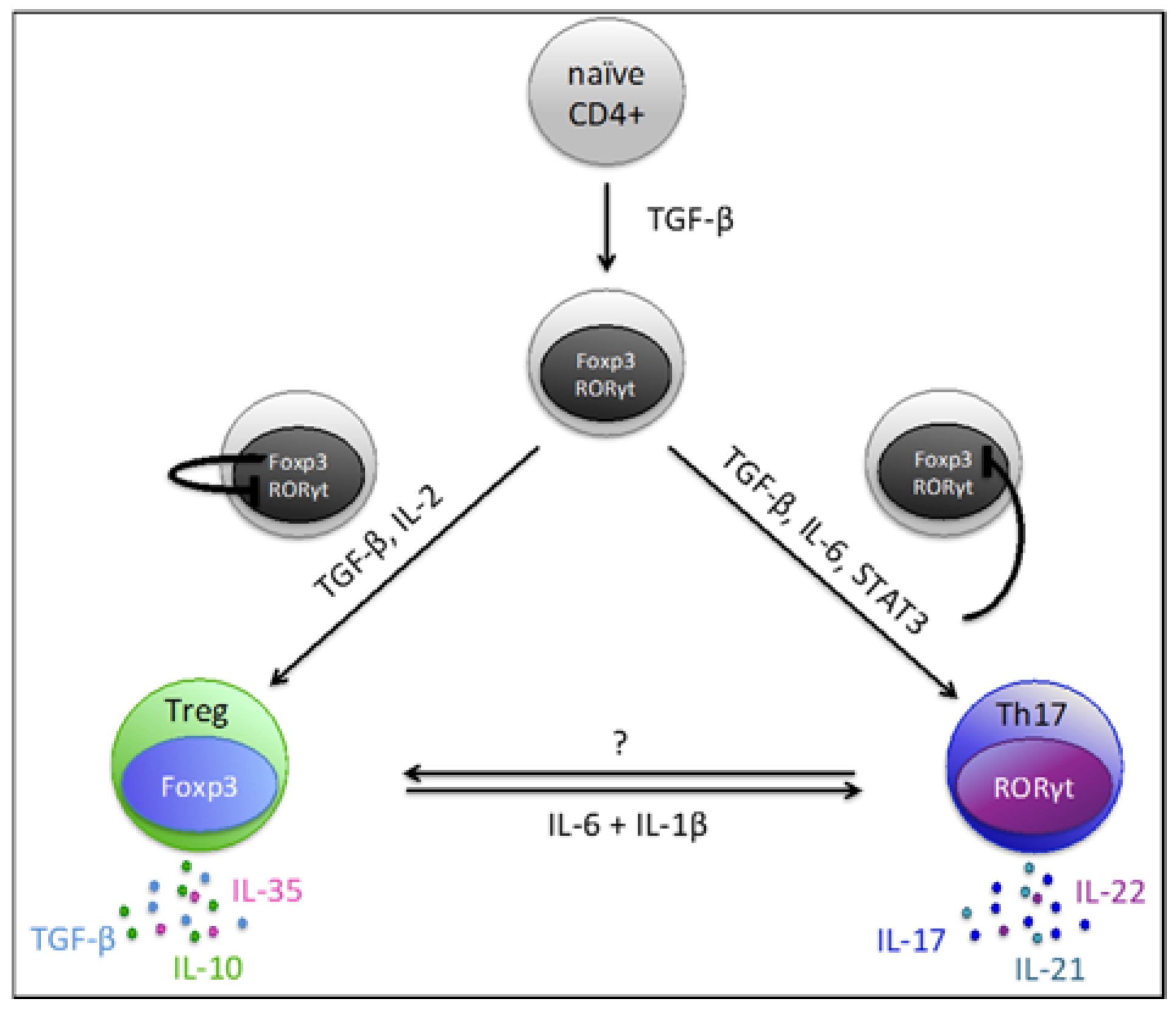
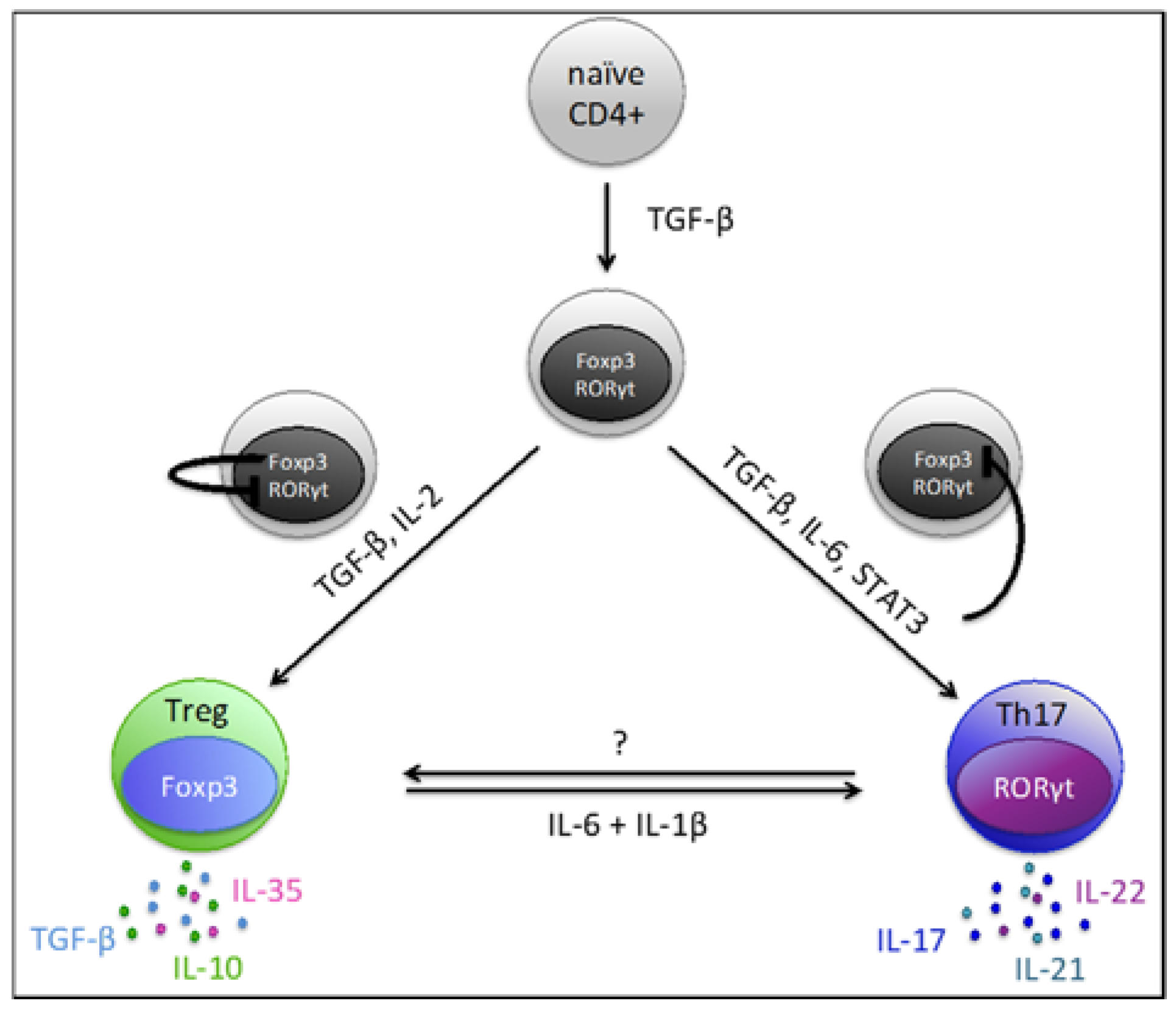
4. Th17/Treg Plasticity and Balance
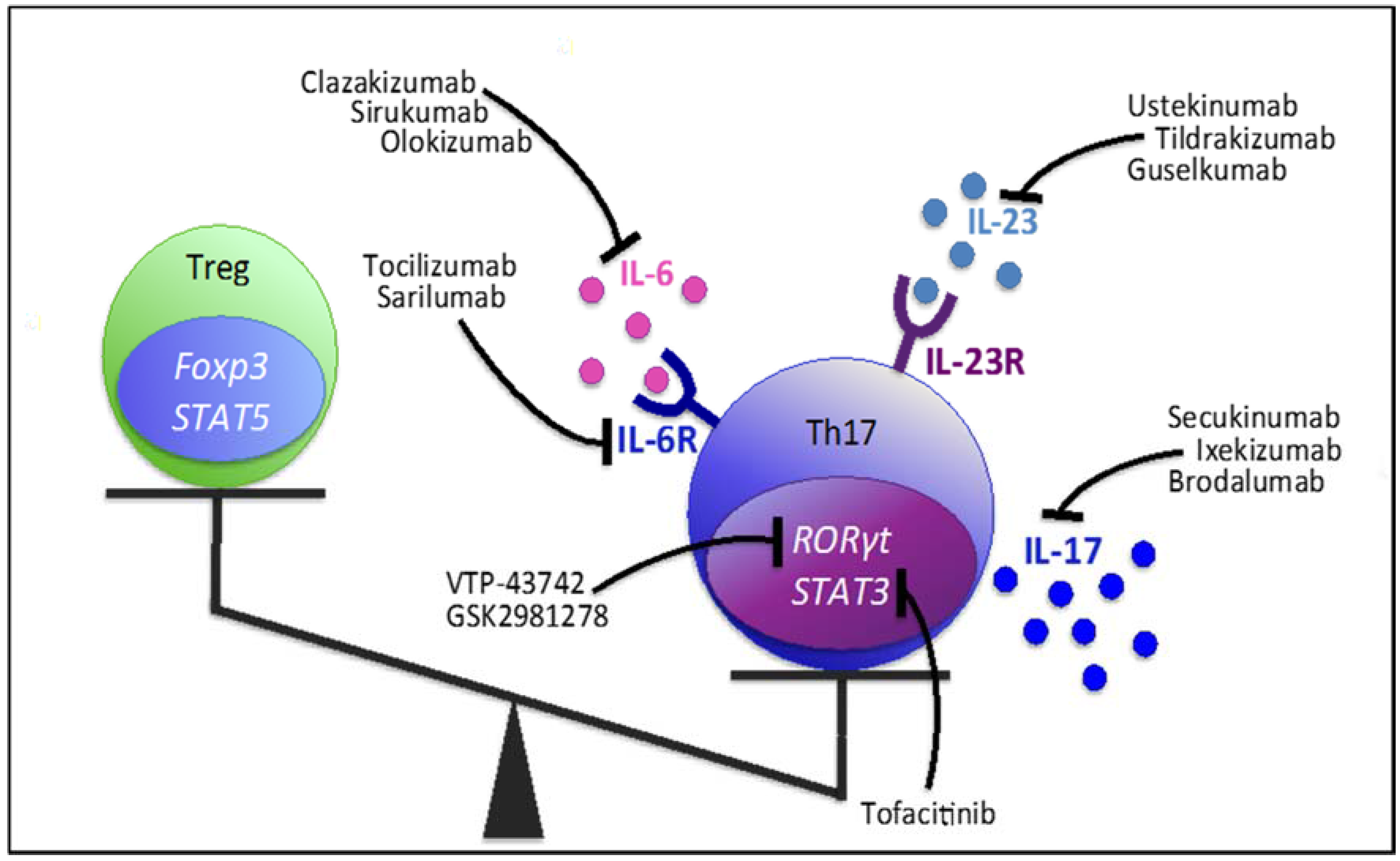
5. Therapeutic Approaches: Molecules Influencing the Th17/Treg Axis
5.1. Targeting Th17-Related Cytokines and Receptors
5.1.1. IL-17
5.1.2. IL-23
5.1.3. IL-6 & IL-6R
5.2. Targeting Transcription Factors
5.2.1. RORγt
5.2.2. STAT3
5.2.3. Foxp3
5.2.4. FoxO1
5.3. Targeting Intracellular Signaling Pathways
5.3.1. ROCK Inhibition
5.3.2. MAPK Inhibition
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Grant, C.R.; Liberal, R.; Mieli-Vergani, G.; Vergani, D.; Longhi, M.S. Regulatory T-cells in autoimmune diseases: Challenges, controversies and—Yet—Unanswered questions. Autoimmun. Rev. 2015, 14, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Yang, J.; Wang, X.; Li, D.; Lv, L.; Li, B. Th17 cells in autoimmune diseases. Front. Med. 2015, 9, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Beringer, A.; Noack, M.; Miossec, P. IL-17 in Chronic Inflammation: From Discovery to Targeting. Trends Mol. Med. 2016, 22, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Waite, J.C.; Skokos, D. Th17 response and inflammatory autoimmune diseases. Int. J. Inflam. 2012, 2012, 819467. [Google Scholar] [CrossRef] [PubMed]
- Hueber, W.; Patel, D.D.; Dryja, T.; Wright, A.M.; Koroleva, I.; Bruin, G.; Antoni, C.; Draelos, Z.; Gold, M.H.; Group, P.S.; et al. Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci. Transl. Med. 2010, 2, 52ra72. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Langley, R.G.; Sigurgeirsson, B.; Abe, M.; Baker, D.R.; Konno, P.; Haemmerle, S.; Thurston, H.J.; Papavassilis, C.; Richards, H.B. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: A randomized, double-blind, placebo-controlled phase II dose-ranging study. Br. J. Dermatol. 2013, 168, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.G.; Elewski, B.E.; Lebwohl, M.; Reich, K.; Griffiths, C.E.M.; Papp, K.; Puig, L.; Nakagawa, H.; Spelman, L.; Sigurgeirsson, B.; et al. Secukinumab in plaque psoriasis—Results of two phase 3 trials. N. Engl. J. Med. 2014, 371, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Prinz, J.C.; Gottlieb, A.B.; Kingo, K.; Sofen, H.; Ruer-Mulard, M.; Singh, V.; Pathan, R.; Papavassilis, C.; Cooper, S. Secukinumab administration by pre-filled syringe: Efficacy, safety and usability results from a randomized controlled trial in psoriasis (FEATURE). Br. J. Dermatol. 2015, 172, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Teitsma, X.M.; Marijnissen, A.K.A.; Bijlsma, J.W.J.; Lafeber, F.P.J.; Jacobs, J.W.G. Tocilizumab as monotherapy or combination therapy for treating active rheumatoid arthritis: A meta-analysis of efficacy and safety reported in randomized controlled trials. Arthritis Res. Ther. 2016, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Turnier, J.L.; Brunner, H.I. Tocilizumab for treating juvenile idiopathic arthritis. Expert Opin. Biol. Ther. 2016, 16, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sun, L.; Fan, X.; Wang, Z.; Cheng, Y.; Zhu, J.; Jin, T. Role of regulatory B cells in neuroimmunologic disorders. J. Neurosci. Res. 2016, 94, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Cosmi, L.; Liotta, F.; Lazzeri, E.; Francalanci, M.; Angeli, R.; Mazzinghi, B.; Santarlasci, V.; Manetti, R.; Vanini, V.; Romagnani, P.; et al. Human CD8+CD25+ thymocytes share phenotypic and functional features with CD4+CD25+ regulatory thymocytes. Blood 2003, 102, 4107–4115. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2009, 8, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Bluestone, J.A.; Abbas, A.K. Natural versus adaptive regulatory T cells. Nat. Rev. Immunol. 2003, 3, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, C.A.; Letterio, J.J.; Thornton, A.M.; McHugh, R.S.; Mamura, M.; Mizuhara, H.; Shevach, E.M. CD4+CD25+ Regulatory T Cells Can Mediate Suppressor Function in the Absence of Transforming Growth Factor β1 Production and Responsiveness. J. Exp. Med. 2002, 196, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Bacchetta, R.; Barzaghi, F.; Roncarolo, M.-G. From IPEX syndrome to FOXP3 mutation: A lesson on immune dysregulation. Ann. N. Y. Acad. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, V.L.; Wilkinson, J.E.; Russell, L.B. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am. J. Pathol. 1991, 138, 1379–1387. [Google Scholar] [PubMed]
- Williams, L.M.; Rudensky, A.Y. Maintenance of the Foxp3-dependent developmental program in mature regulatory T cells requires continued expression of Foxp3. Nat. Immunol. 2007, 8, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.A.; Manlove, L.S.; Farrar, M.A. Interleukin-2 and STAT5 in regulatory T cell development and function. JAK-STAT 2013, 2, e23154. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Hamann, A.; Klugewitz, K.; Austrup, F.; Jablonski-Westrich, D. Activation induces rapid and profound alterations in the trafficking of T cells. Eur. J. Immunol. 2000, 30, 3207–3218. [Google Scholar] [CrossRef]
- De Boer, O.J.; van der Loos, C.M.; Teeling, P.; van der Wal, A.C.; Teunissen, M.B. Immunohistochemical Analysis of Regulatory T Cell Markers FOXP3 and GITR on CD4+CD25+ T Cells in Normal Skin and Inflammatory Dermatoses. J. Histochem. Cytochem. 2007, 55, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, G.; Shufesky, W.J.; Tokita, D.; Morelli, A.E.; Thomson, A.W.; Raimondi, G.; Shufesky, W.J.; Tokita, D.; Morelli, A.E.; Thomson, A.W. Regulated compartmentalization of programmed cell death-1 discriminates CD4+CD25+ resting regulatory T cells from activated T cells. J. Immunol. 2006, 176, 2808–2816. [Google Scholar] [CrossRef] [PubMed]
- Garcia Santana, C.A.; Tung, J.W.; Gulnik, S. Human Treg cells are characterized by low/negative CD6 expression. Cytom. A 2014, 85, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Salgado, F.J.; Pérez-Díaz, A.; Villanueva, N.M.; Lamas, O.; Arias, P.; Nogueira, M. CD26: A negative selection marker for human Treg cells. Cytom. A 2012, 81, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Starke, M.; Di Mitri, D.; Borsellino, G.; Battistini, L.; Rötzschke, O.; Falk, K. CD49d provides access to “untouched” human Foxp3+ Treg free of contaminating effector cells. Blood 2009, 113, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.S.; Wang, J.; Shen, D.F.; Yuan, X.L.; Dong, P.; Li, M.X.; Xue, J.; Zhang, F.M.; Ge, H.L.; Xu, D. CD4+CD25+CD127low/- regulatory T cells express Foxp3 and suppress effector T cell proliferation and contribute to gastric cancers progression. Clin. Immunol. 2009, 131, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hartigan-O’Connor, D.J.; Poon, C.; Sinclair, E.; McCune, J.M. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J. Immunol. Methods 2007, 319, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming Growth Factor-β Controls Development, Homeostasis, and Tolerance of T Cells by Regulatory T Cell-Dependent and -Independent Mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Liggitt, D.; Rudensky, A.Y. Cellular Mechanisms of Fatal Early-Onset Autoimmunity in Mice with the T Cell-Specific Targeting of Transforming Growth Factor-β Receptor. Immunity 2006, 25, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Flavell, R.A. Contextual Regulation of Inflammation: A Duet by Transforming Growth Factor-β and Interleukin-10. Immunity 2008, 28, 468–476. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Flavell, R.A. TGF-β: A Master of All T Cell Trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson, W.R.; et al. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity 2008, 28, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, A.; Samstein, R.M.; Treuting, P.; Liang, Y.; Pils, M.C.; Heinrich, J.M.; Jack, R.S.; Wunderlich, F.T.; Brüning, J.C.; Müller, W.; et al. Interleukin-10 Signaling in Regulatory T Cells Is Required for Suppression of Th17 Cell-Mediated Inflammation. Immunity 2011, 34, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Gagliani, N.; Esplugues, E.; O’Connor, W.; Huber, F.J.; Chaudhry, A.; Kamanaka, M.; Kobayashi, Y.; Booth, C.J.; Rudensky, A.Y.; et al. Th17 Cells Express Interleukin-10 Receptor and Are Controlled by Foxp3- and Foxp3+ Regulatory CD4+ T Cells in an Interleukin-10-Dependent Manner. Immunity 2011, 34, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Wadwa, M.; Klopfleisch, R.; Adamczyk, A.; Frede, A.; Pastille, E.; Mahnke, K.; Hansen, W.; Geffers, R.; Lang, K.S.; Buer, J.; et al. IL-10 downregulates CXCR3 expression on Th1 cells and interferes with their migration to intestinal inflammatory sites. Mucosal Immunol. 2016, 9, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Turovskaya, O.; Kim, G.; Madan, R.; Karp, C.L.; Kronenberg, M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat. Immunol. 2009, 10, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Chaturvedi, V.; Henderson, A.L.; Giacomin, P.R.; Guy, C.; Bankoti, J.; Finkelstein, D.; Forbes, K.; Workman, C.J.; Brown, S.A.; et al. IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol. 2010, 11, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Collison, L.W.; Workman, C.J.; Kuo, T.T.; Boyd, K.; Wang, Y.; Vignali, K.M.; Cross, R.; Sehy, D.; Blumberg, R.S.; Vignali, D.A.A. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature 2007, 450, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, S.; Billmeier, U.; Mcheldlidze, T.; Blumberg, R.S.; Neurath, M.F. Interleukin-35 Mediates Mucosal Immune Responses That Protect Against T-Cell–Dependent Colitis. Gastroenterology 2011, 141, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17–producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Korn, T.; Oukka, M.; Kuchroo, V.K. Induction and effector functions of T(H)17 cells. Nature 2008, 453, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Torchinsky, M.B.; Blander, J.M. T helper 17 cells: Discovery, function, and physiological trigger. Cell. Mol. Life Sci. 2010, 67, 1407–1421. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human Th-17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγT. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, K.; Yoshida, H.; Wakabayashi, Y.; Chinen, T.; Saeki, K.; Nakaya, M.; Takaesu, G.; Hori, S.; Yoshimura, A.; Kobayashi, T. Foxp3 Inhibits RORγt-mediated IL-17A mRNA Transcription through Direct Interaction with RORγt. J. Biol. Chem. 2008, 283, 17003–17008. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jager, A.; Strom, T.B.; Oukka, M.; Kuchroo, V.K. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 2007, 448, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Zhou, L.; Littman, D.R. Transcriptional regulation of Th17 cell differentiation. Semin. Immunol. 2007, 19, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.J.; Boniface, K.; Chan, J.R.; McKenzie, B.S.; Blumenschein, W.M.; Mattson, J.D.; Basham, B.; Smith, K.; Chen, T.; Morel, F.; et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007, 8, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Santarlasci, V.; Capone, M.; Peired, A.; Frosali, F.; Crome, S.Q.; Querci, V.; Fambrini, M.; Liotta, F.; Levings, M.K.; et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur. J. Immunol. 2010, 40, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Cosmi, L.; Liotta, F.; Maggi, E.; Romagnani, S. Defining the human T helper 17 cell phenotype. Trends Immunol. 2012, 33, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.; Maggi, L.; Micheletti, A.; Lazzeri, E.; Tamassia, N.; Cosmi, L.; Lunardi, C.; Annunziato, F.; Romagnani, S.; Marco, A.; et al. Evidence for a cross-talk between human neutrophils and Th17 cells Evidence for a cross-talk between human neutrophils and Th17 cells. Blood 2010, 115, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Anderson, D.E.; Baecher-Allan, C.; Hastings, W.D.; Bettelli, E.; Oukka, M.; Kuchroo, V.K.; Hafler, D.A. IL-21 and TGF-β are required for differentiation of human TH17 cells. Nature 2008, 454, 350–352. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.D.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.M.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Wang, S.C.; Li, J. Interleukin 22, a potential therapeutic target for rheumatoid arthritis. J. Rheumatol. 2012, 39, 2220–2221. [Google Scholar] [CrossRef] [PubMed]
- Stephen-Victor, E.; Fickenscher, H.; Bayry, J. IL-26: An Emerging Proinflammatory Member of the IL-10 Cytokine Family with Multifaceted Actions in Antiviral, Antimicrobial, and Autoimmune Responses. PLoS Pathog. 2016, 12, 4–9. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain Th-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Laurence, A.; Yang, X.; Tato, C.M.; Mandy, J.; Konkel, J.; Ramos, H.L.; Wei, L.; Davidson, T.; Grainger, J.; et al. Generation of Pathogenic Th17 Cells in the Absence of TGF-β Signaling. Nature 2010, 467, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.; Tian, Q.; Dong, C. Critical regulation of early Th17 cell differentiation by IL-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Regev, A.; Kuchroo, V. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yosef, N.; Gaublomme, J.; Wu, C.; Lee, Y.; Clish, C.B.; Kaminski, J.; Xiao, S.; Zu Horste, G.M.; Pawlak, M.; et al. CD5L/AIM Regulates Lipid Biosynthesis and Restrains Th17 Cell Pathogenicity. Cell 2015, 163, 1413–1427. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Nurieva, R.; Martinez, G.J.; Kang, H.S.; Pappu, B.P.; Shah, B.; Chang, S.H.; Schluns, K.S.; Watowich, S.S.; Feng, X.; et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 2008, 29, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Deknuydt, F.; Bioley, G.; Valmori, D.; Ayyoub, M. IL-1β and IL-2 convert human Treg into TH17 cells. Clin. Immunol. 2009, 131, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.G.; Wang, J.; Horwitz, D.A. Cutting Edge: Foxp3+CD4+CD25+ Regulatory T Cells Induced by IL-2 and TGF-β Are Resistant to Th17 Conversion by IL-6. J. Immunol. 2008, 180, 7112–7116. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Vesely, M.C.A.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limón, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Farahnik, B.; Beroukhim, K.; Abrouk, M.; Nakamura, M.; Zhu, T.H.; Singh, R.; Lee, K.; Bhutani, T.; Koo, J. Brodalumab for the Treatment of Psoriasis: A Review of Phase III Trials. Dermatol. Ther. (Heidelb.) 2016, 6, 111–124. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Mease, P.J.; Kirkham, B.; Kavanaugh, A.; Ritchlin, C.T.; Rahman, P.; Van Der Heijde, D.; Landewe, R.; Conaghan, P.G.; Gottlieb, A.B.; et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 386, 1137–1146. [Google Scholar] [CrossRef]
- Mease, P.J.; McInnes, I.B.; Kirkham, B.; Kavanaugh, A.; Rahman, P.; van der Heijde, D.; Landewé, R.; Nash, P.; Pricop, L.; Yuan, J.; et al. Secukinumab Inhibition of Interleukin-17A in Patients with Psoriatic Arthritis. N. Engl. J. Med. 2015, 373, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Sieper, J.; Braun, J.; Baraliakos, X.; Dougados, M.; Emery, P.; Deodhar, A.; Porter, B.; Martin, R.; Andersson, M.; et al. Secukinumab, an Interleukin-17A Inhibitor, in Ankylosing Spondylitis. N. Engl. J. Med. 2015, 373, 2534–2548. [Google Scholar] [CrossRef] [PubMed]
- Baeten, D.; Baraliakos, X.; Braun, J.; Sieper, J.; Emery, P.; van der Heijde, D.; McInnes, I.; van Laar, J.M.; Landewé, R.; Wordsworth, P.; et al. Anti-interleukin-17A monoclonal antibody secukinumab in treatment of ankylosing spondylitis: A randomised, double-blind, placebo-controlled trial. Lancet 2013, 382, 1705–1713. [Google Scholar] [CrossRef]
- Burmester, G.R.; Durez, P.; Shestakova, G.; Genovese, M.C.; Schulze-Koops, H.; Li, Y.; Wang, Y.A.; Lewitzky, S.; Koroleva, I.; Berneis, A.A.; et al. Association of HLA-DRB1 alleles with clinical responses to the anti-interleukin-17A monoclonal antibody secukinumab in active rheumatoid arthritis. Rheumatology 2016, 55, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Aelion, J.A.; Lee, S.-H.; Codding, C.E.; Kellner, H.; Ikawa, T.; et al. One-year Efficacy and Safety Results of Secukinumab in Patients With Rheumatoid Arthritis: Phase II, Dose-finding, Double-blind, Randomized, Placebo-controlled Study. J. Rheumatol. 2014, 41, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Greenwald, M.; Cho, C.-S.; Berman, A.; Jin, L.; Cameron, G.S.; Benichou, O.; Xie, L.; Braun, D.; Berclaz, P.-Y.; et al. A Phase II Randomized Study of Subcutaneous Ixekizumab, an Anti-Interleukin-17 Monoclonal Antibody, in Rheumatoid Arthritis Patients Who Were Naive to Biologic Agents or Had an Inadequate Response to Tumor Necrosis Factor Inhibitors. Arthritis Rheumatol. 2014, 66, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.A.; Churchill, M.; Flores-Suarez, L.; Cardiel, M.H.; Wallace, D.; Martin, R.; Phillips, K.; Kaine, J.L.; Dong, H.; Salinger, D.; et al. A phase Ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res. Ther. 2013, 15, R164. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Durez, P.; Richards, H.B.; Supronik, J.; Dokoupilova, E.; Mazurov, V.; Aelion, J.A.; Lee, S.-H.; Codding, C.E.; Kellner, H.; et al. Efficacy and safety of secukinumab in patients with rheumatoid arthritis: A phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann. Rheum. Dis. 2013, 72, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Pavelka, K.; Chon, Y.; Newmark, R.; Lin, S.-L.; Baumgartner, S.; Erondu, N. A Study to Evaluate the Safety, Tolerability, and Efficacy of Brodalumab in Subjects with Rheumatoid Arthritis and an Inadequate Response to Methotrexate. J. Rheumatol. 2015, 42, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Targan, S.R.; Feagan, B.G.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Blosch, C.; Newmark, R.; Zhang, N.; Chon, Y.; Lin, S.-L.; et al. A randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and efficacy of AMG 827 in subjects with moderate to severe crohn’s disease. Gastroenterology 2012, 143, e26. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, W., Jr.; Zenewicz, L.A.; Flavell, R.A. The dual nature of T(H)17 cells: Shifting the focus to function. Nat. Immunol. 2010, 11, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kuchroo, V. Defining the functional states of Th17 cells. F1000Research 2015, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Fitch, E.; Harper, E.; Skorcheva, I.; Kurtz, S.E.; Blauvelt, A. Pathophysiology of psoriasis: Recent advances on IL-23 and Th17 cytokines. Curr. Rheumatol. Rep. 2010, 9, 461–467. [Google Scholar] [CrossRef]
- Lee, E.; Trepicchio, W.L.; Oestreicher, J.L.; Pittman, D.; Wang, F.; Chamian, F.; Dhodapkar, M.; Krueger, J.G. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J. Exp. Med. 2004, 199, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.A.; Langrish, C.L.; Chen, Y.; Blumenschein, W.; McClanahan, T.; Kastelein, R.A.; Sedgwick, J.D.; Cua, D.J. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J. Exp. Med. 2003, 198, 1951–1957. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, K.; Kanda, T.; Takaishi, M.; Shiga, T.; Miyoshi, K.; Nakajima, H.; Kamijima, R.; Tarutani, M.; Benson, J.M.; Elloso, M.M.; et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J. Immunol. Res. 2011, 186, 4481–4489. [Google Scholar]
- Leonardi, C.L.; Kimball, A.B.; Papp, K.A.; Yeilding, N.; Guzzo, C.; Wang, Y.; Li, S.; Dooley, L.T.; Gordon, K.B. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 2008, 371, 1665–1674. [Google Scholar] [CrossRef]
- Papp, K.A.; Langley, R.G.; Lebwohl, M.; Krueger, G.G.; Szapary, P.; Yeilding, N.; Guzzo, C.; Hsu, M. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 2008, 371, 1675–1684. [Google Scholar] [CrossRef]
- Mcinnes, I.B.; Kavanaugh, A.; Gottlieb, A.B.; Puig, L.; Rahman, P.; Ritchlin, C.; Brodmerkel, C.; Li, S.; Wang, Y. Efficacy and safety of ustekinumab in patients with active psoriatic arthritis: 1 year results of the phase 3, multicentre, double-blind, placebo-controlled PSUMMIT 1 trial. Lancet 2013, 382, 780–789. [Google Scholar] [CrossRef]
- Kavanaugh, A.; Ritchlin, C.; Rahman, P.; Puig, L.; Gottlieb, A.B.; Li, S.; Wang, Y.; Noonan, L.; Brodmerkel, C.; Song, M.; et al. Ustekinumab, an anti-IL-12/23 p40 monoclonal antibody, inhibits radiographic progression in patients with active psoriatic arthritis: Results of an integrated analysis of radiographic data from the phase 3, multicentre, randomised, double-blind, placebo-c. Ann. Rheum. Dis. 2014, 73, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, A.; Puig, L.; Gottlieb, A.B.; Ritchlin, C.; You, Y.; Li, S.; Song, M.; Randazzo, B.; Rahman, P.; Mcinnes, I.B. Efficacy and safety of ustekinumab in psoriatic arthritis patients with peripheral arthritis and physician-reported spondylitis: Post-hoc analyses from two phase III, multicentre, double-blind, placebo-controlled studies (PSUMMIT-1/PSUMMIT-2). Ann. Rheum. Dis. 2016, 75, 1984–1988. [Google Scholar] [CrossRef] [PubMed]
- Ritchlin, C.; Rahman, P.; Kavanaugh, A.; Mcinnes, I.B.; Puig, L.; Li, S.; Wang, Y.; Shen, Y.; Doyle, M.K.; Mendelsohn, A.M.; et al. Efficacy and safety of the anti-IL-12/23 p40 monoclonal antibody, ustekinumab, in patients with active psoriatic arthritis despite conventional non-biological and biological anti-tumour necrosis factor therapy: 6-month and 1-year results of the controlled. Ann. Rheum. Dis. 2014, 73, 990–999. [Google Scholar] [CrossRef] [PubMed]
- Segal, B.M.; Constantinescu, C.S.; Raychaudhuri, A.; Kim, L.; Fidelus-Gort, R.; Kasper, L.H. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: A phase II, double-blind, placebo-controlled, randomised, dose-ranging study. Lancet 2008, 7, 796–804. [Google Scholar] [CrossRef]
- Gordon, K.B.; Langley, R.G.; Gottlieb, A.B.; Papp, K.A.; Krueger, G.G.; Strober, B.E.; Williams, D.A.; Gu, Y.; Valdes, J.M. A phase III, randomized, controlled trial of the fully human IL-12/23 mAb briakinumab in moderate-to-severe psoriasis. J. Investig. Dermatol. 2012, 132, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.G.; Ferris, L.K.; Menter, A.; Wagner, F.; White, A.; Visvanathan, S.; Lalovic, B.; Aslanyan, S.; Wang, E.E.L.; Hall, D.; et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: Safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial. J. Allergy Clin. Immunol. 2015, 136, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Audia, S.; Janikashvili, N.; Ciudad, M.; Trad, M.; Fraszczak, J.; Ornetti, P.; Maillefert, J.F.; Miossec, P.; Bonnotte, B. Brief report: Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2499–2503. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Hashizume, M.; Kaneko, Y.; Yoshimoto, K.; Nishina, N.; Takeuchi, T. Peripheral blood CD4+CD25+CD127low regulatory T cells are significantly increased by tocilizumab treatment in patients with rheumatoid arthritis: Increase in regulatory T cells correlates with clinical response. Arthritis Res. Ther. 2015, 17, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, Z.; Li, L.; Liu, X. Interleukin-6 (IL-6) Receptor Antagonist Protects Against Rheumatoid Arthritis. Med. Sci. Monit. 2016, 22, 2113–2118. [Google Scholar] [CrossRef] [PubMed]
- Tada, Y.; Ono, N.; Suematsu, R.; Tashiro, S.; Sadanaga, Y.; Tokuda, Y.; Ono, Y.; Nakao, Y.; Maruyama, A.; Ohta, A.; et al. The balance between Foxp3 and Ror-γt expression in peripheral blood is altered by tocilizumab and abatacept in patients with rheumatoid arthritis. BMC Musculoskelet. Disord. 2016, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Pesce, B.; Soto, L.; Sabugo, F.; Wurmann, P.; Cuchacovich, M.; López, M.N.; Sotelo, P.H.; Molina, M.C.; Aguillón, J.C.; Catalán, D. Effect of interleukin-6 receptor blockade on the balance between regulatory T cells and T helper type 17 cells in rheumatoid arthritis patients. Clin. Exp. Immunol. 2013, 171, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Park, W.; Park, S.H.; Shim, S.C.; Baek, H.J.; Yoo, D.H.; Kim, H.A.; Lee, S.K.; Leee, Y.J.; Park, Y.E.; et al. Low baseline interleukin-17A levels are associated with better treatment response at 12 weeks to tocilizumab therapy in rheumatoid arthritis patients. J. Immunol. Res. 2015, 2015, 487230. [Google Scholar] [CrossRef] [PubMed]
- Strand, V.; Kosinski, M.; Chen, C.; Joseph, G.; Rendas-baum, R.; Graham, N.M.H.; Van Hoogstraten, H.; Bayliss, M.; Fan, C.; Huizinga, T.; et al. Sarilumab plus methotrexate improves patient-reported outcomes in patients with active rheumatoid arthritis and inadequate responses to methotrexate: Results of a phase III trial. Arthritis Res. Ther. 2016, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Genovese, M.C.; Fleischmann, R.; Kivitz, A.J.; Rell-Bakalarska, M.; Martincova, R.; Fiore, S.; Rohane, P.; Van Hoogstraten, H.; Garg, A.; Fan, C.; et al. Sarilumab Plus Methotrexate in Patients with Active Rheumatoid Arthritis and Inadequate Response to Methotrexate. Results of a Phase III Study. Arthritis Rheumatol. 2015, 67, 1424–1437. [Google Scholar] [CrossRef] [PubMed]
- Boyapati, A.; Msihid, J.; Fiore, S.; van Adelsberg, J.; Graham, N.M.H.; Hamilton, J.D. Sarilumab plus methotrexate suppresses circulating biomarkers of bone resorption and synovial damage in patients with rheumatoid arthritis and inadequate response to methotrexate: A biomarker study of MOBILITY. Arthritis Res. Ther. 2016, 18, 225. [Google Scholar] [CrossRef] [PubMed]
- Sieper, J.; Braun, J.; Kay, J.; Badalamenti, S.; Radin, A.R.; Jiao, L.; Fiore, S.; Momtahen, T.; Yancopoulos, G.D.; Stahl, N.; et al. Sarilumab for the treatment of ankylosing spondylitis: Results of a Phase II, randomised, double-blind, placebo-controlled study (ALIGN). Ann. Rheum. Dis. 2015, 74, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Mease, P.; Mysler, E.; Takeuchi, T.; Drescher, E.; Berman, A.; Xing, J.; Zilberstein, M.; Banerjee, S.; Emery, P. The Efficacy and Safety of Subcutaneous Clazakizumab in Patients with Moderate-to-Severe Rheumatoid Arthritis and an Inadequate Response to Methotrexate. Arthritis Rheumatol. 2015, 67, 2591–2600. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Gottlieb, A.B.; Berman, A.; Drescher, E.; Xing, J.; Wong, R.; Banerjee, S. The Efficacy and Safety of Clazakizumab, an Anti-Interleukin-6 Monoclonal Antibody, in a Phase IIb Study of Adults With Active Psoriatic Arthritis. Arthritis Rheumatol. 2016, 68, 2163–2173. [Google Scholar] [CrossRef] [PubMed]
- Cook, D.N.; Kang, H.S.; Jetten, A.M. Retinoic acid orphan receptors (RORs): Regulatory functions in immunity, development, circadian rhythm and metabolism. Nucl. Recept. Res. 2015, 2, 101185. [Google Scholar] [CrossRef] [PubMed]
- Fujita-Sato, S.; Ito, S.; Isobe, T.; Ohyama, T.; Wakabayashi, K.; Morishita, K.; Ando, O.; Isono, F. Structural basis of digoxin that antagonizes RORγt receptor activity and suppresses Th17 cell differentiation and interleukin (IL)-17 production. J. Biol. Chem. 2011, 286, 31409–31417. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.R.; Leung, M.W.L.; Huang, P.; Ryan, D.A.; Michael, R.; Malapaka, R.R.V.; Chow, J.; Manel, N.; Ciofani, M.; Kim, S.V.; et al. Digoxin and its derivatives suppress Th17 cell differentiation by antagonizing RORγt activity. Nature 2011, 472, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Baek, S.; Lee, J.; Lee, J.; Lee, D.; Park, M.; Cho, M.; Park, S.; Kwok, S. Digoxin ameliorates autoimmune arthritis via suppression of Th17 differentiation. Int. Immunopharmacol. 2015, 26, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.-Y.; Lee, J.J.; Lee, D.-G.; Park, M.-K.; Lee, J.J.; Kwok, S.-K.; Cho, M.; Park, S.-H. Ursolic acid ameliorates autoimmune arthritis via suppression of Th17 and B cell differentiation. Acta Pharmacol. Sin. 2014, 35, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Yosef, N.; Yang, J.; Wang, Y.; Zhou, L.; Zhu, C.; Wu, C.; Baloglu, E.; Schmidt, D.; Ramesh, R.; et al. Small-molecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 2014, 40, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Skepner, J.; Trocha, M.; Ramesh, R.; Qu, X.A.; Schmidt, D.; Baloglu, E.; Lobera, M.; Davis, S.; Nolan, M.A.; Thaddeus, J.; et al. In vivo regulation of gene expression and T helper type 17 differentiation by RORγt inverse agonists. Immunology 2014, 145, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Skepner, J.; Ramesh, R.; Trocha, M.; Schmidt, D.; Baloglu, E.; Lobera, M.; Carlson, T.; Hill, J.; Orband-Miller, L.A.; Barnes, A.; et al. Pharmacologic Inhibition of RORγt Regulates Th17 Signature Gene Expression and Suppresses Cutaneous Inflammation In Vivo. J. Immunol. 2014, 192, 2564–2575. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, M.; Ishizaki, M.; Suzuki, K.; Isobe, T.; Shimozato, T.; Sano, S. Oral administration of a novel RORγt antagonist attenuates psoriasis-like skin lesion of two independent mouse models through neutralization of IL-17. J. Dermatol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.H.; Peredo, C.E.; Takeda, Y.; Bui, T.; Neil, J.; Rickard, D.; Millerman, E.; Therrien, J.P.; Nicodeme, E.; Brusq, J.M.; et al. Development of a topical treatment for psoriasis targeting RORγ: From bench to skin. PLoS ONE 2016, 11, e0147979. [Google Scholar] [CrossRef] [PubMed]
- Gege, C. Retinoid-related orphan receptor gamma t (RORγt) inhibitors from Vitae Pharmaceuticals (WO2015116904) and structure proposal for their Phase I candidate VTP-43742. Expert Opin. Ther. Pat. 2016, 26, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Nie, J.; Gao, Y.; Xu, P.; Sun, Q.; Yang, J.; Han, L. Reciprocal regulation of RORγt acetylation and function by p300 and HDAC1. Sci. Rep. 2015, 5, 16355. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.S.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnolzer, M.; et al. SIRT1 deacetylates RORγt and enhances Th17 cell generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, J.; Han, L.; Zhao, K.; Wu, Q.; Bao, L.; Li, Z.; Lv, L.; Li, B. TRAF5-mediated Lys-63-linked polyubiquitination plays an essential role in positive regulation of RORγt in promoting IL-17A expression. J. Biol. Chem. 2015, 290, 29086–29094. [Google Scholar] [CrossRef] [PubMed]
- Rutz, S.; Kayagaki, N.; Phung, Q.T.; Eidenschenk, C.; Noubade, R.; Wang, X.; Lesch, J.; Lu, R.; Newton, K.; Huang, O.W.; et al. Deubiquitinase DUBA is a post-translational brake on interleukin-17 production in T cells. Nature 2015, 518, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Kathania, M.; Khare, P.; Zeng, M.; Cantarel, B.; Zhang, H.; Ueno, H.; Venuprasad, K. Itch inhibits IL-17-mediated colon inflammation and tumorigenesis by ROR-γt ubiquitination. Nat. Immunol. 2016, 17, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Wang, F.; Ma, J.; Sen, S.; Zhang, J.; Gwack, Y.; Zhou, Y.; Sun, Z. Ubiquitination of RORγt at Lysine 446 Limits Th17 Differentiation by Controlling Coactivator Recruitment. J. Immunol. 2016, 197, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Yang, J.; Wang, X.; Wu, Q.; Yin, S.; Li, Z.; Zhang, J.; Xing, Y.; Chen, Z.; Tsun, A.; et al. The E3 deubiquitinase USP17 is a positive regulator of retinoic acid-related orphan nuclear receptor γt (RORγt) in Th17 cells. J. Biol. Chem. 2014, 289, 25546–25555. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Hodge, J.A.; Kawabata, T.T.; Krishnaswami, S.; Clark, J.D.; Telliez, J.; Dowty, M.E.; Menon, S.; Lamba, M.; Zwillich, S.; Hodge, J.A.; et al. The mechanism of action of tofacitinib—An oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2016, 34, 318–328. [Google Scholar] [PubMed]
- Meyer, D.M.; Jesson, M.I.; Li, X.; Elrick, M.M.; Funckes-shippy, C.L.; Warner, J.D.; Gross, C.J.; Dowty, M.E.; Ramaiah, S.K.; Hirsch, J.L.; et al. Anti-inflammatory activity and neutrophil reductions mediated by the JAK1/JAK3 inhibitor, CP-690,550, in rat adjuvant-induced arthritis. J. Inflamm. 2010, 7, 41. [Google Scholar] [CrossRef] [PubMed]
- Boyle, D.L.; Soma, K.; Hodge, J.; Kavanaugh, A.; Mandel, D.; Mease, P.; Shurmur, R.; Singhal, A.K.; Wei, N.; Rosengren, S.; et al. The JAK inhibitor tofacitinib suppresses synovial JAK1-STAT signalling in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Ghoreschi, K.; Jesson, M.I.; Li, X.; Jamie, L.; Ghosh, S.; Alsup, J.W.; Warner, J.D.; Tanaka, M.; Steward-tharp, S.M.; Gadina, M.; et al. Modulation of Innate and Adaptive Immune Responses by Tofacitinib (CP-690,550). J. Immunol. 2011, 186, 4234–4243. [Google Scholar] [CrossRef] [PubMed]
- Bachelez, H.; van de Kerkhof, P.C.M.; Strohal, R.; Kubanov, A.; Valenzuela, F.; Lee, J.; Yakusevich, V. Tofacitinib versus etanercept or placebo in moderate-to-severe chronic plaque psoriasis: A phase 3 randomised non-inferiority trial. Lancet 2015, 386, 552–561. [Google Scholar] [CrossRef]
- Valenzuela, F.; Paul, C.; Mallbris, L.; Tan, H.; Papacharalambous, J.; Valdez, H.; Mamolo, C. Tofacitinib versus etanercept or placebo in patients with moderate to severe chronic plaque psoriasis: Patient-reported outcomes from a Phase 3 study. JEADV 2016, 30, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Krueger, J.G.; Feldman, S.R. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: Long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J. Am. Acad. Dermatol. 2016, 74, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; McGarry, T.; Orr, C.; McCormick, J.; Veale, D.J.; Fearon, U. Tofacitinib regulates synovial inflammation in psoriatic arthritis, inhibiting STAT activation and induction of negative feedback inhibitors. Ann. Rheum. Dis. 2016, 75, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Izzo, R.; Bevivino, G.; Monteleone, G. Tofacitinib for the treatment of ulcerative colitis. Expert Opin. Investig. Drugs 2016, 8, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Grisham, M.; Hodge, J.; Telliez, J. JAK inhibition using tofacitinib for inflammatory bowel disease treatment: A hub for multiple inflammatory cytokines. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G155–G162. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kwok, S.K.; Lim, M.A.; Kim, E.K.; Ryu, J.G.; Kim, S.M.; Oh, H.J.; Ju, J.H.; Park, S.H.; Kim, H.Y.; et al. La STA-21, a promising STAT-3 inhibitor that reciprocally regulates Th17 and Treg cells, inhibits osteoclastogenesis in mice and humans and alleviates autoimmune inflammation in an experimental model of rheumatoid arthritis. Arthritis Rheumatol. 2014, 66, 918–929. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yan, S.; Chen, J.; Luo, X.; Li, P. JAK1-STAT3 blockade by JAK inhibitor SHR0302 attenuates inflammatory responses of adjuvant-induced arthritis rats and decreases Th17 and total B cells. Jt. Bone Spine 2016, 83, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.J.; Lee, J.; Lee, S.Y.; Kim, E.K.; Moon, Y.M.; Jung, Y.O.; Park, S.H.; Cho, M.L. EGCG attenuates autoimmune arthritis by inhibition of STAT3 and HIF-1α pathway with Th17/Treg control. PLoS ONE 2014, 9, e86062. [Google Scholar]
- Astry, B.; Venkatesha, S.H.; Laurence, A.; Christensen-Quick, A.; Garzino-demo, A.; Frieman, M.B.; O’Shea, J.J.; Moudgil, K.D. Celastrol, a Chinese herbal compound, controls autoimmune inflammation by altering the balance of pathogenic and regulatory T cells in the target organ. Clin. Immunol. 2015, 157, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Jhun, J.Y.; Moon, S.J.; Yoon, B.Y.; Byun, J.K.; Kim, E.K.; Yang, E.J.; Ju, J.H.; Hong, Y.S.; Min, J.K.; Park, S.H.; et al. La Grape seed proanthocyanidin extract-mediated regulation of STAT3 proteins contributes to Treg differentiation and attenuates inflammation in a murine model of obesity-associated arthritis. PLoS ONE 2013, 8, e78843. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.J.; Park, J.S.; Woo, Y.J.; Lim, M.A.; Kim, S.M.; Lee, S.Y.; Kim, E.K.; Lee, H.J.; Lee, W.S.; Park, S.H.; et al. Rebamipide suppresses collagen-induced arthritis through reciprocal regulation of Th17/Treg cell differentiation and heme oxygenase 1 induction. Arthritis Rheumatol. 2014, 66, 874–885. [Google Scholar] [CrossRef] [PubMed]
- Park, M.K.; Park, J.S.; Park, E.M.; Lim, M.A.; Kim, S.M.; Lee, D.G.; Baek, S.Y.; Yang, E.J.; Woo, J.W.; Lee, J.; et al. Halofuginone ameliorates autoimmune arthritis in mice by regulating the balance between Th17 and treg cells and inhibiting osteoclastogenesis. Arthritis Rheumatol. 2014, 66, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tian, T.; Gao, J.; Liu, X.; Hou, H.; Cao, R.; Li, B.; Quan, M.; Guo, L. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J. Neuroimmunol. 2016, 292, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T. Saka Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Ohata, J.; Miura, T.; Johnson, T.A.; Hori, S.; Ziegler, S.F.; Kohsaka, H. Enhanced efficacy of regulatory T cell transfer against increasing resistance, by elevated Foxp3 expression induced in arthritic murine hosts. Arthritis Rheum. 2007, 56, 2947–2956. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lopes, J.E.; Chong, M.M.W.; Ivanov, I.I.; Min, R.; Victora, G.D.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits Th17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Quaglino, P.; Bergallo, M.; Ponti, R.; Barberio, E.; Cicchelli, S.; Buffa, E.; Comessatti, A.; Costa, C.; Terlizzi, M.E.; Astegiano, S.; et al. Th1, Th2, Th17 and regulatory T cell pattern in psoriatic patients: Modulation of cytokines and gene targets induced by etanercept treatment and correlation with clinical response. Dermatology 2011, 223, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Lina, C.; Conghua, W.; Nan, L.; Ping, Z. Combined treatment of etanercept and MTX reverses Th1/Th2, Th17/Treg imbalance in patients with rheumatoid arthritis. J. Clin. Immunol. 2011, 31, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.D.; Merrill, P.T.; Jaffe, G.J.; Dick, A.D.; Kurup, S.K.; Sheppard, J.; Schlaen, A.; Pavesio, C.; Cimino, L.; van Calster, J.; et al. Adalimumab for prevention of uveitic flare in patients with inactive non-infectious uveitis controlled by corticosteroids (VISUAL II): A multicentre, double-masked, randomised, placebo-controlled phase 3 trial. Lancet 2016, 6736, 1–10. [Google Scholar] [CrossRef]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef]
- Emery, P. Optimizing outcomes in patients with rheumatoid arthritis and an inadequate response to anti-TNF treatment. Rheumatology 2012, 51, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Sansom, D.M.; Walker, L.S.K. The role of CD28 and cytotoxic T-lymphocyte antigen-4 (CTLA-4) in regulatory T-cell biology. Immunol. Rev. 2006, 212, 131–148. [Google Scholar] [CrossRef] [PubMed]
- Bonelli, M.; Göschl, L.; Blüml, S.; Karonitsch, T.; Hirahara, K.; Ferner, E.; Steiner, C.W.; Steiner, G.; Smolen, J.S.; Scheinecker, C. Abatacept (CTLA-4Ig) treatment reduces T cell apoptosis and regulatory T cell suppression in patients with rheumatoid arthritis. Rheumatology 2016, 55, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Sfikakis, P.P.; Souliotis, V.L.; Fragiadaki, K.G.; Moutsopoulos, H.M.; Boletis, J.N.; Theofilopoulos, A.N. Increased expression of the FoxP3 functional marker of regulatory T cells following B cell depletion with rituximab in patients with lupus nephritis. Clin. Immunol. 2007, 123, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Floess, S.; Freyer, J.; Siewert, C.; Baron, U.; Olek, S.; Polansky, J.; Schlawe, K.; Chang, H.D.; Bopp, T.; Schmitt, E.; et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007, 5, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.; Zhang, N.; van der Touw, W.; Ding, Y.; Ju, W.; Bottinger, E.P.; Reid, S.P.; Levy, D.E.; Bromberg, J.S. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 2009, 182, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Amjadi, P.; Green, P.; Read, J.E.; Brennan, F.; Gregory, B.; Williams, R.O. Methotrexate restores regulatory T cell function through demethylation of the FoxP3 upstream enhancer in patients with rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.-J.; Baker, J.; Leveson-Gower, D.B.; Smith, A.T.; Sega, E.I.; Negrin, R.S. Rapamycin and IL-2 reduce lethal acute graft-versus-host disease associated with increased expansion of donor type CD4+CD25+Foxp3+ regulatory T cells. Blood 2011, 118, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Golovina, T.N.; Mikheeva, T.; Brusko, T.M.; Blazar, B.R.; Bluestone, J.A.; Riley, J.L. Retinoic acid and rapamycin differentially affect and synergistically promote the ex vivo expansion of natural human T regulatory cells. PLoS ONE 2011, 6, e15868. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tao, R.; Hancock, W.W. Using histone deacetylase inhibitors to enhance Foxp3(+) regulatory T-cell function and induce allograft tolerance. Immunol. Cell Biol. 2009, 87, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, L.; Predina, J.; Han, R.; Beier, U.H.; Wang, L.C.; Kapoor, V.; Bhatti, T.R.; Akimova, T.; Singhal, S.; et al. Inhibition of p300 impairs Foxp3+ T regulatory cell function and promotes antitumor immunity. Nat. Med. 2013, 19, 1173–1177. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- De Zoeten, E.F.; Wang, L.; Butler, K.; Beier, U.H.; Akimova, T.; Sai, H.; Bradner, J.E.; Mazitschek, R.; Kozikowski, A.P.; Matthias, P.; et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3+ T-regulatory cells. Mol. Cell. Biol. 2011, 31, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Akimova, T.; Liu, Y.; Wang, L.; Hancock, W.W. Histone/protein deacetylases control Foxp3 expression and the heat shock response of T-regulatory cells. Curr. Opin. Immunol. 2011, 23, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jiao, J.; Wang, L.; O’Brien, S.; Newick, K.; Wang, L.C.S.; Falkensammer, E.; Liu, Y.; Han, R.; Kapoor, V.; et al. HDAC5 controls the functions of Foxp3+ T-regulatory and CD8+ T cells. Int. J. Cancer 2016, 138, 2477–2486. [Google Scholar] [CrossRef] [PubMed]
- Lainé, A.; Martin, B.; Luka, M.; Mir, L.; Auffray, C.; Lucas, B.; Bismuth, G. Foxo1 Is a T Cell−Intrinsic Inhibitor of the RORγt-Th17 Program. J. Immunol. 2015, 195, 1791–1803. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, K.; Gonzalez-Martin, A.; Kim, B.; Jin, H.; Jin, W.; Xu, W.; Sabouri-Ghomi, M.; Xu, S.; Zheng, P.; Xiao, C.; et al. The MicroRNA-183–96–182 Cluster Promotes T Helper 17 Cell Pathogenicity by Negatively Regulating Transcription Factor Foxo1 Expression. Immunity 2016, 44, 1284–1298. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Ping, C.; Shang, X.; Tian, J.; Zhao, S.; Li, L.; Fang, S.; Sun, W.; Zhao, Y.; Li, Z.; et al. MicroRNA 182 inhibits CD4+CD25+Foxp3+ Treg differentiation in experimental autoimmune encephalomyelitis. Clin. Immunol. 2016, 173, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, Y.; Gu, L. Dehydroabietic acid reverses TNF-α-induced the activation of FOXO1 and suppression of TGF-β1/Smad signaling in human adult dermal fibroblasts. Int. J. Clin. Exp. Pathol. 2014, 7, 8616–8626. [Google Scholar] [PubMed]
- Liu, Y.; Luo, W.; Chen, S. Comparison of gene expression profiles reveals aberrant expression of FOXO1, Aurora A/B and EZH2 in lesional psoriatic skins. Mol. Biol. Rep. 2011, 38, 4219–4224. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Su, X.; Yang, X.; Hu, C.; Li, B.; Lv, Y.; Shuai, Y.; Jing, H.; Deng, Z.; Jin, Y. TNF-α Inhibits FoxO1 by Upregulating miR-705 to Aggravate Oxidative Damage in Bone Marrow- Derived Mesenchymal Stem Cells during Osteoporosis. Stem Cells 2016, 34, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, M.R.; Evans, J.G.; Singh, A.; Moore, S.; Warnes, G.; Isenberg, D.A.; Mauri, C.; Wca, L. Compromised Function of Regulatory T Cells in Rheumatoid Arthritis and Reversal by Anti-TNFα Therapy. J. Exp. Med. 2004, 200, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xia, L.; Lu, J.; Xiao, W. Infliximab Reduces the Frequency of Interleukin 17-Producing Cells and the Amounts of Interleukin 17 in Patients With Rheumatoid Arthritis. J. Investig. Med. 2010, 58, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Aravena, O.; Pesce, B.; Soto, L.; Orrego, N.; Sabugo, F.; Wurmann, P.; Molina, M.C.; Alfaro, J.; Cuchacovich, M.; Aguillón, J.C.; et al. Anti-TNF therapy in patients with rheumatoid arthritis decreases Th1 and Th17 cell populations and expands IFN-γ-producing NK cell and regulatory T cell subsets. Immunobiology 2011, 216, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, A.L.; Kitz, A.; Wu, C.; Lowther, D.E.; Rodriguez, D.M.; Vudattu, N.; Deng, S.; Herold, K.C.; Kuchroo, V.K.; Kleinewietfeld, M.; et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J. Clin. Investig. 2015, 125, 4212–4222. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.T.; Liao, W.; Dadi, S.; Toure, A.; Li, M.O. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature 2016, 529, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkhurst, C.N.; Newberry, K.M.; et al. A validated regulatory network for Th17 cell specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.S.; Gupta, S.; Chang, E.; Song, L.; Stirzaker, R.A.; Liao, J.K.; Bhagat, G.; Pernis, A.B. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Investig. 2010, 120, 3280–3295. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Xu, H.; Liang, L.; Zhan, Z.; Yang, X.; Yu, X.; Ye, Y.; Sun, L. Antiinflammatory effect of Rho kinase blockade via inhibition of NF-kappaB activation in rheumatoid arthritis. Arthritis Rheum. 2008, 58, 3366–3376. [Google Scholar] [CrossRef] [PubMed]
- Isgro, J.; Gupta, S.; Jacek, E.; Pavri, T.; Duculan, R.; Kim, M.; Kirou, K.A.; Salmon, J.E.; Pernis, A.B. Enhanced Rho-Associated Protein Kinase Activation in Patients With Systemic Lupus Erythematosus. Arthritis Rheum. 2013, 65, 1592–1602. [Google Scholar] [CrossRef] [PubMed]
- Zanin-Zhorov, A.; Weiss, J.M.; Nyuydzefe, M.S.; Chen, W.; Scher, J.U.; Mo, R. Selective oral ROCK2 inhibitor down-regulates IL-21 and IL-17 secretion in human T cells via STAT3-dependent mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 16814–16819. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.; Paz, K.; Du, J.; Reichenbach, D.K.; Taylor, P.A.; Panoskaltsis-mortari, A.; Vulic, A.; Luznik, L.; Macdonald, K.K.P.; Hill, G.R.; et al. Targeted Rho-associated kinase 2 inhibition suppresses murine and human chronic GVHD through a Stat3-dependent mechanism. Blood 2016, 127, 2144–2155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, X.; Ding, Z.; Yang, X.; Zhang, H. The Therapeutic Potential of Rho Kinase Inhibitor Fasudil Derivative FaD-1 in Experimental Autoimmune Encephalomyelitis. J. Mol. Neurosci. 2015, 55, 725–732. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yu, J.; Xin, Y.; Feng, L.; Chai, Z.; Liu, J.; Zhang, H. Protective effect of a novel Rho kinase inhibitor WAR-5 in experimental autoimmune encephalomyelitis by modulating inflammatory response and neurotrophic factors. Exp. Mol. Pathol. 2015, 99, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Joetham, A.; Ohnishi, H.; Okamoto, M.; Takeda, K.; Schedel, M.; Domenico, J.; Dakhama, A.; Gelfand, E.W. Loss of T Regulatory Cell Suppression following Signaling through Glucocorticoid-induced Tumor Necrosis Receptor (GITR) Is Dependent on c-Jun N-terminal Kinase Activation. J. Biol. Chem. 2012, 287, 17100–17108. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shi, Y.; Yang, M.; Ma, J.; Tian, J.; Chen, J.; Mao, C.; Jiao, Z.; Ko, K.; Lu, L. Glucocorticoid-Induced Tumor Necrosis Factor Receptor Family-Related Protein Exacerbates Collagen-Induced Arthritis by Enhancing the Expansion of Th17 Cells. Am. J. Pathol. 2012, 180, 1059–1067. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Tian, J.; Ma, J.; Wang, J.; Qi, C.; Rui, K.; Wang, Y.; Xu, H.; Lu, L.; Wang, S. GITRL modulates the activities of p38 MAPK and STAT3 to promote Th17 cell differentiation in autoimmune arthritis. Oncotarget 2015, 7. [Google Scholar] [CrossRef]
- Li, L.; Wen, W.; Jia, R.; Li, Y.; Liu, X.; Sun, X. GITRL is associated with increased autoantibody production in patients with rheumatoid arthritis. Clin. Rheumatol. 2016, 35, 2195–2202. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.B.; Cheng, T.; Chindalore, V.; Damjanov, N.; Burgos-vargas, R.; Delora, P.; Zimany, K.; Travers, H.; Caulfield, J.P. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009, 60, 335–344. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fasching, P.; Stradner, M.; Graninger, W.; Dejaco, C.; Fessler, J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules 2017, 22, 134. https://doi.org/10.3390/molecules22010134
Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules. 2017; 22(1):134. https://doi.org/10.3390/molecules22010134
Chicago/Turabian StyleFasching, Patrizia, Martin Stradner, Winfried Graninger, Christian Dejaco, and Johannes Fessler. 2017. "Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders" Molecules 22, no. 1: 134. https://doi.org/10.3390/molecules22010134
APA StyleFasching, P., Stradner, M., Graninger, W., Dejaco, C., & Fessler, J. (2017). Therapeutic Potential of Targeting the Th17/Treg Axis in Autoimmune Disorders. Molecules, 22(1), 134. https://doi.org/10.3390/molecules22010134