2. Results and Discussion
Compound
1 was obtained as a yellow solid. The quasi-molecular ion at
m/
z 355.1550 [M + H]
+ obtained by HRESIMS indicated the molecular formula of
1 was C
21H
22O
5 with 11 degrees of unsaturation. The
13C-NMR spectrum combined with the DEPT-135 spectrum showed 21 signals (
Table 1), assigned to ten sp
2 quaternary carbons [including one ketone carbonyl (δ
C 197.5), nine aromatic/olefinic carbons (δ
C 162.8, 162.4, 154.5, 147.1, 138.5, 133.2, 124.8, 116.8, and 113.6), five sp
2 methine carbons (δ
C 137.6, 121.8, 119.5, 116.8, and 109.3), one sp
3 oxygenated methine carbon (δ
C 103.4), one sp
3 methylene carbon (δ
C 27.8), and four methyl carbons (δ
C 56.9, 25.8, 21.8, and 17.8). In the
1H-NMR spectrum of
1 (
Table 1), the characteristic signals of two exchangeable protons (δ
H 13.62 (1H, s) and 11.46 (1H, s)), five aromatic or olefinic protons (δ
H 7.33 (1H, d,
J = 8.3 Hz), 6.94 (1H, s), 6.87 (1H, s), 6.58 (1H, d,
J = 8.3 Hz), and 5.33 (1H, br t,
J = 7.3 Hz), one sp
3 oxygenated methine proton (δ
H 5.64 (1H, s)), one sp
3 methylene (δ
H 3.34 (2H, d,
J = 7.3 Hz)), and four methyl protons (δ
H 3.57 (3H, s), 2.37 (3H, s), 1.77 (3H, br s), and 1.73 (3H, br s)) were observed. The non-exchangeable proton resonances were associated with the directly attached carbon atoms in the HSQC experiment (
Table 1). The analysis of the
1H-
1H COSY experiment and the coupling values of the protons indicated the presence of two subunits (C-7–C-8 and C-1′–C-2′) as shown in
Figure 2. Combined with the analysis of the
1H-
1H COSY experiment and molecular formula, the HMBC correlations from H
3-1 to C-2/C-3/C-15, from H-3 to C-1/C-5/C-13/C-15, from H-5 to C-3/C-6/C-13/5-O
CH
3, from H-7 to C-6/C-9/C-11, from H-8 to C-6/C-10, from H
2-1′ to C-8/C-9/C-10, from H
3-4′ to C-2′/C-3′/C-5′, from H
3-5′ to C-2′/C-3′/C-4′, from 5-OC
H3 to C-5, from 10-O
H to C-9/C-10/C-11, and from 14-O
H to C-13/C-14/C-15 revealed the planer structure of
1 (
Figure 2). On the basis of the above analyses, the planar structure of
1 (
Figure 2) was established, and it was named arugosin K. The assignments of all proton and carbon resonances are provided in
Table 1.
The specific rotation value of
1 was close to zero, indicating that
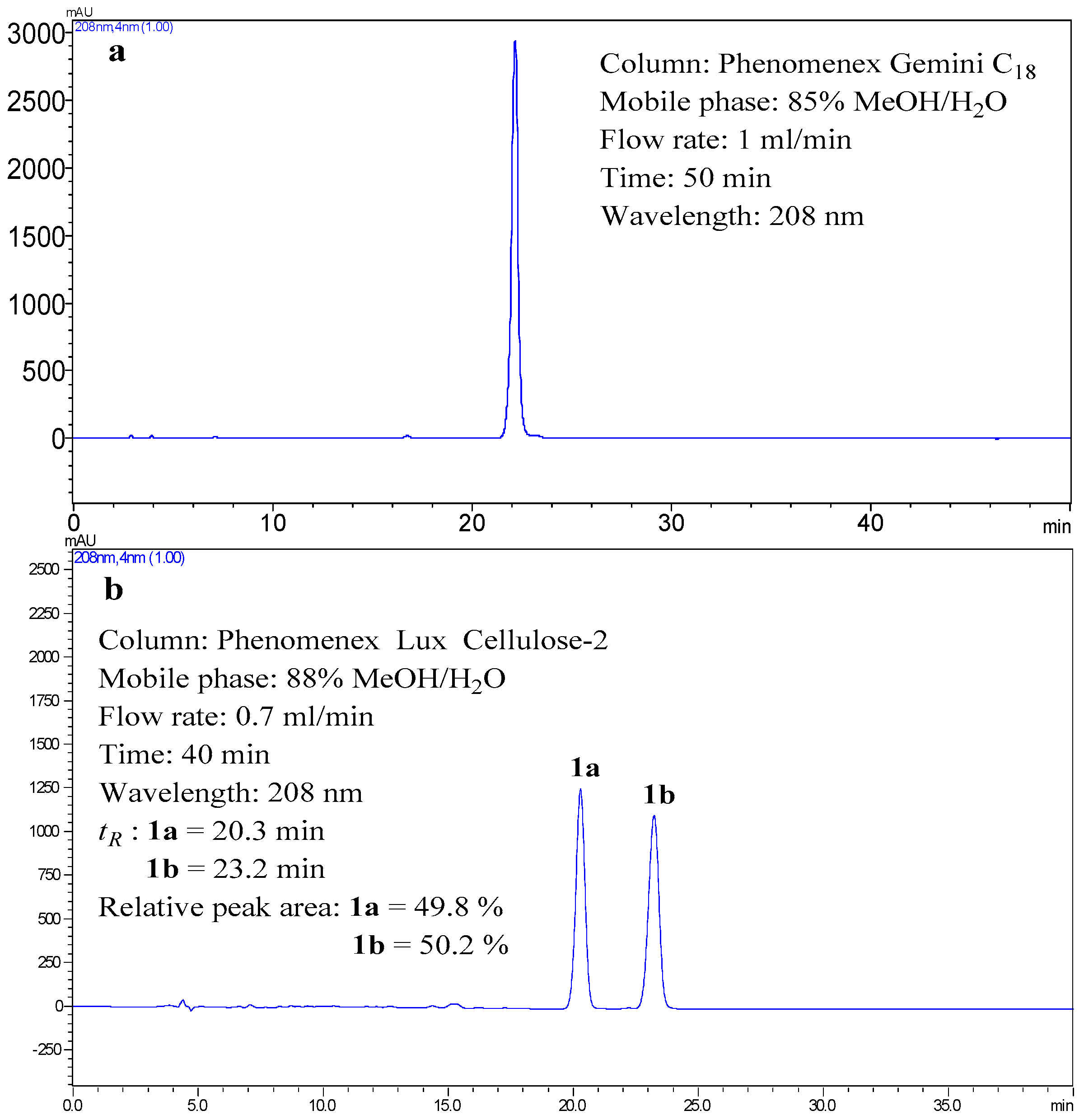
1 was a racemic mixture. Compound
1 displayed two peaks in a Phenomenex Lux Cellulose-2 chiral column (
Figure 3), which confirmed the above deduction. Therefore, (+)-
1a (
tR = 20.3 min) and (−)-
1b (
tR = 23.2 min) were isolated from
1 by a Phenomenex Lux Cellulose-2 chiral column using CH
3OH/H
2O (88/12,
v/
v) as eluent at a flow rate of 0.7 mL/min. The ECD spectra and
values of
1a (
+27.8 (
c 0.5, CHCl
3)) and
1b (
−27.0 (
c 0.5, CHCl
3)) showed their enantiomeric relationship.
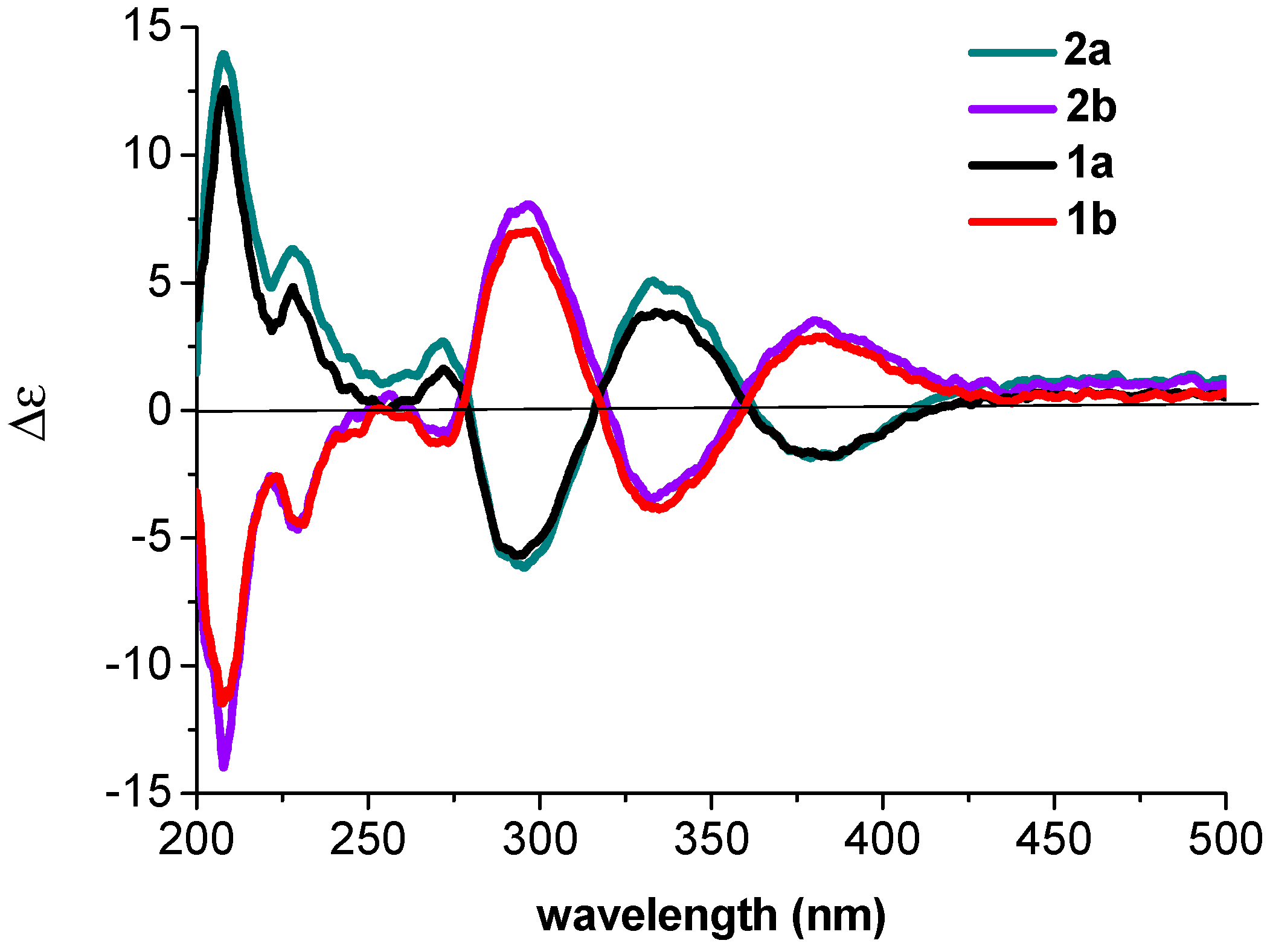
The absolute configurations of
1a and
1b were determined by quantum chemical ECD calculations at the APFD/6-311++G(2d,p) level. The predicted ECD curves of (5
S)-
1 and (5
R)-
1 (
Figure 4) were in good accordance with the experimental ECD for
1a and
1b, respectively. Therefore, the absolute configurations of
1a and
1b were identified as 5
S and 5
R, respectively.
Compound
2 was obtained as a yellow solid. The quasi-molecular ion at
m/
z 369.1695 [M + H]
+ by HRESIMS indicated the molecular formula of
2 was C
22H
24O
5 with 11 degrees of unsaturation. Except for the loss of an oxygenated methyl at δ
C 56.9 (5-O
CH
3) and the appearance of an additional oxygenated methylene at δ
C 65.3 (C-1′′) and methyl at δ
C 14.8 (C-2′′), the NMR data of
2 (
Table 1) were similar to those of
1 (
Table 1), which indicated that the 5-OCH
3 in
1 was replaced by 5-OCH
2CH
3 in
2. This deduction was supported by the
1H-
1H COSY correlations of Ha-1′′/Hb-1′′ and H
3-2′′ and the key HMBC correlations from H-5 to C-1′′, from Ha-1′′/Hb-1′′ to C-5/C-2′′, and from H
3-2′′ to C-1′′. On the basis of the exhaustive analysis of 2D NMR data (
1H-
1H COSY, HSQC, and HMBC) (
Table S2, Supplementary Materials), the planar structure of
2, named arugosin L, was established (
Figure 1).
Since
1 and
2 coexist in the same strain,
2 was also a racemic mixture, and it displayed two peaks in a Phenomenex Lux Cellulose-2 chiral column (
Figure S1, Supplementary Materials). Therefore, (+)-
2a (
tR = 22.6 min) and (−)-
2b (
tR = 26.6 min) were isolated from
2 by a Phenomenex Lux Cellulose-2 chiral column using CH
3OH/H
2O (88/12,
v/
v) as eluent at a flow rate of 0.7 mL/min. The ECD spectra and
values of
2a (
+27.4 (
c 0.5, CHCl
3)) and
2b (
−26.8 (
c 0.5, CHCl
3)) showed their enantiomeric relationship. Since the ECD data of 2a were similar to that of
1a (
Figure 5), the absolute configuration of
2a was determined as 5
S (
Figure 5). The similar observation was found between
2b and
1b. Therefore, the absolute configurations of
2a and
2b were assigned as 5
S and 5
R, respectively.
Compound
3 was obtained as a yellow solid. The quasi-molecular ion at
m/
z 355.1541 [M + H]
+ by HRESIMS indicated the molecular formula of
3 was C
21H
22O
5 with 11 degrees of unsaturation, which indicated that
3 was the isomer of
1. Furthermore, the NMR data of
3 (
Table 1) were similar to those of
1, which suggested that
3 and
1 had the similar skeleton. The analysis of the
1H-
1H COSY experiment and the coupling values of the protons indicated the presence of two subunits (C-7–C-8 and C-1′–C-2′) as shown in
Figure 2. Combined with the analysis of the
1H–
1H COSY experiment and molecular formula, the HMBC correlations (
Figure 2) from H
3-1 to C-2/C-3/C-15, from H-3 to C-1/C-5/C-13/C-15, from H-5 to C-3/C-10/C-13/5-O
CH
3, from H-7 to C-6/C-9/C-11, from H-8 to C-6/C-10, from Ha-1′/Hb-1′ to C-8/C-9/C-10, from H
3-4′ to C-2′/C-3′/C-5′, from H
3-5′ to C-2′/C-3′/C-4′, from 5-OC
H3 to C-5, from 6-O
H to C-6/C-7/C-11, and from 14-O
H to C-13/C-14/C-15 confirmed the planar structure of
3, which is named arugosin M.
Since
1 and
3 coexist in the same strain,
3 was also a racemic mixture, and it displayed two peaks in a Phenomenex Lux Cellulose-2 chiral column (
Figure S2, Supplementary Materials). Therefore, (+)-
3a (
tR = 20.4 min) and (−)-
3b (
tR = 22.1 min) were isolated from
3 by a Phenomenex Lux Cellulose-2 chiral column using CH
3OH/H
2O (88/12,
v/
v) as eluent at a flow rate of 0.7 mL/min. The ECD spectra and
values of
3a (
+28.4 (
c 0.5, CHCl
3)) and
3b (
−27.6 (
c 0.5, CHCl
3)) showed their enantiomeric relationship. The absolute configurations of
3a and
3b were determined by quantum chemical ECD calculations at the APFD/6-311++g(2d,p) level. The predicted ECD curves of (5
S)-
3 and (5
R)-
3 were in good accordance with the experimental ECD for
3a and
3b, respectively (
Figure 6). Therefore, the absolute configurations of
3a and
3b were identified as 5
S and 5
R, respectively.
Compounds
4 and
5 were obtained as mixtures of yellow solids. The quasi-molecular ion at
m/
z 363.1213 [M + Na]
+ by HRESIMS indicated the molecular formulas of
4 and
5 were C
20H
20O
5, with 11 degrees of unsaturation. The NMR data of the mixtures presented two sets of signals, assigned to
4 and
5 respectively (
Table 2). The molecular formula of
4 was fourteen atomic mass units less than
3. Except for the loss of an oxygenated methyl at δ
C 57.3 (5-O
CH
3), the
1H NMR and
13C-NMR data of
4 (
Table 2) were similar to those of
3 (
Table 1), which indicated that the 5-O
CH
3 in
3 was replaced by the hydroxyl in
4. On the basis of the exhaustive analysis of 2D NMR data (
1H-
1H COSY, HSQC, and HMBC) (
Table S4, Supplementary Materials), the planar structure of
4 was established, named arugosin N.
The molecular formula of
5 was fourteen atomic mass units less than
1. Except for the loss of an oxygenated methyl at δ
C 56.9 (5-O
CH
3), the
1H-NMR and
13C-NMR data of
5 (
Table 2) were similar to those of
1 (
Table 1), which indicated that the 5-O
CH
3 in
1 was replaced by the hydroxyl in
5. On the basis of the exhaustive analysis of 2D NMR data (
1H-
1H COSY, HSQC, and HMBC) (
Table S5, Supplementary Materials), the planar structure of
5 was established as the known compound, 1,6,10-trihydroxy-8-methyl-2-(3-methyl-2-butenyl)-dibenz[
b,
e]oxepin-11(6
H)-one [
7].
The C-5 positions of
4 and
5 were hemiacetal linkage, which were unstable and easy to split. Therefore,
4 and
5 can self-interconvert (
Figure 7) and exist as inseparable tautomeric mixtures in Nature.
Compound
6 was obtained as a yellow solid. The ESI-MS (positive):
m/
z 255 [M + H]
+; ESI-MS (negative):
m/
z 253 [M − H]
− indicated the molecular weight was 254. It was identified as chrysophanol by comparing its data (
1H- and
13C-NMR, MS) with literature values [
16].
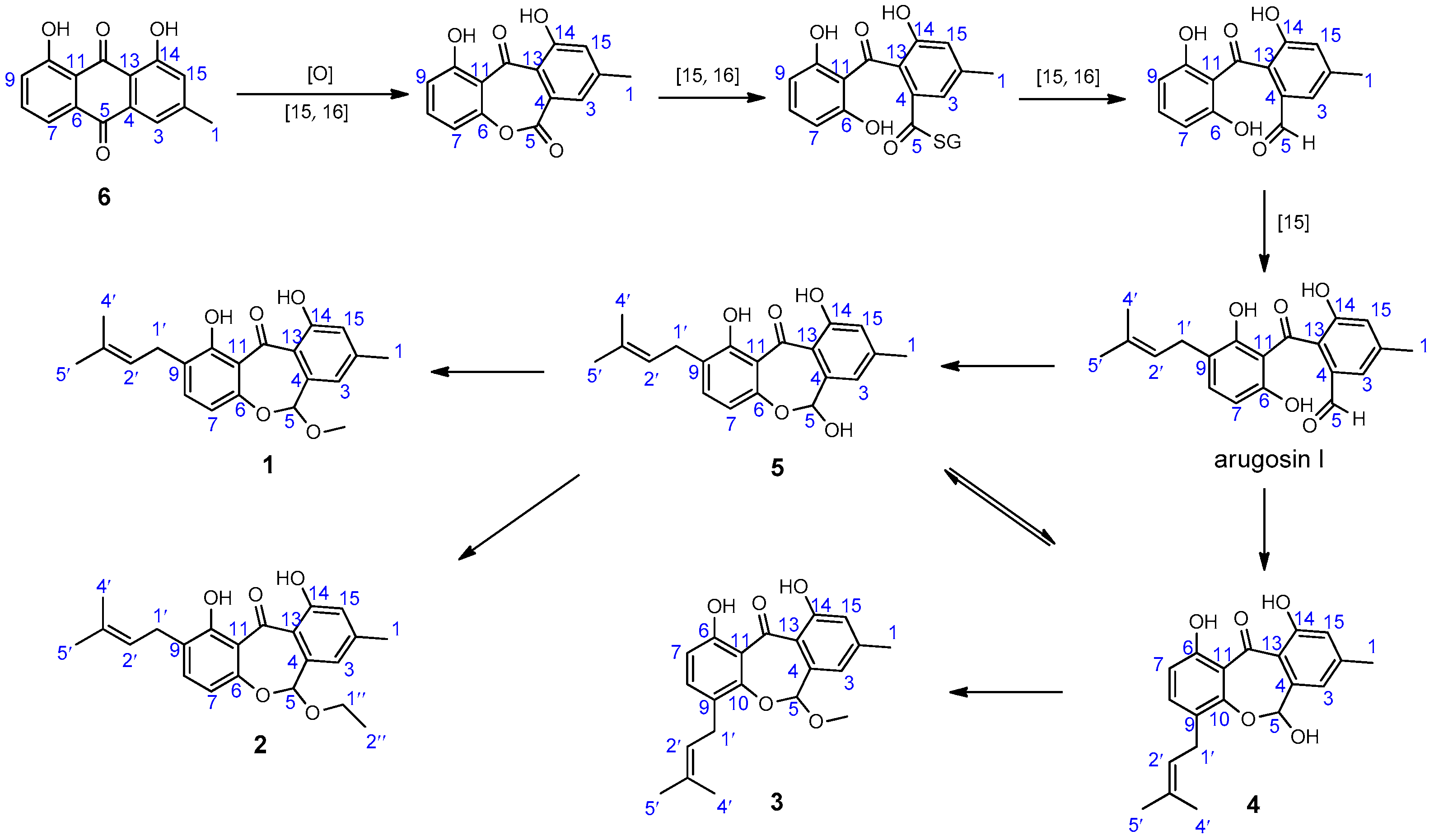
Based on the structural features of compounds
1–
6 and literature investigation, a plausible biosynthetic pathway of them was proposed (
Scheme 1). Chrysophanol (
6) undergoes oxidization [
17,
18], ring-cleavage [
17,
18], reduction [
17,
18], and isoprentenylation to yield arugosin I [
17], which may be the precursor of arugosin N (
4) and 1,6,10-trihydroxy-8-methyl-2-(3-methyl-2-butenyl)-dibenz[
b,e]oxepin-11(6
H)-one (
5). Then, arugosin K (
1) and arugosin L (
2) may originate from
5 by methylation and ethylation, respectively. Arugosin M (
3) may originate from
4 by methylation.
3. Materials and Methods
3.1. Chemicals
Methanol (MeOH) was purchased from Yuwang Industrial Co. Ltd. (Yucheng, China). Cyclohexane, ethyl acetate (EtOAc), and petroleum ether (PE) were purchased from Tianjin Fuyu Fine Chemical Co. Ltd. (Tianjin, China).
3.2. General Experimental Procedures
Optical rotations were measured on a P1020 digital polarimeter (Jasco International Co. Ltd., Tokyo, Japan). UV data were recorded using a Jasco V-550 UV/Vis spectrometer. IR data were recorded on a Jasco FT/IR-480 plus spectrometer. ECD spectrum was recorded on a Jasco J-810 spectrophotometer using MeOH as the solvent. The ESI-MS spectra were recorded on a Finnigan LCQ Advantage MAX mass spectrometer (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The HR-ESI-MS spectra were obtained on a Micromass Q-TOF mass spectrometer (Waters Corporation, Milford, MA, USA). The NMR spectra were measured with Bruker AV-400 and Bruker AV-600 spectrometers (Bruker BioSpin Group, Faellanden, Switzerland) using the solvent signals (CDCl3: δH 7.26/δC 77.0) as internal standard. The analytical HPLC was performed on a Dionex HPLC system equipped with an Ultimate 3000 pump, an Ultimate 3000 diode array detector (DAD), an Ultimate 3000 Column Compartment, an Ultimate 3000 autosampler (Thermo Fisher Scientific, Inc.) using a Gemini C18 column (4.6 mm × 250 mm, 5 μm) (Phenomenex Inc., Los Angeles, CA, USA). The semi-preparative HPLC was performed on a Shimadzu LC-6-AD Liquid Chromatography with an SPD-20A Detector using a Phenomenex Gemini C18 column (Phenomenex Inc.) (10.0 mm × 250 mm, 5 μm). The chiral HPLC was performed on a Shimadzu LC-6-AD Liquid Chromatography with an SPD-20A Detector using a Phenomenex Lux Cellulose-2 chiral column (4.6 mm × 250 mm, 3 μm).
3.3. Fungus Material
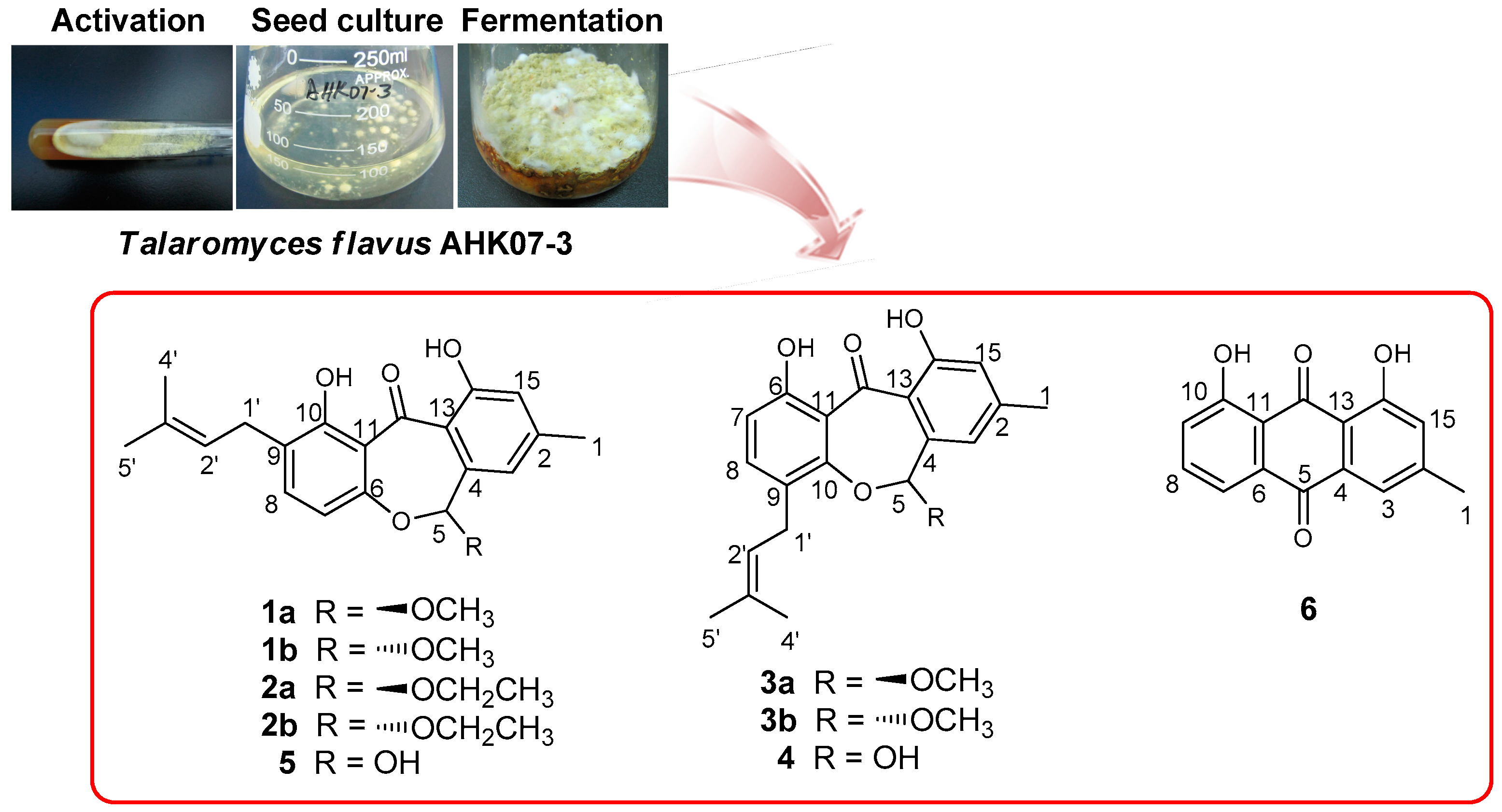
The strain AHK07-3 was isolated from the soil, collected at the wetland of Ahongkou, Sinkiang Province, China. The strain was identified as Talaromyces flavus based on the morphological characters and gene sequence analysis. The ribosomal internal transcribed spacer (ITS) and the 5.8S rRNA gene sequences (ITS1-5.8S-ITS2) of the strain have been deposited at GenBank as KX011167. The fungus was cultured on slants of potato dextrose agar at 25 °C for 5 days. Agar plugs were used to inoculate four Erlenmeyer flasks (250 mL), each containing 100 mL of potato dextrose broth. Four flasks of the inoculated media were incubated at 25 °C on a rotary shaker at 200 rpm for 5 days to prepare the seed culture. Fermentation was carried out in 20 Erlenmeyer flasks (500 mL), each containing 70 g of rice. Distilled H2O (105 mL) was added to each flask, and the rice was soaked overnight before autoclaving at 120 °C for 30 min. After cooling to room temperature, each flask was inoculated with 5.0 mL of the seed culture containing mycelia and incubated at room temperature for 50 days.
3.4. Extraction and Isolation
The culture was extracted with EtOAc (3 × 5000 mL). Removal of EtOAc under reduced pressure yielded a crude extract (19.7 g) that was dissolved in 90% v/v aqueous MeOH (400 mL). The methanolic solution was further extracted with cyclohexane (400 mL) to afford a cyclohexane fraction C (4.6 g). Fraction C was subjected to silica gel (200–300 mesh) column chromatography initially eluting with cyclohexane (500 mL) and then with MeOH (500 mL) to afford subfractions CC and CM. Subfraction CC (3.3 g) was further separated by open silica gel column chromatography eluting with cyclohexane–EtOAc (100:0 (150 mL), 99:1 (150 mL), 98:2 (150 mL), 97:3 (150 mL), 96:4 (150 mL), 95:5 (150 mL), 94:6 (150 mL), 93:7 (150 mL), 92:8 (150 mL), 91:9 (150 mL), 90:10 (150 mL), 50:50 (150 mL), and 0:100 (150 mL), v/v) to yield four sub-subfractions (CC1 to CC4). Sub-subfraction CC2 (2.8 g) was subjected to open silica gel column chromatography eluting with PE (petroleum ether)–EtOAc (100:0 (100 mL), 99:1 (100 mL), 98:2 (100 mL), 97:3 (100 mL), 96:4 (100 mL), 95:5 (100 mL), 94:6 (100 mL), 93:7 (100 mL), 92:8 (100 mL), 91:9 (100 mL), 90:10 (100 mL), 50:50 (100 mL) and 0:100 (100 mL), v/v) to yield four sub-subfractions (CC2a to CC2d). Sub-subfraction CC2b (1.1 g) was isolated by semi-preparative HPLC purification using MeOH–H2O (88:12, v/v) at a flow rate of 3 mL/min to yield 1 (617.4 mg), 2 (2.6 mg), and 3 (24.7 mg). Sub-subfraction CC2c (0.6 g) was purified by semi-preparative HPLC purification using MeOH–H2O (90:10, v/v) at a flow rate of 3 mL/min to yield 6 (1.1 mg). Sub-subfraction CC4 (220.1 mg) was isolated by semi-preparative HPLC purification using MeOH–H2O (80:20, v/v) at a flow rate of 3 mL/min to yield the mixtures of 4 and 5 (12.5 mg). Enantioseparation of 1–3 was carried out on a Phenomenex Lux Cellulose-2 chiral column using CH3OH/H2O (88/12, v/v) as mobile phase.
3.5. Spectroscopic Data of Isolated Compounds
(±)-Arugosin K (
1): yellow solid; UV (MeOH) λ
max (log ε) 205 (3.75), 221 (3.63), 304 (3.35), 359 (3.29) nm; IR (KBr) ν
max 3438, 2920, 1708, 1619, 1590, 1421, 1206, 1052 cm
−1; ESI-MS (positive):
m/
z 355 [M + H]
+, ESI-MS (negative):
m/
z 353 [M − H]
−; HRESIMS (positive)
m/z 355.1550 [M + H]
+ (calcd. for C
21H
23O
5, 355.1545);
1H- and
13C-NMR data see
Table 1.
(+)-S-Arugosin K (1a): +27.8 (c 0.5, CHCl3); ECD (c 8.5 × 10−5 M, MeOH) λmax (Δε) 208 (+12.50), 294 (−5.71), 334 (+3.76), 382 (−1.77) nm.
(−)-R-Arugosin K (1b): −27.0 (c 0.5, CHCl3); ECD (c 8.5 × 10−5 M, MeOH) λmax (Δε) 208 (−11.38), 295 (+6.96), 334 (−3.88), 382 (+2.82) nm.
(±)-Arugosin L (
2): yellow solid; UV (MeOH) λ
max (log ε) 206 (3.70), 222 (3.57), 304 (3.30), 359 (3.23) nm; IR (KBr) ν
max 3440, 2916, 1624, 1590, 1422, 1206, 1057 cm
−1; ESI-MS (positive):
m/
z 369 [M + H]
+; ESI-MS (negative):
m/
z 367 [M − H]
−; HRESIMS (positive)
m/
z 369.1695 [M + H]
+ (calcd. for C
22H
25O
5, 369.1702);
1H- and
13C-NMR data see
Table 1.
(+)-S-Arugosin L (2a): +27.4 (c 0.5, CHCl3); ECD (c 6.8 × 10−5 M, MeOH) λmax (Δε) 208 (+13.90), 295 (−6.07), 335 (+5.07), 382 (−1.91) nm.
(−)-R-Arugosin L (2b): −26.8 (c 0.5, CHCl3); ECD (c 6.8 × 10−5 M, MeOH) λmax (Δε) 208 (−13.79), 295 (+7.98), 335 (−3.44), 382 (+3.40) nm.
(±)-Arugosin M (
3): yellow solid; UV (MeOH) λ
max (log ε) 206 (3.85), 221 (3.73), 299 (3.42), 357 (3.36) nm; IR (KBr) ν
max 3441, 2913, 1620, 1464, 1202, 1050 cm
−1; ESI-MS (positive):
m/
z 355 [M + H]
+, ESI-MS (negative):
m/
z 353 [M − H]
−; HRESIMS (positive)
m/z 355.1541 [M + H]
+ (calcd. for C
21H
23O
5, 355.1545);
1H- and
13C-NMR data see
Table 1.
(+)-S-Arugosin M (3a): +28.4 (c 0.5, CHCl3); ECD (c 8.5 × 10−5 M, MeOH) λmax (Δε) 209 (+14.24), 290 (−6.29), 334 (+6.67), 381 (−3.30) nm.
(−)-R-Arugosin M (3b): −27.6 (c 0.5, CHCl3); ECD (c 8.5 × 10−5 M, MeOH) λmax (Δε) 209 (−12.84), 292 (+7.26), 334 (−5.93), 381 (+4.27) nm.
Arugosin N (
4): 1,6,10-trihydroxy-8-methyl-2-(3-methyl-2-butenyl)-dibenz[
b,
e]oxepin-11(6
H)-one (
5): yellow solid; UV (MeOH) λ
max (log ε) 206 (3.75), 222 (3.65), 286 (3.29), 356 (3.16) nm; IR (KBr) ν
max 3443, 2917, 1628, 1589, 1420, 1353, 1173, 1052 cm
−1; ESI-MS (positive):
m/
z 341 [M + H]
+,
m/
z 363 [M + Na]
+; ESI-MS (negative):
m/
z 339 [M − H]
−; HRESIMS (positive)
m/
z 363.1213 [M + Na]
+ (calcd. for C
20H
20O
5Na, 363.1208);
1H- and
13C-NMR data see
Table 2.
3.6. ECD Calculation
Before molecular initial 3D structures were generated with CORINA version 3.4 (Molecular Networks GmbH, Erlangen, Germany), the molecules of (5
S)-
1, (5
R)-
1, (5
S)-
3, and (5
R)-
3 were converted into SMILES codes, respectively. Conformer databases were generated in CONFLEX version 7.0 (CONFLEX Corporation, Tokyo, Japan) using the MMFF94s force-field, with an energy window for acceptable conformers (ewindow) of 5 kcal·mol
−1 above the ground state, a maximum number of conformations per molecule (maxconfs) of 100, and an RMSD cutoff (rmsd) of 0.5 Å. Then each conformer of the acceptable conformers was optimized with HF/6-31G(d) method in Gaussian09 [
19]. Further optimization at the APFD/6-31G(d) level led the dihedral angles to be got. After that, eight lowest energy conformers of (5
S)-
1 and (5
R)-
1 (see
Supplementary Materials Table S6 and Figure S3) were found out. Six lowest energy conformers of (5
S)-
3 and (5
R)-
3 (see
Supplementary Materials Table S7 and Figure S4) were found out. The optimized conformers were taken for the ECD calculations, which were performed with Gaussian09 (APFD/6-311++G(2d,p)). The solvent effect was taken into account by the polarizable-conductor calculation model (IEFPCM, methanol as the solvent). Comparisons of the experimental and calculated spectra were done with the software SpecDis [
20,
21]. It was also used to apply a UV shift to the ECD spectra, Gaussian broadening of the excitations, and Boltzmann weighting of the spectra.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}