3. Materials and Methods
3.1. General Information
All commercially available materials were used as received.
1H-NMR spectra were recorded at 400 MHz and
13C-NMR spectra at 100 MHz. IR spectra were recorded with neat samples and signals are labelled as strong (s), medium (m) or weak (w). HRMS were recorded in ESI mode and only molecular masses are reported.
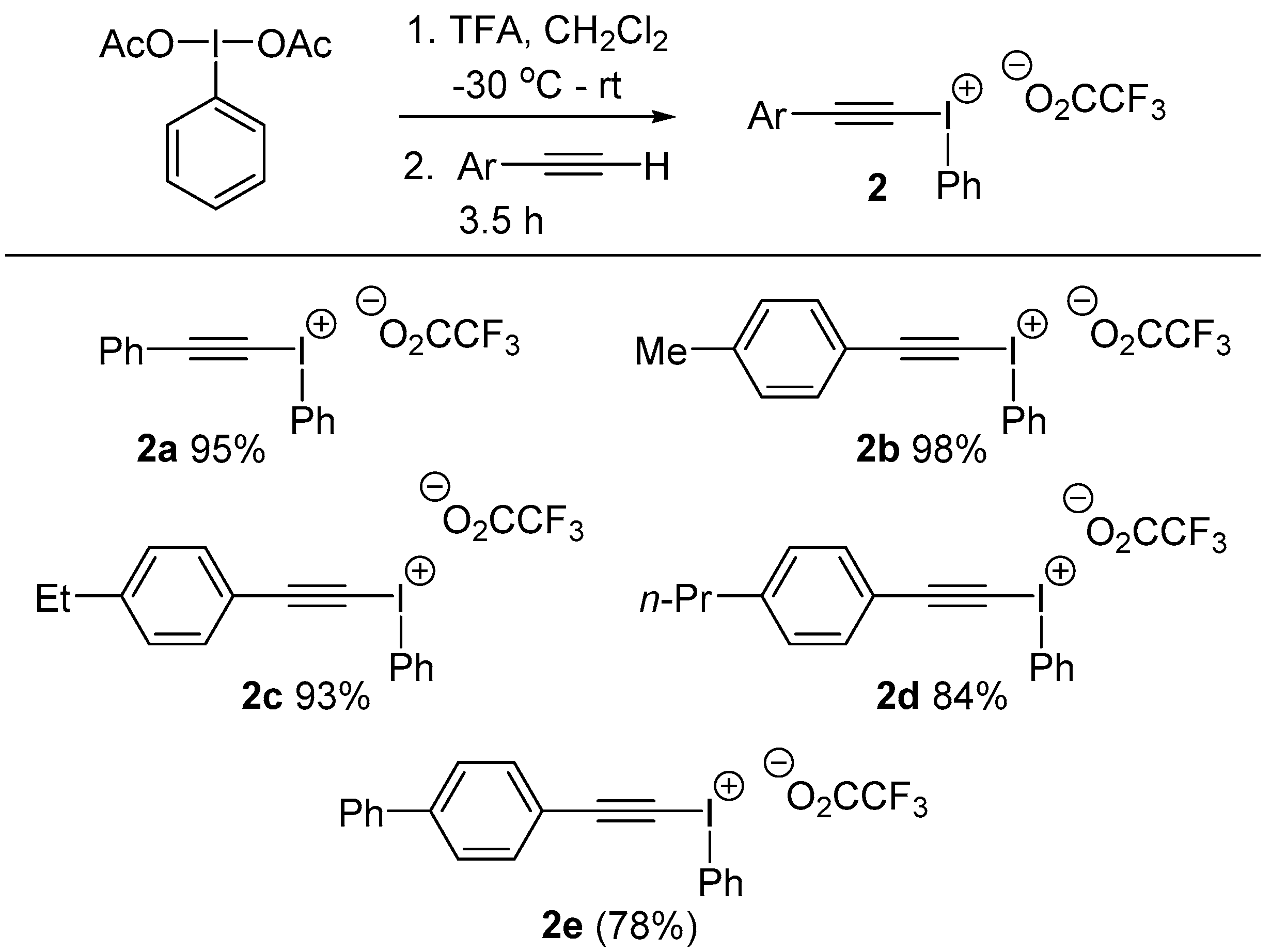
2a and
2b were prepared according to the method of Carroll and co-workers and the data matched the literature values [
23].
3aa was prepared following Ochiai’s protocol [
14].
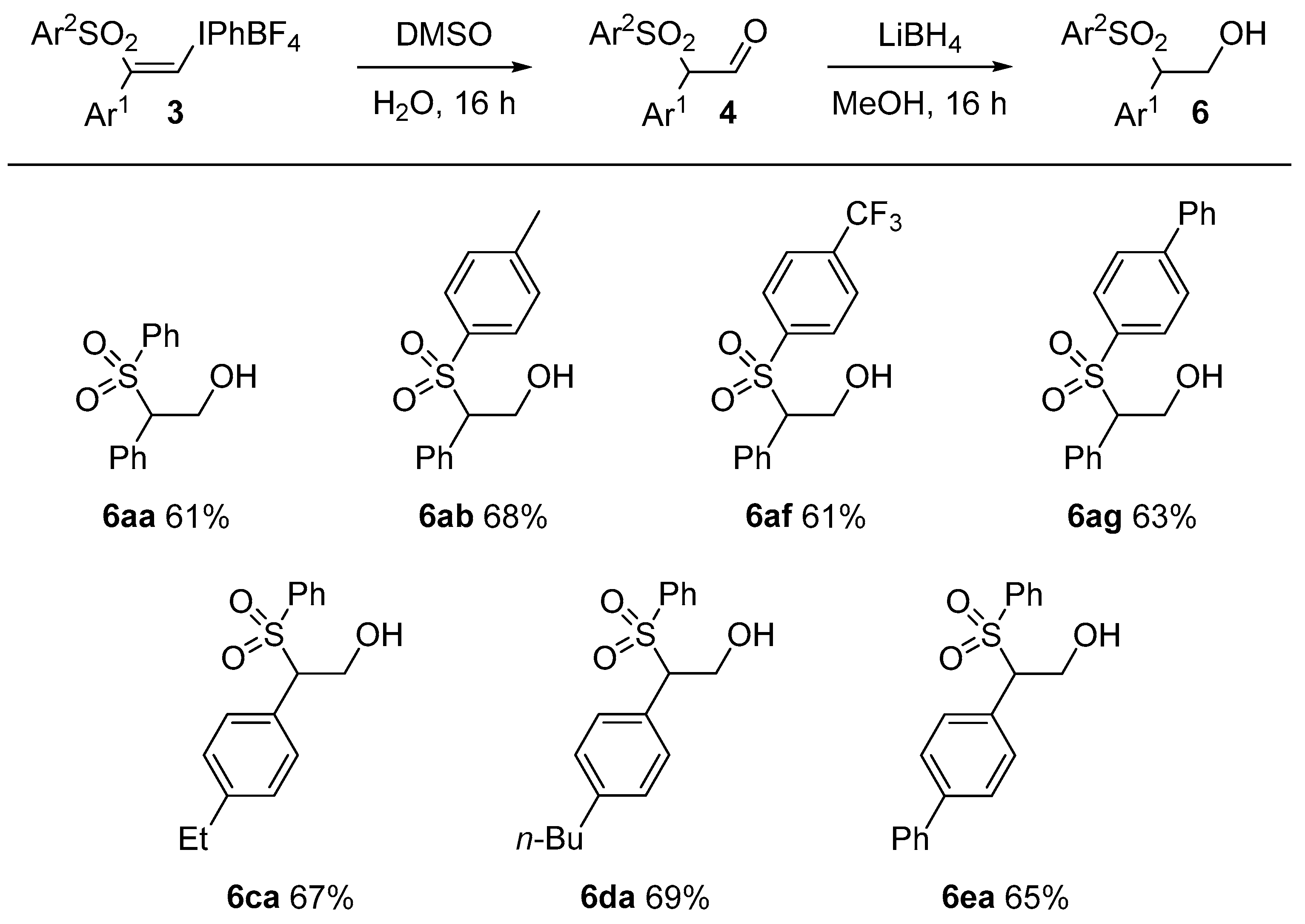
6aa and
6ab are known compounds [
24].
3.2. General Procedure for Alkynyl(phenyl)iodonium Trifluoroacetate 2 Formation: Preparation of ((4-Ethylphenyl)ethynyl)(phenyl)iodonium 2,2,2-trifluoroacetate 2c
Following Carroll’s procedure [
23], trifluoroacetic acid (1.4 mL, 19 mmol, 2 equiv.) was added dropwise to a stirred solution of diacetoxyiodobenzene (3 g, 9.3 mmol, 1 equiv.) in CH
2Cl
2 (60 mL) at −30 °C. The mixture was stirred for 30 min, then warmed to room temperature and stirred for a further hour. 1-Ethyl-4-ethynyl benzene (1.3 mL, 9.3 mmol, 1 equiv.) was added dropwise and the resulting mixture was stirred in darkness at room temperature for 3.5 h. The solution was concentrated in vacuo to around 30 mL then diethyl ether (20 mL) and petroleum ether (40 mL) were added which initiated crystallization of the product. After cooling in a freezer (−20 °C) for 48 h, the solid was filtered and dried in vacuo to provide
2c as a white crystalline solid (3.8 g, 93%). Melting point: 81–83 °C. IR: 1656 (s), 1417 (m), 1124 (s), 989 (m), 720 (s) cm
−1.
1H-NMR: δ 1.22 (3H, t,
J = 7.7 Hz), 2.67 (2H, q,
J = 7.6 Hz), 7.20 (2H, d,
J = 8.1 Hz), 7.39 (2H, d,
J = 8.1 Hz), 7.49 (2H, t,
J = 7.9 Hz), 7.60 (1H, t,
J = 7.5 Hz) 8.15 (2H, d,
J = 8.0 Hz).
13C-NMR: δ 15.5, 29.3, 44.8, 105.0, 115.8 (1C, q,
J = 292 Hz), 114.4, 117.3, 117.7, 121.1, 128.6 (2C), 132.2, 132.4 (2C), 133.3 (2C), 133.7 (2C), 148.1, 162.9 (1C, q,
J = 38 Hz). HRMS:
m/
z calcd. for [M − OCOCF
3]
+ C
18H
18I
+ 333.0135 found 333.0140.
((4-Butylphenyl)ethynyl)(phenyl)iodonium 2,2,2-trifluoroacetate, 2d: Melting point: 83–85 °C. IR: 1665 (s), 1417 (m), 1183 (s), 991.2 (m), 721 (s) cm−1. 1H-NMR: δ 0.91 (3H, t, J = 7.3 Hz), 1.33 (2H, hex, J = 7.3 Hz), 1.57 (2H, pen, J = 7.7 Hz), 2.63 (2H, t, J = 7.6 Hz), 7.18 (2H, d, J = 7.9 Hz), 7.39 (2H, d, J = 7.9 Hz), 7.50 (2H, t, J = 7.9Hz), 7.60 (1H, t, J = 7.4 Hz), 8.16 (2H, d, J = 8.4 Hz). 13C-NMR: δ 14.2, 22.6, 33.6, 36.0, 44.9, 105.0, 115.7 (1C, q, J = 291 Hz), 117.7, 121.0, 129.1 (2C), 132.1 (2C), 132.1 (2C), 132.4, 133.1 (2C), 133.6 (2C), 146.8, 162.9 (1C, q, J = 38 Hz). HRMS: m/z calcd. for [M − OCOCF3]+ C16H14I+ 361.448 found 361.01463.
([1,1′-Biphenyl]-4-ylethynyl)(phenyl)iodonium 2,2,2-trifluoroacetate, 2e: This compound is unstable and complete characterization could not be obtained. 1H-NMR: δ (2H, d, J = 6.9 Hz), 7.33–7.61 (5H, m), 7.62–7.89 (5H, m), 8.17 (2H, d, J = 7.7 Hz). HRMS: m/z calcd. for [M − OCOCF3]+ C20H14I+ 381.0135 found 381.0132.
3.3. General Procedure for Alkenyl(phenyl)iodonium Tetrafluoroborate 3 Formation: Preparation of (Z)-Phenyl(2-phenyl-2-tosylvinyl)iodonium Tetrafluoroborate, 3ab
Following Ochiai’s procedure [
14], phenyl(phenylethynyl)iodonium trifluoroacetate (0.50 g, 1.2 mmol, 1 equiv.) dissolved in MeOH (5 mL) was added to a solution of 4-methylbenzene sulfinic acid (0.20 g, 1.3 mmol, 1.1 equiv.) in MeOH (2 mL) at 0 °C under a N
2 atmosphere. The reaction mixture was stirred for 45 min then poured into saturated aqueous sodium tetrafluoroborate solution (5 mL). A white precipitate was formed which was filtered off and washed with CH
2Cl
2 (2 × 8 mL). The combined organic layers were washed with water, dried over magnesium sulfate, filtered and concentrated in vacuo. The residue was a clear, colourless, viscous oil. A 1:1 mixture of diethyl ether and petroleum ether (5 + 5 mL) was added to the residue, then the mixture was swirled and the solvent removed by decantation. This was repeated until a white solid was formed. This crude product was recrystallized from the minimum amount of CH
2Cl
2 with slow addition of petroleum ether. (
Z)-Phenyl(2-phenyl-2-tosylvinyl)iodonium tetrafluoroborate
3ab was isolated as a white microcrystalline solid (0.54 g, 83%). Melting point: 125–127 °C. IR: 1535 (w), 1294 (m), 1135 (m), 1076 (s), 627 (s) cm
−1.
1H-NMR: δ 2.40 (3H, s), 7.28 (2H, d,
J = 7.9 Hz), 7.36 (2H, t,
J = 7.6 Hz), 7.40–7.49 (3H, m), 7.63–7.73 (4H, m), 7.82 (1H, t,
J = 7.6 Hz), 8.33 (1H, s), 8.40 (2H, d,
J = 7.9 Hz).
13C-NMR: δ 22.5, 116.9, 117.1, 129.7 (2C), 129.9 (2C), 130.5 (2C), 131.7 (2C), 131.8 (2C), 133.1 (2C), 133.7, 134.1 (2C), 137.6, 146.9, 148.1. HRMS:
m/
z calcd. for [M − BF
4]
+ C
21H
18IO
2S
+ 461.0067 found 461.0079.
(Z)-(2-((2-Fluorophenyl)sulfonyl)-2-phenylvinyl)(phenyl)iodonium tetrafluoroborate, 3ac: Melting point: 186–188 °C. IR: 1597 (w), 1474 (m), 1317 (m), 1022 (m), 522 (s) cm−1. 1H-NMR: δ 7.29–7.38 (4H, m), 7.40–7.46 (2H, t, m), 7.59 (1H, t, J = 9.6 Hz), 7.67 (2H, t, J = 7.8 Hz), 7.74–7.93 (3H, m), 8.40 (2H, d, J = 7.8 Hz), 8.55 (1H, s). 13C-NMR: δ 116.9, 118.4, 119.1 (1C, d, J = 20 Hz), 124.4 (1C, d, J = 13 Hz), 127.2, 130.1 (2C), 130.4 (2C), 131.3, 132.1 (1C, d, J = 5.4 Hz), 133.2 (2C), 134.2, 137.7 (2C), 140.5 (1C, d, J = 9.8 Hz), 146.6, 159.1, 161.6. HRMS: m/z calcd. for [M − BF4]+ C20H15FIO2S+ 464.9816 found 464.9826.
(Z)-Phenyl(2-phenyl-2-((4-(trifluoromethyl)phenyl)sulfonyl)vinyl)iodonium tetrafluoroborate, 3ad: Melting point: 184–186 °C. IR: 1317 (s), 1129 (s), 1117 (m), 1059 (s), 641 (s) cm−1. 1H-NMR: δ 7.29 (2H, d, J = 7.8 Hz), 7.38 (2H, t, J = 7.6 Hz), 7.46 (1H, t, J = 7.6 Hz), 7.68 (2H, t, J = 7.8 Hz), 7.82 (1H, t, J = 7.4 Hz), 8.1 (4H, S), 8.39 (2H, d, J = 8.6 Hz), 8.50 (1H, s). 13C-NMR: 117.1, 119.2, 124.3 (1C, q, J = 274.2 Hz), 128.3 (1C, d, J = 3.3 Hz), 130.1 (2C), 130.7 (2C), 130.8 (2C), 131.3. 131.9, 133.0 (2C), 134.3 (2C), 136.1 (1C, q, J = 33 Hz), 137.6 (2C), 140.6, 145.8. HRMS: m/z calcd. for [M − BF4]+ C21H15F3IO2S+ 514.9784 found 514.9807.
(Z)-(2-([1,1′-Biphenyl]-4-ylsulfonyl)-2-phenylvinyl)(phenyl)iodonium tetrafluoroborate, 3ae: Melting point: 162–164 °C. IR: 1596 (w), 1484 (m), 1289 (m), 1050 (m), 674 (s) cm−1. 1H-NMR: δ 7.33 (2H, d, J = 7.2 Hz), 7.38 (2H, t, J = 7.5 Hz), 7.42–7.58 (4H, m), 7.67 (2H, t, J = 7.9 Hz), 7.77 (2H, d, J = 7.8 Hz), 7.82 (1H, t, J = 7.5 Hz), 7.88 (2H, d, J = 8.2 Hz), 7.96 (2H, d, J = 8.5 Hz), 8.39 (1H, s), 8.41(2H, d, J = 7.8 Hz). 13C-NMR: δ 116.9, 117.7, 128.6 (2C), 129.1 (2C), 130.0 (2C), 130.4 (2C), 130.5 (2C), 130.6 (2C), 131.8 (2C), 131.9, 133.1, 134.1(2C), 135.3, 137.6(2C), 138.7, 146.7, 148.1. HRMS: m/z calcd. for [M − BF4]+ C26H20IO2S+ 523.0223 found 523.0236.
(Z)-(2-((5-Chlorothiophen-2-yl)sulfonyl)-2-phenylvinyl)(phenyl)iodonium tetrafluoroborate, 3af: Melting point: 187–189 °C. IR: 1399 (w), 1310 (w), 639 (s), 628 (m), 523 (m) cm−1. 1H-NMR: δ 7.35 (2H, d, J = 7.6 Hz), 7.39–7.46 (3H, m), 7.51 (1H, t, J = 7.4 Hz), 7.65 (2H, t, J = 7.8 Hz), 7.73–7.83 (2H, m), 8.36 (3H, d, J = 9.4 Hz). 13C-NMR: 116.9, 117.2, 130.2 (2C), 130.9 (2C), 131.6, 131.9, 132.2, 133.2 (2C), 134.2, 134.6, 137.6 (2C), 139.0, 142.4, 147.3. HRMS: m/z calcd. for [M − BF4]+ C18H13ClIO2S2+ 486.9085 found 486.9091.
(Z)-Phenyl(2-(phenylsulfonyl)-2-(p-tolyl)vinyl)iodonium, 3ba: Melting point: 125–127 °C. IR: 1535 (m), 1303 (m), 1136 (m), 1059 (s), 651 (s) cm−1. 1H-NMR: δ 2.26 (3H, s), 7.17 (4H, t, J = 6.9 Hz), 7.61–7.72 (4H, m), 7.77–7.89 (4H, m), 8.29 (1H, S), 8.39 (2H, d, J = 7.8 Hz). 13C-NMR: δ 22.07, 116.9, 128.9, 129.6 (2C), 130.4 (2C), 130.5 (2C), 131.2 (2C), 133.1(2C), 134.1, 136.8 (3C), 136.9, 137.6, 141.8, 146.7. HRMS: m/z calcd for [M − BF4]+ C21H18IO2S+ 461.0067 found 461.0082.
(Z)-(2-(4-Ethylphenyl)-2-(phenylsulfonyl)vinyl)(phenyl)iodonium, 3ca: Melting point: 122–124 °C. IR: 1526 (m), 1299 (m), 1134 (m), 1060 (s), 620 (s) cm−1. 1H-NMR: δ 1.10 (3H, t, J = 7.5 Hz), 2.56 (2H, q, J = 7.8 Hz), 7.20 (4H, s), 7.67 (4H, t, J = 7.6 Hz), 7.85–7.97 (4H, m), 8.31 (1H, S), 8.39 (2H, d, J = 3.8 Hz). 13C-NMR: δ 16.4, 29.1, 116.9, 117.0, 129.1, 129.3 (2C), 129.5 (2C), 130.5 (2C), 131.2 (2C), 133.1 (2C), 134.1, 136.7, 136.9, 137.6 (2C), 146.7, 147.9. HRMS: m/z calcd. for [M − BF4]+ C22H20IO2S+ 475.0223 found 475.0247.
(Z)-(2-(4-Butylphenyl)-2-(phenylsulfonyl)vinyl)(phenyl)iodonium tetrafluoroborate, 3da: Melting point: 120–122 °C. IR: 1526 (m), 1293 (m), 1130 (s), 1048 (s), 569 (s) cm−1. 1H-NMR: δ 0.84 (3H, t, J = 7.5 Hz), 1.21 (2H, hex, J = 7.5 Hz), 1.77 (2H, pen, J = 7.5 Hz), 2.49–2.55 (2H, m), 7.18 (4H, s), 7.66 (4H, q, J = 7.2 Hz), 7.81 (4H, t, J = 8.6 Hz) 8.31 (1H, S), 8.39 (2H, d, J = 3.9 Hz). 13C-NMR: δ 14.9, 22.7, 33.9, 35.6, 116.8, 116.9, 129.01, 129.5, 129.8 (2C), 130.4 (2C), 131.1 (2C), 133.03 (2C), 134.1, 136.7 (2C), 136.9, 137.6 (2C), 146.5, 146.7. HRMS: m/z calcd. for [M − BF4]+ C24H24IO2S+ 503.0536 found 503.0549.
(Z)-(2-([1,1′-Biphenyl]-4-yl)-2-(phenylsulfonyl)vinyl)(phenyl)iodonium, 3ea: Melting point: 164–166 °C. IR: 1501 (w), 1310 (m), 1132 (m), 1073 (m), 533 (s) cm−1. 1H-NMR: δ 7.40 (3H, t, J = 8.5 Hz), 7.47 (2H, t, J = 7.6 Hz), 7.62–7.73 (8H, m), 7.83 (2H, t, J = 7.2 Hz), 7.80 (2H, d, J = 7.8 Hz), 8.41 (3H, t, J = 7.3 Hz). 13C-NMR: δ 117.0, 117.8, 127.9 (2C), 128.0 (2C), 129.5, 129.6 (2C), 130.3 (2C), 130.7, 131.13 (2C), 131.3 (2C), 131.9, 133.1 (2C), 134.1, 136.8, 137.1, 137.6, 139.7, 143.2, 146.3. HRMS: m/z calcd. for [M − BF4]+ C26H20IO2S+ 523.0223 found 523.0207.
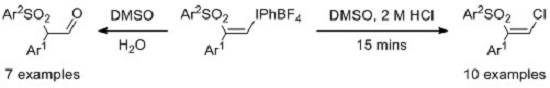
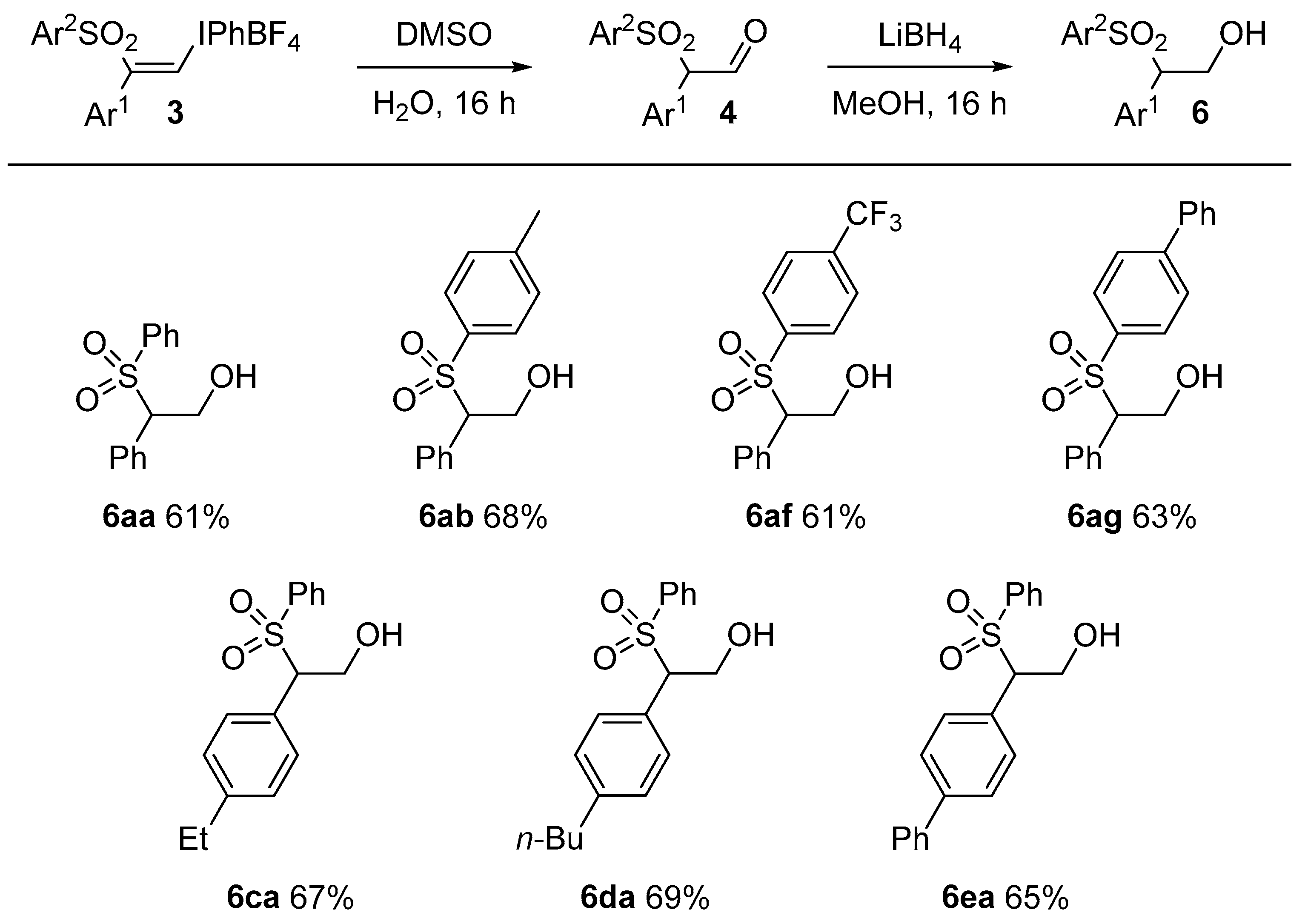
3.4. General Procedure for Formation of Alcohols 6: Preparation of 2-phenyl-2-(phenylsulfonyl)ethan-1-ol, 6aa
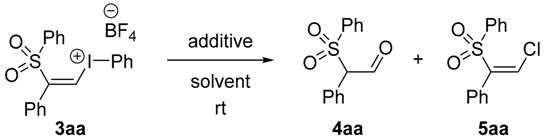
(Z)-Phenyl(2-phenyl-2-(phenylsulfonyl)vinyl)iodonium tetrafluoroborate, 3aa (50 mg, 0.094 mmol, 1 equiv.) was dissolved in DMSO (0.5 mL) at room temperature. Deionized water (20 μL) was added and the mixture was stirred overnight. Water (2.5 mL) was added to the mixture and the solution was shaken and then extracted with ethyl acetate (2 × 5 mL). The organic layer was dried with anhydrous magnesium sulfate, filtered and concentrated under vacuum. The crude product was dissolved in methanol (2 mL) and LiBH4 (2 mg, 0.094 mmol, 1 equiv.) was added in one portion. The mixture was stirred overnight and then extracted with diethyl ether (2 × 5 mL) and washed with water (5 mL). The residue was purified by flash chromatography (5:1 petrolem ether/EtOAc) which furnished 6aa as a colorless oil (0.015 g, 61%). IR: 3521 (w), 2428 (s), 1235 (m), 567 (w) cm−1. 1H-NMR: δ 2.77 (OH, dd, J = 8.7, 4.8 Hz), 4.16 (1H, ddd, J = 13, 8.7, 4.5 Hz), 4.38 (1H, dd, J = 7.9, 4.7 Hz), 4.66 (1H, ddd, J = 12, 7.9, 4.5 Hz), 7.06 (2H, d, J = 7.4 Hz), 7.22–7.30 (3H, m), 7.34 (1H, t, J = 7.4 Hz), 7.59–7.70 (4H, m). 13C-NMR: δ 61.5, 73.3, 126.1, 126.2, 129.2 (2C), 129.8 (2C), 129.9 (2C), 130.0 (2C), 130.6, 140.7. HRMS: m/z calcd. for [M + H]+ C14H15O3S+ 263.0736 found 263.0701.
2-Phenyl-2-tosylethan-1-ol, 6ab: IR: 3521 (w), 2428 (s), 1235 (m), 567 (w) cm−1. 1H-NMR: δ 2.40 (3H, s), 2.99 (OH, br), 4.08 (1H, ddd, J = 13, 7.8, 4.9 Hz), 4.33 (1H, dd, J = 8.2, 4.5 Hz), 4.61 (1H, t, J = 10 Hz), 7.03 (2H, d, J = 7.9 Hz ), 7.19 (2H, d, J = 7.4 Hz), 7.21–7.27 (2H, m), 7.38 (2H, d, J = 8.2 Hz). 13C-NMR: δ 21.9, 61.7, 73.0, 128.9 (2C), 129.3 (2C), 129.4 (2C), 129.7 (2C), 129.9 (2C), 131.3, 134.0, 145.4. HRMS: m/z calcd. for [M + H]+ C15H17O3S+ 277.0893 found 277.0876.
2-Phenyl-2-((4-(trifluoromethyl)phenyl)sulfonyl)ethan-1-ol, 6af: IR: 2359 (w), 1319 (s), 1172 (s), 712 (m) cm−1. 1H-NMR: δ 2.79 (OH, br), 4.16 (1H, d, J = 12 Hz), 4.38 (1H, dd, J = 7.9, 4.7 Hz), 4.66 (1H, dd, J = 12, 7.9 Hz), 7.06 (2H, d, J = 7.5 Hz ), 7.27 (2H, t, J = 7.4 Hz), 7.34 (1H, t, J = 7.4 Hz), 7.61–7.70 (4H, m). 13C-NMR: δ61.4, 73.3, 123.4 (1C, q, J = 271.4 Hz), 126.2 (1C, d, J = 3.4 Hz), 129.2 (2C), 129.8 (2C), 129.9 (2C), 130.0 (2C), 130.6, 135.7, 136.0, 140.8. HRMS: m/z calcd. for [M + NH4]+ C15H17F3NO3S+ 348.0876 found 348.0877.
2-([1,1′-Biphenyl]-4-ylsulfonyl)-2-phenylethan-1-ol, 6ag: IR: 3565 (w), 2359 (s), 2250 (m), 650 (w) cm−1. 1H-NMR: δ 2.92–3.00 (OH, m), 4.07–4.19 (1H, m), 4.39 (1H, dd, J = 8.3, 4.5 Hz), 4.61–4.71 (1H, m), 7.08 (1H, d, J = 7.5 Hz), 7.26 (4H, t, J = 7.2 Hz), 7.33 (1H, t, J = 7.2 Hz), 7.38–7.52 (3H, m), 7.53–7.65 (5H, m). 13C-NMR: δ 61.7, 73.2, 127.6 (2C), 127.7 (2C), 129.0 (2C), 129.1, 129.4 (2C), 129.6, 129.9 (2C), 130.0 (2C), 131.2, 135.5, 139.2, 147.2. HRMS: m/z calcd. for [M + H]+ C20H22O3S+ 356.1315 found 356.1322.
2-(4-Ethylphenyl)-2-(phenylsulfonyl)ethan-1-ol, 6ca: IR: 2561 (s), 1345 (m), 1227 (m), 1162 (s), 767 (m) cm−1. 1H-NMR: δ 1.19 (3H, t, J = 7.7), 2.61 (2H, q, J = 7.7 Hz), 2.91 (OH, dd, J = 9.0 HZ, 4.7 Hz), 4.09 (1H, ddd, J = 13, 9, 5 Hz), 4.33 (1H, dd, J = 8.2, 4.3 Hz), 4.59 (1H, ddd, J = 12, 8.0, 4.0 Hz), 6.94 (2H, d, J = 8.1 Hz ), 7.07 (2H, d, J = 7.8 Hz), 7.41 (2H, t, J = 7.8 Hz), 7.53 (2H, d, J = 7.6 Hz), 7.59 (1H, t, J = 7.4 Hz). 13C-NMR: δ 15.8, 28.9, 61.7, 72.9, 128.2, 128.5 (2C), 129.1 (2C), 129.4 (2C), 129.8 (2C), 134.3 (2C), 137.1, 145.9. HRMS: m/z calcd. for [M + NH4]+ C16H22NO3S+ 308.1315 found 308.1318.
2-(4-Butylphenyl)-2-(phenylsulfonyl)ethan-1-ol, 6da: IR: 2341 (s), 1446 (m), 1304 (m), 1142 (s), 687.6 (m) cm−1. 1H-NMR: δ 0.91 (3H, t, J = 7.3 Hz), 1.24–1.35 (2H, m), 1.54 (2H, pent, J = 7.4 Hz), 2.56 (2H, t, J = 7.6 Hz), 2.99 (OH, br), 4.09 (1H, dd, J = 8.1, 4.4 Hz), 4.33 (1H, dd, J = 7.9, 4.6 Hz), 4.59 (1H, dd, J = 12, 8.1 Hz), 6.92 (2H, d, J = 7.9 Hz), 7.04 (2H, d, J = 7.4 Hz), 7.39 (2H, t, J = 7.8 Hz), 7.51 (2H, d, J = 3.8 Hz), 7.60 (1H, t, J = 7.4 Hz). 13C-NMR: δ 14.2, 22.5, 33.7, 35.6, 61.6, 72.8 128.1, 129.0 (3C), 128.9, 129.4, 130.4, 129.7, 134.2, 137.1, 144.5. HRMS: m/z calcd. for [M + NH4]+ C18H26NO3S+ 336.1628 found 336.1630.
2-([1, 1′-Biphenyl]-4-yl)-2-(phenylsulfonyl)ethan-1-ol, 6ea: IR: 3568 (w), 2358 (s), 2248 (m), 647 (w) cm−1. 1H-NMR: δ 4.1–4.2 (1H, m), 4.40 (1H, dd, J = 8.1, 4.5 Hz), 4.65 (1H, dd, J = 12, 8.1 Hz), 7.11 (2H, d, J = 7.7 Hz), 7.37 (1H, t, J = 7.3 Hz), 7.40–7.51 (6H, m), 7.52–7.64 (5H, m). 13C-NMR: δ 61.6, 72.8, 127.4 (2C), 127.6 (2C), 128.2, 129.2 (2C), 129.3 (2C), 129.4 (2C), 130.1 (2C), 130.4, 134.4, 137.2, 140.4, 142.4. HRMS: m/z calcd. for [M + Na]+ C20H18NaO3S+ 361.0869 found 361.0869.
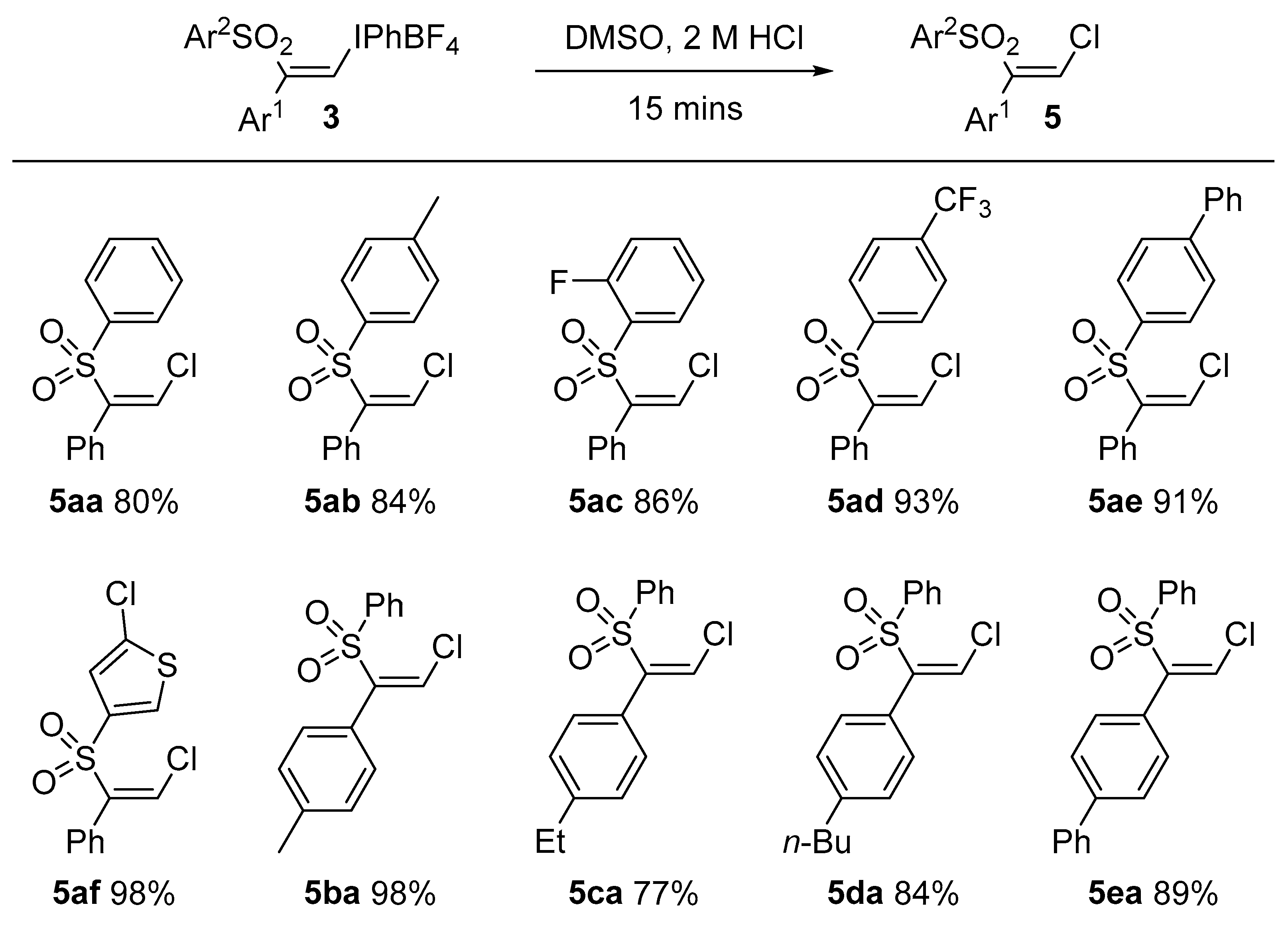
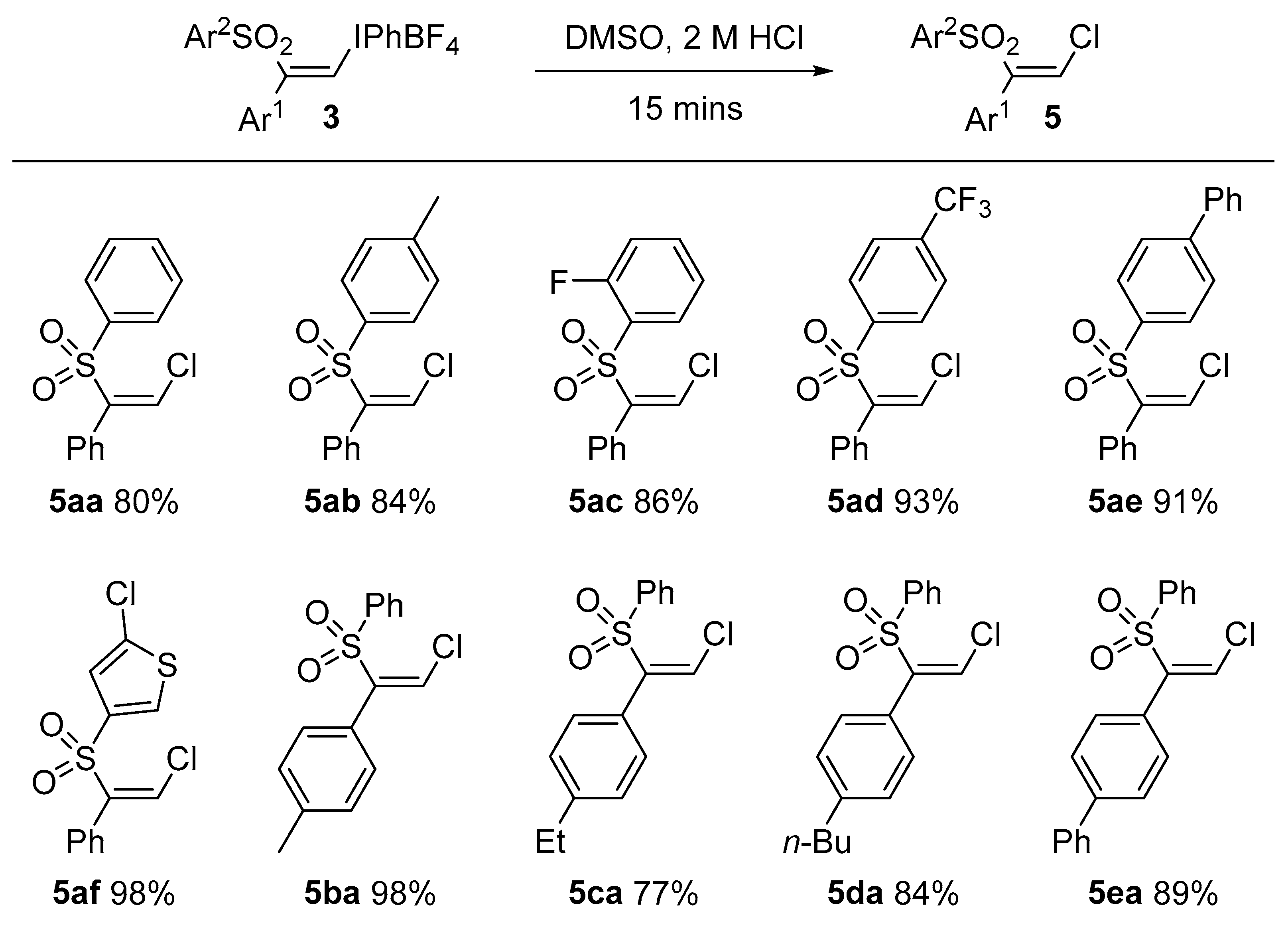
3.5. General Procedure for Formation of Vinyl Chlorides 5: Preparation of (Z)-(2-Chloro-1-(phenylsulfonyl)vinyl)benzene, 5aa
(Z)-Phenyl(2-phenyl-2-(phenylsulfonyl)vinyl)iodonium tetrafluoroborate 3aa (0.050 g, 0.094 mmol) was dissolved in DMSO (0.5 mL) at room temperature. HCl (2 N, 50 μL) was added and the mixture was stirred for 15 min. Brine (5 mL) was added to the mixture and the solution was shaken. This was extracted with ethyl acetate (2 × 10 mL) and washed with water (5 mL). The organic layer was dried with anhydrous magnesium sulfate, filtered and concentrated under vacuum. The residue was purified by flash chromatography (20:1 petrolem ether/EtOAc) which furnished 5aa as a colorless oil (0.026 g, 80%). IR: 1586 (w), 1444 (m), 1320 (m), 1147 (s), 628 (s) cm−1. 1H-NMR: δ 6.73 (1H, s), 7.26 (2H, d, J = 7.3 Hz), 7.34 (2H, t, J = 7.2 Hz), 7.41 (1H, t, J = 7.2 Hz), 7.49 (2H, t, J =7.8 Hz), 7.62 (1H, t, J = 7.4 Hz), 7.83 (2H, d, J = 7.6 Hz). 13C-NMR: δ 128.7 (2C), 128.8 (2C), 129.2, 129.4 (2C), 130.0, 130.5, 133.2 (2C), 134.2, 140.5, 145.6. HRMS: m/z calcd. for [M + H]+ C14H11ClO2S+ 279.0232 found 279.0241.
(Z)-1-((2-Chloro-1-phenylvinyl)sulfonyl)-4-methylbenzene, 5ab: IR: 1578 (m), 1443 (m), 1146 (s), 1083 (m), 754 (s) cm−1. 1H-NMR: δ 2.42 (3H, s), 6.69 (1H, s), 7.26 (4H, d, J = 16 Hz), 7.33 (2H, t, J = 7.4 Hz), 7.35 (1H, t, J = 7.4 Hz), 7.70 (2H, d, J = 7.7 Hz). 13C-NMR: δ 22.7, 128.7 (2C), 128.8, 128.9 (2C), 129.9 (3C), 130.5, 133.3 (2C), 137.5, 145.2, 145.7. HRMS: m/z calcd. for [M + H]+ C15H14ClO2S+ 293.0398 found 293.0390.
(Z)-1-((2-Chloro-1-phenylvinyl)sulfonyl)-2-fluorobenzene, 5ac: IR: 1599 (w), 1264 (m), 1146 (m), 1076 (s), 770 (m) cm−1. 1H-NMR: δ 6.82 (1H, s), 7.16 (1H, t, J = 9.0 Hz), 7.29 (1H, t, J = 7.6 Hz), 7.32–7.43 (5H, m), 7.56–7.65 (1H, m), 7.99 (1H, dd, J = 7.5, 1.6 Hz). 13C-NMR: δ 117.2 (1C, d, J = 20 Hz), 124.7 (1C, d, J = 3.8 Hz), 128.8 (3C), 129.4 (1C, d, J = 1.2 Hz), 130.1 (2C), 130.4 (1C, d, J = 1.2 Hz), 131.3, 132.5, 136 (1C, d, J = 8.5 Hz), 145.9, 160.0 (1C, d, J = 256 Hz). HRMS: m/z calcd. for [M + NH4]+ C14H14ClFNO2S+ 314.0412 found 314.0410.
(Z)-1-((2-Chloro-1-phenylvinyl)sulfonyl)-4-(trifluoromethyl)benzene, 5ad: IR: 1591 (m), 1320 (s), 1125 (s), 1085 (s), 714 (m) cm−1. 1H-NMR: δ 6.80 (1H, s), 7.27 (2H, d, J = 8.5 Hz), 7.33–7.47 (3H, m), 7.76 (2H, d, J = 7.9 Hz), 7.96 (2H, d, J = 7.97 Hz). 13C-NMR: δ 123.5 (1C, q, J = 274 Hz), 126.4, 128.9 (2C), 129.1 (2C), 130.3 (2C), 130.5 (2C), 132.6, 135.8 (1C, q, J = 33 Hz), 143.9, 144.9. HRMS: m/z calcd. for [M + NH4]+ C15H14ClF3NO2S+ 364.0380 found 364.0375.
(Z)-4-((2-Chloro-1-phenylvinyl)sulfonyl)-1,1′-biphenyl, 5ae: IR: 1592 (w), 1329 (m), 1148 (m), 1085 (w), 581 (m) cm−1. 1H-NMR: δ 6.75 (1H, s), 7.30 (2H, d, J = 3.7 Hz), 7.36 (2H, t, J = 7.6 Hz), 7.42 (2H, t, J = 7.8 Hz), 7.48 (2H, t, J = 7.6 Hz), 7.60 (2H, d, J = 7.6 Hz), 7.70 (2H, d, J = 8.3 Hz), 7.88 (2H, d, J = 8.3 Hz). 13C-NMR: δ 127.7 (2C), 127.9 (2C), 128.8 (2C), 129.1, 129.2 (2C), 129.4, 129.5 (2C), 130.0, 130.6 (2C), 133.2, 138.9, 139.4, 145.6, 147.1. HRMS: m/z calcd. for [M + H]+ C20H19ClNO2S+ 372.0820 found 372.0816.
(Z)-2-Chloro-5-((2-chloro-1-phenylvinyl)sulfonyl)thiophene, 5af: IR: 1402 (m), 1332 (s), 1146 (s), 990 (m), 605 (s) cm−1. 1H-NMR: δ 6.78 (1H, s), 6.93 (1H, d, J = 3.9 Hz), 7.29 (2H, d, J = 7.9 Hz), 7.38 (2H, t, J = 7.7 Hz) 7.40–7.46 (2H, m). 13C-NMR: δ 127.4, 128.8 (2C), 129.8, 130.2, 130.5 (2C), 132.6, 134.6, 138.9, 140.5, 145.2. HRMS: m/z calcd. for [M + NH4]+ C12H12Cl2NO2S2 335.9681 found 335.9684.
(Z)-1-(2-Chloro-1-(phenylsulfonyl)vinyl)-4-methylbenzene, 5ba: IR: 1578 (m), 1443 (m), 1146 (s), 1083 (m), 754 (s) cm−1. 1H-NMR: δ 2.36 (3H, s), 6.70 (1H, s), 7.15 (4H, s), 7.49 (2H, t, J = 7.5 Hz), 7.61(1H, t, J = 7.5 Hz), 7.83 (2H, d, J = 7.5 Hz). 13C-NMR: δ 21.7, 128.6 (2C), 128.9, 129.3 (2C), 129.5 (2C), 130.3, 130.4 (2C), 134.1, 140.2, 140.6, 145.5. HRMS: m/z calcd. for [M + NH4]+ C15H17ClNO2S+ 310.0663 found 310.0663.
(Z)-1-(2-Chloro-1-(phenylsulfonyl)vinyl)-4-ethylbenzene, 5ca: IR: 2359 (s), 1709 (m), 1332 (m), 1149 (s), 1084 (m), 722 (m) cm−1. 1H-NMR: δ 1.23 (3H, t, J = 7.7 Hz), 2.65 (2H, q, J = 7.6 Hz), 6.70 (1H, s), 7.18 (4H, s), 7.49 (2H, t, J = 7.7 Hz), 7.61(1H, t, J = 7.6 Hz), 7.83 (2H, d, J = 7.7 Hz). 13C-NMR: δ 15.6, 28.9, 128.3 (2C), 128.6 (2C), 128.9, 129.2 (2C), 130.4, 130.5 (2C), 134.1, 140.6, 145.6, 146.4. HRMS: m/z calcd. for [M + NH4]+ C16H19ClNO2S+ 324.0820 found 324.0822.
(Z)-1-(2-Chloro-1-(phenylsulfonyl)vinyl)-4-propylbenzene, 5da: IR: 2359 (s), 1709 (m), 1332 (m), 1149 (s), 1084 (m), 722 (m) cm−1. 1H-NMR: δ 0.93 (3H, t, J = 7.3 Hz), 1.34 (2H, hex, J = 7.3 Hz), 1.59 (2H, q, J = 7.6 Hz), 2.61 (2H, t, J = 7.9 Hz), 6.70 (1H, s), 7.10–7.20 (4H, m), 7.48 (2H, t, J = 7.6 Hz), 7.60 (1H, t, J = 7.4 Hz), 7.83 (2H, d, J = 8.1 Hz). 13C-NMR: δ 15.6, 28.9, 128.3 (2C), 128.6 (2C), 128.9, 129.2 (2C), 130.4, 130.5 (2C), 134.1, 140.6, 145.6, 146.4. HRMS: m/z calcd. for [M + H]+ C18H20ClO2S 335.0867 found 335.0866.
(Z)-4-(2-Chloro-1-(phenylsulfonyl)vinyl)-1,1′-biphenyl, 5ea: IR: 1481 (w), 1318 (m), 1180 (m), 1004 (w), 648 (s) cm−1. 1H-NMR: δ 6.78 (1H, s), 7.32–7.41 (3H, m), 7.42–7.54 (4H, m), 7.55–7.67 (5H, m), 7.88 (2H, d, J = 7.6 Hz). 13C-NMR: δ 127.4 (2C), 127.5 (2C), 128.2, 128.6 (2C), 129.3 (2C), 129.3 (2C), 129.4, 130.9 (2C), 132.0, 134.2, 140.3, 140.5, 142.9, 145.3. HRMS: m/z calcd. for [M + NH4]+ C20H19ClNO2S+ 372.0820 found 372.0821.





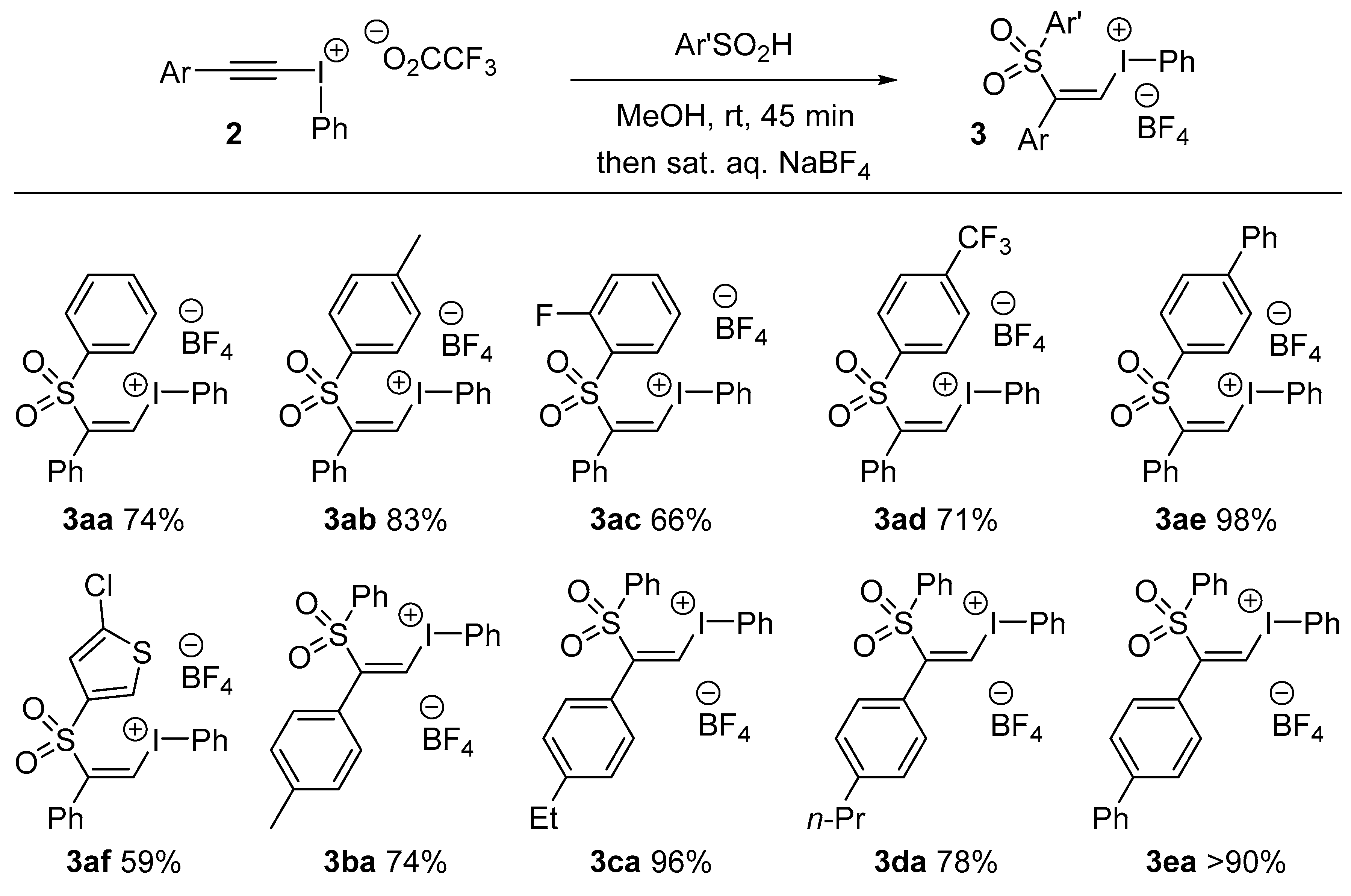

{kind=link}
{kind=link}
{kind=link}
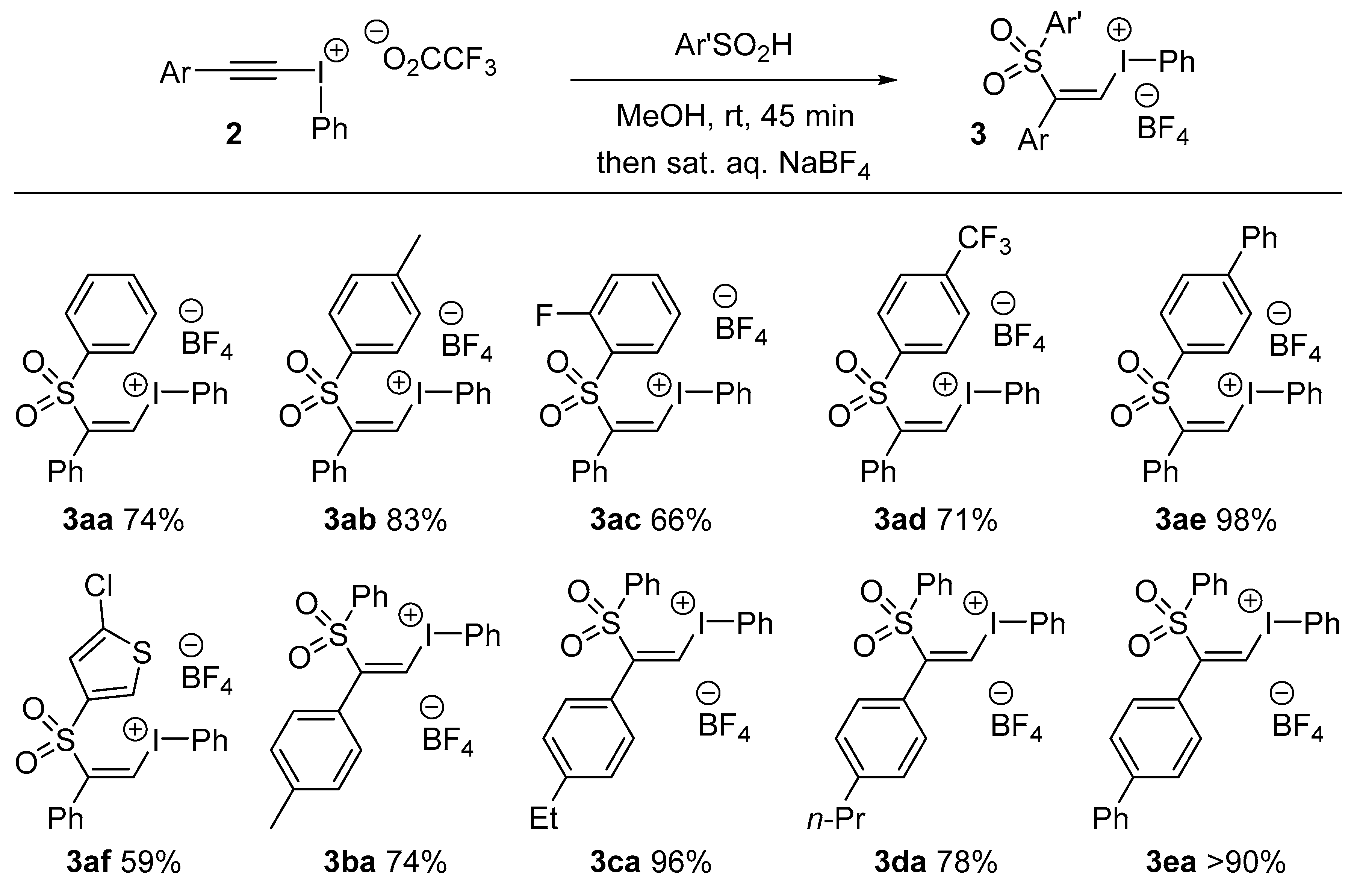{kind=link}
{kind=link}
{kind=link}