
Process of Fragment-Based Lead Discovery—A Perspective from NMR

Abstract
: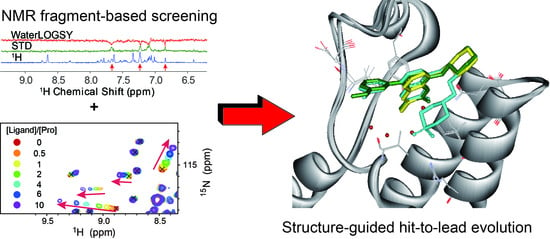
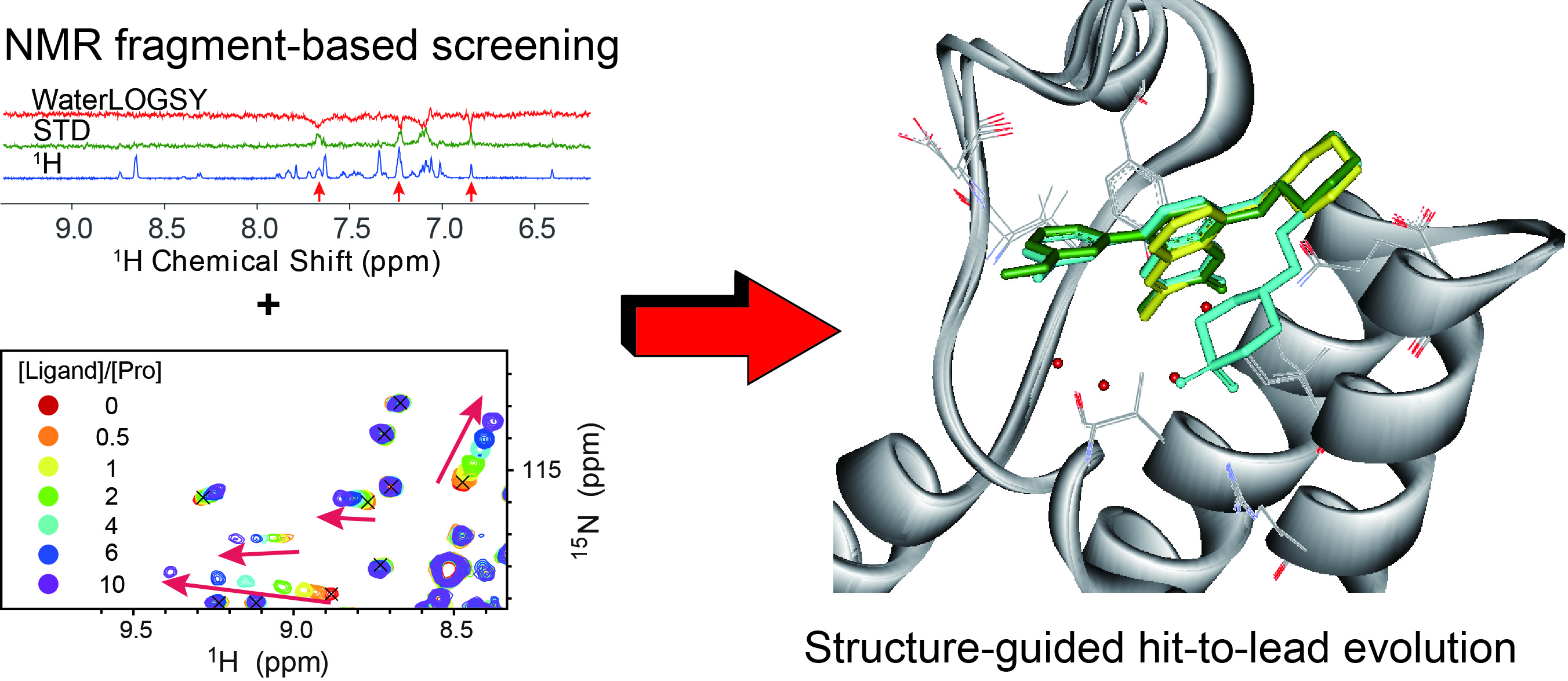{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Prioritization of Targets
3. NMR Fragment Library
4. NMR Methods for Fragment-Based Screening
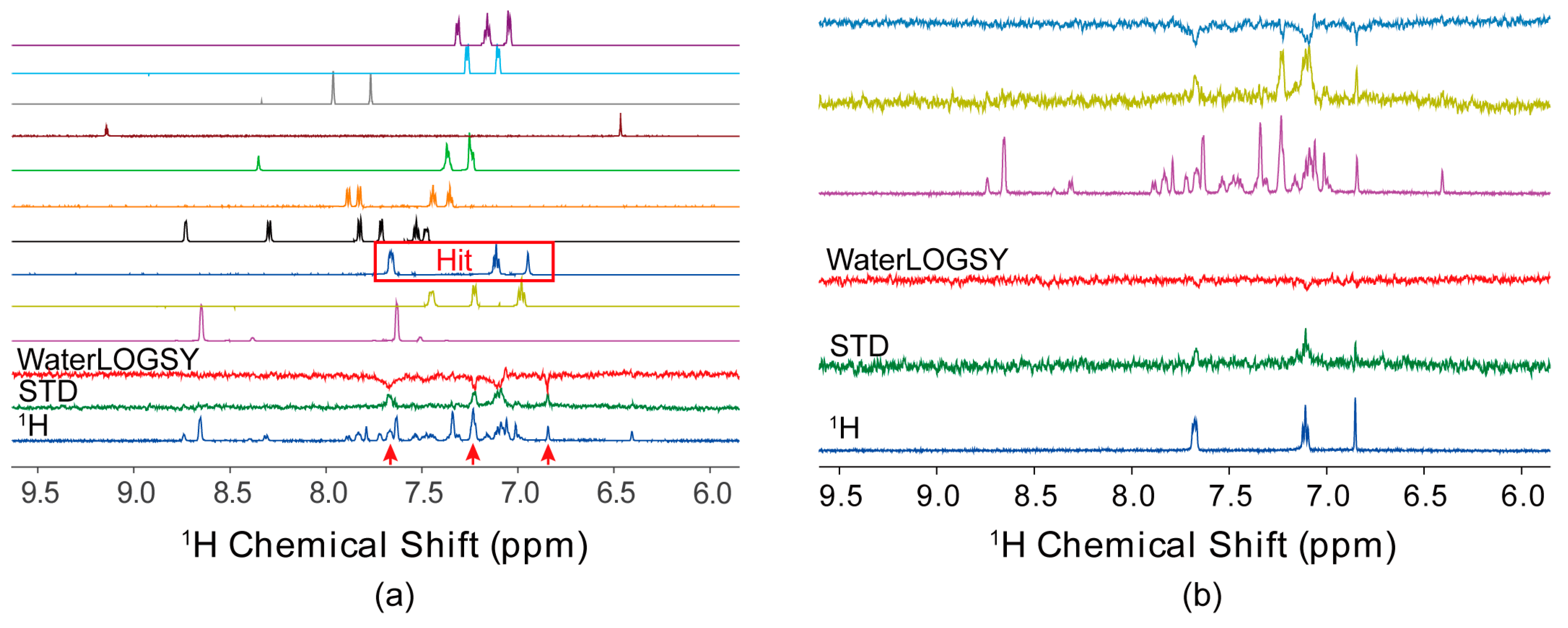
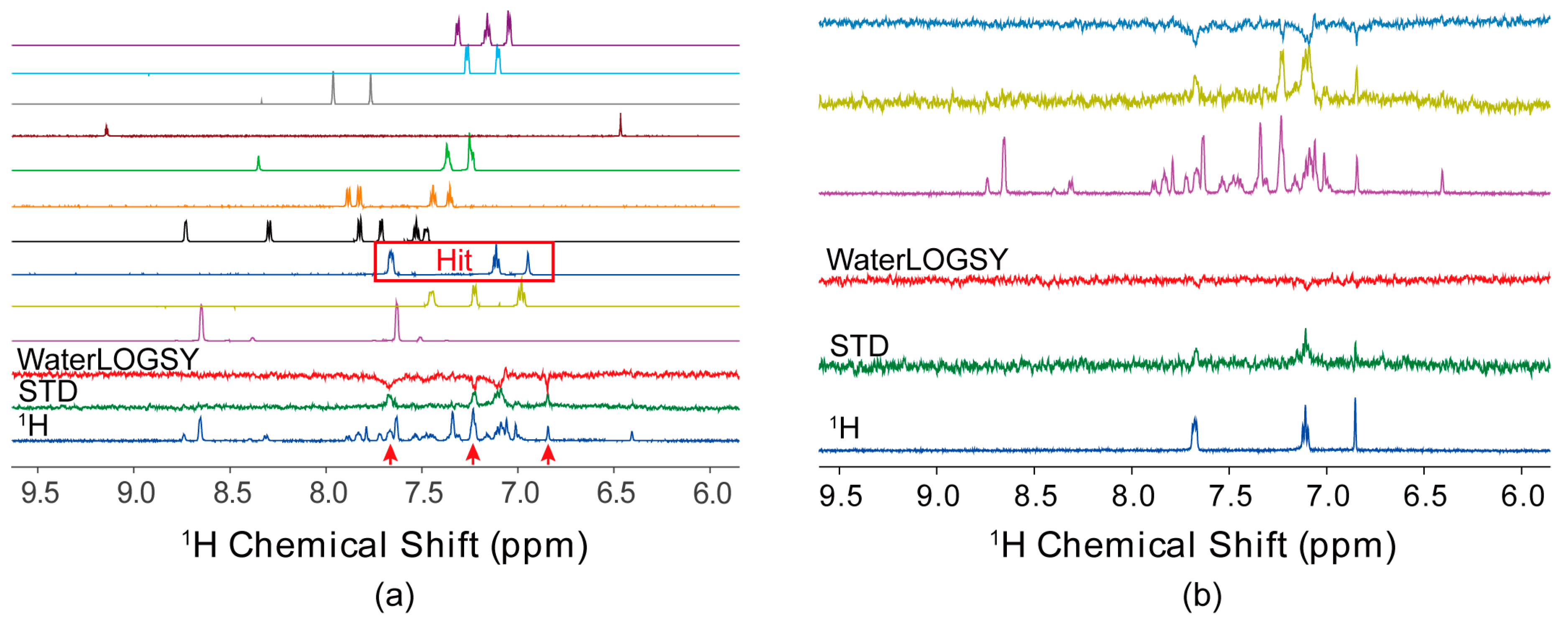
4.1. Ligand-Observed NMR Fragment-Based Screening
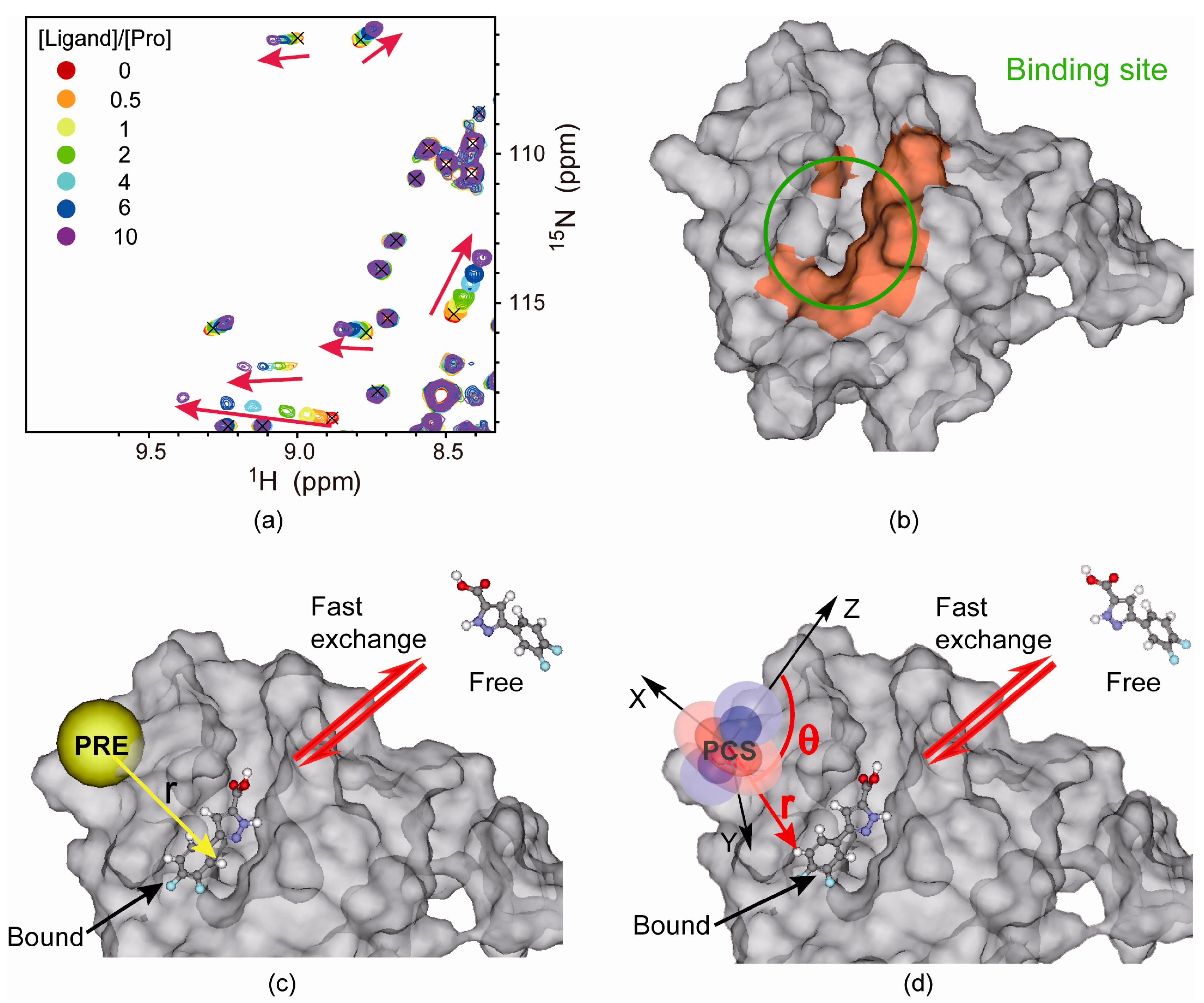
4.2. Protein-Observed NMR Fragment-Based Screening
5. Generation of Structural Models from Sparse NMR Restraints
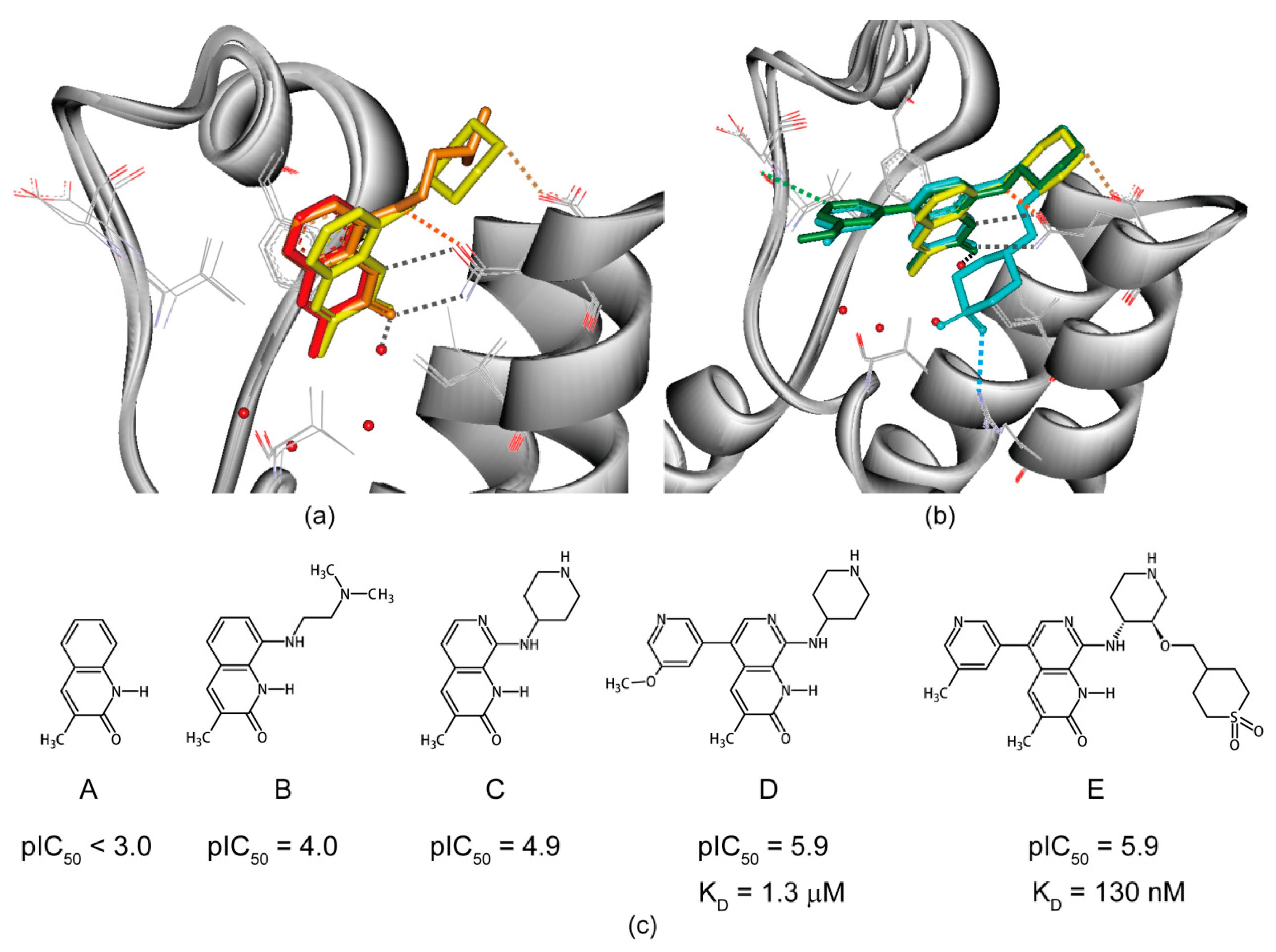
6. Structure-Guided Hit-to-Lead Evolution
7. Summary and Outlook
Acknowledgments
Conflicts of Interest
References
- Shuker, S.B.; Hajduk, P.J.; Meadows, R.P.; Fesik, S.W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Fragment-based lead discovery grows up. Nat. Rev. Drug Discov. 2013, 12, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Petros, A.M.; Dinges, J.; Augeri, D.J.; Baumeister, S.A.; Betebenner, D.A.; Bures, M.G.; Elmore, S.W.; Hajduk, P.J.; Joseph, M.K.; Landis, S.K.; et al. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J. Med. Chem. 2006, 49, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N. Eng. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Orita, M.; Warizaya, M.; Amano, Y.; Ohno, K.; Niimi, T. Advances in fragment-based drug discovery platforms. Exp. Opin. Drug Discov. 2009, 4, 1125–1144. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.W.; Verdonk, M.L.; Rees, D.C. Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 2012, 33, 224–232. [Google Scholar] [CrossRef] [PubMed]
- Fink, T.; Reymond, J.-L. Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: Assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J. Chem. Inf. Model. 2007, 47, 342–353. [Google Scholar] [PubMed]
- Fink, T.; Bruggesser, H.; Reymond, J.L. Virtual exploration of the small-molecule chemical universe below 160 daltons. Angew. Chem. Int. Ed. 2005, 44, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Hesterkamp, T.; Whittaker, M. Fragment-based activity space: Smaller is better. Curr. Opin. Chem. Biol. 2008, 12, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Burrows, J.N.; Griffen, E. Lead discovery and the concepts of complexity and lead-likeness in the evolution of drug candidates. Compr. Med. Chem. II 2006, 4, 435–458. [Google Scholar]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Abad-Zapatero, C. Ligand efficiency indices for effective drug discovery. Exp. Opin. Drug Discov. 2007, 2, 469–488. [Google Scholar] [CrossRef] [PubMed]
- Neumann, T.; Junker, H.D.; Schmidt, K.; Sekul, R. Spr-based fragment screening: Advantages and applications. Curr. Top. Med. Chem. 2007, 7, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Schade, M. NMR fragment screening: Advantages and applications. IDrugs 2006, 9, 110–113. [Google Scholar] [PubMed]
- Schade, M.; Oschkinat, H. NMR fragment screening: Tackling protein–protein interaction targets. Curr. Opin. Drug Discov. Dev. 2005, 8, 365–373. [Google Scholar]
- Harner, M.J.; Frank, A.O.; Fesik, S.W. Fragment-based drug discovery using NMR spectroscopy. J. Biomol. NMR 2013, 56, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Jhoti, H.; Cleasby, A.; Verdonk, M.; Williams, G. Fragment-based screening using X-ray crystallography and NMR spectroscopy. Curr. Opin Chem. Biol. 2007, 11, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Klein, J.; Mayer, M.; Meinecke, R.; Moller, H.; Neffe, A.; Schuster, O.; Wulfken, J.; Ding, Y.; Knaie, O.; et al. Saturation transfer difference NMR spectroscopy for identifying ligand epitopes and binding specificities. Leucoc. Traffick. 2004, 44, 149–167. [Google Scholar]
- Lucas, L.H.; Price, K.E.; Larive, C.K. Epitope mapping and competitive binding of hsa drug site II ligands by NMR diffusion measurements. J. Am. Chem. Soc. 2004, 126, 14258–14266. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fogliatto, G.; Stewart, A.; Veronesi, M.; Stockman, B. Waterlogsy as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.; Walters, M.A. Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.E.; Ehebauer, M.T.; Pukala, T.; Marsh, M.; Blundell, T.L.; Venkitaraman, A.R.; Abell, C.; Hyvoenen, M. Using a fragment-based approach to target protein–protein interactions. ChemBioChem 2013, 14, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.F.; Pannifer, A. Using fragment-based technologies to target protein–protein interactions. Curr. Pharm. Des. 2012, 18, 4685–4696. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for high-hanging fruit in drug discovery at protein–protein interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Magee, T.V. Progress in discovery of small-molecule modulators of protein–protein interactions via fragment screening. Bioorg. Med. Chem. Lett. 2015, 25, 2461–2468. [Google Scholar] [CrossRef] [PubMed]
- Arkin, M.R.; Tang, Y.; Wells, J.A. Small-molecule inhibitors of protein–protein interactions: Progressing toward the reality. Chem. Biol. 2014, 21, 1102–1114. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Wisniewski, J.A.; Ji, H. Hot spot-based design of small-molecule inhibitors for protein–protein interactions. Bioorg. Med. Chem. Lett. 2014, 24, 2546–2554. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of bet bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 339–358. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. Bet bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of bet bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.-W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of bet recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H.; Fairlamb, A.H. Target assessment for antiparasitic drug discovery. Trends Parasit. 2007, 23, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Hann, M.M.; Leach, A.R.; Harper, G. Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 2001, 41, 856–864. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ‘rule of three’ for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Hajduk, P.J. Fragment-based drug design: How big is too big? J. Med. Chem. 2006, 49, 6972–6976. [Google Scholar] [PubMed]
- Gao, J.; Ma, R.; Wang, W.; Wang, N.; Sasaki, R.; Snyderman, D.; Wu, J.; Ruan, K. Automated NMR fragment based screening identified a novel interface blocker to the LARG/RhoA complex. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Caronni, D.; Mongelli, N.; Veronesi, M.; Vulpetti, A. NMR-based quality control approach for the identification of false positives and false negatives in high throughput screening. Curr. Drug Discov. Technol. 2006, 3, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Navratilova, I.; Hopkins, A.L. Fragment screening by surface plasmon resonance. ACS Med. Chem. Lett. 2010, 1, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, C.A.; Hopkins, A.L.; Navratilova, I. Fragment screening by spr and advanced application to gpcrs. Prog. Biophys. Mol. Biol. 2014, 116, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Campos-Olivas, R. NMR screening and hit validation in fragment based drug discovery. Curr. Top. Med. Chem. 2011, 11, 43–67. [Google Scholar] [CrossRef] [PubMed]
- Chilingaryan, Z.; Yin, Z.; Oakley, A.J. Fragment-based screening by protein crystallography: Successes and pitfalls. Int. J. Mol. Sci. 2012, 13, 12857–12879. [Google Scholar] [CrossRef] [PubMed]
- Caliandro, R.; Belviso, D.B.; Aresta, B.M.; de Candia, M.; Altomare, C.D. Protein crystallography and fragment-based drug design. Future Med. Chem. 2013, 5, 1121–1140. [Google Scholar] [CrossRef] [PubMed]
- Recht, M.I.; Nienaber, V.; Torres, F.E. Fragment-based screening for enzyme inhibitors using calorimetry. Methods Enzym. 2016, 567, 47–69. [Google Scholar]
- Wienken, C.J.; Baaske, P.; Rothbauer, U.; Braun, D.; Duhr, S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef] [PubMed]
- Seidel, S.A.I.; Wienken, C.J.; Geissler, S.; Jerabek-Willemsen, M.; Duhr, S.; Reiter, A.; Trauner, D.; Braun, D.; Baaske, P. Label-free microscale thermophoresis discriminates sites and affinity of protein–ligand binding. Angew. Chem. Int. Ed. 2012, 51, 10656–10659. [Google Scholar] [CrossRef] [PubMed]
- Nierode, G.; Kwon, P.S.; Dordick, J.S.; Kwon, S.-J. Cell-based assay design for high-content screening of drug candidates. J. Microb. Biotech. 2016, 26, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Wagner, G. NMR studies of protein interactions. Curr. Opin. Struct. Biol. 2006, 16, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.; Meyer, B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 2001, 123, 6108–6117. [Google Scholar] [CrossRef] [PubMed]
- Begley, D.W.; Moen, S.O.; Pierce, P.G.; Zartler, E.R. Saturation transfer difference NMR for fragment screening. Curr. Prot. Chem. Biol. 2013, 5, 251–268. [Google Scholar]
- Hu, J.; Eriksson, P.O.; Kerna, G. Aroma waterlogsy: A fast and sensitive screening tool for drug discovery. Mag. Res. Chem. 2010, 48, 909–911. [Google Scholar] [CrossRef] [PubMed]
- Gossert, A.D.; Henry, C.; Blommers, M.J.J.; Jahnke, W.; Fernandez, C. Time efficient detection of protein–ligand interactions with the polarization optimized po-waterlogsy NMR experiment. J. Biomol. NMR 2009, 43, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Olejniczak, E.T.; Fesik, S.W. One-dimensional relaxation- and diffusion-edited NMR methods for screening compounds that bind to macromolecules. J. Am. Chem. Soc. 1997, 119, 12257–12261. [Google Scholar] [CrossRef]
- Stebbins, J.L.; Jung, D.W.; Leone, M.; Zhang, X.K.; Pellecchia, M. A structure-based approach to retinoid x receptor-α inhibition. J. Biol. Chem. 2006, 281, 16643–16648. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Fagerness, P.E.; Hadden, D.T.A.; Sarver, R.W.; Stockman, B.J. Fluorine-NMR experiments for high-throughput screening: Theoretical aspects, practical considerations, and range of applicability. J. Am. Chem. Soc. 2003, 125, 7696–7703. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Flocco, M.; Veronesi, M.; Stockman, B.J. Fluorine-NMR competition binding experiments for high-throughput screening of large compound mixtures. Comb. Chem. High Throughput Screen. 2002, 5, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part II. Experimental aspects. Concepts Magn. Reson. 1998, 10, 197–237. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part 1. Basic theory. Concepts Magn. Reson. 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Jahnke, W.; Rudisser, S.; Zurini, M. Spin label enhanced NMR screening. J. Am. Chem. Soc. 2001, 123, 3149–3150. [Google Scholar] [CrossRef] [PubMed]
- Vanwetswinkel, S.; Heetebrij, R.J.; van Duynhoven, J.; Hollander, J.G.; Filippov, D.V.; Hajduk, P.J.; Siegal, G. Tins, target immobilized NMR screening: An efficient and sensitive method for ligand discovery. Chem. Biol. 2005, 12, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Dalvit, C.; Mongelli, N.; Papeo, G.; Giordano, P.; Veronesi, M.; Moskau, D.; Kummerle, R. Sensitivity improvement in 19F NMR-based screening experiments: Theoretical considerations and experimental applications. J. Am. Chem. Soc. 2005, 127, 13380–13385. [Google Scholar] [CrossRef] [PubMed]
- Fattorusso, R.; Jung, D.W.; Crowell, K.J.; Forino, M.; Pellecchia, M. Discovery of a novel class of reversible non-peptide caspase inhibitors via a structure-based approach. J. Med. Chem. 2005, 48, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Frommlet, A.; Perez, M.; Cobas, C.; Blechschmidt, A.; Dominguez, S.; Lingel, A. Fast and efficient fragment-based lead generation by fully automated processing and analysis of ligand-observed NMR binding data. J. Med. Chem. 2016, 59, 3303–3310. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spec. 2013, 73, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Williams, J.G.; Campbell, S.L. Protein–protein interaction analysis by nuclear magnetic resonance spectroscopy. Methods Mol. Biol. 2004, 261, 79–92. [Google Scholar] [PubMed]
- Chi, S.W.; Muto, Y.; Inoue, M.; Kim, I.; Sakamoto, H.; Shimura, Y.; Yokoyama, S.; Choi, B.S.; Kim, H. Chemical shift perturbation studies of the interactions of the second RNA-binding domain of the drosophila sex-lethal protein with the transformer pre-mRNA polyuridine tract and 3′ splice-site sequences. Eur. J. Biochem. 1999, 260, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Schanda, P.; Kupce, E.; Brutscher, B. Sofast-hmqc experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 2005, 33, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Medek, A.; Hajduk, P.J.; Mack, J.; Fesik, S.W. The use of differential chemical shifts for determining the binding site location and orientation of protein-bound ligands. J. Am. Chem. Soc. 2000, 122, 1241–1242. [Google Scholar] [CrossRef]
- Gonzalez-Ruiz, D.; Gohlke, H. Steering protein–ligand docking with quantitative NMR chemical shift perturbations. J. Chem. Inf. Model. 2009, 49, 2260–2271. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.; Powers, R. Rapid protein–ligand costructures using chemical shift perturbations. J. Am. Chem. Soc. 2008, 130, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Li, D.W.; Levy, L.A.; Gabel, S.A.; Lebetkin, M.S.; DeRose, E.F.; Wall, M.J.; Howell, E.E.; London, R.E. Interligand overhauser effects in type II dihydrofolate reductase. Biochemistry 2001, 40, 4242–4252. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, Z.; Stebbins, J.L.; Zhang, X.; Hoffman, R.; Moore, A.; Pellecchia, M. A fragment-based approach for the discovery of isoform-specific p38α inhibitors. ACS Chem. Biol. 2007, 2, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Sledz, P.; Silvestre, H.L.; Hung, A.W.; Ciulli, A.; Blundell, T.L.; Abell, C. Optimization of the interligand overhauser effect for fragment linking: Application to inhibitor discovery against mycobacterium tuberculosis pantothenate synthetase. J. Am. Chem. Soc. 2010, 132, 4544–4545. [Google Scholar] [CrossRef] [PubMed]
- Rega, M.F.; Wu, B.; Wei, J.; Zhang, Z.; Cellitti, J.F.; Pellecchia, M. SAR by interligand nuclear overhauser effects (iloes) based discovery of acylsulfonamide compounds active against Bcl-xL and Mcl-1. J. Med. Chem. 2011, 54, 6000–6013. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.; Scheraga, H.A. Use of the transferred nuclear overhauser effect to determine the conformations of ligands bound to proteins. Acc. Chem. Res. 1994, 27, 257–264. [Google Scholar] [CrossRef]
- Clore, G.M.; Iwahara, J. Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 2009, 109, 4108–4139. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.; Schwieters, C.D.; Clore, G.M. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature 2007, 449, 1078–1082. [Google Scholar]
- Tang, C.; Iwahara, J.; Clore, G.M. Visualization of transient encounter complexes in protein–protein association. Nature 2006, 444, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Gochin, M.; Zhou, G.; Phillips, A.H. Paramagnetic relaxation assisted docking of a small indole compound in the HIV-1 gp41 hydrophobic pocket. ACS Chem. Biol. 2011, 6, 267–274. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz, L.; Nguyen, T.H.; Ozawa, K.; Shin, J.; Graham, B.; Huber, T.; Otting, G. Binding of low molecular weight inhibitors promotes large conformational changes in the dengue virus NS2B-NS3 protease: Fold analysis by pseudocontact shifts. J. Am. Chem. Soc. 2011, 133, 19205–19215. [Google Scholar] [CrossRef] [PubMed]
- Pintacuda, G.; John, M.; Su, X.-C.; Otting, G. NMR structure determination of protein–ligand complexes by lanthanide labeling. Acc. Chem. Res. 2007, 40, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Otting, G. Protein NMR using paramagnetic ions. Ann. Rev. Biophys. 2010, 39, 387–405. [Google Scholar] [CrossRef] [PubMed]
- Saio, T.; Ogura, K.; Shimizu, K.; Yokochi, M.; Burke, T.R., Jr.; Inagaki, F. An NMR strategy for fragment-based ligand screening utilizing a paramagnetic lanthanide probe. J. Biomol. NMR 2011, 51, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.-Y.; Keizers, P.H.J.; Liu, W.-M.; Loehr, F.; Skinner, S.P.; Heeneman, E.A.; Schwalbe, H.; Ubbink, M.; Siegal, G. Small-molecule binding sites on proteins established by paramagnetic NMR spectroscopy. J. Am. Chem. Soc. 2013, 135, 5859–5868. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Salum, L.B.; Abraham, D.J. Structure-based drug design strategies in medicinal chemistry. Curr. Top. Med. Chem. 2009, 9, 771–790. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Greer, J. A decade of fragment-based drug design: Strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Huth, J.R.; Park, C.; Petros, A.M.; Kunzer, A.R.; Wendt, M.D.; Wang, X.L.; Lynch, C.L.; Mack, J.C.; Swift, K.M.; Judge, R.A.; et al. Discovery and design of novel HSP90 inhibitors using multiple fragment-based design strategies. Chem. Biol. Drug Des. 2007, 70, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barile, E.; Pellecchia, M. NMR-based approaches for the identification and optimization of inhibitors of protein–protein interactions. Chem. Rev. 2014, 114, 4749–4763. [Google Scholar] [CrossRef] [PubMed]
- Philpott, M.; Yang, J.; Tumber, T.; Fedorov, O.; Uttarkar, S.; Filippakopoulos, P.; Picaud, S.; Keates, T.; Felletar, I.; Ciulli, A.; et al. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol. Biosyst. 2011, 7, 2899–2908. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-W.; Dean, A.W.; Woolven, J.M.; Bamborough, P. Fragment-based discovery of bromodomain inhibitors part 1: Inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2012, 55, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, D.; Chen, T.; Wang, Y.; Miao, Z.; Xu, Y.; Chen, W.; Wang, X.; Li, Y.; Du, Z.; et al. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. J. Med. Chem. 2013, 56, 3833–3851. [Google Scholar] [CrossRef] [PubMed]
- Vidler, L.R.; Brown, N.; Knapp, S.; Hoelder, S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012, 55, 7346–7359. [Google Scholar] [CrossRef] [PubMed]
- Harner, M.J.; Chauder, B.A.; Phan, J.; Fesik, S.W. Fragment-based screening of the bromodomain of ATAD2. J. Med. Chem. 2014, 57, 9687–9692. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Petros, A.M.; Fedorov, O.; Xu, J.; Knapp, S. Structure-based approaches towards identification of fragments for the low-druggability ATAD2 bromodomain. MedChemComm 2014, 5, 1843–1848. [Google Scholar] [CrossRef]
- Demont, E.H.; Chung, C.-W.; Furze, R.C.; Grandi, P.; Michon, A.-M.; Wellaway, C.; Barrett, N.; Bridges, A.M.; Craggs, P.D.; Diallo, H.; et al. Fragment-based discovery of low-micromolar ATAD2 bromodomain inhibitors. J. Med. Chem. 2015, 58, 5649–5673. [Google Scholar] [CrossRef] [PubMed]
- Bamborough, P.; Chung, C.-W.; Furze, R.C.; Grandi, P.; Michon, A.-M.; Sheppard, R.J.; Barnett, H.; Diallo, H.; Dixon, D.P.; Douault, C.; et al. Structure-based optimization of naphthyridones into potent ATAD2 bromodomain inhibitors. J. Med. Chem. 2015, 58, 6151–6178. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, R.; Wang, P.; Wu, J.; Ruan, K. Process of Fragment-Based Lead Discovery—A Perspective from NMR. Molecules 2016, 21, 854. https://doi.org/10.3390/molecules21070854
Ma R, Wang P, Wu J, Ruan K. Process of Fragment-Based Lead Discovery—A Perspective from NMR. Molecules. 2016; 21(7):854. https://doi.org/10.3390/molecules21070854
Chicago/Turabian StyleMa, Rongsheng, Pengchao Wang, Jihui Wu, and Ke Ruan. 2016. "Process of Fragment-Based Lead Discovery—A Perspective from NMR" Molecules 21, no. 7: 854. https://doi.org/10.3390/molecules21070854
APA StyleMa, R., Wang, P., Wu, J., & Ruan, K. (2016). Process of Fragment-Based Lead Discovery—A Perspective from NMR. Molecules, 21(7), 854. https://doi.org/10.3390/molecules21070854