
Effects of Brassicaceae Isothiocyanates on Prostate Cancer

 ,
,  ,
, 

Abstract
:
1. Introduction
2. Biological Activity of Glucosinolates and ITCs: Their Role in Cancer Prevention
3. Role of ITC in Cancer Epigenetics
4. In Vitro Studies
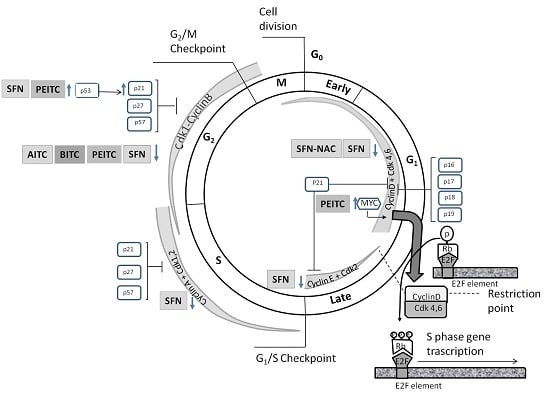
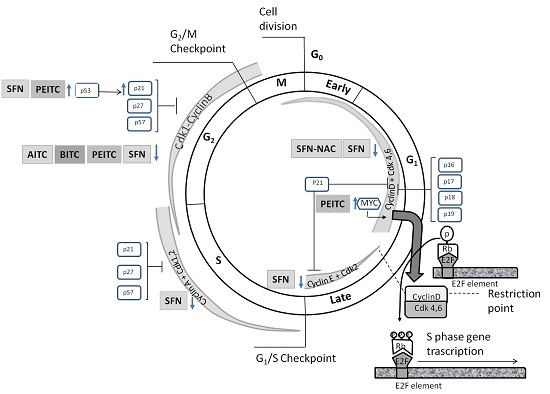
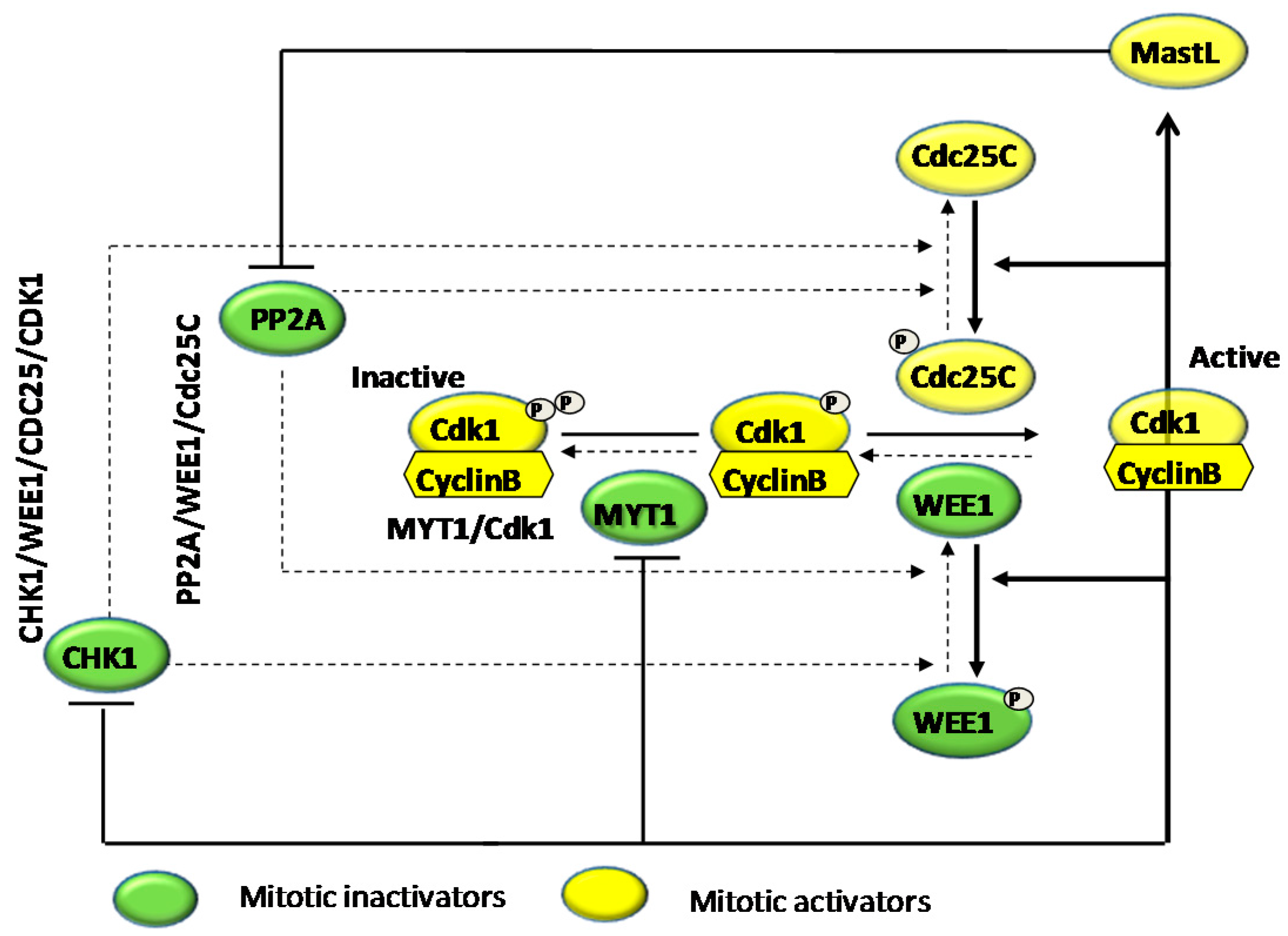
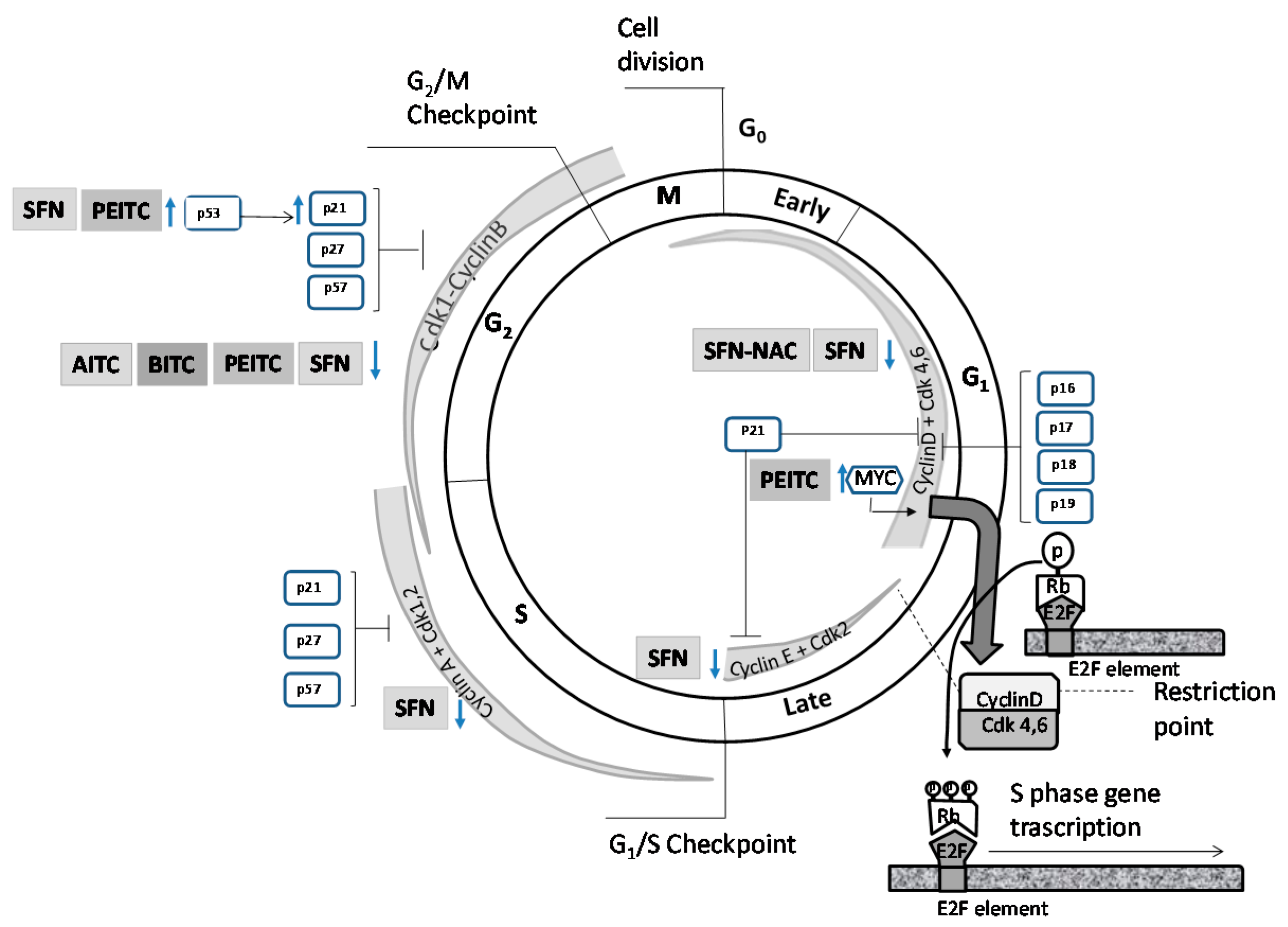
4.1. Effects on Cell Cycle Proteins: Cell Cycle Arrest
4.2. Induction of Apoptosis
4.2.1. Intrinsic and Extrinsic Pathway
4.2.2. Anti-Apoptotic/pro-Apoptotic Proteins
4.2.3. Inhibitor of Apoptosis Proteins (IAPs)
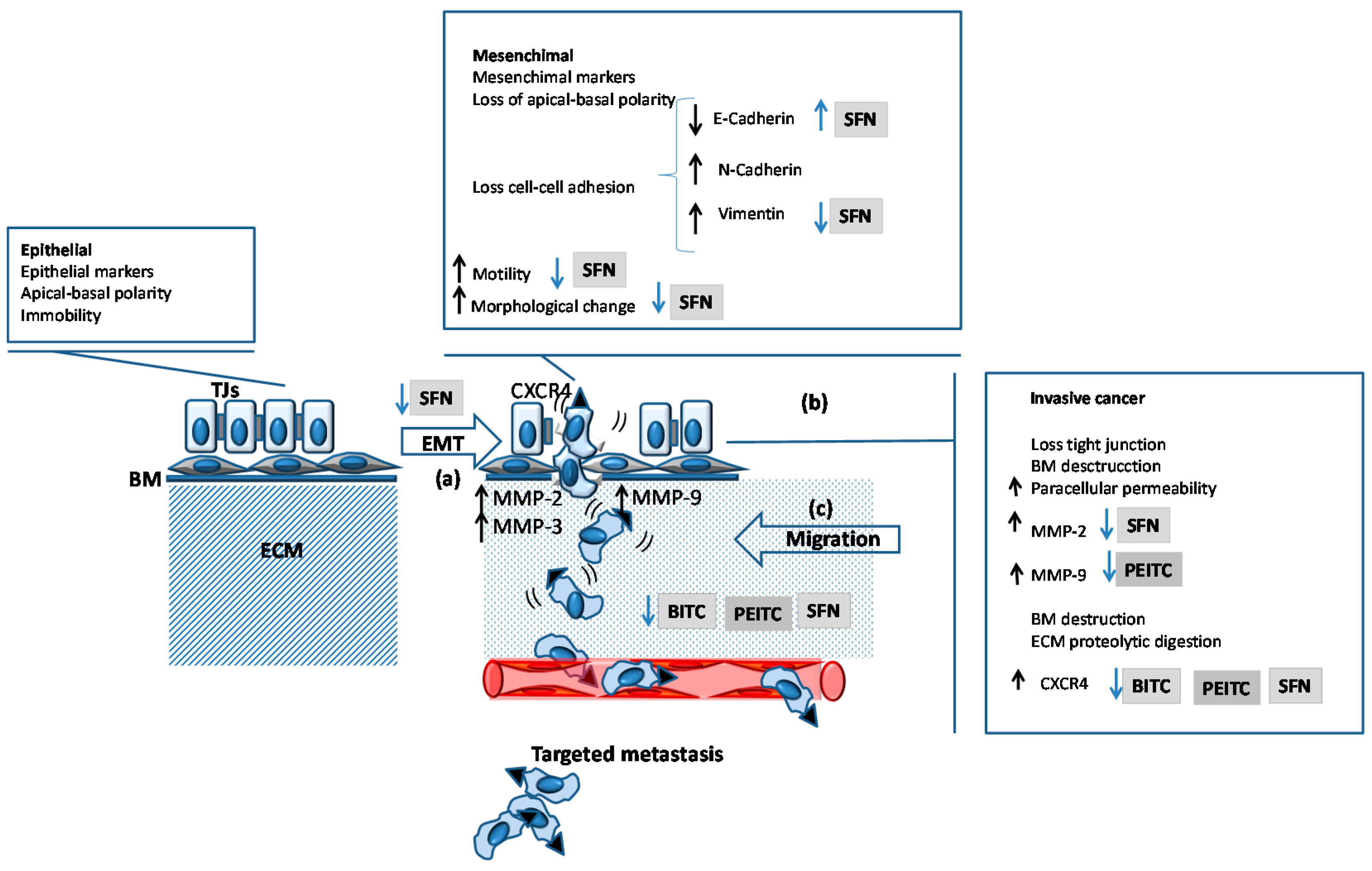
4.3. Inhibition of Migration and Metastasis
4.4. ADT Resistance
5. In Vivo Studies
- (i)
- the dose of ITCs: the necessary concentration of ITC might not have been administered in vivo, thus being required a more intensive dosing regimen to elicit a response;
- (ii)
- the metabolism of ITCs: the exposure of cultured tumor cells to ITCs can lead to a very high intracellular accumulation of them, which may not be possible in vivo due to the rapid excretion of the conjugates of ITCs;
- (iii)
- the activation of in vivo (but not in vitro) mechanisms to counteract the anticancer effect of the compounds (p.e. induction of prosurvival pathways, increased expression of IAPs, etc.).
5.1. Prevention of Cancer Development in Animal Models
5.2. Suppression of Cancer Cell Viability in Association with Apoptosis and/or Autophagy Induction
5.2.1. Cellular Proliferation
5.2.2. Apoptosis
5.2.3. Autophagy
5.3. Inhibition of Metastasis
5.4. Inhibition of Angiogenesis
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Higdon, J.V.; Delage, B.; Williams, D.E.; Dashwood, R.H. Cruciferous vegetables and human cancer risk: Epidemiologic evidence and mechanistic basis. Pharmacol. Res. 2007, 55, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Herr, I.; Büchler, M.W. Dietary constituents of broccoli and other cruciferous vegetables: Implications for prevention and therapy of cancer. Cancer Treat. Rev. 2010, 36, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, G.R.; Heaney, R.K.; Mullin, W.J. Glucosinolates and their breakdown products in food and food plants. Crit. Rev. Food Sci. Nutr. 1983, 18, 123–201. [Google Scholar] [CrossRef] [PubMed]
- Cartea, M.E.; Rodríguez, V.M.; de Haro, A.; Velasco, P.; Ordás, A. Variation of glucosinolates and nutritional value in nabicol (Brassica napus pabularia group). Euphytica 2008, 159, 111–122. [Google Scholar] [CrossRef]
- Cartea, M.E.; de Haro, A.; Obregón, S.; Soengas, P.; Velasco, P. Glucosinolate variation in leaves of Brassica rapa crops. Plant Foods Hum. Nutr. 2012, 67, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.W.; Zalcmann, A.T.; Talalay, P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 2001, 56, 5–51. [Google Scholar] [CrossRef]
- Mithen, R.F.; Dekker, M.; Verkerk, R.; Rabot, S.; Johnson, I.T. The nutritional significance, biosynthesis and bioavailability of glucosinolates in human foods. J. Sci. Food Agric. 2000, 80, 967–984. [Google Scholar] [CrossRef]
- Talalay, P.; Fahey, J.W. Phytochemicals from cruciferous plants protect against cancer by modulating carcinogen metabolism. J. Nutr. 2001, 131 (Suppl. 11), 3027S–3033S. [Google Scholar] [PubMed]
- Traka, M.; Mithen, R.F. Glucosinolates, isothiocyanates and human health. Phytochem. Rev. 2009, 8, 269–282. [Google Scholar] [CrossRef]
- Manchali, S.; Murthy, K.N.C.; Patil, B.S. Crucial facts about health benefits of popular cruciferous vegetables. J. Funct. Foods 2012, 4, 94–106. [Google Scholar] [CrossRef]
- Wattenberg, L.W. Inhibition of carcinogenic effects of polycyclic hydrocarbons by benzyl isothiocyanate and related compounds. J. Natl. Cancer Inst. 1977, 58, 395–398. [Google Scholar] [PubMed]
- Powolny, A.A.; Bommareddy, A.; Hahm, E.R.; Normolle, D.P.; Beumer, J.H.; Nelson, J.B.; Singh, S.V. Chemopreventative potential of the cruciferous vegetable constituent phenethyl isothiocyanate in a mouse model of prostate cancer. J. Natl. Cancer Inst. 2011, 103, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Srivastava, S.K. Antitumor activity of phenethyl isothiocyanate in HER2-positive breast cancer models. BMC Med. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, D.T.; Goldbohm, R.A.; van Poppel, G.; Verhagen, H.; van den Brandt, P.A. Epidemiological studies on brassica vegetables and cancer risk. Cancer Epidemiol. Biomark. Prev. 1996, 5, 733–748. [Google Scholar]
- Graham, S.; Dayal, H.; Swanson, M.; Mittelman, A.; Wilkinson, G. Diet in the epidemiology of cancer of the colon and rectum. J. Natl. Cancer Inst. 1978, 61, 709–714. [Google Scholar] [PubMed]
- Gupta, P.; Kim, B.; Kim, S.H.; Srivastava, S.K. Molecular targets of isothiocyanates in cancer: Recent advances. Mol. Nutr. Food Res. 2014, 58, 1685–1707. [Google Scholar] [CrossRef] [PubMed]
- Kirsh, V.A.; Peters, U.; Mayne, S.T.; Subar, A.F.; Chatterjee, N.; Johnson, C.C.; Hayes, R.B. Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial. Prospective study of fruit and vegetable intake and risk of prostate cancer. J. Natl. Cancer Inst. 2007, 99, 1200–1209. [Google Scholar] [CrossRef] [PubMed]
- Juge, N.; Mithen, R.F.; Traka, M. Molecular basis for chemoprevention by sulforaphane: A comprehensive review. Cell Mol. Life Sci. 2007, 64, 1105–1127. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, F.; Paredes-Gonzalez, X.; Kong, A.T. Dietary glucosinolates sulforaphane, phenethyl isothiocyanate, indole-3-carbinol/3,3′-diindolylmethane: Anti-oxidative stress/inflammation, Nrf2, epigenetics/epigenomics and in vivo cancer chemopreventive efficacy. Curr. Pharmacol. Rep. 2015, 1, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Valgimigli, L.; Iori, R. Antioxidant and pro-oxidant capacities of ITCs. Environ. Mol. Mutagen. 2009, 50, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, A.M.; Huang, T.H.; Toland, A.E. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin. Cancer Biol. 2009, 19, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Basse, C.; Arock, M. The increasing roles of epigenetics in breast cancer: Implications for pathogenicity, biomarkers, prevention and treatment. Int. J. Cancer 2015, 137, 2785–2794. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Aziz, K.; Tran, P.T.; Gorgoulis, V.G.; Yang, E.S.; Georgakilas, A.G. Epigenetic inactivation of DNA repair in breast cancer. Cancer Lett. 2014, 342, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A.; et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, S5–S24. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Hsu, A.; Buchanan, A.; Palomera-Sanchez, Z.; Beaver, L.M.; Houseman, E.A.; Williams, D.E.; Dashwood, R.H.; Ho, E. Effects of sulforaphane and 3,3′-diindolylmethane on genome-wide promoter methylation in normal prostate epithelial cells and prostate cancer cells. PLoS ONE 2014, 9, e86787. [Google Scholar] [CrossRef] [PubMed]
- Denis, H.; Ndlovu, M.N.; Fuks, F. Regulation of mammalian DNA methyltransferases: A route to new mechanisms. EMBO Rep. 2011, 12, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Morey Kinney, S.R.; Smiraglia, D.J.; James, S.R.; Moser, M.T.; Foster, B.A.; Karpf, A.R. Stage-specific alterations of DNA methyltransferase expression, DNA hypermethylation, and DNA hypomethylation during prostate cancer progression in the transgenic adenocarcinoma of mouse prostate model. Mol. Cancer Res. 2008, 6, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.K.; Wu, C.Y.; Chang, J.W.; Juan, L.J.; Hsu, H.S.; Chen, C.Y.; Lu, Y.Y.; Tang, Y.A.; Yang, Y.C.; Yang, P.C.; et al. Dysregulation of p53/Sp1 control leads to DNA methyltransferase-1 overexpression in lung cancer. Cancer Res. 2010, 70, 5807–5817. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Chijiwa, T.; Okamura, T.; Akashi, K.; Fukumaki, Y.; Niho, Y.; Sasaki, H. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, F.; Yang, L.; Guo, C.; Wan, R.; Ke, A.; Xu, L.; Hu, G.; Xu, X.; Shen, J.; et al. Expression of DNMT1 and DNMT3a are regulated by GLI1 in human pancreatic cancer. PLoS ONE 2011, 6, e27684. [Google Scholar] [CrossRef] [PubMed]
- Etoh, T.; Kanai, Y.; Ushijima, S.; Nakagawa, T.; Nakanishi, Y.; Sasako, M.; Kitano, S.; Hirohashi, S. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am. J. Pathol. 2004, 164, 689–699. [Google Scholar] [CrossRef]
- Sproul, D.; Gilbert, N.; Bickmore, W.A. The role of chromatin structure in regulating the expression of clustered genes. Nat. Rev. Genet. 2005, 6, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M. Histone acetylation and an epigenetic code. Bioessays 2000, 22, 836–845. [Google Scholar] [CrossRef]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef] [PubMed]
- Byler, S.; Sarkar, S. Do epigenetic drug treatments hold the key to killing cancer progenitor cells? Epigenomics 2014, 6, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Ong, T.P.; Moreno, F.S.; Ross, S.A. Targeting the epigenome with bioactive food components for cancer prevention. J. Nutrigenet. Nutrigenom. 2011, 4, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.I.; Aumsuwan, P.; Khan, I.A.; Walker, L.A.; Dasmahapatra, A.K. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem. Res. Toxicol. 2012, 25, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Royston, K.J.; Tollefsbol, T.O. The epigenetic impact of cruciferous vegetables on cancer prevention. Curr. Pharmacol. Rep. 2015, 1, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Li, Y.; Shukla, S.; Tollefsbol, T.O. Bioactive dietary supplements reactivate ER expression in ER-negative breast cancer cells by active chromatin modifications. PLoS ONE 2012, 7, e37748. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, C. Epigenetic impact of dietary isothiocyanates in cancer chemoprevention. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Singh, K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis 2012, 33, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Forster, T.; Rausch, V.; Zhang, Y.; Isayev, O.; Heilmann, K.; Schoensiegel, F.; Liu, L.; Nessling, M.; Richter, K.; Labsch, S.; et al. Sulforaphane counteracts aggressiveness of pancreatic cancer driven by dysregulated Cx43-mediated gap junctional intercellular communication. Oncotarget 2014, 5, 1621–1634. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Kidane, A.I.; Yu, T.W.; Dashwood, W.M.; Bisson, W.H.; Löhr, C.V.; Ho, E.; Williams, D.E.; Dashwood, R.H. HDAC turnover, CtIP acetylation and dysregulated DNA damage signaling in colon cancer cells treated with sulforaphane and related dietary isothiocyanates. Epigenetics 2013, 8, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Chew, Y.C.; Eckert, R.L. Sulforaphane suppresses polycomb group protein level via a proteasome-dependent mechanism in skin cancer cells. Mol. Pharmacol. 2011, 80, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Chew, Y.C.; Adhikary, G.; Wilson, G.M.; Xu, W.; Eckert, R.L. Sulforaphane induction of p21(Cip1) cyclin-dependent kinase inhibitor expression requires p53 and Sp1 transcription factors and is p53-dependent. J. Biol. Chem. 2012, 287, 16168–16178. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.Y.; Zhang, C.; Lee, J.H.; Shu, L.; Wu, T.Y.; Khor, T.O.; Conney, A.H.; Lu, Y.P.; Kong, A.N. Requirement and epigenetics reprogramming of Nrf2 in suppression of tumor promoter TPA-induced mouse skin cell transformation by sulforaphane. Cancer Prev. Res. 2014, 7, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Chiao, J.W. Prostate cancer chemopreventive activity of phenethyl isothiocyanate through epigenetic regulation (review). Int. J. Oncol. 2010, 37, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.H.; Melchini, A.; Mithen, R.F. Sulforaphane and prostate cancer interception. Drug Discov. Today 2014, 19, 1488–1492. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.; Wong, C.P.; Yu, Z.; Williams, D.E.; Dashwood, R.H.; Ho, E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin. Epigenet. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Meeran, S.M.; Patel, S.N.; Tollefsbol, T.O. Sulforaphane causes epigenetic repression of hTERT expression in human breast cancer cell lines. PLoS ONE 2010, 5, e11457. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Hardin, K.; Wang, R.; Dashwood, R.H.; Ho, E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2006, 27, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Dashwood, W.M.; Orner, G.A.; Ho, E.; Dashwood, R.H. Sulforaphane inhibits histone deacetylase in vivo and suppresses tumorigenesis in Apc-minus mice. FASEB J. 2006, 20, 506–508. [Google Scholar] [PubMed]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, A.; Schwartzman, J.; Deng, V.; Alumkal, J. Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc. Natl. Acad. Sci. USA 2009, 106, 16663–16668. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer. 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Huggins, C.; Stevens, R.E., Jr.; Hodges, C.V. Studies on prostatic cancer. II. The effects of castration on advanced carcinoma of the prostate gland. Arch. Surg. 1941, 43, 209–223. [Google Scholar] [CrossRef]
- Marrocco, D.L.; Tilley, W.D.; Bianco-Miotto, T.; Evdokiou, A.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; Butler, L.M. Suberoylanilide hydroxamic acid (vorinostat) represses androgen receptor expression and acts synergistically with an androgen receptor antagonist to inhibit prostate cancer cell proliferation. Mol. Cancer Ther. 2007, 6, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Meng, S.; Wang, H.; Bali, P.; Bai, W.; Li, B.; Atadja, P.; Bhalla, K.N.; Wu, J. Chemical ablation of androgen receptor in prostate cancer cells by the histone deacetylase inhibitor LAQ824. Mol. Cancer Ther. 2005, 4, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Liu, X.M.; Chiao, J.W. Repression of androgen receptor in prostate cancer cells by phenethyl isothiocyanate. Carcinogenesis 2006, 27, 2124–2132. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, D.; Ahmed, T.; Chung, F.L.; Conaway, C.; Chiao, J.W. Targeting cell cycle machinery as a molecular mechanism of sulforaphane in prostate cancer prevention. Int. J. Oncol. 2004, 24, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Chiao, J.W.; Wu, H.; Ramaswamy, G.; Conaway, C.C.; Chung, F.L.; Wang, L.; Liu, D. Ingestion of an isothiocyanate metabolite from cruciferous vegetables inhibits growth of human prostate cancer cell xenografts by apoptosis and cell cycle arrest. Carcinogenesis 2004, 25, 1403–1408. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Liu, X.M.; Fang, Y.; Dai, W.; Chiao, F.B.; Puccio, G.M.; Feng, J.; Liu, D.; Chiao, J.W. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int. J. Oncol. 2008, 33, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, R.; Toyooka, S.; Toyooka, K.O.; Virmani, A.K.; Zöchbauer-Müller, S.; Farinas, A.J.; Minna, J.D.; McConnell, J.; Frenkel, E.P.; Gazdar, A.F. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin. Cancer Res. 2002, 8, 514–519. [Google Scholar] [PubMed]
- Woodson, K.; Gillespie, J.; Hanson, J.; Emmert-Buck, M.; Phillips, J.M.; Linehan, W.M.; Tangrea, J.A. Heterogeneous gene methylation patterns among pre-invasive and cancerous lesions of the prostate: A histopathologic study of whole mount prostate specimens. Prostate 2004, 60, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Beklemisheva, A.; Liu, X.M.; Ferrari, A.C.; Feng, J.; Chiao, J.W. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol. Carcinog. 2007, 46, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Srinidhi, S.; Vishwanatha, J.K. Oncogenic activation in prostate cancer progression and metastasis: Molecular insights and future challenges. J. Carcinog. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, D.; You, Z. In vitro and in vivo model systems used in prostate cancer research. J. Biol. Methods 2015, 2. [Google Scholar] [CrossRef] [PubMed]
- Simone, C.B., 2nd; John-Aryankalayil, M.; Palayoor, S.T.; Makinde, A.Y.; Cerna, D.; Falduto, M.T.; Magnuson, S.R.; Coleman, C.N. mRNA expression profiles for prostate cancer following fractionated irradiation are influenced by p53 status. Transl. Oncol. 2013, 6, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.; Blando, J.; Silver, E.; Beltran, L.; Sessler, J.; DiGiovanni, J. 6-Shogaol from dried ginger inhibits growth of prostate cancer cells both in vitro and in vivo through inhibition of STAT3 and NF-κB signaling. Cancer Prev. Res. 2014, 7, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.A.; Kornbluth, S. Cdc25 and Wee1: Analogous opposites? Cell Div. 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- Clarke, J.D.; Dashwood, R.H.; Ho, E. Multi-targeted prevention of cancer by sulforaphane. Cancer Lett. 2008, 269, 291–304. [Google Scholar] [CrossRef] [PubMed]
- De Witt Hamer, P.C.; Mir, S.E.; Noske, D.; van Noorden, C.J.; Würdinger, T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clin. Cancer Res. 2011, 17, 4200–4207. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.S.; Hashimoto, Y.; Ma, Z.Q.; Tsai, M.J.; Tsai, S.Y. Overexpression of Cdc25B, an androgen receptor coactivator, in prostate cancer. Oncogene 2003, 22, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Ozen, M.; Ittmann, M. Increased expression and activity of CDC25C phosphatase and an alternatively spliced variant in prostate cancer. Clin. Cancer Res. 2005, 11, 4701–4706. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.T.; Han, H.Y.; Leung, S.C.; Yuen, H.F.; Chau, C.W.; Guo, Z.; Qiu, Y.; Chan, K.W.; Wang, X.; Wong, Y.C.; et al. CDC25A functions as a novel Ar corepressor in prostate cancer cells. J. Mol. Biol. 2009, 385, 446–445. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Wang, Y.W.; Murray, D. Ionizing radiation-induced responses in human cells with differing TP53 status. Int. J. Mol. Sci. 2013, 14, 22409–22435. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Martinez, L.A.; Sikes, C.R.; Johnston, D.A.; Stephens, L.C.; McDonnell, T.J.; Logothetis, C.J.; Trapman, J.; Pisters, L.L.; Ordoñez, N.G.; et al. The association of p21((WAF-1/CIP1)) with progression to androgen-independent prostate cancer. Clin. Cancer Res. 2002, 8, 775–781. [Google Scholar] [PubMed]
- De Luca, P.; Moiola, C.P.; Zalazar, F.; Gardner, K.; Vazquez, E.S.; de Siervi, A. BRCA1 and p53 regulate critical prostate cancer pathways. Prostate Cancer Prostatic. Dis. 2013, 16, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Srivastava, S.K.; Lew, K.L.; Zeng, Y.; Hershberger, P.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis 2003, 24, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Proteasome-mediated degradation of cell division cycle 25C and cyclin-dependent kinase 1 in phenethyl isothiocyanate-induced G2-M-phase cell cycle arrest in PC-3 human prostate cancer cells. Mol. Cancer Ther. 2004, 3, 567–575. [Google Scholar] [PubMed]
- Hwang, E.S.; Lee, H.J. Effects of phenylethyl isothiocyanate and its metabolite on cell-cycle arrest and apoptosis in LNCaP human prostate cancer cells. Int. J. Food Sci. Nutr. 2010, 61, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.Y.; Huang, Y.T.; Yu, C.S.; Ko, Y.C.; Wu, S.H.; Ji, B.C.; Yang, J.S.; Yang, J.L.; Hsia, T.C.; Chen, Y.Y.; et al. Phenethyl isothiocyanate (PEITC) promotes G2/M phase arrest via p53 expression and induces apoptosis through caspase- and mitochondria-dependent signaling pathways in human prostate cancer DU 145 cells. Anticancer Res. 2011, 31, 1691–1702. [Google Scholar] [PubMed]
- Qu, Y.; Oyan, A.M.; Liu, R.; Hua, Y.; Zhang, J.; Hovland, R.; Popa, M.; Liu, X.; Brokstad, K.A.; Simon, R.; et al. Generation of prostate tumor-initiating cells is associated with elevation of reactive oxygen species and IL-6/STAT3 signaling. Cancer Res. 2013, 73, 7090–7100. [Google Scholar] [CrossRef] [PubMed]
- Herman-Antosiewicz, A.; Xiao, H.; Lew, K.L.; Singh, S.V. Induction of p21 protein protects against sulforaphane-induced mitotic arrest in LNCaP human prostate cancer cell line. Mol. Cancer Ther. 2007, 6, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Herman-Antosiewicz, A.; Singh, A.V.; Lew, K.L.; Srivastava, S.K.; Kamath, R.; Brown, K.D.; Zhang, L.; Baskaran, R. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J. Biol. Chem. 2004, 279, 25813–25822. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.D.; Li, G.; Hu, H.; Jiang, C.; Kang, K.S.; Lee, Y.S.; Kim, S.H.; Lu, J. Involvement of c-Jun N-terminal kinase in G2/M arrest and caspase-mediated apoptosis induced by sulforaphane in DU145 prostate cancer cells. Nutr. Cancer. 2005, 52, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.C.; Huang, Y.T.; Wu, P.P.; Ji, B.C.; Yang, J.S.; Yang, J.L.; Chiu, T.H.; Chueh, F.S.; Chung, J.G. The roles of AIF and Endo G in the apoptotic effects of benzyl isothiocyanate on DU 145 human prostate cancer cells via the mitochondrial signaling pathway. Int. J. Oncol. 2011, 38, 787–796. [Google Scholar] [PubMed]
- Sakao, K.; Desineni, S.; Hahm, E.R.; Singh, S.V. Phenethyl isothiocyanate suppresses inhibitor of apoptosis family protein expression in prostate cancer cells in culture and in vivo. Prostate 2012, 72, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Gong, A.; He, M.; Krishna Vanaja, D.; Yin, P.; Karnes, R.J.; Young, C.Y. Phenethyl isothiocyanate inhibits STAT3 activation in prostate cancer cells. Mol. Nutr. Food Res. 2009, 53, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Beaver, L.M.; Buchanan, A.; Sokolowski, E.I.; Riscoe, A.N.; Wong, C.P.; Chang, J.H.; Löhr, C.V.; Williams, D.E.; Dashwood, R.H.; Ho, E. Transcriptome analysis reveals a dynamic and differential transcriptional response to sulforaphane in normal and prostate cancer cells and suggests a role for Sp1 in chemoprevention. Mol. Nutr. Food Res. 2014, 58, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Chiao, J.W.; Chung, F.L.; Kancherla, R.; Ahmed, T.; Mittelman, A.; Conaway, C.C. Sulforaphane and its metabolite mediate growth arrest and apoptosis in human prostate cancer cells. Int. J. Oncol. 2002, 20, 631–636. [Google Scholar] [PubMed]
- Clarke, J.D.; Hsu, A.; Yu, Z.; Dashwood, R.H.; Ho, E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells vs. hyperplastic and cancerous prostate cells. Mol. Nutr. Food Res. 2011, 55, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Portt, L.; Norman, G.; Clapp, C.; Greenwood, M.; Greenwood, M.T. Anti-apoptosis and cell survival: A review. Biochim. Biophys. Acta 2011, 1813, 238–259. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.F.; Tsai, T.F.; Liao, P.C.; Lin, Y.H.; Lin, Y.C.; Chen, H.E.; Chou, K.Y.; Hwang, T.I. Benzyl isothiocyanate induces protective autophagy in human prostate cancer cells via inhibition of mTOR signaling. Carcinogenesis 2013, 34, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Xiao, D.; Lew, K.L.; Dhir, R.; Singh, S.V. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis 2004, 25, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Lew, K.L.; Xiao, H.; Herman-Antosiewicz, A.; Xiao, D.; Brown, C.K.; Singh, S.V. d,l-Sulforaphane-induced cell death in human prostate cancer cells is regulated by inhibitor of apoptosis family proteins and Apaf-1. Carcinogenesis 2007, 28, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Wiczk, A.; Hofman, D.; Konopa, G.; Herman-Antosiewicz, A. Sulforaphane, a cruciferous vegetable-derived isothiocyanate, inhibits protein synthesis in human prostate cancer cells. Biochim. Biophys. Acta 2012, 1823, 1295–1305. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Powolny, A.A.; Moura, M.B.; Kelley, E.E.; Bommareddy, A.; Kim, S.H.; Hahm, E.R.; Normolle, D.; van Houten, B.; Singh, S.V. Phenethyl isothiocyanate inhibits oxidative phosphorylation to trigger reactive oxygen species-mediated death of human prostate cancer cells. J. Biol. Chem. 2010, 285, 26558–26569. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Gan, N.; Cheema, A.; Dakshanamurthy, S.; Wang, X.; Yang, D.C.; Chung, F.L. Cancer preventive isothiocyanates induce selective degradation of cellular alpha- and beta-tubulins by proteasomes. J. Biol. Chem. 2009, 284, 17039–17051. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Srivastava, S.K.; Choi, S.; Lew, K.L.; Antosiewicz, J.; Xiao, D.; Zeng, Y.; Watkins, S.C.; Johnson, C.S.; Trump, D.L.; et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J. Biol. Chem. 2005, 280, 19911–19924. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Berriguete, G.; Torrealba, N.; Ortega, M.A.; Martínez-Onsurbe, P.; Olmedilla, G.; Paniagua, R.; Guil-Cid, M.; Fraile, B.; Royuela, M. Prognostic value of inhibitors of apoptosis proteins (IAPs) and caspases in prostate cancer: Caspase-3 forms and XIAP predict biochemical progression after radical prostatectomy. BMC Cancer 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.P.; Lin, C.Y.; Hsiao, P.H.; Wang, H.D.; Sheng Jiang, S.; Hsu, J.M.; Jim, W.T.; Chen, M.; Kung, H.J.; Chuu, C.P. Difference in protein expression profile and chemotherapy drugs response of different progression stages of LNCaP sublines and other human prostate cancer cells. PLoS ONE 2013, 8, e82625. [Google Scholar] [CrossRef] [PubMed]
- Raffo, A.J.; Perlman, H.; Chen, M.W.; Day, M.L.; Streitman, J.S.; Buttyan, R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Res. 1995, 55, 4438–4445. [Google Scholar] [PubMed]
- Lin, Y.; Fukuchi, J.; Hiipakka, R.A.; Kokontis, J.M.; Xiang, J. Up-regulation of Bcl-2 is required for the progression of prostate cancer cells from an androgen-dependent to an androgen-independent growth stage. Cell Res. 2007, 17, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.J.; Sesterhenn, I.A.; Mostofi, F.K.; McLeod, D.G.; Srivastava, S.; Moul, J.W. Elevated levels of apoptosis regulator proteins p53 and bcl-2 are independent prognostic biomarkers in surgically treated clinically localized prostate cancer. J. Urol. 1996, 156, 1511–1516. [Google Scholar] [CrossRef]
- Matsushima, H.; Kitamura, T.; Goto, T.; Hosaka, Y.; Homma, Y.; Kawabe, K. Combined analysis with Bcl-2 and P53 immunostaining predicts poorer prognosis in prostatic carcinoma. J. Urol. 1997, 158, 2278–2283. [Google Scholar] [CrossRef]
- Yong, W.P.; Innocenti, F.; Ratain, M.J. The role of pharmacogenetics in cancer therapeutics. Br. J. Clin. Pharmacol. 2006, 62, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Gyrd-Hansen, M.; Meier, P. IAPs: From caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 2010, 10, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Berezovskaya, O.; Schimmer, A.D.; Glinskii, A.B.; Pinilla, C.; Hoffman, R.M.; Reed, J.C.; Glinsky, G.V. Increased expression of apoptosis inhibitor protein XIAP contributes to anoikis resistance of circulating human prostate cancer metastasis precursor cells. Cancer Res. 2005, 65, 2378–2386. [Google Scholar] [CrossRef] [PubMed]
- Seligson, D.B.; Hongo, F.; Huerta-Yepez, S.; Mizutani, Y.; Miki, T.; Yu, H.; Horvath, S.; Chia, D.; Goodglick, L.; Bonavida, B. Expression of X-linked inhibitor of apoptosis protein is a strong predictor of human prostate cancer recurrence. Clin. Cancer Res. 2007, 13, 6056–6063. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.L.; Kyprianou, N. Role of androgens and the androgen receptor in epithelial-mesenchymal transition and invasion of prostate cancer cells. FASEB J. 2010, 24, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, J.T.; Henry, M.D. Epithelial-to-mesenchymal transition in prostate cancer: Paradigm or puzzle? Nat. Rev. Urol. 2011, 8, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.Y.; Lee, W.S.; Jung, J.H.; Hong, S.H.; Park, C.; Kim, H.J.; Kim, G.Y.; Hwang, H.J.; Kim, G.S.; Jung, J.M.; et al. Flavonoids from Orostachys japonicus A. Berger inhibit the invasion of LnCaP prostate carcinoma cells by inactivating Akt and modulating tight junctions. Int. J. Mol. Sci. 2013, 14, 18407–18420. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Gopalan, A.; Leversha, M.A.; Satagopan, J.M.; Zhou, Q.; Al-Ahmadie, H.A.; Fine, S.W.; Eastham, J.A.; Scardino, P.T.; Scher, H.I.; Tickoo, S.K.; et al. TMPRSS2-ERG gene fusion is not associated with outcome in patients treated by prostatectomy. Cancer Res. 2009, 69, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Kandagatla, P.; Singareddy, R.; Kropinski, A.; Sheng, S.; Cher, M.L.; Chinni, S.R. Androgens induce functional CXCR4 through ERG factor expression in TMPRSS2-ERG fusion-positive prostate cancer cells. Transl. Oncol. 2010, 3, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, J.J.; Ng, B.H.; Smits, M.M.; Wang, J.; Jasavala, R.J.; Martinez, H.D.; Lee, J.; Alston, J.J.; Misonou, H.; Trimmer, J.S.; Wright, M.E. Androgen receptor and chemokine receptors 4 and 7 form a signaling axis to regulate CXCL12-dependent cellular motility. BMC Cancer 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Sakao, K.; Vyas, A.R.; Chinni, S.R.; Amjad, A.I.; Parikh, R.; Singh, S.V. CXCR4 is a novel target of cancer chemopreventative isothiocyanates in prostate cancer cells. Cancer Prev. Res. 2015, 8, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Labsch, S.; Liu, L.; Bauer, N.; Zhang, Y.; Aleksandrowicz, E.; Gladkich, J.; Schönsiegel, F.; Herr, I. Sulforaphane and TRAIL induce a synergistic elimination of advanced prostate cancer stem-like cells. Int. J. Oncol. 2014, 44, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Zhou, Y.; Tian, H.; Yang, G.; Li, C.; Geng, Y.; Wu, S.; Wu, W. Sulforaphane inhibits invasion by phosphorylating ERK1/2 to regulate E-cadherin and CD44v6 in human prostate cancer DU145 cells. Oncol. Rep. 2015, 34, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.G.; Gao, W.Q. The Notch pathway in prostate development and cancer. Differentiation 2008, 76, 699–716. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Banerjee, S.; Kong, D.; Ahmad, A.; Nogueira, V.; Hay, N.; Sarkar, F.H. Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth, migration and invasion, and induces apoptosis via inactivation of Akt, mTOR, and NF-kappaB signaling pathways. J. Cell Biochem. 2010, 109, 726–736. [Google Scholar] [PubMed]
- Bin Hafeez, B.; Adhami, V.M.; Asim, M.; Siddiqui, I.A.; Bhat, K.M.; Zhong, W.; Saleem, M.; Din, M.; Setaluri, V.; Mukhtar, H. Targeted knockdown of Notch1 inhibits invasion of human prostate cancer cells concomitant with inhibition of matrix metalloproteinase-9 and urokinase plasminogen activator. Clin. Cancer Res. 2009, 15, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.R.; Chandra-Kuntal, K.; Desai, D.; Amin, S.; Singh, S.V. Notch activation is dispensable for d, l-sulforaphane-mediated inhibition of human prostate cancer cell migration. PLoS ONE 2012, 7, e44957. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Sehrawat, A.; Sakao, K.; Hahm, E.R.; Singh, S.V. Notch activation by phenethyl isothiocyanate attenuates its inhibitory effect on prostate cancer cell migration. PLoS ONE 2011, 6, e26615. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, B.; Beresford, M.; Bowen, R.; Mitchard, J.; Chalmers, A.D. Searching for prostate cancer stem cells: Markers and methods. Stem Cell Rev. 2013, 9, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Ojo, D.; Lin, X.; Wong, N.; Gu, Y.; Tang, D. Prostate cancer stem-like cells contribute to the development of castration-resistant prostate cancer. Cancers 2015, 7, 2290–2308. [Google Scholar] [CrossRef] [PubMed]
- Vander Griend, D.J.; Litvinov, I.V.; Isaacs, J.T. Conversion of androgen receptor signaling from a growth suppressor in normal prostate epithelial cells to an oncogene in prostate cancer cells involves a gain of function in c-Myc regulation. Int. J. Biol. Sci. 2014, 10, 627–642. [Google Scholar] [CrossRef] [PubMed]
- Sankpal, U.T.; Goodison, S.; Abdelrahim, M.; Basha, R. Targeting Sp1 transcription factors in prostate cancer therapy. Med. Chem. 2011, 7, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Meng, Y.; Liu, N.; Wen, X.F.; Yang, T. Insights into chemoresistance of prostate cancer. Int. J. Biol. Sci. 2015, 11, 1160–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Jiang, B.; Gao, F.H. Small molecule inhibitors of STAT3 for cancer therapy. Curr. Med. Chem. 2011, 18, 4012–4018. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Thaper, D.; Zoubeidi, A. The multifaceted roles of STAT3 signaling in the progression of prostate cancer. Cancers 2014, 6, 829–859. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.E.; Karras, J.G.; Murphy, T.F.; Barton, A.; Huang, H.F. Signal transducer and activator of transcription 3 (STAT3) activation in prostate cancer: Direct STAT3 inhibition induces apoptosis in prostate cancer lines. Mol. Cancer Ther. 2004, 3, 11–20. [Google Scholar] [PubMed]
- Yue, P.; Turkson, J. Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 2009, 18, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Pencik, J.; Schlederer, M.; Gruber, W.; Unger, C.; Walker, S.M.; Chalaris, A.; Marié, I.J.; Hassler, M.R.; Javaheri, T.; Aksoy, O.; et al. STAT3 regulated ARF expression suppresses prostate cancer metastasis. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: Direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar] [PubMed]
- Aaronson, D.S.; Muller, M.; Neves, S.R.; Chung, W.C.; Jayaram, G.; Iyengar, R.; Ram, P.T. An androgen-IL-6-Stat3 autocrine loop re-routes EGF signal in prostate cancer cells. Mol. Cell Endocrinol. 2007, 270, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Siegmund, S.A.; Sommer, J.; Scheller, J.; Rose-John, S.; Garbers, C. ID: 204: Interleukin-6-Induced and constitutive activation of signal transducer and activator of transcription 3 relies on protein kinase II activity. Cytokine 2015, 76, 101–102. [Google Scholar] [CrossRef]
- Hahm, E.R.; Singh, S.V. Sulforaphane inhibits constitutive and interleukin-6-induced activation of signal transducer and activator of transcription 3 in prostate cancer cells. Cancer Prev. Res. 2010, 3, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Edlind, M.P.; Hsieh, A.C. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J. Androl. 2014, 16, 378–386. [Google Scholar] [PubMed]
- Khor, T.O.; Keum, Y.S.; Lin, W.; Kim, J.H.; Hu, R.; Shen, G.; Xu, C.; Gopalakrishnan, A.; Reddy, B.; Zheng, X.; et al. Combined inhibitory effects of curcumin and phenethyl isothiocyanate on the growth of human PC-3 prostate xenografts in immunodeficient mice. Cancer Res. 2006, 66, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Ganapathy, S.; Srivastava, R.K. Sulforaphane enhances the therapeutic potential of TRAIL in prostate cancer orthotopic model through regulation of apoptosis, metastasis, and angiogenesis. Clin Cancer Res. 2008, 14, 6855–6866. [Google Scholar] [CrossRef] [PubMed]
- Hudson, T.S.; Perkins, S.N.; Hursting, S.D.; Young, H.A.; Kim, Y.S.; Wang, T.C.; Wang, T.T. Inhibition of androgen-responsive LNCaP prostate cancer cell tumor xenograft growth by dietary phenethyl isothiocyanate correlates with decreased angiogenesis and inhibition of cell attachment. Int. J. Oncol. 2012, 40, 1113–1121. [Google Scholar] [PubMed]
- Xiao, D.; Zeng, Y.; Choi, S.; Lew, K.L.; Nelson, J.B.; Singh, S.V. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous vegetable-derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res. 2005, 11, 2670–2679. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Lew, K.L.; Zeng, Y.; Xiao, H.; Marynowski, S.W.; Dhir, R.; Singh, S.V. Phenethyl isothiocyanate-induced apoptosis in PC-3 human prostate cancer cells is mediated by reactive oxygen species-dependent disruption of the mitochondrial membrane potential. Carcinogenesis 2006, 27, 2223–2234. [Google Scholar] [CrossRef] [PubMed]
- Sakao, K.; Hahm, E.R.; Singh, S.V. In vitro and in vivo effects of phenethyl isothiocyanate treatment on vimentin protein expression in cancer cells. Nutr Cancer. 2013, 65 (Suppl. 1), 61–67. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.V.; Warin, R.; Xiao, D.; Powolny, A.A.; Stan, S.D.; Arlotti, J.A.; Zeng, Y.; Hahm, E.R.; Marynowski, S.W.; Bommareddy, A.; et al. Sulforaphane inhibits prostate carcinogenesis and pulmonary metastasis in TRAMP mice in association with increased cytotoxicity of natural killer cells. Cancer Res. 2009, 69, 2117–2125. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.L.; Morse, M.A.; Eklind, K.I.; Lewis, J. Quantitation of human uptake of the anticarcinogen phenethyl isothiocyanate after a watercress meal. Cancer Epidemiol Biomark. Prev. 1992, 1, 383–388. [Google Scholar]
- Xiao, D.; Singh, S.V. Phenethyl isothiocyanate sensitizes androgen-independent human prostate cancer cells to docetaxel-induced apoptosis in vitro and in vivo. Pharm. Res. 2010, 27, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Barve, A.; Khor, T.O.; Hao, X.; Keum, Y.S.; Yang, C.S.; Reddy, B.; Kong, A.N. Murine prostate cancer inhibition by dietary phytochemicals—Curcumin and phenyethylisothiocyanate. Pharm. Res. 2008, 25, 2181–2189. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Tong, P.; Dashwood, W.M.; Dashwood, R.H.; Ho, E. Sulforaphane retards the growth of human PC-3 xenografts and inhibits HDAC activity in human subjects. Exp. Biol. Med. (Maywood) 2007, 232, 227–234. [Google Scholar] [PubMed]
- Bommareddy, A.; Hahm, E.R.; Xiao, D.; Powolny, A.A.; Fisher, A.L.; Jiang, Y.; Singh, S.V. Atg5 regulates phenethyl isothiocyanate-induced autophagic and apoptotic cell death in human prostate cancer cells. Cancer Res. 2009, 69, 3704–3712. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.H.; Spinks, C.A.; Doleman, J.F.; Melchini, A.; Ball, R.Y.; Mills, R.D.; Mithen, R. F The dietary isothiocyanate sulforaphane modulates gene expression and alternative gene splicing in a PTEN null preclinical murine model of prostate cancer. Mol. Cancer. 2010, 9. [Google Scholar] [CrossRef] [PubMed]
- Xiao, D.; Singh, S.V. p66Shc is indispensable for phenethyl isothiocyanate-induced apoptosis in human prostate cancer cells. Cancer Res. 2010, 70, 3150–3158. [Google Scholar] [CrossRef] [PubMed]
- Li, R.W.; Li, C.; Wang, T.T. Transcriptomic alterations in human prostate cancer cell LNCaP tumor xenograft modulated by dietary phenethyl isothiocyanate. Mol. Carcinog. 2013, 52, 426–437. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Lim, D.Y.; Kwon, G.T.; Kim, J.H.; Huang, Z.; Song, H.; Oh, Y.S.; Kang, Y.H.; Lee, K.W.; Dong, Z.; et al. Benzyl isothiocyanate inhibits prostate cancer development in the transgenic adenocarcinoma mouse prostate (TRAMP) model, which is associated with the induction of cell cycle G1 arrest. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.K.; Xiao, D.; Lew, K.L.; Hershberger, P.; Kokkinakis, D.M.; Johnson, C.S.; Trump, D.L.; Singh, S.V. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits growth of PC-3 human prostate cancer xenografts in vivo. Carcinogenesis 2003, 24, 1665–1670. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, O.J. Molecular and prognostic markers in prostate cancer. A study of cell-cycle regulators, angiogenesis and candidate markers. APMIS Suppl. 2008, 123, 5–62. [Google Scholar] [PubMed]
- Liang, J.; Slingerland, J.M. Multiple roles of the PI3K/PKB (Akt) pathway in cell cycle progression. Cell Cycle 2003, 2, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Srinivasula, S.M.; Ashwell, J.D. IAPs: What’s in a name? Mol. Cell. 2008, 30, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, D.; Qiu, Y. Role of PARP-1 in prostate cancer. Am. J. Clin. Exp. Urol. 2015, 3, 1–12. [Google Scholar] [PubMed]
- Choi, K.S. Autophagy and cancer. Exp. Mol. Med. 2012, 44, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Nandana, S.; Chung, L.W. Prostate cancer progression and metastasis: Potential regulatory pathways for therapeutic targeting. Am. J. Clin. Exp. Urol. 2014, 2, 92–101. [Google Scholar] [PubMed]
- Ganguly, S.S.; Li, X.; Miranti, C.K. The host microenvironment influences prostate cancer invasion, systemic spread, bone colonization, and osteoblastic metastasis. Front Oncol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Lomax-Browne, H.J.; Carter, T.M.; Kinch, C.E.; Hall, D.M. Molecular interactions in cancer cell metastasis. Acta Histochem. 2010, 112, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Tenta, R.; Katopodis, H.; Chatziioannou, A.; Pilalis, E.; Calvo, E.; Luu-The, V.; Labrie, F.; Kolisis, F.; Koutsilieris, M. Microarray analysis of survival pathways in human PC-3 prostate cancer cells. Cancer Genom. Proteom. 2007, 4, 309–318. [Google Scholar]
- Abbott, A. Cancer: The root of the problem. Nature 2006, 442, 742–743. [Google Scholar] [CrossRef] [PubMed]
- Bono, A.V.; Celato, N.; Cova, V.; Salvadore, M.; Chinetti, S.; Novario, R. Microvessel density in prostate carcinoma. Prostate Cancer Prostatic. Dis. 2002, 5, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Mukherji, D.; Temraz, S.; Wehbe, D.; Shamseddine, A. Angiogenesis and anti-angiogenic therapy in prostate cancer. Crit. Rev. Oncol. Hematol. 2013, 87, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.A.; Powolny, A.; Giovannucci, E.; Liao, Z.; Kenfield, S.A.; Shen, R.; Stampfer, M.J.; Clinton, S.K. Prospective study of prostate tumor angiogenesis and cancer-specific mortality in the health professionals follow-up study. J. Clin. Oncol. 2009, 27, 5627–5633. [Google Scholar] [CrossRef] [PubMed]
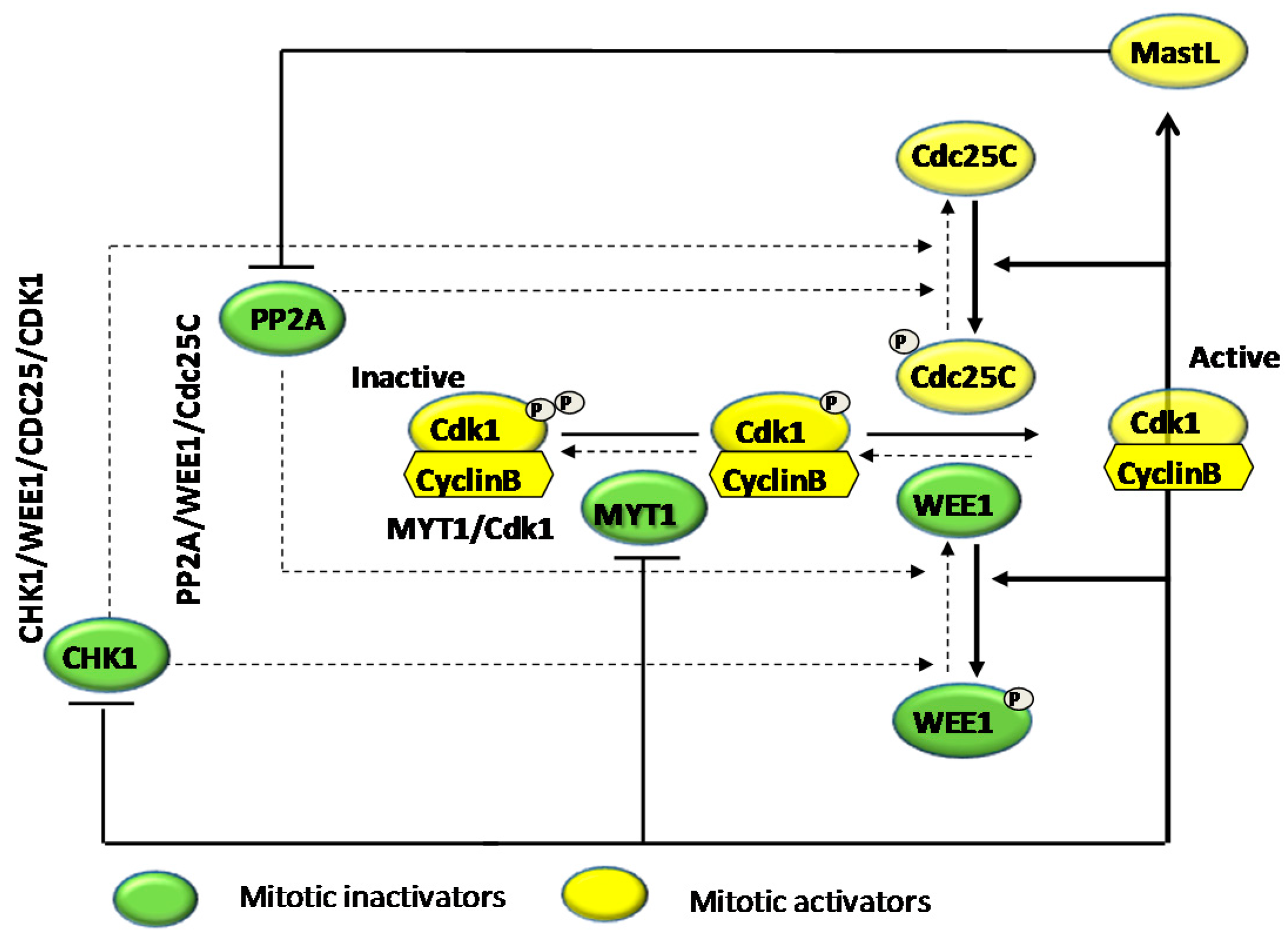
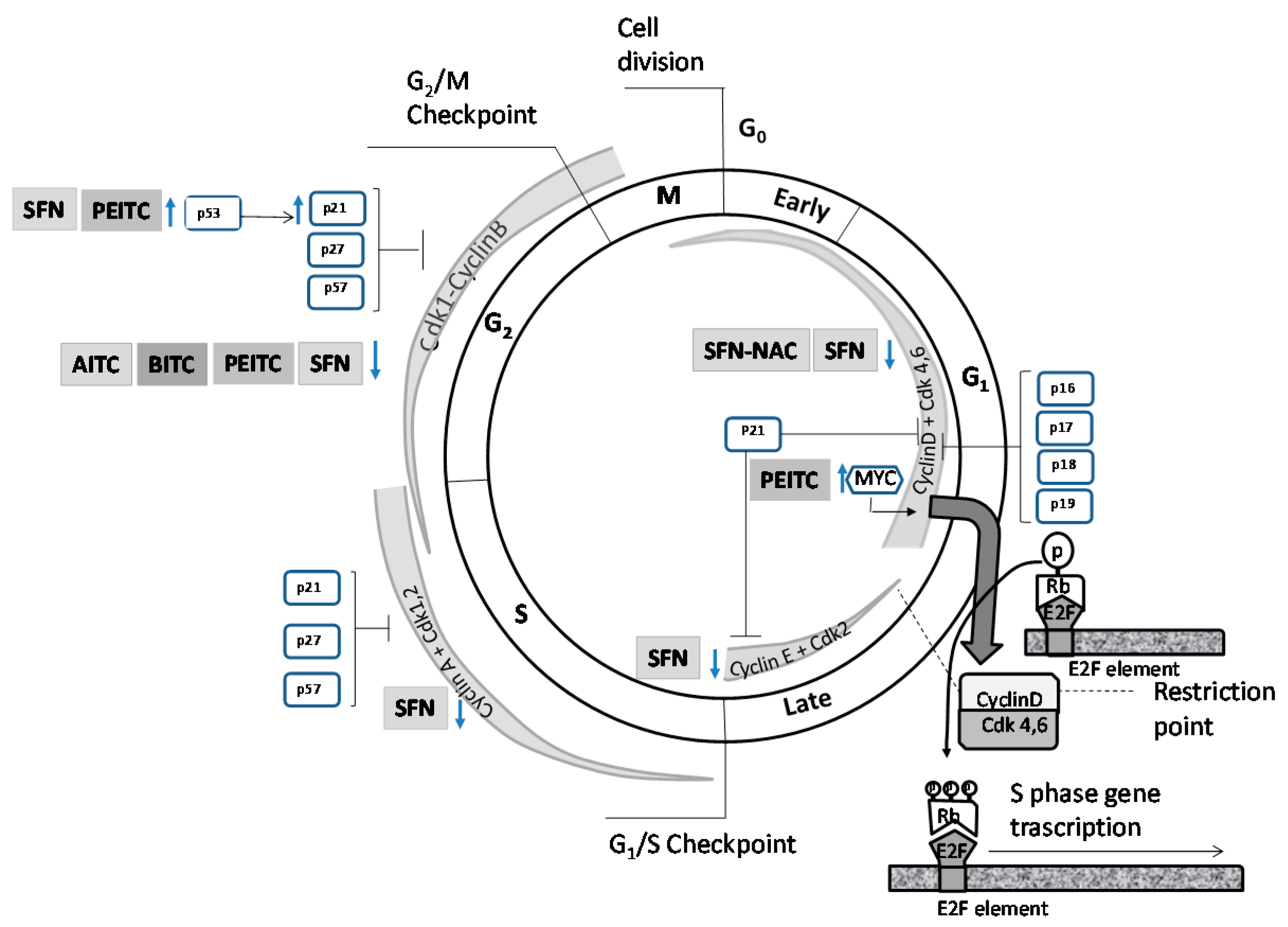
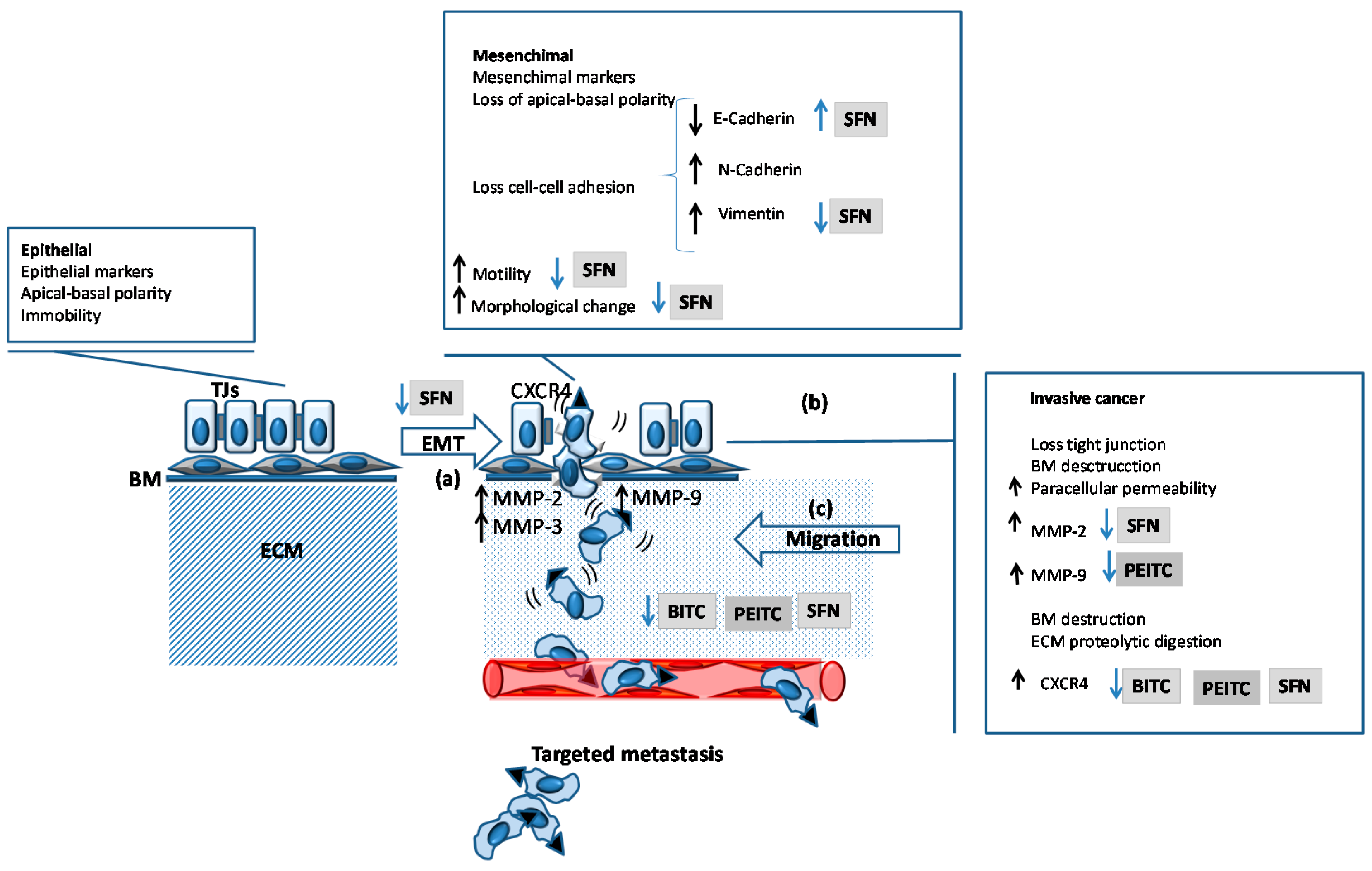

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hydrolysis Products | ||||
|---|---|---|---|---|
| Glucosinolate | Isothiocyanates (ITCs) | Nitriles | Indoles | Crops or Species |
| Aliphatic | ||||
| Glucoraphanin | Sulforaphane (SFN) | Sulforaphane nitrile | Broccoli | |
| Sinigrin | Allyl isothiocyanate (AITC) | Allyl nitrile | Kale, cabbage, Brussels sprouts, cauliflower | |
| Glucoiberin | Iberin | Kale, cabbage, cauliflower | ||
| Glucoerucin | Erucin | Cabbage, broccoli | ||
| Indolic | ||||
| Glucobrassicin | Indole-3-carbinol (I3C) | Kale, cabbage, broccoli, Brussels sprouts, cauliflower | ||
| Aromatic | ||||
| Gluconasturtiin | Phenethyl isothiocyanate (PEITC) | Water cress (Nasturtium officinalis), white mustard (Sinapis alba), turnips | ||
| Glucotropaeolin | Benzyl isothiocyanate (BITC) | Indian cress or garden nasturtium or (Tropaeolum majus) | ||
| ITC | Cells | Factor | Regulatory Partner(s) | Substrate | Effect | Ref. |
|---|---|---|---|---|---|---|
| AITC | LNCaP | ↓ Cdk1 | ↓ Cyclin B1 | ↓ Cdc25B | G2/M phase arrest | [83] |
| ↓ Cdc25C | ||||||
| AITC | PC3 | ↓ Cdk1 | ns | ↓ Cdc25B | G2/M phase arrest | [83] |
| ↓ Cdc25C | ||||||
| BITC | DU 145 | ns | ↓ Cyclin B1 | ↓ Cdc25C | G2/M phase arrest | [91] |
| ↑ WEE1 | ||||||
| PEITC-NAC | LNCaP | ↓ Cdk1 | ↓ Cyclin B1 | ns | G2/M phase arrest | [85] |
| PEITC | LNCaP | ↓ Cdk1 | ↓ Cyclin B1 | ns | G2/M phase arrest | [85] |
| PEITC | PC3 | ↓ c-Myc | ns | ↓ p21 mRNA and protein levels | G0/G1 phase arrest | [66] |
| PEITC | DU 145 | ↓ Cdk1 | ns | ↓ Cdc25C | G2/M phase arrest | [86] |
| ↑ p53 | ||||||
| ↑ WEE1 | ||||||
| PEITC | PC3 | ↓ Cdk1 | ns | ↓ Cdc25C | G2/M phase arrest | [84] |
| PEITC | LNCaP | ↓ Cdk1 | ↓ Cyclin B1 | ns | G2/M phase arrest | [85] |
| SFN-NAC | LNCaP | ns | ↓ Cyclin D1 | ns | G1 phase arrest | [95] |
| SFN | LNCaP | ↓ Cdk1 | ↓ Cyclin B1 | ↓ Cdc25C | G2/M phase arrest | [88] |
| ns | ns | ↑ p21 | G2/M phase arrest | [88] | ||
| ↑ p53 | ||||||
| ↓ Cdk4 | ns | ns | S phase arrest | [88] | ||
| ↓ Cdk6 | ns | ns | S phase arrest | [88] | ||
| SFN | BPH1, PC3 | ns | ns | ↑ p21 mRNA and protein levels | G2/M phase arrest | [96] |
| SFN | PrEC | ns | ns | ≈ p21 mRNA protein levels | ≈ G2/M phase | [96] |
| ITC | Cells | Factor | Ref. |
|---|---|---|---|
| AITC | PC3, LNCaP | ↓ Bcl-2 | [83] |
| AITC | LNCaP | ↓ Bcl-XL | [83] |
| BITC | PC3 | ↓ Bcl-2 | [98] |
| PEITC | PC3 | ↓ Bcl-2, ↓ Bcl-XL (+) caspase-8, caspase-9 pathways | [84] |
| PEITC | DU 145 | (+) caspase-8-, caspase-9-, caspase-3 -dependent pathways | [86] |
| PEITC | LNCaP, PC3 | (+) Bcl-2, (-) complex III activity | [92] |
| PEITC | PC3 | ↓ XIAP, ↓ survivin | [92] |
| PITC | DU 145 | ↓ Bcl-2, ↑ Bax (+) caspase 3 | [86] |
| SFN | PC3 | ↑ Bax, ↓ Bcl-2, modified Bax:Bcl-2 ratio | [99] |
| SFN | DU 145 | ↓ Bcl-2 | [99] |
| SFN | LNCaP, PC3 | (+) Bax | [100] |
| SFN | PC3 | ↑ Apaf-1, (+) transcriptional E2F1 | [100] |
| SFN | PC3 | ↓ Survivin | [101] |
| SFN | DU 145 | (+) Caspase | [64] |
| SFN | PC3 | (+) Caspase | [99] |
| SFN | PC3 | ↑ Bid, ↑ Smac/Diablo, ↑ ICAD, ↑ cytochrome c, ↑ c-IAP1, ↑ HSP27, ↑ Lamin A/C; ↑ BRE | [94] |
| SFN | PrEC, LNCaP, PC3 | ↑ Bax, ↑ MEK4, ↓ Lamin3 | [94] |
| SFN | LNCaP | ↓ Bim, ↓ Bmf | [94] |
| SFN | PrEC, PC3 | ↓ ASK1 | [94] |
| SFN | PrEC | ↓ cytochrome c, ↓ c-IAP1, ↓ HSP27 | [94] |
| SFN | PC3, BPH1 | (+) Multicaspase | [96] |
| SFN | BPH1 | ↓ HDAC2 | [96] |
| SFN | BPH1, LNCaP, PC3 | (−) HDAC, ↓ HDAC3, ↓ HDAC6 | [96] |
| SFN | PC3 | ↓ Survivin | [101] |
| SFN | BPH1, LNCaP | ↓ HDAC4 | [96] |
| SFN | PC3 | ↓ Bid, ↓ Smac/Diablo, ↓ ICAD | [94] |
| ITC | Factor | In Vitro | In Vivo |
|---|---|---|---|
| PEITC | Bak | ↑ [149] | ↓ [150] |
| PEITC | Bcl-XL | ↓ [84,149] | ↑ [150] |
| PEITC | Vimentin | ↑ [151] | ↓ [151] |
| SFN | Bcl-XL | ≈ [99] | ↓ [147] |
| SFN | Bid | ≈ [99] | ↑ [152] |
| ITC | Factor | Model (Cell Line) | Ref. |
|---|---|---|---|
| BITC | ↓ Ki-67 | TRAMP | [161] |
| BITC | ↓ Cyclin D1 | TRAMP | [161] |
| BITC | ↓ Cyclin A | TRAMP | [161] |
| BITC | ↓ Cdk2 | TRAMP | [161] |
| PEITC | ↓ Akt | TRAMP | [155] |
| PEITC | ↓ FKHR | TRAMP | [155] |
| PEITC | ↑ IGFBP3 | Subcutaneous xenograft (LNCaP) | [160] |
| PEITC | ≈ Ki-67 | Subcutaneous xenograft (LNCaP) | [148] |
| PEITC | ↓ Ki-67 | TRAMP | [12] |
| PEITC | ≈ PCNA | Subcutaneous xenograft (LNCaP) | [148] |
| PEITC | ↓ PCNA | TRAMP | [155] |
| PEITC | ↓ PCNA | Subcutaneous xenograft (PC3) | [154] |
| PEITC | ↓ PDK1 | TRAMP | [155] |
| PEITC-NAC | ↓ Cyclin D1 | Subcutaneous xenograft (PC3) | [65] |
| PEITC-NAC | ↓ Cyclin E | Subcutaneous xenograft (PC3) | [65] |
| PEITC-NAC | ↑ p21 | Subcutaneous xenograft (PC3) | [65] |
| PEITC-NAC | ↑ p27 | Subcutaneous xenograft (PC3) | [65] |
| PEITC-NAC | ↓ pRb | Subcutaneous xenograft (PC3) | [65] |
| SFN | ↓ COX-2 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ Cyclin D1 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ IL-6 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ IL-8 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ Ki-67 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ Ki-67 | CAM xenograft (PC3) | [123] |
| SFN | ↓ PCNA | TRAMP | [152] |
| SFN | ↓ PCNA | Orthotopic assay (PC3) | [147] |
| ITC | Factor | Model (Cell Line) | Ref. |
|---|---|---|---|
| AITC | ≈ Bax | Subcutaneous xenograft (PC3) | [162] |
| AITC | ↓ Bcl-2 | Subcutaneous xenograft (PC3) | [162] |
| AITC | ≈ Bcl-XL | Subcutaneous xenograft (PC3) | [162] |
| AITC | ↑ Bid | Subcutaneous xenograft (PC3) | [162] |
| AITC | ≈ Clusterin | TRAMP | [12] |
| PEITC | ↑ Bad | TRAMP | [155] |
| PEITC | ≈ Bad | Subcutaneous xenograft (PC3) | [146] |
| PEITC | ↓ Bak | Subcutaneous xenograft (PC3) | [150] |
| PEITC | ≈ Bak | Subcutaneous xenograft (PC3) | [154] |
| PEITC | ↑ Bax | Subcutaneous xenograft (PC3) | [150] |
| PEITC | ≈ Bax | Subcutaneous xenograft (LNCaP) | [148] |
| PEITC | ≈ Bax | Subcutaneous xenograft (PC3) | [154] |
| PEITC | ↓ Bcl-2 | Subcutaneous xenograft (PC3) | [154] |
| PEITC | ↑ Bcl-XL | Subcutaneous xenograft (PC3) | [150] |
| PEITC | ↑ Bid | Subcutaneous xenograft (PC3) | [150] |
| PEITC | ↓ Bok | Subcutaneous xenograft (PC3) | [150] |
| PEITC | ≈ Caspasa 3 | Subcutaneous xenograft (LNCaP) | [148] |
| PEITC | ↑ Caspase-3 | TRAMP | [155] |
| PEITC | ↑ Caspase-3 | Subcutaneous xenograft (PC3) | [146] |
| PEITC | ↓ Clusterin | TRAMP | [12] |
| PEITC | ↓ GSK3βα | Subcutaneous xenograft (PC3) | [146] |
| PEITC | ↓ IκBα | Subcutaneous xenograft (PC3) | [146] |
| PEITC | ↓ IKKβα | Subcutaneous xenograft (PC3) | [146] |
| PEITC | ≈ p66Shc | Subcutaneous xenograft (PC3) | [159] |
| PEITC | ↑ PARP | Subcutaneous xenograft (PC3) | [146] |
| PEITC | ↓ Pin1 | Subcutaneous xenograft (PC3) | [159] |
| PEITC | ↑ RANBP1 | Subcutaneous xenograft (LNCaP) | [160] |
| PEITC | ↓ Survivin | TRAMP | [92] |
| PEITC | ↓ XIAP | TRAMP | [92] |
| PEITC | ≈ XIAP | Subcutaneous xenograft (PC3) | [154] |
| PEITC-NAC | ↑ PARP | Subcutaneous xenograft (PC3) | [65] |
| SFN | ↑ Bad | TRAMP | [152] |
| SFN | ↑ Bak | Orthotopic assay (PC3) | [147] |
| SFN | ↑ Bak | TRAMP | [152] |
| SFN | ↑ Bax | Orthotopic assay (PC3) | [147] |
| SFN | ↑ Bax | Subcutaneous xenograft (PC3) | [99,156] |
| SFN | ↑ Bax | TRAMP | [152] |
| SFN | ↓ Bcl-2 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ Bcl-2 | Subcutaneous xenograft (PC3) | [99] |
| SFN | ≈ Bcl-XL | Subcutaneous xenograft (PC3) | [99] |
| SFN | ↓ Bcl-XL | Orthotopic assay (PC3) | [147] |
| SFN | ↑ Bid | Subcutaneous xenograft (PC3) | [99] |
| SFN | ↑ Bid | TRAMP | [152] |
| SFN | ↑ Caspase 3 | CAM xenograft (PC3) | [123] |
| SFN | ↑ Caspase-3 | Orthotopic assay (PC3) | [147] |
| SFN | ↑ Caspase-8 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ Clusterin | TRAMP | [12] |
| SFN | ↓ Mcl-1 | TRAMP | [152] |
| SFN | ↑ PARP | TRAMP | [152] |
| ITC | Factor | Model (Cell Line) | Ref. |
|---|---|---|---|
| PEITC | ↑ LC3 | Subcutaneous xenograft (PC3) | [157] |
| PEITC | ↑ LC3 | TRAMP | [12] |
| PEITC | ↓ p62 | TRAMP | [12] |
| ITC | Factor | Model (Cell Line) | Reference |
|---|---|---|---|
| PEITC | ↑ E-cadherin | TRAMP | [12] |
| PEITC | ↑ Fibronectin 1 | Subcutaneous xenograft (LNCaP) | [160] |
| PEITC | ↓ Integrin β6 | Subcutaneous xenograft (LNCaP) | [160] |
| PEITC | ↑ Notch2 | Subcutaneous xenograft (PC3) | [129] |
| PEITC | ↑ Notch2 | TRAMP | [129] |
| PEITC | ↓ Vimentin | TRAMP | [151] |
| PEITC | ↓ CSC markers * | Subcutaneous xenograft (PC3) | [122] |
| SFN | ↓ CSC markers * | CAM xenograft (PC3) | [123] |
| SFN | ≈ E-cadherin | TRAMP | [152] |
| SFN | ↓ MMP-2 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ MMP-7 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ MMP-9 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ MMP-14 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ NF-κB | Orthotopic assay (PC3) | [147] |
| SFN | ↓ TGF-β1 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ uPAR | Orthotopic assay (PC3) | [147] |
| ITC | Factor | Model (Cell Line) | Reference |
|---|---|---|---|
| PEITC | ≈ PECAM-1/CD31 | TRAMP | [12] |
| PEITC | ↓ PECAM-1/CD31 | Subcutaneous xenograft (LNCaP) | [148] |
| PEITC | ≈ VEGF | Subcutaneous xenograft (LNCaP) | [148] |
| SFN | ≈ PECAM-1/CD31 | TRAMP | [152] |
| SFN | ↓ Akt | Orthotopic assay (PC3) | [147] |
| SFN | ↓ ERK1/2 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ FOXO3a | Orthotopic assay (PC3) | [147] |
| SFN | ↓ HIF-1 α | Orthotopic assay (PC3) | [147] |
| SFN | ↓ IL-6 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ IL-8 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ TGF-β1 | Orthotopic assay (PC3) | [147] |
| SFN | ↓ VEGF | Orthotopic assay (PC3) | [147] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novío, S.; Cartea, M.E.; Soengas, P.; Freire-Garabal, M.; Núñez-Iglesias, M.J. Effects of Brassicaceae Isothiocyanates on Prostate Cancer. Molecules 2016, 21, 626. https://doi.org/10.3390/molecules21050626
Novío S, Cartea ME, Soengas P, Freire-Garabal M, Núñez-Iglesias MJ. Effects of Brassicaceae Isothiocyanates on Prostate Cancer. Molecules. 2016; 21(5):626. https://doi.org/10.3390/molecules21050626
Chicago/Turabian StyleNovío, Silvia, María Elena Cartea, Pilar Soengas, Manuel Freire-Garabal, and María Jesús Núñez-Iglesias. 2016. "Effects of Brassicaceae Isothiocyanates on Prostate Cancer" Molecules 21, no. 5: 626. https://doi.org/10.3390/molecules21050626
APA StyleNovío, S., Cartea, M. E., Soengas, P., Freire-Garabal, M., & Núñez-Iglesias, M. J. (2016). Effects of Brassicaceae Isothiocyanates on Prostate Cancer. Molecules, 21(5), 626. https://doi.org/10.3390/molecules21050626