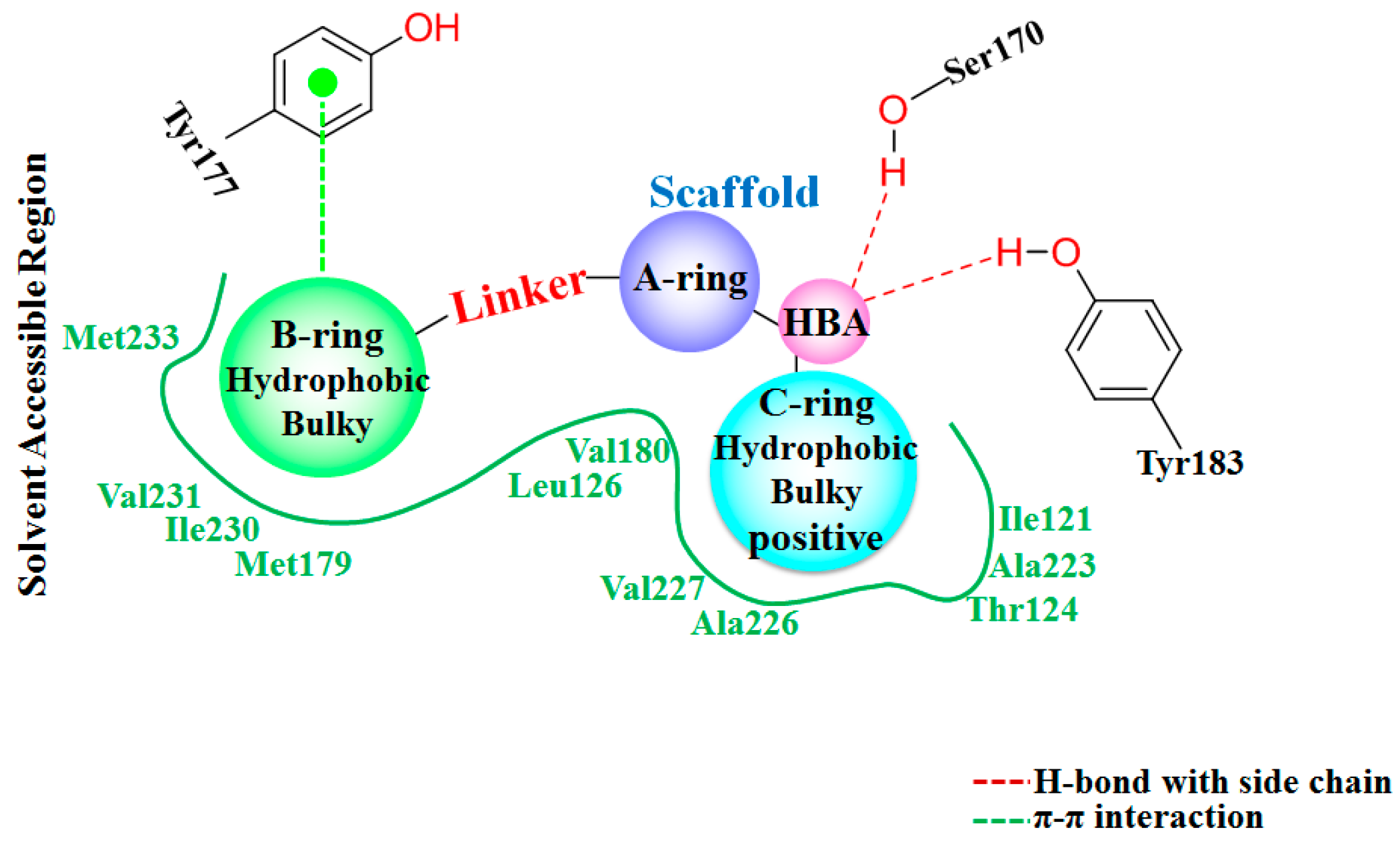
3.1. Docking Analysis
In this study, flexible docking calculations were performed on inhibitors of 11β-HSD1 to predict their potential binding conformations using the Surflex-Dock procedure. As shown in
Figure S2a in the Supplementary Materials, the obtained docking pose of
1 was similar to its experimental conformation in the co-crystal structure with a root mean-square deviation (RMSD) of 0.39 Å, indicating the high reliability of Surflex-Dock in reproducing the experimental binding mode of 11β-HSD1 inhibitors.
Figure S2b displays all 40 inhibitors docked into the binding pocket of 11β-HSD1. Although the spatial positions of the scaffolds were not kept in the alignment, all molecules share a similar binding mode.
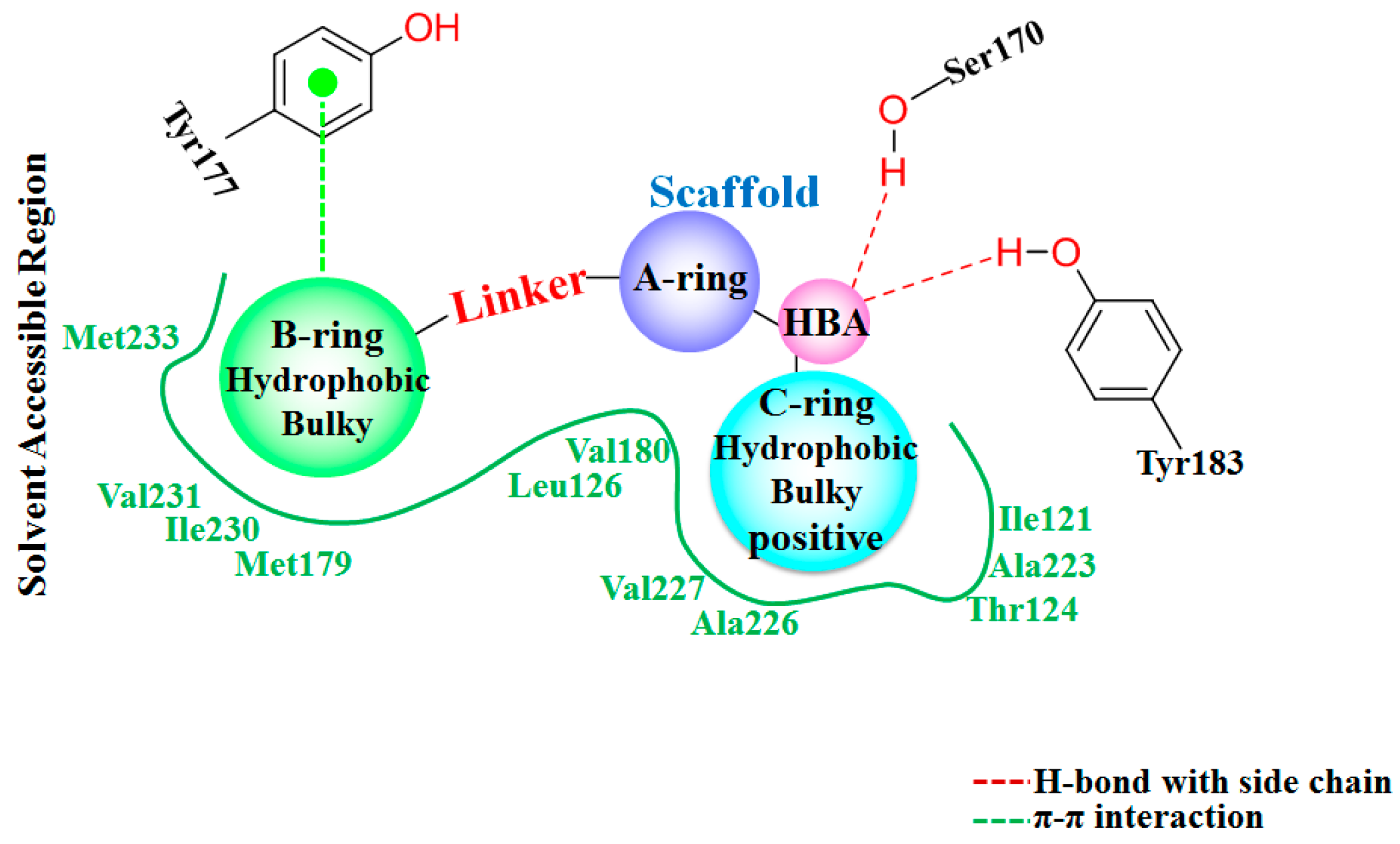
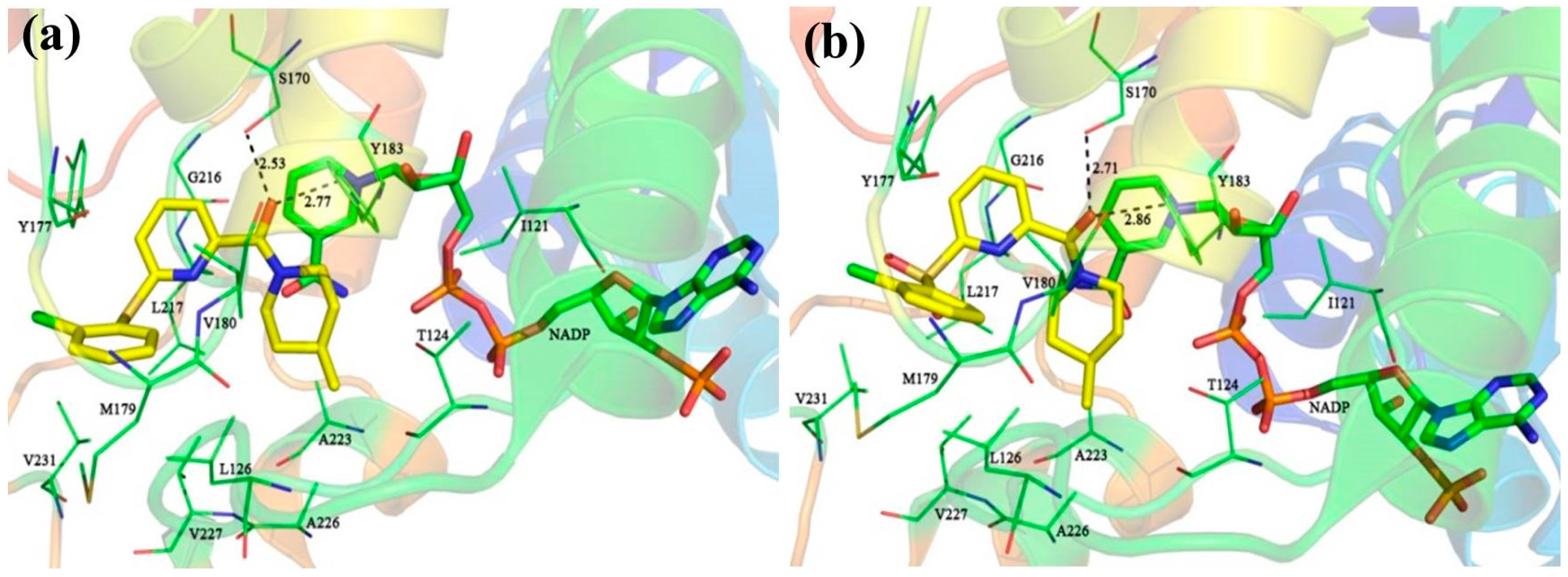
The detailed binding mode of
1 in 11β-HSD1 is depicted in
Figure S2c. Its amide carbonyl oxygen forms two hydrogen bonds (black dashes) with the side chain hydroxyl groups of residues Ser170 and Tyr183 with distances of 2.67 Å and 2.70 Å, respectively. The 3-fluoro-4-methylphenyl moiety is embedded in a hydrophobic cavity composed of Leu126, Tyr177, Met179, Val180, and Val231. It was further observed that both the 3-fluoro-4-methylphenyl moiety and the central pyridine core of
1 may have strong parallel π-π interactions with the side chain phenyl ring of Tyr177. Moreover, the central pyridine core is also parallel to the backbone of Gly216 and Leu217 with van der Waals interaction. The 3,3-dimethylpiperidinyl moiety is surrounded by the hydrophobic side chains of Ile121, Thr124, Tyr183, Ala223, Ala226, and Val227.
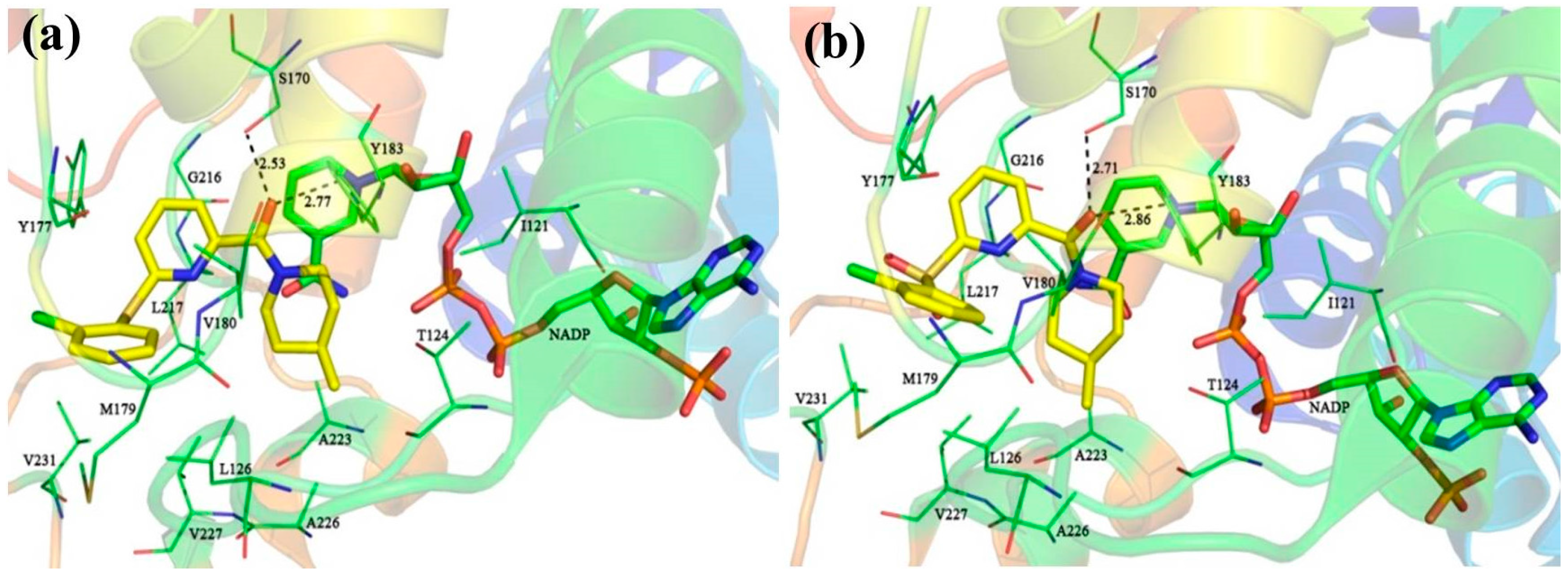
For comparison, compounds
5,
11,
14, and
39 were selected for detailed analyses of their binding mechanism on 11β-HSD1. The binding modes of these four compounds combined with the main residues of 11β-HSD1 are displayed in
Figure 1 and
Figure S3 in the Supplementary Materials. Particularly, the amide carbonyl oxygens of
5,
11,
14 and N1 and N2 atoms of
39 form hydrogen bonds with the side chain hydroxyl groups of residues Ser170 and Tyr183. Thus, these two residues may provide crucial hydrophilic interactions with an inhibitor binding to 11β-HSD1. In addition, as compared to the highly active compound
1, compound
5 contains a less hydrophobic 2-chlorophenyl moiety and a 4-methylpiperidinyl group resulting in a smaller hydrophobic space-filling in the binding pocket (
Figure S3a). This result would explain why compound
5 has lower bioactivity than
1. As for compound
11 or
14 (
Figure 1a,b), a relatively weak T-shaped π-π interaction is formed between the 2-chlorophenyl group and the side chain phenyl ring of Tyr177 due to a single heteroatom linker (S or SO
2) between the central pyridine core and the 2-chlorophenyl moiety. Moreover, compound
14 with a strong electron withdrawing linkage (SO
2) is detrimental to 11β-HSD1 potency. On the other hand, inactive compound
39 has a different binding mode in comparison to
1. Typically, the 2-methyl-4-chlorophenyl substituent of
39 approaches to a solvent exposed area of the binding site and is close to the hydrophobic residue Tyr177 with a weak T-shaped π-π interaction. The
N-acetylpiperidin-4-yl moiety is oriented to another side of the binding pocket, reducing the hydrophobic interactions with the side chain of Ile121 (
Figure S3b). Moreover, its polar acetyl substituent is also disfavored for interacting with 11β-HSD1. Due to the fact that the pocket is limited by residues Ile121, Thr124, and Tyr183, a modest steric hindrance could be beneficial for a ligand binding to 11β-HSD1. According to various bioactivities of these compounds, it may be hypothesized that favored hydrophobic and hydrophilic interactions would be beneficial to improve the inhibitory potency of a ligand.
3.2. Statistical Results of the CoMFA and CoMSIA Models
The statistical parameters of the generated CoMFA and CoMSIA models are summarized in
Table 1. The best CoMFA model gave a cross-validated correlation coefficient
q2 of 0.656 with an ONC of 5, a non-cross-validated
r2 of 0.964, SEE of 0.286, and
F value of 132.964. The corresponding field contributions of parameters were 58.6% of steric fields and 41.4% of electrostatic fields, indicating a greater influence of steric fields on the bioactivity. Such a prediction would be consistent with the results from the MD simulations discussed later.
The CoMSIA model was generated based on five fields, steric field (S), electrostatic field (E), hydrophobic field (H), H-bond donor (D), and H-bond acceptor (A), giving a cross-validated
q2 of 0.523 and a non-cross-validated
r2 of 0.936 with an ONC of 6. The corresponding field contributions were 10.4% (S), 26.4% (E), 27.1% (H), 18.0% (D), and 18.1% (A), respectively. The electrostatic and hydrophobic contributions shared large parts for the inhibitory activity for 11β-HSD1 in the generated CoMSIA model. It was noticed that steric contribution in the CoMSIA model was much lower than that in the CoMFA model. As we know, the CoMFA method is restricted to electrostatic fields with Lennard-Jones and Coulomb potentials. This may introduce errors in scaling, alignment sensitivity, and interpretation of contours. However, the CoMSIA method has been developed to make usage of hydrophobic fields in addition to the electrostatic fields. It can improve these inherent deficiencies arising from the CoMFA method [
56]. Accordingly, there is 27.1% of hydrophobic field in addition to steric field (10.4%) in the generated CoMSIA model. In our previous docking study, it has been recognized that the hydrogen bond and hydrophobic interaction would play significant roles in the protein-ligand binding affinity. It also proves that the hydrophobic properties are important in the design of 11β-HSD1 inhibitors. The predictive capabilities of the generated CoMFA and CoMSIA models were further evaluated using the test set of compounds excluded from the construction of 3D-QSAR models. The experimental and predicted pIC
50 values and residuals defined as the experimental minus the predicted pIC
50 values are summarized in
Table S2 in the Supplementary Materials. It was found that the predicted values of molecules in both training and test sets were close to the actual pIC
50 values without deviation of more than one logarithmic unit. The correlations between the experimental and model-predicted bioactivities of all compounds are shown in
Figure S4 in the Supplementary Materials. The predictive correlation coefficients
r2pred of the CoMFA and CoMSIA models were 0.938 and 0.923, respectively. The high
r2pred values indicate a good predictive ability of our generated 3D-QSAR models.
3.3. Interpretation of the CoMFA and CoMSIA Models Based on Receptor
The CoMFA and CoMSIA results were graphically described by the field contribution maps using the STDEV*COEFF field type.
Figure 2 and
Figure 3 display the contour maps derived from the generated CoMFA and CoMSIA models, respectively. Compound
1 was labeled on the map as a template for visualization to explore the structure-activity relationships of these 11β-HSD1 inhibitors. The contour analyses have been performed by dividing the total molecular area into three subdivisions (
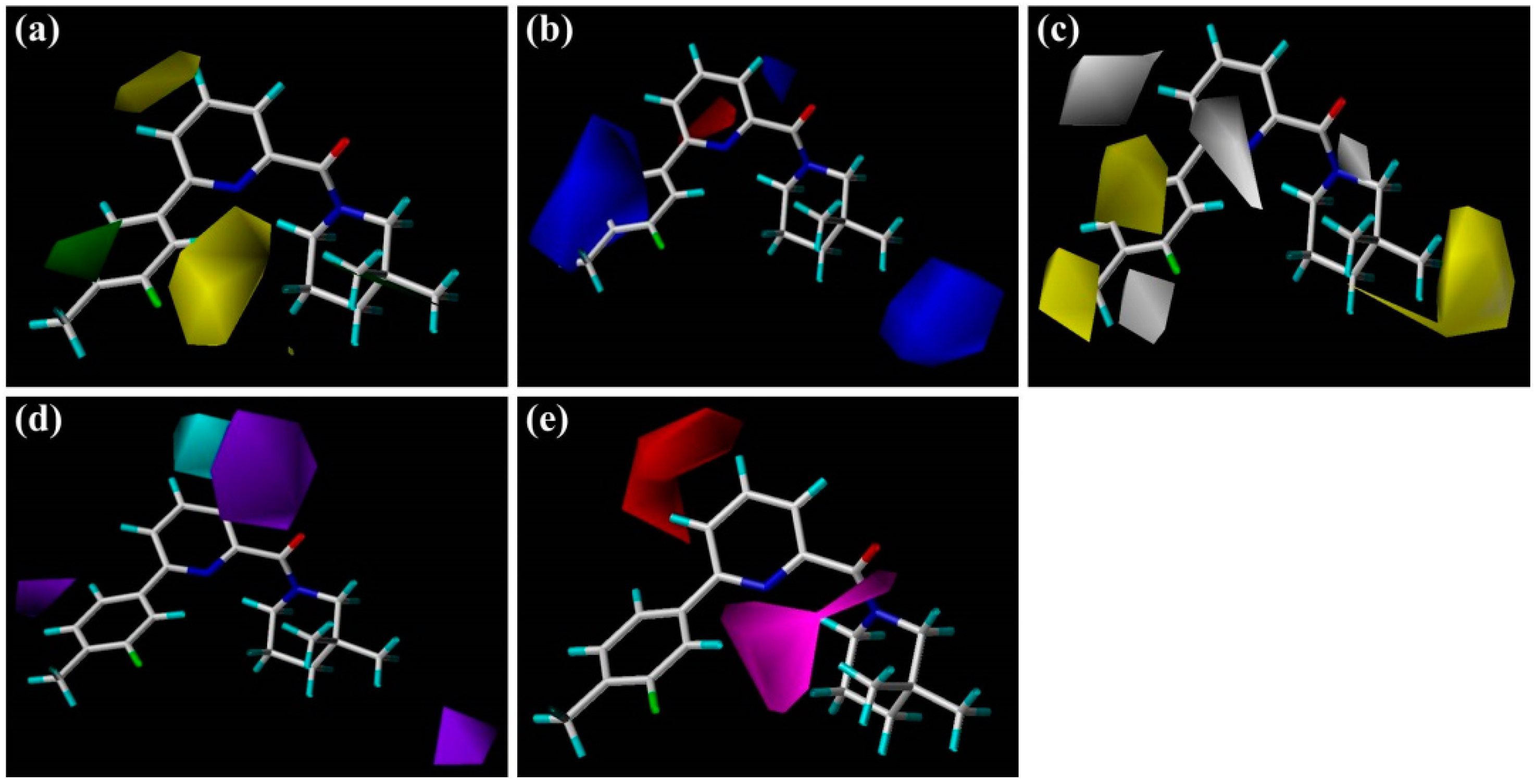
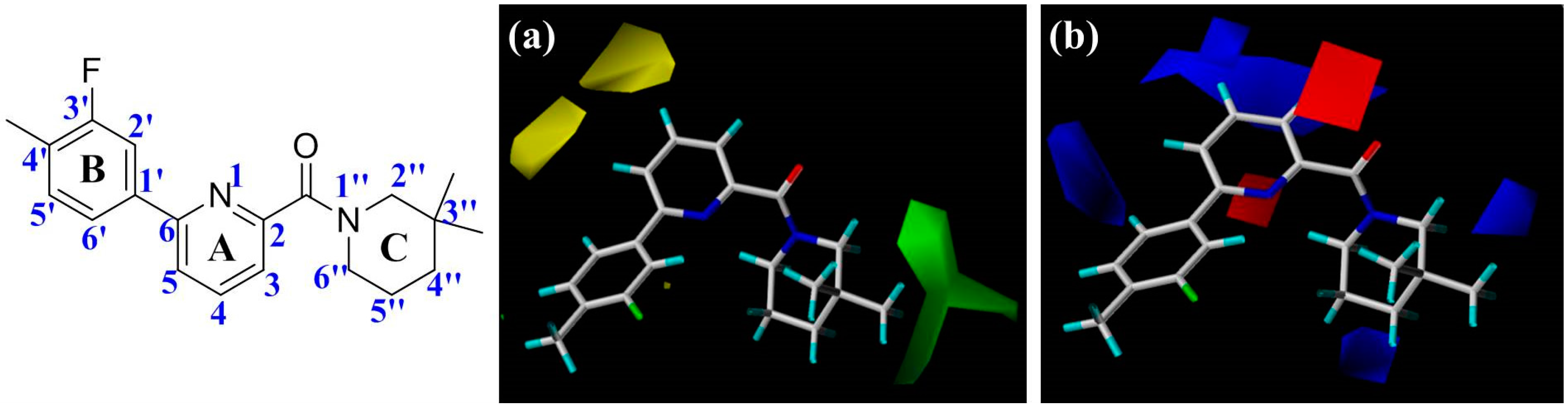
Figure 2), including the central pyridine or TZP core (A-ring), the distal aromatic ring (B-ring), and the aliphatic moiety (C-ring).
In
Figure 2a, the contour map of the steric field of CoMFA model, a large green contour is close to 3″-position of C-ring, indicating that this area would prefer a bulky substituent rather than a small branch. According to the docking simulations, C-ring was pointed toward a hydrophobic pocket composed of residues Ile121, Thr124, Tyr183, Ala223, Ala226, and Val227. Moreover, this region could also be demonstrated in the hydrophobic contour map of the CoMSIA model with a large yellow polyhedron at the same position (
Figure 3c), indicating that a hydrophobic group is favorable for bioactivity. This observation in either CoMFA or CoMSIA model is consistent with the experimental data that compound
2 with a 3″,3″-dimethyl group is more active than
5 with a smaller 4′-methyl substituent. Compounds
19–
23 with diverse substituents on C-ring reflected the similar trend of bioactivity. Especially, compound
22 showed more than 800-fold higher bioactivity against 11β-HSD1 in comparison with
19. On the other hand, there are two yellow moderate polyhedrons a little far away from the 4- and 5-positions of A-ring in the CoMFA model, indicating that bulky substituents in these positions may result in a decreased activity. Actually, our docking simulations indicated that the 4- or 5-position of
1′s A-ring would point to a limited space composed of residues Leu171, Gly216, Leu217, and Met233. Therefore, bulky substituents in these places would be disadvantageous to the biological activities.
The electrostatic contour map of CoMFA model can be seen clearly from the
Figure 2b. Since compounds
16 and
20–
22, which possess a nitrogen atom at the 4-position of A-ring, exhibited potent inhibitory activities, a small blue contour area is obtained near this site. Moreover, compound
30 with an electron-donating NH group among the ‘three-atom’ sulfonamide linker, which could be superimposed on the 4-position of
1′s A-ring, has higher bioactivity than
29 with a ‘two-atom’ sulfone linker in the corresponding position. Another large blue contour is found around the 3- and 4-positions of A-ring. This contour could be used to explain why compound
18 with its nitrogen atom in pyridine ring is less potent than
15. Besides, the blue contour near 6′-position of B-ring indicates that the considerable activity differences with an order of
9 >
15 >
10 could be due to the electronegativity of the corresponding linkers −SCH
2−, −SO
2CH
2−, and −OCH
2−. While two small blue polyhedrons at the 3″- and 4″-positions of C-ring reveal that an electropositive group at these positions would be beneficial for the binding affinity, like compounds
16 vs.
24,
34 vs.
37, and
35 vs.
36. Conversely, a red region appears around the carbonyl oxygen of amide or the nitrogen atom at the 1-position of the TZP core, highlighting that an electronegative group is preferable at this position. This is in agreement with the docking results in which the carbonyl oxygen atom of highly active compound
1 accepts hydrogen bonds from the side chain hydroxyl groups of Ser170 and Tyr183. Another red region around the 1-position of the A-ring represents that electronegative group is favorable for enhancing bioactivity.
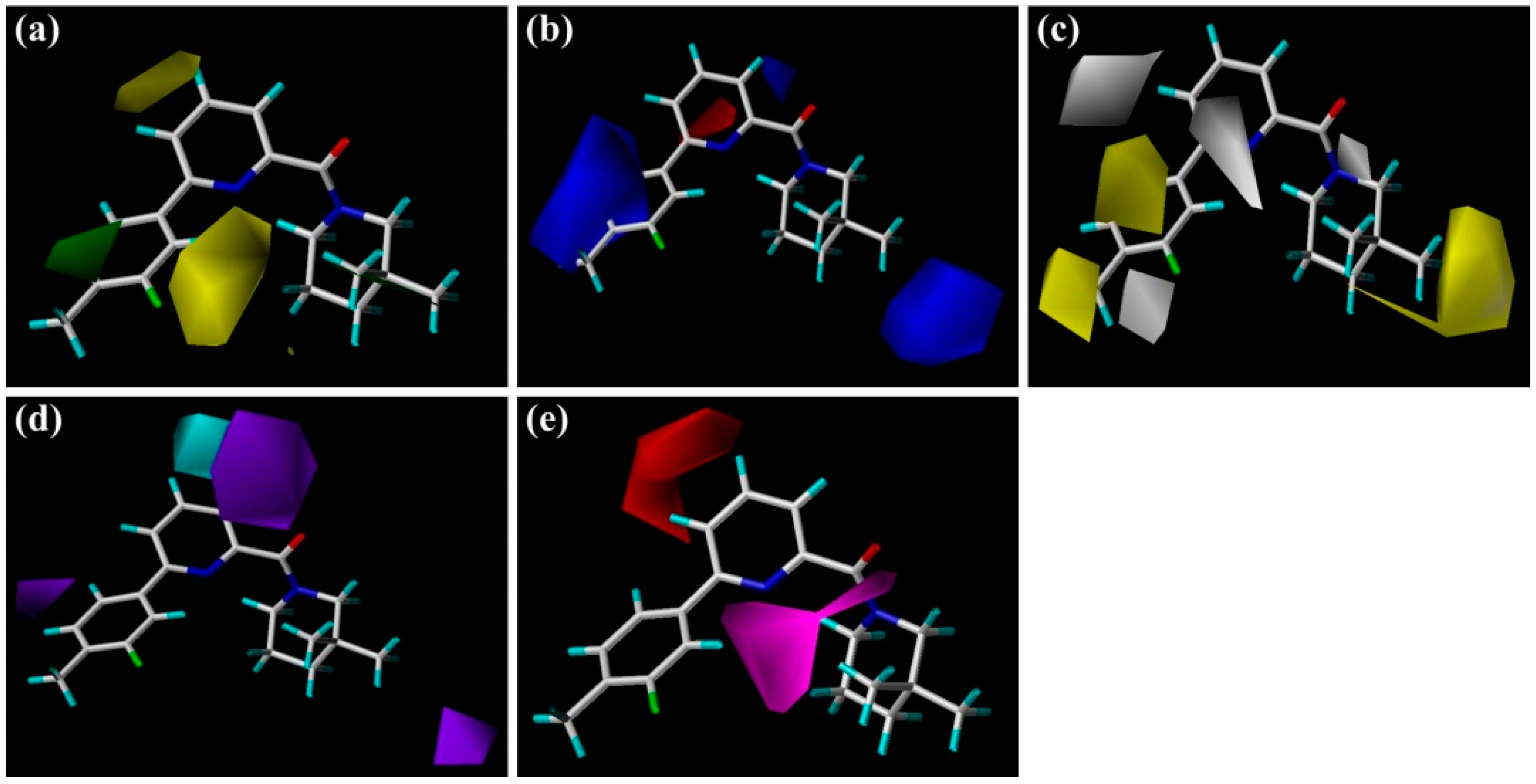
The steric and electrostatic contour maps of the CoMSIA model are depicted in
Figure 3a,b, respectively. These contour maps were found to be nearly identical to the corresponding CoMFA contours except some minor differences. As shown in
Figure 3a, there is a small green contour beside the 5′-position of
1′s B-ring, suggesting that introduction of a hydrophobic substituent at this region would be beneficial to improve the inhibitory activity of a ligand. For instance, the activities of compounds
9 and
10 are higher than those of
11 and
12. As compared with linker atoms between A-ring and B-ring in
11 (−S−) and
12 (−O−), the longer linkers of
9 (−SCH
2−) and
10 (−OCH
2−) may extend their phenyl ring B to the 5′-position of
1′s B-ring, making
9 and
10 more similar to
1 in shape. In fact, the B-ring of
1 was positioned at the edge of the binding pocket of 11β-HSD1 and extended to the solvent in the docking-simulated structural model of complex 11β-HSD1-
1 (
Figure S2c), thus it would allow a bulky substituent. In comparison, compound
11 or
12 has its aromatic ring B pointing to the medium-sized yellow contour near the 2′-position of
1′s B-ring, suggesting that a steric substituent at this position degrades the biological activity of the molecule. Furthermore, compound
6 with 2′-NO
2 or
8 with 2′-phenyl also showed slightly lower bioactivity than
1. As for the electrostatic field (
Figure 3b), three blue contours and one red contour are presented in similar sites like those in the CoMFA electrostatic map (
Figure 2b). Since these contours were discussed above in the CoMFA model, our following discussion will focus on the CoMSIA hydrophobic contours as well as H-bond donor and acceptor contours.
As illustrated in the CoMSIA hydrophobic contour map (
Figure 3c), the most parts of B-ring and 3″-position of C-ring are surrounded by yellow contours, suggesting that hydrophobic features at these positions are crucial for the inhibitory activity. Above docking results also disclosed that the 4′-substituted group of B-ring may interact with the hydrophobic residues Met179 and Val231. For example, compound
3 with 4′-Cl or
4 with 4′-OCF
3 held high potency for 11β-HSD1 but showed slightly lower activity than compound
1 with 3′-F-4′-CH
3. Meanwhile, compounds
25,
30,
32, and
33, bearing a suitable linker between A-ring and B-ring, made them be similar to
1 in molecular shape and exhibited higher activities than analog
28 with an oxygen linker. Furthermore, the 3″-position of C-ring would likely to occupy a sub-hydrophobic pocket composed of hydrophobic residues, like Ile121, Leu126, Val180, Tyr183, Ala223, and Ala226. Together, with the green contours in steric field map discussed above, it could be inferred that bulky and hydrophobic groups at these positions would enhance the inhibitory activity. On the other hand, two gray contours near the 1-position of A-ring and 1″-position of C-ring are congruent with the results obtained from the generated CoMFA model, showing that an electronegative atom would be desirable for an inhibitor. Another two gray areas appear at the 5-positon of A-ring and 3′-positon of B-ring, indicating that modifications of these parts with some hydrophilic substitutions could increase the bioactivity.
Figure 3d,e depict the graphical interpretation of the H-bond donor and H-bond acceptor features in the CoMSIA model, respectively. In
Figure 3d, a cyan polyhedron is presented near the 4-position of
1′s A-ring, suggesting that an H-bond donor would be favored to interact with the highly electronegative center. In fact, the NH group of sulfonamide linker in compound
30 was found to be superimposed on the 4-position of
1′s A-ring, so it showed much higher potency than
29 with a sulfone linker. On the other hand, compound
31 has its NH group in sulfonamide linker pointing to the purple region presented around the 3-position of
1′s A-ring. A piece of small purple contour near the 6′-position of B-ring may explain the lower activity of compound
13 with an NH group than
5. Another small purple contour little far away from the 3″-position of C-ring is accordance with the decreased activity of compound
24 in comparison with
23. As for H-bond acceptor contour maps (
Figure 3e), a large magenta contour area covers the scaffold of compound
1 from the carbonyl oxygen of amide or N1-positon of the TZP core to the 2′-position of B-ring, indicating that groups with a hydrogen bond acceptor would be favored at the corresponding positions. Our docking simulations have also shown that the carbonyl oxygen atom or the nitrogen atoms of the TZP core form key hydrogen bonds with the side chain hydroxyl groups of Ser170 and Tyr183, which were considered as significant hydrogen bond interactions in the binding mode. This could be verified by the satisfactory potency of compound
6 bearing a nitro group at the 2′-position of B-ring. On the other hand, the medium-sized red contour beside the 4-position of A-ring reveals that an H-bond acceptor group there would be disadvantageous to the inhibitory activity.
3.4. MESP, HOMO-LUMO, and Mulliken Atomic Charges Analyses
To understand surface electronic properties of 11β-HSD1 inhibitors and to comprehend the pharmacophoric features required for binding, two highly active compounds (1 and 32), and two low activity compounds (14 and 39) selected from the docking study were analyzed based on MESPs, HOMO-LUMO parameters and dipole moments. In addition, Mulliken population analyses of studied inhibitors were also calculated to get more detailed insights at the electronic level.
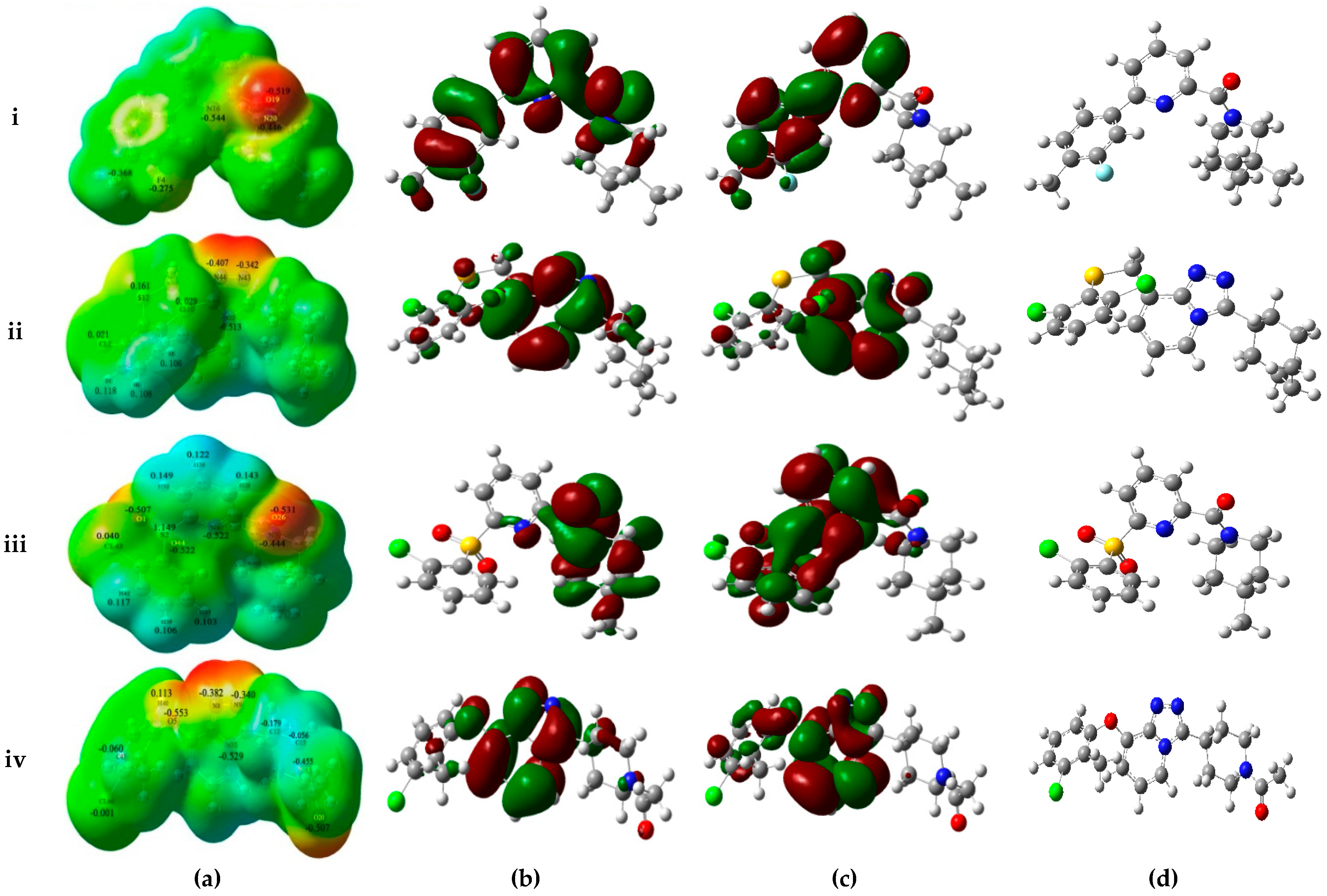
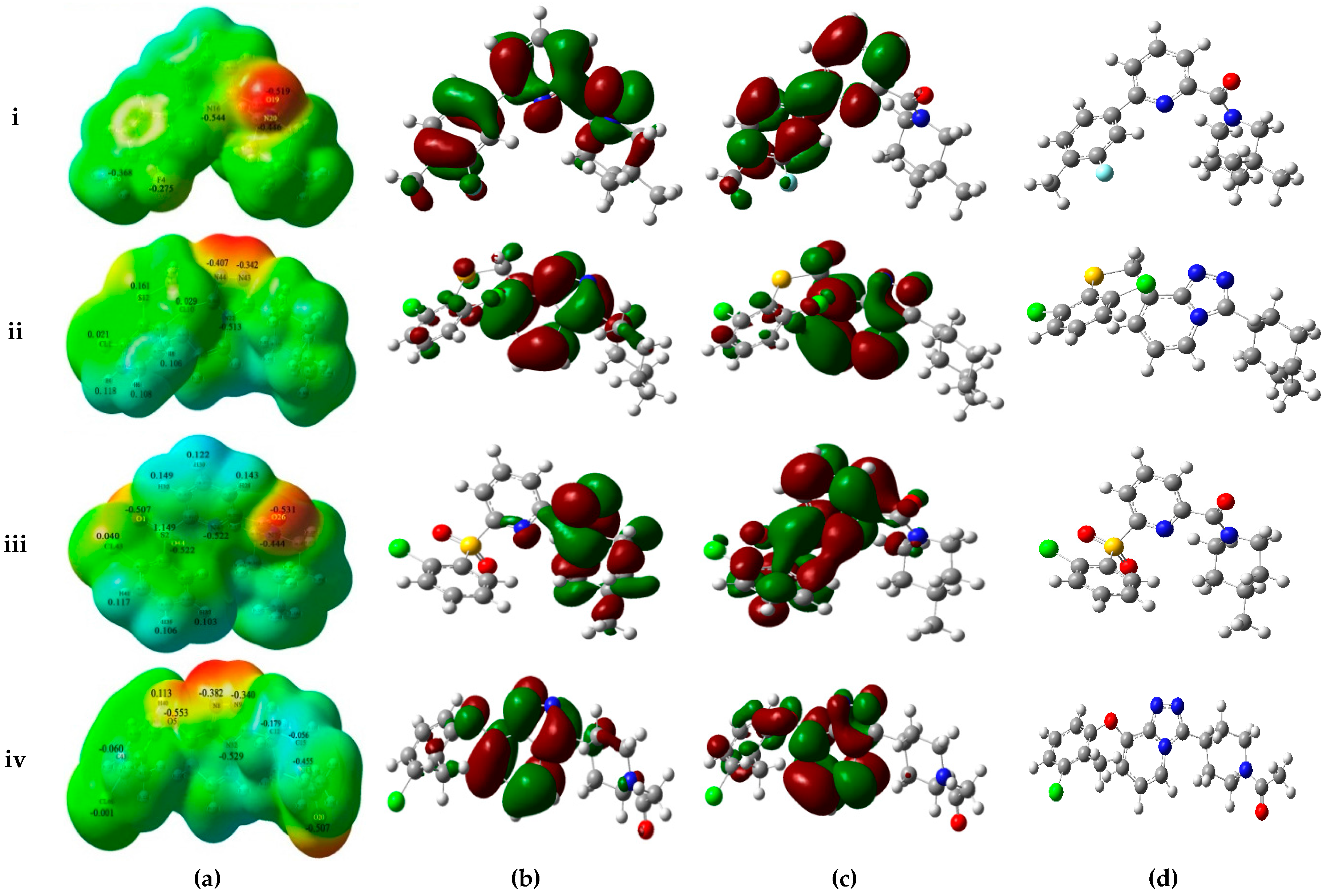
As shown in
Figure 4a, MESPs plotted on these four compounds reveal that the amide carbonyl oxygen atoms or the nitrogen atoms of the TZP core represent most electronegative potential regions (deep red color) favored for electrophilic attack. These results are in accordance with the medium-sized red contour in CoMFA model and purple and magenta contours in the CoMSIA model. Besides, previous docking results have also indicated the involvement of these areas in the important H-bond interactions with the key residues Ser170 and Tyr183. On the other hand, an apparent red region was observed near the electron-attracting oxygen atoms O1 and O44 of the sulfone linker in the low active compound
14 with an average Mulliken charge of −0.515, which does not fit with the blue contour around the 6′-position of B-ring in the electrostatic maps of 3D-QSAR models. Similarly, compound
39 shows strong electronegative potentials near the linker oxygen atom O5 with the Mulliken charge of −0.553, which is in conflict with the red contour in the H-bond acceptor contour of CoMSIA model. In addition, the carbonyl oxygen O20 of
39 exhibits the strongest electronegative potential (Mulliken charge: −0.507), which is also not in accordance with the large blue contour in the CoMSIA model. Thus, MESPs plotted over the low active compounds
14 and
39 indicated that their molecular electronic properties do not fit well with the required electrostatic features predicted by above generated 3D-QSAR models.
The frontier orbitals HOMO and LUMO, which are quantum chemical descriptors, were calculated for these four molecules. The HOMO energy is closely related to reactivity to electrophilic attack while LUMO energy is closely related to reactivity to nucleophilic attack. Thus, the eigenvalues of HOMO and LUMO and their energy gap reflect the biological activity of the molecule. Usually, the decrease in the HOMO and LUMO energy gap explains the eventual charge transfer interaction taking place within the molecule under the influence of an external electric field. As shown in
Figure 4b,c, the HOMO and LUMO sites were plotted onto the molecular surfaces of compounds
1,
32,
14, and
39, respectively. The distributions of HOMO and LUMO sites are distinct in the active and low active compounds. In the active compound
1, both HOMO and LUMO orbitals are overlapped significantly at A-ring and B-ring, while the carbonyl oxygen atom of amide and some positions of C-ring possess HOMO orbitals. Such extensive overlapping frontier orbitals indicated the highly reactive nature of the active compound. The HOMO orbital of
14 is located only at the carbonyl oxygen atom and C-ring. By comparing active
32 and low active
39, the TZP core possesses both HOMO and LUMO orbitals, whereas B-ring and the linker between A-ring and B-ring have the obviously different electronic features. The computed quantum chemical descriptors HOMO and LUMO values of these four molecules in both gas and solvation (water) phases are summarized in
Table S3 in the Supplementary Materials. For this analysis, because of the small values of both HOMO and LUMO energies (−0.24 to −0.21 eV and −0.08 to −0.04 eV), electron transfer and exchange may be equally possible, making these compounds very reactive. The HOMO-LUMO gap of the active compounds is slightly higher (~0.006 eV) when compared with that of low active compounds. Additionally, the dipole moments of these four compounds are also computed in both gas and solvation (water) phase (
Table S3 in the Supplementary Materials). Higher values of dipole moments are obtained in the solvent phases (3.71 to 8.63 Debye) compared to the gas phase (3.39 to 6.09 Debye). This is due to the intermolecular interactions between the solvent and the studied compound. On the other hand, dipole moment represents a direct measure of the electron distribution in a molecule, which may give some insight on the degree of hydrophobicity/hydrophilicity of the compound. In particular, the two active compounds (
1 and
32) show higher dipole moments than another two low active inhibitors (
14 and
39), thus compounds
1 and
32 have much stronger hydrophobic interactions with the binding pocket than those of
14 and
39.
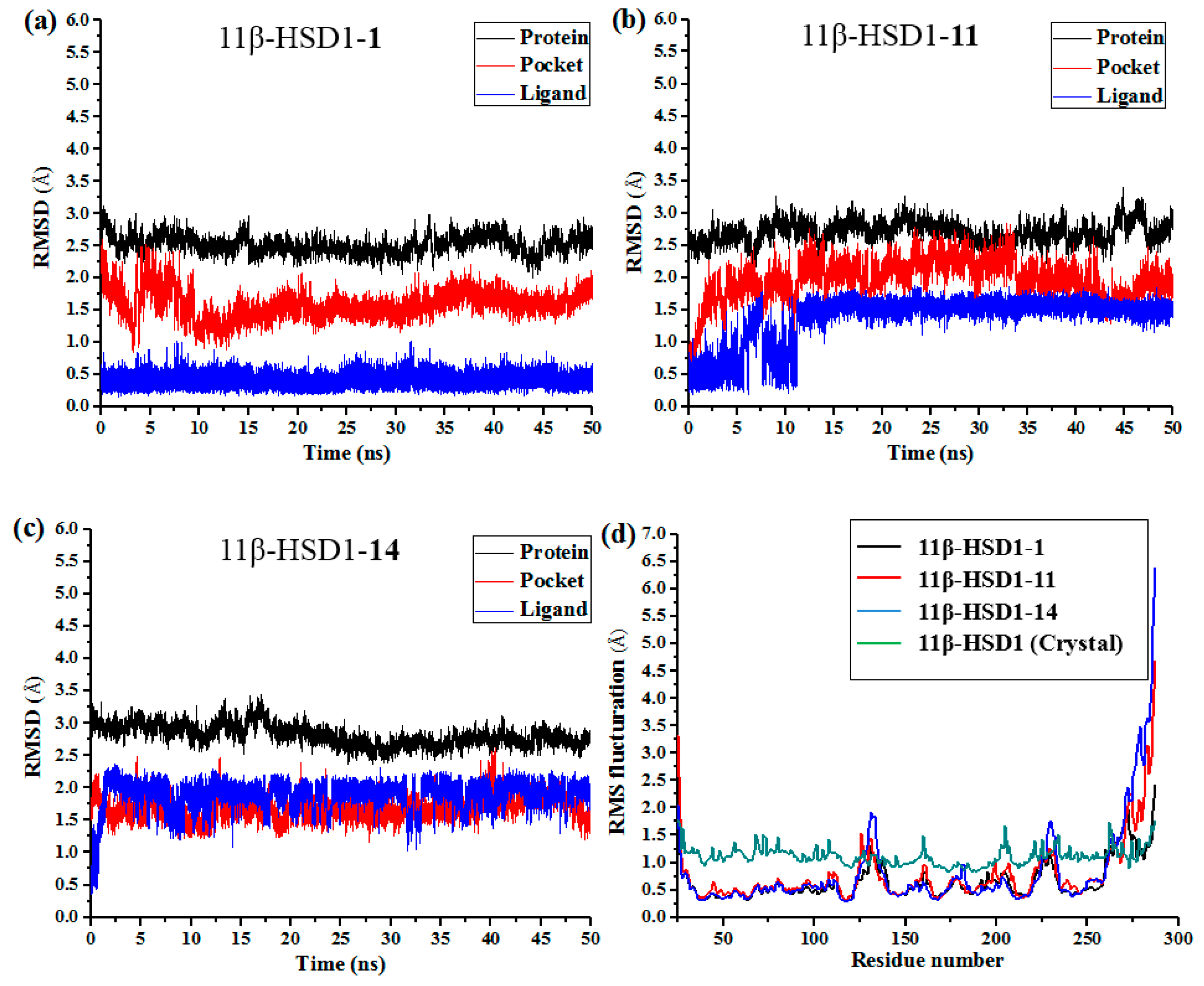
3.6. Molecular Dynamics Simulations
Although docking simulations provide a good starting for further calculations with the purpose of predicting the binding modes, the solvent effect on the whole system and the potential conformational changes are not fully taken into account. Therefore, molecular dynamics simulations were undertaken to investigate the stability of the enzyme-inhibitor complex in aqueous solution.
The docking-simulated structural models of compounds
1,
11, and
14 with 11β-HSD1, respectively, were applied as starting structures for MD simulations to analyze the structural requirements for the inhibitory activity and compare the binding modes between the inhibitors and 11β-HSD1. The root-mean-square deviations (RMSDs) for all heavy atoms of each ligand, backbone atoms of whole protein, and backbone atoms of residues in the binding pocket within 5 Å around the ligand were analyzed to explore the dynamic stabilities of all systems. The RMSDs of each system relative to their starting structures are plotted in
Figure 6. As depicted in the plots, all three systems are in a relatively stable equilibrium in most of the time along the MD simulations procedures of 50 ns. Thus, it is reasonable to further calculate the binding free energies and free energy decomposition based on the conformations extracted from last 4 ns MD simulations trajectories. It was noticed that compounds
11 and
14 with lower biological activities spent longer time to reach equilibrium (
Figure 6b,c). In addition, among all three complexes,
11 and
14 showed higher fluctuations than the most active compound
1, with RMSD values of ~1.5 Å and ~2.0 Å for ligands, respectively, indicating that these two compounds would have some unstable binding mode in their complexes with 11β-HSD1.
In the meantime, the residue flexibility was also examined by analyzing the root mean square fluctuation (RMSF) of Cα atoms of protein in each complex. For comparison, the corresponding values of RMSF obtained from the co-crystal structure of 11β-HSD1-
1 are also shown in
Figure 6d. The protein structures of these three systems with different inhibitors share similar RMSF distributions and trends of dynamics features. It shows that the residues with higher flexibilities distribute near the N- and C-terminal parts. The active site, including residues Ile121, Thr124, Leu126, Ser170-Ala172, Tyr177-Val180, Tyr183, Leu215-Ile218, Thr222-Ala223, Ile230-Val231, and Met233, exhibits rigid behavior. It also suggests that all inhibitors have similar interaction mechanism with 11β-HSD1. Accordingly, the observations were in agreement with the experimental X-ray crystallographic data, indicating the reliability of our MD results. By comparing the RMSF fluctuations of the binding pocket in the three complexes, it would be rational to conjecture that compound
1 has more stable interactions with 11β-HSD than the other two inhibitors, while
14 shows relatively higher structural mobility among the three complexes. Overall, these analyses of binding stabilization are consistent with the experimental activities.
3.7. Comparison of 3D-QSAR, Molecular Docking, and MD Simulation Results
In order to further investigate the receptor-ligand interactions in the binding process, we compared the conformations of complexes during the MD simulations with the initial structures obtained from docking simulations. Clustering grouping was performed to analyze the structural variations of each complex during the MD simulations. Based on the pairwise similarity measured by RMSD with the
means algorithm [
57], five clusters were produced from the trajectory frames using the
ptraj module of AMBER12. The representative conformations were then extracted from the cluster and compared with the above docking-simulated structures. In general, the conformation of the binding pocket and the inhibitors were found to be stable during the MD simulations, suggesting the rationality and validity of the docking models. However, some differences could still be observed between the MD simulated structures and the docked models. The program LIGPLOT [
58] was used to generate 2D receptor-ligand interaction diagrams.
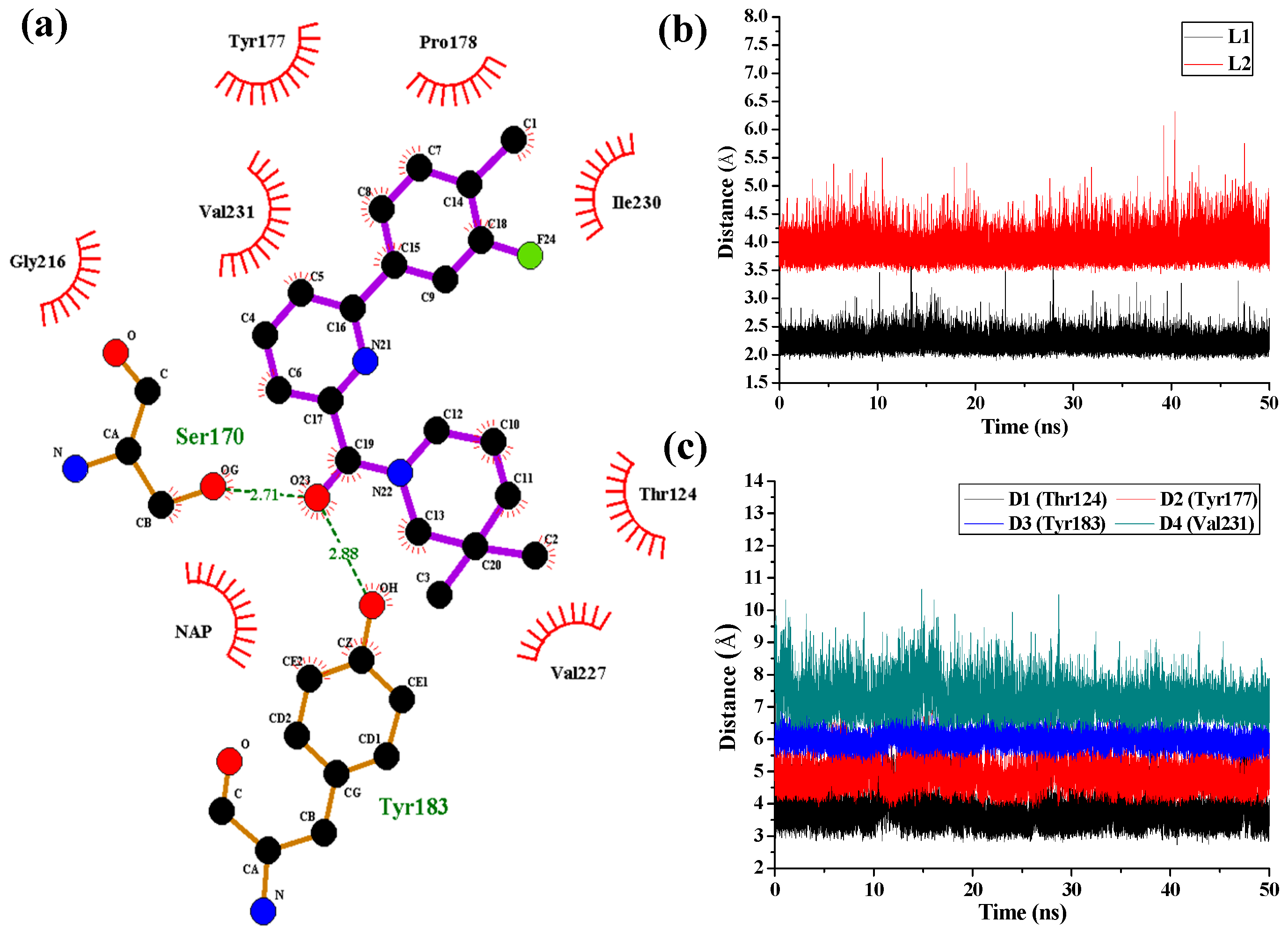
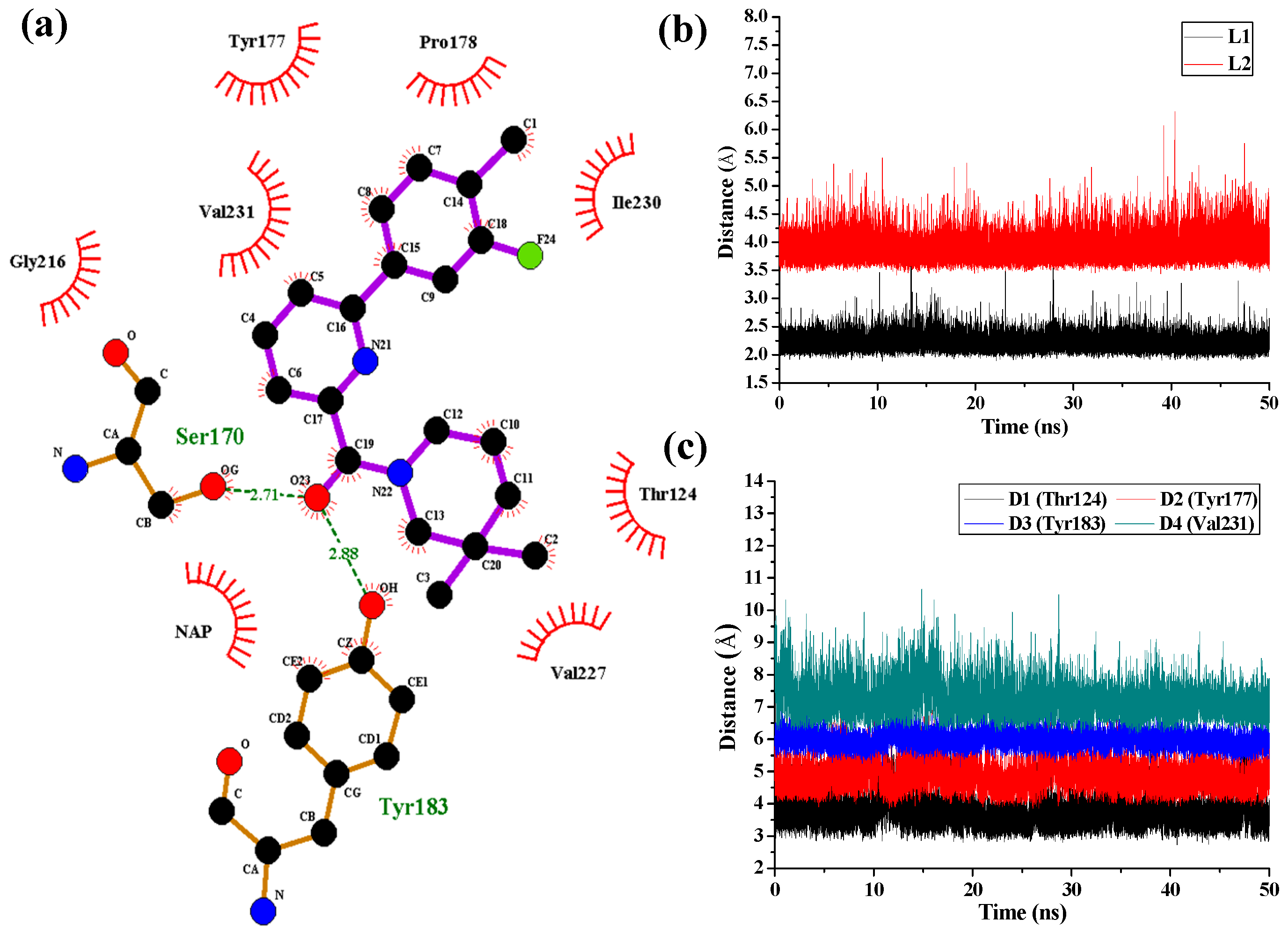
Figure 7 shows the interaction features between the compound
1 and 11β-HSD1.
The H-bond interactions with key residues in the active site along with H-bond length, angle and their occupancies for the three MD-simulated enzyme-inhibitor systems are listed in
Table S4 in the Supplementary Materials. In the MD-simulated 11β-HSD1-
1 complex (
Figure 7a), two stable H-bonds were formed for
1 to interact with adjacent residues around the binding pocket. One H-bond is formed between the carbonyl oxygen atom and the side chain hydroxyl group of Ser170 with an average distance of 2.71 Å and occupancy of 99.41%. Another one is formed between the carbonyl oxygen atom and the side chain phenolic hydroxyl group of Tyr183 with an average distance of 2.88 Å and occupancy of 99.43%.
Figure 7b shows the H-bond lengths vs. time evolution. It can be seen that these two H-bonds were stable during MD simulations. In fact, above 3D-QSAR models showed a red contour in the electrostatic map of generated CoMFA model and a magenta region in the H-bond acceptor feature contours of generated CoMSIA model, suggesting that a highly electronegative atom is favorable for 11β-HSD1 bioactivity. Furthermore, we noticed that there were several hydrophobic contacts between the 3-fluoro-4-methylphenyl moiety and dimethylpiperidinyl moiety of compound
1 and residues Thr124, Tyr177, Pro178, Tyr183, Leu217, Val227, Ile230, and Val231, indicating that the hydrophobic interactions may also play critical roles in an inhibitor binding to 11β-HSD1. The interactions identified by MD-simulated models were consistent with the results from our generated steric contour maps in 3D-QSAR models, in which there were two green regions located around 5′-position of B-ring and 3′′-position of C-ring. Furthermore, our docking results indicated that the 3″,3″-dimethyl group of
1′s C-ring remained into the hydrophobic region surrounded by the residues Ile121, Thr124, Val180, and Tyr183, and the space of the substituents on the B-ring are also limited by the residues Met179 and Val231. The stabilities of the hydrophobic interactions between 11β-HSD1 and
1 may be estimated by examining the time dependences of the related mass-center (CM) distances. As shown in
Figure 7c, D1 and D3 represent the distances between the dimethylpiperidinyl ring of
1 and the main chains of Thr124 and Tyr183, respectively. D2 and D4 represent the distances between the 3-fluoro-4-methylphenyl moiety of
1 and the main chains of residues Tyr177 and Val231, respectively. The stability of the interatomic distances reveals that these hydrophobic interactions are also favorable to stabilize the binding of
1 to 11β-HSD1.
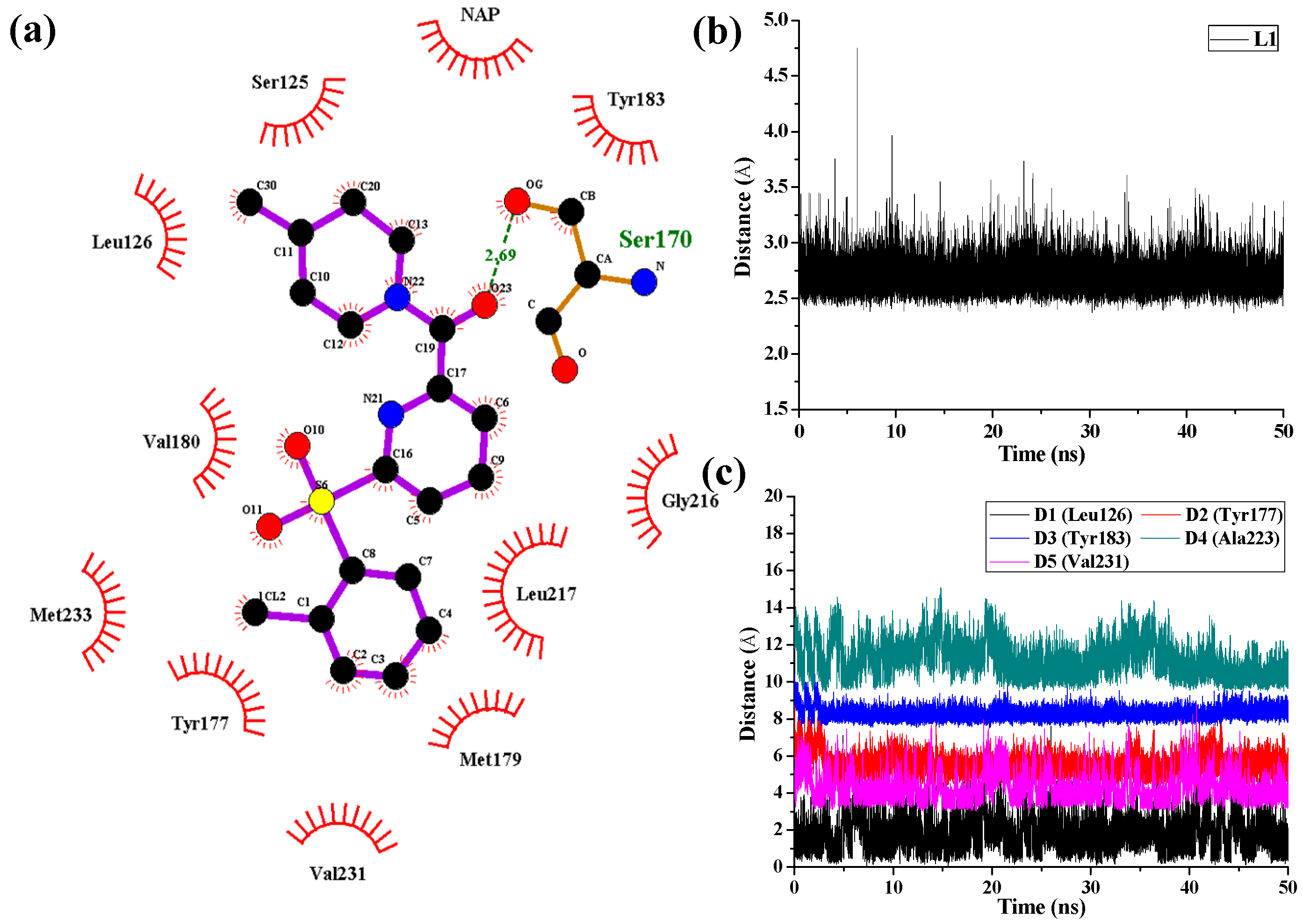
Figure S5 in the Supplementary Materials describes the interaction mode between
11 and 11β-HSD1. As shown in
Figure S5a, the amide carbonyl oxygen engaged in one H-bond with the critical residue Ser170 with an average distance of 2.69 Å and occupancy of 99.38%. It is worth noting that the residue Val231 has weaker hydrophobic interaction with
11 at the beginning (
Figure S5c). This is due to the fact that the 2-chlorine phenyl of
11 has undergone several movements during the MD simulation.
As shown in
Figure 8 in the case of 11β-HSD1-
14 complex, D1, D3, and D4 represent the distances between the 4-methylpiperidinyl ring of
14 and the main chains of Leu126, Tyr183, and Ala223, respectively. D2 and D5 represent the distances between the 2-chlorine phenyl moiety of
14 and the main chains of residues Tyr177 and Val231, respectively. As results, only one H-bond was found to be formed between the amide carbonyl oxygen and the side chain of Ser170 with an average distance of 2.69 Å and occupancy of 99.78%, while the hydrophobic interactions between
14 and its surrounding residues are relatively weaker. Moreover, the hydrogen bonding interactions between Tyr183 and compounds
11 and
14 were disappeared after MD simulation, indicating that the hydrogen bonding interactions between Tyr183 and these two inhibitors are relatively weak which led to decreases in their bioactivities. From the results discussed above, the ligands and the protein were both treated in a flexible way and the solvation effect was also taken into consideration during the MD simulations. However, the docking simulations ignored the flexibility of the protein when the ligands bound to the receptor. Therefore, MD simulations would not only confirm the rationality and effectiveness of the molecular docking and the 3D-QSAR models but also point out their defects in molecular modeling.
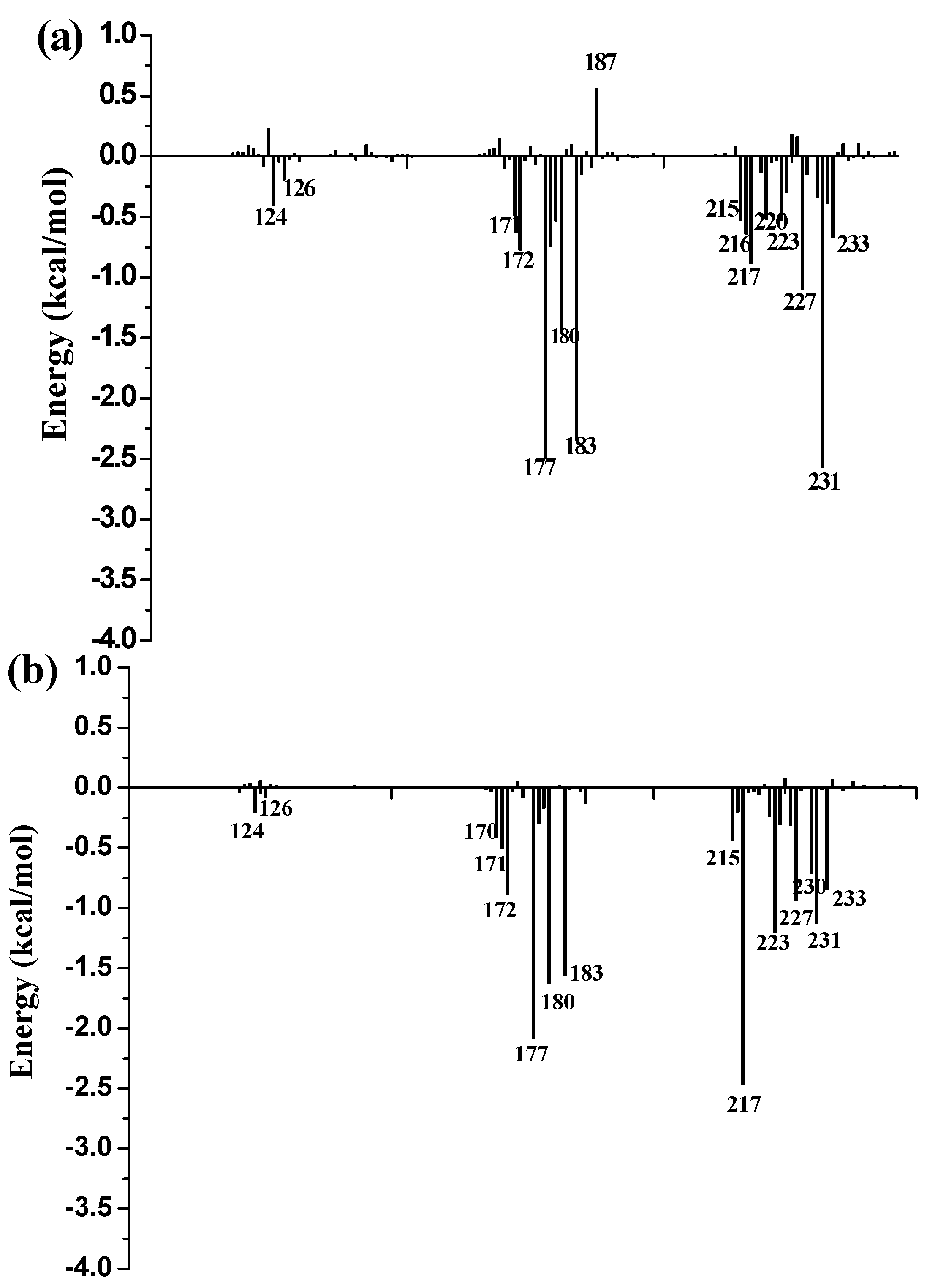
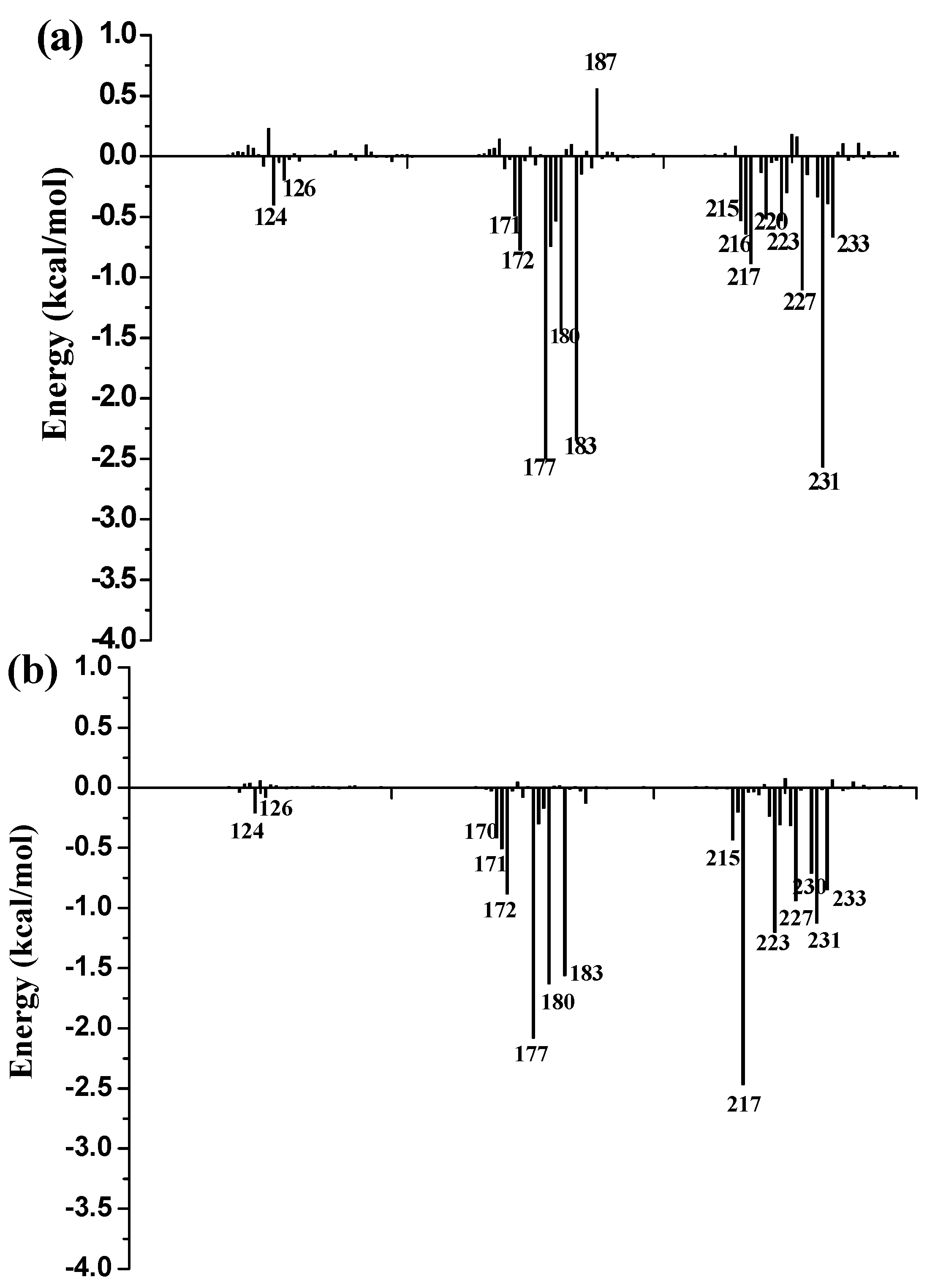
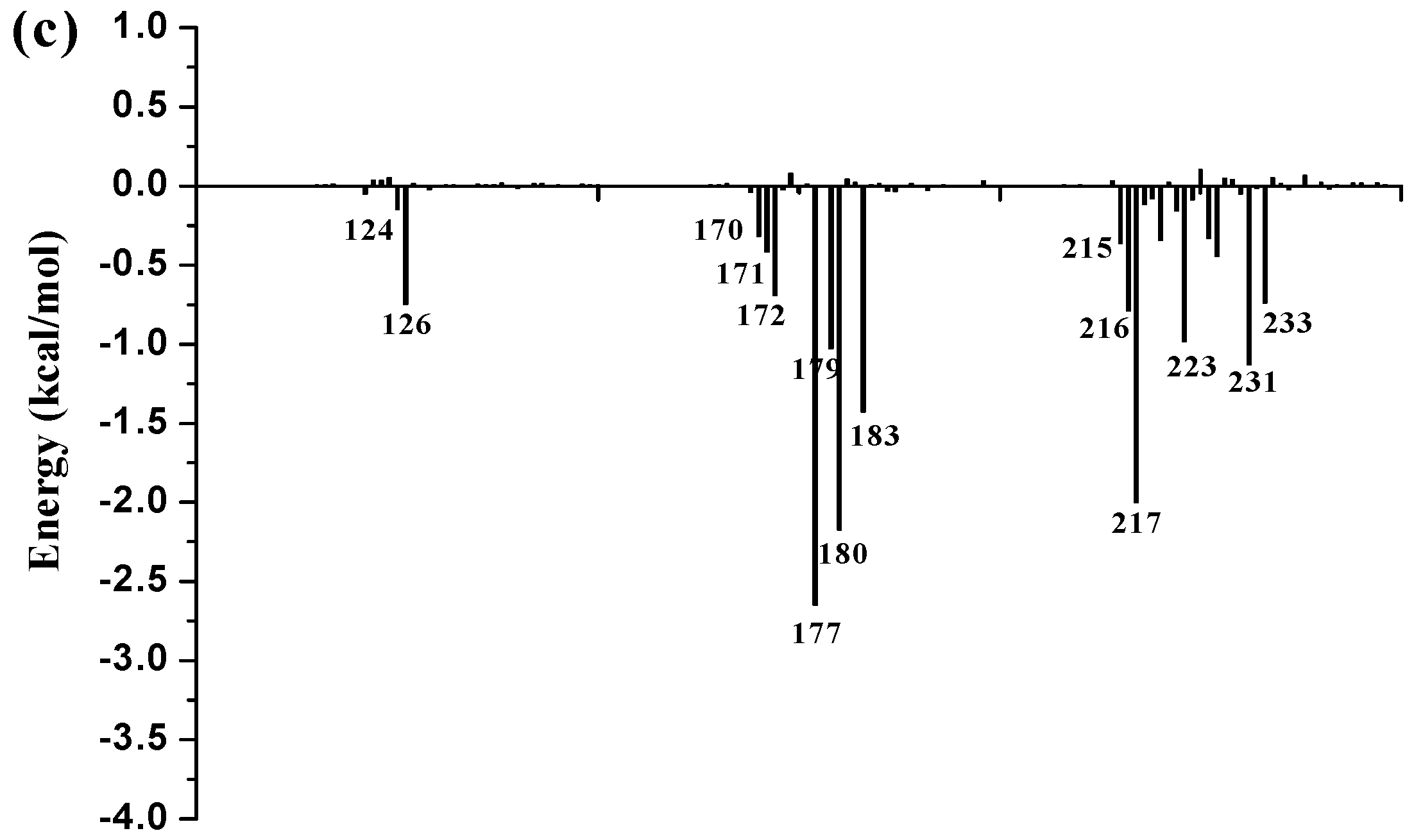
3.9. Free Energy Decomposition
To further identify key residues affecting the binding process, binding free energy was subsequently decomposed into the contribution of each residue by using the MM/GBSA method. The detailed interaction information of above three complexes is shown in
Figure 9, and the energy contributions of the key residues for each inhibitor binding to 11β-HSD1 are compared in
Figure S6 in the Supplementary Materials. As illustrated in
Figure 9, major favorable energy contributions originate predominately from residues Leu171, Ala172, Tyr177, Val180, Tyr183, Leu215, Gly216, Leu217, Ala223, Val231, and Met233.
Most of the key residues are hydrophobic enough to form strong van der Waals interactions with the inhibitors. Particular attention had been paid to those residues with relatively large differences in the contribution to binding free energies. Typically, the residues Ser170 and Tyr183, which have H-bond interactions with the inhibitor as discussed above, showed different interaction contributions. Ser170 has a little more energy contributions to
11 (−0.42 ± 0.02 kcal/mol) and
14 (−0.32 ± 0.02 kcal/mol) than
1 (−0.03 ± 0.02 kcal/mol), another residue Tyr183, which only has an H-bond interaction with compound
1 during MD simulations, has more energy contributions to
1 (−2.35 ± 0.02 kcal/mol) than
11 (−1.56 ± 0.02 kcal/mol) and
14 (−1.43 ± 0.02 kcal/mol). Among the resides in the binding pocket of 11β-HSD1, residues Thr124, Tyr183, Val227, and Val231 have stronger interactions with compound
1 in comparison with aforementioned residues, while residues Leu126, Met179, and Val180 interact with compound
14 differently from the other inhibitors. The residue Thr124 showed higher energy contributions to
1 (−0.40 ± 0.02 kcal/mol) binding to 11β-HSD1 in comparison with
11 (−0.21 ± 0.02 kcal/mol) and
14 (−0.05 ± 0.02 kcal/mol) since the side chain of Thr124 has interaction with the dimethylpiperidyl moiety of
1. Among the hydrophobic residues in the binding pocket of 11β-HSD1, the residue Val231 contributes more than twice to interact with
1 (−2.57 ± 0.02 kcal/mol) than
11 (−1.12 ± 0.02 kcal/mol) and
14 (−1.13 ± 0.02 kcal/mol). This observation is consistent with the docking result that compound
1 with the 3-fluoro-4-methylphenyl moiety has strong hydrophobic interaction with the side chain of residue Val231. Therefore, we may safely conclude that Thr124, Tyr183, and Val 231 could be the key residues for inhibitors binding to 11β-HSD1. In order to gain insight into the energy contributions of the important residues mentioned above and to represent the results more intuitively, we divided each residue′s contribution into polar and nonpolar components (
Figure S6 in the Supplementary Materials). As shown in
Figure S6a, it is further confirmed that the van der Waals interactions and the nonpolar solvation energies regarding residues Thr124, Ala172, Tyr177, Pro178, Val227, and Val231 provide dominant forces for the binding process and are responsible for the difference between the binding free energies of the three complexes. As illustrated in
Figure S6b, the total electrostatic and polar solvation energies of Ser170 and Val180 participate in yielding energetic differences for compound
14 from the other compounds. In general, Ser170 has significantly hydrophilic energy contributions for these three compounds, while Tyr183 has a favorable hydrophobic contribution to the binding of an inhibitor since its aromatic ring. In the meantime, Tyr183 also shows greater hydrophilic energy contributions for these three compounds. Therefore, Tyr183 plays a more critical role in forming H-bond interaction with an active inhibitor than Ser170. Taken together, we may conclude that the H-bond interactions with Ser170 and Tyr183, and the hydrophobic interactions with Thr124, Tyr177, Tyr183, Val227, and Val231 play important roles in improving the 11β-HSD1 inhibitory activity.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}