
Betulin Phosphonates; Synthesis, Structure, and Cytotoxic Activity

 ,
,
Abstract
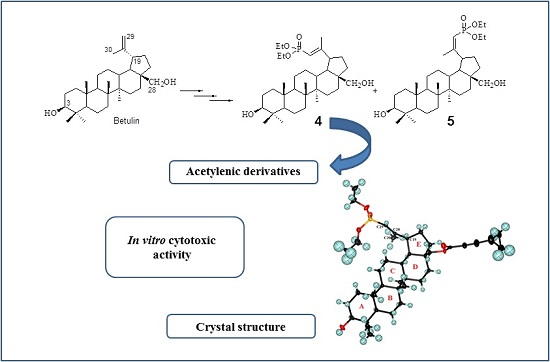
:
1. Introduction
2. Results and Discussion
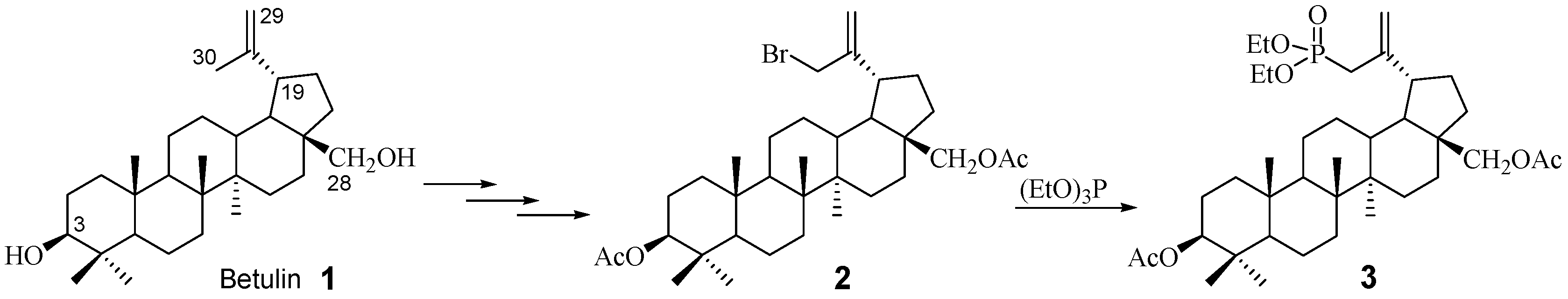
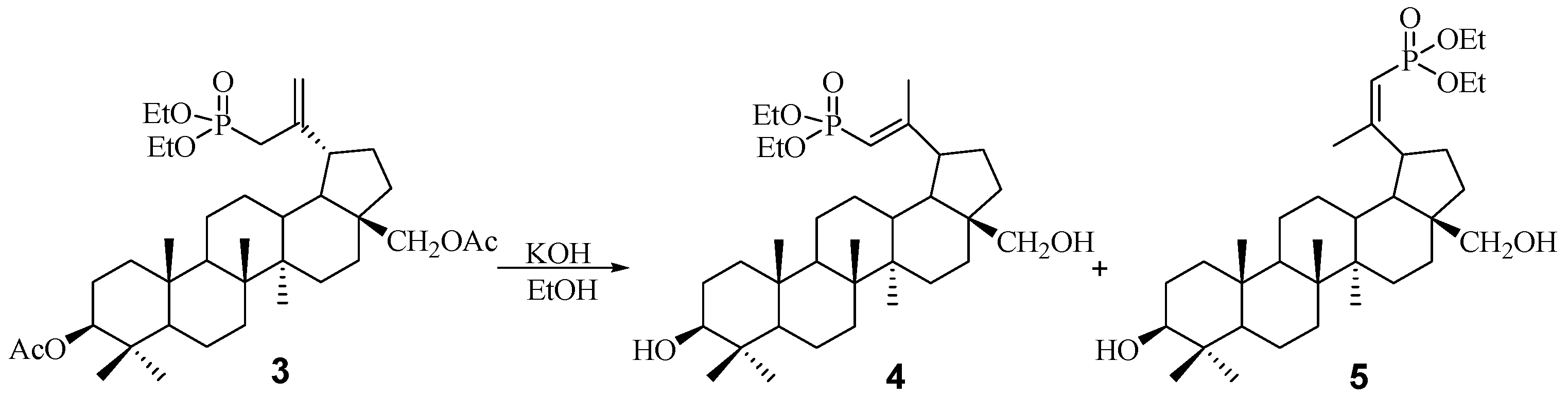
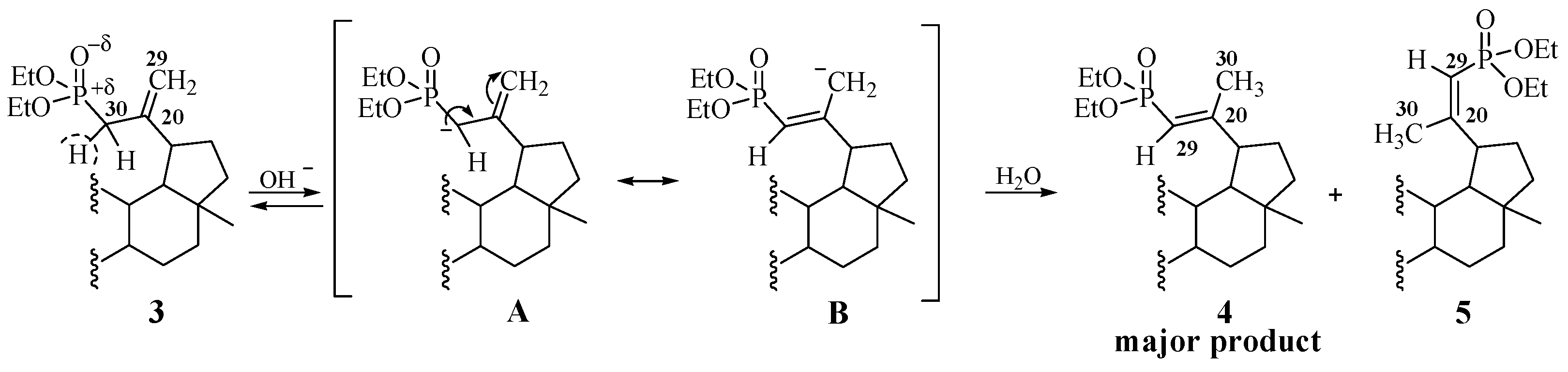
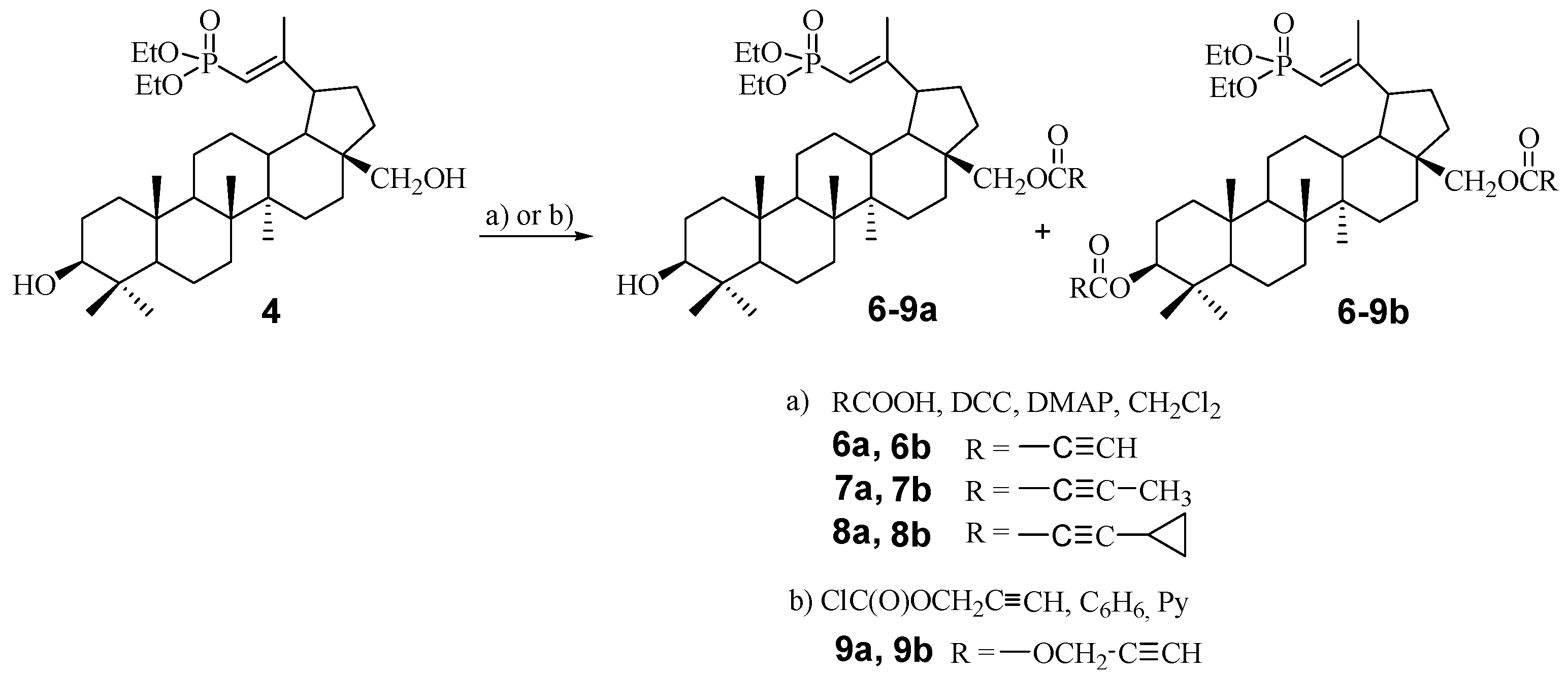
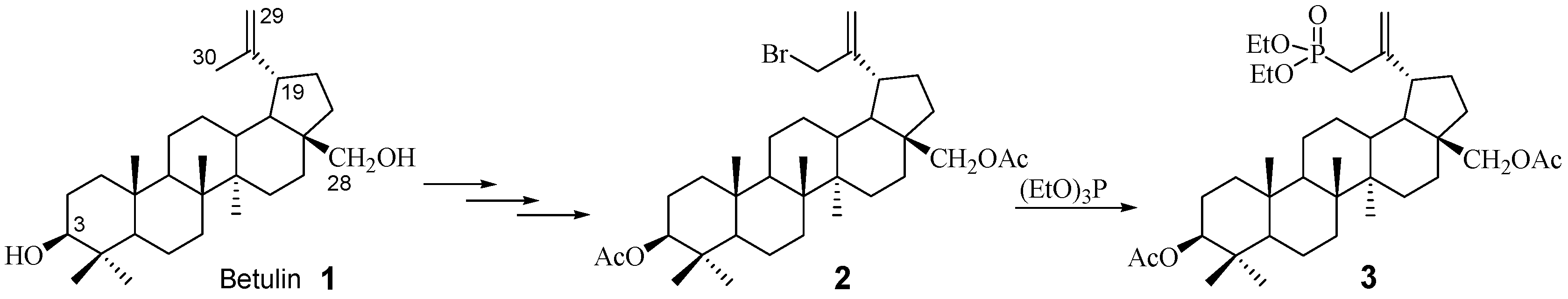
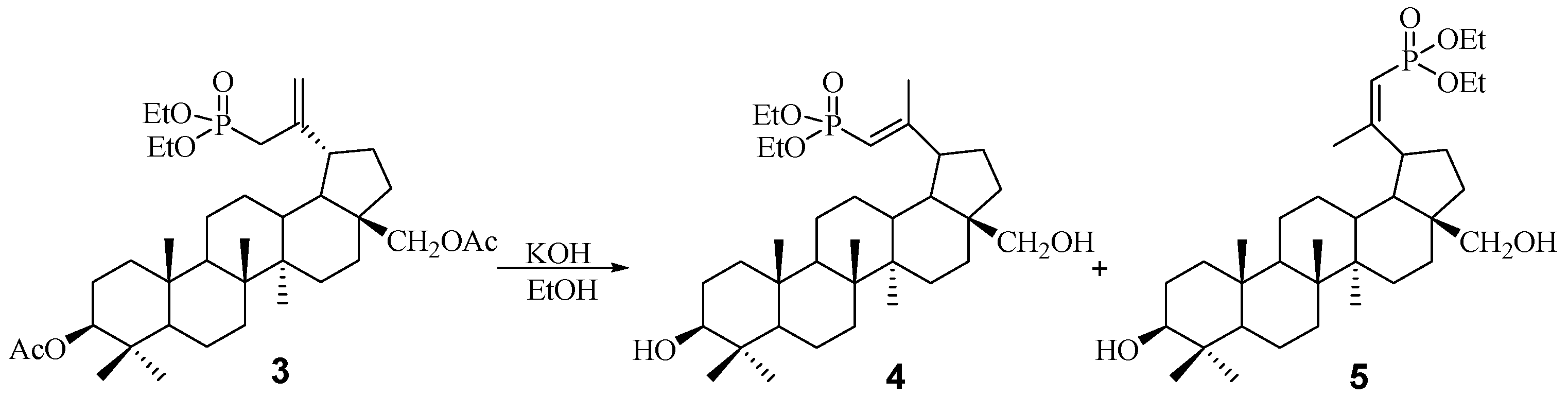
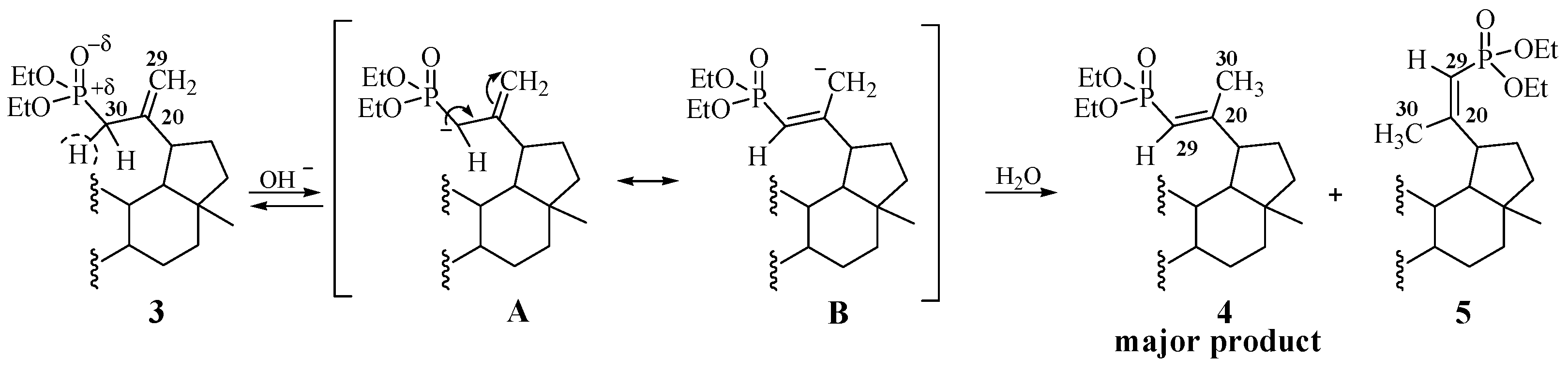
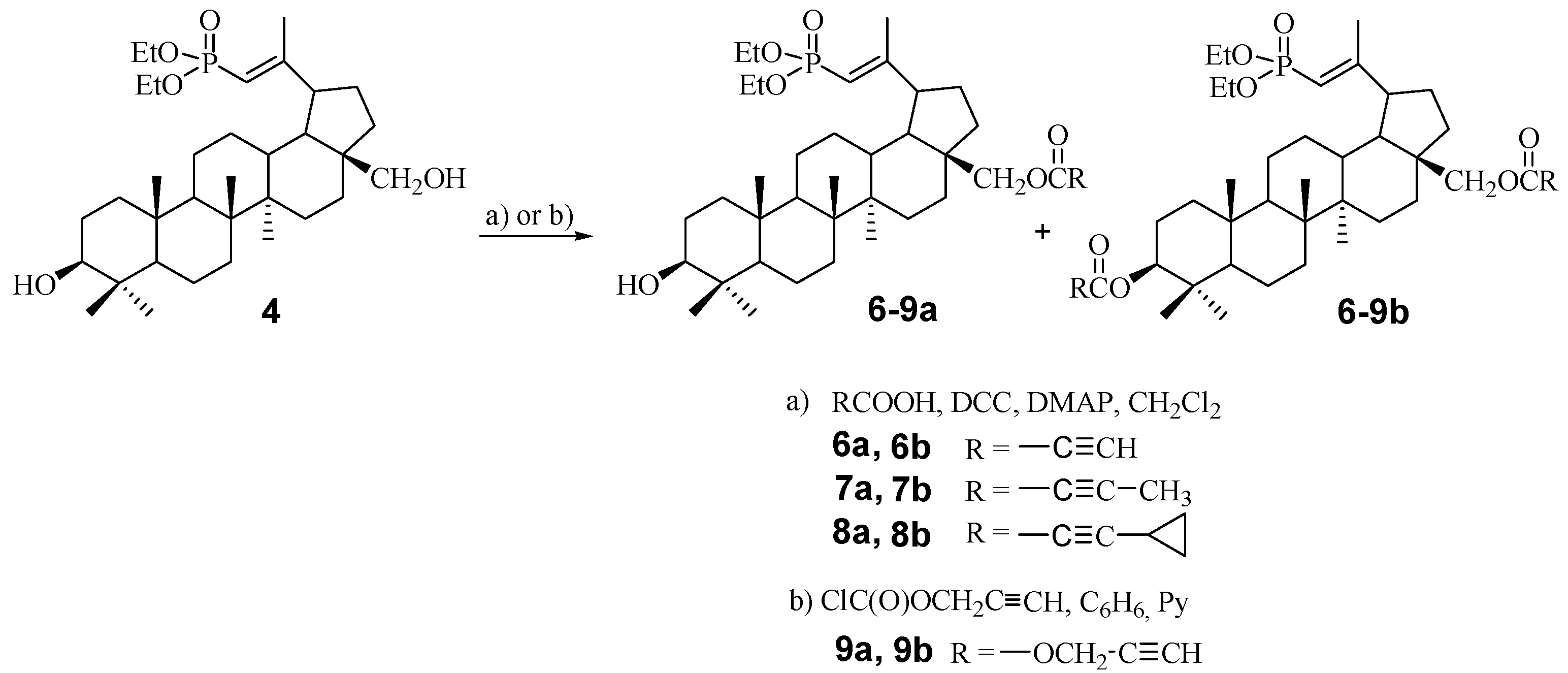
2.1. Chemistry
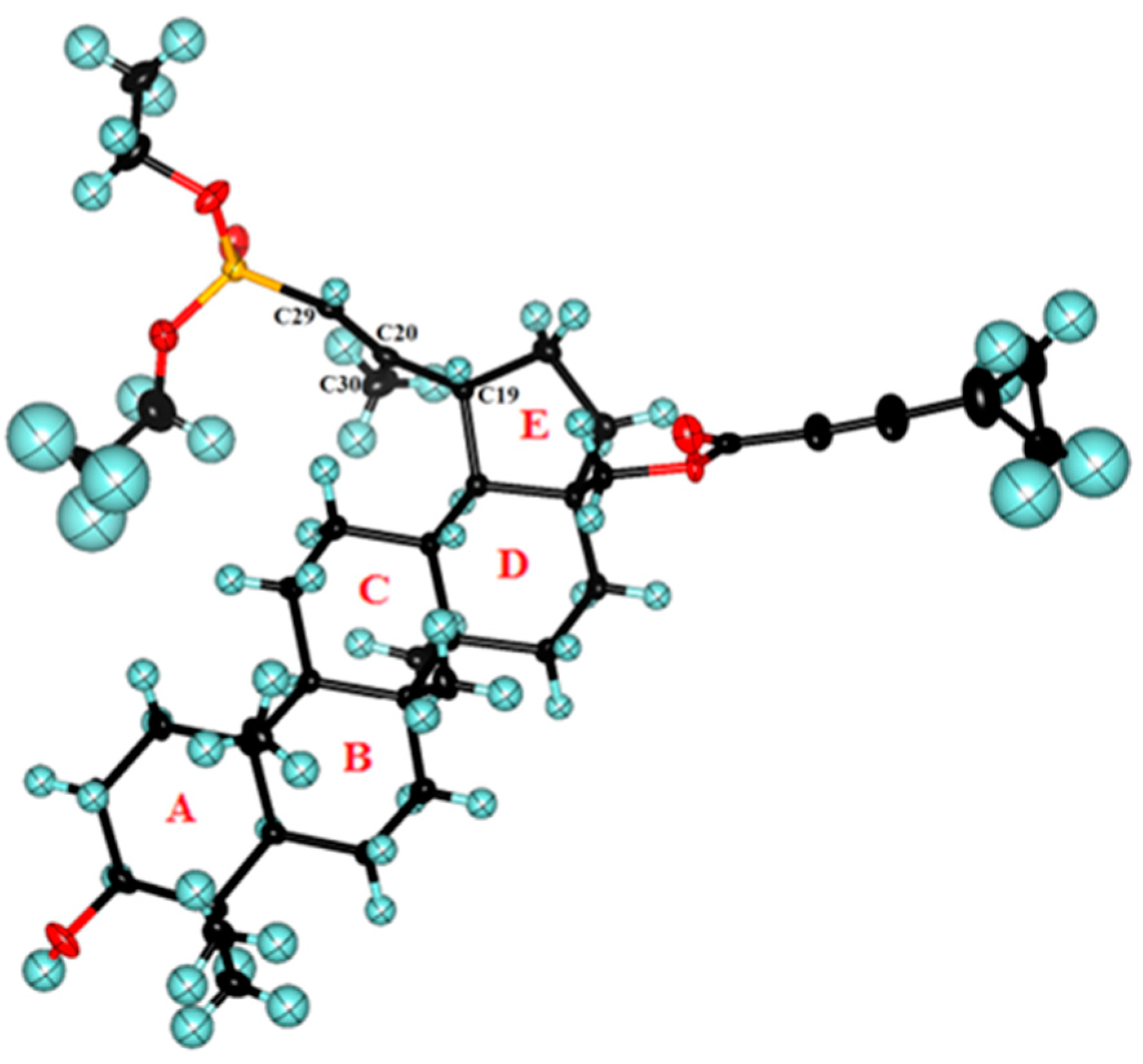
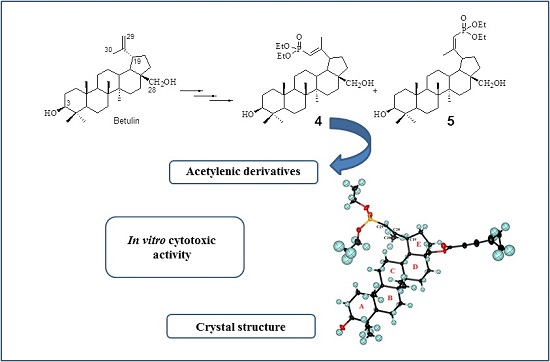
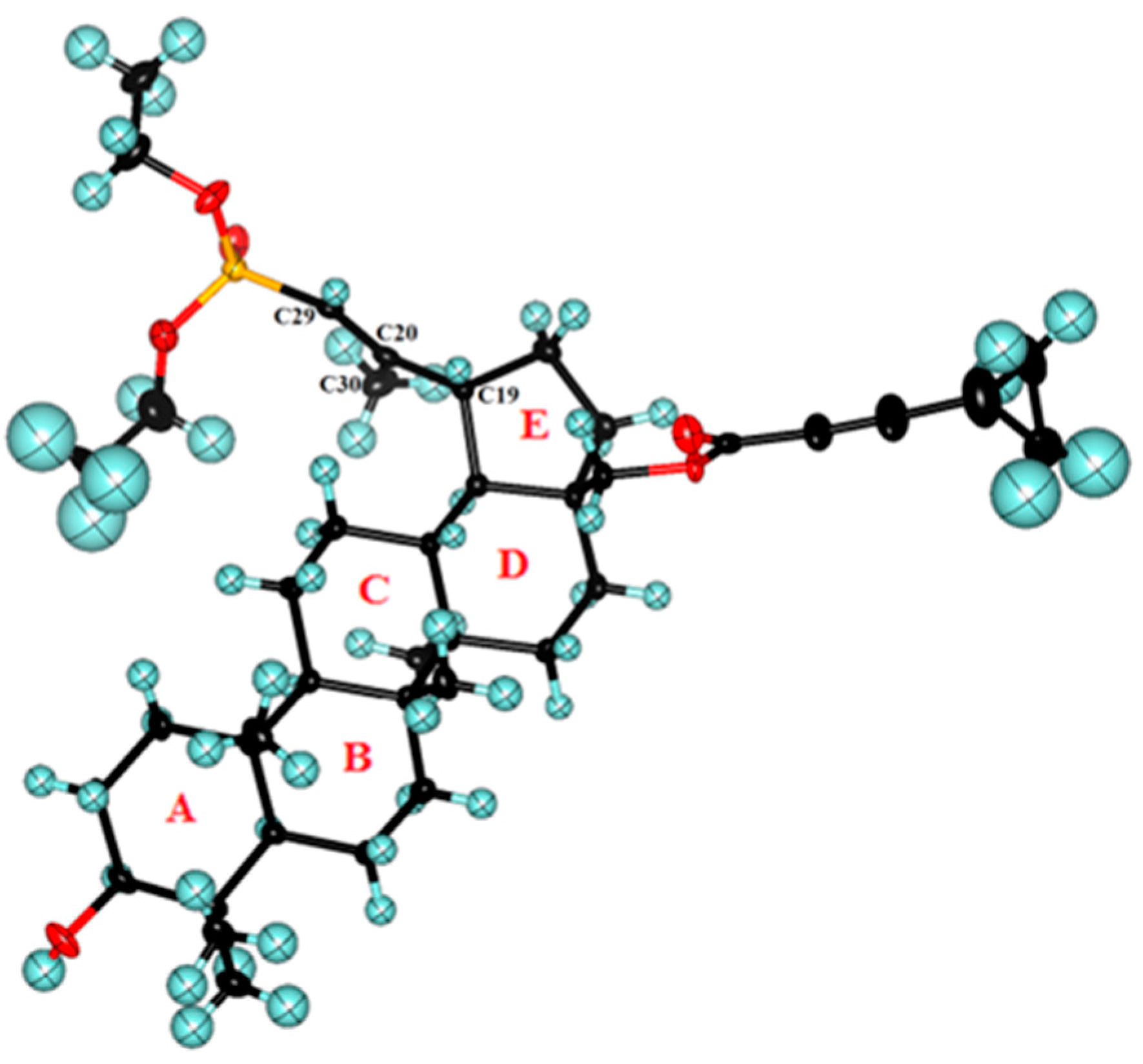
2.2. Crystal Structure of 29-Diethoxyphosphoryl-28-cyclopropylpropynoyloxy-lup-20E(29)-en-3β-ol 8a
2.3. Cytotoxic Activity
3. Materials and Methods
3.1. General Techniques
3.2. Procedure for the Synthesis of 3β,28-Diacetoxy-30-diethoxyphosphoryl-lup-20(29)-ene 3
3.3. Procedure for the Hydrolysis of 3β,28-Diacetoxy-30-diethoxyphosphoryl-lup-20(29)-ene 3
3.4. General Procedure for the Synthesis of Acetylenic Derivatives of Betulin 6a–8a and 6b–8b
3.5. Procedure for the Synthesis of Propagyloxycarbonyl Derivatives of Betulin 9a and 9b
3.6. Crystal Structure Determination
3.7. Antiproliferative Assay in Vitro
3.7.1. Cell culture
3.7.2. Biological Activity of Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alakurtti, S.; Mäkelä, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Boryczka, S.; Bębenek, E.; Wietrzyk, J.; Kępińska, K.; Jastrzębska, M.; Kusz, J.; Nowak, M. Synthesis, structure and cytotoxic activity of new acetylenic derivatives of betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R.; Nitsche, C.; Sczepek, R.; Schwarz, S.; Siewert, B. Synthesis of antitumor-active betulinic acid-derived hydroxypropargylamines by copper-catalyzed Mannich reactions. Arch. Pharm. Chem. Life Sci. 2013, 346, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Majeed, R.; Sangwan, P.L.; Chinthakindi, P.K.; Khan, I.; Dangroo, N.A.; Thota, N.; Hamid, A.; Sharma, P.R.; Saxena, A.K.; Koul, S. Synthesis of 3-O-propargylated betulinic acid and its 1,2,3-triazoles as potential apoptotic agents. Eur. J. Med. Chem. 2013, 63, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Engel, R. Phosphonates as analogues of natural phosphates. Chem. Rev. 1977, 77, 349–367. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, H.; Davrazou, F.; Kutateladze, T.G.; Shi, X.; Gozani, O.; Prestwich, G.D. Stabilized phosphatidylinositol-5-phosphate analogues as ligands for the nuclear protein ING2: Chemistry, biology, and molecular modeling. J. Am. Chem. Soc. 2007, 129, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.L.; Lin, T.C.; Wang, S.Y.; Shie, J.J.; Tsai, K.C.; Cheng, Y.S.E.; Jan, J.T.; Lin, C.J.; Fang, J.M.; Wong, C.H. Tamiphosphor monoesters as effective anti-influenza agents. Eur. J. Med. Chem. 2014, 81, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, M.; Rao, K.U.M.; Sundar, C.S.; Reddy, N.B.; Nayak, S.K.; Reddy, C.S. Green Synthesis and bioactivity of 2-amino-4H-chromen-4-yl-phosphonates. Chem. Pharm. Bull. 2012, 60, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-B.; Liu, J.-B.; Dou, D.-L.; Song, F.-B.; Li, L.-Y.; Xi, Z. Synthesis and screening of novel inositol phosphonate derivatives for anticancer functions in vitro. Chin. Chem. Lett. 2015, 26, 329–333. [Google Scholar] [CrossRef]
- Abdou, W.M.; Shaddy, A.A. The development of bisphosphonates for therapeutic uses, and bisphosphonate structure-activity consideration. Arkivoc 2009, 2009, 143–182. [Google Scholar]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154–9218. [Google Scholar] [CrossRef] [PubMed]
- Sun, I.-C.; Wang, H.-K.; Kashiwada, Y.; Shen, J.-K.; Cosentino, L. M.; Chen, C.-H.; Yang, L.-M.; Lee, K.-H. Anti-AIDS Agents. 34. Synthesis and structure-activity relationships of betulin derivatives as anti-HIV agents. J. Med. Chem. 1998, 41, 4648–4657. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, S.; Heiska, T.; Kiriazis, A.; Sacerdoti-Sierra, N.; Jaffe, C.L.; Yli-Kauhaluoma, J. Synthesis and anti-leishmanial activity of heterocyclic betulin derivatives. Bioorg. Med. Chem. 2010, 18, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Quin, D.; Williams, A.J. Phosphonic and phosphinic acids and derivatives. In Practical Interpretation of P-31 NMR Spectra and Computer-Assisted Structure Verification; Advanced Chemistry Development, Inc.: Toronto, ON, Canada, 2004; p. 30. [Google Scholar]
- Kiddle, J.J.; Babler, J.H. A facile route to allylic phosphonates via base-catalyzed isomerization of the corresponding vinyl phosphonates. J. Org. Chem. 1993, 58, 3572–3574. [Google Scholar] [CrossRef]
- Rowe, B.J.; Spilling, C.D. Stereospecific Pd(0)-catalyzed arylation of an allylic hydroxy phosphonate derivative: Formal synthesis of (S)-(+)-ar-Turmerone. J. Org. Chem. 2003, 68, 9502–9505. [Google Scholar]
- Malla, R.K.; Ridenour, J.N.; Spilling, C.D. Relay cross metathesis reactions of vinylphosphonates. Beilstein J. Org. Chem. 2014, 10, 1933–1941. [Google Scholar] [CrossRef] [PubMed]
- Solberghe, G.F.; Markó, I.E. Regio- and stereocontrolled preparation of α-substituted phosphonocrotonate derivatives. Tetrahedron Lett. 2002, 43, 5061–5065. [Google Scholar] [CrossRef]
- Principato, B.; Maffei, M.; Siv, C.; Buono, G.; Peiffer, G. Palladium catalysed allylic acetoxylation of dialkyl ally1 phosphonates. Tetrahedron 1996, 52, 2087–2096. [Google Scholar] [CrossRef]
- Nalewajek, D.; Soriano, D.S. Preparation of Vinylphosphonate Diesters. US Patent 4582652, 30 December 1986. [Google Scholar]
- Probst, M.F.; Modro, A.M.; Modro, T.A. 3-Phosphorylated 1,5-hexadienes as precursors for vinylic or 3-silanylvinylic phosphonates. Can. J. Chem. 1997, 75, 1131–1135. [Google Scholar] [CrossRef]
- Quntar, A.A.; Melman, A.; Srebnik, M. Selective Preparation of (E)-3-oxo-1-alkenyl-phosphonates by insertion of acyl chlorides and nitriles into zirconacycles. J. Org. Chem. 2002, 67, 3769–3772. [Google Scholar] [CrossRef] [PubMed]
- Konno, T.; Kinugawa, R.; Morigaki, A.; Ishihara, T. An efficient protocol for the stereoselective construction of multisubstituted fluorine-containing alkenes. A palladium-catalyzed bisstannylation of fluorinated internal alkynes. J. Org. Chem. 2009, 74, 8456–8459. [Google Scholar] [CrossRef] [PubMed]
- Boutagy, J.; Thomas, R. Olefin synthesis with organic phosphonate carbanions. Chem. Rev. 1974, 74, 87–99. [Google Scholar] [CrossRef]
- Corey, E.J.; Cane, D.E. The synthesis of olefins from β-hydroxyphosphonamides. Stereo-chemistry and extension to the formation of conjugated dienes. J. Org. Chem. 1969, 34, 3053–3057. [Google Scholar] [CrossRef]
- Modro, M.; Modro, T.A. The prototropic equilibria in pentenylphosphonic systems. Can. J. Chem. 1988, 66, 1541–1545. [Google Scholar] [CrossRef]
- Boryczka, S.; Michalik, E.; Jastrzębska, M.; Kusz, J.; Zubko, M.; Bębenek, E. X-ray crystal structure of betulin–DMSO solvate. J. Chem. Crystallogr. 2012, 42, 345–351. [Google Scholar] [CrossRef]
- Boryczka, S.; Bębenek, E.; Jastrzębska, M.; Kusz, J.; Zubko, M. Crystal structure of betulinic acid-DMSO solvate. Z. Kristallogr. 2012, 227, 379–384. [Google Scholar] [CrossRef]
- Spackman, M.A.; Joshua, J.; McKinnon, J.J. Fingerprinting intermolecular interactions in molecular crystals. Cryst. Eng. Commun. 2002, 4, 378–392. [Google Scholar] [CrossRef]
- Jelsch, C.; Ejsmont, K.; Huder, L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Guillot, B. MoProViewer: A molecule viewer for the MoPro charge density analysis program. Acta Cryst. 2012, A68, s204. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Jelsch, C.; Guillot, B.; Lagoutte, A.; Lecomte, C. Advances in proteins and small molecules. charge density refinement methods using software MoPro. J. Appl. Cryst. 2005, 38, 38–54. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
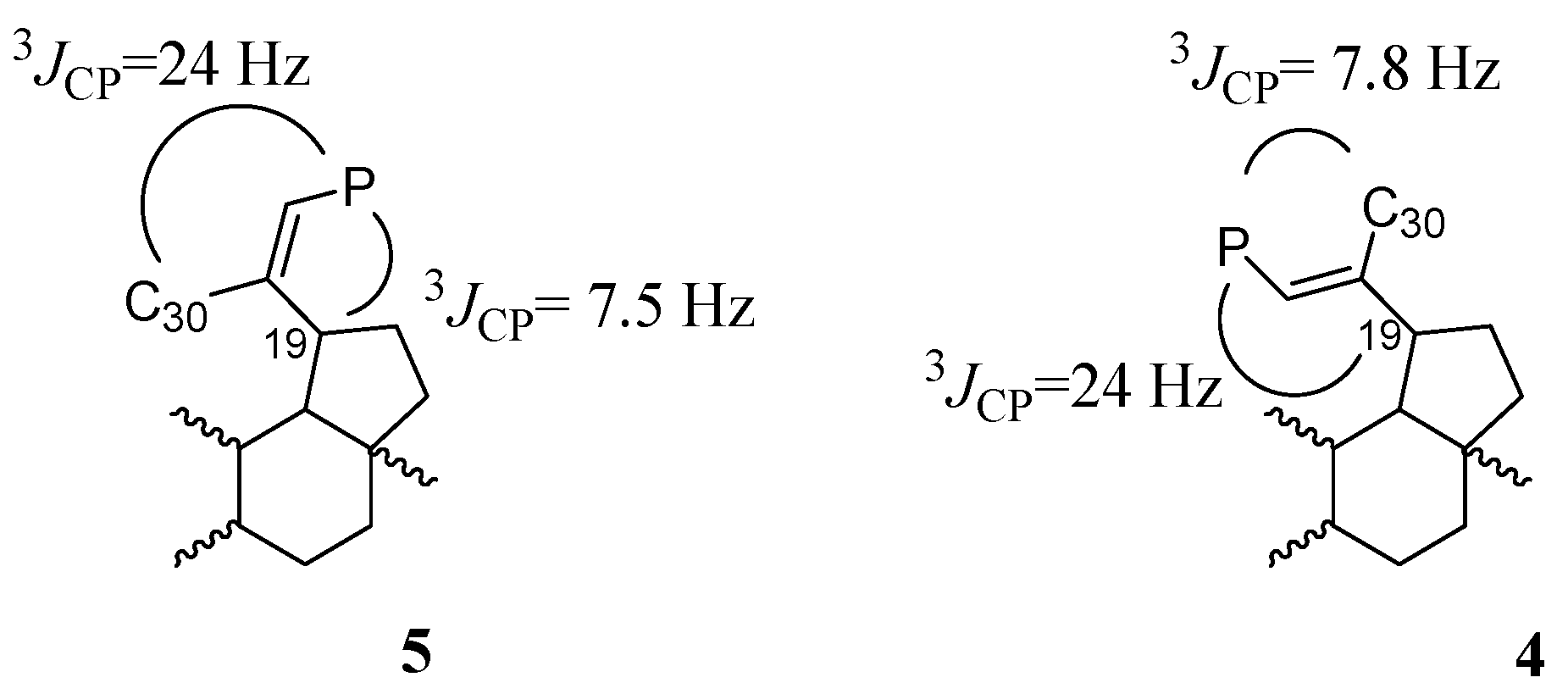{kind=link}
{kind=link}
{kind=link}
| Atom Type | Ho | C | O | Hc | P |
|---|---|---|---|---|---|
| % Surface | 1.77 | 13.23 | 7.49 | 76.43 | 1.08 |
| Ho | 0.0 | ||||
| C | 0.1 | 1.0 | contacts X···Y (%) | ||
| O | 1.8 | 1.7 | 0.0 | ||
| Hc | 1.8 | 21.9 | 11.3 | 58.2 | |
| P | 0.0 | 0.0 | 0.0 | 2.2 | 0.0 |
| Ho | 0.00 | ||||
| C | 0.30 | 0.60 | enrichment of contacts | ||
| O | 6.54 | 0.89 | 0.00 | ||
| Hc | 0.62 | 1.11 | 1.00 | 0.99 | |
| P | 0.04 | 0.03 | 0.00 | 1.30 | 0.00 |
| Compound | Cytotoxic Activity IC50 (µg/mL) | ||
|---|---|---|---|
| T47D | SNB-19 | C32 | |
| Betulin 1 | 77.87 ± 4.71 | 23.52 ± 2.02 | 34.91 ± 5.37 |
| 3 | 58.00 ± 3.36 | 51.88 ± 0.76 | 2.15 ± 0.19 |
| 4 | 6.23 ± 0.53 | 5.92 ± 0.42 | 5.09 ± 0.1 |
| 5 | 0.89 ± 0.08 | 4.80 ± 0.09 | 5.25 ± 0.51 |
| 6a | 0.44 ± 0.08 | 0.27 ± 0.09 | 0.38 ± 0.05 |
| 6b | 2.84 ± 0.57 | 0.60 ± 0.06 | 2.71 ± 0.52 |
| 7a | 9.63 ± 0.58 | 7.52 ± 0.28 | 4.96 ± 0.07 |
| 7b | 13.92 ± 3.42 | 7.58 ± 1.00 | 4.9 ± 0.26 |
| 8a | 8.18 ± 0.56 | 2.20 ± 0.78 | 4.69 ± 0.72 |
| 8b | 79.52 ± 1.92 | 40.41 ± 6.85 | 9.44 ± 0.22 |
| 9a | 49.38 ± 6.12 | 24.50 ± 12.62 | 5.95 ± 0.35 |
| 9b | Neg | 0.49 ± 0.05 | 51.67 ± 4.76 |
| cisplatin | 55.95 ± 4.55 | 5.29 ± 0.12 | 4.29 ± 0.65 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrobak, E.; Bębenek, E.; Kadela-Tomanek, M.; Latocha, M.; Jelsch, C.; Wenger, E.; Boryczka, S. Betulin Phosphonates; Synthesis, Structure, and Cytotoxic Activity. Molecules 2016, 21, 1123. https://doi.org/10.3390/molecules21091123
Chrobak E, Bębenek E, Kadela-Tomanek M, Latocha M, Jelsch C, Wenger E, Boryczka S. Betulin Phosphonates; Synthesis, Structure, and Cytotoxic Activity. Molecules. 2016; 21(9):1123. https://doi.org/10.3390/molecules21091123
Chicago/Turabian StyleChrobak, Elwira, Ewa Bębenek, Monika Kadela-Tomanek, Małgorzata Latocha, Christian Jelsch, Emmanuel Wenger, and Stanisław Boryczka. 2016. "Betulin Phosphonates; Synthesis, Structure, and Cytotoxic Activity" Molecules 21, no. 9: 1123. https://doi.org/10.3390/molecules21091123
APA StyleChrobak, E., Bębenek, E., Kadela-Tomanek, M., Latocha, M., Jelsch, C., Wenger, E., & Boryczka, S. (2016). Betulin Phosphonates; Synthesis, Structure, and Cytotoxic Activity. Molecules, 21(9), 1123. https://doi.org/10.3390/molecules21091123