
Modulation of Neural Network Activity through Single Cell Ablation: An in Vitro Model of Minimally Invasive Neurosurgery



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
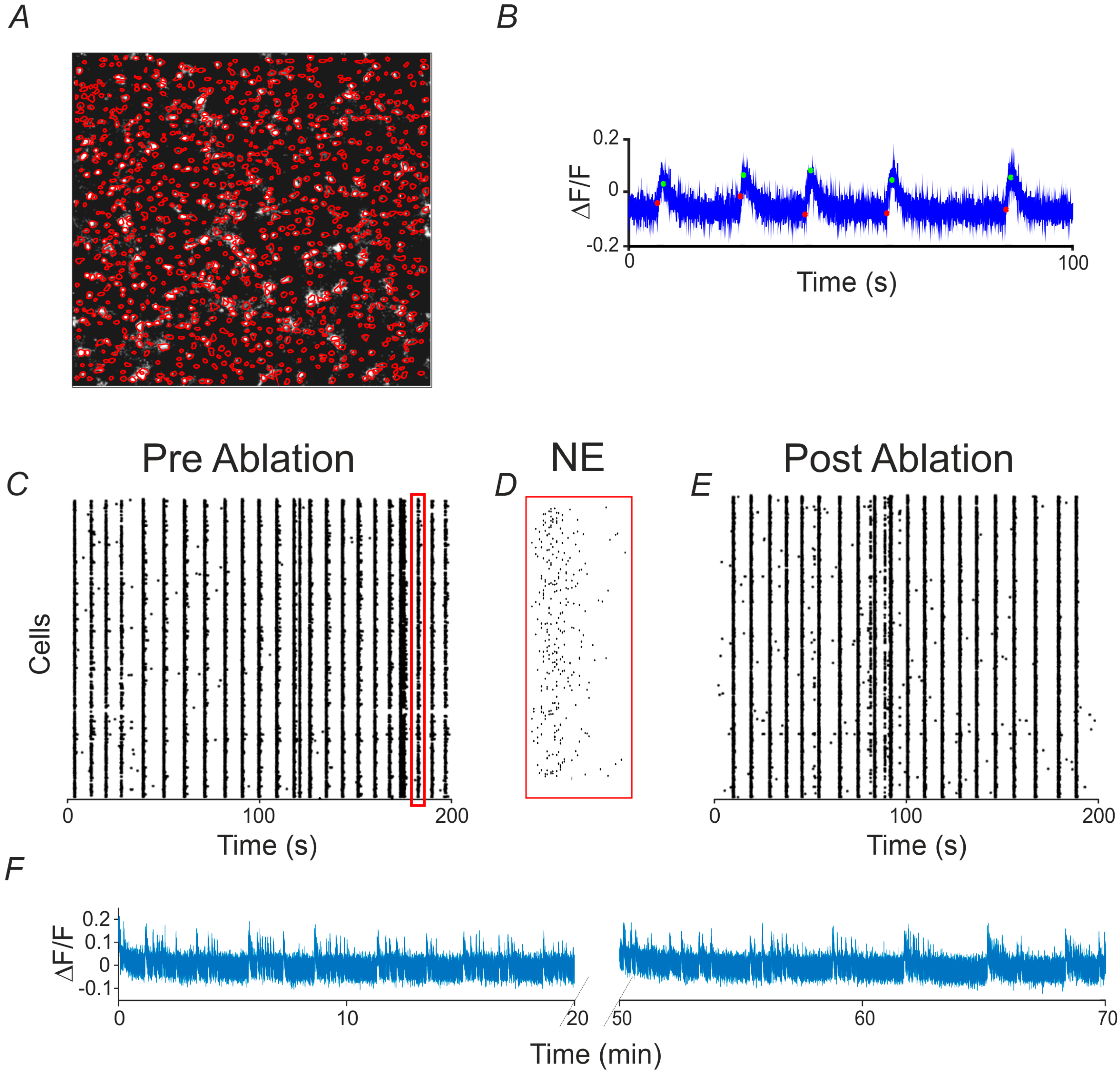
2.1. Long-Term Calcium Imaging
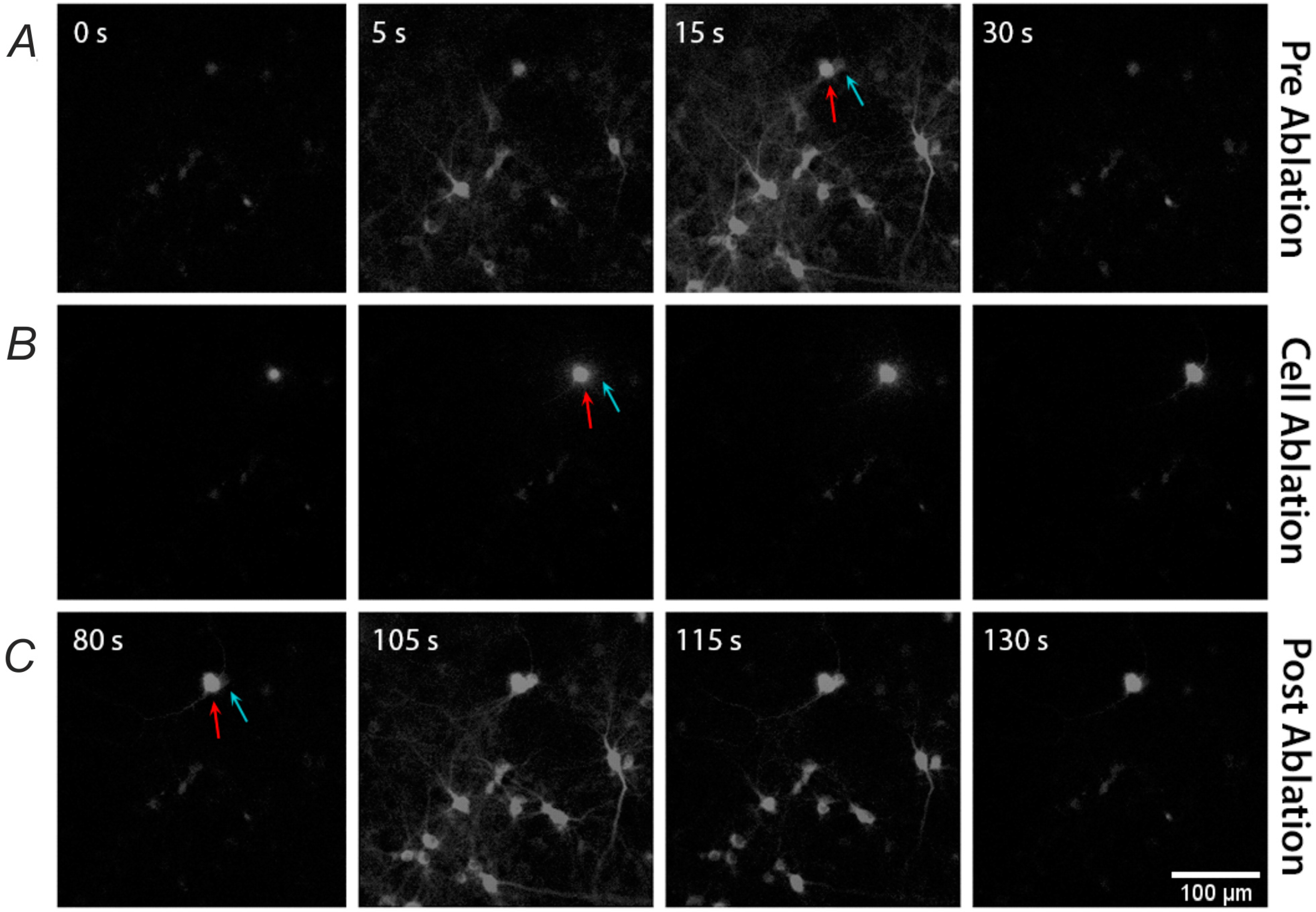
2.2. Single-Cell Ablation
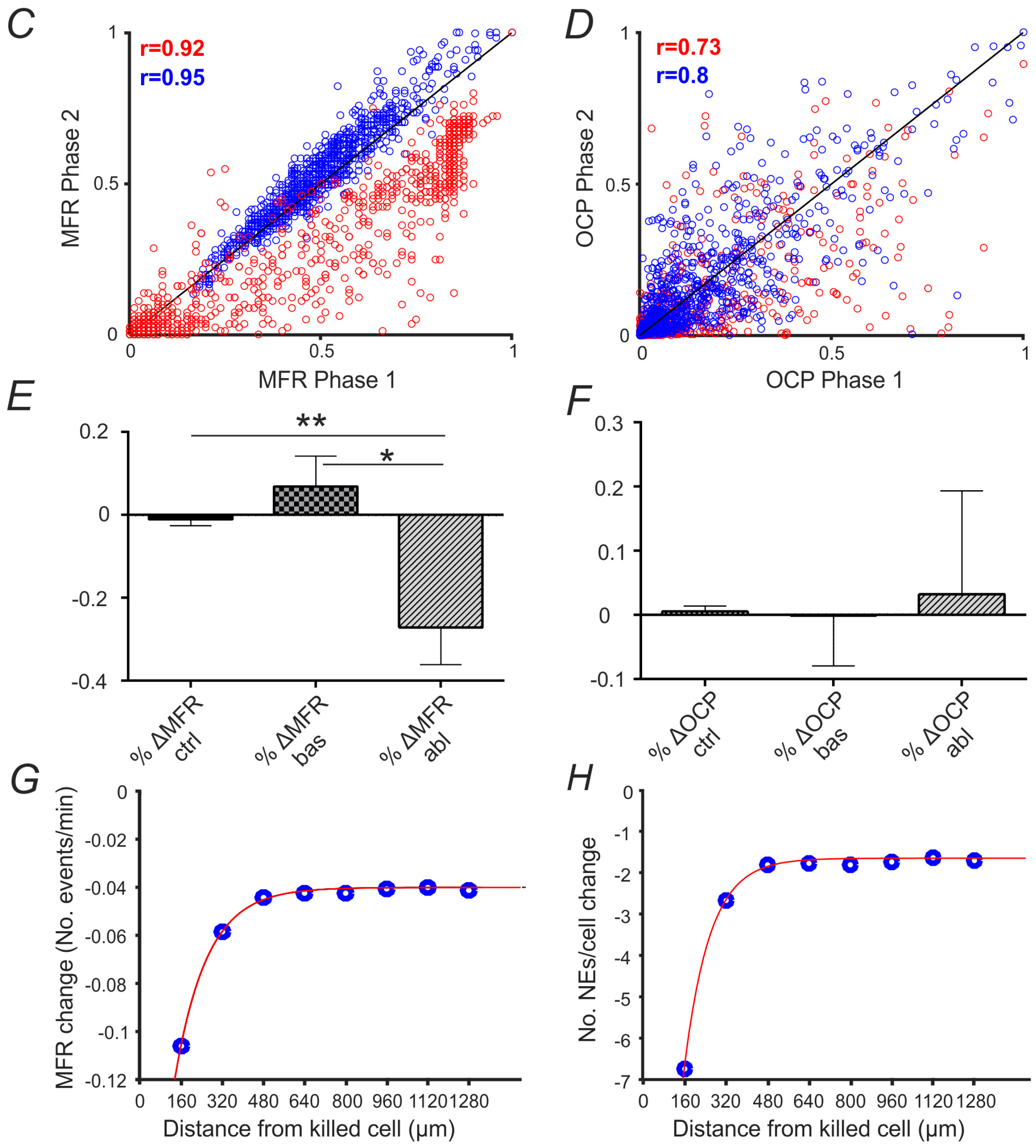
2.3. Mean Firing Rate and Connectivity Degree at Single Cell and Network Level
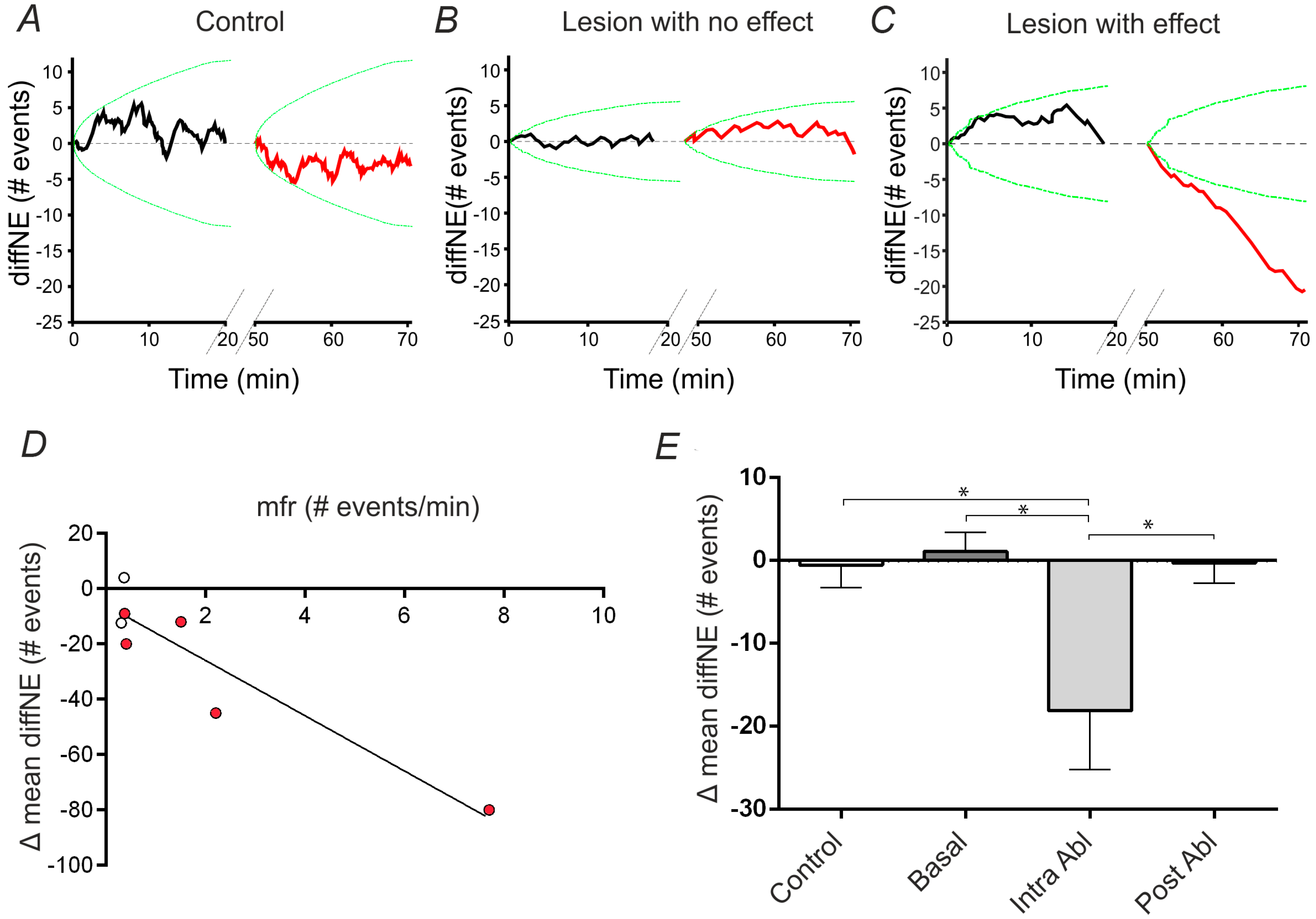
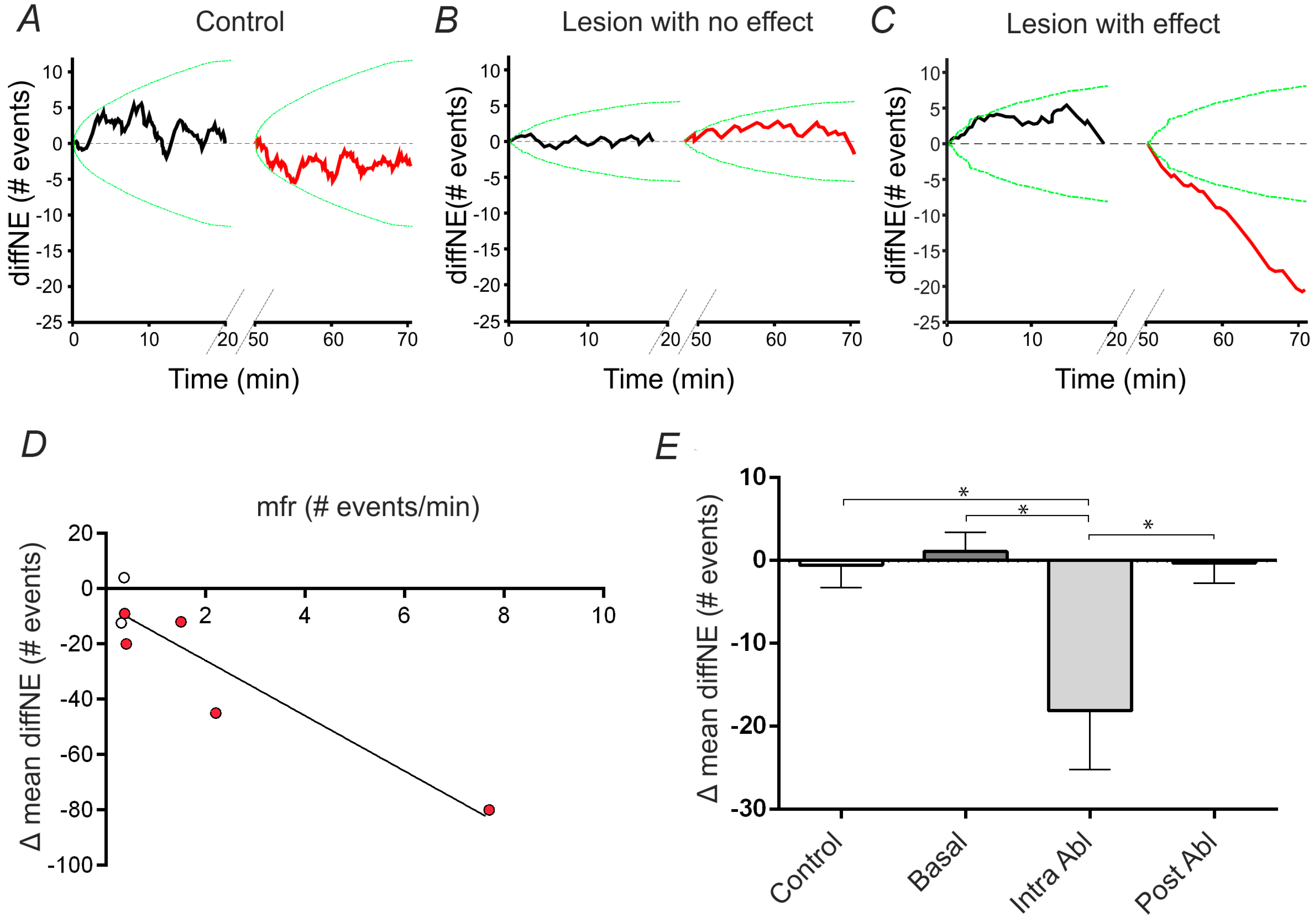
2.4. Analysis and Quantification of Single Cell Surgery Efficiency
3. Discussion
4. Materials and Methods
4.1. Optical Setup
4.2. Cell Cultures
4.3. Adeno Associated Viral Particle Infection
4.4. Long-Term Calcium Imaging Protocol
4.5. Analysis of Network Dynamics
4.6. Directed Functional Connectivity, Mean Firing Rate and Network Events
4.7. Impact on Global Network Dynamics
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vogel, A.; Noack, J.; Hüttman, G.; Paltauf, G. Mechanisms of femtosecond laser nanosurgery of cells and tissues. Appl. Phys. B 2005, 81, 1015–1047. [Google Scholar] [CrossRef]
- Diaspro, A. Confocal and Two-Photon Microscopy: Foundations, Applications and Advances; Wiley: Hoboken, NJ, USA, 2001. [Google Scholar]
- Eklof-Ljunggren, E.; Haupt, S.; Ausborn, J.; Dehnisch, I.; Uhlen, P.; Higashijima, S.-I.; El Manira, A. Origin of excitation underlying locomotion in the spinal circuit of zebrafish. Proc. Natl. Acad. Sci. USA 2012, 109, 5511–5516. [Google Scholar] [CrossRef] [PubMed]
- Habibey, R.; Golabchi, A.; Latifi, S.; Difato, F.; Blau, A. Microchannel device for selective laser dissection, long-term microelectrode array electrophysiology and imaging of confined axonal projections. Lab Chip 2015, 15, 4578–4590. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, J.B.; Higgins, M.L.; Lucas, J.H.; Gross, G.W. In vitro simulation of neural trauma by laser. J. Neuropathol. Exp. Neurol. 1985, 44, 268–284. [Google Scholar] [CrossRef] [PubMed]
- Soloperto, A.; Palazzolo, G.; Tsushima, H.; Chieregatti, E.; Vassalli, M.; Difato, F. Laser nano-neurosurgery from gentle manipulation to nano-incision of neuronal cells and scaffolds: An advanced neurotechnology tool. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [PubMed]
- Gomis-Rüth, S.; Wierenga, C.J.; Bradke, F. Plasticity of polarization: Changing dendrites into axons in neurons integrated in neuronal circuits. Curr. Biol. 2008, 18, 992–1000. [Google Scholar] [CrossRef] [PubMed]
- Go, M.A.; Choy, J.M.C.; Colibaba, A.S.; Redman, S.; Bachor, H.-A.; Stricker, C.; Daria, V.R. Targeted pruning of a neuron’s dendritic tree via femtosecond laser dendrotomy. Sci. Rep. 2016, 6, 19078. [Google Scholar] [CrossRef] [PubMed]
- Allegra Mascaro, A.L.; Cesare, P.; Sacconi, L.; Grasselli, G.; Mandolesi, G.; Maco, B.; Knott, G.W.; Huang, L.; de Paola, V.; Strata, P.; et al. In vivo single branch axotomy induces GAP-43-dependent sprouting and synaptic remodeling in cerebellar cortex. Proc. Natl. Acad. Sci. USA 2013, 110, 10824–10829. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Mohanty, S.; Gomez-Godinez, V.; Shi, L.Z.; Liaw, L.-H.; Miotke, J.; Meyer, R.L.; Berns, M.W. Neuronal growth cones respond to laser-induced axonal damage. J. R. Soc. Interface 2012, 9, 535–547. [Google Scholar] [CrossRef] [PubMed]
- Difato, F.; Dal Maschio, M.; Marconi, E.; Ronzitti, G.; Maccione, A.; Fellin, T.; Berdondini, L.; Chieregatti, E.; Benfenati, F.; Blau, A. Combined optical tweezers and laser dissector for controlled ablation of functional connections in neural networks. J. Biomed. Opt. 2011, 16, 051306. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Karthikeyan, K.; Chirvi, S.; Davé, D.P. Neuro-optical microfluidic platform to study injury and regeneration of single axons. Lab Chip 2009, 9, 2409–2612. [Google Scholar] [CrossRef] [PubMed]
- Uchugonova, A.; Breunig, H.G.; Ana BaKonig, K.; Bueckle, R.; Isemann, A.; Tempea, G.; König, K.; Bueckle, R.; Isemann, A.; Tempea, G. Targeted transfection of stem cells with sub-20 femtosecond laser pulses. Opt. Express 2008, 16, 9357–9364. [Google Scholar] [CrossRef] [PubMed]
- Canty, A.J.; Teles-Grilo Ruivo, L.M.; Nesarajah, C.; Song, S.; Jackson, J.S.; Little, G.E.; de Paola, V. Synaptic elimination and protection after minimal injury depend on cell type and their prelesion structural dynamics in the adult cerebral cortex. J. Neurosci. 2013, 33, 10374–10383. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Miller, J.H. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004, 5, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Difato, F.; Tsushima, H.; Pesce, M.; Benfenati, F.; Blau, A.; Chieregatti, E. The formation of actin waves during regeneration after axonal lesion is enhanced by BDNF. Sci. Rep. 2011, 1, 183. [Google Scholar] [CrossRef] [PubMed]
- Canty, A.J.; Huang, L.; Jackson, J.S.; Little, G.E.; Knott, G.; Maco, B.; de Paola, V. In Vivo single neuron axotomy triggers axon regeneration to restore synaptic density in specific cortical circuits. Nat. Commun. 2013, 4, 2038. [Google Scholar] [CrossRef] [PubMed]
- El Bejjani, R.; Hammarlund, M. Notch signaling inhibits axon regeneration rachid. Neuron 2012, 73, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Samara, C.; Rohde, C.B.; Gilleland, C.L.; Norton, S.; Haggarty, S.J.; Yanik, M.F. Large-scale in vivo femtosecond laser neurosurgery screen reveals small-molecule enhancer of regeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 18342–18347. [Google Scholar] [CrossRef] [PubMed]
- Yanik, M.F.; Cinar, H.; Cinar, H.N.; Chisholm, A.D.; Jin, Y.; Ben-Yakar, A. Neurosurgery: Functional regeneration after laser axotomy. Nature 2004, 432, 822. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hayes, J.A.; Picardo, M.C.D.; del Negro, C.A. Automated cell-specific laser detection and ablation of neural circuits in neonatal brain tissue. J. Physiol. 2013, 591, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.A.; Wang, X.; del Negro, C.A. Cumulative lesioning of respiratory interneurons disrupts and precludes motor rhythms in vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 8286–8291. [Google Scholar] [CrossRef] [PubMed]
- Kalb, J.; Egelhaaf, M.; Kurtz, R. Robust integration of motion information in the fly visual system revealed by single cell photoablation. J. Neurosci. 2006, 26, 7898–7906. [Google Scholar] [CrossRef] [PubMed]
- Packer, A.M.; Russell, L.E.; Dalgleish, H.W.P.; Häusser, M. Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo. Nat. Methods 2015, 12, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Hagihara, K.M.; Murakami, T.; Yoshida, T.; Tagawa, Y.; Ohki, K. Neuronal activity is not required for the initial formation and maturation of visual selectivity. Nat. Neurosci. 2015, 18, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Wulff, P.; Wisden, W. Dissecting neural circuitry by combining genetics and pharmacology. Trends Neurosci. 2005, 28, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Fellin, T.; Halassa, M.M. Spatial light modu-lators for complex spatio-temporal illumination of neuronal networks. Neuronal Netw. Anal. 2012, 67, 61–68. [Google Scholar]
- Nishimura, N.; Schaffer, C.B.; Friedman, B.; Tsai, P.S.; Lyden, P.D.; Kleinfeld, D. Targeted insult to subsurface cortical blood vessels using ultrashort laser pulses: Three models of stroke. Nat. Methods 2006, 3, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, P.; Difato, F.; Massobrio, P.; Breschi, G.L.; Pasquale, V.; Levi, T.; Goldin, M.; Bornat, Y.; Tedesco, M.; Bisio, M.; et al. In vitro large-scale experimental and theoretical studies for the realization of bi-directional brain-prostheses. Front. Neural Circuits 2013, 7, 40. [Google Scholar] [CrossRef] [PubMed]
- Chase, A. Traumatic brain injury increases the risk of Parkinson disease. Nat. Rev. Neurol. 2015, 11, 184. [Google Scholar] [CrossRef] [PubMed]
- Fleminger, S.; Oliver, D.L.; Lovestone, S.; Rabe-Hesketh, S.; Giora, A. Head injury as a risk factor for Alzheimer’s disease: The evidence 10 years on; a partial replication. J. Neurol. Neurosurg. Psychiatry 2003, 74, 857–862. [Google Scholar] [CrossRef] [PubMed]
- Tilve, S.; Difato, F.; Chieregatti, E. Cofilin 1 activation prevents the defects in axon elongation and guidance induced by extracellular alpha-synuclein. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hoy, C.L.; Ferhanoglu, O.; Yildirim, M.; Karajanagi, S.S.; Chan, K.M.C.; Kobler, J.B.; Zeitels, S.M.; Ben-Yakar, A. Clinical ultrafast laser surgery: Recent advances and future directions. IEEE J. Sel. Top. Quantum Electron. 2014, 20, 242–255. [Google Scholar] [CrossRef]
- Van Dam, G.M.; Themelis, G.; Crane, L.M.A.; Harlaar, N.J.; Pleijhuis, R.G.; Kelder, W.; Sarantopoulos, A.; de Jong, J.S.; Arts, H.J.G.; van der Zee, A.G.J.; et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-α targeting: First in-human results. Nat. Med. 2011, 17, 1315–1319. [Google Scholar] [CrossRef] [PubMed]
- Medvid, R.; Ruiz, A.; Komotar, R.J.; Jagid, J.R.; Ivan, M.E.; Quencer, R.M.; Desai, M.B. Current applications of MRI-guided laser interstitial thermal therapy in the treatment of brain neoplasms and epilepsy: A radiologic and neurosurgical overview. Am. J. Neuroradiol. 2015, 36, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Cash, S.S.; Hochberg, L.R. The emergence of single neurons in clinical neurology. Neuron 2015, 86, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Spruston, N.; Schiller, Y.; Stuart, G.; Sakmann, B. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 1995, 268, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Leary, S.P.; Liu, C.Y.; Apuzzo, M.L.J. Toward the emergence of nanoneurosurgery: Part III—Nanomedicine: Targeted nanotherapy, nanosurgery, and progress toward the realization of nanoneurosurgery. Neurosurgery 2006, 58, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.C.; Hawkes, E.; Keller, C.G.; Sretavan, D.W. Axon repair: Surgical application at a subcellular scale. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Zeigler, M.B.; Chiu, D.T. Single-cell nanosurgery. Methods Mol. Biol. 2013, 991, 139–148. [Google Scholar] [PubMed]
- Grundfest, W.S.; Litvack, I.F.; Goldenberg, T.; Sherman, T.; Morgenstern, L.; Carroll, R.; Fishbein, M.; Forrester, J.; Margitan, J.; McDermid, S. Pulsed ultraviolet lasers and the potential for safe laser angioplasty. Am. J. Surg. 1985, 150, 220–226. [Google Scholar] [CrossRef]
- Berns, M.; Aist, J.; Edwards, J.; Strahs, K.; Girton, J.; McNeill, P.; Rattner, J.; Kitzes, M.; Hammer-Wilson, M.; Liaw, L.; et al. Laser Microsurgery in Cell and Developmental Biology. Science 1981, 213, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.S.C.; Lin, A.P.; Koerte, I.K.; Pasternak, O.; Liao, H.; Merugumala, S.; Bouix, S.; Shenton, M.E. Neuroimaging in repetitive brain trauma. Alzheimer’s Res. Ther. 2014, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, P.; Goldin, M.; Picardo, M.A.; Jorquera, I.; Cattani, A.; Bianconi, G.; Represa, A.; Ben-Ari, Y.; Cossart, R. GABAergic hub neurons orchestrate synchrony in developing hippocampal networks. Science 2009, 326, 1419–1424. [Google Scholar] [CrossRef] [PubMed]
- Yaron, P.; Menahem, S.; Elisha, M. Network synchronization in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2016, 113, 3341–3346. [Google Scholar]
- Flusberg, B.A.; Nimmerjahn, A.; Cocker, E.D.; Mukamel, E.A.; Barretto, R.P.J.; Ko, T.H.; Burns, L.D.; Jung, J.C.; Schnitzer, M.J. High-speed, miniaturized fluorescence microscopy in freely moving mice. Nat. Methods 2008, 5, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Witte, S.; Negrean, A.; Lodder, J.C.; de Kock, C.P.J.; Testa Silva, G.; Mansvelder, H.D.; Louise Groot, M. Label-free live brain imaging and targeted patching with third-harmonic generation microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 5970–5975. [Google Scholar] [CrossRef] [PubMed]
- Richards, L.M.; Towle, E.L.; Fox, D.J.; Dunn, A.K. Intraoperative laser speckle contrast imaging with retrospective motion correction for quantitative assessment of cerebral blood flow. Neurophotonics 2014, 1, 015006. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.T.; Dsouza, A.V.; Davis, S.C.; Olson, J.D.; Paulsen, K.D.; Roberts, D.W.; Pogue, B.W. Review of fluorescence guided surgery visualization and overlay techniques. Biomed. Opt. Express 2015, 6, 3765–3782. [Google Scholar] [CrossRef] [PubMed]
- Ben-Yakar, A.; Hoy, C. Targeted transfection by femtosecond laser. SPIE Newsroom 2008, 418, 290–291. [Google Scholar]
- Juhasz, T.; Djotyan, G.; Loesel, F.H.; Kurtz, R.M.; Horvath, C.; Bille, J.F.; Mourou, G. Applications of femtosecond lasers in corneal surgery. Laser Phys. 2000, 10, 495–500. [Google Scholar]
- Beal, E. Epilepsy: Single-neuron activity in epilepsy. Nat. Rev. Neurol. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Aviv, M.S.; Pesce, M.; Tilve, S.; Chieregatti, E.; Zalevsky, Z.; Difato, F. Motility flow and growth-cone navigation analysis during in vitro neuronal development by long-term bright-field imaging. J. Biomed. Opt. 2013, 18, 111415. [Google Scholar] [CrossRef] [PubMed]
- Difato, F.; Schibalsky, L.; Benfenati, F.; Blau, A. Integration of optical manipulation and electrophysiological tools to modulate and record activity in neural networks. Int. J. Optomechatron. 2011, 5, 191–216. [Google Scholar] [CrossRef]
- Frega, M.; Pasquale, V.; Tedesco, M.; Marcoli, M.; Contestabile, A.; Nanni, M.; Bonzano, L.; Maura, G.; Chiappalone, M. Cortical cultures coupled to Micro-Electrode arrays: A novel approach to perform in vitro excitotoxicity testing. Neurotoxicol. Teratol. 2012, 34, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Crépel, V.; Aronov, D.; Jorquera, I.; Represa, A.; Ben-Ari, Y.; Cossart, R. A Parturition-associated nonsynaptic coherent activity pattern in the developing hippocampus. Neuron 2007, 54, 105–120. [Google Scholar] [CrossRef] [PubMed]
- Takano, H.; McCartney, M.; Ortinski, P.I.; Putt, M.E.; Coulter, D.A. Deterministic and stochastic neuronal contributions to distinct synchronous CA3 network bursts. J. Neurosci. 2012, 32, 4743–4754. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.



© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soloperto, A.; Bisio, M.; Palazzolo, G.; Chiappalone, M.; Bonifazi, P.; Difato, F. Modulation of Neural Network Activity through Single Cell Ablation: An in Vitro Model of Minimally Invasive Neurosurgery. Molecules 2016, 21, 1018. https://doi.org/10.3390/molecules21081018
Soloperto A, Bisio M, Palazzolo G, Chiappalone M, Bonifazi P, Difato F. Modulation of Neural Network Activity through Single Cell Ablation: An in Vitro Model of Minimally Invasive Neurosurgery. Molecules. 2016; 21(8):1018. https://doi.org/10.3390/molecules21081018
Chicago/Turabian StyleSoloperto, Alessandro, Marta Bisio, Gemma Palazzolo, Michela Chiappalone, Paolo Bonifazi, and Francesco Difato. 2016. "Modulation of Neural Network Activity through Single Cell Ablation: An in Vitro Model of Minimally Invasive Neurosurgery" Molecules 21, no. 8: 1018. https://doi.org/10.3390/molecules21081018
APA StyleSoloperto, A., Bisio, M., Palazzolo, G., Chiappalone, M., Bonifazi, P., & Difato, F. (2016). Modulation of Neural Network Activity through Single Cell Ablation: An in Vitro Model of Minimally Invasive Neurosurgery. Molecules, 21(8), 1018. https://doi.org/10.3390/molecules21081018