Experiments involving moisture and/or air sensitive components were performed under a positive pressure of nitrogen in oven-dried glassware equipped with a rubber septum inlet. Dried solvents and liquid reagents were transferred by oven-dried syringes or hypodermic syringe cooled to ambient temperature in a desiccator. Reaction mixtures were stirred in 10 mL sample vials with Teflon-coated magnetic stirring bars unless otherwise stated. Moisture in non-volatile reagents/compounds was removed in high vacuum by means of an oil pump and subsequent purging with nitrogen. Solvents were removed in vacuo under ~30 mmHg and heated with a water bath at 30–35 °C using a rotary evaporator with an aspirator. The condenser was cooled with running water at 0 °C.
All experiments were monitored by analytical thin layer chromatography (TLC). TLC was performed on pre-coated plates, 60 F254. After elution, each plate was visualized under UV illumination at 254 nm for UV-active material. Further visualization was achieved by staining with KMnO4, ceric molybdate, or anisaldehyde solution. For those using the aqueous stains, the TLC plates were heated on a hot plate.
Columns for flash chromatography (FC) contained 200–300 mesh silica gel. Columns were packed as slurry of silica gel in petroleum ether and equilibrated solution using the appropriate solvent system. The elution was assisted by applying pressure of about 2 atm with an air pump.
Enantiomeric excesses were determined by chiral high-performance liquid chromatography (HPLC) analysis. UV detection was monitored at 254 nm, 230 nm, and 210 nm at the same time. HPLC samples were dissolved in HPLC grade isopropanol (IPA), unless otherwise stated.
All commercial reagents were purchased with the highest purity grade. They were used without further purification unless specified. All solvents used, mainly petroleum ether (PE) and ethyl acetate (EtOAc), were distilled. Anhydrous DCM was freshly distilled from CaH2 and stored under N2 atmosphere. tert-butylbenzene was freshly distilled from sodium/benzophenone before use. All compounds synthesized were stored in a −20 °C freezer and light-sensitive compounds were protected with aluminum foil.
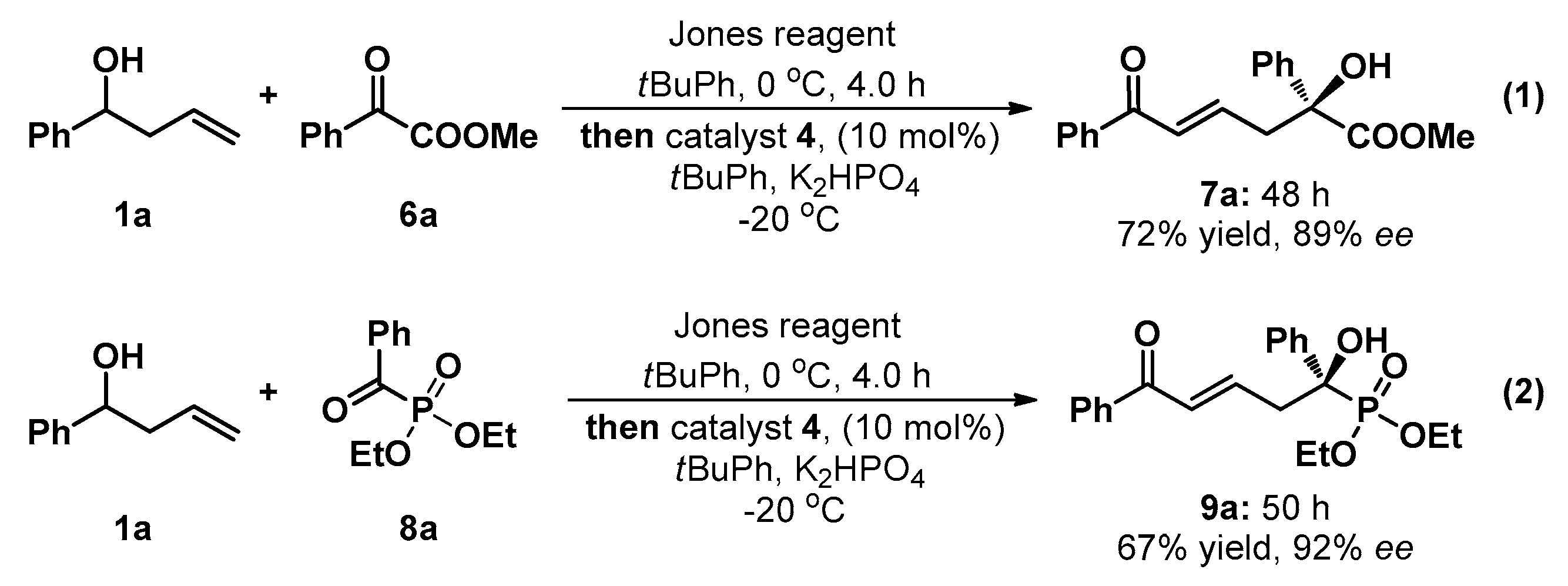
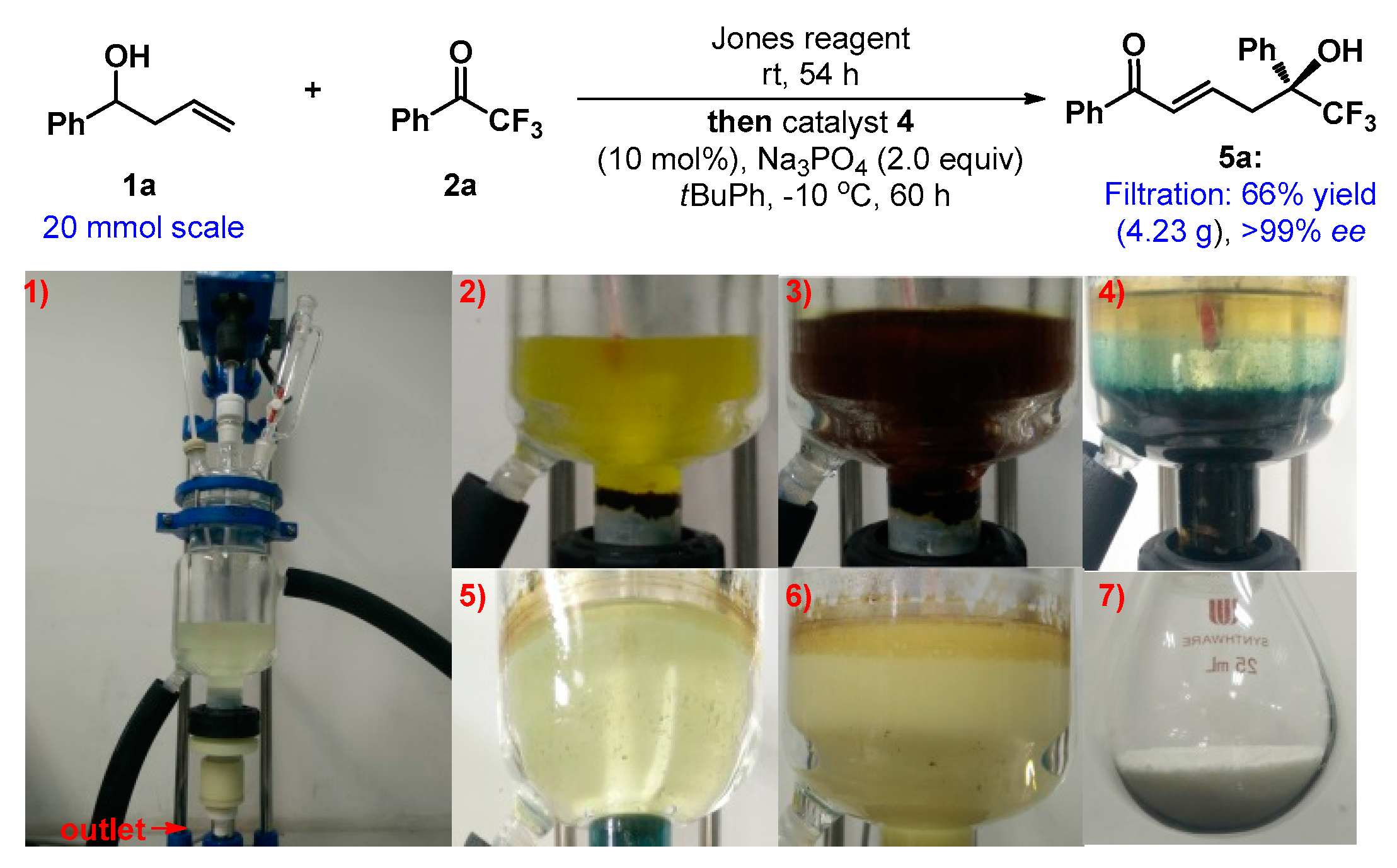
3.2. General Experimental Procedure for the One-Pot Tandem Direct Asymmetric Vinylogous Aldol Reaction of Homoallylic Alcohol 1a to Acyclic Activated Ketones (6a and 8a)
Jones reagent (0.45 mL) was added dropwise to a solution of homoallic alcohol 1a (0.4 mmol, 4 equiv.) in tert-butylbenzene (800 μL) at 0 °C over a period of 4 h. When the reaction was completed (monitored by TLC), the sample was stilled for a moment, and the aqueous phase was released. The reaction mixture was stirred at −20 °C for 10 min. Potassium phosphate anhydrous (0.2 mmol, 2.0 equiv.), catalyst 4 (0.01 mmol, 0.1 equiv.), and 6a/8a (0.1 mmol, 1.0 equiv.) were added sequentially (10 min interval). The reaction mixture was stirred at −20 °C and monitored by TLC. Upon complete consumption of acyclic activated ketones 6a/8a, the reaction mixture was directly loaded onto a short silica column, followed by gradient elution with PE/EA mixture (20/1–1/1 ratio). Removing the solvent in vacuum afforded products 7a/9a.
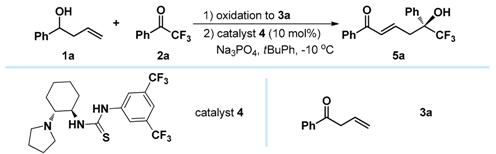
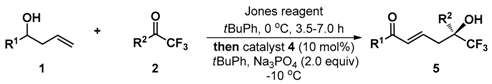
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1,5-diphenylhex-2-en-1-one (5a): White solid, Mp 133.7–135.2 °C; 25.6 mg (0.1 mmol), 80% yield; 95% ee; −36.4 (c 2.47, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.75 (d, J = 7.4 Hz, 2H), 7.59–7.51 (m, 3H), 7.46–7.33 (m, 5H), 6.91 (d, J = 15.5 Hz, 1H), 6.76–6.66 (m, 1H), 3.25–3.05 (m, 2H), 3.16 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ 190.4, 140.6, 137.1, 135.8, 133.0, 130.7, 128.9, 128.6 (two peaks), 128.5, 126.3, 125.2 (q, J = 284.0 Hz), 77.7 (q, J = 28.2 Hz), 39.0; 19F-NMR (376 MHz, CDCl3) δ −79.55; HRMS (ESI) m/z 343.0928 [M + Na+], calc. for C18H15F3O2Na 343.0922. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 10.2 min (minor) and 11.2 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1-phenyl-5-(4-(trifluoromethyl)phenyl)hex-2-en-1-one (5b): White solid, Mp 88.2–90.9 °C; 33.8 mg (0.1 mml), 87% yield; 92% ee; –26.3 (c 2.47, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.75–7.66 (m, 6H), 7.51 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.8Hz, 2H), 6.92 (d, J = 15.5 Hz, 1H), 6.75–6.65 (m, 1H), 3.59 (s, 1H), 3.17 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 190.3, 140.0, 139.7, 136.9, 133.3, 131.8, 131.3, 130.9, 130.6, 130.4, 129.2 (two peaks), 128.6, 128.2, 127.0, 126.8, 125.6 (two peaks), 125.5 (two peaks), 125.4, 123.0, 122.0, 118.4, 77.2, 76.8, 76.4, 76.0, 39.0; 19F-NMR (376 MHz, CDCl3) δ −62.77, −79.37; HRMS (ESI) m/z 411.0797 [M + Na+], calc. for C19H14F6O2Na 411.0796. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 10.9 min (minor) and 15.4 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-(4-fluorophenyl)-5-hydroxy-1-phenylhex-2-en-1-one (5c): White solid, Mp 107.0–108.3 °C; 24.0 mg (0.1 mmol), 71% yield; 94% ee; −31.0 (c 2.42, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.77 (d, 2H), 7.78–7.52 (m, 3H), 7.41 (t, J = 7.6 Hz, 2H), 7.09 (t, J = 8.64 Hz, 2H), 6.92 (d, J = 15.5 Hz, 1H), 6.76–6.66 (m, 1H), 3.47 (s, 1H), 3.22–3.04 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 190.3, 164.5 , 161.2, 140.3, 137.0, 133.2, 131.8, 131.7, 130.7, 128.6 (two peaks), 128.5 (two peaks), 128.4 (two peaks), 125.1 (q, J = 283.4 Hz), 123.2, 115.6, 115.4, 76.4 (q, J = 28.4 Hz), 38.9; 19F-NMR (376 MHz, CDCl3) δ −79.80, −112.84; HRMS (ESI) m/z 361.0829 [M + Na+], calc. for C18H14F4O2Na 361.0828. The ee was determined by HPLC analysis. CHIRALPAK IB-3 (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 11.2 min (major) and 12.6 min (minor).
(−)-(S,E)-5-(4-Chlorophenyl)-6,6,6-trifluoro-5-hydroxy-1-phenylhex-2-en-1-one (5d): White solid, Mp 110.4–111.6 °C; 34.8 mg (0.1 mmol), 98% yield; 94% ee; −46.6 (c 3.41, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.76 (d, J = 7.32 Hz, 2H), 7.53–7.50 (m, 3H), 7.44–7.37 (m, 4H), 6.91 (d, J = 15.51 Hz, 1H), 6.75–6.65 (m, 2H), 3.51 (s, 1H), 3.21–3.04 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 190.3, 140.1, 137.0, 135.0, 134.5, 133.2, 130.8, 128.8, 128.6 (two peaks), 127.9, 125.0 (q, J = 284.3 Hz), 76.5 (q, J = 28.5 Hz ), 38.9; 19F-NMR (376 MHz, CDCl3) δ −79.66; HRMS (ESI) m/z 377.0531 [M + Na+], calc. for C18H14ClF3O2Na 377.0532. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 6.8 min (minor) and 9.1 min (major).
(−)-(S,E)-5-(4-Bromophenyl)-6,6,6-trifluoro-5-hydroxy-1-phenylhex-2-en-1-one (5e): White solid, Mp 114.2–115.3 °C; 31.9 mg (0.1 mmol); 80% yield; 95% ee; −54.5 (c 1.42, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.75 (d, J = 7.4 Hz, 2H), 7.56–7.53 (m, 3H), 7.47–7.39 (m, 4H), 6.90 (d, J = 15.5 Hz, 1H), 6.74–6.65 (m, 1H), 3.50 (s, 1H), 3.20–3.03 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 190.3, 140.1, 137.0, 135.1, 133.2, 131.7, 130.8, 128.6 (two peaks), 128.2 (two peaks), 126.8, 124.9 (q, J = 284.2 Hz), 123.3, 76.5 (q, J = 28.4 Hz), 38.9; 19F-NMR (376 MHz, CDCl3) δ −79.64; HRMS (ESI) m/z 423.0009 [M + Na+], calc. for C18H14BrF3O2Na 423.0006. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 7.0 min (minor) and 12.3 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-(3-fluorophenyl)-5-hydroxy-1-phenylhex-2-en-1-one (5f): Colorless oil; 32.1 mg (0.1 mmol), 95% yield; 91% ee; −41.5 (c 2.56, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.78 (d, J = 7.3 Hz, 2H), 7.55 (t, J = 7.4 Hz, 2H), 7.45–7.32 (m, 5H), 7.08 (t, J = 7.4 Hz, 1H), 6.93 (d, J = 15.5 Hz, 1H), 6.75–6.65 (m, 1H), 3.25 (s, 1H), 3.21–3.04 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 190.3 (two peaks), 164.5, 161.2, 140.1, 140.0, 138.6, 138.5, 137.0, 133.2, 130.8, 130.2, 130.1, 128.6 (two peaks), 125.0 (q, J = 284.0 Hz), 122.0 (two peaks), 116.0, 115.7, 114.2, 113.8 (two peaks), 76.2 (two peaks), 75.8 (two peaks), 39.0; 19F-NMR (376 MHz, CDCl3) δ −79.51, −111.68; HRMS (ESI) m/z 339.1004 [M + H+], calc. for C18H15F4O2 339.1008. The ee was determined by HPLC analysis. CHIRALPAK IB-3 (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 11.3 min (major) and 13.2 min (minor).
(−)-(S,E)-5-(3-Chlorophenyl)-6,6,6-trifluoro-5-hydroxy-1-phenylhex-2-en-1-one (5g): Colorless oil; 33.3 mg (0.1 mmol), 94% yield; 90% ee; −35.6 (c 3.04, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.77 (d, J = 7.3 Hz, 2H), 7.62 (s, 1H), 7.55 (t, J = 7.4 Hz, 1H), 7.47–7.32 (m, 5H), 6.92 (d, J = 15.5 Hz, 1H), 6.74–6.64 (m, 1H), 3.54 (s, 1H), 3.21–3.05 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 190.4, 140.0, 138.1, 137.0, 134.7, 133.2, 130.9, 129.8, 129.1, 128.7, 128.6, 126.9, 126.8, 124.9 (q, J = 284.1 Hz), 124.6 (two peaks), 76.4 (q, J = 28.4 Hz), 39.0; 19F-NMR (376 MHz, CDCl3) δ −79.48; HRMS (ESI) m/z 377.0533 [M + Na+], calc. for C18H14ClF3O2 377.0532. The ee was determined by HPLC analysis. CHIRALPAK IA (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 95/05; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 9.8 min (major) and 11.8 min (minor).
(−)-(S,E)-6,6,6-Trifluoro-5-(2-fluorophenyl)-5-hydroxy-1-phenylhex-2-en-1-one (5h): White solid, Mp 106.9–107.2 °C; 24.0 mg (0.1 mmol), 71% yield; 99% ee; −26.5 (c 2.25, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.77–7.74 (m, 2H), 7.70–7.64 (m, 1H), 7.53 (t, J = 7.4 Hz, 1H), 7.42–7.34 (m, 3H), 7.21 (td, J = 7.9, 1.1 Hz, 1H), 7.13–7.06 (m, 1H), 7.00 (d, J = 15.5 Hz, 1H), 6.85–6.75 (m, 1H), 3.76 (d, J = 5.0 Hz, 1H), 3.60–3.52 (m, 1H), 3.08–3.00 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 190.5, 161.7, 158.4, 141.0, 137.2, 133.0, 131.4, 131.3, 130.4, 130 (two peaks), 128.6, 128.5, 124.9 (qd, J = 284.4, 1.7 Hz), 124.5 (two peaks), 122.4, 122.3, 116.8, 116.4, 76.2 (qd, J = 30, 3.6 Hz), 37.5, 37.4; 19F-NMR (376 MHz, CDCl3) δ −80.65, −80.69, −111.46, −111.50; HRMS (ESI) m/z 339.1004 [M + H+], calc. for C18H15F4O2 339.1008. The ee was determined by HPLC analysis. CHIRALPAK IA (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 95/05; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 9.4 min (major) and 11.9 min (minor).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1-phenyl-5-(p-tolyl)hex-2-en-1-one (5i): White solid, Mp 109.2–110.6 °C; 20.1 mg (0.1 mmol), 60% yield; 95% ee; −51.2 (c 1.32, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.78–7.75 (m, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.46–7.38 (m, 4H), 7.22 (d, J = 8.1 Hz, 2H), 6.91 (d, J = 15.5 Hz, 1H), 6.76–6.66 (m, 1H), 3.23–3.02 (m, 2H), 2.97 (d, J = 5.9 Hz, 1H), 2.37 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 190.3, 140.6, 138.8, 137.2, 133.0, 132.9, 130.7, 129.3, 128.6, 128.5, 127.1, 125.2 (q, J = 284.2 Hz), 76.7 (q, J = 28.2 Hz), 39.0, 21.0; 19F-NMR (376 MHz, CDCl3) δ −79.74; HRMS (ESI) m/z 357.1077 [(M + Na+], calc. for C19H17F3O2Na 357.1078. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 6.7 min (minor) and 10.8 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1-phenyl-5-(m-tolyl)hex-2-en-1-one (5j): White solid, Mp 78.5–80.4 °C; 22.4 mg (0.1 mmol), 67% yield; 94% ee; −45.8 (c 1.84, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.78–7.74 (m, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.43–7.28 (m, 5H), 7.19 (d, J = 7.2 Hz, 1H), 6.92 (d, J = 15.5 Hz, 1H), 6.76–6.66 (m, 1H), 3.24–3.06 (m, 2H), 3.02, (s, 1H) 2.38 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 190.4, 140.7, 138.3, 137.2, 135.8, 133.0, 130.8, 129.7, 128.7, 128.6, 128.5, 127.0 (two peaks), 125.2 (q, J = 284.0 Hz), 123.4 (two peaks), 77.7 (q, J = 28.2 Hz), 39.1, 21.6; 19F-NMR (376 MHz, CDCl3) δ −79.99; HRMS (ESI) m/z 357.1087 [M + Na+], calc. for C19H17F3O2Na 357.1078. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 9.5 min (minor) and 11.0 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-5-(naphthalen-2-yl)-1-phenylhex-2-en-1-one (5k): White solid, Mp 163.1–164.9 °C; 25.9 mg (0.1 mmol), 70% yield; 91% ee; −40.1 (c 0.87, CHCl3); 1H-NMR (300 MHz, acetone-d6) δ 8.30 (s, 1H), 8.00–7.92 (m, 3H), 7.83 (d, J = 8.8 Hz, 1H), 7.72 (d, J = 7.9 Hz, 2H), 7.58–7.51 (m, 3H), 7.36 (t, J = 7.7 Hz, 2H), 7.15 (d, J = 15.5 Hz, 1H), 6.76–6.67 (m, 1H), 6.13 (s, 1H), 3.63–3.56 (m, 1H), 3.31–3.24 (m, 1H); 13C-NMR (75 MHz, acetone-d6) δ 190.1, 141.7, 138.4, 135.2, 134.0, 133.9, 133.6, 131.1, 129.4, 129.3, 129.2, 128.8, 128.4, 127.8, 127.6, 127.3, 126.9 (q, J = 284.2 Hz), 77.5 (q, J = 27.5 Hz), 38.8; 19F-NMR (376 MHz, CDCl3) δ −79.23; HRMS (ESI) m/z 371.1257 [M + H+], calc. for C22H18F3O2 371.1259. The ee was determined by HPLC analysis. CHIRALPAK IE (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 95/05; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 11.8 min (minor) and 12.7 min (major).
(−)-(R,E)-6,6,6-Trifluoro-5-hydroxy-1-phenyl-5-(thiophen-2-yl)hex-2-en-1-one (5l): White solid, Mp 77.2–82.2 °C; 27.1 mg (0.1 mmol), 83% yield; 92% ee; −26.6 (c 2.26, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.81 (d, J = 7.3 Hz, 2H), 7.55 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.5 Hz, 2H), 7.36 (d, J = 5.1 Hz, 1H), 7.16 (d, J = 3.4 Hz, 1H), 7.04 (dd, J = 4.9, 3.9 Hz, 1H), 6.94 (d, J = 15.5 Hz, 1H), 6.87–6.78 (m, 1H), 3.58 (s, 1H), 3.13 (s, 1H), 3.10 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ 190.4, 140.2, 140.0, 137.1, 133.1, 130.8, 128.7, 128.6, 127.3, 126.6, 126.1, 124.6 (q, J = 283.9 Hz), 76.4 (q, J = 29.7 Hz), 40.0; 19F-NMR (376 MHz, CDCl3) δ −80.58; HRMS (ESI) m/z 349.0493 [M + Na+], calc. for C16H13F3O2SNa 349.0486. The ee was determined by HPLC analysis. CHIRALPAK IB-3 (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 10.4 min (major) and 11.8 min (minor).
(+)-(R,E)-5-Hydroxy-1-phenyl-5-(trifluoromethyl)hept-2-en-1-one (5m): Colorless oil; 16.3 mg (0.1 mmol), 60% yield; 91% ee; +12.4 (c 0.74, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.94–7.92 (m, 2H), 7.58 (t, J = 7.3 Hz, 1H), 7.48 (t, J = 7.5 Hz, 2H), 7.08–6.93 (m, 2H), 2.72 (d, J = 5.07 Hz, 2H), 2.36 (s, 1H), 1.81 (q, J = 7.6 Hz, 2H), 1.04 (t, J = 7.6 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ 190.2, 141.2, 137.4, 133.0, 129.8, 128.6 (two peaks), 126.2 (q, J = 284.9 Hz), 75.4 (q, J = 26.9 Hz), 36.5, 27.0, 7.2; 19F-NMR (376 MHz, CDCl3) δ −79.51; HRMS (ESI) m/z 273.1103 [M + H+], calc. for C14H16F3O2 273.1102. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 7.0 min (major) and 7.7 min (minor).
(−)-(S,E)-6,6,6-Trifluoro-1-(4-fluorophenyl)-5-hydroxy-5-phenylhex-2-en-1-one (5n): White solid, Mp 106.7–108.0 °C; 27.7 mg (0.1 mmol), 82% yield; 95% ee; −30.2 (c 2.30, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.79–7.72 (m, 2H), 7.57 (d, J = 7.0 Hz, 2H), 7.45–7.36 (m, 3H), 7.10–7.03 (m, 2H), 6.86 (d, J = 15.5 Hz, 1H), 6.75–6.65 (m, 1H), 3.27 (s, 1H), 3.25–3.17 (m, 1H), 3.12–3.04 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 188.89, 167.4, 164.0, 140.9, 135.9, 133.5, 133.4, 131.3, 131.2, 130.3, 128.9, 128.6, 126.3, 125.2 (q, J = 284.1 Hz), 115.8, 115.5, 76.7 (q, J = 28.1 Hz), 39.0; 19F-NMR (376 MHz, CDCl3) δ −79.56, −105.09; HRMS (ESI) m/z 339.1015 [M + H+], calc. for C18H15F4O2 339.1008. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 8.0 min (minor) and 9.9 min (major).
(−)-(S,E)-1-(4-Chlorophenyl)-6,6,6-trifluoro-5-hydroxy-5-phenylhex-2-en-1-one (5o): White solid, Mp 90.1–91.5 °C; 30.5 mg (0.1 mmol), 86% yield; 96% ee; −37.2 (c 2.60, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.67–7.63 (m, 2H), 7.57 (d, J = 6.8 Hz, 2H), 7.47–7.34 (m, 5H), 6.83 (d, J = 15.5 Hz, 1H), 6.75–6.65 (m, 1H), 3.34 (s, 1H), 3.24–3.17 (m, 1H), 3.12–3.04 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 189.3, 141.3, 139.5, 135.8, 135.4, 130.2, 130.0, 128.9, 128.8, 128.6, 126.30 (two peaks), 125.2 (q, J = 284.2 Hz), 76.7 (q, J = 28.2 Hz), 39.0; HRMS (ESI) m/z 355.0720[M + H+], calc. for C18H15ClF3O2 355.0713. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 11.0 min (minor) and 16.7 min (major).
(−)-(S,E)-6,6,6-Trifluoro-1-(3-fluorophenyl)-5-hydroxy-5-phenylhex-2-en-1-one (5p): White solid, Mp 87.1–88.9 °C; 31.1 mg (0.1 mmol), 92% yield; 96% ee; −27.5 (c 1.32, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.58–7.50 (m, 3H), 7.46–7.35 (m, 5H), 7.23–7.20 (m, 1H), 6.84 (d, J = 15.6 Hz, 1H), 6.77–6.70 (m, 1H), 3.22 (m, 1H), 3.08 (m, 1H), 2.87 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ 189.0, 164.4, 161.1, 141.3, 139.4, 139.3, 135.8, 130.4, 130.3, 130.2, 129.0, 128.7, 126.3, 125.2 (q, J = 283.9 Hz), 124.3 (two peaks), 120.2, 119.9, 115.6, 115.3, 39.1; HRMS (ESI) m/z 339.1010 [M + H+], calc. for C18H15F4O2 339.1008. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 7.6 min (minor) and 8.6 min (major).
(−)-(S,E)-1-(3-Chlorophenyl)-6,6,6-trifluoro-5-hydroxy-5-phenylhex-2-en-1-one (5q): Colorless oil; 28.4 mg (0.1 mmol), 80% yield; 95% ee; −38.2 (c 1.82, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.70 (t, J = 1.8 Hz, 1H), 7.59 (t, J = 8.2 Hz, 3H), 7.51–7.31 (m, 5H), 6.83 (d, J = 15.6 Hz, 1H), 6.76–6.66 (m, 1H), 3.25–3.18 (m, 1H), 3.12–3.05 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 189.0, 141.5, 138.7, 135.7, 134.8, 132.9, 130.3, 129.8, 129.0, 128.6, 126.6, 126.2 (two peaks), 125.2 (q, J = 284.0 Hz), 76.2, 39.0; HRMS (ESI) m/z 355.0703 [M + H+], calc. for C18H15ClF3O2 355.0713. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 7.7 min (minor) and 8.8 min (major).
(−)-(S,E)-6,6,6-Trifluoro-1-(2-fluorophenyl)-5-hydroxy-5-phenylhex-2-en-1-one (5r): White solid, Mp 74.0–75.9 °C; 22.0 mg (0.1 mmol), 65% yield; 88% ee; −40.5 (c 1.86, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.64–7.55 (m, 3H), 7.52–7.35 (m, 4H), 7.20–7.05 (m, 2H), 6.83 (dd, J = 15.5, 3.0 Hz, 1H), 6.73–6.64 (m, 1H), 3.23–3.16 (m, 1H), 3.07–3.03 (m, 1H), 2.96 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ 188.4, 162.9, 159.5, 140.6, 135.8, 134.3, 134.2, 134.0, 133.9, 131.0, 130.9, 128.9, 128.5, 126.3 (two peaks), 125.1 (q, J = 284.2 Hz), 124.5, 124.4, 116.6, 116.3, 76.6 (q, J = 28.3 Hz), 38.9; HRMS (ESI) m/z 339.1009 [M + H+], calc. for C18H15F4O2 339.1008. The ee was determined by HPLC analysis. CHIRALPAK IA (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 95/05; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 9.3 min (major) and 11.1 min (minor).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-5-phenyl-1-(p-tolyl)hex-2-en-1-one (5s): White solid, Mp 106.2–107.7 °C; 30.8 mg (0.1 mmol), 92% yield; 95% ee; −33.2 (c 1.69, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.67 (d, J = 8.2 Hz, 2H), 7.58 (d, J = 7.2 Hz, 2H), 7.45–7.38 (m, 3H), 7.20 (d, J = 8.0 Hz, 2H), 6.74–6.64 (m, 1H), 6.78–6.61 (m, 1H), 3.24–3.04 (m, 3H), 2.39 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 189.8, 143.9, 140.0, 136.0, 134.6, 130.8, 129.2, 128.8 (two peaks), 126.3 (two peaks), 125.2 (q, J = 284.1 Hz), 76.7, 39.0, 21.6; HRMS (ESI) m/z 335.1261 [(M + H+], calc. for C19H18F3O2 335.1259. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 13.6 min (minor) and 19.3 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-5-phenyl-1-(m-tolyl)hex-2-en-1-one (5t): Colorless oil; 30.4 mg (0.1 mmol), 91% yield; 94% ee; −40.7 (c 2.46, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.59–7.53 (m, 4H), 7.45–7.28 (m, 5H), 6.89 (d, J = 15.5 Hz, 1H), 6.74–6.64 (m, 1H), 3.25–3.18 (m, 1H), 3.15 (s, 1H), 3.12–3.04 (m, 1H), 2.36 (s, 3H); 13C-NMR (75 MHz, CDCl3) δ 190.6, 140.2, 138.4, 137.2, 135.9, 133.8, 131.0, 130.9, 129.2, 128.8, 128.6, 128.4, 126.3 (two peaks), 125.8, 125.2 (q, J = 284.0 Hz), 76.7 (q, J = 28.2 Hz), 39.0, 21.3; HRMS (ESI) m/z 335.1257 [M + H+], calc. for C19H18F3O2 335.1259. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 10.5 min (minor) and 11.9 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1-(4-methoxyphenyl)-5-phenylhex-2-en-1-one (5u): White solid, Mp 107.7–109.2 °C; 25.9 mg (0.1 mmol), 74% yield; 95% ee; −39.6 (c 2.20, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.77 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 7.0 Hz, 2H), 7.43–7.35 (m, 3H), 6.93–6.86 (m, 3H), 6.73–6.63 (m, 1H), 3.85 (s, 3H), 3.33 (s, 1H), 3.23–3.16 (m, 1H), 3.11–3.04 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 188.6, 163.6, 139.4, 136.0, 131.0, 130.6, 130.0, 128.8, 128.5, 127.1, 126.4 (two, peaks), 125.2 (q, J = 284.0 Hz), 113.8, 76.7 (q, J = 28.1 Hz), 55.4, 38.9; HRMS (ESI) m/z 351.1212 [M + H+], calc. for C19H18F3O2 351.1208. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 12.1 min (minor) and 15.0 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1-(3-methoxyphenyl)-5-phenylhex-2-en-1-one (5v): Colorless oil; 27.7 mg (0.1 mmol), 79% yield; 94% ee; −36.3 (c 2.76, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.57 (d, J = 7.1 Hz, 2H), 7.45–7.38 (m, 3H), 7.33–7.31 (m, 3H), 7.11–7.06 (m, 1H), 6.89 (d, J = 15.5 Hz, 1H), 6.77–6.67 (m, 1H), 3.81 (s, 3H), 3.25–3.18 (m, 1H), 3.12–3.04 (m, 2H); 13C-NMR (75 MHz, CDCl3) δ 190.0, 159.8, 140.5, 138.6, 135.9, 130.8, 129.5, 128.9, 128.6, 126.3, 125.2 (q, J = 284.2 Hz), 121.3, 119.7, 112.8, 76.7 (q, J = 28.2 Hz), 55.4, 39.0; HRMS (ESI) m/z 351.1211 [M + H+], calc. for C19H18F3O3 351.1208. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 17.7 min (minor) and 19.8 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1-(2-methoxyphenyl)-5-phenylhex-2-en-1-one (5w): Colorless oil; 15.4 mg (0.1 mmol), 44% yield; 73% ee; −38.3 (c 1.17, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.57 (d, J = 7.0 Hz, 2H), 7.48–7.34 (m, 5H), 7.00–6.91 (m, 2H), 6.83 (d, J = 15.6 Hz, 1H), 6.59–6.48 (m, 1H), 3.83 (s, 3H), 3.18–3.11 (m, 1H), 3.08–3.01 (m, 1H), 2.93 (s, 1H); 13C-NMR (75 MHz, CDCl3) δ 192.0, 158.1, 137.8, 136.2, 136.1, 133.3, 130.5, 128.8, 128.5, 128.2, 126.3, 125.1 (q, J = 283.9 Hz), 120.8, 111.5, 76.4 (q, J = 28.1 Hz), 55.7, 38.8; HRMS (ESI) m/z 351.1204 [M + H+], calc. for C19H18F3O3 351.1208. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 95/05; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 19.3 min (minor) and 22.1 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-1-(naphthalen-2-yl)-5-phenylhex-2-en-1-one (5x): White solid, Mp 91.4–93.2 °C; 29.6 mg (0.1 mmol), 80% yield; 93% ee; −28.0 (c 3.13, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 8.20 (s, 1H), 7.87–7.80 (m, 4H), 7.63–7.37 (m, 7H), 7.04 (d, J = 15.5 Hz, 1H), 6.83–6.73 (m, 1H), 3.43 (d, J = 3.3 Hz, 1H), 3.30–3.22 (m, 1H), 3.17–3.10 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 190.3, 140.5, 136.0, 135.5, 134.4, 132.3, 130.8, 130.4, 129.5, 128.9, 128.6, 128.5 (two peaks), 126.7, 126.4 (two peaks), 125.2 (q, J = 283.9 Hz), 76.7 (q, J = 28.1 Hz), 38.9; HRMS (ESI) m/z 371.1258 [M + H+], calc. for C22H18F3O2 371.1259. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 12.5 min (minor) and 20.9 min (major).
(−)-(S,E)-6,6,6-Trifluoro-5-hydroxy-5-phenyl-1-(thiophen-2-yl)hex-2-en-1-one (5y): White solid, Mp 126.3–128.0 °C; 27.1 mg (0.1 mmol), 83% yield; 98% ee; −44.6 (c 2.46, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.64–7.56 (m, 4H), 7.44–7.34 (m, 3H), 7.11–7.08 (m, 1H), 6.84 (d, J = 15.3 Hz, 1H), 6.80–6.73 (m, 1H), 3.26–3.18 (m, 2H), 3.11–3.05 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 181.7, 144.4, 139.9, 135.9, 134.4, 132.5, 130.1, 128.9, 128.5, 128.2, 126.3, 125.2 (q, J = 284.1 Hz), 76.6 (q, J = 28.3 Hz), 38.8; HRMS (ESI) m/z 327.0666 [M + H+], calc. for C16H14F3O2S 327.0667. The ee was determined by HPLC analysis. CHIRALPAK IB (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 90/10; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 30.6 min (minor) and 31.5 min (major).
(+)-(S,E)-Methyl 2-hydroxy-6-oxo-2,6-diphenylhex-4-enoate (7a): Colorless oil; 24.2 mg (0.1 mmol), 78% yield; 89% ee; +5.8 (c 1.27, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.85–7.82 (m, 2H), 7.63–7.52 (m, 3H), 7.46–7.30 (m, 5H), 6.92–6.90 (m, 2H), 3.82 (s, 4H), 3.22–3.15 (m, 1H), 3.06–3.00 (m, 1H); 13C-NMR (75 MHz, CDCl3) δ 190.7, 174.5, 142.6, 140.8, 137.6, 132.7, 129.9, 128.6, 128.5 (two peaks), 128.2, 125.3, 78.0, 53.5, 43.0; HRMS (ESI) m/z 333.1092 [M + H+], calc. for C19H18O4Na 333.1103. The ee was determined by HPLC analysis. CHIRALPAK IC (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 20.7 min (minor) and 23.6 min (major).
(−)-(R,E)-Diethyl (1-hydroxy-5-oxo-1,5-diphenylpent-3-en-1-yl)phosphonate (9a): White solid; Mp 130.1‒131.9 °C; 26.0 mg (0.1 mmol), 67% yield; 92% ee; −16.1 (c 1.13, CHCl3); 1H-NMR (300 MHz, CDCl3) δ 7.73–7.70 (m, 2H), 7.76–7.59 (m, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 7.5 Hz, 4H), 7.31 (dd, J = 7.2, 1.6 Hz, 1H), 6.86–6.70 (m, 2H), 4.17–4.07 (m, 2H), 4.04–3.89 (m, 1H), 3.87–3.76 (m, 1H), 3.63 (d, J = 6.7 Hz, 1H), 3.21 (t, J = 7.1 Hz, 2H), 3.21 (t, J = 7.1 Hz, 2H), 1.27 (t, J = 7.0 Hz, 3H), 1.17 (t, J = 7.1 Hz, 3H); 13C-NMR (75 MHz, CDCl3) δ 190.8, 142.4, 142.2, 138.1, 137.5, 132.6, 130.3, 128.6, 128.4, 128.3, 128.2, 127.7, 127.6, 126.1 (two peaks), 76.6, 74.5, 63.7, 63.6, 63.5, 63.4, 41.4, 41.3, 16.4, 16.3 (two peaks), 16.2; HRMS (ESI) m/z 411.1336 [M + Na+], calc. for C21H25O5P 411.1337. The ee was determined by HPLC analysis. CHIRALPAK IC (4.6 mm i.d. × 250 mm); Hexane/2-propanol = 80/20; flow rate 1.0 mL/min; 25 °C; 254 nm; retention time: 19.2 min (minor) and 22.4 min (major).





{kind=link}
{kind=link}