Regulation of T Cell Activation and Differentiation by Extracellular Vesicles and Their Pathogenic Role in Systemic Lupus Erythematosus and Multiple Sclerosis



{kind=link}
Abstract
:1. Introduction
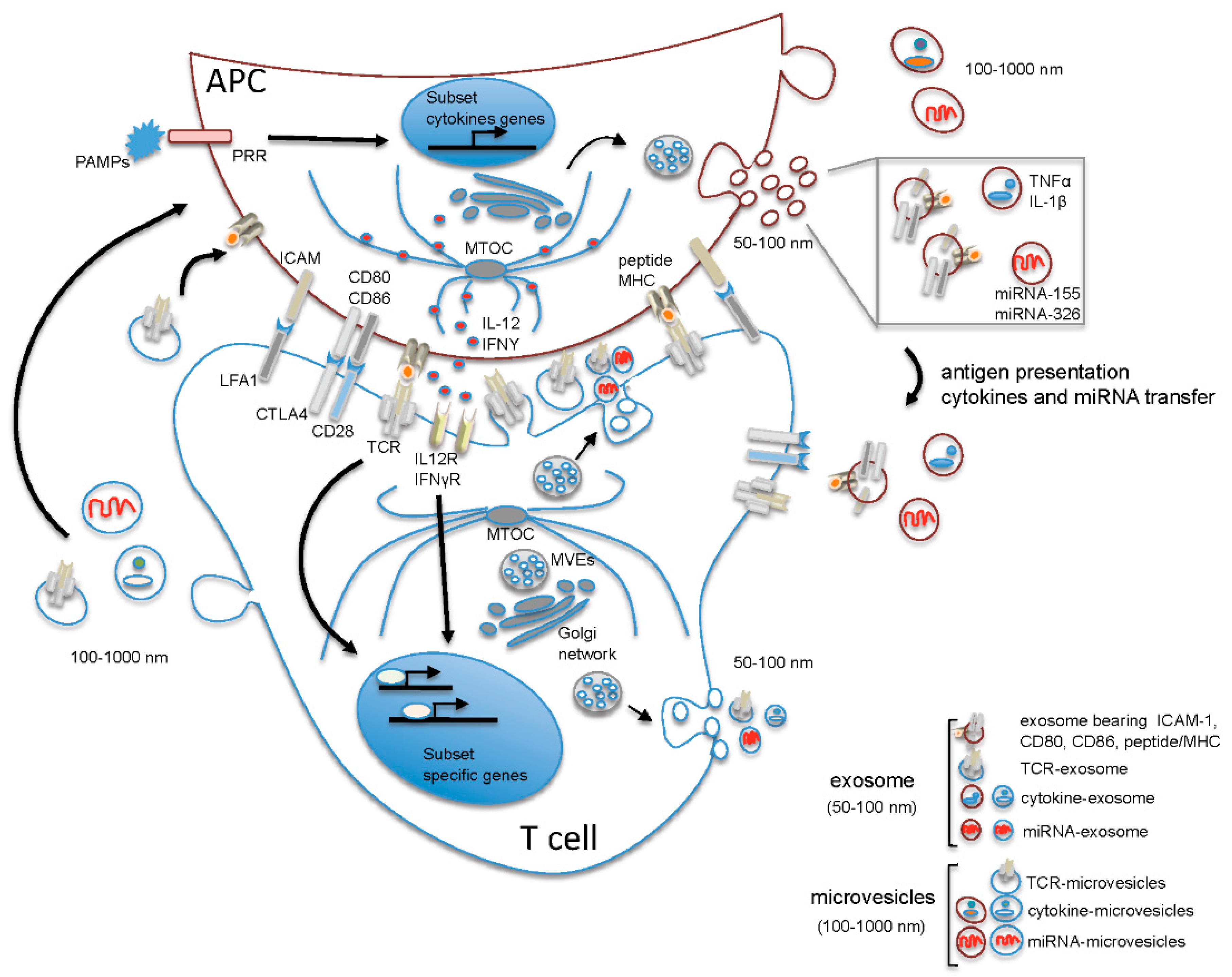
2. The Microenvironment of the Immune Synapse Cleft Controls Helper T Cell Differentiation
3. Role of EVs in Th17/Treg Cell Differentiation and Function
4. Contribution of EVs to Autoimmune Disease
4.1. Systemic Lupus Erythematosus
4.2. Multiple Sclerosis
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J.M.; Ramirez-Santana, C.; Alzate, M.A.; Molano-Gonzalez, N.; Rojas-Villarraga, A. The Autoimmune Ecology. Front. Immunol. 2016, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Mercadante, E.R.; Lorenz, U.M. Breaking Free of Control: How Conventional T Cells Overcome Regulatory T Cell Suppression. Front. Immunol. 2016, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell. Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Yamane, H.; Paul, W.E. Early signaling events that underlie fate decisions of naive CD4(+) T cells toward distinct T-helper cell subsets. Immunol. Rev. 2013, 252, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Van Panhuys, N. TCR Signal Strength Alters T-DC Activation and Interaction Times and Directs the Outcome of Differentiation. Front. Immunol. 2016, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Van Panhuys, N.; Klauschen, F.; Germain, R.N. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity 2014, 41, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Pulecio, J.; Petrovic, J.; Prete, F.; Chiaruttini, G.; Lennon-Dumenil, A.M.; Desdouets, C.; Gasman, S.; Burrone, O.R.; Benvenuti, F. Cdc42-mediated MTOC polarization in dendritic cells controls targeted delivery of cytokines at the immune synapse. J. Exp. Med. 2010, 207, 2719–2732. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.A.; Irvine, D.J.; Schreiber, R.; Glimcher, L.H. A role for the immunological synapse in lineage commitment of CD4 lymphocytes. Nature 2004, 431, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Lillemeier, B.F.; Kuhns, M.S.; Chen, D.S.; Davis, M.M. T cells use two directionally distinct pathways for cytokine secretion. Nat. Immunol. 2006, 7, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Shiner, E.K.; Holbrook, B.C.; Alexander-Miller, M.A. CD4+ T cell subset differentiation and avidity setpoint are dictated by the interplay of cytokine and antigen mediated signals. PLoS ONE 2014, 9, e100175. [Google Scholar] [CrossRef] [PubMed]
- Purvis, H.A.; Stoop, J.N.; Mann, J.; Woods, S.; Kozijn, A.E.; Hambleton, S.; Robinson, J.H.; Isaacs, J.D.; Anderson, A.E.; Hilkens, C.M. Low-strength T-cell activation promotes Th17 responses. Blood 2010, 116, 4829–4837. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.O.; Lee, J.B.; Chang, J. Cholera Toxin Promotes Th17 Cell Differentiation by Modulating Expression of Polarizing Cytokines and the Antigen-Presenting Potential of Dendritic Cells. PLoS ONE 2016, 11, e0157015. [Google Scholar] [CrossRef] [PubMed]
- Benvenuti, F. The Dendritic Cell Synapse: A Life Dedicated to T Cell Activation. Front. Immunol. 2016, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Chiaruttini, G.; Piperno, G.M.; Jouve, M.; De Nardi, F.; Larghi, P.; Peden, A.A.; Baj, G.; Müller, S.; Valitutti, S.; Galli, T.; et al. The SNARE VAMP7 Regulates Exocytic Trafficking of Interleukin-12 in Dendritic Cells. Cell. Rep. 2016, 14, 2624–2636. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, N.; Lankar, D.; Faure, F.; Regnault, A.; Dumont, C.; Raposo, G.; Hivroz, C. TCR activation of human T cells induces the production of exosomes bearing the TCR/CD3/zeta complex. J. Immunol. 2002, 168, 3235–3241. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, K.; Llodrá, J.; Roth, E.W.; Tsai, J.; Gordo, S.; Wucherpfennig, K.W.; Kam, L.C.; Stokes, D.L.; Dustin, M.L. Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 2014, 507, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 282. [Google Scholar] [CrossRef] [PubMed]
- Mittelbrunn, M.; Vicente-Manzanares, M.; Sánchez-Madrid, F. Organizing polarized delivery of exosomes at synapses. Traffic 2015, 16, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Nicco, C.; Lombard, B.; Véron, P.; Raposo, G.; Batteux, F.; Amigorena, S.; Théry, C. ICAM-1 on exosomes from mature dendritic cells is critical for efficient naive T-cell priming. Blood 2005, 106, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Duban, L.; Segura, E.; Véron, P.; Lantz, O.; Amigorena, S. Indirect activation of naïve CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002, 3, 1156–1162. [Google Scholar] [CrossRef] [PubMed]
- Chaput, N.; Théry, C. Exosomes: immune properties and potential clinical implementations. Semin. Immunopathol. 2011, 33, 419–440. [Google Scholar] [CrossRef]
- Greening, D.W.; Gopal, S.K.; Xu, R.; Simpson, R.J.; Chen, W. Exosomes and their roles in immune regulation and cancer. Semin. Cell. Dev. Biol. 2015, 40, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 2009, 30, 636–645. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Coomes, S.M.; Pelly, V.S.; Czieso, S.; Papayannopoulos, V.; Tolmachova, T.; Seabra, M.C.; Wilson, M.S. MicroRNA-containing T-regulatory-cell-derived exosomes suppress pathogenic T helper 1 cells. Immunity 2014, 41, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Smyth, L.A.; Ratnasothy, K.; Tsang, J.Y.; Boardman, D.; Warley, A.; Lechler, R.; Lombardi, G. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur. J. Immunol. 2013, 43, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhang, W.; Yang, F.; Yu, L.; Yu, Z.; Pan, J.; Wang, L.; Cao, X.; Wang, J. Immunosuppressive exosomes from TGF-β1 gene-modified dendritic cells attenuate Th17-mediated inflammatory autoimmune disease by inducing regulatory T cells. Cell. Res. 2012, 22, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Shen, Y.; Guo, D.; Yang, D.; Liu, J.; Fei, X.; Yang, Y.; Zhang, B.; Lin, Z.; Yang, F.; Wang, X.; Wang, K.; Wang, J.; Cai, Z. EpCAM-dependent extracellular vesicles from intestinal epithelial cells maintain intestinal tract immune balance. Nat. Commun. 2016, 7, 13045. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yang, F.; Jiang, L.; Chen, Y.; Wang, K.; Xu, F.; Wei, Y.; Cao, X.; Wang, J.; Cai, Z. Exosomes with membrane-associated TGF-β1 from gene-modified dendritic cells inhibit murine EAE independently of MHC restriction. Eur. J. Immunol. 2013, 43, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Yamada, N.; Kuranaga, Y.; Kumazaki, M.; Shinohara, H.; Taniguchi, K.; Akao, Y. Colorectal cancer cell-derived extracellular vesicles induce phenotypic alteration of T cells into tumor-growth supporting cells with transforming growth factor-β1-mediated suppression. Oncotarget 2016, 7, 27033–27043. [Google Scholar] [CrossRef] [PubMed]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef] [PubMed]
- Mrizak, D.; Martin, N.; Barjon, C.; Jimenez-Pailhes, A.S.; Mustapha, R.; Niki, T.; Guigay, J.; Pancré, V.; de Launoit, Y.; Busson, P.; Moralès, O.; Delhem, N. Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory T cells. J. Natl. Cancer Inst. 2015, 107, 363. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of breast cancer cells mediated by transforming growth factor-β in exosomes from cancer cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [PubMed]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [PubMed]
- Baumjohann, D.; Ansel, K.M. MicroRNA-mediated regulation of T helper cell differentiation and plasticity. Nat. Rev. Immunol. 2013, 13, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Garo, L.P.; Murugaiyan, G. Contribution of MicroRNAs to autoimmune diseases. Cell. Mol. Life Sci. 2016, 73, 2041–2051. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Yang, Z.; Lu, N. From pathogenesis to clinical application: insights into exosomes as transfer vectors in cancer. J. Exp. Clin. Cancer Res. 2016, 35, 156. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.B.; Zhang, H.; Cai, T.T.; Liu, Y.N.; Ni, J.J.; He, J.; Peng, J.Y.; Chen, Q.Y.; Mo, H.Y.; Cui, J.; et al. Exosomal miR-24–3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J. Pathol. 2016, 240, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.B.; Li, Z.L.; Luo, D.H.; Huang, B.J.; Chen, Y.S.; Zhang, X.S.; Cui, J.; Zeng, Y.X.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439–5452. [Google Scholar] [CrossRef] [PubMed]
- Murugaiyan, G.; Beynon, V.; Mittal, A.; Joller, N.; Weiner, H.L. Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 2011, 187, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, Y.; Cui, W.; Li, M.; Li, B.; Guo, L. MicroRNA-155 modulates Th1 and Th17 cell differentiation and is associated with multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2014, 266, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.F.; Thai, T.H.; Calado, D.P.; Chaudhry, A.; Kubo, M.; Tanaka, K.; Loeb, G.B.; Lee, H.; Yoshimura, A.; Rajewsky, K.; Rudensky, A.Y. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity 2009, 30, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Huffaker, T.B.; Hu, R.; Runtsch, M.C.; Bake, E.; Chen, X.; Zhao, J.; Round, J.L.; Baltimore, D.; O’Connell, R.M. Epistasis between microRNAs 155 and 146a during T cell-mediated antitumor immunity. Cell. Rep. 2012, 2, 1697–1709. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Csak, T.; Momen-Heravi, F.; Lippai, D.; Kodys, K.; Catalano, D.; Satishchandran, A.; Ambros, V.; Szabo, G. Biodistribution and function of extracellular miRNA-155 in mice. Sci Rep. 2015, 5, 10721. [Google Scholar] [CrossRef] [PubMed]
- Mycko, M.P.; Cichalewska, M.; Machlanska, A.; Cwiklinska, H.; Mariasiewicz, M.; Selmaj, K.W. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2012, 109, E1248–E1257. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Liu, C.; Kang, J.; Zhao, G.; Ye, Z.; Huang, S.; Li, Z.; Wu, Z.; Pei, G. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 2009, 10, 1252–1259. [Google Scholar] [CrossRef]
- Nouraee, N.; Khazaei, S.; Vasei, M.; Razavipour, S.F.; Sadeghizadeh, M.; Mowla, S.J. MicroRNAs contribution in tumor microenvironment of esophageal cancer. Cancer Biomark. 2016, 16, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Buzas, E.I.; György, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Turpin, D.; Truchetet, M.E.; Faustin, B.; Augusto, J.F.; Contin-Bordes, C.; Brisson, A.; Blanco, P.; Duffau, P. Role of extracellular vesicles in autoimmune diseases. Autoimmun. Rev. 2016, 15, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Strauss, L.; Wieckowski, E.; Czystowska, M.; Albers, A.; Wang, Y.; Zeidler, R.; Lang, S.; Whiteside, T.L. Tumor-derived microvesicles in sera of patients with head and neck cancer and their role in tumor progression. Head Neck 2009, 31, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.D.; Gercel-Taylor, C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol. Oncol. 2008, 110, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.T.; Østergaard, O.; Stener, L.; Iversen, L.V.; Truedsson, L.; Gullstrand, B.; Jacobsen, S.; Heegaard, N.H. Increased IgG on cell-derived plasma microparticles in systemic lupus erythematosus is associated with autoantibodies and complement activation. Arthritis Rheum. 2012, 64, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Østergaard, O.; Nielsen, C.T.; Iversen, L.V.; Tanassi, J.T.; Knudsen, S.; Jacobsen, S.; Heegaard, N.H. Unique protein signature of circulating microparticles in systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 2680–2690. [Google Scholar] [CrossRef] [PubMed]
- Duffau, P.; Seneschal, J.; Nicco, C.; Richez, C.; Lazaro, E.; Douchet, I.; Bordes, C.; Viallard, J.F.; Goulvestre, C.; Pellegrin, J.L.; Weil, B.; Moreau, J.F.; Batteux, F.; Blanco, P. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Sci. Transl. Med. 2010, 2, 47ra63. [Google Scholar] [CrossRef] [PubMed]
- Ullal, A.J.; Reich, C.F.; Clowse, M.; Criscione-Schreiber, L.G.; Tochacek, M.; Monestier, M.; Pisetsky, D.S. Microparticles as antigenic targets of antibodies to DNA and nucleosomes in systemic lupus erythematosus. J. Autoimmun. 2011, 36, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Sally, B.; D’Agati, V.; Martinez-Ortiz, W.; Özçakar, Z.B.; David, J.; Rashidfarrokhi, A.; Yeste, A.; Panea, C.; Chida, A.S.; et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- Wilber, A.; O’Connor, T.P.; Lu, M.L.; Karimi, A.; Schneider, M.C. Dnase1l3 deficiency in lupus-prone MRL and NZB/W F1 mice. Clin. Exp. Immunol. 2003, 134, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.T.; Østergaard, O.; Rekvig, O.P.; Sturfelt, G.; Jacobsen, S.; Heegaard, N.H. Galectin-3 binding protein links circulating microparticles with electron dense glomerular deposits in lupus nephritis. Lupus 2015, 24, 1150–1160. [Google Scholar] [CrossRef] [PubMed]
- Ulivieri, C.; Fanigliulo, D.; Masi, G.; Savino, M.T.; Gamberucci, A.; Pelicci, P.G.; Baldari, C.T. p66Shc is a negative regulator of FcεRI-dependent signaling in mast cells. J. Immunol. 2011, 186, 5095–5106. [Google Scholar] [CrossRef] [PubMed]
- Finetti, F.; Pellegrini, M.; Ulivieri, C.; Savino, M.T.; Paccagnini, E.; Ginanneschi, C.; Lanfrancone, L.; Pelicci, P.G.; Baldari, C.T. The proapoptotic and antimitogenic protein p66SHC acts as a negative regulator of lymphocyte activation and autoimmunity. Blood 2008, 111, 5017–5027. [Google Scholar] [CrossRef] [PubMed]
- Masi, G.; Mercati, D.; Vannuccini, E.; Paccagnini, E.; Riparbelli, M.G.; Lupetti, P.; Pelicci, P.G.; Baldari, C.T.; Ulivieri, C. p66Shc regulates vesicle-mediated secretion in mast cells by affecting F-actin dynamics. J. Leukoc. Biol. 2014, 95, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kebir, H.; Kreymborg, K.; Ifergan, I.; Dodelet-Devillers, A.; Cayrol, R.; Bernard, M.; Giuliani, F.; Arbour, N.; Becher, B.; Prat, A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 2007, 13, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- Tsuge, M.; Yasui, K.; Ichiyawa, T.; Saito, Y.; Nagaoka, Y.; Yashiro, M.; Yamashita, N.; Morishima, T. Increase of tumor necrosis factor-alpha in the blood induces early activation of matrix metalloproteinase-9 in the brain. Microbiol. Immunol. 2010, 54, 417–424. [Google Scholar]
- Minagar, A.; Long, A.; Ma, T.; Jackson, T.H.; Kelley, R.E.; Ostanin, D.V.; Sasaki, M.; Warren, A.C.; Jawahar, A.; Cappell, B.; Alexander, J.S. Interferon (IFN)-beta 1a and IFN-beta 1b block IFN-gamma-induced disintegration of endothelial junction integrity and barrier. Endothelium 2003, 10, 299–307. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, K.; Zhang, X.; Zhang, J.; Xu, B. ATP Induces Disruption of Tight Junction Proteins via IL-1 Beta-Dependent MMP-9 Activation of Human Blood-Brain Barrier In Vitro. Neural. Plast. 2016, 2016, 8928530. [Google Scholar] [PubMed]
- Marcos-Ramiro, B.; Oliva Nacarino, P.; Serrano-Pertierra, E.; Blanco-Gelaz, M.A.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Tuñón, A.; López-Larrea, C.; Millán, J.; et al. Microparticles in multiple sclerosis and clinically isolated syndrome: effect on endothelial barrier function. BMC Neurosci. 2014, 15, 110. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Jy, W.; Jimenez, J.J.; Sheremata, W.A.; Mauro, L.M.; Mao, W.W.; Horstman, L.L.; Ahn, Y.S. Elevated plasma endothelial microparticles in multiple sclerosis. Neurology 2001, 56, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Sáenz-Cuesta, M.; Irizar, H.; Castillo-Triviño, T.; Muñoz-Culla, M.; Osorio-Querejeta, I.; Prada, A.; Sepúlveda, L.; López-Mato, M.P.; López de Munain, A.; Comabella, M.; et al. Circulating microparticles reflect treatment effects and clinical status in multiple sclerosis. Biomark. Med. 2014, 8, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Sbai, O.; Ould-Yahoui, A.; Ferhat, L.; Gueye, Y.; Bernard, A.; Charrat, E.; Mehanna, A.; Risso, J.J.; Chauvin, J.P.; Fenouillet, E.; et al. Differential vesicular distribution and trafficking of MMP-2, MMP-9, and their inhibitors in astrocytes. Glia 2010, 58, 344–366. [Google Scholar] [CrossRef] [PubMed]
- Verderio, C.; Muzio, L.; Turola, E.; Bergami, A.; Novellino, L.; Ruffini, F.; Riganti, L.; Corradini, I.; Francolini, M.; Garzetti, L.; et al. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 2012, 72, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Pravettoni, E.; Colombo, A.; Schenk, U.; Möller, T.; Matteoli, M.; Verderio, C. Astrocyte-derived ATP induces vesicle shedding and IL-1 beta release from microglia. J. Immunol. 2005, 174, 7268–7277. [Google Scholar] [CrossRef] [PubMed]
- Perry, D.M.; Newcomb, B.; Adada, M.; Wu, B.X.; Roddy, P.; Kitatani, K.; Siskind, L.; Obeid, L.M.; Hannun, Y.A. Defining a role for acid sphingomyelinase in the p38/interleukin-6 pathway. J. Biol Chem. 2014, 289, 22401–22412. [Google Scholar] [CrossRef] [PubMed]
- Lopes Pinheiro, M.A.; Kroon, J.; Hoogenboezem, M.; Geerts, D.; van Het Hof, B.; van der Pol, S.M.; van Buul, J.D.; de Vries, H.E. Acid Sphingomyelinase-Derived Ceramide Regulates ICAM-1 Function during T Cell Transmigration across Brain Endothelial Cells. J. Immunol. 2016, 196, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Carandini, T.; Colombo, F.; Finardi, A.; Casella, G.; Garzetti, L.; Verderio, C.; Furlan, R. Microvesicles: What is the Role in Multiple Sclerosis? Front. Neurol. 2015, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Grapp, M.; Wrede, A.; Schweizer, M.; Hüwel, S.; Galla, H.J.; Snaidero, N.; Simons, M.; Bückers, J.; Low, P.S.; Urlaub, H.; et al. Choroid plexus transcytosis and exosome shuttling deliver folate into brain parenchyma. Nat. Commun. 2013, 4, 2123. [Google Scholar] [CrossRef] [PubMed]
- Balusu, S.; Van Wonterghem, E.; De Rycke, R.; Raemdonck, K.; Stremersch, S.; Gevaert, K.; Brkic, M.; Demeestere, D.; Vanhooren, V.; Hendrix, A.; et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol. Med. 2016, 8, 1162–1183. [Google Scholar] [CrossRef] [PubMed]
- Wheway, J.; Latham, S.L.; Combes, V.; Grau, G.E. Endothelial microparticles interact with and support the proliferation of T cells. J. Immunol. 2014, 193, 3378–3387. [Google Scholar] [CrossRef] [PubMed]
- Potolicchio, I.; Carven, G.J.; Xu, X.; Stipp, C.; Riese, R.J.; Stern, L.J.; Santambrogio, L. Proteomic analysis of microglia-derived exosomes: metabolic role of the aminopeptidase CD13 in neuropeptide catabolism. J. Immunol 2005, 175, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Jagot, F.; Davoust, N. Is It worth Considering Circulating microRNAs in Multiple Sclerosis? Front. Immunol 2016, 7, 129. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Ridder, K.; Keller, S.; Dams, M.; Rupp, A.K.; Schlaudraff, J.; Del Turco, D.; Starmann, J.; Macas, J.; Karpova, D.; Devraj, K.; et al. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol. 2014, 12, e1001874. [Google Scholar] [CrossRef] [PubMed]
- Jarmalavičiūtė, A.; Pivoriūnas, A. Exosomes as a potential novel therapeutic tools against neurodegenerative diseases. Pharmacol. Res. 2016, 113, 816–822. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulivieri, C.; Baldari, C.T. Regulation of T Cell Activation and Differentiation by Extracellular Vesicles and Their Pathogenic Role in Systemic Lupus Erythematosus and Multiple Sclerosis. Molecules 2017, 22, 225. https://doi.org/10.3390/molecules22020225
Ulivieri C, Baldari CT. Regulation of T Cell Activation and Differentiation by Extracellular Vesicles and Their Pathogenic Role in Systemic Lupus Erythematosus and Multiple Sclerosis. Molecules. 2017; 22(2):225. https://doi.org/10.3390/molecules22020225
Chicago/Turabian StyleUlivieri, Cristina, and Cosima T. Baldari. 2017. "Regulation of T Cell Activation and Differentiation by Extracellular Vesicles and Their Pathogenic Role in Systemic Lupus Erythematosus and Multiple Sclerosis" Molecules 22, no. 2: 225. https://doi.org/10.3390/molecules22020225
APA StyleUlivieri, C., & Baldari, C. T. (2017). Regulation of T Cell Activation and Differentiation by Extracellular Vesicles and Their Pathogenic Role in Systemic Lupus Erythematosus and Multiple Sclerosis. Molecules, 22(2), 225. https://doi.org/10.3390/molecules22020225