
Boronic Acid Group: A Cumbersome False Negative Case in the Process of Drug Design



Abstract
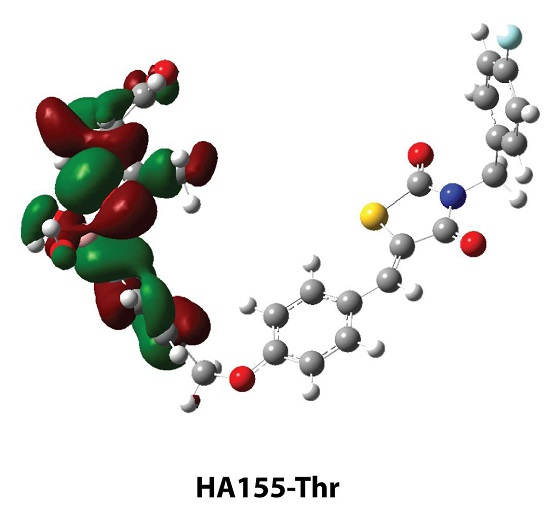
:
1. Introduction
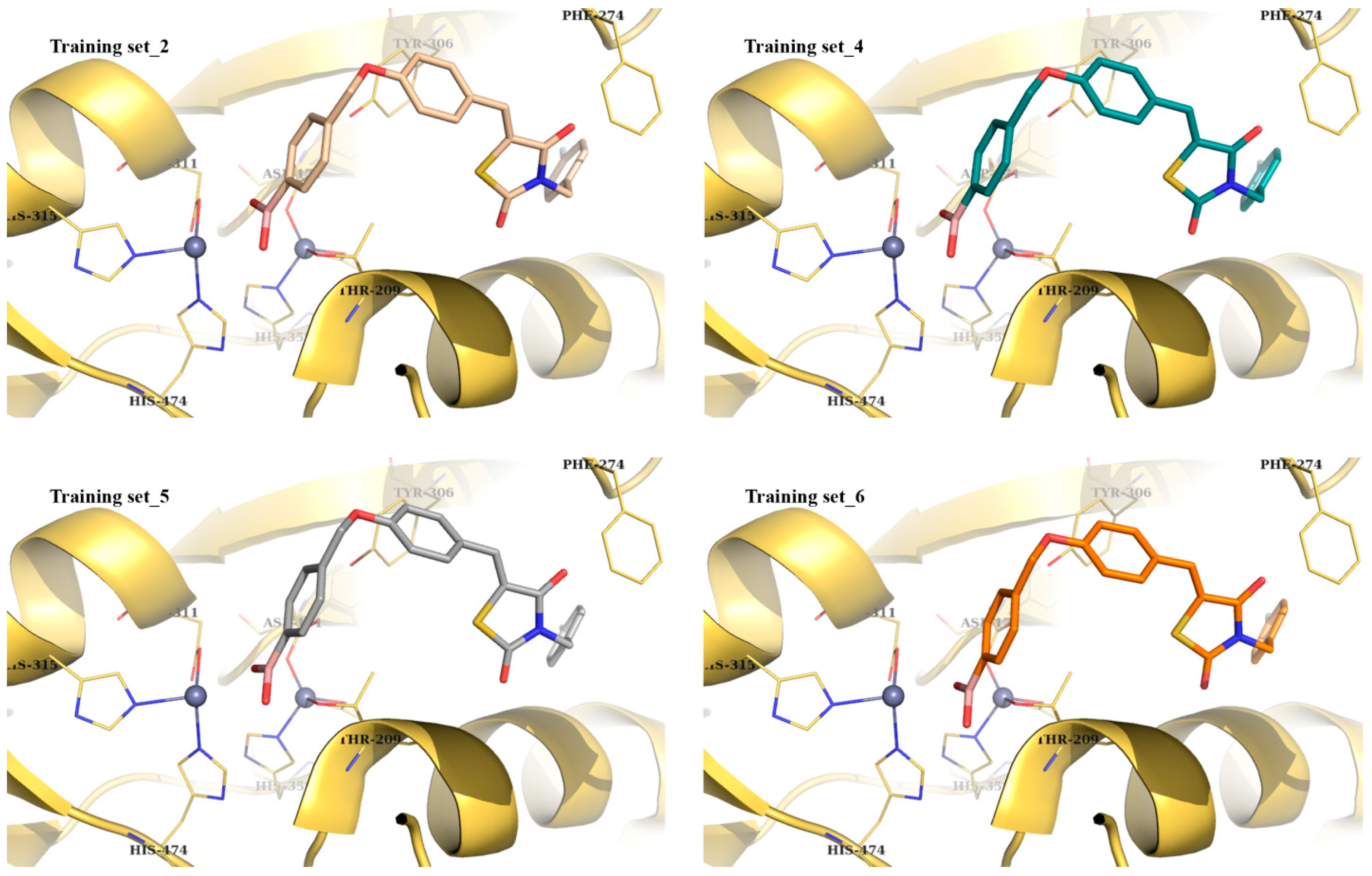
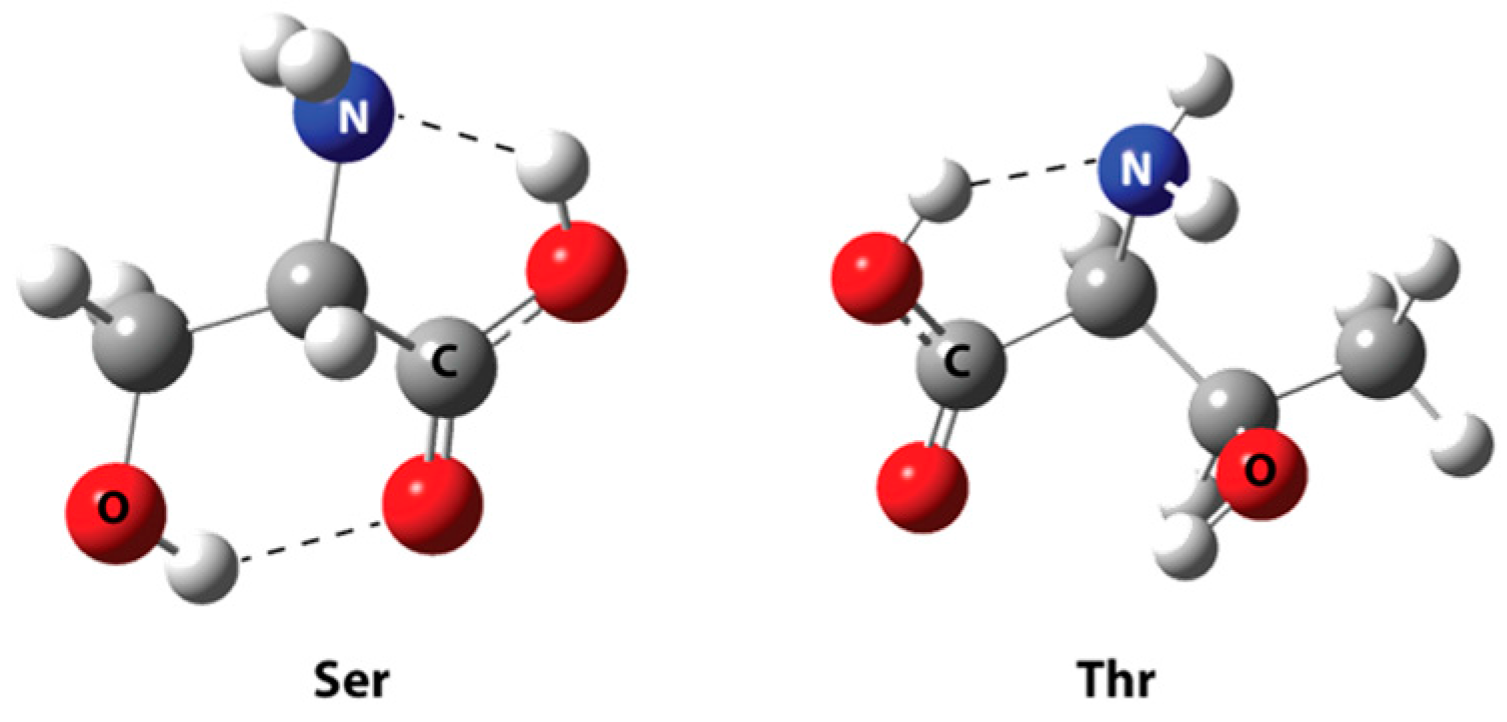
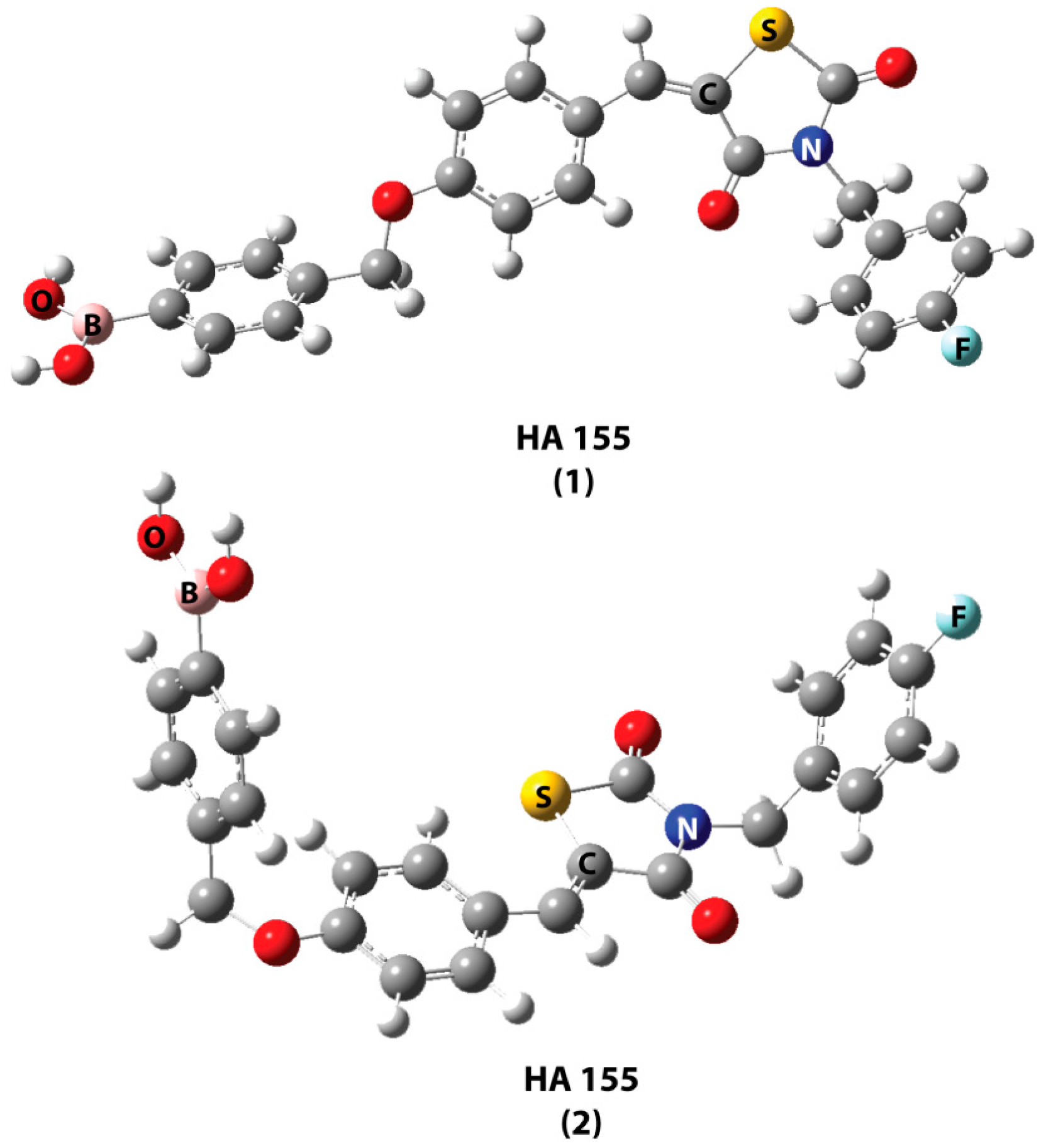
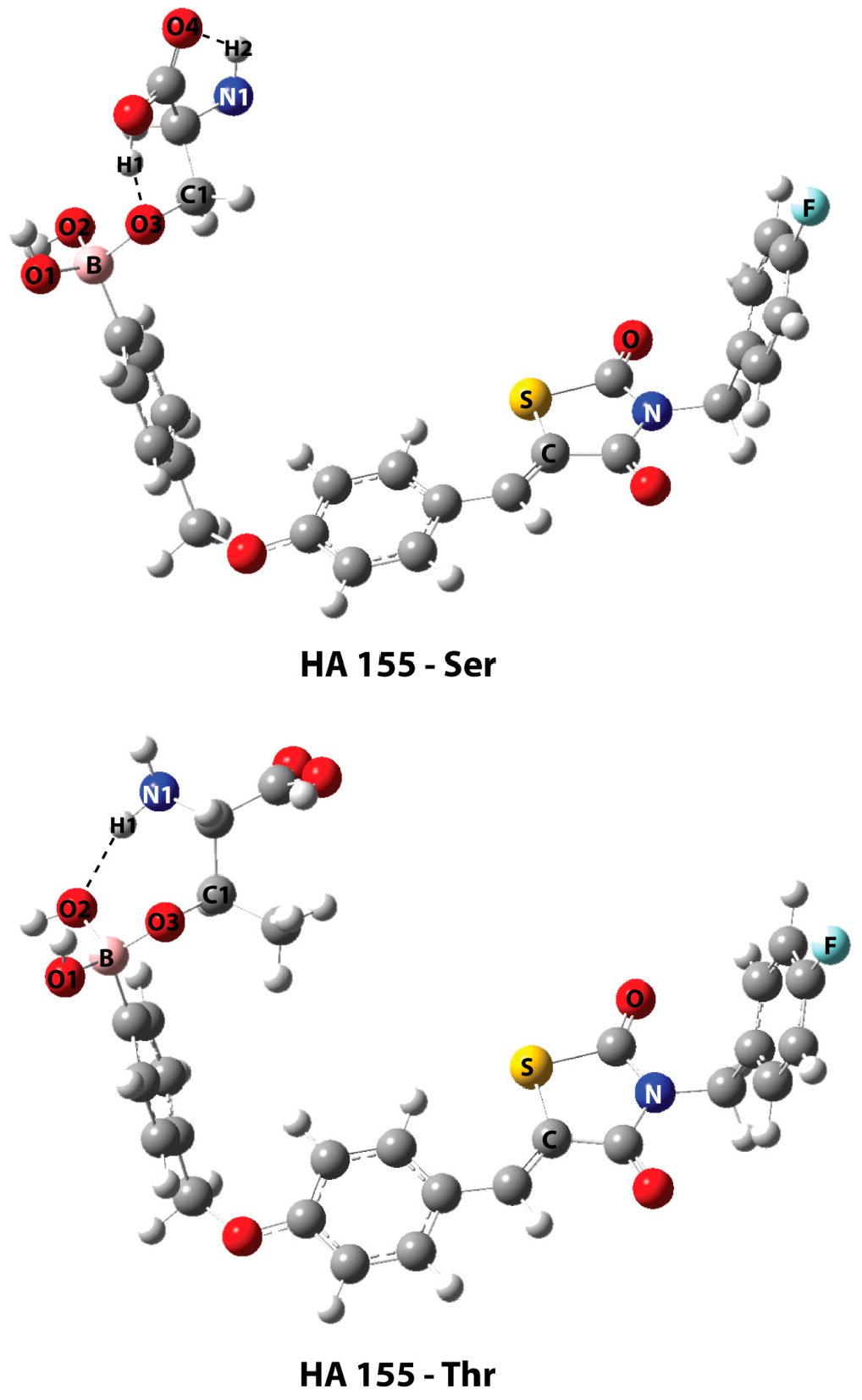
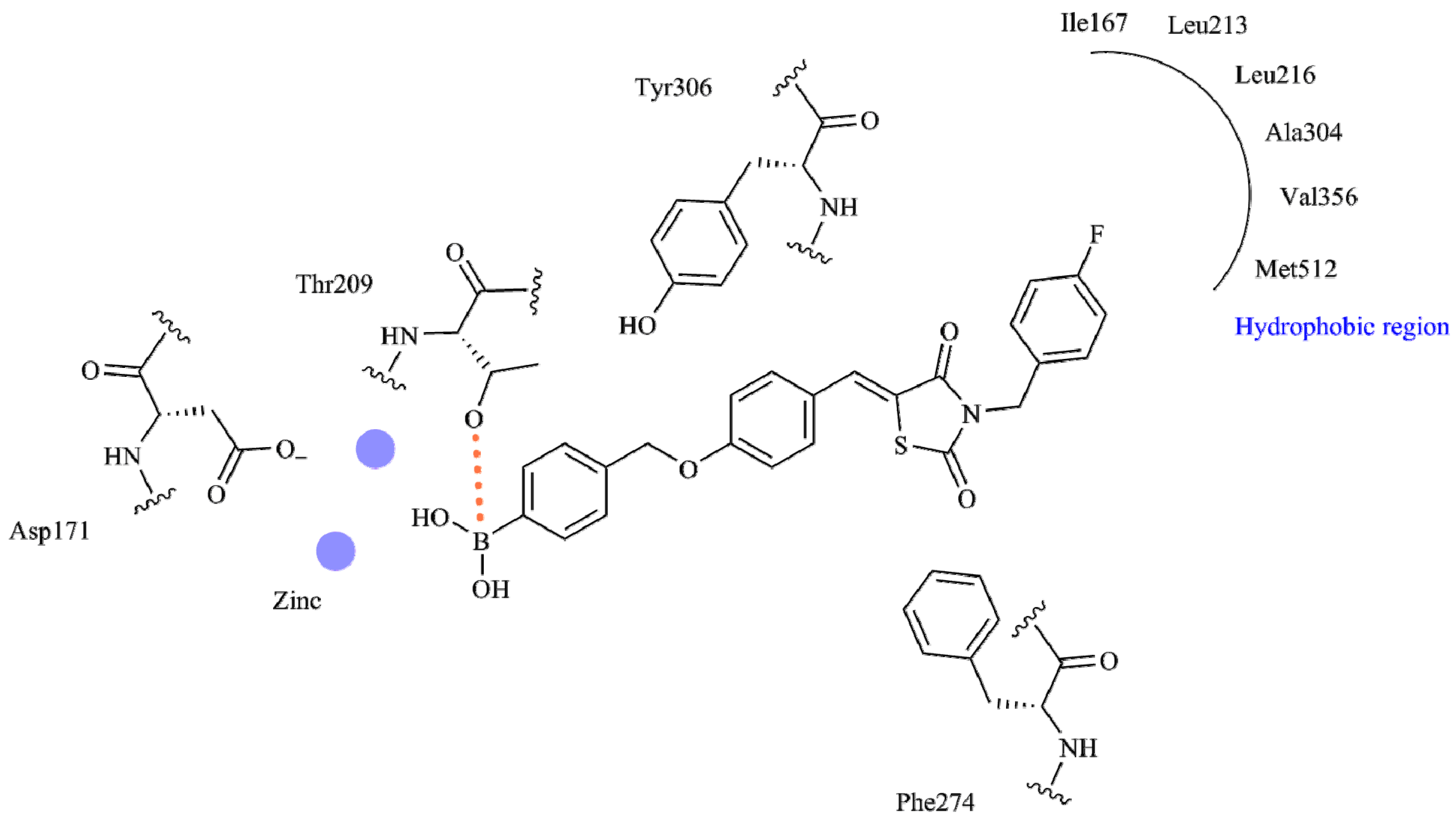
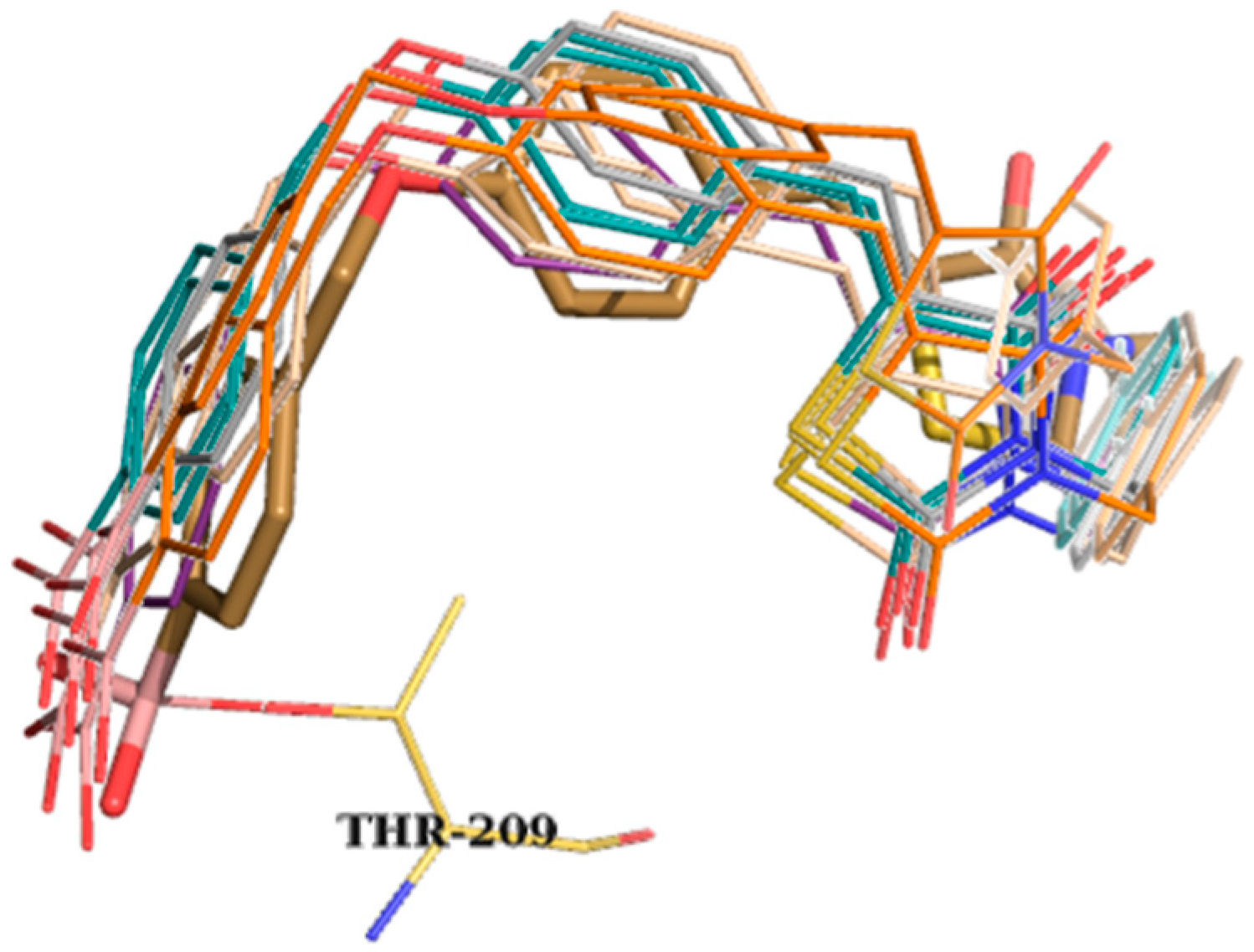
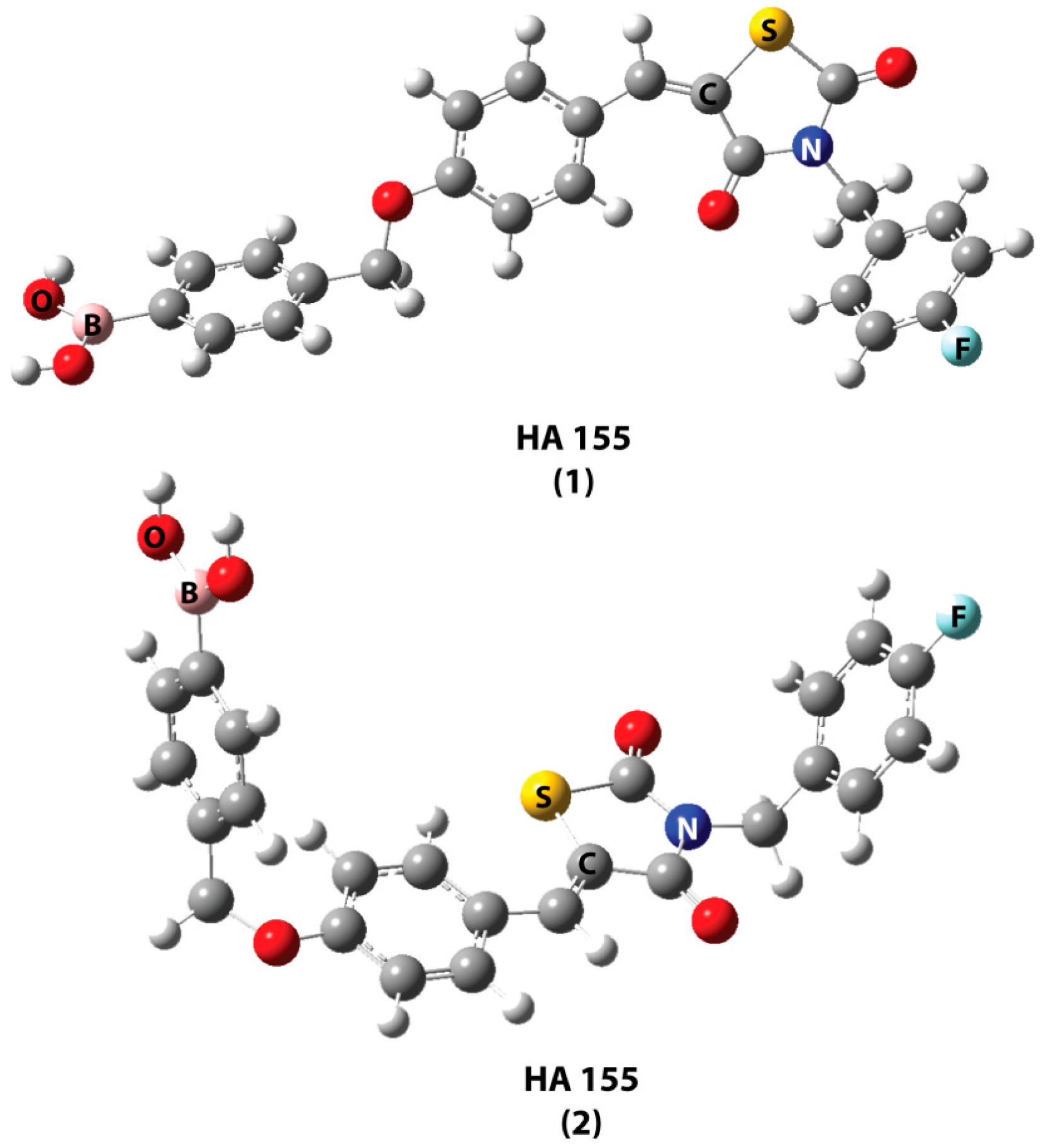
2. Results and Discussion
3. Computational Details
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2D | two-dimensional |
| 3D | three-dimensional |
| ATX | autotaxin |
| B3LYP | Becke three-parameter Lee-Yang-Par |
| CPU | central processing unit |
| DFT | density functional theory |
| HB | hydrogen bond |
| HOMO | highest occupied molecular orbital |
| HTS | high-throughput screening |
| LPA | lysophosphatidic acid |
| LPC | lysophosphatidylcholine |
| NBO | natural bond orbital |
| PDB | protein data bank |
| PDE | phosphodiesterase |
| PPI | protein-protein interaction |
| RAM | random access memory |
| QSAR | quantitative structure-activity relationship |
| XRD | X-ray diffraction |
References
- Ballatore, C.; Huryn, D.M.; Smith, A.B., 3rd. Carboxylic acid (bio)isosteres in drug design. ChemMedChem 2013, 8, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Behnke, M.; Chen, S.; Cruickshank, A.A.; Dick, L.R.; Grenier, L.; Klunder, J.M.; Ma, Y.T.; Plamondon, L.; Stein, R.L. Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998, 8, 333–338. [Google Scholar] [CrossRef]
- Lesnikowski, Z.J. Recent developments with boron as a platform for novel drug design. Expert Opin. Drug. Discov. 2016, 11, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Smoum, R.; Al-Quntar, A.A.; Abu Ali, H.; Pergament, I.; Srebnik, M. Natural occurrence of boron-containing compounds in plants, algae and microorganisms. Plant Sci. 2002, 163, 931–942. [Google Scholar] [CrossRef]
- Trippier, P.C.; McGuigan, C. Boronic acids in medicinal chemistry: Anticancer, antibacterial and antiviral applications. MedChemComm 2010, 1, 183–198. [Google Scholar] [CrossRef]
- Das, B.C.; Thapa, P.; Karki, R.; Schinke, C.; Das, S.; Kambhampati, S.; Banerjee, S.K.; Van Veldhuizen, P.; Verma, A.; Weiss, L.M.; et al. Boron chemicals in diagnosis and therapeutics. Future Med. Chem. 2013, 5, 653–676. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Aligaga, K.; Bermejo-Bescos, P.; Martin-Aragon, S.; Csaky, A.G. Discovery of alkenylboronic acids as neuroprotective agents affecting multiple biological targets involved in Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2013, 23, 426–429. [Google Scholar] [CrossRef] [PubMed]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crepin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.; Ovaa, H. Chemical evolution of autotaxin inhibitors. Chem. Rev. 2012, 112, 2593–2603. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.; Hendrickx, L.J.; van Tol, R.J.; Hausmann, J.; Perrakis, A.; Ovaa, H. Structure-based design of novel boronic acid-based inhibitors of autotaxin. J. Med. Chem. 2011, 54, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.; Dong, A.; van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; van Tilburg, E.W.; Schuurman, K.; van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.; van Meeteren, L.A.; Egan, D.A.; van Tilburg, E.W.; Moolenaar, W.H.; Ovaa, H. Discovery and optimization of boronic acid based inhibitors of autotaxin. J. Med. Chem. 2010, 53, 4958–4967. [Google Scholar] [CrossRef] [PubMed]
- Akama, T.; Baker, S.J.; Zhang, Y.K.; Hernandez, V.; Zhou, H.; Sanders, V.; Freund, Y.; Kimura, R.; Maples, K.R.; Plattner, J.J. Discovery and structure-activity study of a novel benzoxaborole anti-inflammatory agent (AN2728) for the potential topical treatment of psoriasis and atopic dermatitis. Bioorg. Med. Chem. Lett. 2009, 19, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Minkkila, A.; Saario, S.M.; Kasnanen, H.; Leppanen, J.; Poso, A.; Nevalainen, T. Discovery of boronic acids as novel and potent inhibitors of fatty acid amide hydrolase. J. Med. Chem. 2008, 51, 7057–7060. [Google Scholar] [CrossRef] [PubMed]
- Knott, K.; Fishovitz, J.; Thorpe, S.B.; Lee, I.; Santos, W.L. N-Terminal peptidic boronic acids selectively inhibit human ClpXP. Org. Biomol. Chem. 2010, 8, 3451–3456. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Soria, G.; Gonzalez-Galvez, G.; Argoud, G.M.; Gerstman, M.; Littlejohn, T.W., 3rd; Schwartz, S.L.; O’Farrell, A.M.; Li, X.; Cherrington, J.M.; Bennett, C.; et al. The dipeptidyl peptidase-4 inhibitor PHX1149 improves blood glucose control in patients with type 2 diabetes mellitus. Diabetes Obes. Metab. 2008, 10, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.S.; Usui, T.; Nabeyama, W.; Morita, H.; Fukuzawa, K.; Nakamura, H. Discovery of boron-conjugated 4-anilinoquinazoline as a prolonged inhibitor of EGFR tyrosine kinase. Org. Biomol. Chem. 2009, 7, 4415–4427. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, A.; McCarthy, K.A.; Kelly, M.A.; Gao, J. Targeting bacteria via iminoboronate chemistry of amine-presenting lipids. Nat. Commun. 2015, 6, 6561. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.E.; Alley, M.R.; Sader, H.S.; Biedenbach, D.J.; Jones, R.N. Potency and spectrum of activity of AN3365, a novel boron-containing protein synthesis inhibitor, tested against clinical isolates of Enterobacteriaceae and nonfermentative Gram-negative bacilli. Antimicrob. Agents Chemother. 2013, 57, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, K.; Tambov, K.V.; Gundla, R.; Manaev, A.V.; Yarovenko, V.; Traven, V.F.; Neamati, N. Discovery of 3-acetyl-4-hydroxy-2-pyranone derivatives and their difluoridoborate complexes as a novel class of HIV-1 integrase inhibitors. Bioorg. Med. Chem. 2008, 16, 8988–8998. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.M.; Jolly, R.A.; Linder, R.J. Boronic Acids and Derivatives-Probing the Structure-Activity Relationships for Mutagenicity. Org. Process Res. Dev. 2015, 19, 1507–1516. [Google Scholar] [CrossRef]
- Katsamakas, S.; Bermperoglou, E.; Hadjipavlou-Litina, D. Considering Autotaxin Inhibitors in Terms of 2D-QSAR and 3D-Mapping- Review and Evaluation. Curr. Med. Chem. 2015, 22, 1428–1461. [Google Scholar] [CrossRef] [PubMed]
- Katsamakas, S.; Hadjipavlou-Litina, D. Boronic Acid Based Inhibitors of Autotaxin: Understanding their Biological Role in Terms of Quantitative Structure Activity Relationships (QSAR). Lett. Drug Des. Discov. 2013, 10, 11–18. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Tsilikounas, E.; Kettner, C.A.; Bachovchin, W.W. Identification of serine and histidine adducts in complexes of trypsin and trypsinogen with peptide and nonpeptide boronic acid inhibitors by proton NMR spectroscopy. Biochemistry 1992, 31, 12839–12846. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, A.A.; Muthusamy, R.; Soliman, M.E. New drug design with covalent modifiers. Expert Opin. Drug Discov. 2016, 11, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Mah, R.; Thomas, J.R.; Shafer, C.M. Drug discovery considerations in the development of covalent inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Gestwicki, J.E. Features of protein-protein interactions that translate into potent inhibitors: Topology, surface area and affinity. Expert Rev. Mol. Med. 2012, 14, e16. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphics System; Version 1.4.1; Schrödinger, LLC: Cambridge, MA, USA; Available online: https://www.pymol.org. (accessed on 6 September 2016).
- McGann, M. FRED pose prediction and virtual screening accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef] [PubMed]
- McGaughey, G.B.; Sheridan, R.P.; Bayly, C.I.; Culberson, J.C.; Kreatsoulas, C.; Lindsley, S.; Maiorov, V.; Truchon, J.F.; Cornell, W.D. Comparison of topological, shape, and docking methods in virtual screening. J. Chem. Inf. Model. 2007, 47, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- McGann, M.R.; Almond, H.R.; Nicholls, A.; Grant, J.A.; Brown, F.K. Gaussian docking functions. Biopolymers 2003, 68, 76–90. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Model. 2012, 52, 2919–2936. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, P.C.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A. 02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z=11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Amovilli, C.; Barone, V.; Cammi, R.; Cancès, E.; Cossi, M.; Mennucci, B.; Pomelli, C.S.; Tomasi, J. Recent Advances in the Description of Solvent Effects with the Polarizable Continuum Model. In Advances in Quantum Chemistry; Academic Press: San Diego, CA, USA, 1998; Volume 32, pp. 227–261. [Google Scholar]
- Tomasi, J.; Persico, M. Molecular-Interactions in Solution—An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
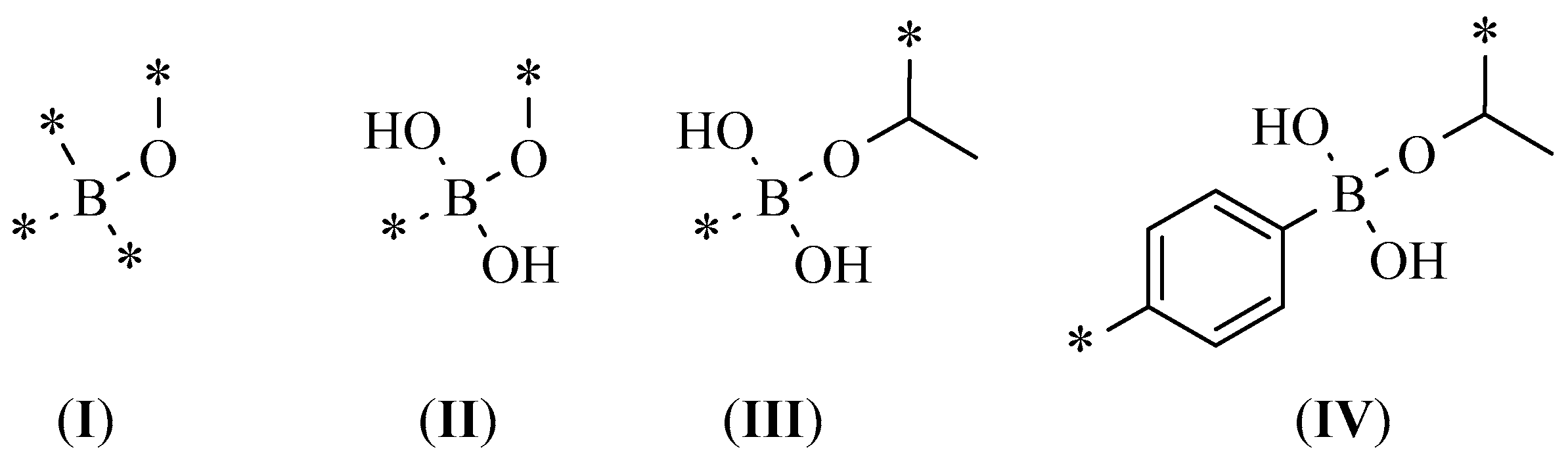
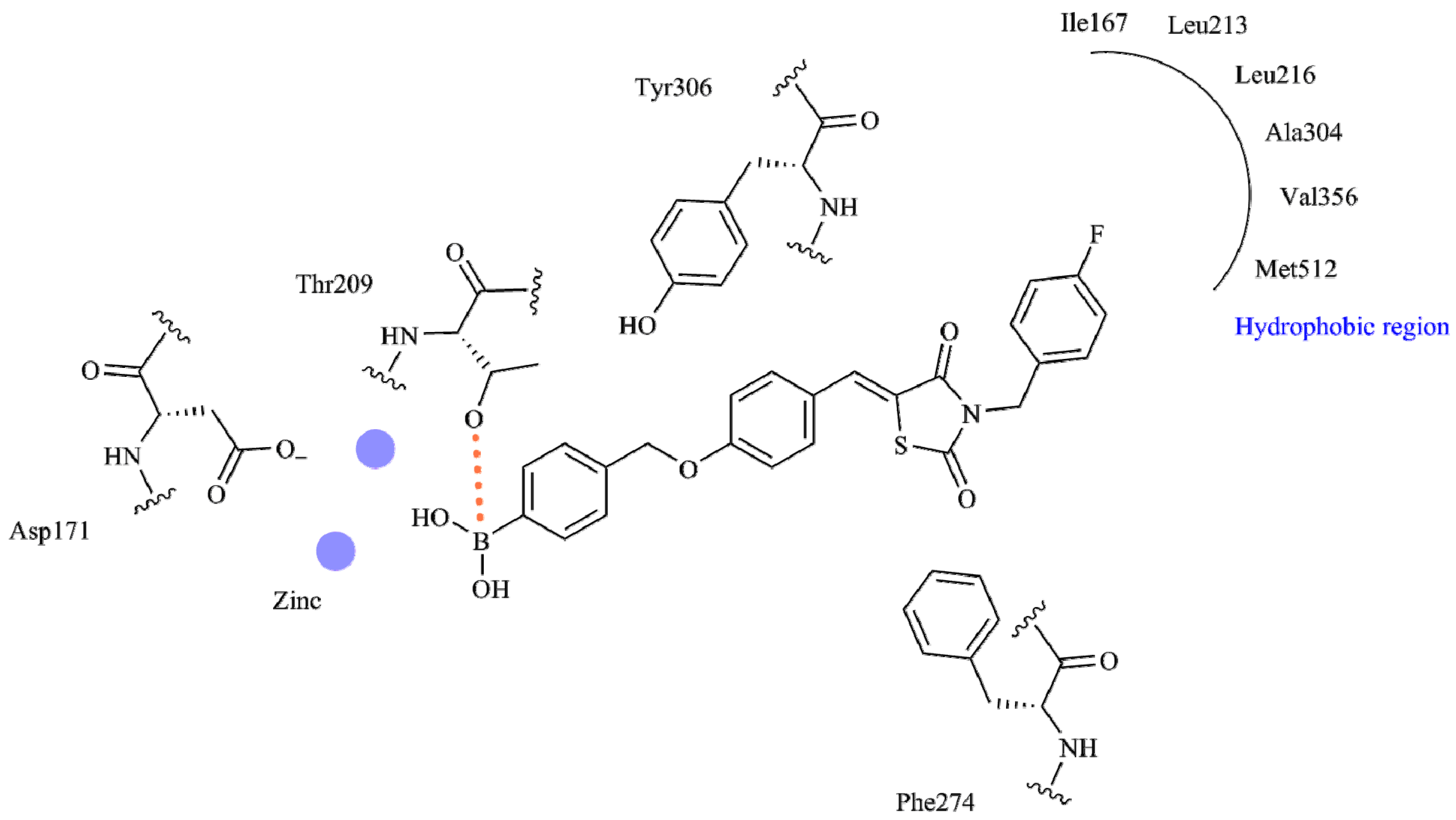
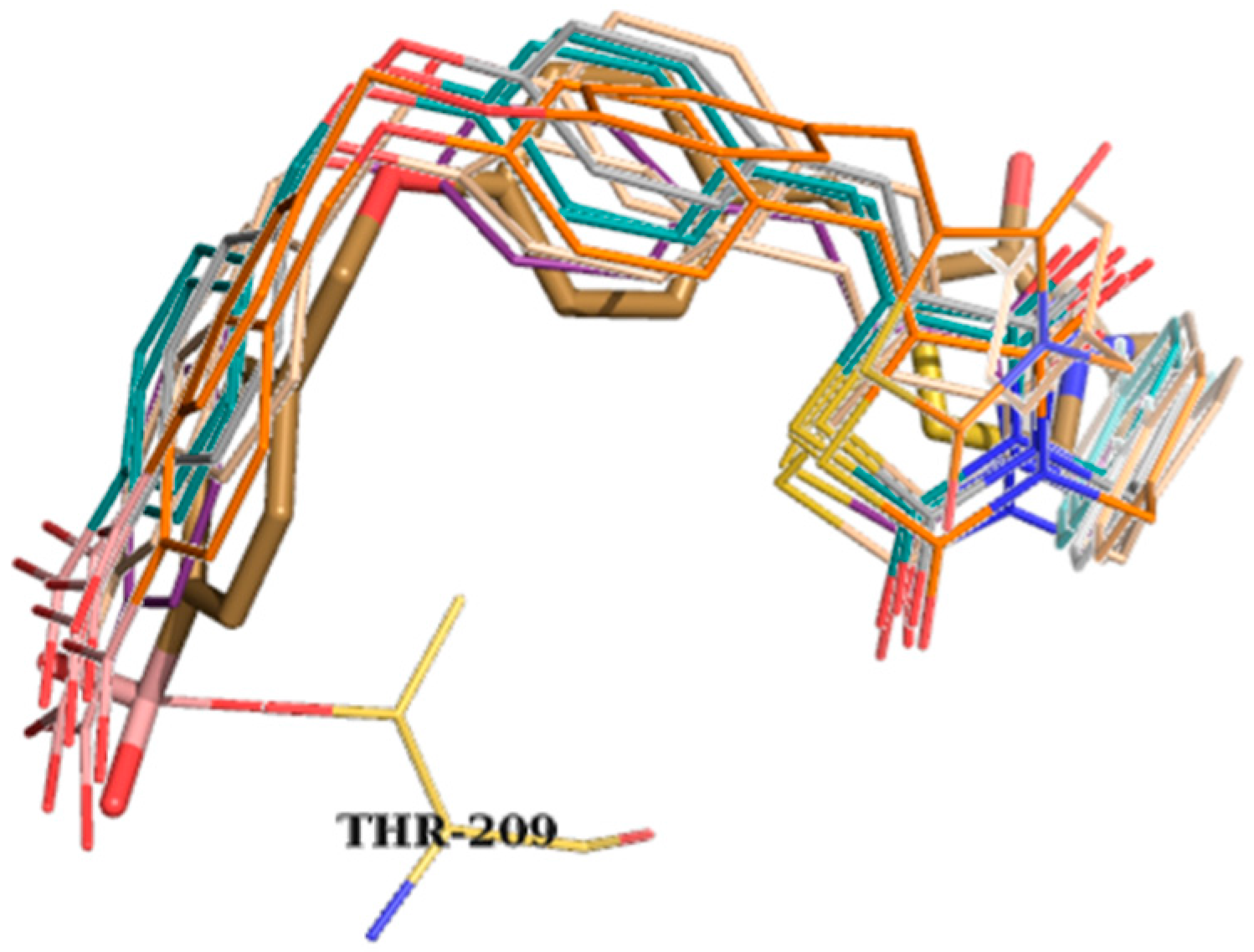

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
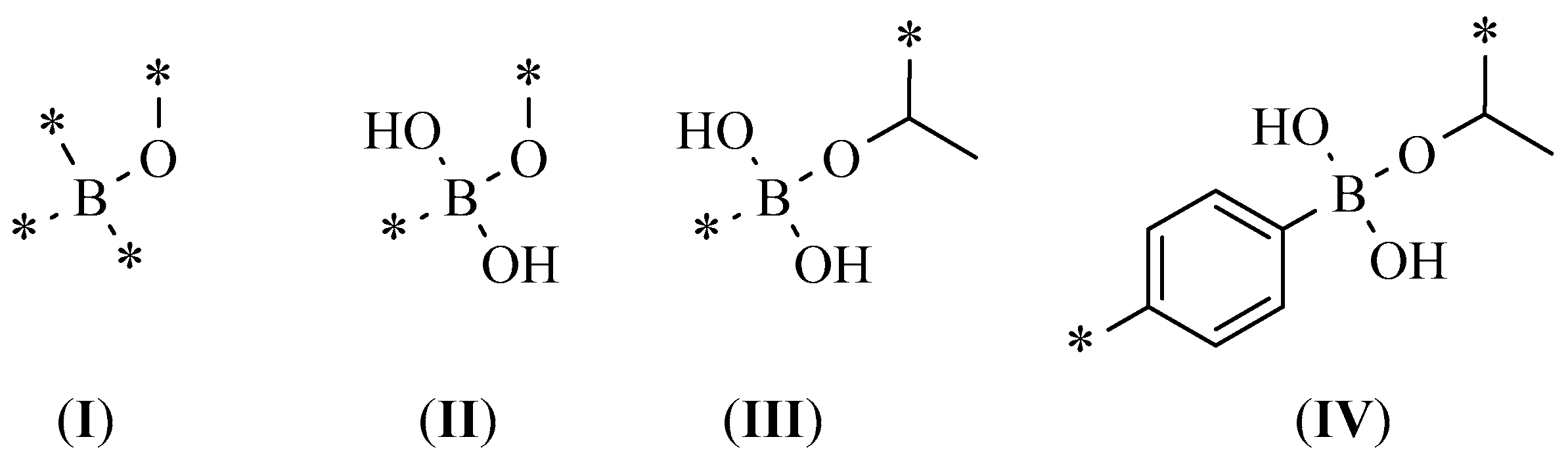{kind=link}
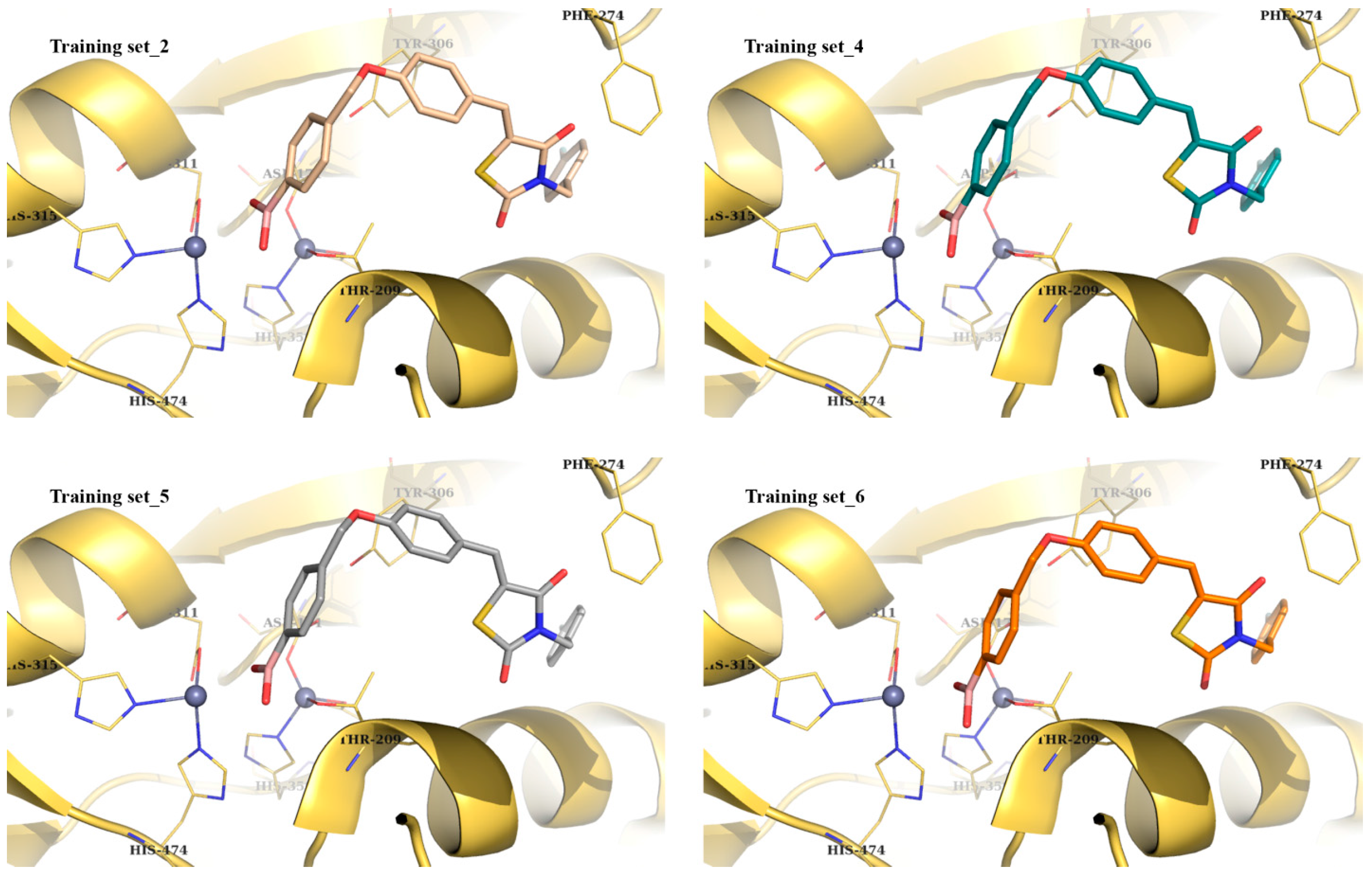
| Best Poses | General Score (%) | HBs Formed (%) | Chemgauss4 Score |
|---|---|---|---|
| # Training set_1 | 68–93 | 87–95 | −5.64 |
| Training set_2 | 40–46 | 82–90 | −9.48 |
| Training set_3 | 20 | 94 | −8.67 |
| Training set_4 | 77–82 | 92–98 | −9.35 |
| Training set_5 | 82–85 | 71–95 | −9.21 |
| Training set_6 | 66–92 | 92–93 | −9.05 |
| * Training set_7 | 7–30 | 55–80 | −12.18 (off position) |
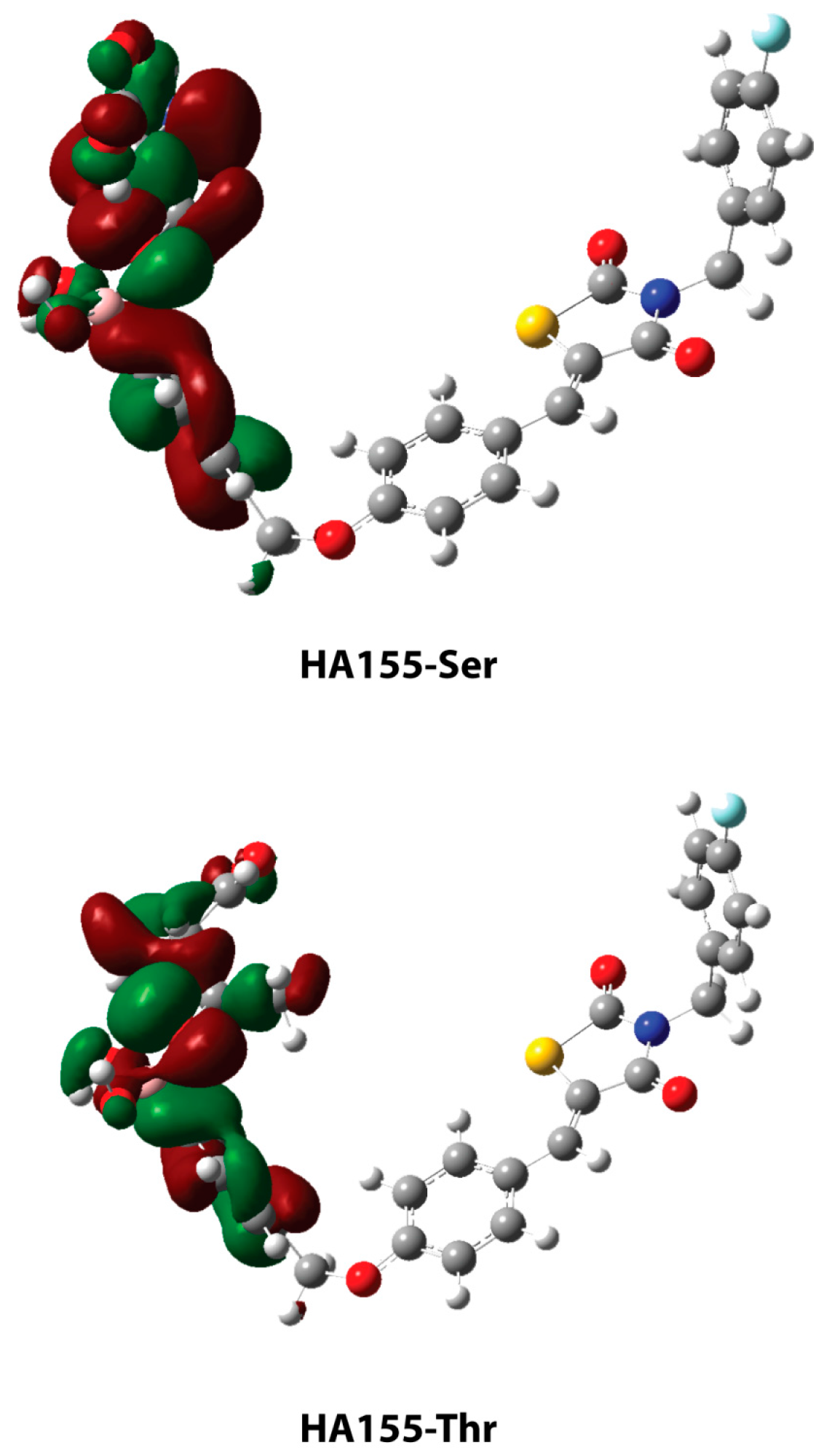
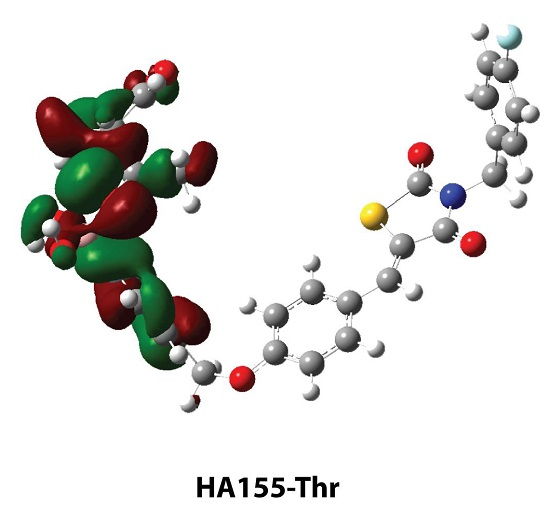
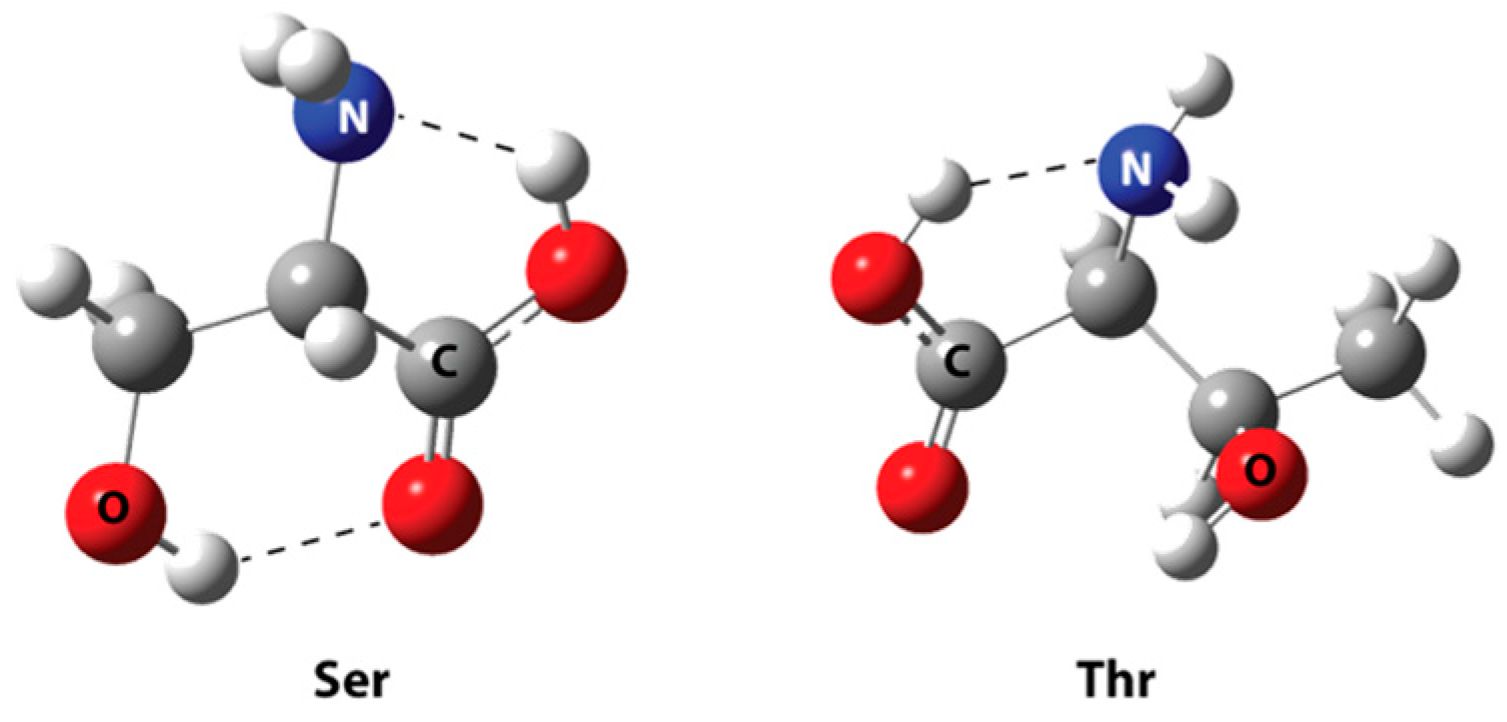
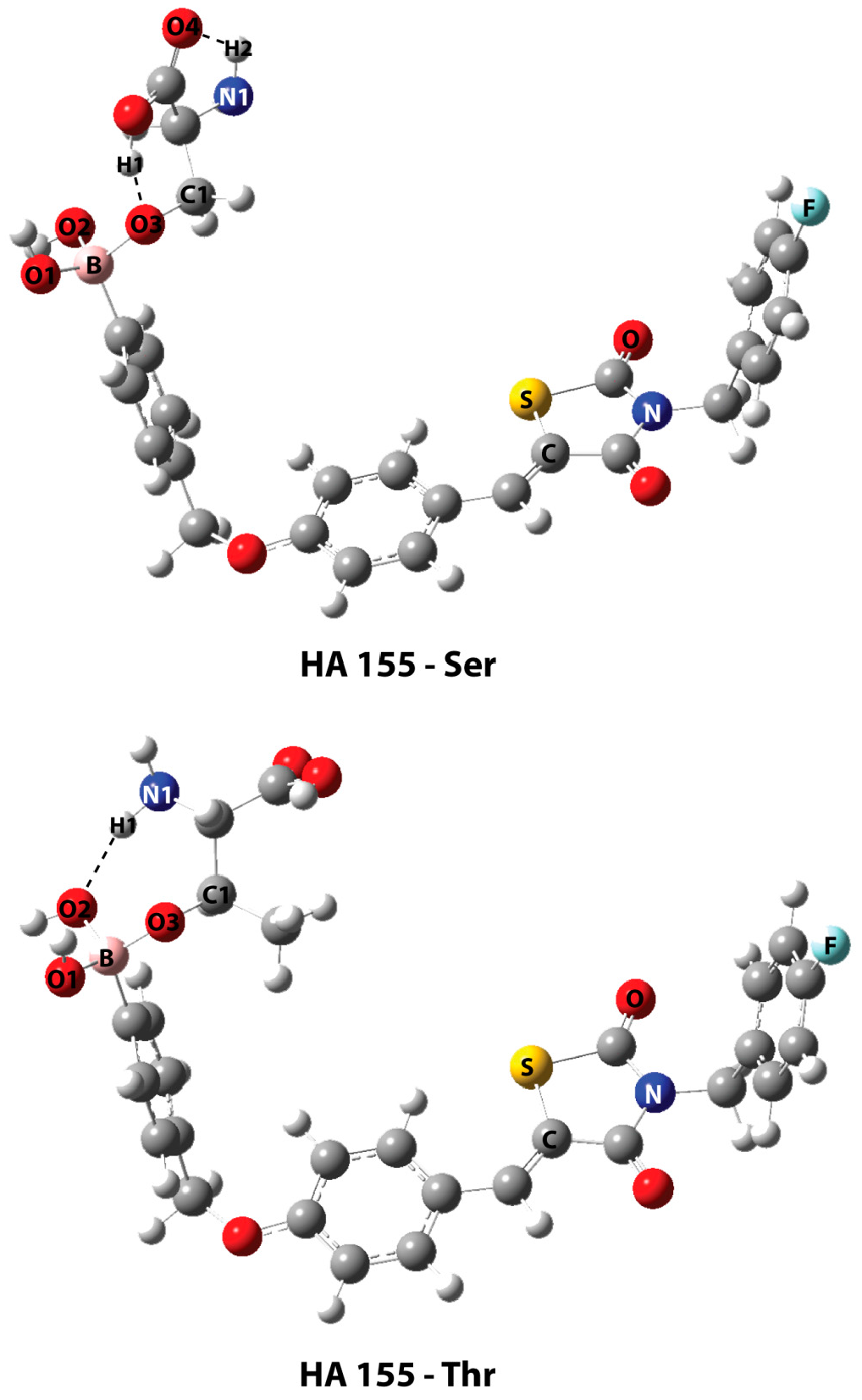
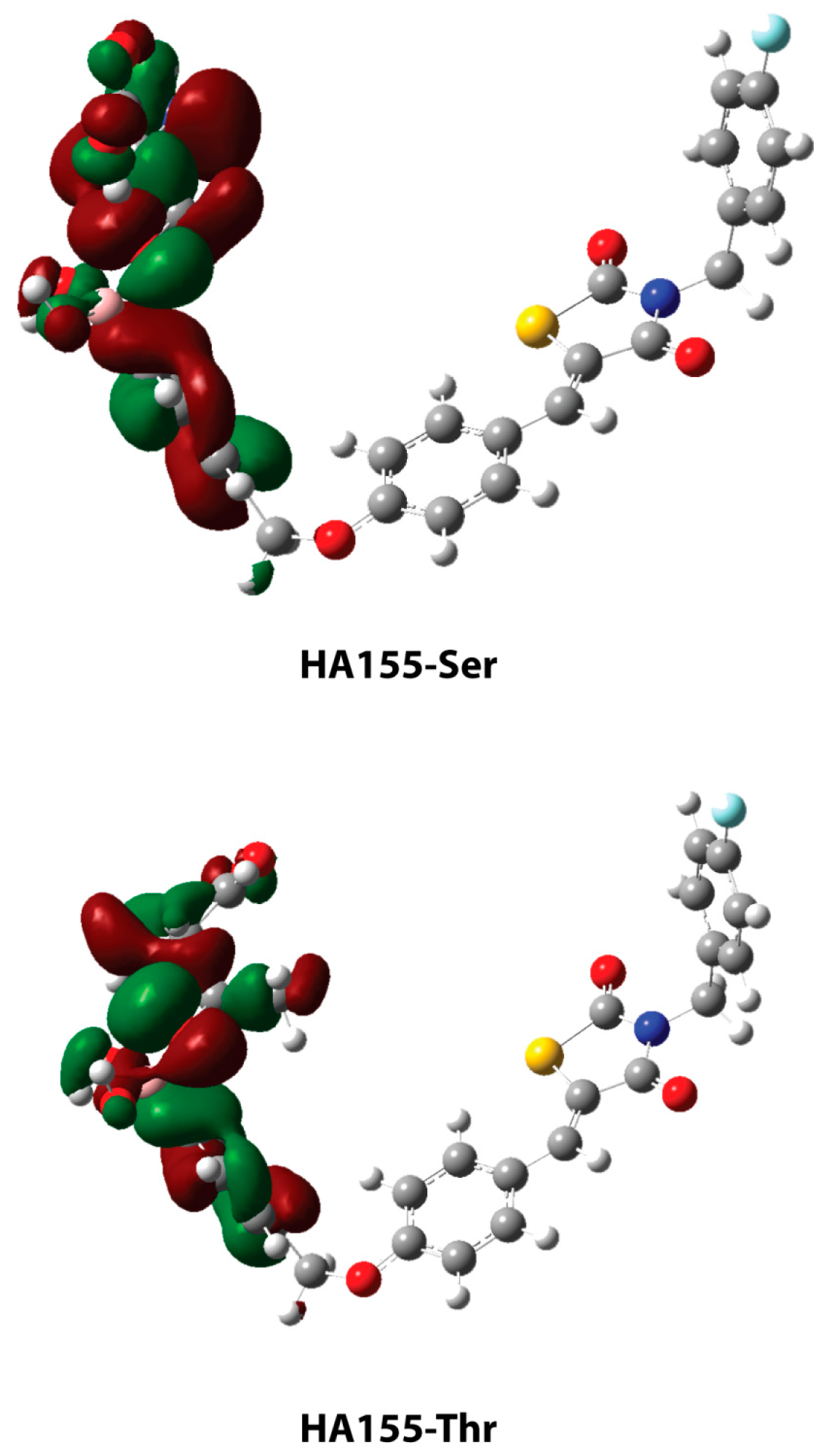
| HA155-Ser | HA155-Thr | |
| Bonds * | ||
| B-O1 | 1.460 | 1.465 |
| B-O2 | 1.484 | 1.496 |
| B-O3 | 1.521 | 1.495 |
| Angles | ||
| B-O3-C1 | 119.6 | 122.8 |
| Intermolecular Hydrogen Bond | ||
| H1…O3 | 1.568 | - |
| H2…O4 | 2.210 | - |
| H1…O2 | - | 1.955 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsamakas, S.; Papadopoulos, A.G.; Hadjipavlou-Litina, D. Boronic Acid Group: A Cumbersome False Negative Case in the Process of Drug Design. Molecules 2016, 21, 1185. https://doi.org/10.3390/molecules21091185
Katsamakas S, Papadopoulos AG, Hadjipavlou-Litina D. Boronic Acid Group: A Cumbersome False Negative Case in the Process of Drug Design. Molecules. 2016; 21(9):1185. https://doi.org/10.3390/molecules21091185
Chicago/Turabian StyleKatsamakas, Sotirios, Anastasios G. Papadopoulos, and Dimitra Hadjipavlou-Litina. 2016. "Boronic Acid Group: A Cumbersome False Negative Case in the Process of Drug Design" Molecules 21, no. 9: 1185. https://doi.org/10.3390/molecules21091185
APA StyleKatsamakas, S., Papadopoulos, A. G., & Hadjipavlou-Litina, D. (2016). Boronic Acid Group: A Cumbersome False Negative Case in the Process of Drug Design. Molecules, 21(9), 1185. https://doi.org/10.3390/molecules21091185