Chemical Incorporation of Chain-Terminating Nucleoside Analogs as 3′-Blocking DNA Damage and Their Removal by Human ERCC1-XPF Endonuclease

Abstract
: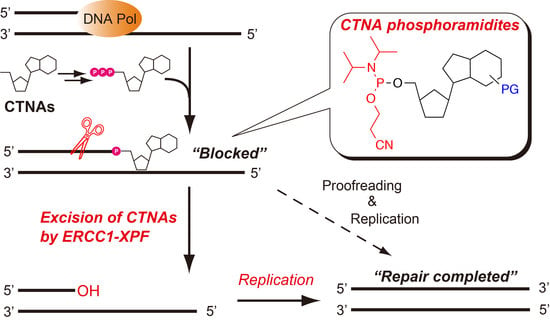
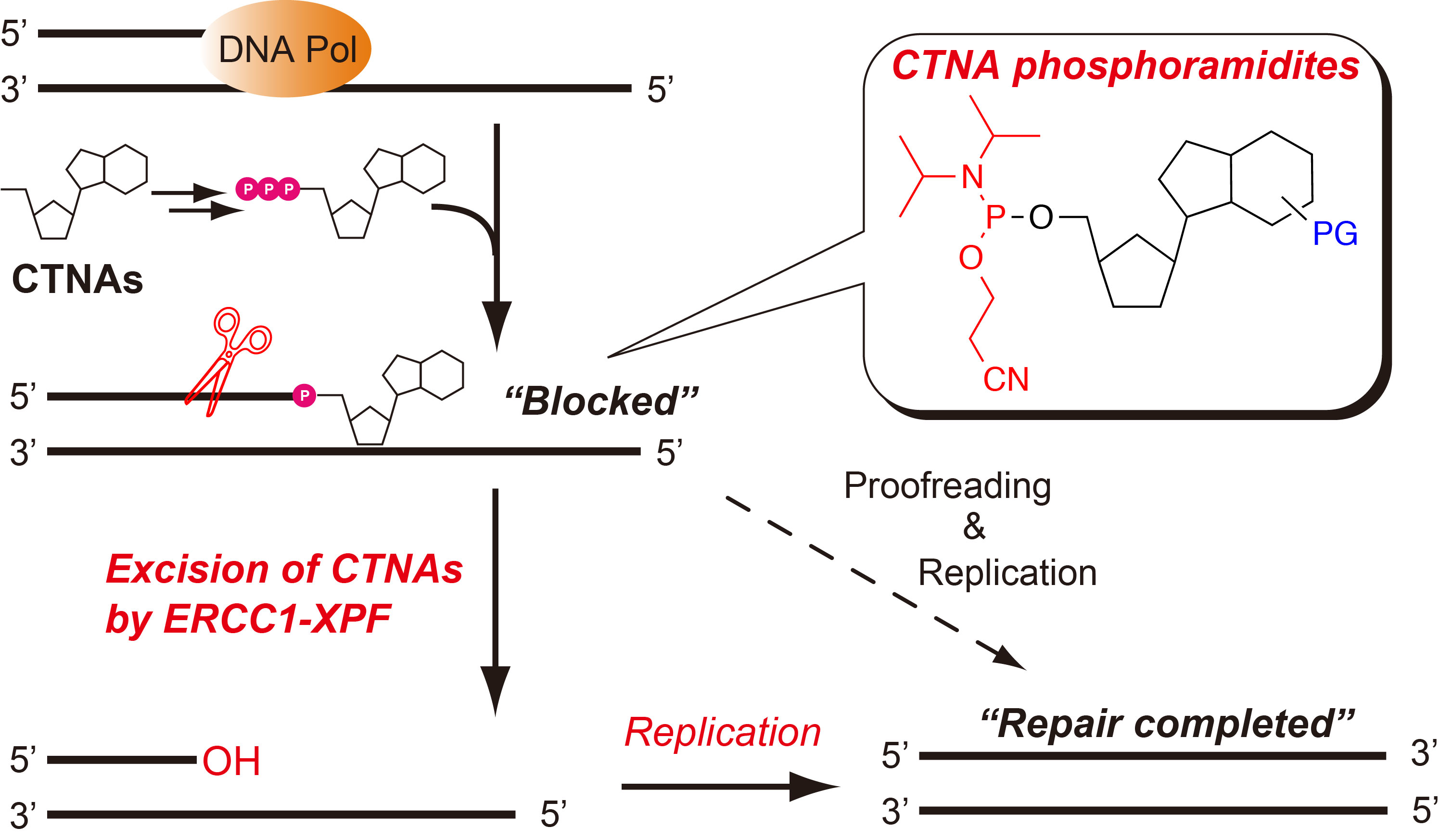{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
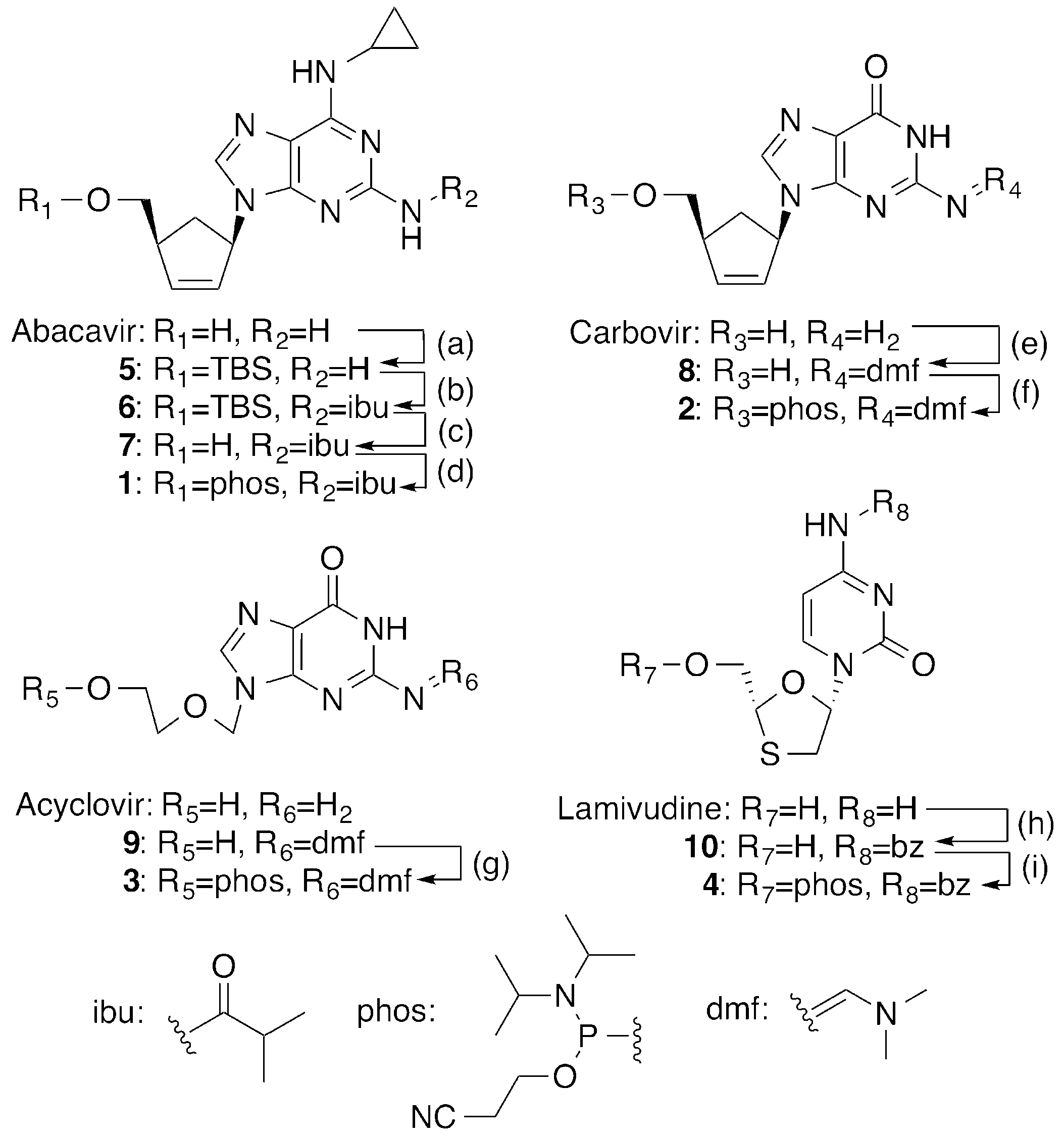{kind=link}
1. Introduction
2. Results
2.1. Synthesis of 5′-Phosphoramidites of CTNAs
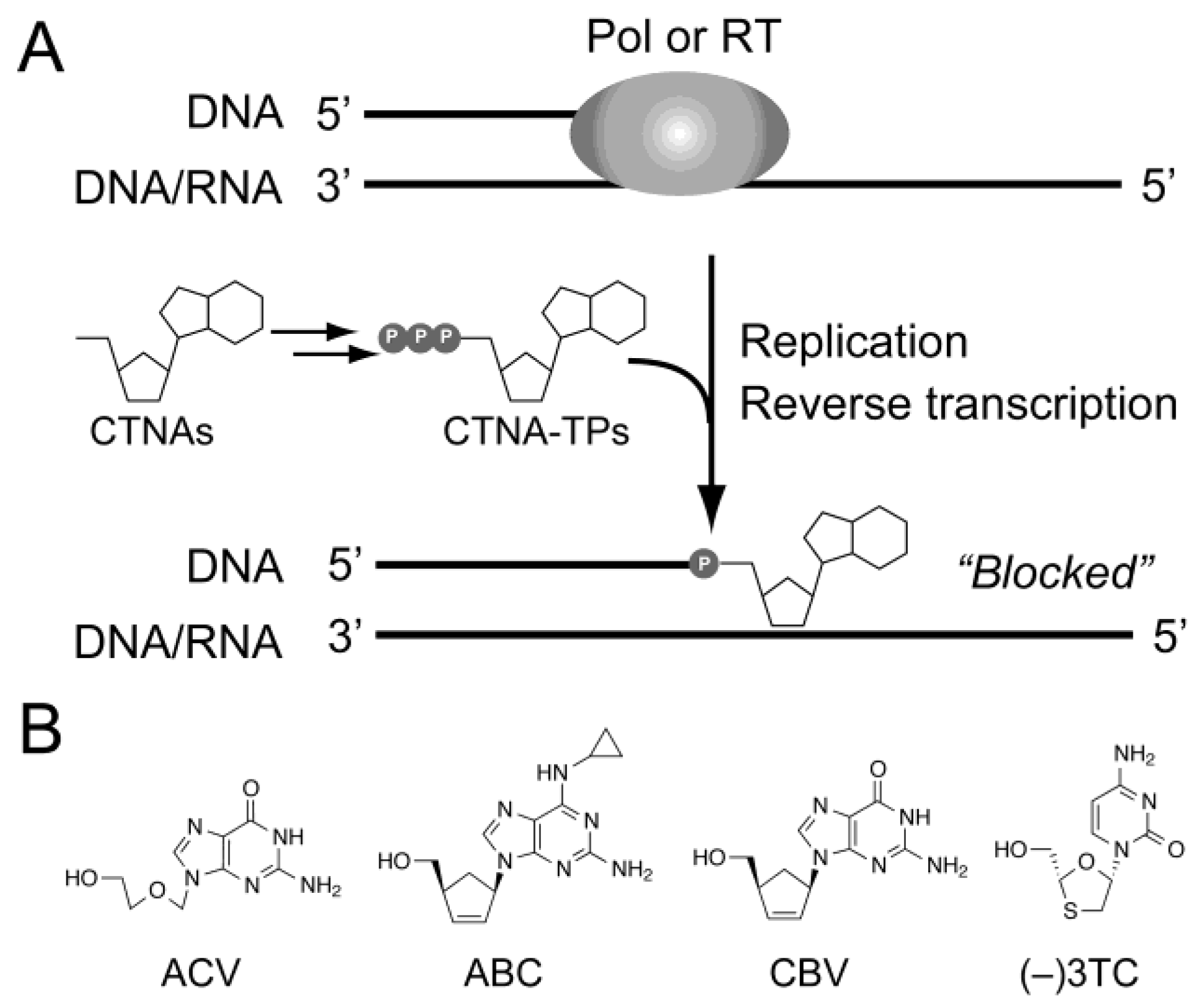
2.2. Chemical Synthesis of Oligonucleotides Containing CTNAs in the 5′ → 3′ Direction
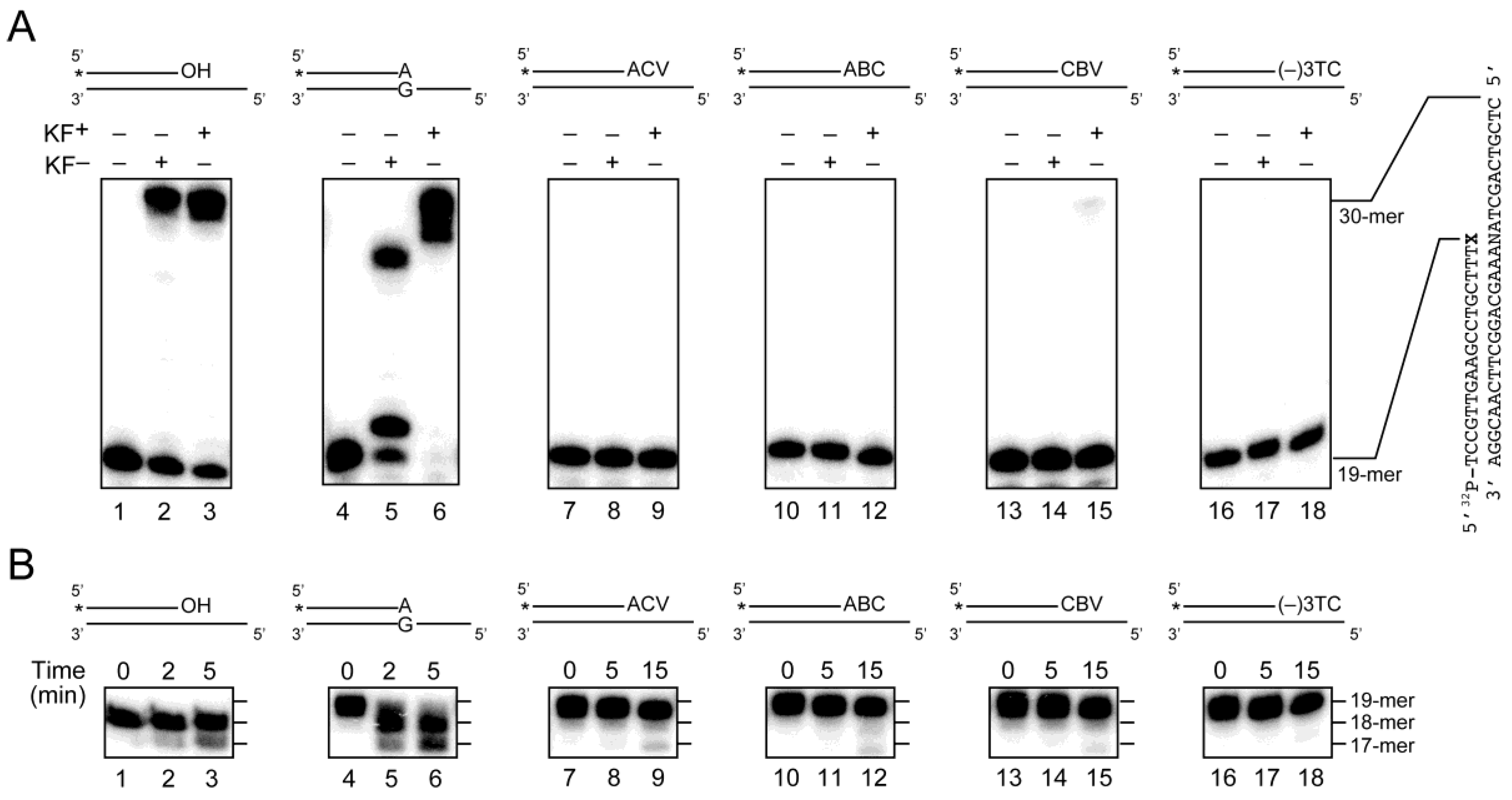
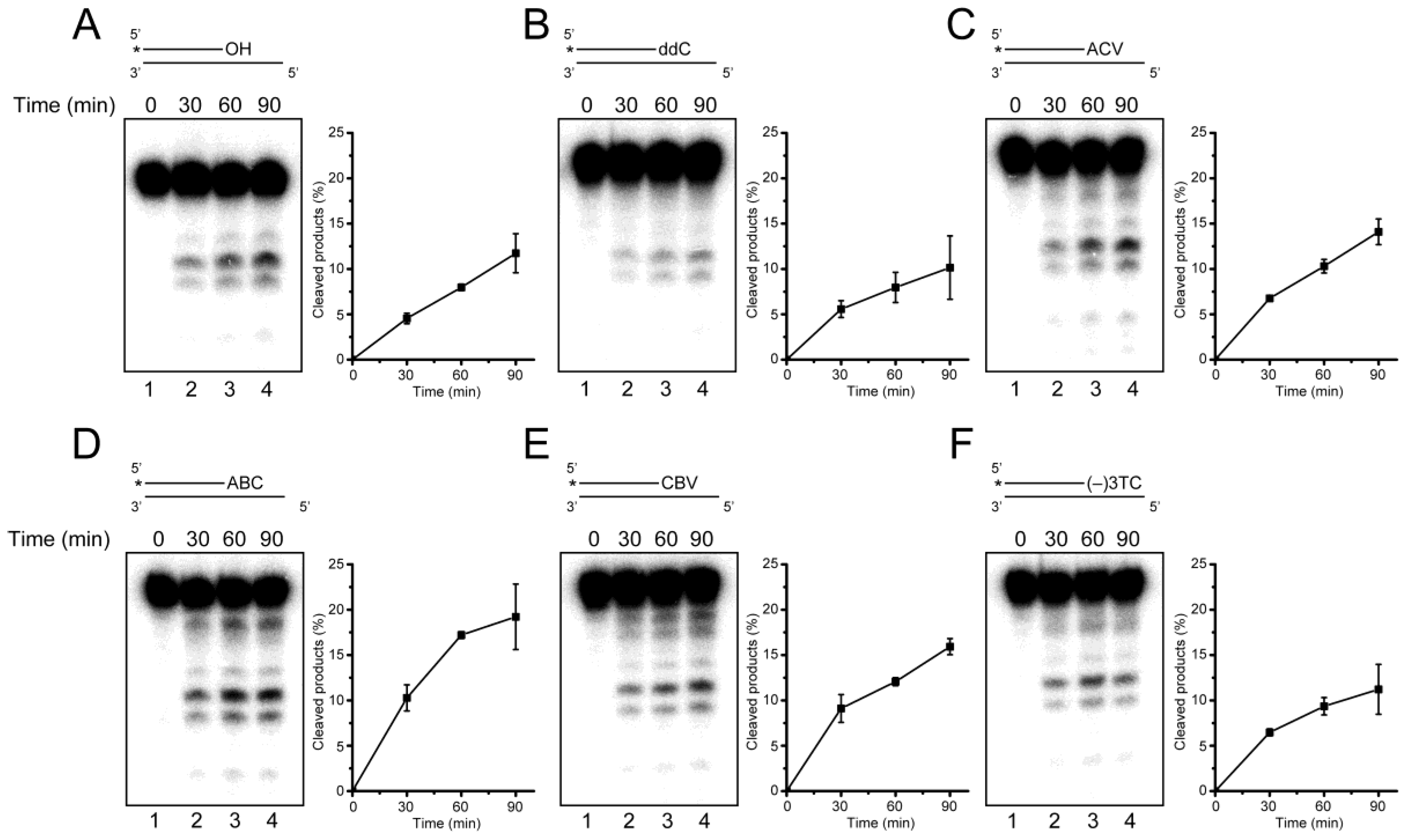
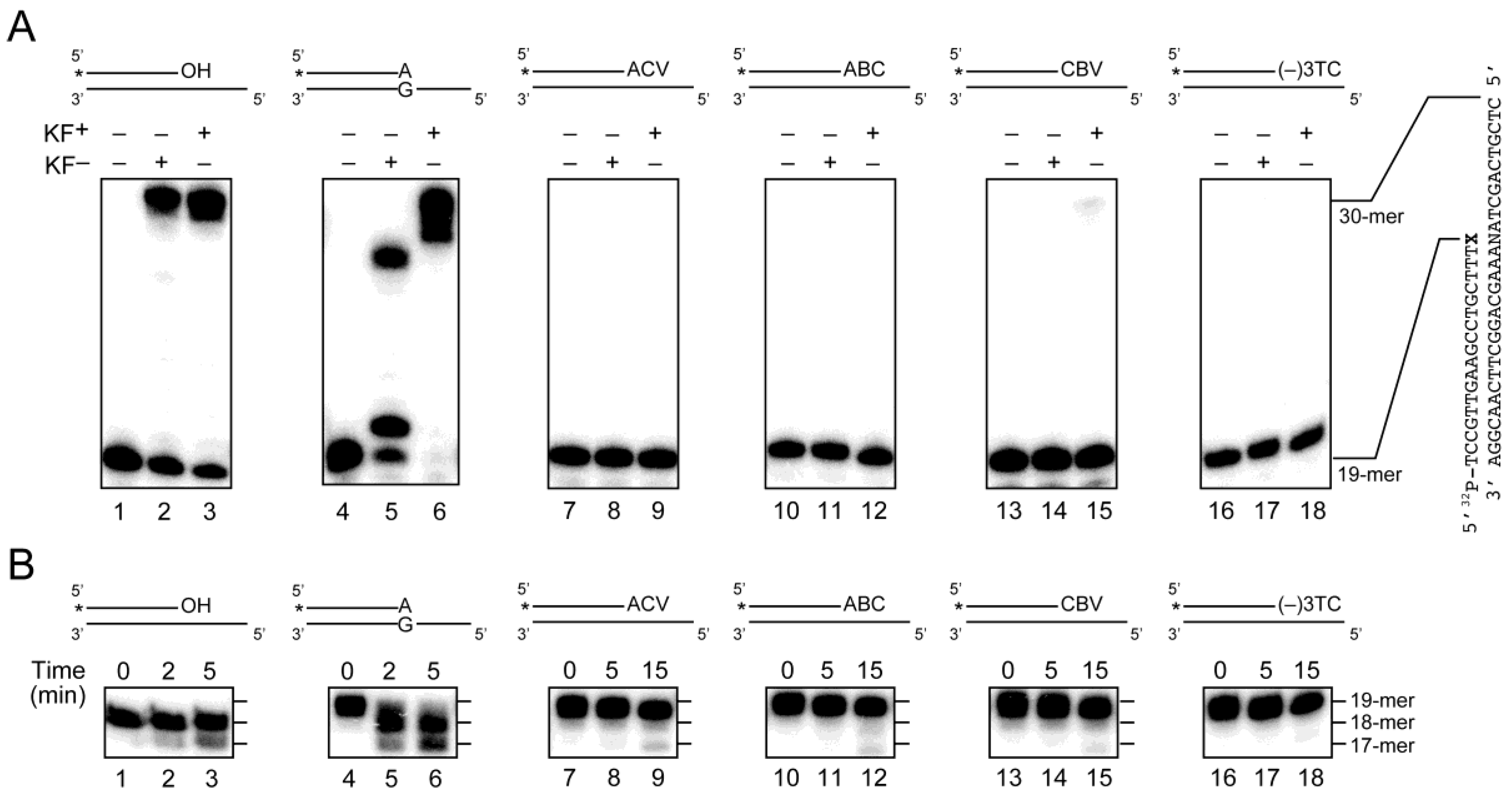
2.3. Primer Extension from the CTNA-Blocking Termini by Klenow Fragments
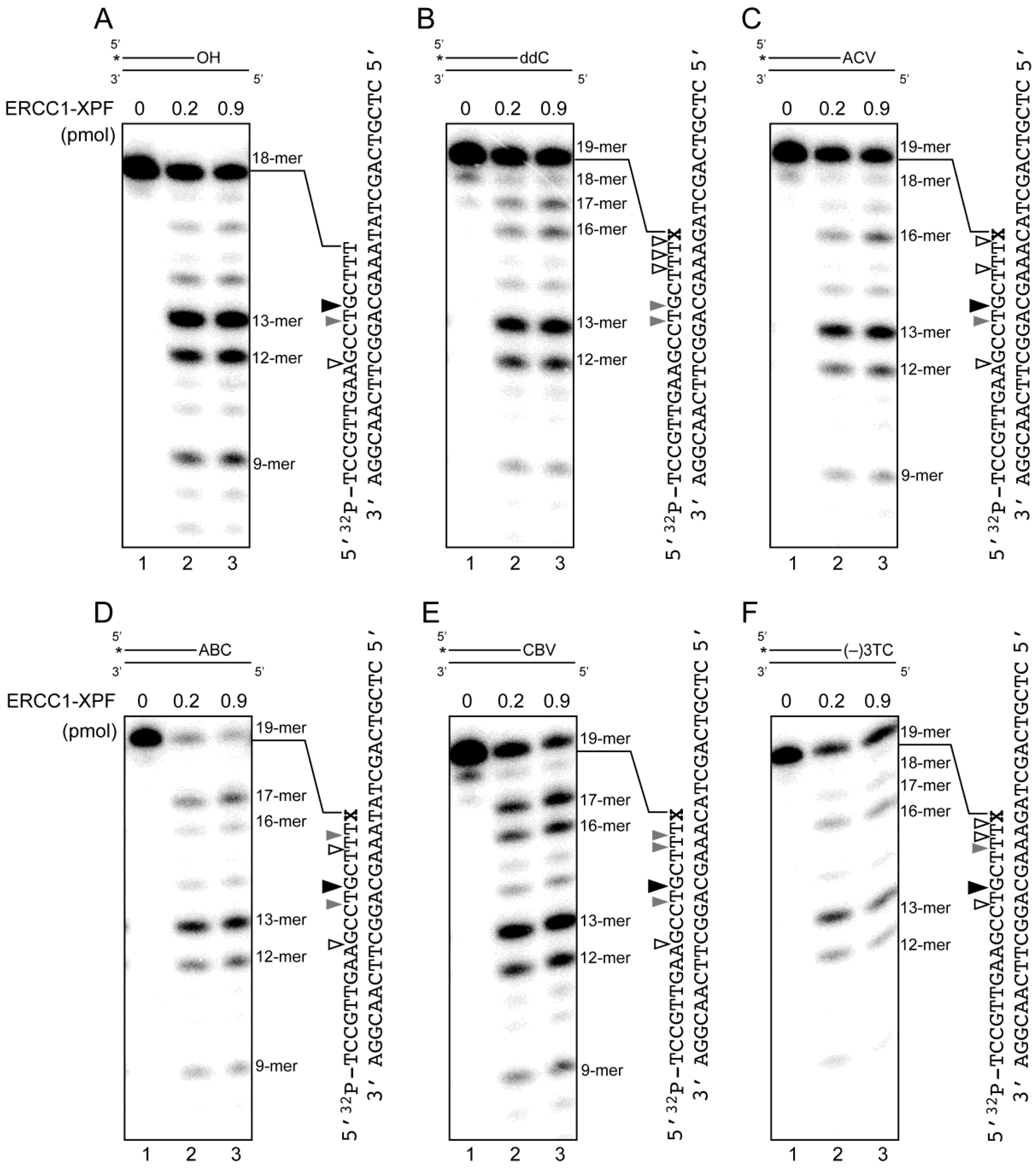
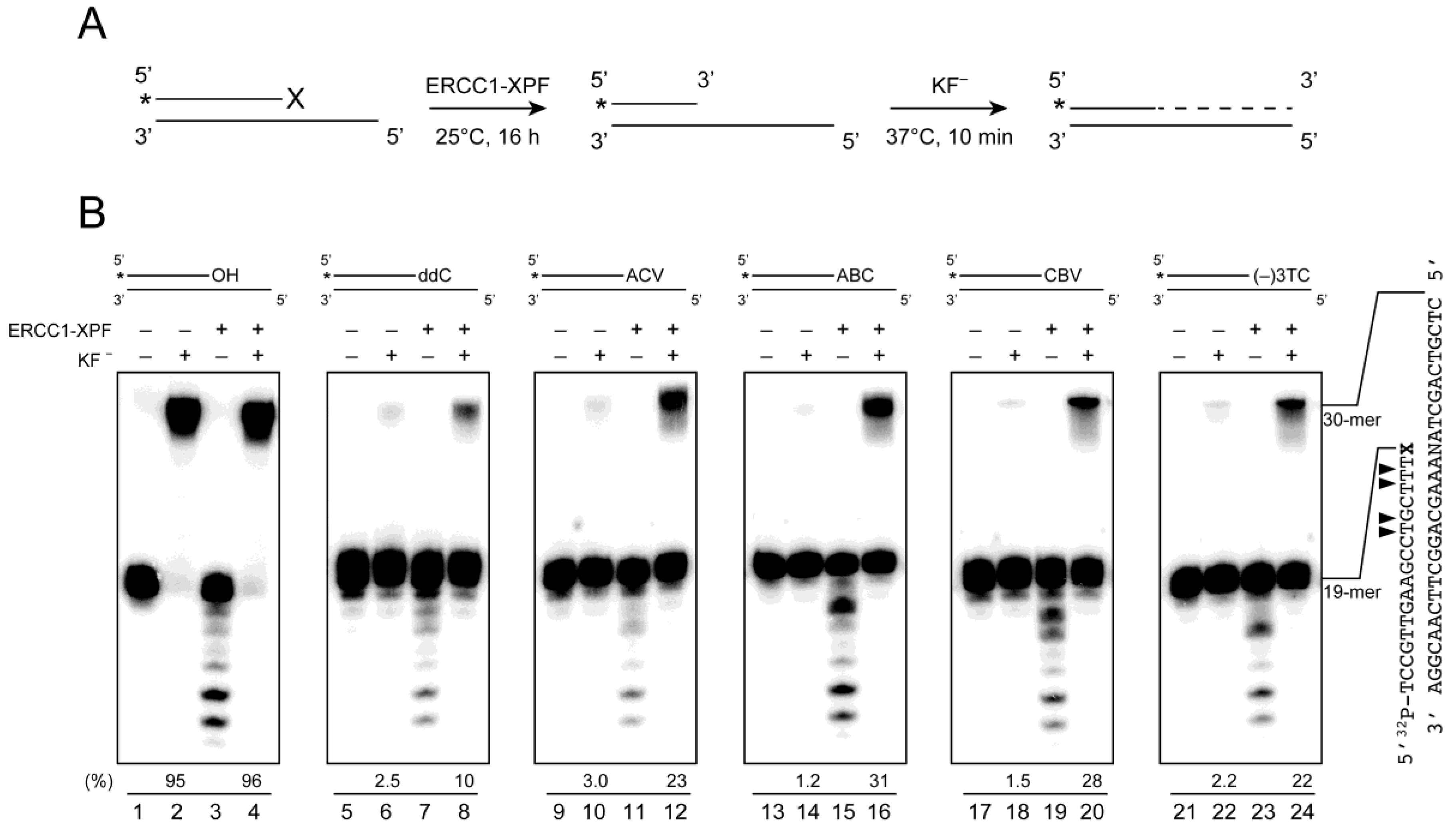
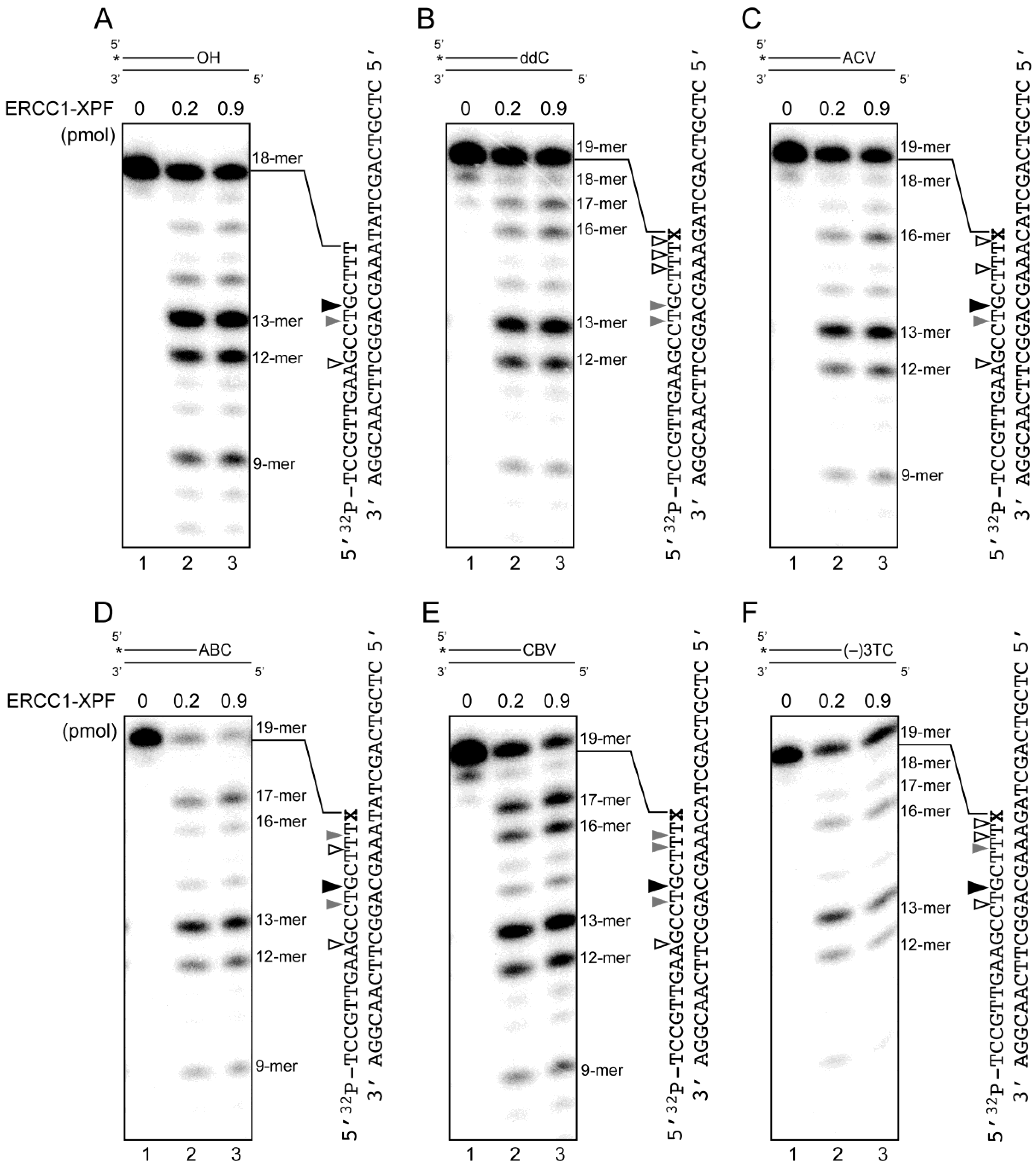
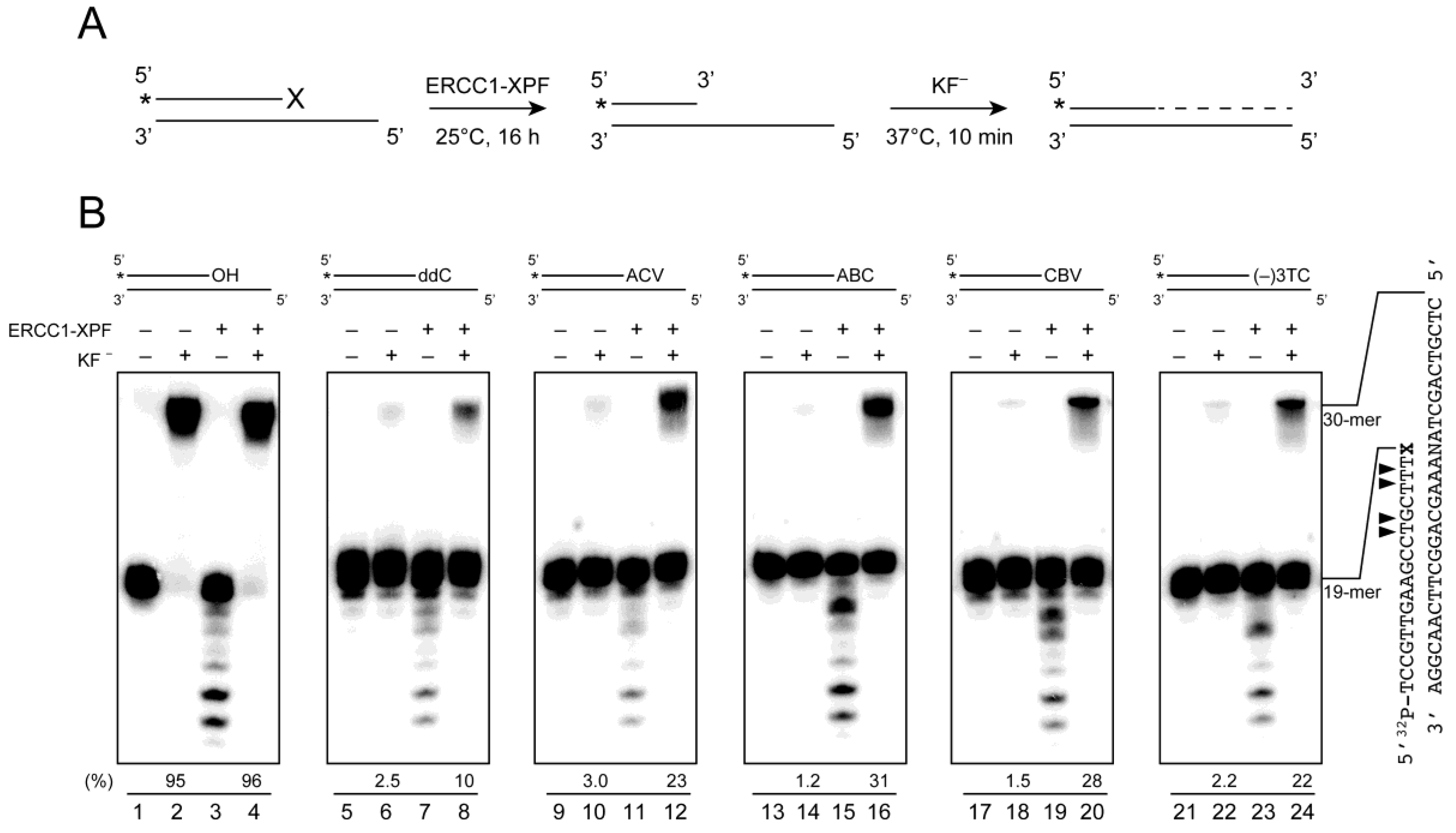
2.4. Removal of 3′-CTNAs by ERCC1-XPF Endonuclease in Vitro
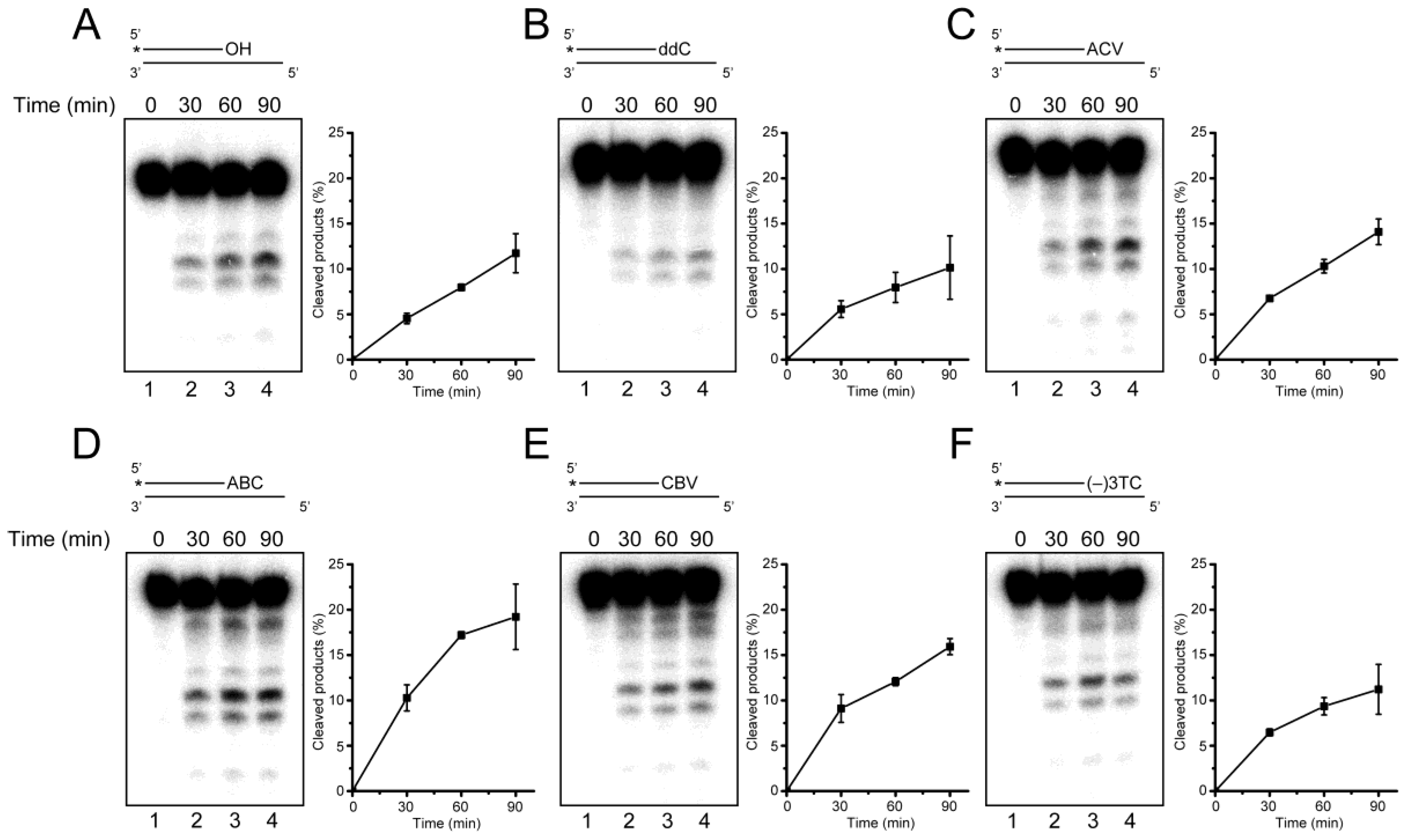
2.5. ERCC1-XPF Associated Repair of CTNA-Induced DNA Damage
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Synthesis of CTNA Phosphoramidites
4.2.1. Synthesis of 5′-O-TBS-abacavir (5)
4.2.2. Synthesis of N2-Isobutyryl-5′-O-TBS-abacavir (6)
4.2.3. Synthesis of N2-Isobutyryl-abacavir (7)
4.2.4. Synthesis of N2-Isobutyryl-abacavir phosphoramidite (1)
4.2.5. Synthesis of N2-DMF-carbovir (8)
4.2.6. Synthesis of N2-DMF-Carbovir phosphoramidite (2)
4.2.7. Synthesis of N2-DMF-Acyclovir phosphoramidite (3)
4.2.8. Synthesis of (−)-β-l-N4-Benzoyl-2’,3′-dideoxy-3′-thiacytidine (10)
4.2.9. Synthesis of N4-Benzoyl-lamivudine phosphoramidite (4)
4.3. Synthesis of Oligonucleotides Containing CTNAs at the 3′-Termini
4.4. General Procedure for the Biochemical Experiments
4.5. Removal of the CTNAs by ERCC1-XPF Endonucleases
4.6. Resumption of DNA Synthesis by DNA Polymerase after the Removal of CTNAs
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 3TC | 2’,3′-dideoxy-3′-thiacytidine |
| ABC | Abacavir |
| ACV | Acyclovir |
| AZT | 3′-deoxy-3′-azidothymidine |
| BSA | Bovine Serum Albumin |
| bz | Benzoyl |
| CBV | Carbovir |
| CTNAs | Chain-terminating Nucleoside Analogs |
| ddC | 2′,3′-dideoxycytidine |
| DIPEA | Diisopropylethylamine |
| dmf | N,N-dimethylformamidyl |
| dNTPs | 2’-deoxynucleoside triphosphates |
| DTT | Dithiothreitol |
| ERCC1-XPF | Excision repair cross complementing protein 1–xeroderma pigmentosum group F |
| FAB-HRMS | Fast atom bombardment-High resolution mass spectroscopy |
| ibu | Isobutyryl |
| KF | Klenow fragments of Escherichia coli DNA polymerase I |
| mtDNA | Mitochondrial DNA |
| NER | Nucleoside excision repair |
| nt | Nucleotide |
| PAGE | Polyacrylamide gel electrophoresis |
| phos | 2-cyanoethyl-N,N-diisopropylphosphoramidyl |
| Polγ | DNA polymerase γ |
| RT | Reverse transcriptase |
| SD | Standard deviation |
| TBS | tert-butyldimethylsilyl |
| TDP1 | Tyrosyl-DNA phosphodiesterase 1 |
| TEA | Triethylamine |
References
- Berdis, A.J. DNA polymerases as therapeutic targets. Biochemistry 2008, 47, 8253–8260. [Google Scholar] [CrossRef] [PubMed]
- Mitsuya, H.; Yarchoan, R.; Broder, S. Molecular targets for AIDS therapy. Science 1990, 249, 1533–1544. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.; Day, B.J.; Copeland, W.C. Mitochondrial toxicity of nrti antiviral drugs: an integrated cellular perspective. Nat. Rev. Drug Discov. 2003, 2, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.E.; Copeland, W.C. Differential incorporation and removal of antiviral deoxynucleotides by human DNA polymerase gamma. J. Biol. Chem. 2001, 276, 23616–23623. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Ray, A.S.; Hanes, J.; Suo, Z.; Colacino, J.M.; Anderson, K.S.; Johnson, K.A. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 2001, 276, 40847–40857. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hanes, J.; Johnson, K.A. Toxicity of nucleoside analogues used to treat AIDS and the selectivity of the mitochondrial DNA polymerase. Biochemistry 2003, 42, 14711–14719. [Google Scholar] [CrossRef] [PubMed]
- Nickel, W.; Austermann, S.; Bialek, G.; Grosse, F. Interactions of azidothymidine triphosphate with the cellular DNA polymerases α, δ, and ε and with DNA primase. J. Biol. Chem. 1992, 267, 848–854. [Google Scholar] [PubMed]
- Pouliot, J.J.; Yao, K.C.; Robertson, C.A.; Nash, H.A. Yeast gene for a Tyr-DNA phosphodiesterase that repairs topoisomerase I complexes. Science 1999, 286, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Dexheimer, T.S.; Maddali, K.; Pommier, Y. Role of tyrosyl-DNA phosphodiesterase (TDP1) in mitochondria. Proc. Natl. Acad. Sci. USA 2010, 107, 19790–19795. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Murai, J.; Dalla, R.I.; Dexheimer, T.S.; Naumova, A.; Gmeiner, W.H.; Pommier, Y. TDP1 repairs nuclear and mitochondrial DNA damage induced by chain-terminating anticancer and antiviral nucleoside analogs. Nucleic Acids Res. 2013, 41, 7793–7803. [Google Scholar] [CrossRef] [PubMed]
- Tada, K.; Kobayashi, M.; Takiuchi, Y.; Iwai, F.; Sakamoto, T.; Nagata, K.; Shinohara, M.; Io, K.; Shirakawa, K.; Hishizawa, M.; et al. Abacavir, an anti-HIV-1 drug, targets TDP1-deficient adult T cell leukemia. Sci. Adv. 2015, 1, e1400203. [Google Scholar] [CrossRef] [PubMed]
- Faletto, M.B.; Miller, W.H.; Garvey, E.P.; Clair, M.H.S.; Daluge, S.M.; Good, S.S. Unique intracellular activation of the potent anti-human immunodeficiency virus agent 1592U89. Antimicrob. Agents Chemother. 1997, 41, 1099–1107. [Google Scholar] [PubMed]
- Vince, R.; Hua, M. Synthesis of carbovir and abacavir from a carbocyclic precursor. In Current Protocols in Nucleic Acid Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; Chapter 14, Unit 14.4. [Google Scholar]
- Kudoh, T.; Fukuoka, M.; Ichikawa, S.; Murayama, T.; Ogawa, Y.; Hashii, M.; Higashida, H.; Kunerth, S.; Weber, K.; Guse, A.H.; et al. Synthesis of stable and cell-type selective analogues of cyclic ADP-ribose, a Ca2+-mobilizing second messenger. Structure-activity relationship of the N1-ribose moiety. J. Am. Chem. Soc. 2005, 127, 8846–8855. [Google Scholar] [CrossRef] [PubMed]
- Saral, R.; Burns, W.H.; Laskin, O.L.; Santos, G.W.; Lietman, P.S. Acyclovir prophylaxis of herpes-simplex-virus infections. N. Engl. J. Med. 1981, 305, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Derudas, M.; Carta, D.; Brancale, A.; Vanpouille, C.; Lisco, A.; Margolis, L.; Balzarini, J.; McGuigan, C. The application of phosphoramidate protide technology to acyclovir confers anti-HIV inhibition. J. Med. Chem. 2009, 52, 5520–5530. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Y.; Johnson, A.A.; Johnson, K.A.; Anderson, K.S. Insights into the molecular mechanism of mitochondrial toxicity by AIDS drugs. J. Biol. Chem. 2001, 276, 23832–23837. [Google Scholar] [CrossRef] [PubMed]
- Bardwell, A.; Bardwell, L.; Tomkinson, A.; Friedberg, E. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-Rad10 DNA endonuclease. Science 1994, 265, 2082–2085. [Google Scholar] [CrossRef] [PubMed]
- Sijbers, A.M.; de Laat, W.L.; Ariza, R.R.; Biggerstaff, M.; Wei, Y.-F.; Moggs, J.G.; Carter, K.C.; Shell, B.K.; Evans, E.; de Jong, M.C.; et al. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 1996, 86, 811–822. [Google Scholar] [CrossRef]
- De Laat, W.L.; Appledoorn, E.; Jaspers, N.G.J.; Hoeijmakers, J.H.J. DNA structural elements required for ERCC1-XPF endonuclease activity. J. Biol. Chem. 1998, 273, 7835–7842. [Google Scholar] [CrossRef] [PubMed]
- Guillet, M.; Boiteux, S. Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2 and Rad1/Rad10 in Saccharomyces cerevisiae. EMBO J. 2002, 21, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.A.; Samson, L.; Bessho, T. Removal of reactive oxygen species-induced 3′-blocked ends by XPF-ERCC1. Chem. Res. Toxicol. 2011, 24, 1876–1881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Regairaz, M.; Seiler, J.A.; Agama, K.K.; Doroshow, J.H.; Pommier, Y. Poly(ADP-ribose) polymerase and XPF-ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011, 39, 3607–3620. [Google Scholar] [CrossRef] [PubMed]
- Takahata, C.; Masuda, Y.; Takedachi, A.; Tanaka, K.; Iwai, S.; Kuraoka, I. Repair synthesis step involving ERCC1-XPF participates in DNA repair of the Top1-DNA damage complex. Carcinogenesis 2015, 36, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.M. Properties of and substrate determinants for the exonuclease activity of human apurinic endonuclease Ape1. J. Mol. Biol. 2003, 330, 1027–1037. [Google Scholar] [CrossRef]
- Jilani, A.; Ramotar, D.; Slack, C.; Ong, C.; Yang, X.M.; Scherer, S.W.; Lasko, D.D. Molecular cloning of the human gene, PNKP, encoding a polynucleotide kinase 3′-phosphatase and evidence for its role in repair of DNA strand breaks caused by oxidative damage. J. Biol. Chem. 1999, 274, 24176–24186. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Tada, M.; Igarashi, S.; Koyama, A.; Date, H.; Yokoseki, A.; Shiga, A.; Yoshida, Y.; Tsuji, S.; Nishizawa, M.; et al. Aprataxin, causative gene product for EAOH/AOA1, repairs DNA single-strand breaks with damaged 3′-phosphate and 3′-phosphoglycolate ends. Nucleic Acids Res. 2007, 35, 3797–3809. [Google Scholar] [CrossRef] [PubMed]
- Fishman-Lobell, J.; Haber, J. Removal of nonhomologous DNA ends in double-strand break recombination: the role of the yeast ultraviolet repair gene RAD1. Science 1992, 258, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Robinson, A.R.; Duensing, A.; van Drunen, E.; Beverloo, H.B.; Weisberg, D.B.; Hasty, P.; Hoeijmakers, J.H.J.; Niedernhofer, L.J. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol. Cell. Biol. 2008, 28, 5082–5092. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples used in this study are available from the authors.
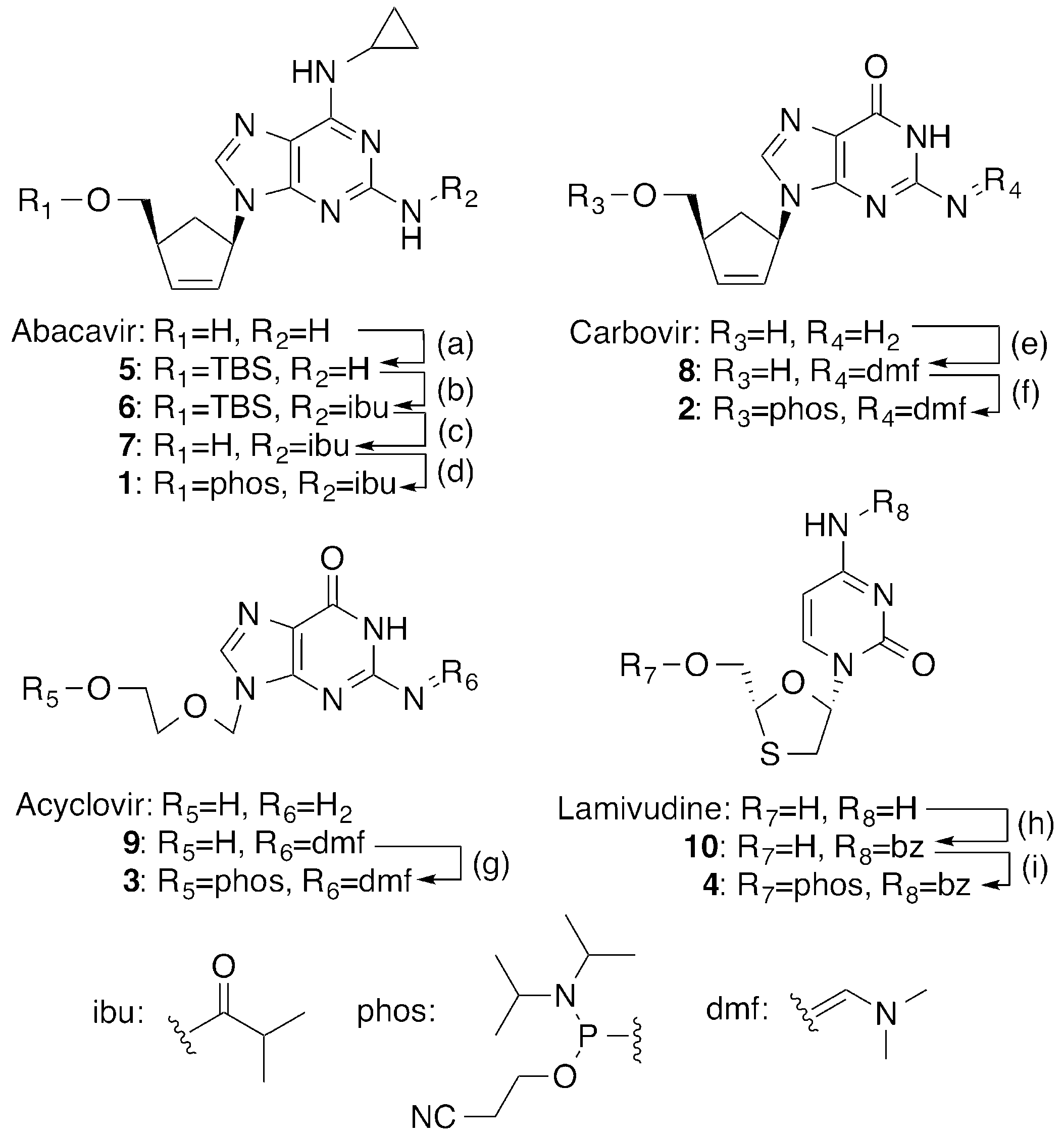
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, J.; Takahata, C.; Kuraoka, I.; Hirota, K.; Iwai, S. Chemical Incorporation of Chain-Terminating Nucleoside Analogs as 3′-Blocking DNA Damage and Their Removal by Human ERCC1-XPF Endonuclease. Molecules 2016, 21, 766. https://doi.org/10.3390/molecules21060766
Yamamoto J, Takahata C, Kuraoka I, Hirota K, Iwai S. Chemical Incorporation of Chain-Terminating Nucleoside Analogs as 3′-Blocking DNA Damage and Their Removal by Human ERCC1-XPF Endonuclease. Molecules. 2016; 21(6):766. https://doi.org/10.3390/molecules21060766
Chicago/Turabian StyleYamamoto, Junpei, Chiaki Takahata, Isao Kuraoka, Kouji Hirota, and Shigenori Iwai. 2016. "Chemical Incorporation of Chain-Terminating Nucleoside Analogs as 3′-Blocking DNA Damage and Their Removal by Human ERCC1-XPF Endonuclease" Molecules 21, no. 6: 766. https://doi.org/10.3390/molecules21060766
APA StyleYamamoto, J., Takahata, C., Kuraoka, I., Hirota, K., & Iwai, S. (2016). Chemical Incorporation of Chain-Terminating Nucleoside Analogs as 3′-Blocking DNA Damage and Their Removal by Human ERCC1-XPF Endonuclease. Molecules, 21(6), 766. https://doi.org/10.3390/molecules21060766