
Stereoselective Reduction of Imines with Trichlorosilane Using Solid-Supported Chiral Picolinamides

 and
and
Abstract
:1. Introduction
2. Results and Discussion
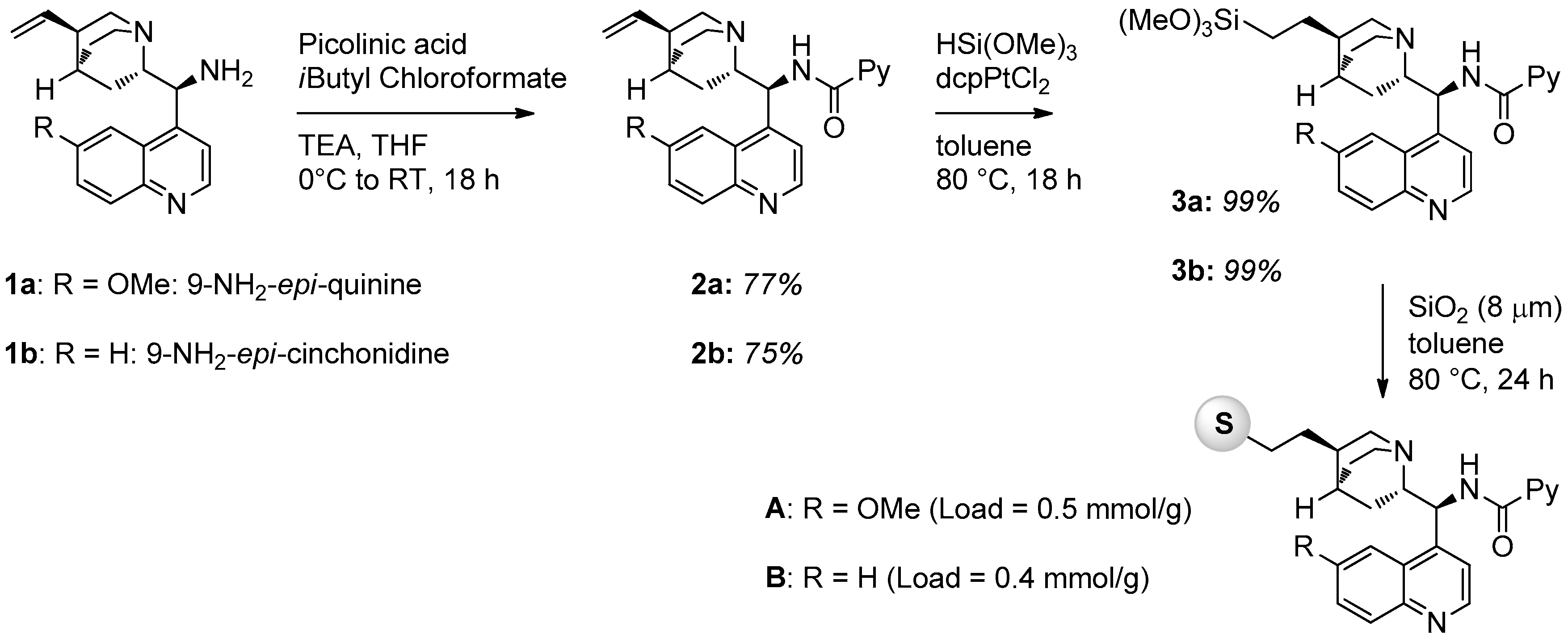
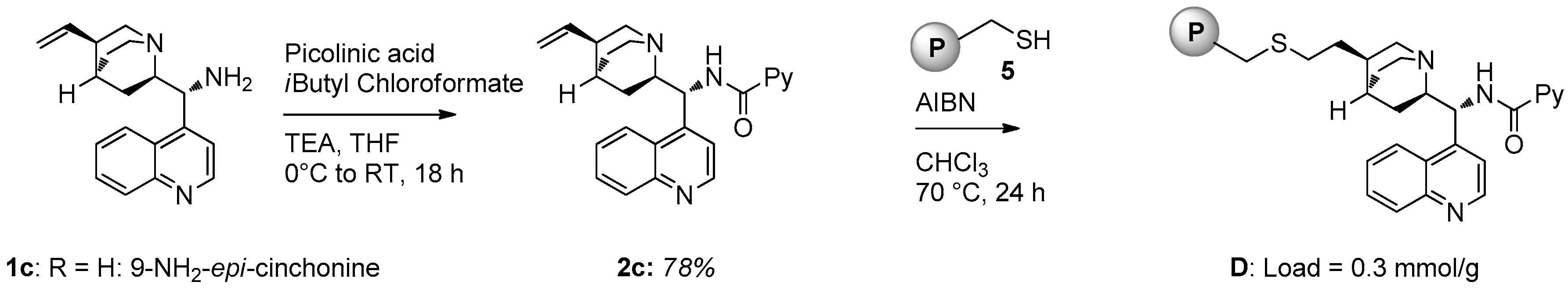
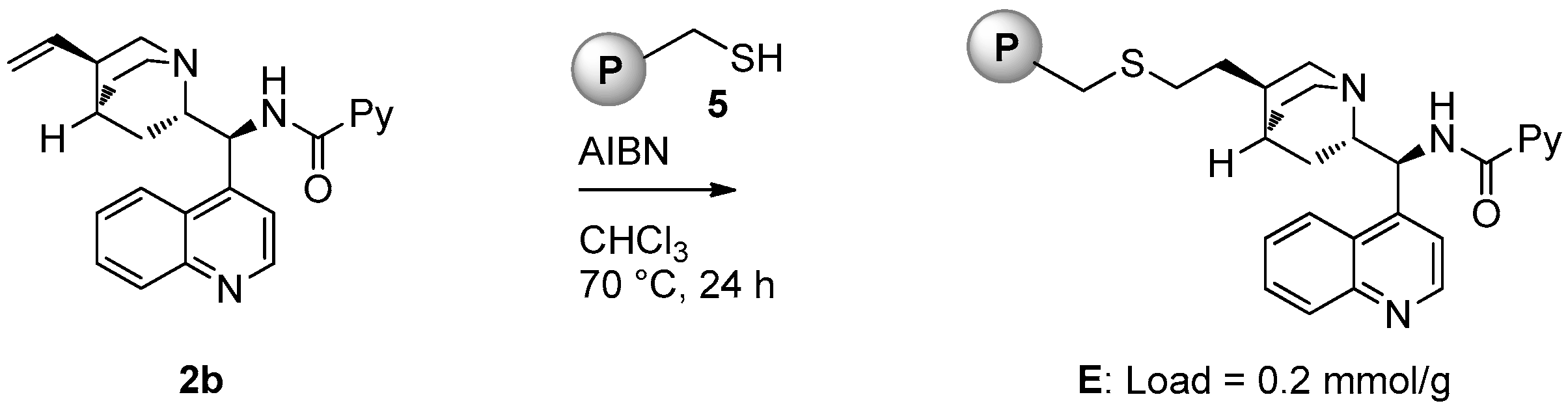
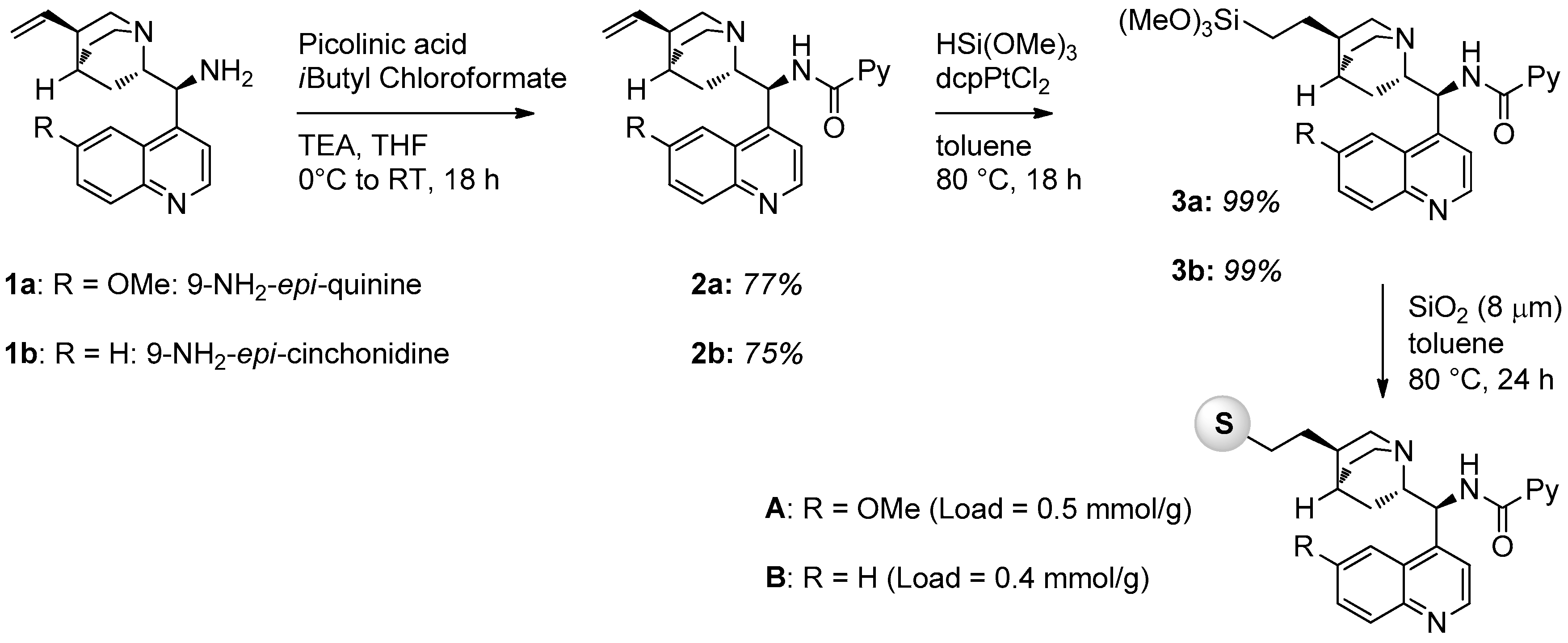
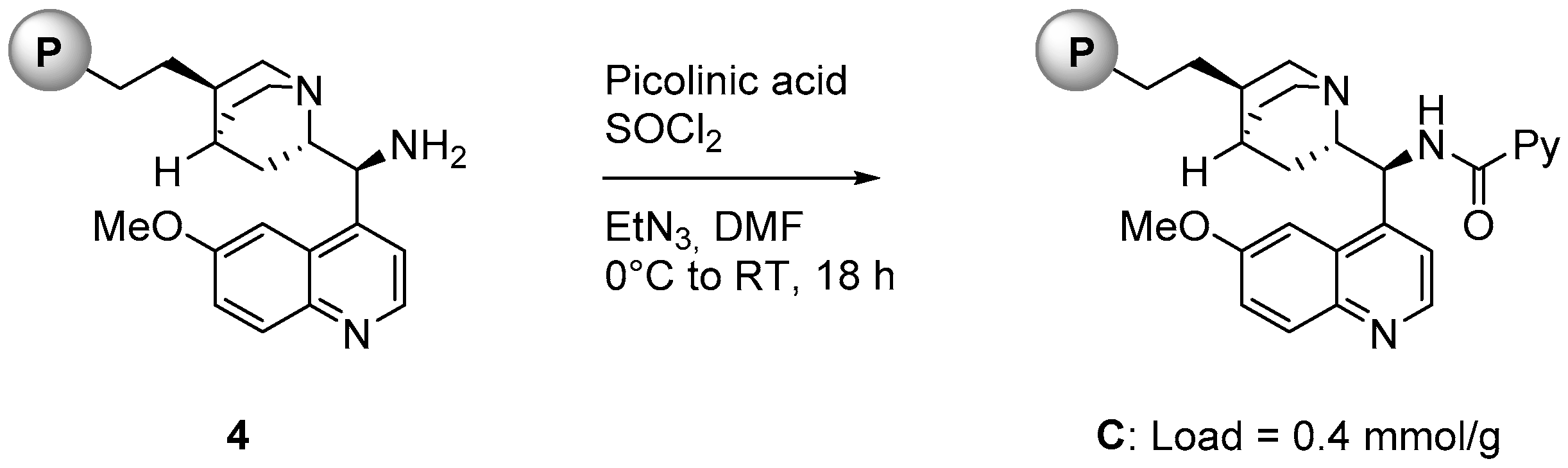
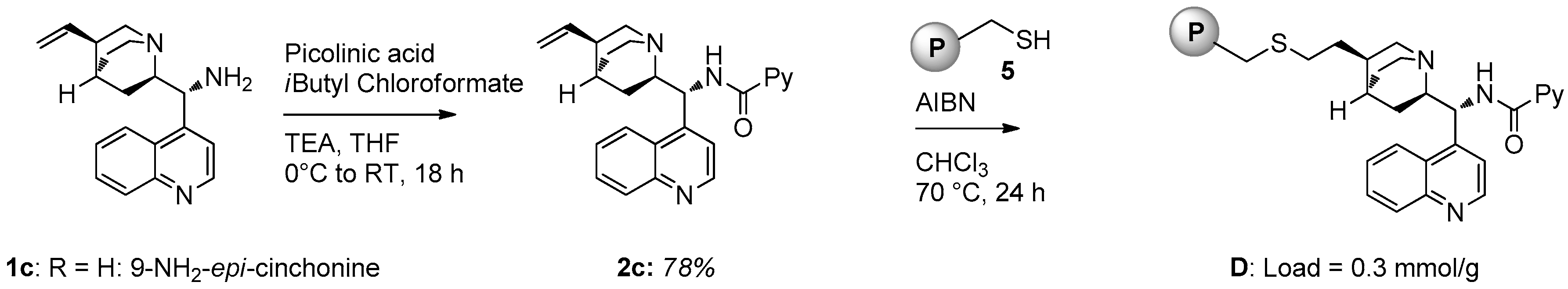
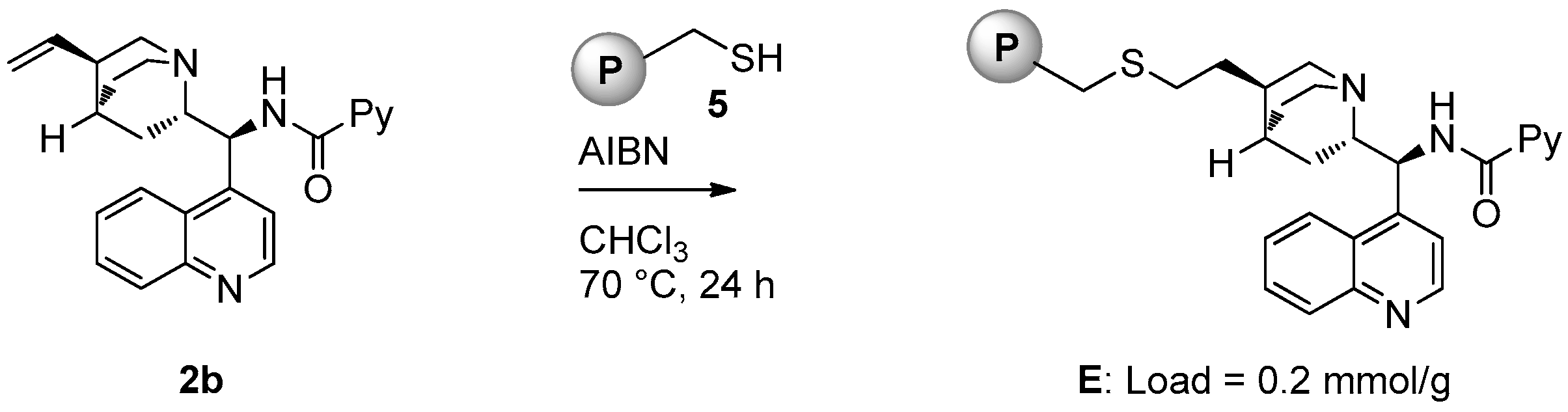
2.1. Catalyst Synthesis
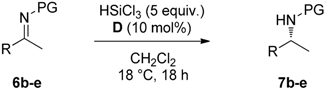
2.2. Stereoselective Reduction of Imines with HSiCl3 in Batch
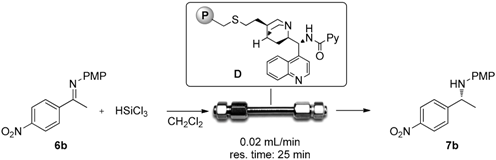
2.3. Continuous Flow Reaction
3. Materials and Methods
3.1. General Information
3.2. Catalysts Synthesis
3.3. Stereoselective Reduction of Imines
3.4. Product Characterization
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Okhuma, T.; Noyori, R. Comprehensive Asymmetric Catalysis; Jacobsen, E.N., Pfaltz, A., Yamamoto, H., Eds.; Springer: New York, NY, USA, 2004. [Google Scholar]
- Benaglia, M. Recoverable and Recyclable Catalysts; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Trindade, A.F.; Gois, P.M.P.; Afonso, C.A.M. Recyclable Stereoselective Catalysts. Chem. Rev. 2009, 109, 418. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N; Itsuno, S. Polymeric Chiral Catalyst Design and Chiral Polymer Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2011. [Google Scholar]
- Puglisi, A.; Benaglia, M.; Chiroli, V. Stereoselective organic reactions promoted by immobilized chiral catalysts in continuous flow systems. Green Chem. 2013, 15, 1790–1813. [Google Scholar] [CrossRef]
- Tsubogo, T.; Ishiwata, T.; Kobayashi, S. Asymmetric Carbon-Carbon Bond Formation under Continuous-Flow Conditions with Chiral Heterogeneous Catalysts. Angew. Chem. Int. Ed. 2013, 52, 6590. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Benaglia, M.; Puglisi, A. Flow Chemistry: Recent Developments in the Synthesis of Pharmaceutical Products. Org. Proc. Res. Dev. 2016, 20, 2–25. [Google Scholar] [CrossRef]
- Cantillo, D.; Kappe, C.O. Immobilized Transition Metals as Catalysts for Cross-Couplings in Continuous Flow—A Critical Assessment of the Reaction Mechanism and Metal Leaching. ChemCatChem 2014, 6, 3286. [Google Scholar] [CrossRef]
- Gursel, I.V.; Noel, T.; Wang, Q.; Hessel, V. Separation/recycling methods for homogeneous transition metal catalysts in continuous flow. Green Chem. 2015, 17, 2012. [Google Scholar] [CrossRef]
- Benaglia, M.; Genoni, A.; Bonsignore, M. Enantioselective organocatalytic reductions. In Stereoselective Organocatalysis: From C-C to C-Heteroatom Bond Formation; Torres, R.R., Ed.; Wiley: Hoboken, NJ, USA, 2012. [Google Scholar]
- Rossi, S.; Benaglia, M.; Massolo, E.; Raimondi, L. Organocatalytic strategies for enantioselective metal-free reductions. Catal. Sci. Technol. 2014, 9, 2708. [Google Scholar] [CrossRef]
- Guizzetti, S.; Benaglia, M. Trichlorosilane-Mediated Stereoselective Reduction of C=N Bonds. Eur. J. Org. Chem. 2010, 5529. [Google Scholar] [CrossRef]
- Jones, S.; Warner, C.J.A. Trichlorosilane mediated asymmetric reductions of the C=N double bon. Org Biomol. Chem. 2012, 10, 2189. [Google Scholar] [CrossRef] [PubMed]
- For a seminal work in this field see: Malkov, A.V.; Mariani, A.; MacDougall, K.N.; Kocovsy, P. Role of Noncovalent Interactions in the Enantioselective Reduction of Aromatic Ketimines with Trichlorosilane. Org. Lett. 2004, 6, 2253. [Google Scholar] [CrossRef] [PubMed]
- For recent works in the field see the following: Ye, J.; Wang, C.; Chen, L.; Wu, X.; Zhou, L.; Sun, J. Chiral Lewis Base-Catalyzed, Enantioselective Reduction of Unprotected β-Enamino Esters with Trichlorosilane. Adv. Synth. Catal. 2016. [Google Scholar] [CrossRef]
- Wang, T.; Di, X.; Wang, C.; Li, Z.; Sun, J. Reductive Hydrazination with Trichlorosilane: A Method for the Preparation of 1,1-Disubstituted Hydrazines. Org. Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Malkov, A.V.; Figlus, M.; Kocovsy, P. Polymer-Supported Organocatalysts: Asymmetric Reduction of Imines with Trichlorosilane Catalyzed by an Amino Acid-Derived Formamide anchored to a Polymer. J. Org. Chem. 2008, 73, 3985. [Google Scholar] [CrossRef] [PubMed]
- Malkov, A.; Figlus, M.; Prestly, M.R.; Rabani, G.; Cooke, G.; Kocovsky, P. Soluble Polymer-Supported Organocatalysts: Asymmetric Reduction of Imines with Trichlorosilane Catalyzed by an Amino Acid Derived Formamide Anchored to a Soluble Polymer. Chem. Eur. J. 2009, 15, 9651–9654. [Google Scholar] [CrossRef] [PubMed]
- Figlus, M.; Caldwell, S.T.; Walas, D.; Yesilba, G.; Cooke, G.; Kocovsy, P.; Malkov, A.V.; Sanyal, A. Dendron-anchored organocatalysts: The asymmetric reduction of imines with trichlorosilane, catalysed by an amino acid-derived formamide appended to a Dendron. Org. Biomol. Chem. 2010, 8, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Benaglia, M.; Coccia, F.; Cozzi, F.; Puglisi, A. Solid Supported 9-Amino-9-deoxy-epi-quinine as Efficient Organocatalyst for Stereoselective Reactions in Batch and Under Continuous Flow Conditions. Adv. Synth. Catal. 2015, 357, 377. [Google Scholar] [CrossRef]
- Attempts to monitor the reaction from 3 to C via solid state NMR were unsuccessful; a complete conversion of the primary amine into the corresponding picolinamide could not be confirmed.
- Braslau, R.; Rivera, F.; Tansakul, C. Reversible crosslinking of polymers bearing pendant or terminal thiol groups prepared by nitroxide-mediated radical polymerization. React. Funct. Polym. 2013, 73, 624. [Google Scholar] [CrossRef]
- The reaction in the presence of polystyrene supported primary amine 4 as catalyst gave amine 7a in 45% yield as a racemic mixture.
- Barrulas, P.C.; Genoni, A.; Benaglia, M.; Burke, A.J. Cinchona-Derived Picolinamides: Effective Organocatalysts for Stereoselective Imine Hydrosilylation. Eur. J. Org. Chem. 2014, 7339. [Google Scholar] [CrossRef]
- Brunner, H.; Bugler, J.; Nuber, B. Preparation of 9-amino-(9-deoxy) cinchona alkaloids. Tetrahedron Asymmetry 1995, 6, 1699. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 7a–e are available from the authors.


| Entry 1 | Catalyst | Yield (%) 2 | ee (%) 3 |
|---|---|---|---|
| 1 | A | 64 | 30 (S) |
| 2 | B | 95 | 62 (S) |
| 3 | C | 95 | 23 (S) |
| 4 | D | 98 | 90 (R) |
| 5 4 | D | 95 | 91 (R) |
| 6 5 | D | 95 | 79 (R) |
| 7 | E | 95 | 63 (S) |

| Entry 1 | R | PG | Yield (%) 2 | ee (%) 3 |
|---|---|---|---|---|
| 1 | 6b: 4-NO2-C6H4- | PMP | 93 | 85 |
| 2 | 6c: 4-F-C6H4- | PMP | 98 | 87 |
| 3 | 6d: 4-CF3-C6H4- | PMP | 97 | 84 |
| 4 | 6e: Naphtyl | Ph | 96 | 85 |
| Entry | Running Time (min) | Yield (%) 1 | ee (%) 2 |
|---|---|---|---|
| 1 | 0–50 | 98 | 47 |
| 3 | 50–75 | 98 | 44 |
| 4 | 75–100 | 93 | 30 |
| 5 | 100–125 | 93 | 25 |
| 6 | 125–150 | 87 | 22 |
| 7 | 150–175 | 86 | 20 |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, S.D.; Porta, R.; Barrulas, P.C.; Puglisi, A.; Burke, A.J.; Benaglia, M. Stereoselective Reduction of Imines with Trichlorosilane Using Solid-Supported Chiral Picolinamides. Molecules 2016, 21, 1182. https://doi.org/10.3390/molecules21091182
Fernandes SD, Porta R, Barrulas PC, Puglisi A, Burke AJ, Benaglia M. Stereoselective Reduction of Imines with Trichlorosilane Using Solid-Supported Chiral Picolinamides. Molecules. 2016; 21(9):1182. https://doi.org/10.3390/molecules21091182
Chicago/Turabian StyleFernandes, Sílvia D., Riccardo Porta, Pedro C. Barrulas, Alessandra Puglisi, Anthony J. Burke, and Maurizio Benaglia. 2016. "Stereoselective Reduction of Imines with Trichlorosilane Using Solid-Supported Chiral Picolinamides" Molecules 21, no. 9: 1182. https://doi.org/10.3390/molecules21091182