
Efficient Synthesis of Fully Substituted Pyrrolidine-Fused 3-Spirooxindoles via 1,3-Dipolar Cycloaddition of Aziridine and 3-Ylideneoxindole

Abstract
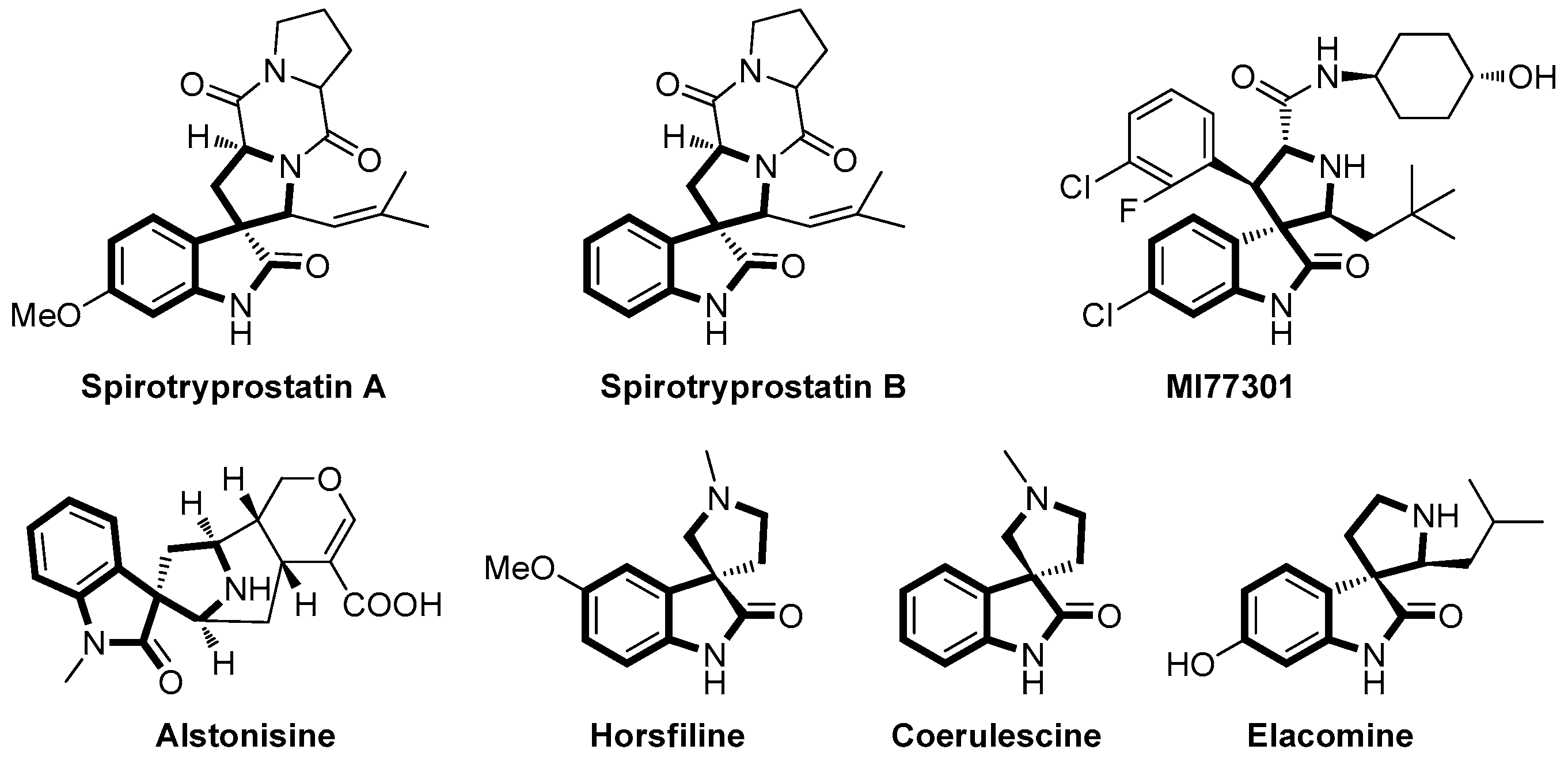
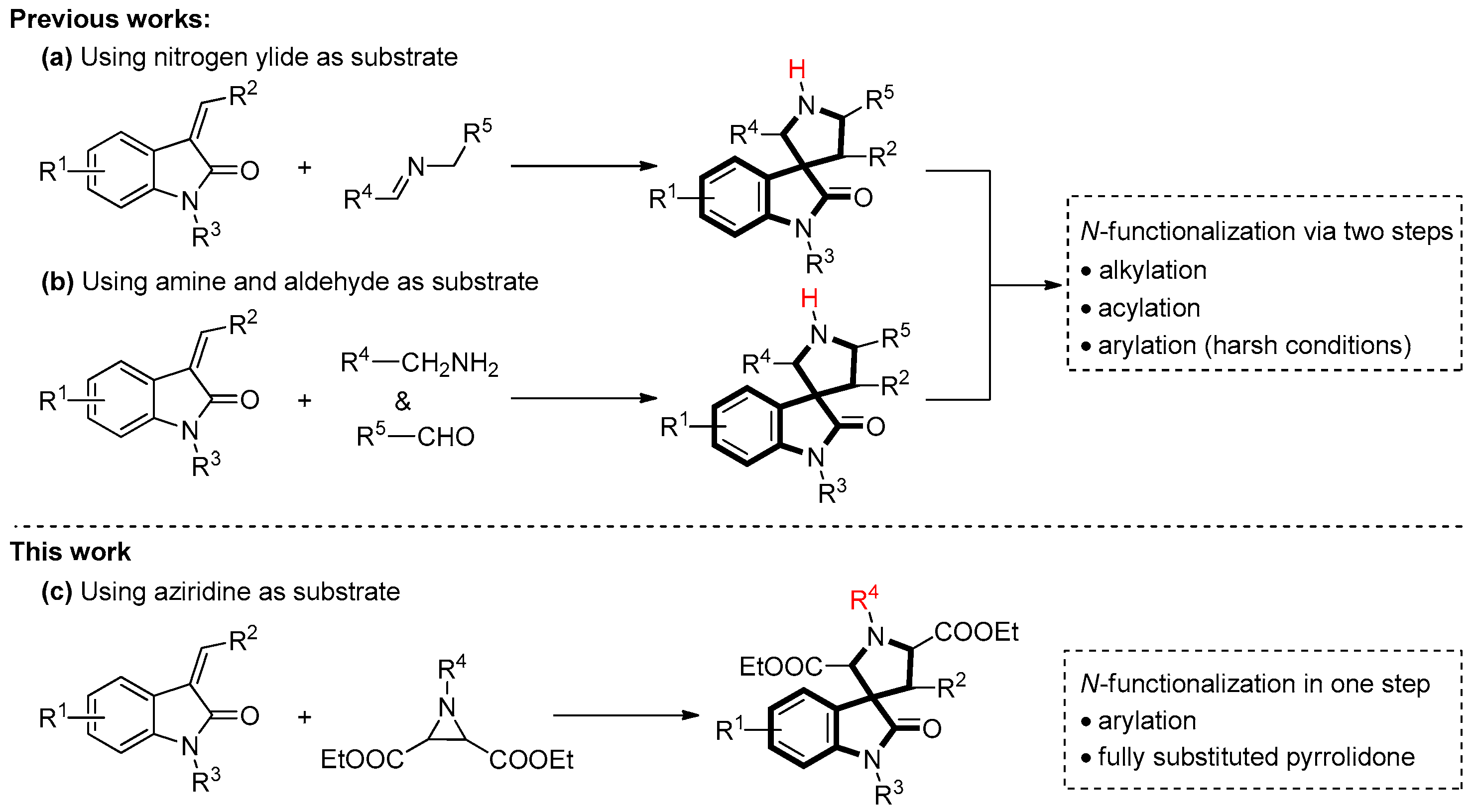
:1. Introduction
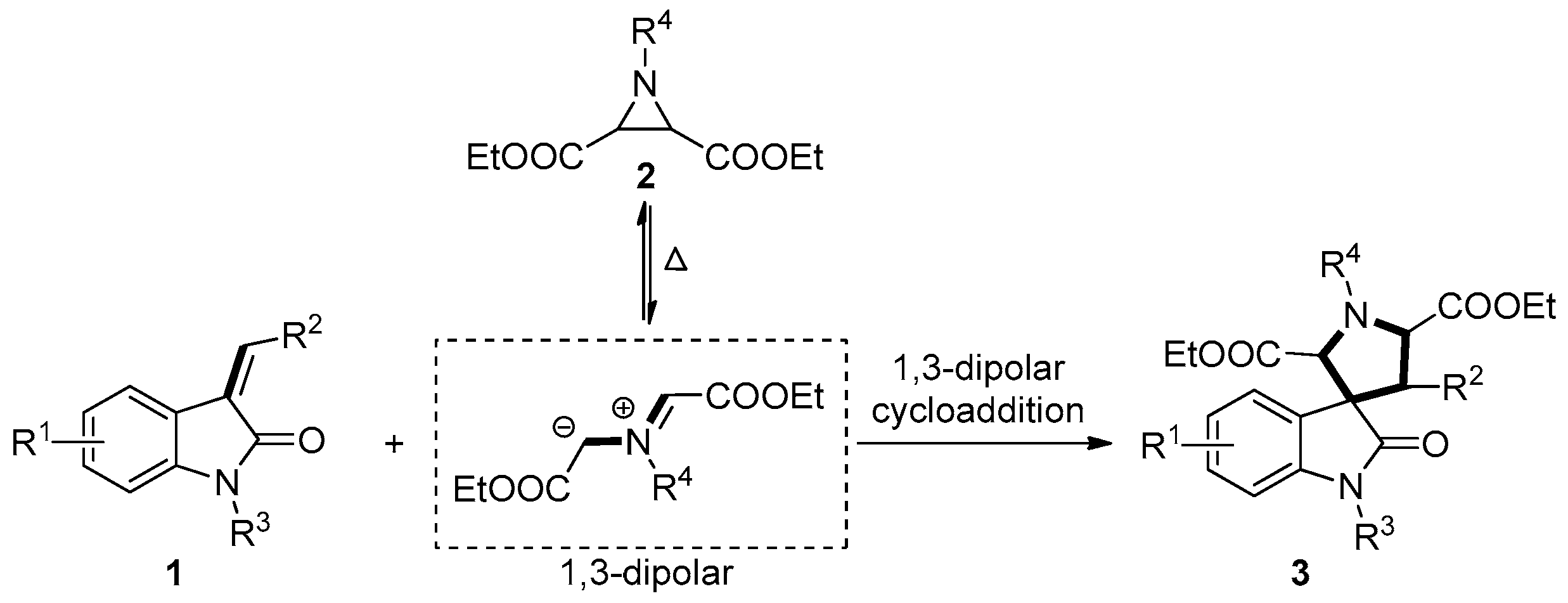
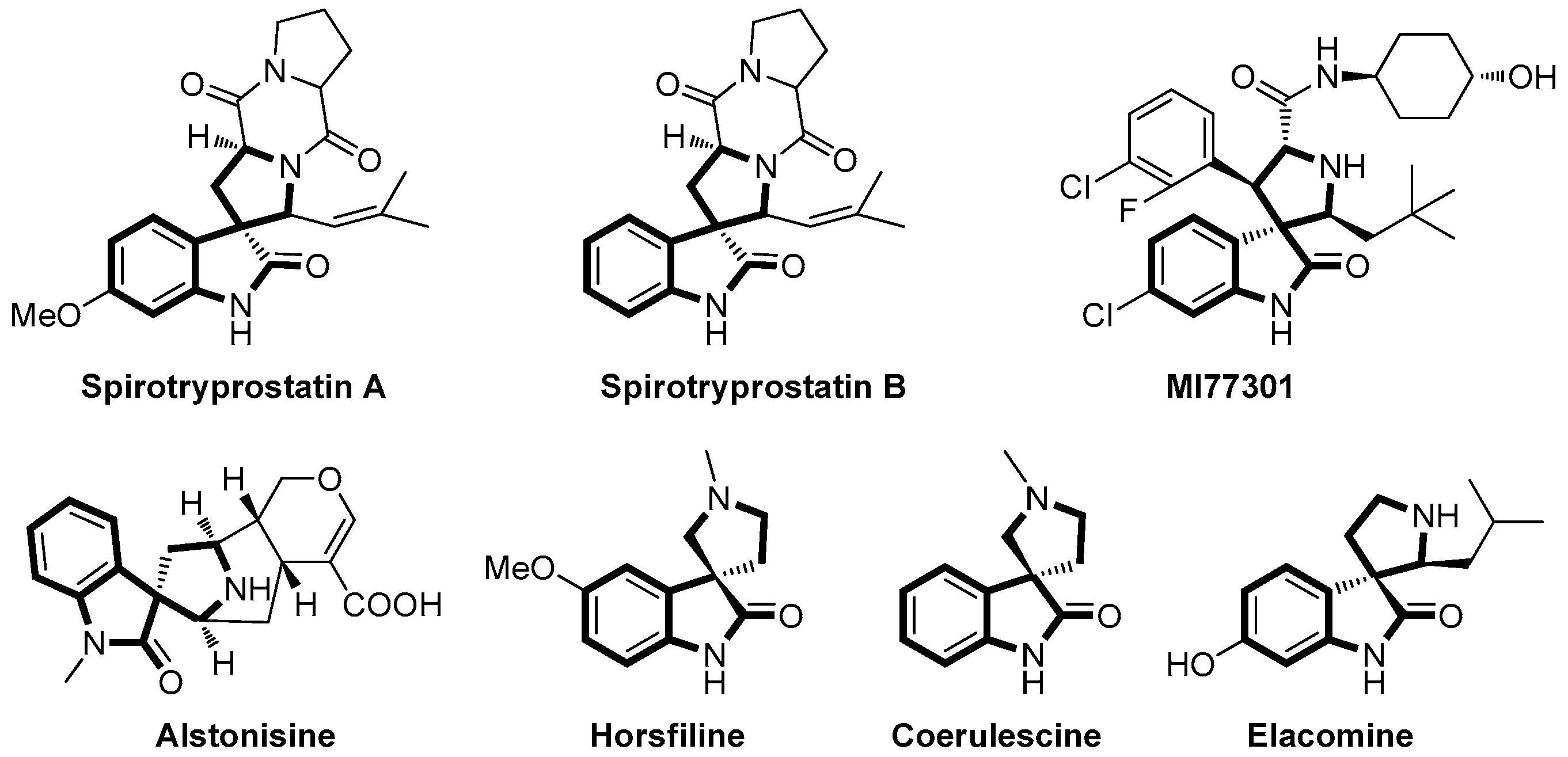
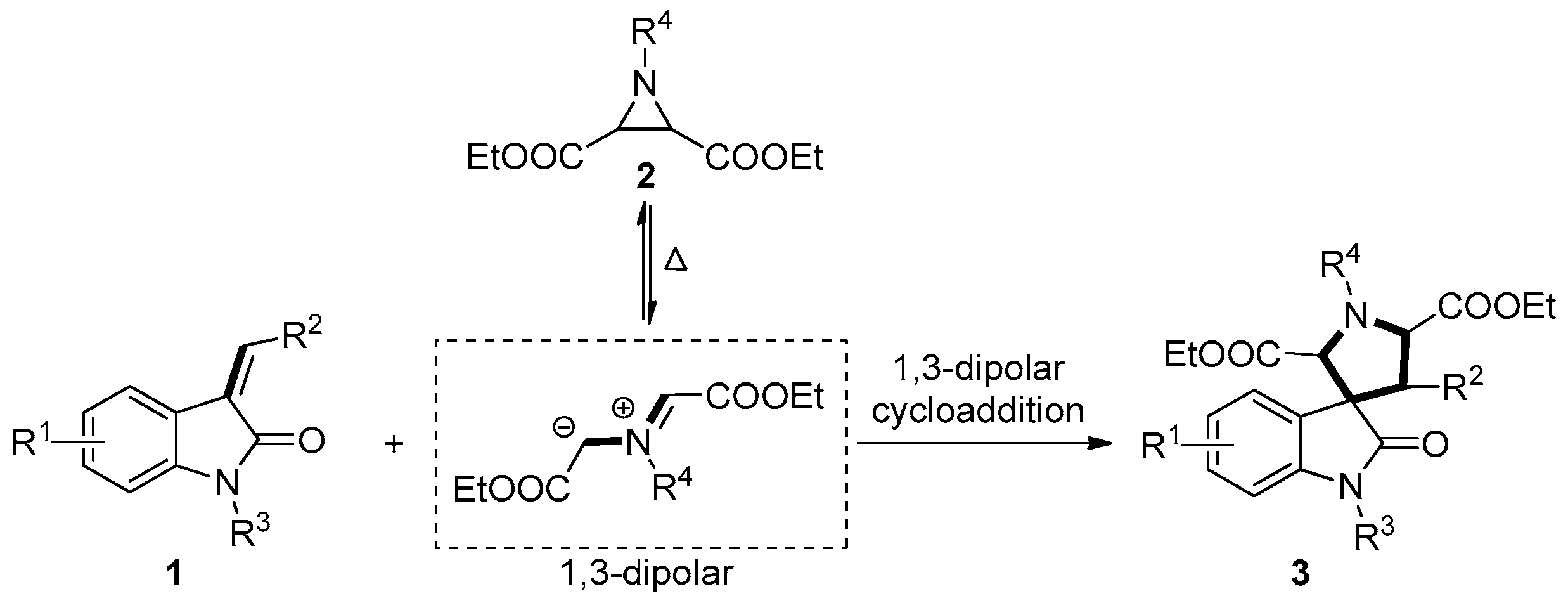
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. General Procedure for the Synthesis of Spirooxindole-Pyrrolidines 3a–t
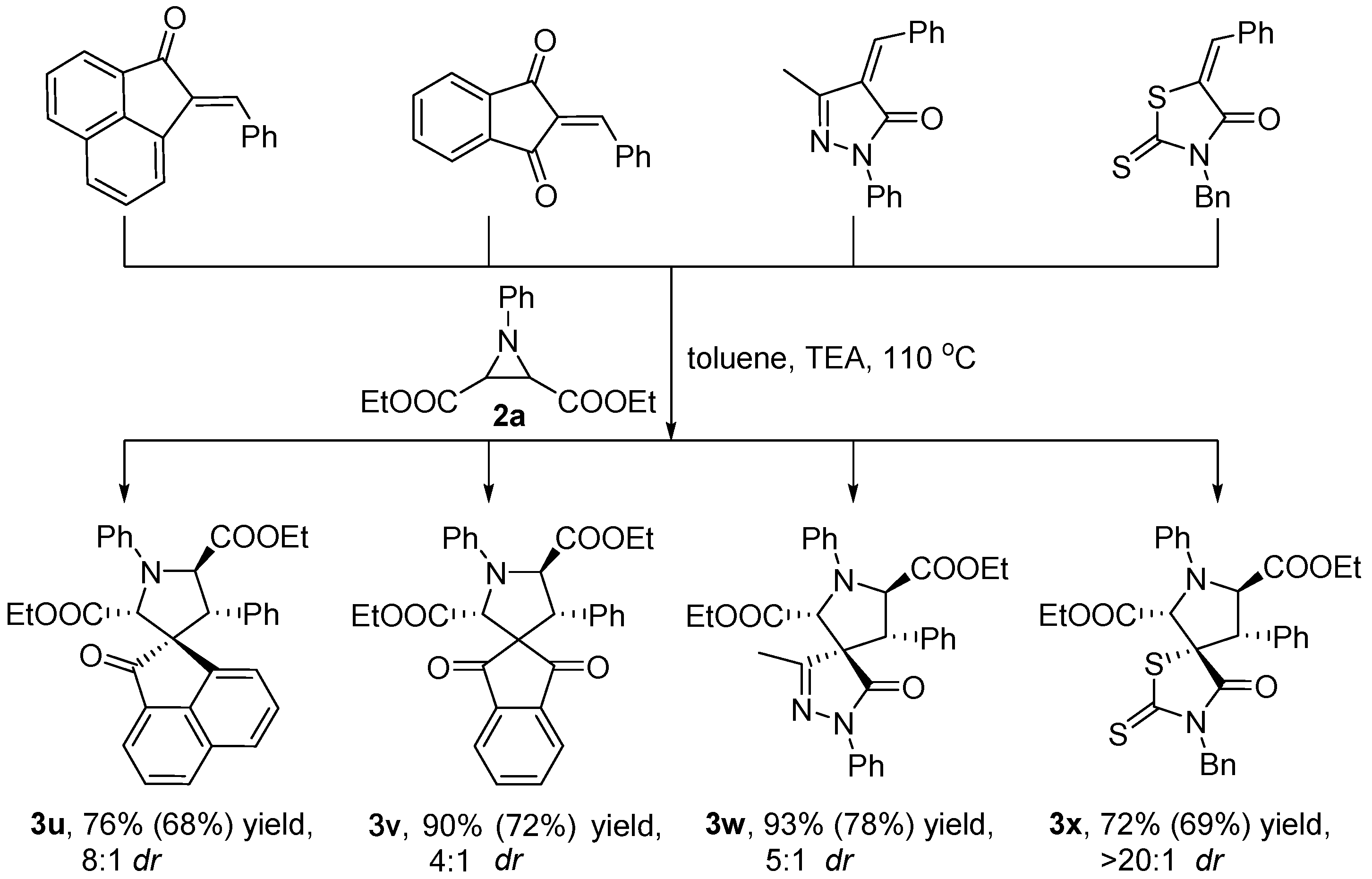
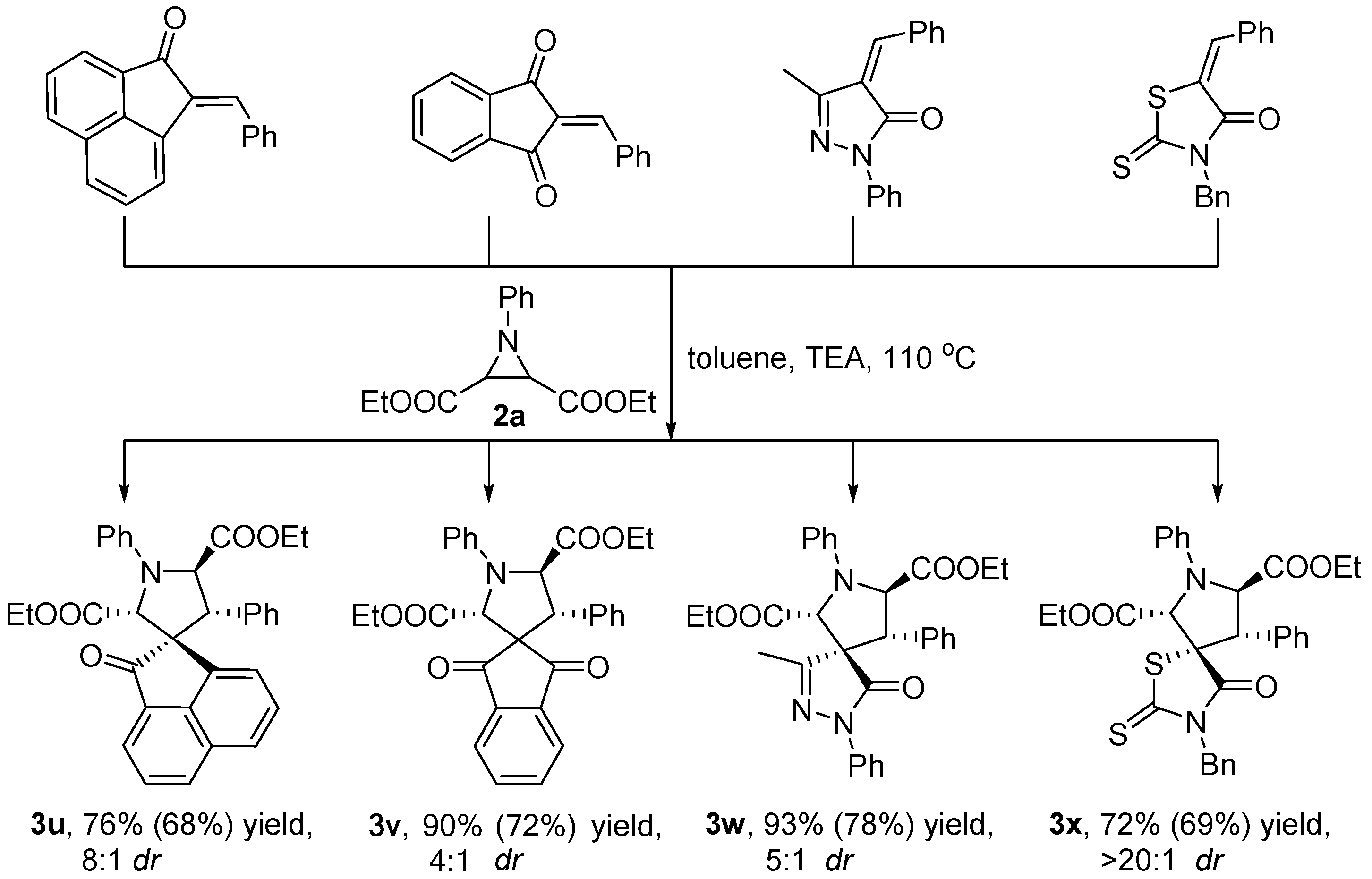
3.2.2. Synthetic Transformations to Access Other Drug-Like Spirocyclic Scaffolds 3u–x
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DBU | 1,8-Diazabicyclo(5.4.0)undec-7-ene |
| DABCO | 1,4-Diazabicyclo(2.2.2)octane |
| TEA | Triethylamine |
References
- Stevens, T.S.; Creighton, E.M.; Gordon, A.B.; MacNicol, M. Degradation of quaternary ammonium salts Part I. J. Chem. Soc. (Resumed) 1928, 3193–3197. [Google Scholar] [CrossRef]
- Pellissier, H. Asymmetric 1,3-dipolar cycloadditions. Tetrahedron 2007, 63, 3235–3285. [Google Scholar] [CrossRef]
- Stanley, L.M.; Sibi, M.P. Enantioselective copper-catalyzed 1,3-dipolar cycloadditions. Chem. Rev. 2008, 108, 2887–2902. [Google Scholar] [CrossRef] [PubMed]
- Kissane, M.; Maguire, A.R. Asymmetric 1,3-dipolar cycloadditions of acrylamides. Chem. Soc. Rev. 2010, 39, 845–883. [Google Scholar] [CrossRef] [PubMed]
- Adrio, J.; Carretero, J.C. Recent advances in the catalytic asymmetric 1,3-dipolar cycloaddition of azomethine ylides. Chem. Commun. 2014, 50, 12434–12446. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Chowdhury, S.; Koley, S. Progress in 1,3-dipolar cycloadditions in the recent decade: an update to strategic development towards the arsenal of organic synthesis. Tetrahedron 2016, 72, 1603–1644. [Google Scholar] [CrossRef]
- Nair, V.; Suja, T.D. Intramolecular 1,3-dipolar cycloaddition reactions in targeted syntheses. Tetrahedron 2007, 63, 12247–12275. [Google Scholar] [CrossRef]
- Nájera, C.; Sansano, J.M. Coinage metal complexes as chiral catalysts for 1,3-dipolar cycloadditions. J. Organomet. Chem. 2014, 771, 78–92. [Google Scholar] [CrossRef]
- Narayan, R.; Potowski, M.; Jia, Z.J.; Antonchick, A.P.; Waldmann, H. Catalytic Enantioselective 1,3-Dipolar Cycloadditions of Azomethine Ylides for Biology-Oriented Synthesis. Acc. Chem. Res. 2014, 47, 1296–1310. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Maruoka, K. Recent Advances of Catalytic Asymmetric 1,3-Dipolar Cycloadditions. Chem. Rev. 2015, 115, 5366–5412. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Cai, G.; Ma, D. A formal (3 + 2) cycloaddition process with nonactivated aziridines to polysubstituted indolizidines. Org. Lett. 2005, 7, 5545–5548. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Zhou, Y.G. AgOAc catalyzed asymmetric (3 + 2) cycloaddition of azomethine ylides with chiral ferrocene derived P,S ligands. Tetrahedron Lett. 2007, 48, 4619–4622. [Google Scholar] [CrossRef]
- Arai, T.; Mishiro, A.; Yokoyama, N.; Suzuki, K.; Sato, H. Chiral Bis(imidazolidine)pyridine-Cu(OTf)2: Catalytic Asymmetric Endo-Selective (3 + 2) Cycloaddition of Imino Esters with Nitroalkenes. J. Am. Chem. Soc. 2010, 132, 5338–5339. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Yokoyama, N.; Mishiro, A.; Sato, H. Catalytic Asymmetric exo′-Selective (3 + 2) Cycloaddition of Iminoesters with Nitroalkenes. Angew. Chem. Int. Ed. 2010, 49, 7895–7898. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.H.; Xue, Z.Y.; Tao, H.Y.; Wang, C.J. Cu(I)/DTBM-BIPHEP-catalyzed exo-selective 1,3-dipolar cycloaddition of azomethine ylides with cis-trifluorocrotonate for asymmetric construction of trifluoromethylated pyrrolidines. Tetrahedron Lett. 2012, 53, 3650–3653. [Google Scholar] [CrossRef]
- Potowski, M.; Bauer, J.O.; Strohmann, C.; Antonchick, A.P.; Waldmann, H. Highly Enantioselective Catalytic (6 + 3) Cycloadditions of Azomethine Ylides. Angew. Chem. Int. Ed. 2012, 51, 9512–9516. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Qiao, G.; Liu, H.; Zhang, L.; Sun, Z.; Xiao, Y.; Guo, H. Brønsted acid-promoted (3 + 3) cycloaddition of azomethine ylides with quinone monoimine: A practical method towards dihydrobenzoxazine derivatives. RSC Adv. 2015, 5, 84290–84294. [Google Scholar] [CrossRef]
- Malatesti, N.; Boa, A.N.; Clark, S.; Westwood, R. 1,3-Dipolar cycloaddition reactions of benzo(b)thiophene 1,1-dioxide with azomethine ylides. Tetrahedron Lett. 2006, 47, 5139–5142. [Google Scholar] [CrossRef]
- Ishii, K.; Kido, M.; Noji, M.; Sugiyama, S. Photoreactions of bicyclic aziridines with alkenes and alkynes: A novel synthetic methodology for 8-azabicyclo (3.2.1) octane derivatives. Org. Biomol. Chem. 2008, 6, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Tong, M.C.; Wang, C.J. Silver Acetate/TF-BiphamPhos-Catalyzed endo-Selective Enantioselective 1,3-Dipolar Cycloaddition of Azomethine Ylides with Vinyl Phenyl Sulfone. Adv. Synth. Catal. 2009, 351, 3101–3106. [Google Scholar] [CrossRef]
- Ribeiro Laia, F.M.; Cardoso, A.L.; Beja, A.M.; Silva, M.R.; Pinho e Melo, T.M.V.D. Reactivity of allenoates towards aziridines: Synthesis of functionalized methylenepyrrolidines and pyrroles. Tetrahedron 2010, 66, 8815–8822. [Google Scholar] [CrossRef]
- Yang, W.L.; Liu, Y.Z.; Luo, S.; Yu, X.; Fossey, J.S.; Deng, W.P. The copper-catalyzed asymmetric construction of a dispiropyrrolidine skeleton via 1,3-dipolar cycloaddition of azomethine ylides to α-alkylidene succinimides. Chem. Commun. 2015, 51, 9212–9215. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.C.; Gupta, A.K.; Wu, M.F.; Liao, J.H.; Lee, G.H. Hetero (6 + 3) cycloaddition of fulvenes with N-alkylidene glycine esters: A facile synthesis of the delavayine and incarvillateine framework. Org. Lett. 2003, 5, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Potowski, M.; Antonchick, A.P.; Waldmann, H. Catalytic asymmetric exo-selective (6 + 3) cycloaddition of iminoesters with fulvenes. Chem. Commun. 2013, 49, 7800–7802. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Liu, H.; Gao, Z.; Zhou, L.; Feng, Y.; Xiao, Y.; Guo, H. Cu(I)-Catalyzed Highly Enantioselective (3 + 3) Cycloaddition between Two Different 1,3-Dipoles, Phthalazinium Dicyanomethanides and Iminoester-Derived Azomethine Ylides. Org. Lett. 2015, 17, 26–29. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ganesan, A. A biomimetic total synthesis of (−)-spirotryprostatin B and related studies. J. Org. Chem. 2000, 65, 4685–4693. [Google Scholar] [CrossRef] [PubMed]
- Sebahar, P.R.; Williams, R.M. The asymmetric total synthesis of (+)- and (−)-spirotryprostatin B. J. Am. Chem. Soc. 2000, 122, 5666–5667. [Google Scholar] [CrossRef]
- Antonchick, A.P.; Schuster, H.; Bruss, H.; Schüermann, M.; Preut, H.; Rauh, D.; Waldmann, H. Enantioselective synthesis of the spirotryprostatin A scaffold. Tetrahedron 2011, 67, 10195–10202. [Google Scholar] [CrossRef]
- Zhang, L.J.; Wang, Y.; Hu, X.Q.; Xu, P.F. Hydrogen-Bonding Network Promoted (3 + 2) Cycloaddition: Asymmetric Catalytic Construction of Spiro-pseudoindoxyl Derivatives. Chem. Asian J. 2016, 11, 834–838. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.B.; Kakeya, H.; Osada, H. Novel mammalian cell cycle inhibitors, spirotryprostatins A and B, produced by Aspergillus fumigatus, which inhibit mammalian cell cycle at G2/M phase. Tetrahedron 1996, 52, 12651–12666. [Google Scholar] [CrossRef]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-spirooxindole natural products as inspirations for the development of potential therapeutic agents. Angew. Chem. Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef] [PubMed]
- Senwar, K.R.; Sharma, P.; Reddy, T.S.; Jeengar, M.K.; Nayak, V.L.; Naidu, V.G.M.; Kamal, A.; Shankaraiah, N. Spirooxindole-derived morpholine-fused-1,2,3-triazoles: Design, synthesis, cytotoxicity and apoptosis inducing studies. Eur. J. Med. Chem. 2015, 102, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yu, D.Q.; Liu, H.M. Spirooxindoles: Promising scaffolds for anticancer agents. Eur. J. Med. Chem. 2015, 97, 673–698. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.M.C.; Neumann, C.S.; Nagayama, S.; Perlstein, E.O.; Schreiber, S.L. A library of spirooxindoles based on a stereoselective three-component coupling reaction. J. Am. Chem. Soc. 2004, 126, 16077–16086. [Google Scholar] [CrossRef] [PubMed]
- Ding, K.; Lu, Y.; Nikolovska-Coleska, Z.; Wang, G.; Qiu, S.; Shangary, S.; Gao, W.; Qin, D.; Stuckey, J.; Krajewski, K.; et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J. Med. Chem. 2006, 49, 3432–3435. [Google Scholar] [CrossRef] [PubMed]
- Shangary, S.; Qin, D.; McEachern, D.; Liu, M.; Miller, R.S.; Qiu, S.; Nikolovska-Coleska, Z.; Ding, K.; Wang, G.; Chen, J.; et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc. Natl. Acad. Sci. USA. 2008, 105, 3933–3938. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, D.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An Optimized Inhibitor of MDM2-p53 Interaction That Induces Complete and Durable Tumor Regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Liu, Y.L.; Zhou, J. Catalytic Asymmetric Synthesis of Oxindoles Bearing a Tetrasubstituted Stereocenter at the C-3 Position. Adv. Synth. Catal. 2010, 352, 1381–1407. [Google Scholar] [CrossRef]
- Singh, G.S.; Desta, Z.Y. Isatins As Privileged Molecules in Design and Synthesis of Spiro-Fused Cyclic Frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef] [PubMed]
- Franz, A.K.; Hanhan, N.V.; Ball-Jones, N.R. Asymmetric Catalysis for the Synthesis of Spirocyclic Compounds. Acs Catal. 2013, 3, 540–553. [Google Scholar] [CrossRef]
- Hong, L.; Wang, R. Recent Advances in Asymmetric Organocatalytic Construction of 3,3′-Spirocyclic Oxindoles. Adv. Synth. Catal. 2013, 355, 1023–1052. [Google Scholar] [CrossRef]
- Santos, M.M.M. Recent advances in the synthesis of biologically active spirooxindoles. Tetrahedron 2014, 70, 9735–9757. [Google Scholar] [CrossRef]
- Antonchick, A.P.; Gerding-Reimers, C.; Catarinella, M.; Schüermann, M.; Preut, H.; Ziegler, S.; Rauh, D.; Waldmann, H. Highly enantioselective synthesis and cellular evaluation of spirooxindoles inspired by natural products. Nat. Chem. 2010, 2, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.L.; Xue, Z.Y.; Tao, H.Y.; Wang, C.J. Catalytic asymmetric 1,3-dipolar cycloaddition of N-unprotected 2-oxoindolin-3-ylidene derivatives and azomethine ylides for the construction of spirooxindole-pyrrolidines. Org. Biomol. Chem. 2011, 9, 1980–1986. [Google Scholar] [CrossRef] [PubMed]
- Awata, A.; Arai, T. Catalytic Asymmetric exo′-Selective (3 + 2) Cycloaddition for Constructing Stereochemically Diversified Spiro[pyrrolidin-3,3′-oxindole]s. Chem. Eur. J. 2012, 18, 8278–8282. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shi, X.M.; Dong, W.P.; Zhu, L.P.; Wang, R. Efficient construction of highly functionalized spiro (γ-butyrolactone-pyrrolidin-3,3′-oxindole) tricyclic skeletons via an organocatalytic 1,3-dipolar cycloaddition. Chem. Commun. 2013, 49, 3458–3460. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Ogawa, H.; Awata, A.; Sato, M.; Watabe, M.; Yamanaka, M. PyBidine-Cu(OTf)2-Catalyzed Asymmetric (3 + 2) Cycloaddition with Imino Esters: Harmony of Cu-Lewis Acid and Imidazolidine-NH Hydrogen Bonding in Concerto Catalysis. Angew. Chem. Int. Ed. 2015, 54, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.H.; Wei, Q.; Luo, S.W.; Xiao, H.; Gong, L.Z. Organocatalytic Synthesis of Spiro(pyrrolidin-3,3′-oxindoles) with High Enantiopurity and Structural Diversity. J. Am. Chem. Soc. 2009, 131, 13819–13825. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Yari, A.; Rezaei, S.J.T.; Taheri, A. Synthesis of novel spiropyrrolidine/pyrrolizine-oxindole scaffolds through 1,3-dipolar cycloadditions. Tetrahedron Lett. 2009, 50, 4724–4726. [Google Scholar] [CrossRef]
- Thangamani, A. Regiospecific synthesis and biological evaluation of spirooxindolopyrrolizidines via (3 + 2) cycloaddition of azomethine ylide. Eur. J. Med. Chem. 2010, 45, 6120–6126. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, L.; Gong, H.; Yan, C.G. Convenient synthesis of functionalized spiro (indoline-3,2′-pyrrolizines) or spiro(indoline-3,3′-pyrrolidines) via multicomponent reactions. Org. Biomol. Chem. 2015, 13, 5905–5917. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Zhang, H.H.; Li, C.; Fan, T.; Shi, F. Catalytic asymmetric chemoselective 1,3-dipolar cycloadditions of an azomethine ylide with isatin-derived imines: diastereo- and enantioselective construction of a spiro(imidazolidine-2,3′-oxindole) framework. Chem. Commun. 2016, 52, 1804–1807. [Google Scholar] [CrossRef] [PubMed]
- Overman, L.E.; Rosen, M.D. Total synthesis of (−)-spirotryprostatin B and three stereoisomers. Angew. Chem. Int. Ed. 2000, 39, 4596–4599. [Google Scholar] [CrossRef]
- Lerchner, A.; Carreira, E.M. First total synthesis of (±)-strychnofoline via a highly selective ring-expansion reaction. J. Am. Chem. Soc. 2002, 124, 14826–14827. [Google Scholar] [CrossRef] [PubMed]
- Meyers, C.; Carreira, E.M. Total synthesis of (−)-spirotryprostatin B. Angew. Chem. Int. Ed. 2003, 42, 694–696. [Google Scholar] [CrossRef] [PubMed]
- Von Nussbaum, F.; Danishefsky, S.J. A rapid total synthesis of spirotryprostatin B: Proof of its relative and absolute stereochemistry. Angew. Chem. Int. Ed. 2000, 39, 2175–2178. [Google Scholar] [CrossRef]
- Edmondson, S.; Danishefsky, S.J.; Sepp-Lorenzino, L.; Rosen, N. Total synthesis of spirotryprostatin A, leading to the discovery of some biologically promising analogues. J. Am. Chem. Soc. 1999, 121, 2147–2155. [Google Scholar] [CrossRef]
- Genin, M.J.; Chidester, C.G.; Rohrer, D.C.; Romero, D.L. Design and synthesis of a conformationally constrained analog of the bis(heteroaryl)piperazine (BHAP) HIV-1 reverse transcriptase inhibitor atevirdine. Bioorg. Med. Chem. Lett. 1995, 5, 1875–1880. [Google Scholar] [CrossRef]
- Ghorai, M.K.; Ghosh, K. Lewis acid mediated nucleophilic ring opening followed by cycloaddition of 2-aryl-N-tosylaziridines with carbonyl compounds: further support towards an SN2-type mechanism. Tetrahedron Lett. 2007, 48, 3191–3195. [Google Scholar] [CrossRef]
- Soeta, T.; Miyamoto, Y.; Fujinami, S.; Ukaji, Y. The Lewis acid-catalyzed (3 + 1 + 1) cycloaddition of azomethine ylides with isocyanides. Tetrahedron 2014, 70, 6623–6629. [Google Scholar] [CrossRef]
- CCDC 1425280 Contains the Supplementary Crystallographic Data for this Paper. These Data Can be Obtained Free of Charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 1223 336033; E-mail: deposit@ccdc.cam.ac.uk).
- Sample Availability: All samples are available from the authors.

| Entry | Solvent | t (h) b | T (°C) c | Additive | Yield (%) d | dr e |
|---|---|---|---|---|---|---|
| 1 | MeCN | 4 | 90 | - | 40 (30) | 3:1 |
| 2 | EtOH | 4 | 90 | - | 33 (25) | 3:1 |
| 3 | toluene | 4 | 90 | - | 45 (36) | 4:1 |
| 4 | 1,4-dioxane | 4 | 90 | - | 35 (26) | 3:1 |
| 5 | DCM | 4 | 90 | - | 37 (28) | 3:1 |
| 6 | toluene | 12 | 70 | - | <10 | - |
| 7 | toluene | 0.5 | 110 | - | 55 (37) | 2:1 |
| 8 | toluene | 0.5 | 110 | CH3COOH | 67 (51) | 3:1 |
| 9 | toluene | 0.5 | 110 | HCOOH | 34 (27) | 4:1 |
| 10 | toluene | 0.5 | 110 | ClCH2COOH | 47 (38) | 4:1 |
| 11 | toluene | 0.5 | 110 | CF3COOH | n.r. | - |
| 12 | toluene | 0.5 | 110 | DBU | 52 (42) | 4:1 |
| 13 | toluene | 0.5 | 110 | DABCO | 65 (49) | 3:1 |
| 14 | toluene | 0.5 | 110 | K2CO3 | 57 (43) | 3:1 |
| 15 | toluene | 0.5 | 110 | TEA | 78 (65) | 5:1 |
| 16 | toluene | 4 | 90 | TEA | 72 (58) | 4:1 |
| 17 f | toluene | 0.5 | 110 | TEA | 66 (53) | 4:1 |

| Entry | R1 | R2 | R3 | R4 | Product | Yield (%) b | dr c |
|---|---|---|---|---|---|---|---|
| 1 | H | COOEt | H | Ph | 3a | 78 (65) | 5:1 |
| 2 | 4-Br | COOEt | H | Ph | 3b | 82 (59) | 2.5:1 |
| 3 | 5-F | COOEt | H | Ph | 3c | 83 (67) | 4:1 |
| 4 | 5-CH3 | COOEt | H | Ph | 3d | 81 (69) | 6:1 |
| 5 | H | COOiPr | H | Ph | 3e | 81 (65) | 4:1 |
| 6 | H | COOPh | H | Ph | 3f | 87 (70) | 4:1 |
| 7 | H | (CN)2 | H | Ph | 3g | 86 (69) | 4:1 |
| 8 | H | Ph | H | Ph | 3h | 63 (61) | >20:1 |
| 9 | H | 4-Br-C6H4 | H | Ph | 3i | 68 (58) | 6:1 |
| 10 | H | 2-F-C6H4 | H | Ph | 3j | 66 (63) | >20:1 |
| 11 | H | 4-NO2-C6H4 | H | Ph | 3k | 62 (52) | 5:1 |
| 12 | H | 4-Me-C6H4 | H | Ph | 3l | 58 (56) | >20:1 |
| 13 | H | 3,4-(MeO)2-C6H4 | H | Ph | 3m | 57 (46) | 4:1 |
| 14 | H | 2-naphthyl | H | Ph | 3n | 55 (53) | >20:1 |
| 15 | H | 2-furyl | H | Ph | 3o | 46 (35) | 3:1 |
| 16 | H | 2-thienyl | H | Ph | 3p | 52 (39) | 3:1 |
| 17 | H | COOEt | Bn | Ph | 3q | 88 (77) | 7:1 |
| 18 | H | COOEt | Boc | Ph | 3r | 82 (71) | 6:1 |
| 19 | H | COOEt | H | 3-Me-C6H4 | 3s | 70 (58) | 5:1 |
| 20 | H | COOEt | H | 4-CF3-C6H4 | 3t | 67 (56) | 5:1 |
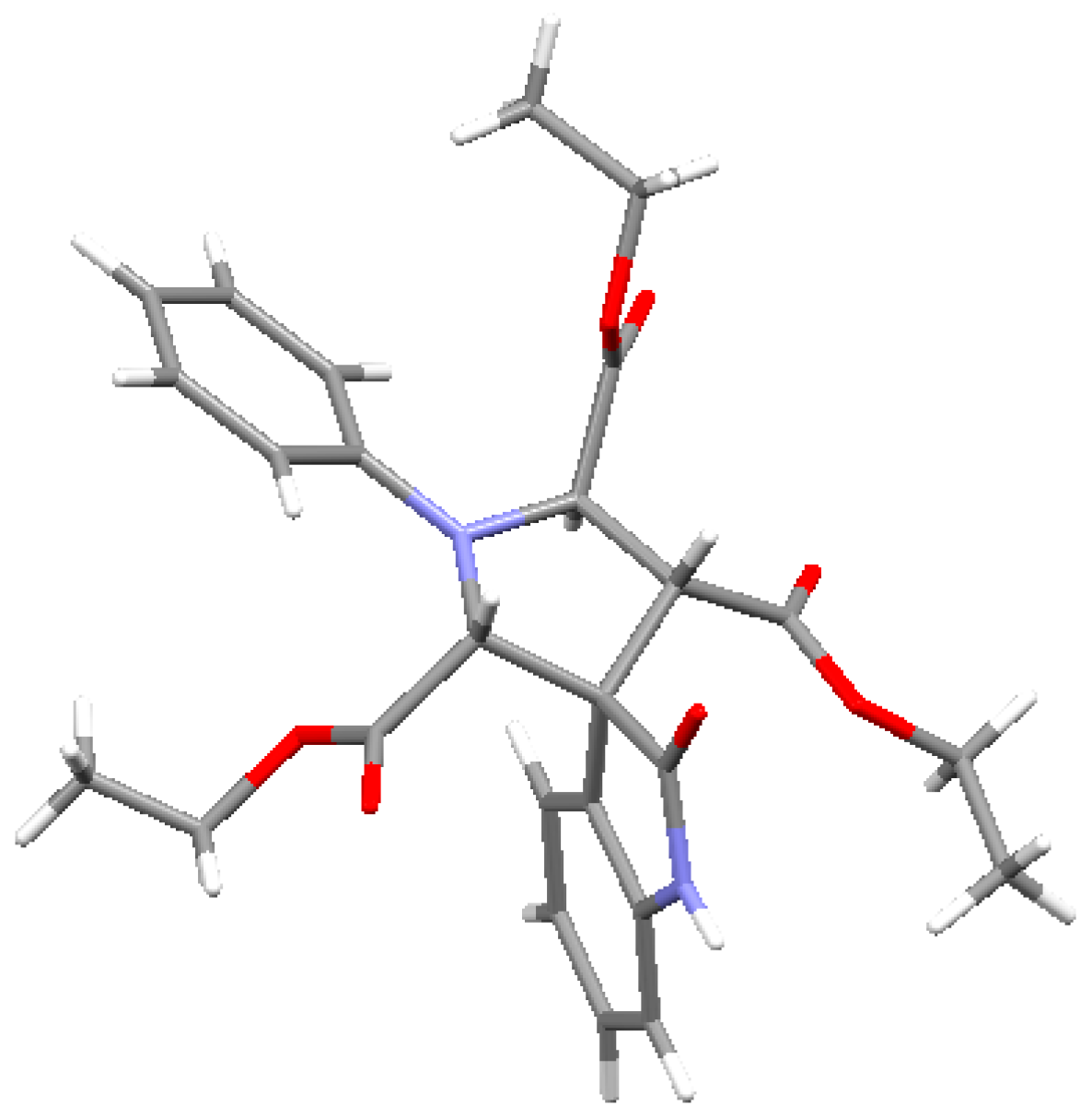
{kind=link}
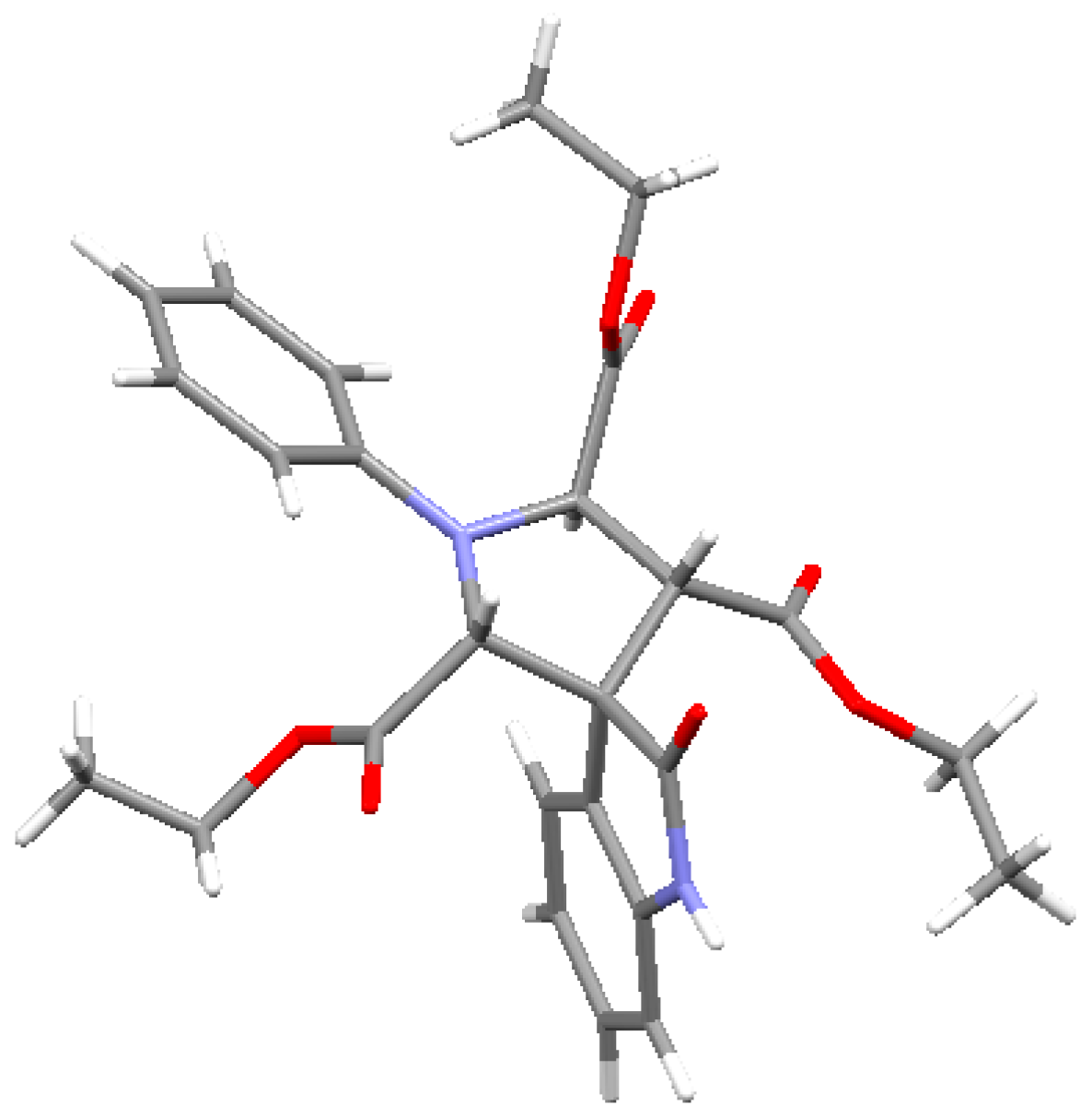{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, W.; Zhao, Q.; Zheng, C.; Zhao, Q.; Guo, L.; Huang, W. Efficient Synthesis of Fully Substituted Pyrrolidine-Fused 3-Spirooxindoles via 1,3-Dipolar Cycloaddition of Aziridine and 3-Ylideneoxindole. Molecules 2016, 21, 1113. https://doi.org/10.3390/molecules21091113
Ren W, Zhao Q, Zheng C, Zhao Q, Guo L, Huang W. Efficient Synthesis of Fully Substituted Pyrrolidine-Fused 3-Spirooxindoles via 1,3-Dipolar Cycloaddition of Aziridine and 3-Ylideneoxindole. Molecules. 2016; 21(9):1113. https://doi.org/10.3390/molecules21091113
Chicago/Turabian StyleRen, Wen, Qian Zhao, Chuan Zheng, Qiong Zhao, Li Guo, and Wei Huang. 2016. "Efficient Synthesis of Fully Substituted Pyrrolidine-Fused 3-Spirooxindoles via 1,3-Dipolar Cycloaddition of Aziridine and 3-Ylideneoxindole" Molecules 21, no. 9: 1113. https://doi.org/10.3390/molecules21091113
APA StyleRen, W., Zhao, Q., Zheng, C., Zhao, Q., Guo, L., & Huang, W. (2016). Efficient Synthesis of Fully Substituted Pyrrolidine-Fused 3-Spirooxindoles via 1,3-Dipolar Cycloaddition of Aziridine and 3-Ylideneoxindole. Molecules, 21(9), 1113. https://doi.org/10.3390/molecules21091113