
Order, Disorder, and Everything in Between



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Dominant Paradigms in Protein Science
2. Defining Intrinsically Disordered Proteins
3. The Subtler Side of Disorder
4. The Thin Line between Order and Disorder
5. The Mechanisms of Disorder
5.1. Entropy
5.2. Accessibility
5.3. Plasticity
6. Disorder-Related Biological Functions
6.1. Signaling
6.2. Regulation
7. Disorder and Protein Evolution
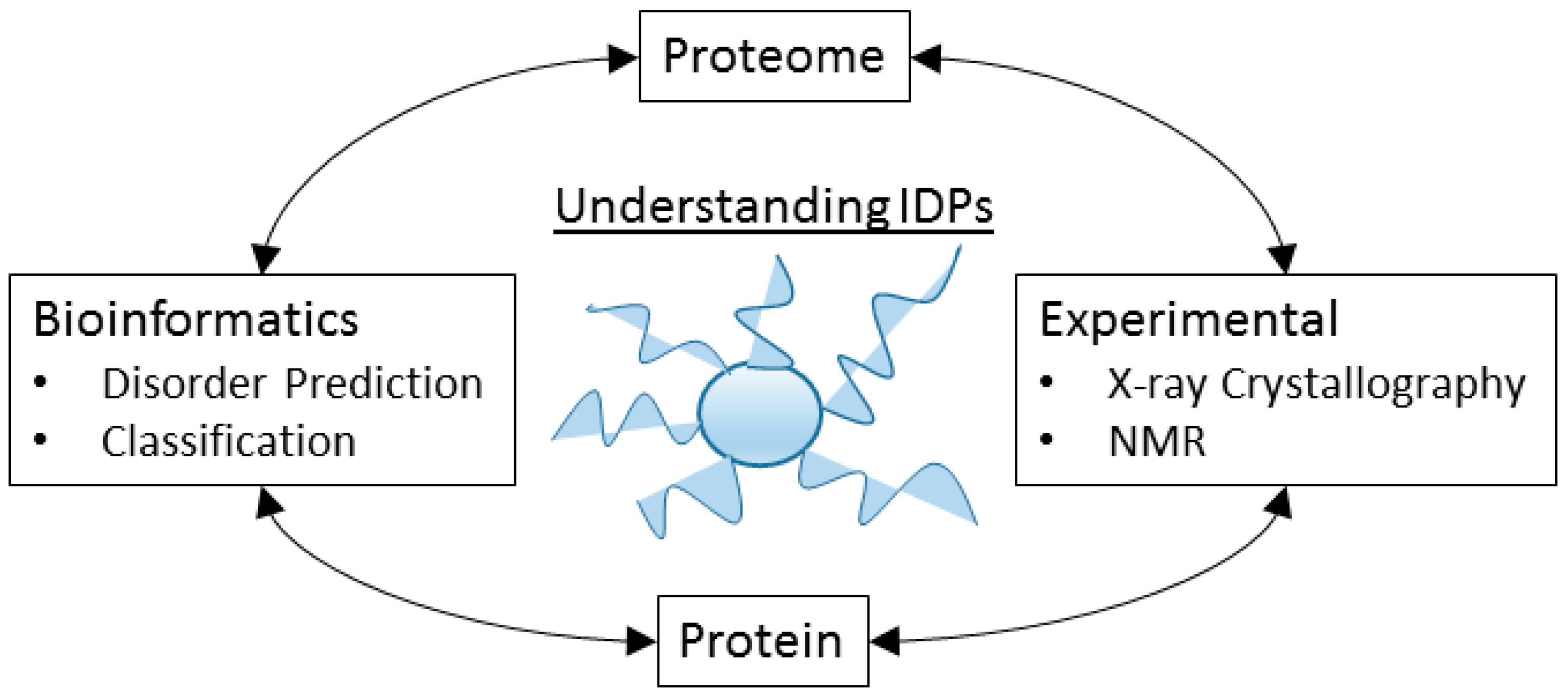
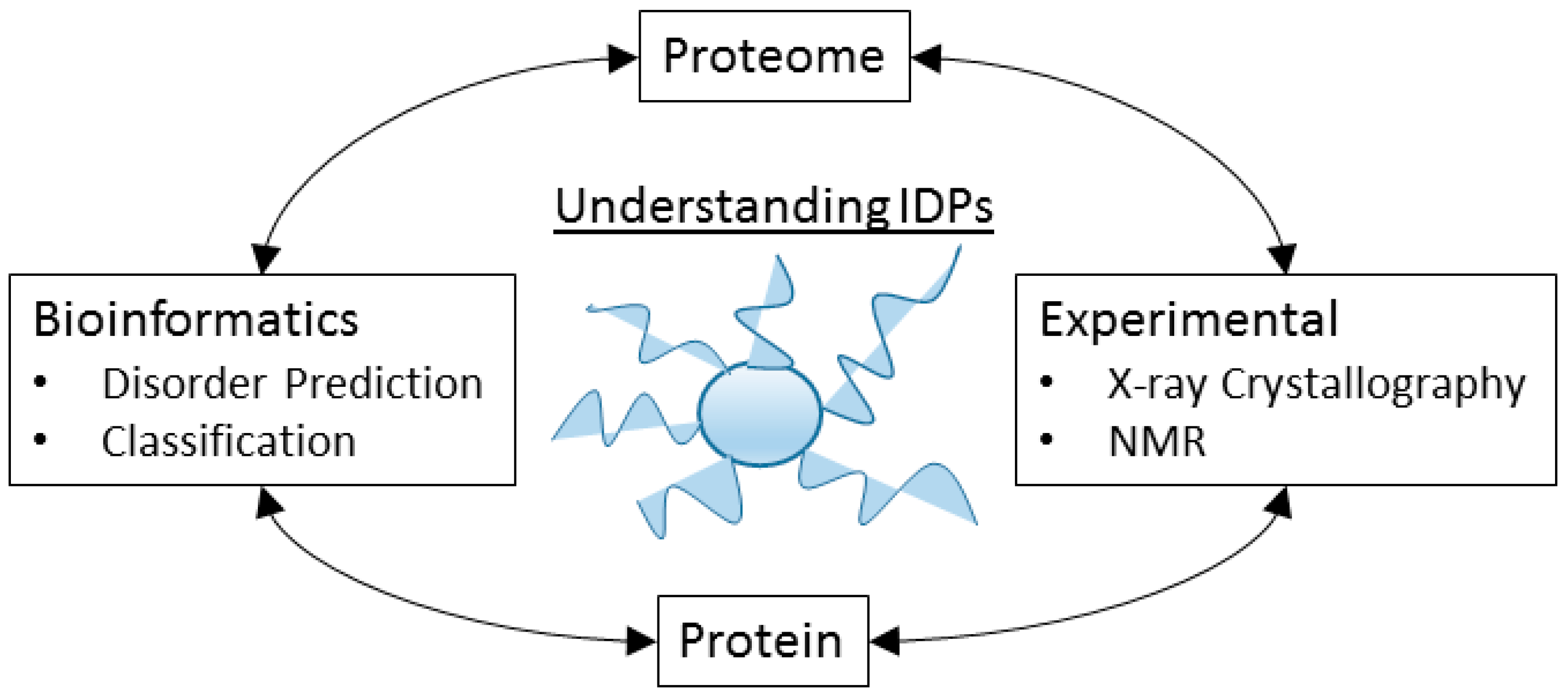
8. The Tools of the Un-Structural Biologist
8.1. Some Experimental Techniques
8.1.1. X-ray Crystallography
8.1.2. Nuclear Magnetic Resonance
8.1.3. Combining Experimental Techniques
8.2. Bioinformatics Analysis
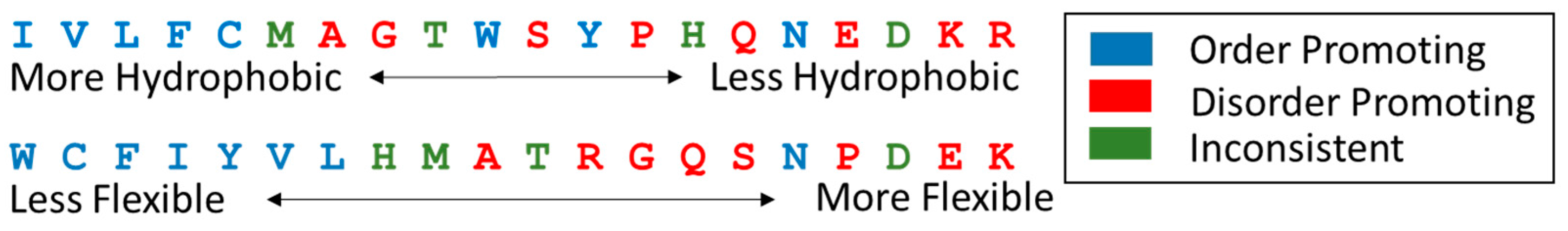
8.2.1. Sequence Characteristics
8.2.2. Disorder Prediction
8.2.3. Classification of Functions
8.2.4. Proteome Level Studies
9. Protein Intrinsic Disorder and Disease
10. Protein Intrinsic Disorder and Drug Design and Discovery
The Story of PTP1B
11. The Field of Protein Intrinsic Disorder
12. Conclusions: Intrinsic Disorder Where You Least Expect It
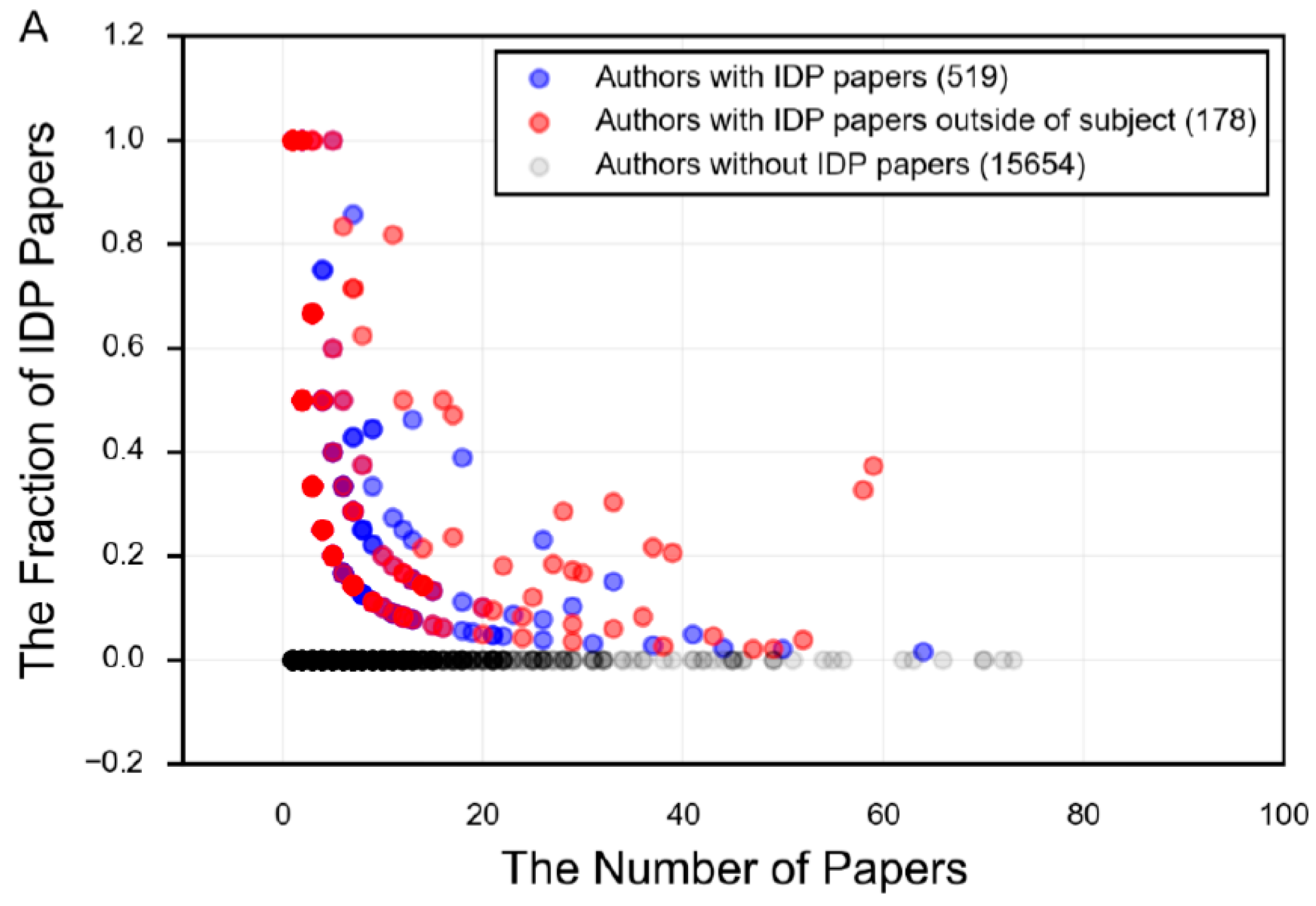
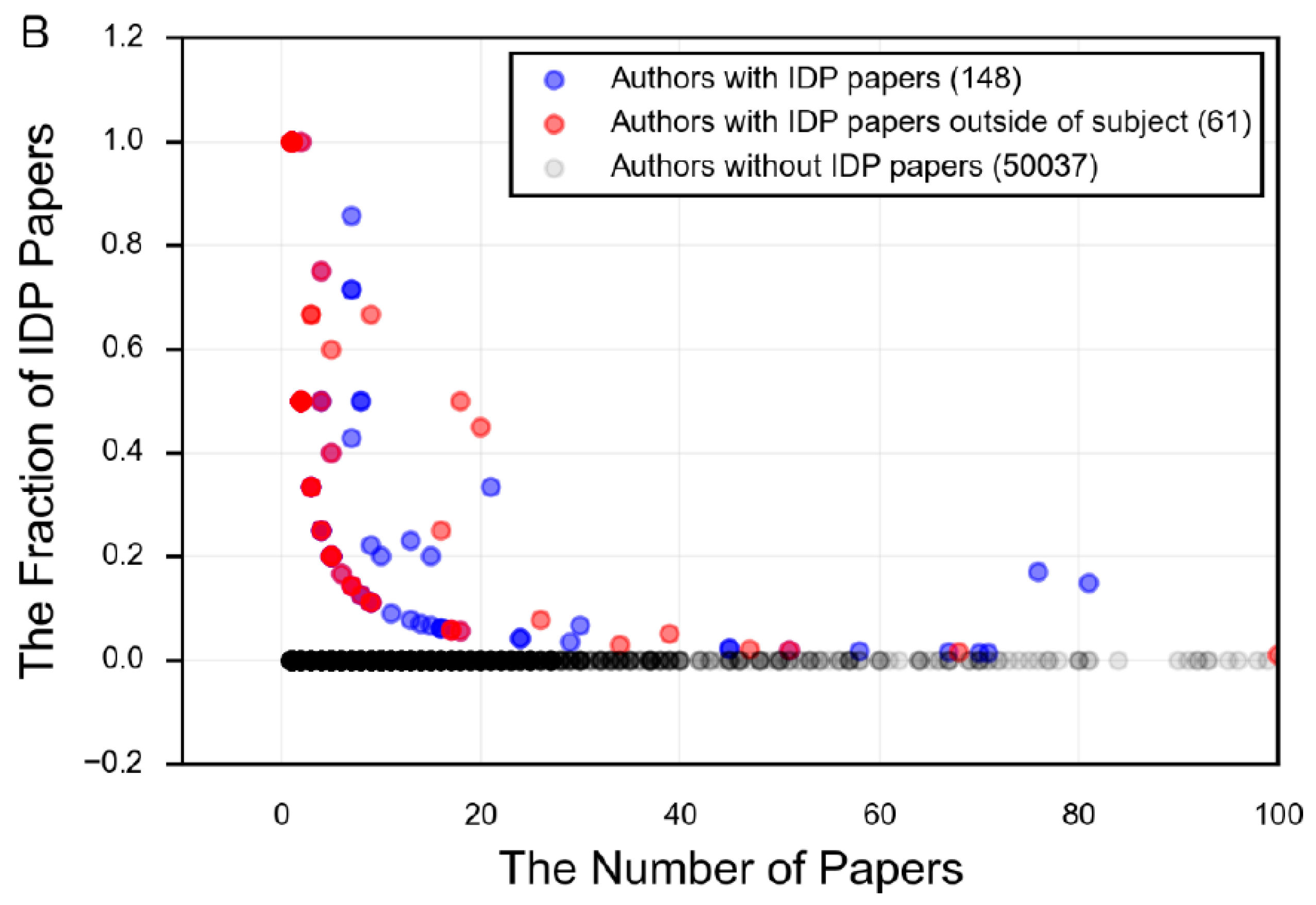
- There is a focus in the IDP literature on mostly or fully disordered proteins, and an incomplete understanding of the diverse mechanisms and functions employed by proteins that have regions of both order and disorder.
- The development and validation of disorder prediction depends on experimental datasets of missing regions from the PDB, however, it is not always clear what the cause and nature of the missing region is.
- There is an increasing number of enzymes with experimentally measured IDPRs in the literature, but no proteome level studies up to this point.
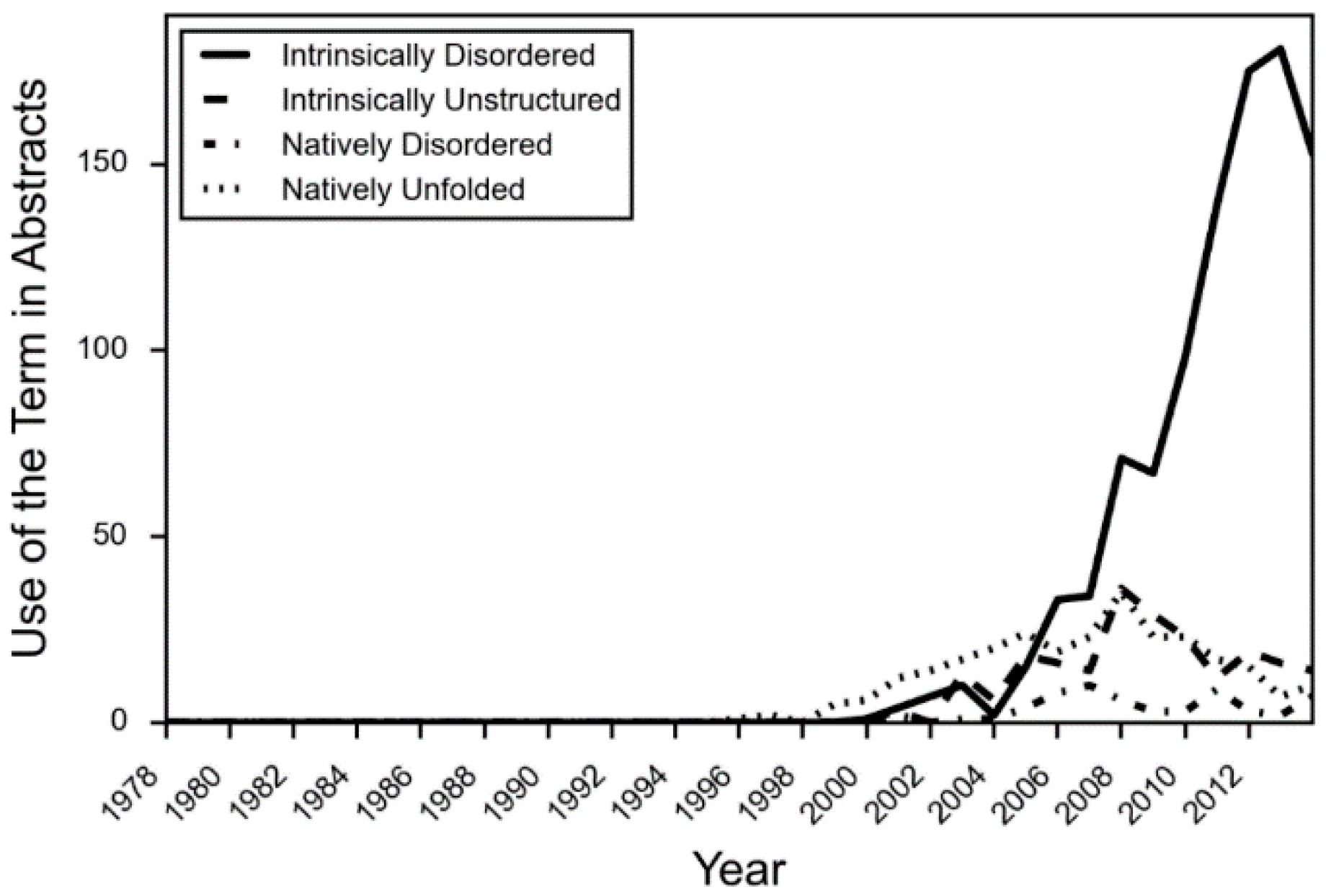
- There is limited acceptance of the language of protein intrinsic disorder outside of those who specialize in studying IDPs.
- Drug design and discovery frequently focuses on the truncated form of the protein, potentially resulting in missed opportunities.
Conflicts of Interest
References
- Tanford, C.; Reynolds, J. Nature’s Robots, a History of Proteins; Oxford University Press: Oxford, UK; New York, NY, USA, 2001. [Google Scholar]
- Jorpes, J.E. Jacob Berzelius, his Life and Work (Trans. B. Steele); Almqvist & Wiksell: Stockholm, Sweden, 1970. [Google Scholar]
- Kunz, H. Emil fischer-unequalled classicist, master of organic chemistry research, and inspired trailblazer of biological chemistry. Angew. Chem. Int. Ed. Engl. 2002, 41, 4439–4451. [Google Scholar] [CrossRef]
- Anfinsen, C.B.; Redfield, R.R.; Choate, W.L.; Page, J.; Carroll, W.R. Studies on the gross structure, cross-linkages, and terminal sequences in ribonuclease. J. Biol. Chem. 1954, 207, 201–210. [Google Scholar] [PubMed]
- Blake, C.C.; Koenig, D.F.; Mair, G.A.; North, A.C.; Phillips, D.C.; Sarma, V.R. Structure of hen egg-white lysozyme. A three-dimensional fourier synthesis at 2 angstrom resolution. Nature 1965, 206, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, H.W.; Hardman, K.D.; Allewell, N.M.; Inagami, T.; Tsernoglou, D.; Johnson, L.N.; Richards, F.M. The structure of ribonuclease-S at 6 a resolution. J. Biol. Chem. 1967, 242, 3749–3753. [Google Scholar] [PubMed]
- Jirgensons, B. Classification of proteins according to conformation. Die Makromol. Chem. 1966, 91, 74–86. [Google Scholar] [CrossRef]
- Herriott, R.M. Isolation, crystallization, and properties of swine pepsinogen. J. Gen. Physiol. 1938, 21, 501–540. [Google Scholar] [CrossRef] [PubMed]
- Jirgensons, B. Optical rotation and viscosity of native and denatured proteins. X. Further studies on optical rotatory dispersion. Arch. Biochem. Biophys. 1958, 74, 57–69. [Google Scholar] [CrossRef]
- Arnone, A.; Bier, C.J.; Cotton, F.A.; Day, V.W.; Hazen, E.E.; Richardson, D.C.; Yonath, A.; Richardson, J.S. A high resolution structure of an inhibitor complex of the extracellular nuclease of Staphylococcus aureus. I. Experimental procedures and chain tracing. J. Biol. Chem. 1971, 246, 2302–2316. [Google Scholar] [PubMed]
- Schweers, O.; Schönbrunn-Hanebeck, E.; Marx, A.; Mandelkow, E. Structural studies of tau protein and alzheimer paired helical filaments show no evidence for β-structure. J. Biol. Chem. 1994, 269, 24290–24297. [Google Scholar] [PubMed]
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T. Nacp, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.W.; Dunker, A.K.; Brown, C.J.; Garner, E.C.; Griswold, M.D. Clusterin, a binding protein with a molten globule-like region. Biochemistry 2001, 40, 11828–11840. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, H.; Rost, B. Loopy proteins appear conserved in evolution. J. Mol. Biol. 2002, 322, 53–64. [Google Scholar] [CrossRef]
- Pullen, R.A.; Jenkins, J.A.; Tickle, I.J.; Wood, S.P.; Blundell, T.L. The relation of polypeptide hormone structure and flexibility to receptor binding: The relevance of X-ray studies on insulins, glucagon and human placental lactogen. Mol. Cell. Biochem. 1975, 8, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Cary, P.D.; Moss, T.; Bradbury, E.M. High-resolution proton-magnetic-resonance studies of chromatin core particles. Eur. J. Biochem. 1978, 89, 475–482. [Google Scholar] [CrossRef] [PubMed]
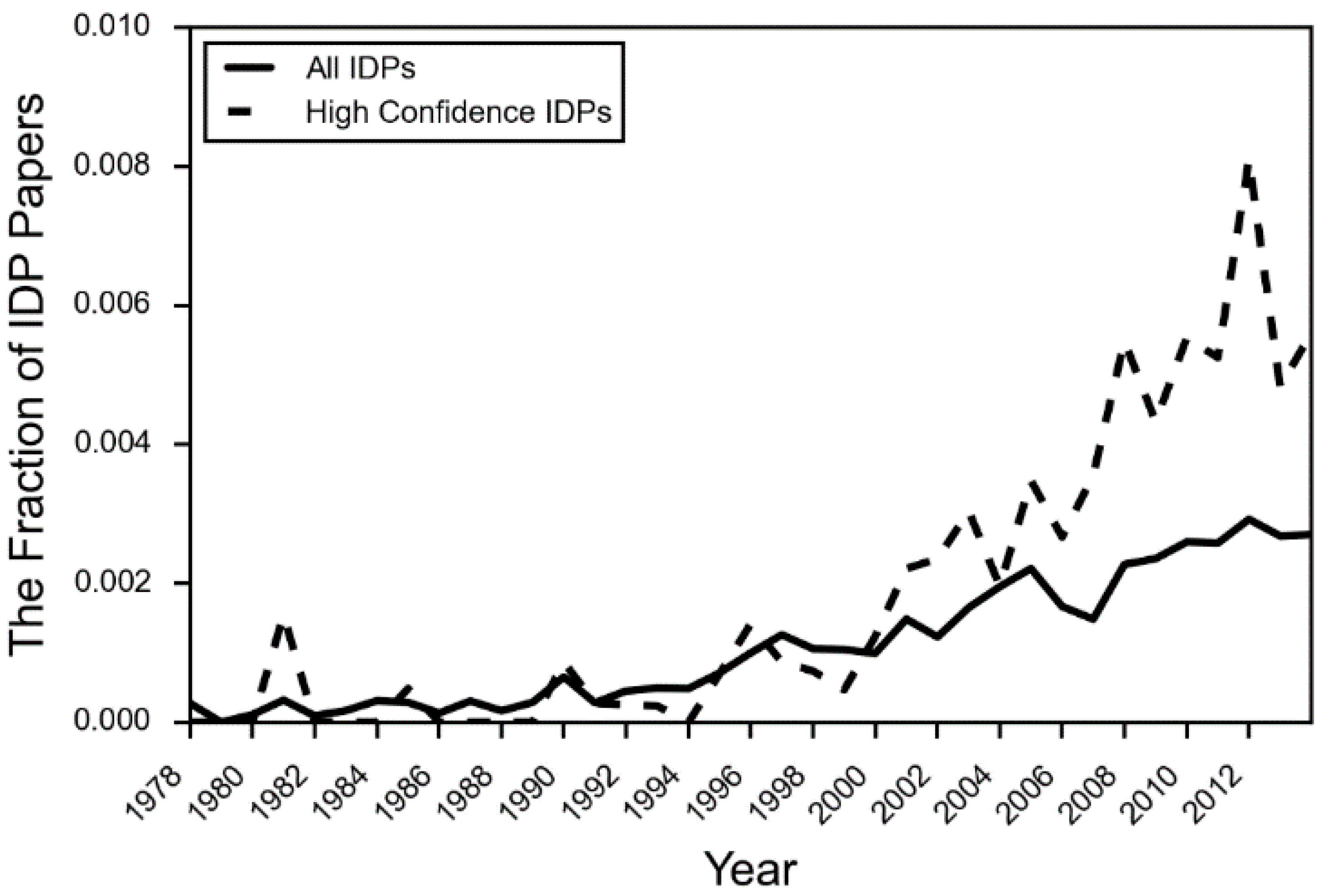
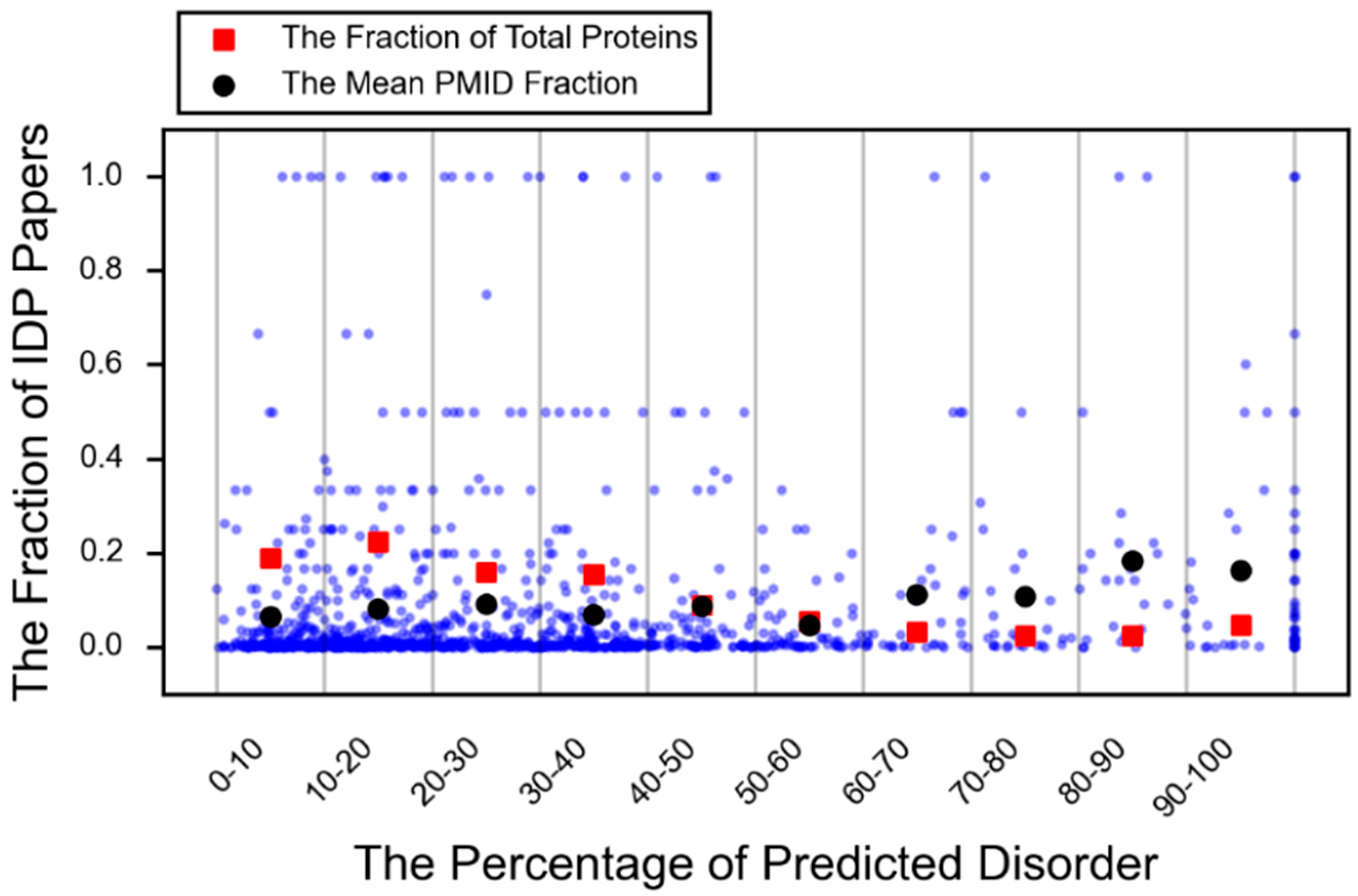
- DeForte, S.; Uversky, V.N. Intrinsically disordered proteins in pubmed: What can the tip of the iceberg tell us about what lies below? RSC Adv. 2016, 6, 11513–11521. [Google Scholar] [CrossRef]
- Tompa, P. Structure and Function of Intrinsically Disordered Proteins; CRC Press: Boca Raton, FL, USA, 2010; p. 331. [Google Scholar]
- Uversky, V.N. Introduction to intrinsically disordered proteins (IDPs). Chem. Rev. 2014, 114, 6557–6560. [Google Scholar] [CrossRef] [PubMed]
- Thomas, W.H.; Weser, U.; Hempel, K. Conformational changes induced by ionic strength and pH in two bovine myelin basic proteins. Hoppe Seyler’s Z Physiol. Chem. 1977, 358, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.A.; Avila, J.; Andreu, J.M. Physicochemical characterization of the heat-stable microtubule-associated protein map2. Eur J. Biochem. 1986, 154, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Oldfield, C.J. Back to the future: Nuclear magnetic resonance and bioinformatics studies on intrinsically disordered proteins. Adv. Exp. Med. Biol. 2015, 870, 1–34. [Google Scholar] [PubMed]
- Jakob, U.; Kriwacki, R.; Uversky, V.N. Conditionally and transiently disordered proteins: Awakening cryptic disorder to regulate protein function. Chem. Rev. 2014, 114, 6779–6805. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Stebbins, C.E. Dynamics of the WPD loop of the Yersinia protein tyrosine phosphatase. Biophys. J. 2006, 91, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, morfs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.F.; Papanayotou, I.; Davis, N.G. The yeast kinase Yck2 has a tripartite palmitoylation signal. Mol. Biol. Cell. 2011, 22, 2702–2715. [Google Scholar] [CrossRef] [PubMed]
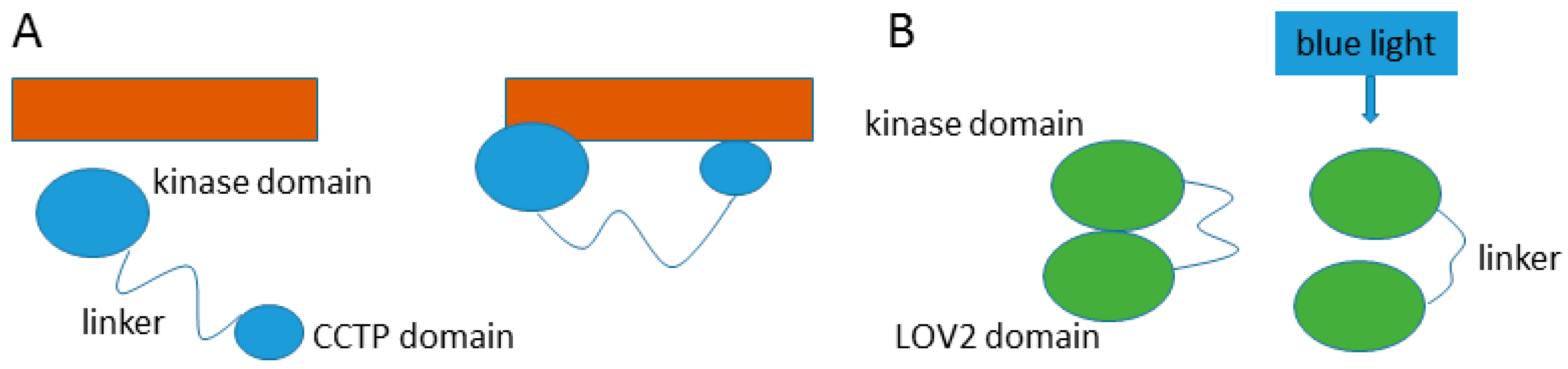
- Takayama, Y.; Nakasako, M.; Okajima, K.; Iwata, A.; Kashojiya, S.; Matsui, Y.; Tokutomi, S. Light-induced movement of the LOV2 domain in an Asp720asn mutant LOV2-kinase fragment of arabidopsis phototropin 2. Biochemistry 2011, 50, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Bentrop, D.; Beyermann, M.; Wissmann, R.; Fakler, B. NMR structure of the “Ball-and-chain” Domain of KCNMB2, the β2-subunit of large conductance Ca2+- and voltage-activated potassium channels. J. Biol. Chem. 2001, 276, 42116–42121. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Belmont, B.J.; Sante, J.M.; Rexach, M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 2007, 129, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Buffa, P.; Manzella, L.; Consoli, M.L.; Messina, A.; Vigneri, P. Modelling of the ABL and ARG proteins predicts two functionally critical regions that are natively unfolded. Proteins 2007, 67, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Manalan, A.S.; Klee, C.B. Activation of calcineurin by limited proteolysis. Proc. Natl. Acad. Sci. USA 1983, 80, 4291–4295. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, N.; McGinnis, K.M.; Park, Y.H.; Gnegy, M.E.; Wang, K.K. Caspase-mediated proteolytic activation of calcineurin in thapsigargin-mediated apoptosis in SH-SY5Y neuroblastoma cells. Arch. Biochem. Biophys. 2000, 379, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Alphey, M.S.; Yu, W.; Byres, E.; Li, D.; Hunter, W.N. Structure and reactivity of human mitochondrial 2,4-dienoyl-CoA reductase: Enzyme-ligand interactions in a distinctive short-chain reductase active site. J. Biol. Chem. 2005, 280, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- VanOudenhove, J.; Anderson, E.; Krueger, S.; Cole, J.L. Analysis of PKR structure by small-angle scattering. J. Mol. Biol. 2009, 387, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, P.; Pancsa, R.; Guharoy, M.; Tompa, P. Functional diversity and structural disorder in the human ubiquitination pathway. PLoS ONE 2013, 8, e65443. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell. Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Pathak, R.R.; Clayman, E.; Xue, B.; Uversky, V.N.; Davé, V. Presence and utility of intrinsically disordered regions in kinases. Mol. Biosyst. 2014, 10, 2876–2888. [Google Scholar] [CrossRef] [PubMed]
- Banci, L.; Bertini, I.; Cefaro, C.; Ciofi-Baffoni, S.; Gajda, K.; Felli, I.C.; Gallo, A.; Pavelkova, A.; Kallergi, E.; Andreadaki, M.; et al. An intrinsically disordered domain has a dual function coupled to compartment-dependent redox control. J. Mol. Biol. 2013, 425, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Bornet, O.; Nouailler, M.; Feracci, M.; Sebban-Kreuzer, C.; Byrne, D.; Halimi, H.; Morelli, X.; Badache, A.; Guerlesquin, F. Identification of a Src kinase SH3 binding site in the C-terminal domain of the human ErbB2 receptor tyrosine kinase. FEBS Lett. 2014, 588, 2031–2036. [Google Scholar] [CrossRef] [PubMed]
- Niklas, K.J.; Bondos, S.E.; Dunker, A.K.; Newman, S.A. Rethinking gene regulatory networks in light of alternative splicing, intrinsically disordered protein domains, and post-translational modifications. Front. Cell. Dev. Biol. 2015, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Erales, J.; Coffino, P. Ubiquitin-independent proteasomal degradation. Biochim. Biophys. Acta 2014, 1843, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Larion, M.; Salinas, R.K.; Bruschweiler-Li, L.; Miller, B.G.; Brüschweiler, R. Order-disorder transitions govern kinetic cooperativity and allostery of monomeric human glucokinase. PLoS Biol. 2012, 10, e1001452. [Google Scholar] [CrossRef] [PubMed]
- Fredrickson, E.K.; Clowes Candadai, S.V.; Tam, C.H.; Gardner, R.G. Means of self-preservation: How an intrinsically disordered ubiquitin-protein ligase averts self-destruction. Mol. Biol. Cell. 2013, 24, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lin, S. Distinct gene number-genome size relationships for eukaryotes and non-eukaryotes: Gene content estimation for dinoflagellate genomes. PLoS ONE 2009, 4, e6978. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Bondos, S.E.; Huang, F.; Oldfield, C.J. Intrinsically disordered proteins and multicellular organisms. Semin Cell. Dev. Biol. 2015, 37, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Pancsa, R.; Tompa, P. Structural disorder in eukaryotes. PLoS ONE 2012, 7, e34687. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.F.; Cao, Z.; Yang, Y.; Wang, C.L.; Su, Z.D.; Zhao, Y.W.; Wang, J.H.; Zhou, Y. Natural protein sequences are more intrinsically disordered than random sequences. Cell. Mol. Life Sci. 2016, 73, 2949–2957. [Google Scholar] [CrossRef] [PubMed]
- Teraguchi, S.; Patil, A.; Standley, D.M. Intrinsically disordered domains deviate significantly from random sequences in mammalian proteins. BMC Bioinform. 2010, 11 (Suppl. 7), S7. [Google Scholar]
- Brown, C.J.; Takayama, S.; Campen, A.M.; Vise, P.; Marshall, T.W.; Oldfield, C.J.; Williams, C.J.; Dunker, A.K. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 2002, 55, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Potenza, E.; di Domenico, T.; Walsh, I.; Tosatto, S.C. Mobidb 2.0: An improved database of intrinsically disordered and mobile proteins. Nucleic Acids Res. 2015, 43, D315–D320. [Google Scholar] [CrossRef] [PubMed]
- Ota, M.; Koike, R.; Amemiya, T.; Tenno, T.; Romero, P.R.; Hiroaki, H.; Dunker, A.K.; Fukuchi, S. An assignment of intrinsically disordered regions of proteins based on nmr structures. J. Struct. Biol. 2013, 181, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. From protein sequence to dynamics and disorder with DynaMine. Nat. Commun. 2013, 4, 2741. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Intrinsically Disordered Protein Analysis, 1st ed.; Humana Press: New York, NY, USA, 2012; Volume 1. [Google Scholar]
- Uversky, V.N.; Dunker, A.K. Intrinsically Disordered Protein Analysis, 1st ed.; Humana Press: New York, NY, USA, 2012; Volume 2. [Google Scholar]
- Uversky, V.N.; Dunker, A.K. Multiparametric analysis of intrinsically disordered proteins: Looking at intrinsic disorder through compound eyes. Anal. Chem. 2012, 84, 2096–2104. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Biophysical methods to investigate intrinsically disordered proteins: Avoiding an “Elephant and blind men” Situation. Adv. Exp. Med. Biol. 2015, 870, 215–260. [Google Scholar] [PubMed]
- Khan, T.; Douglas, G.M.; Patel, P.; Nguyen Ba, A.N.; Moses, A.M. Polymorphism analysis reveals reduced negative selection and elevated rate of insertions and deletions in intrinsically disordered protein regions. Genome Biol. Evol. 2015, 7, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Smithers, B.; Oates, M.E.; Gough, J. Splice junctions are constrained by protein disorder. Nucleic Acids Res. 2015, 43, 4814–4822. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, P.; Guharoy, M.; Tompa, P. Bioinformatics approaches for predicting disordered protein motifs. Adv. Exp. Med. Biol. 2015, 870, 291–318. [Google Scholar] [PubMed]
- Varadi, M.; Vranken, W.; Guharoy, M.; Tompa, P. Computational approaches for inferring the functions of intrinsically disordered proteins. Front. Mol. Biosci. 2015, 2, 45. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Simon, I.; Dosztányi, Z. Prediction and analysis of intrinsically disordered proteins. Methods Mol. Biol. 2015, 1261, 35–59. [Google Scholar] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Williams, R.M.; Obradovi, Z.; Mathura, V.; Braun, W.; Garner, E.C.; Young, J.; Takayama, S.; Brown, C.J.; Dunker, A.K. The protein non-folding problem: Amino acid determinants of intrinsic order and disorder. Pac. Symp. Biocomput. 2001, 89–100. [Google Scholar]
- Vihinen, M.; Torkkila, E.; Riikonen, P. Accuracy of protein flexibility predictions. Proteins 1994, 19, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Kumari, B.; Kumar, R.; Kumar, M. Low complexity and disordered regions of proteins have different structural and amino acid preferences. Mol. Biosyst. 2015, 11, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J. Proteome Res. 2006, 5, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Moesa, H.A.; Wakabayashi, S.; Nakai, K.; Patil, A. Chemical composition is maintained in poorly conserved intrinsically disordered regions and suggests a means for their classification. Mol. Biosyst. 2012, 8, 3262–3273. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, Y.; Wang, X.; Liu, W.; Rong, L.; Bao, J. An overview of predictors for intrinsically disordered proteins over 2010–2014. Int. J. Mol. Sci. 2015, 16, 23446–23462. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell. Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyy, B.; Kryshtafovych, A.; Moult, J.; Tramontano, A.; Fidelis, K. Assessment of protein disorder region predictions in casp10. Proteins 2014, 82 (Suppl. 2), 127–137. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Kinoshita, K. Prdos: Prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 2007, 35, W460–W464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Faraggi, E.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Zhou, Y. Spine-d: Accurate prediction of short and long disordered regions by a single neural-network based method. J. Biomol. Struct. Dyn. 2012, 29, 799–813. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Cozzetto, D. Disopred3: Precise disordered region predictions with annotated protein-binding activity. Bioinform. 2015, 31, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Martin, A.J.; di Domenico, T.; Tosatto, S.C. Espritz: Accurate and fast prediction of protein disorder. Bioinform. 2012, 28, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmok, V.; Tompa, P.; Simon, I. Iupred: Web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinform. 2005, 21, 3433–3434. [Google Scholar] [CrossRef] [PubMed]
- Dosztányi, Z.; Csizmók, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Sickmeier, M.; Hamilton, J.A.; LeGall, T.; Vacic, V.; Cortese, M.S.; Tantos, A.; Szabo, B.; Tompa, P.; Chen, J.; Uversky, V.N.; et al. Disprot: The database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2015, 43, D364–D368. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Mizianty, M.J.; Kurgan, L. Genome-scale prediction of proteins with long intrinsically disordered regions. Proteins 2014, 82, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Mizianty, M.J.; Filipow, P.L.; Uversky, V.N.; Kurgan, L. Rapid: Fast and accurate sequence-based prediction of intrinsic disorder content on proteomic scale. Biochim. Biophys. Acta 2013, 1834, 1671–1680. [Google Scholar] [CrossRef] [PubMed]
- Cilia, E.; Pancsa, R.; Tompa, P.; Lenaerts, T.; Vranken, W.F. The dynamine webserver: Predicting protein dynamics from sequence. Nucleic Acids Res. 2014, 42, W264–W270. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. Pondr-fit: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.P.; Bujnicki, J.M. MetaDisorder: A meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinform. 2012, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Kurgan, L. Accurate prediction of disorder in protein chains with a comprehensive and empirically designed consensus. J. Biomol. Struct. Dyn. 2014, 32, 448–464. [Google Scholar] [CrossRef] [PubMed]
- Vucetic, S.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Flavors of protein disorder. Proteins 2003, 52, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Cozzetto, D.; Jones, D.T. The contribution of intrinsic disorder prediction to the elucidation of protein function. Curr. Opin. Struct. Biol. 2013, 23, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Consortium, G.O. Gene ontology consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A. The enzyme database in 2000. Nucleic Acids Res. 2000, 28, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally abundant exceptions: Comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically disordered proteins in human diseases: Introducing the d2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef] [PubMed]
- Gsponer, J.; Futschik, M.E.; Teichmann, S.A.; Babu, M.M. Tight regulation of unstructured proteins: From transcript synthesis to protein degradation. Science 2008, 322, 1365–1368. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front. Aging Neurosci. 2015, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Davé, V.; Iakoucheva, L.M.; Malaney, P.; Metallo, S.J.; Pathak, R.R.; Joerger, A.C. Pathological unfoldomics of uncontrolled chaos: Intrinsically disordered proteins and human diseases. Chem. Rev. 2014, 114, 6844–6879. [Google Scholar] [CrossRef] [PubMed]
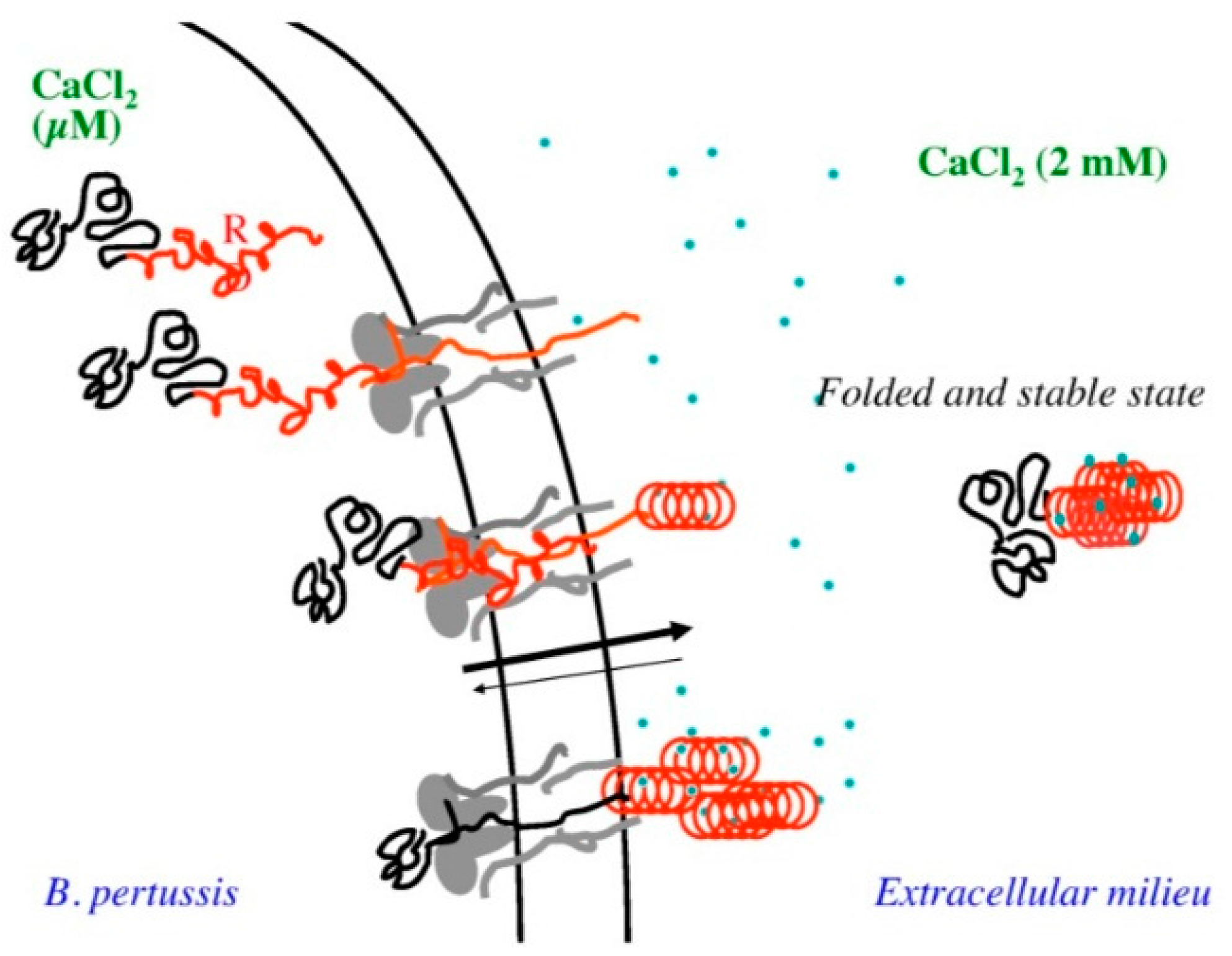
- Sotomayor-Pérez, A.C.; Ladant, D.; Chenal, A. Disorder-to-order transition in the CyaA toxin RTX domain: Implications for toxin secretion. Toxins (Basel) 2015, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Berson, A.; Soreq, H. It all starts at the ends: Multifaceted involvement of C- and N-terminally modified cholinesterases in Alzheimer’s disease. Rambam Maimonides Med. J. 2010, 1, e0014. [Google Scholar] [CrossRef] [PubMed]
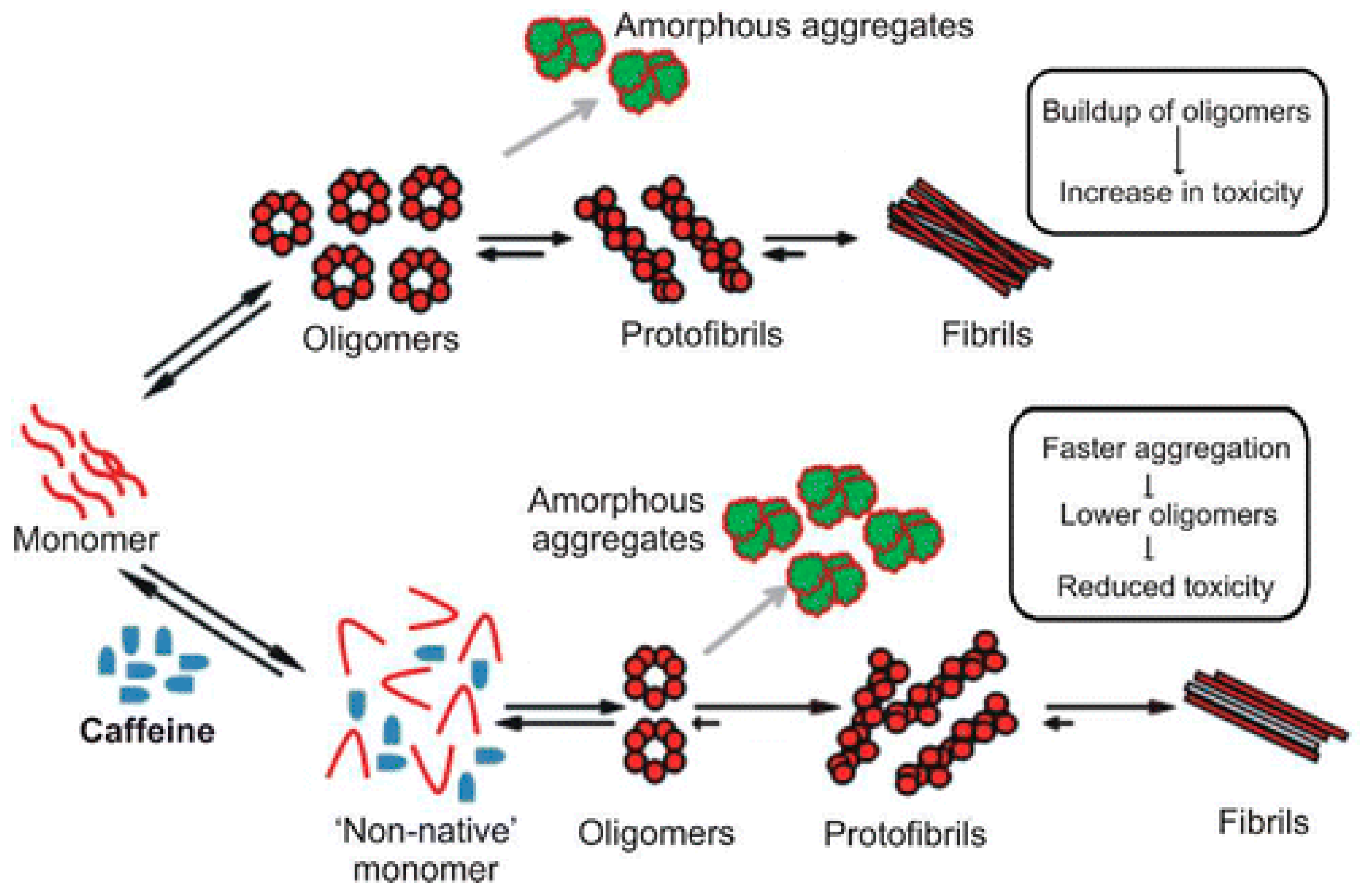
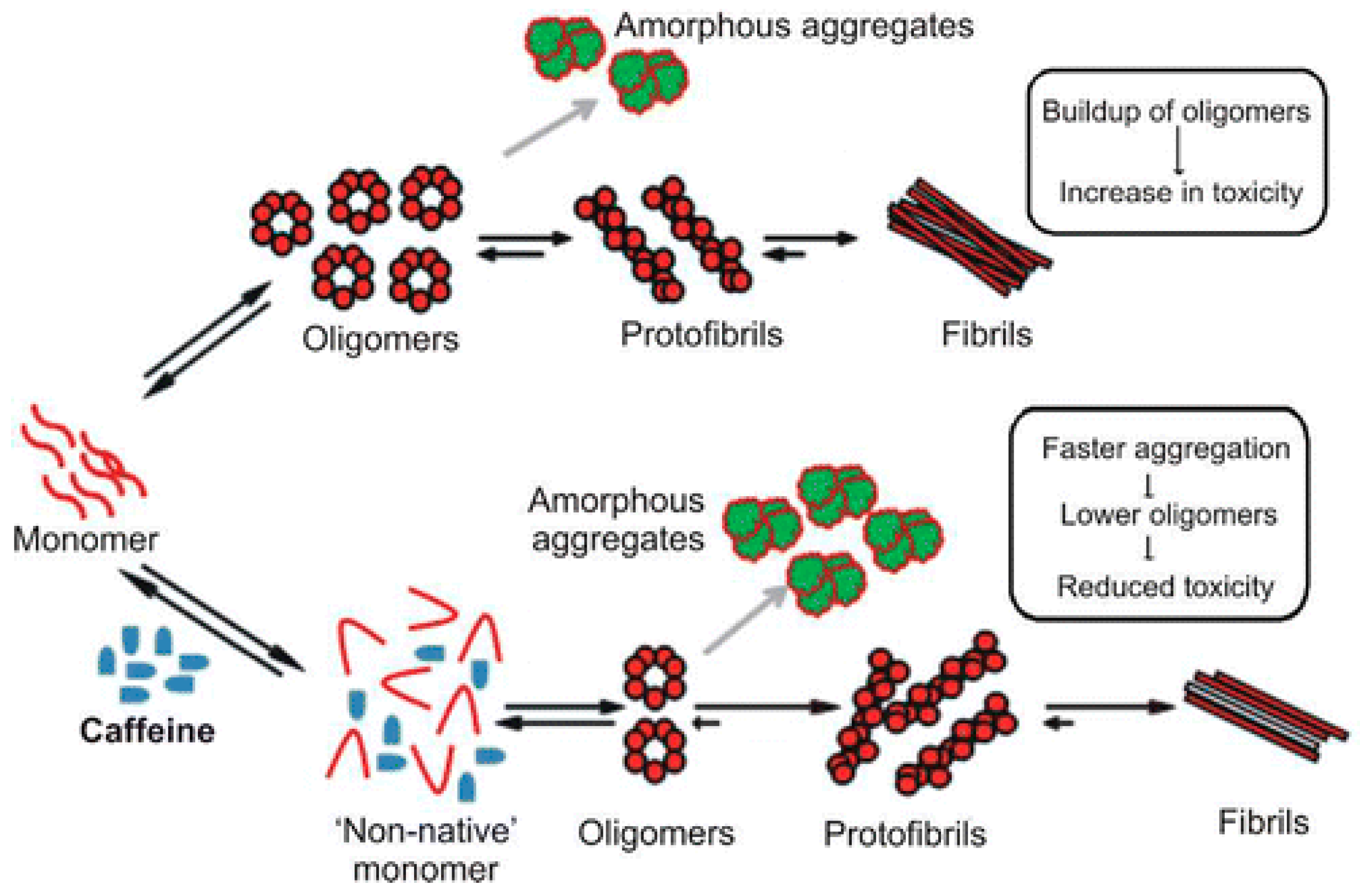
- Kardani, J.; Roy, I. Understanding caffeine’s role in attenuating the toxicity of α-synuclein aggregates: Implications for risk of Parkinson’s disease. ACS Chem. Neurosci. 2015, 6, 1613–1625. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.M.; Page, R.; Blackledge, M.; et al. Targeting the disordered c terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K.; Diltz, C.D.; Fischer, E.H. Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 1988, 263, 6722–6730. [Google Scholar] [PubMed]
- Chernoff, J.; Schievella, A.R.; Jost, C.A.; Erikson, R.L.; Neel, B.G. Cloning of a cdna for a major human protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. USA 1990, 87, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Tiganis, T.; Tonks, N.K.; Charbonneau, H. The noncatalytic C-terminal segment of the T cell protein tyrosine phosphatase regulates activity via an intramolecular mechanism. J. Biol. Chem. 1997, 272, 29322–29329. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. FEBS J. 2013, 280, 346–378. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.N.; Tonks, N.K. Protein tyrosine phosphatase-based therapeutics: Lessons from ptp1b. Top. Curr. Genet. 2004, 5, 201–230. [Google Scholar]
- Lantz, K.A.; Hart, S.G.; Planey, S.L.; Roitman, M.F.; Ruiz-White, I.A.; Wolfe, H.R.; McLane, M.P. Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity (Silver Spring) 2010, 18, 1516–1523. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeForte, S.; Uversky, V.N. Order, Disorder, and Everything in Between. Molecules 2016, 21, 1090. https://doi.org/10.3390/molecules21081090
DeForte S, Uversky VN. Order, Disorder, and Everything in Between. Molecules. 2016; 21(8):1090. https://doi.org/10.3390/molecules21081090
Chicago/Turabian StyleDeForte, Shelly, and Vladimir N. Uversky. 2016. "Order, Disorder, and Everything in Between" Molecules 21, no. 8: 1090. https://doi.org/10.3390/molecules21081090
APA StyleDeForte, S., & Uversky, V. N. (2016). Order, Disorder, and Everything in Between. Molecules, 21(8), 1090. https://doi.org/10.3390/molecules21081090