3. Materials and Methods
All reactions in organic medium were performed in standard oven dried glassware under an inert atmosphere of nitrogen using freshly distilled solvents stored over molecular sieves. Solvents were deoxygenated when necessary by bubbling nitrogen through the solution. All reagents were used as supplied without prior purification and obtained from Sigma-Aldrich Chemical Co. (Toronto, ON, Canada) Reactions were monitored by analytical thin-layer chromatography (TLC) using silica gel 60 F254 precoated plates (E. Merck, Darmstadt, Germany) and compounds were visualized by 254 nm light and/or by dipping into a mixture of sulfuric acid and methanol in water or into a mixture of KMnO4 and K2CO3 in water followed by gentle warming with a heat-gun. Purifications were performed by flash column chromatography using silica gel from Canadian Life Science (60 Å, 40–63 µm) (Peterborough, ON, Canada) with the indicated eluent. 1H-NMR and 13C-NMR spectra were recorded at 300 and/or 600 MHz and 75 and/or 150 MHz, respectively, on a Bruker spectrometer (300 MHz and 600 MHz) (Milton, ON, Canada) and Varian spectrometer (600 MHz) (Milton, ON, Canada). All NMR spectra were measured at 25 °C in indicated deuterated solvents. Proton and carbon chemical shifts (δ) are reported in ppm and coupling constants (J) are reported in Hertz (Hz). The resonance multiplicity in the 1H-NMR spectra are described as “s” (singlet), “d” (doublet), “t” (triplet), and “m” (multiplet) and broad resonances are indicated by “broad”. Residual protic solvent of CDCl3 (1H, δ 7.27 ppm; 13C, δ 77.0 ppm (central resonance of the triplet)), D2O (1H, δ 4.80 ppm and 30.9 ppm for CH3 of Acetone for 13C spectra), MeOD (1H, δ3.30 ppm and 13C, δ 49.0 ppm). Two-dimensional homonuclear correlation 1H-1H COSY experiments were used to confirm NMR peak assignments. Fourier transform infrared (FTIR) spectra were obtained with Thermo-scientific, Nicolet model 6700 (Mississauga, ON, Canada) equipped with Alternate Total Reflection (ATR). The absorptions are given in wave numbers (cm−1). Accurate mass measurements (HRMS) were performed on a LC-MSD-TOF instrument from Agilent Technologies (Santa Clara, CA, USA) in positive electrospray mode. Either protonated molecular ions [M + nH]n+ or adducts [M + nX]n+ (X = Na, K, NH4) were used for empirical formula confirmation. Gel Permeation Chromatography (GPC) was performed using chloroform and THF as the eluent, at 40 °C with a 1 mL/min flow rate on a Viscotek VE 2001 GPCmax (SEC System) (Viscotek/Malvern Instruments Ltd, Worcestershire, UK) with Wyatt DSP/Dawn EOS (Wyatt Technology, Santa Barbara, CA, USA) and refractive index RI/LS system as detectors (Wyatt Technology, Santa Barbara, CA, USA). In addition, 2 PLGel mixed B LS (10 μm, 300 × 7.5 mm) and Light Scattering-Multiangle Laser Light Scattering (LS-MALLS) detection with performances verified with polystyrene 100 kDa and 2000 kDa were used to determine the number-average molecular weight (Mn) and polydispersity index (MW/Mn). Calculations were performed with Zimm Plot (model).
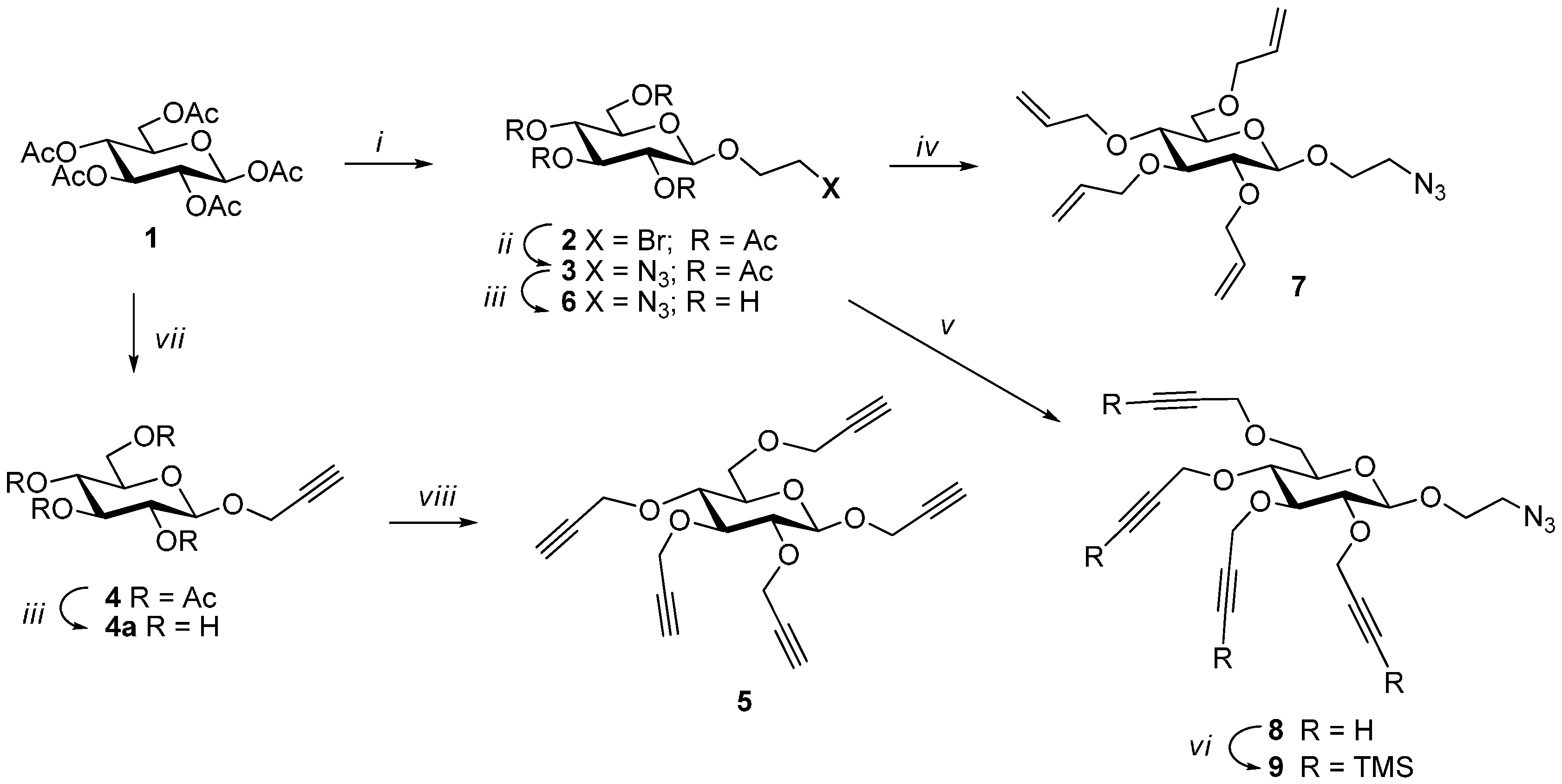
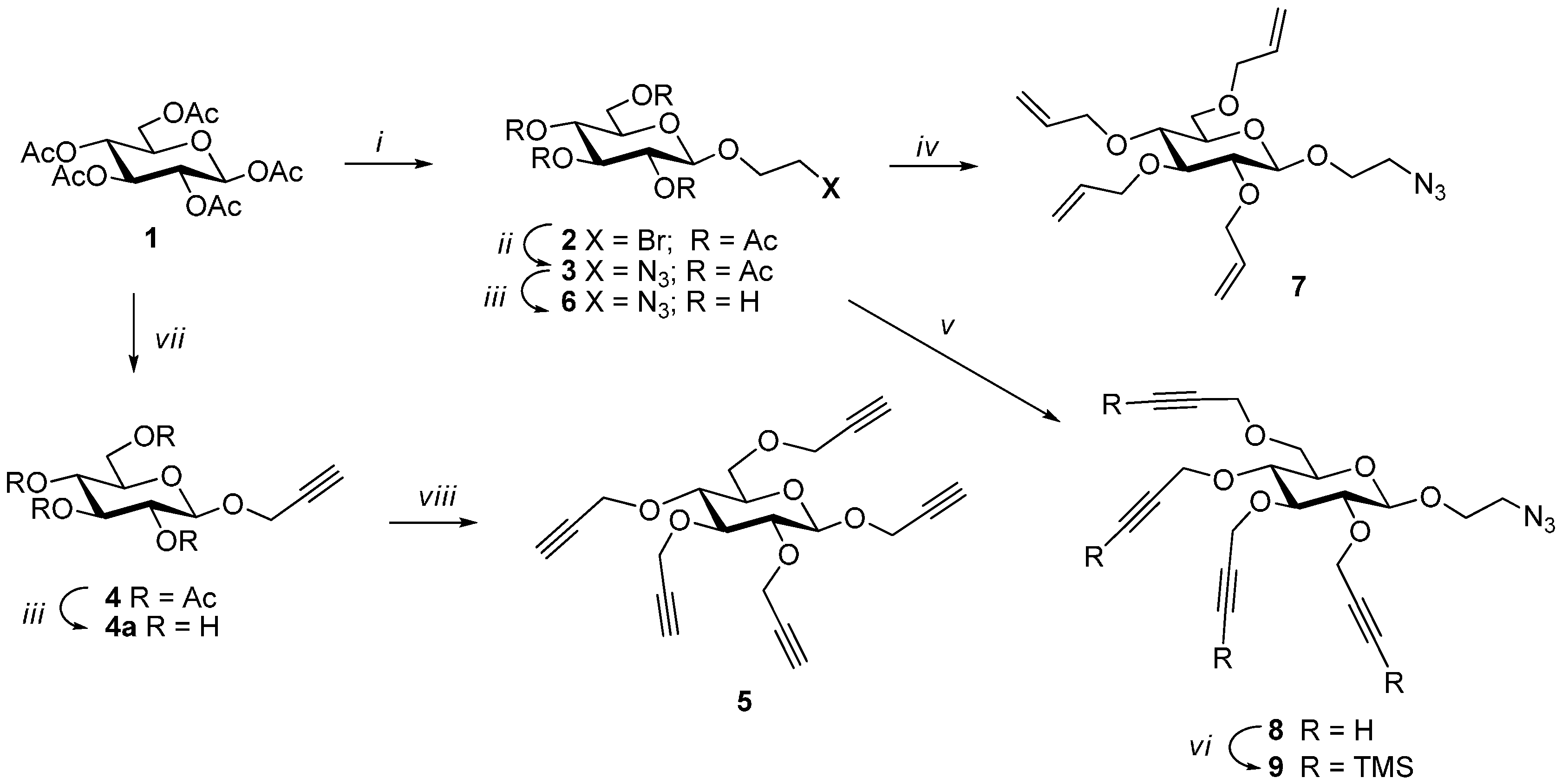
Syntheses of 1,2,3,4,6-penta-O-acetyl-β-d-glucopyranose (1), 2-bromoethyl 2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (2), 2-azidoethyl 2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (3), 2-azidoethyl 2,3,4,6-tetra-O-allyl-β-d-glucopyranoside (7) were performed following the literature procedures in good yields.
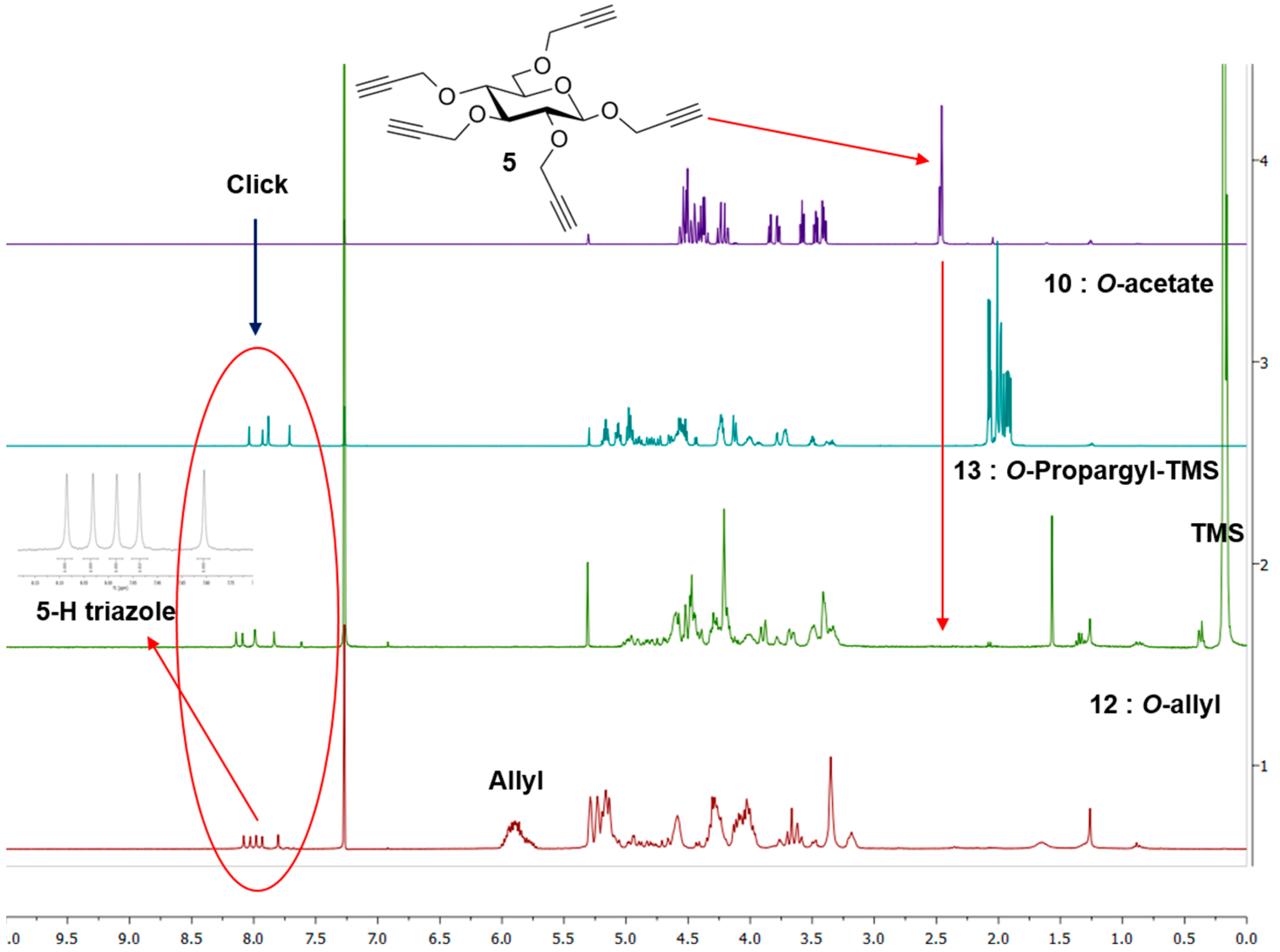
Propargyl 2,3,4,6-tetra-O-propargyl-β-d-glucopyranoside (5). To a solution of prop-2-ynyl-β-d-glucopyranoside (4a) (310 mg, 1.42 mmol) in DMF (5 mL) cooled to 0 °C was added sodium hydride (60% in oil, 530 mg, 13.2 mmol). The mixture was stirred for 30 min at this temperature. Propargyl bromide (3 mL, 20.2 mmol) was added dropwise. The mixture was stirred at 0–5 °C for 1 h. It was quenched with NH4Cl solution. It was extracted with ethyl acetate and washed with brine. The extract was dried over sodium sulfate and evaporated under vacuum. The crude product was chromatographed over silica gel (hexane-ethyl acetate (0–30%) as eluent) yielding 5 as light yellow oil (500 mg, 1.35 mmol, 95%). Rf = 0.22 (Hexanes/EtOAc 2:1); 1H-NMR (600 MHz, CDCl3): δ (ppm) 4.59–4.32 (m, 9H, H-1 and OCH2), 4.28–4.17 (m, 2H), 3.84 (dd, 1H, 3J6a,5 = 1.9 Hz, 3J6a,6b = 10.8 Hz, H-6a), 3.77 (dd, 1H, 3J6b,5 = 4.7 Hz, 3J6b,6a = 10.8 Hz, H-6b), 3.58 (dd, 1H, 3J3,2 = 8.8 Hz, 3J3,4 = 9.5 Hz, H-3), 3.51–3.37 (m, 3H) and 2.51–2.41 (m, 5H). 13C-NMR (75 MHz, CDCl3): δ (ppm) 100.7 (C-1), 83.2, 80.9, 80.0, 79.9, 79.7, 79.5, 78.6, 75.9, 75.1, 74.7, 74.4, 74.3, 74.2, 74.1, 68.3, 60.2, 60.0, 59.3, 58.6, 55.9. FT-IR: νmax (neat)/cm−1 3289 (br, C≡CH) and 2119 (≡CH). ESI+-HRMS: [M + H]+ calcd for C21H23O6, 371.1467; found: 371.1480.
2-Azidoethyl 2,3,4,6-tetra-O-propargyl-β-d-glucopyranoside (8). 2-azidoethyl 2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (300 mg, 0.72 mmol) was dissolved in dry methanol (6 mL) and NaOMe (1.1 M) was added slowly to bring pH to 9. The mixture was stirred at room temperature for 2.5 h. Acidic resin (IR-120) was added to neutralize the base. The mixture was filtered and evaporated to obtain fully deprotected glucoside derivative. Before being used in the next step, it was placed under vacuum for 1 h. It was dissolved in DMF (3 mL) and the solution was cooled to 0 °C. Sodium hydride (60% in oil, 240 mg, 6.0 mmol) was added. The mixture was stirred at 0 °C for 15 min. Propargyl bromide (80% in toluene, 1.5 mL, 10.1 mmol) was added dropwise. The mixture was stirred at 0 °C for 1 h. Saturated NH4Cl solution was added to quench the reaction. It was extracted with ethyl acetate. The extract was washed with brine, dried over MgSO4 and evaporated. 2-Azidoethyl 2,3,4,6-tetra-O-propargyl-β-d-glucopyranoside (8) was obtained by flash chromatography over silica gel (hexane-ethyl acetate (0–30%) as eluent) as light yellow oil (250 mg, 0.62 mmol, 87%). 1H-NMR (300 MHz, CDCl3) δ (ppm) 4.65–4.50 (m, 3H), 4.50–4.37 (m, 3H), 4.34 (d, J = 7.8 Hz, 1H), 4.31–4.16 (m, 2H), 4.04 (ddd, J = 10.6, 5.4, 3.8 Hz,1H), 3.86 (dd, J = 10.7, 1.8 Hz, 1H), 3.79 (d, J = 4.6 Hz, 1H), 3.77–3.65 (m, 1H), 3.63–3.54 (m, 1H), 3.52–3.35 (m, 5H), 2.53–2.42 (m, 4H); 13C NMR (150 MHz, CDCl3) δ (ppm) 103.1, 83.3, 81.2, 80.0, 79.9, 79.8, 79.5, 76.1, 74. 7, 74.4, 74.3, 74.30, 74.2, 68.5, 68.2, 60.3, 60.0, 59.5, 58.7, 50.9. FT-IR: νmax (neat)/cm−1 3290s (br, C≡CH), 2104 (azide). ESI+-HRMS: [M + NH4]+ calcd for C20H27N4O6, 419.1915; found: 419.1925.
2-Azidoethyl 2,3,4,6-tetra-O-trimethylsilylpropargyl-β-d-glucopyranoside (9) A mixture of 2-azidoethyl 2,3,4,6-tetra-O-propargyl-β-d-glucopyranoside (8) (700 mg, 1.74 mmol), AgCl (300 mg, 2.1 mmol) and DBU (3.5 g, 23 mmol) in DCM (25 mL) was brought to 40 °C. Chlorotrimethyl silane (3.1 mL, 24.4 mmol) was added dropwise. It was stirred at 45 °C for 18 h. The reaction mixture was diluted with DCM/H2O (300 mL, 2:1 v/v) and extracted with DCM (200 mL). The extract was washed with water (50 mL) and with brine (50 mL), dried and evaporated. The crude material was passed through a column of silica gel (hexane-ethyl acetate (0–10%) as eluent) to obtain 2-azidoethyl 2,3,4,6-tetra-O-trimethylsilylpropargyl-β-d-glucopyranoside (9) as colorless oil (1.0 g, 1.45 mmol, 83%). 1H-NMR (300 MHz, CDCl3) δ (ppm) 4.63–4.28 (m, 7H), 4.22 (s, 2H), 4.03 (ddd, J = 10.6, 5.5, 4.0 Hz, 1H), 3.93 (d, J = 10.0 Hz, 1H), 3.76–3.60 (m, 2H), 3.60–3.39 (m, 5H), 3.33 (dd, J = 8.9, 7.9 Hz, 1H), 0.19 (s, 35H); 13C-NMR (150 MHz, CDCl3) δ (ppm) 103.1, 102.1, 102.0, 101.8, 101.5, 91.5, 91.2, 91.1, 90.9, 83.7, 81.7, 75.9, 74.5, 69.3, 68.2, 61.0, 60.5, 60.4, 59.5, 50.9, −0.1. FT-IR: νmax (neat)/cm−1 2176 (substituted propargyl) and 2103 (azide). ESI+-HRMS: [M + NH4]+ calcd for C32H59N4O6Si4, 707.3487; found: 707.3506.
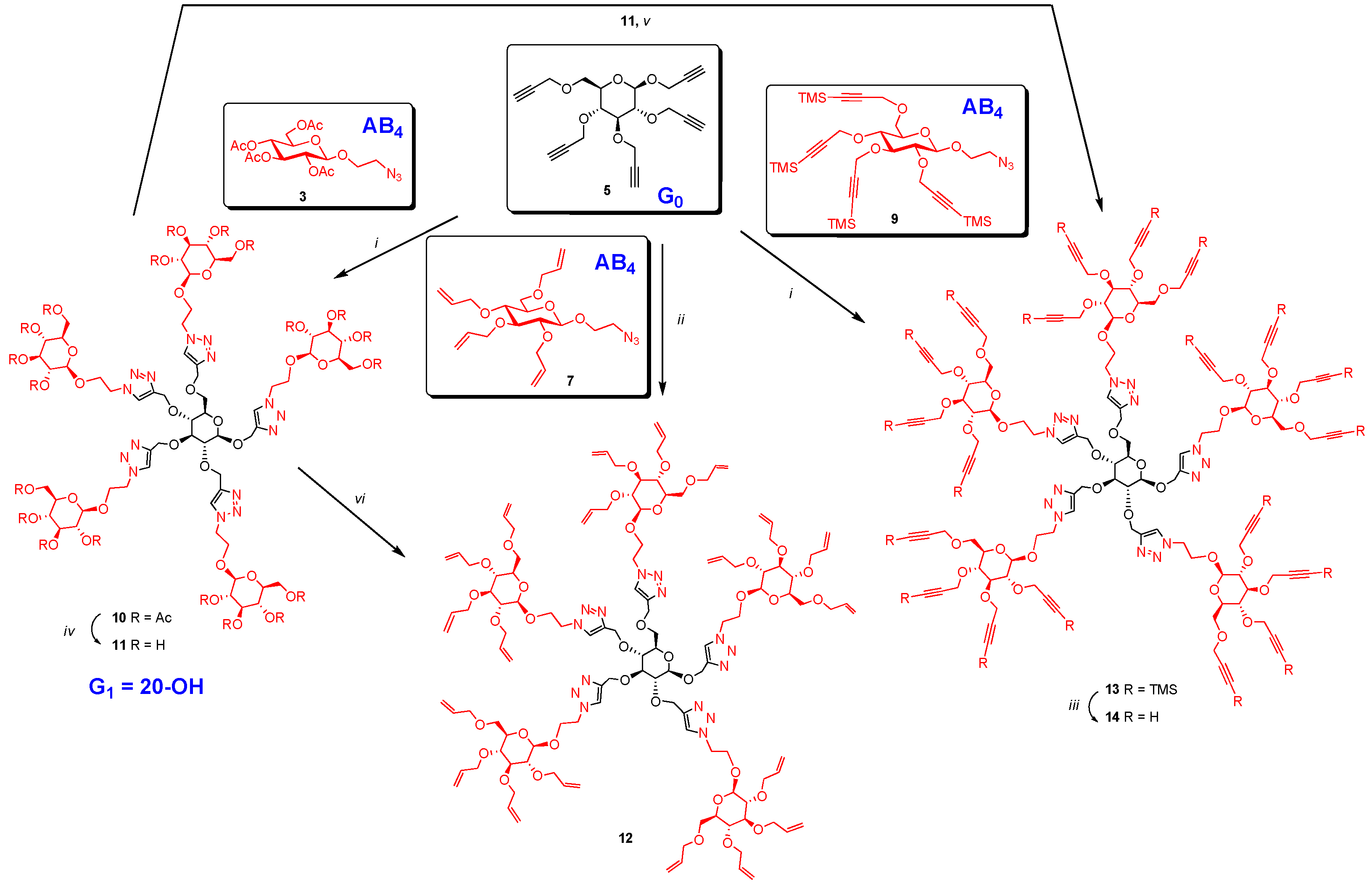
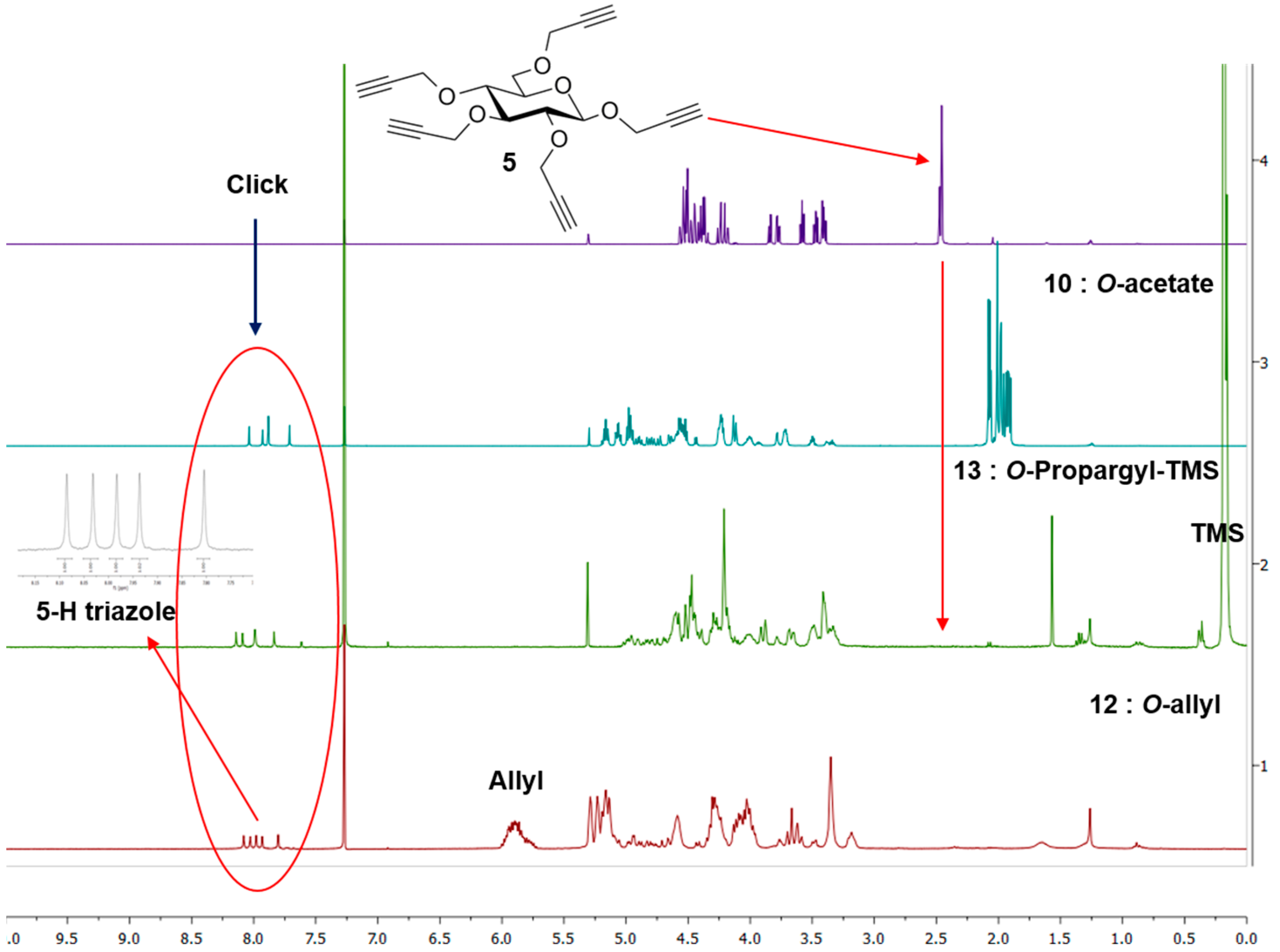
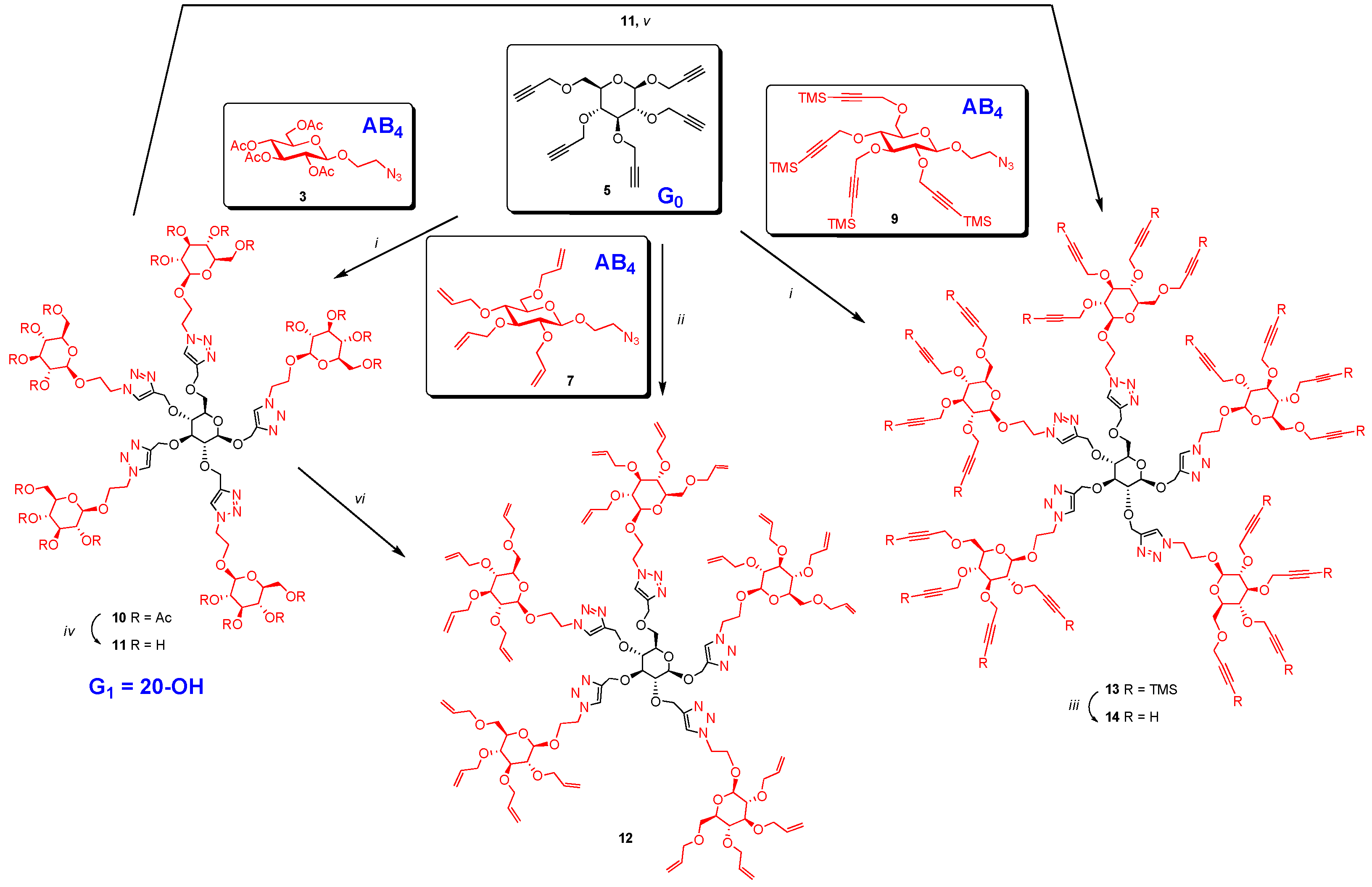
Synthesis of 10. A mixture of 5 (13 mg, 0.035 mmol), 2-azidoethyl 2,3,4,6-tetra-O-acetyl-β-d-glucopyranoside (3) (97 mg, 0.233 mmol) and CuI·P(OEt)3 (7 mg, 0.02 mmol) in dry toluene (2 mL) was held at 70 °C with stirring for 20 h. It was cooled and diluted with ethyl acetate (100 mL). The solution was washed with EDTA (2 × 5 mL) and with a mixture of aqueous NaHCO3 and brine (10 mL), dried and evaporated. The crude material was passed through a column of silica gel with hexane-ethyl acetate (1:1) and 0–6% MeOH in DCM as eluents yielding 10 as colorless oil (73 mg, 0.03 mmol, 85%). 1H-NMR (600 MHz, CDCl3) δ (ppm) 8.04 (s, 1H), 7.93 (s, 1H), 7.88 (s, 2H), 7.71 (s, 1H), 5.22–5.13 (m,5H), 5.06 (td, J = 9.7, 5.4 Hz, 5H), 4.99–4.95 (m, 8H), 4.92–4.87 (m, 2H), 4.82 (d, J = 12.6 Hz, 1H), 4.78 (d, J = 12.0 Hz, 1H), 4.73 (d, J = 12.5 Hz, 1H), 4.69–4.48 (m, 18H), 4.44 (d, J = 7.8 Hz, 1H), 4.26–4.21 (m, 11H), 4.16–4.10 (m, 5H), 4.05–3.89 (m, 5H), 3.78 (br s, 2H), 3.76–3.69 (m, 5H), 3.55–3.43 (m, 2H), 3.43–3.29 (m, 2H), 2.37–1.71 (m, 60H); 13C-NMR (150 MHz, CDCl3) δ (ppm) 170.6, 170.1, 169.4, 169.3, 169.2, 144.8, 144.7, 144.8, 144.5, 144.3, 124.8, 124.7, 124.6, 124.5, 124.2, 102.3, 100.6, 100.5, 100.4, 83.9, 81.7, , 74.4, 72.6, 72.5, 72.5, 72.48, 72.4, 71.9, 71.8, 70.8, 70.7, 68.8, 68.3, 68.2, 67.8, 67.7, 67.6, 66.4, 65.7, 65.7, 64.5, 62.8, 61.7, 61.72, 61.69, 53.4, 49.8, 49.76, 49.70, 20.7, 20.6, 20.5. ESI+-HRMS: [M + 2H]2+ calcd for C101H139N15O56, 1229.4246; found: 1229.4255.
Synthesis of 11. To a solution of 10 (40 mg, 0.016 mmol) in methanol-H2O (10:1) (3 mL) was added sodium methoxide in methanol (0.59 M) slowly to bring pH to 9. It was stirred for 18 h at room temperature. It was stirred with acidic resin (IR-120) to bring the pH around 7. The resin was filtered off and filtrate was evaporated, dissolved in water and lyophilized to give 11 as a fluffy solid (26 mg, 0.16 mmol, 98%). 1H-NMR (600 MHz, D2O) δ (ppm) 8.14 (s, 1H), 8.13 (s, 1H), 8.05 (s, 1H), 8.04 (s, 1H), 8.01 (s, 1H), 4.97 (d, J = 12.7 Hz, 1H), 4.83 (ddd, J = 12.5, 8.9, 3.4 Hz, 4H), 4.74–4.58 (m, 16H), 4.59–4.50 (m, 1H), 4.48–4.37 (m, 6H), 4.35–4.23 (m, 6H), 4.15–4.02 (m, 6H), 3.91–3.84 (m, 6H), 3.74–3.61 (m, 9H), 3.60–3.55 (m, 1H), 3.52 (dd, J = 15.8, 6.8 Hz, 1H), 3.49–3.37 (m, 12H), 3.36–3.27 (m, 5H), 3.26–3.16 (m, 5H); 13C-NMR (150 MHz, D2O + acetond-d6) δ (ppm) 144.4, 144.1, 144.1, 144.0, 126.6, 126.5, 126.3, 126.2, 126.1, 103.0, 102.9, 102. 8, 102.1, 83.4, 81.3, 77.1, 76.5, 76.2, 76.1, 73.9, 73.5, 73.4, 73.4, 72.6, 70.1, 70.1, 68.6, 68.5, 68.3, 66.0, 65.3, 65.2, 63.8, 63.0, 62.7, 61.3, 50.9, 50.8, 50.8, 28.8. ESI+-HRMS: [M + H]+ calcd for C61H98N15O36, 1616.6320; found: 1616.6293.
Synthesis of 12. Procedure A: To a solution of 11 (12 mg, 0.007 mmol) in dry DMF (3 mL) at 0 °C was added sodium hydride (60% in oil, 80 mg, 2 mmol). It was stirred at this temperature for 1 h and then at room temperature for another 2 h. The mixture was cooled back to 0 °C and allyl bromide (200 µL 2.3 mmol) was added slowly. After the addition the mixture was stirred at room temperature for 18 h. Saturated NH4Cl was added to quench the reaction. It was extracted with EtOAc (50 mL). The extract was washed with brine (5 mL), dried over MgSO4 and evaporated. The crude material was chromatographed over silica gel using hexane-EtOAc (3:2) and 5% to 10% methanol in DCM as eluents yielding perallylated product 12 as colorless oil (14 mg, 0.058 mmol, 77%).
Procedure B: Compound 12 could also be obtained by treating 5 (15 mg, 0.04 mmol) and 7 (135 mg, 0.33 mmol) under CuAAC reaction condition using Cu(I)·P(OEt)3 (8 mg, 0.02 mmol) as catalyst in toluene (2.4 mL) by irradiating under microwave at 70 °C for 4 h. Product 12 was isolated by column chromatography after usual work-up in 80% yield (78 mg, 0.032 mmol). 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.08, 8.03, 7.98, 7.93 and 7.80 (5 × s, 5H), 5.98–5.72 (m, 20H), 5.29–5.06 (m, 39H), 4.99–4.75 (m, 8H), 4.71–4.53 (m, 13H), 4.43–4.41 (m, 2H), 4.35–4.19 (m, 24H), 4.13–3.96 (m, 29H), 3.76–3.58 (m, 12H), 3.49–3.47 (m, 3H), 3.35 (broad s, 17H), 3.18 (broad s, 6H); 13C-NMR (150 MHz, CDCl3) δ (ppm) 144.7, 135.2, 134.9, 134.8, 134.7, 134.6, 124.5, 117.2, 117.0, 116.9, 116.7, 103.2, 84.0, 81.2, 74.7, 74.4, 73.8, 73.5, 72.4, 68.7, 67.8, 50.1, 32.0, 29.7, 22.7. ESI+-HRMS: [M + 2H]2+ calcd for C121H179N15O36, 1209.6288; found: 1209.6329.
Synthesis of 13. A mixture of 2-azidoethyl 2,3,4,6-tetra-O-trimethylsilylpropargyl-β-d-glucopyranoside (9) (50 mg, 0.07 mmol), 5 (3.5 mg, 0.01 mmol), CuI·P(OEt)3 (3.5 mg, 0.01 mmol) and N,N-diisopropylethylamine (DIPEA) (30 µL, 0.17 mmol) in dry toluene (2 mL) was held at 65–70 °C with stirring for 20 h. The mixture was cooled and diluted with ethyl acetate (100 mL). It was washed with EDTA (3 × 5 mL), NaHCO3 solution (2 × 5 mL) and brine (5 mL). The extract was dried over MgSO4 and evaporated. The crude was purified by column chromatography over silica gel (hexane-ethyl acetate (1:1) and 2%–5% MeOH in toluene as eluents) yielding TMS-protected dendrimer 13 (32 mg, 0.009 mmol, 90%). 1H-NMR (300 MHz, CDCl3) δ (ppm) 8.14 (s, 1H), 8.09 (s, 1H), 7.99 (s, 2H), 7.84 (s, 1H), 5.03–4.74 (m, 9H), 4.73–4.36 (m, 35H), 4.36–4.11 (m, 30H), 4.07–3.85 (m, 11H), 3.84–3.55 (m, 9H), 3.54–3.29 (m, 24H), 0.17 (broad signal, 180H); 13C-NMR (150 MHz, CDCl3) δ (ppm) 144.8, 144.7, 144.5, 144.4, 144.2, 124. 7, 124.6, 124.4, 124.3, 124.0, 103.1, 103.0, 102.901, 102.0, 101.90, 101.80, 101.40, 91.2, 91.1, 91.0, 83.60, 81.1, 81.0, 80.9, 77.4, 75.9, 75.8, 74.5, 74.3, 68.9, 68.0, 67.9, 66.5, 65.9, 65.7, 64.6, 62.9, 61.0, 60.6, 60.2, 60.1, 60.0, 59.5, 50.0, 49.9, 49.8, 49.8, 30.9, 29.7, −0.1. FT-IR: νmax (neat)/cm−1 2177 (substituted propargyl).
Synthesis of 14. To a solution of 13 (49 mg, 0.013 mmol) in THF (2 mL) was added TBAF (1 M in THF, 300 µL, 0.3 mmol) and 4 drops of glacial acetic acid (adjusted to pH 7). The mixture was stirred at room temperature for 5 h. It was evaporated and dissolved in EtOAc (50 mL). The solution was washed with saturated NH4Cl solution (5 mL) and brine (5 mL) and dried over MgSO4. It was evaporated and passed through a column of silica gel (hexane-ethyl acetate (1:1) and 5%–10% MeOH in toluene as eluents) yielding perpropargylated dendrimer 14 as light yellow oil (26 mg, 0.011 mmol, 85%).
Synthesis of 14 by direct alkylation: To a solution of 11 (193 mg, 0.119 mmol) in DMF (15 mL) was added propargyl bromide (650 µL, 4.37 mmol). It was cooled to 0 °C. Four portions of NaH (60% in oil, 60 mg each, 1.5 mmol each) was added at an interval of one hour. After the additions, the ice-bath was removed and the mixture was stirred at room temperature for 18 h. The reaction was quenched with saturated NH4Cl solution and extracted with EtOAc (150 mL). The extract was washed with brine and dried over MgSO4. The solvent was removed and the crude product was passed through a column of silica gel (using hexane-EtOAc (1:1) and 0–10% MeOH in DCM as eluents) yielding mainly fully propargylated (230 mg) (slightly impure) with a small amount of partially propargylated (57 mg) product. Partially propargylated fraction was further propargylated using the above procedure with propargyl bromide (0.1 mL, 1.1 mmol) and three portions of sodium hydride (60% in oil, 10 mg each, 0.25 mmol each) in THF (4 mL). The product thus obtained was combined with slightly impure fraction (230 mg) and passed through a column of silica gel using hexane-EtOAc (1:1) and 4% methanol in DCM as eluents yielding perpropargylated product, 14 as light yellow oil (198 mg, 0.08 mmol, 70%). 1H-NMR (600 MHz, CDCl3) δ (ppm) 8.15 (s,1H), 8.11 (s, 1H), 8.05 (s, 1H), 8.01 (s, 1H), 7.89 (s, 1H), 5.01–4.97 (m, 3H), 4.93–4.88 (m, 2H), 4.82 (dd, J = 11.8, 8.9 Hz, 2H), 4.75 (d, J = 12.4 Hz, 1H), 4.71–4.56 (m, 14H), 4.54–4.37 (m, 25H), 4.35–4.10 (m, 36H), 4.04–3.97 (m, 6H), 3.86–3.79 (m, 6H), 3.78–3.71 (m, 8H), 3.54–3.48 (m, 8H), 3.47–3.29 (m, 20H), 2.60–2.40 (m, 20H); 13C-NMR (150 MHz, CDCl3) δ (ppm) 144.79, 144.72, 144.26, 128.97, 128.16, 125.23, 124.90, 124.84, 124.60, 102.98, 102.19, 84.01, 83.34, 75.01, 74.95, 74.50, 74.44, 73.98, 68.39, 67.96, 65.73, 64.64, 62.86, 60.19, 59.93, 59.92, 59.34, 49.88. FT-IR: νmax (neat)/cm−1 3283s (br, C≡CH), 2118 (≡CH). ESI+-HRMS: [M + 2H]2+ calcd for C121H139N15O36, 1189.4779; found: 1189.4764.
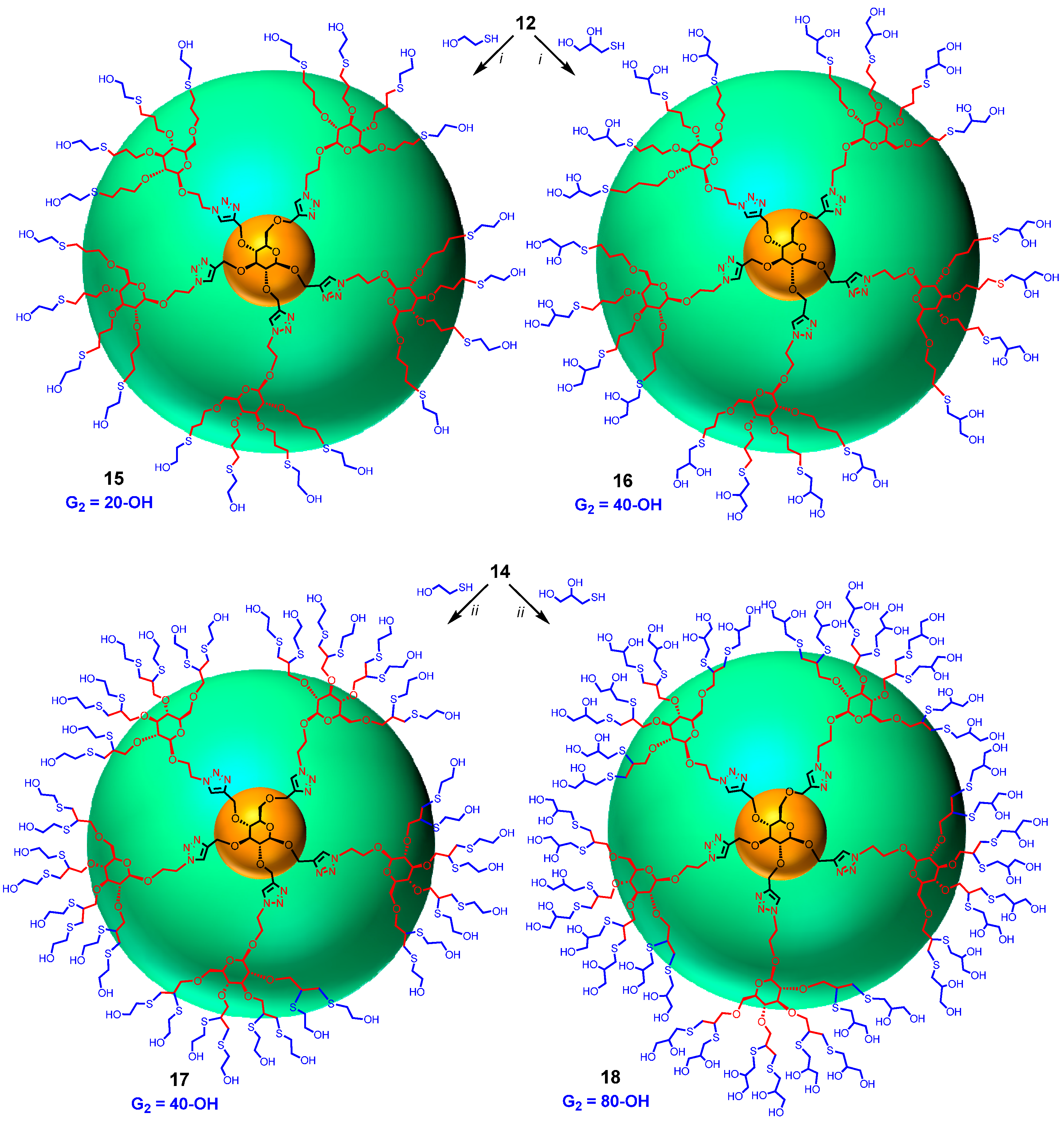
Synthesis of 15. A solution of perallylated intermediate 12 (8 mg, 0.003 mmol), mercaptoethanol (28 mg, 0.036 mmol, 120 eq.) and a catalytic amount of AIBN in dioxane (2.0 mL) was stirred at reflux for 1 h. The mixture was then diluted with H2O, dialysed against water over night, and lyophilized to yield polyol G2 (15) as light yellow oil (5.6 mg, 0.0014 mmol, 47%). 1H-NMR (600 MHz, D2O) δ (ppm) 8.20–8.04 (5H, m), 5.05–4.86 (7H, m), 4.74–4.58 (13H, m), 4.49–4.21 (13H, m), 4.17–3.97 (20H, m), 3.92–3.56 (73H, m), 3.54–3.23(26H, m), 3.21–2.92 (27H, m), 2.92–2.57 (47H, m), 2.56–2.41(5H, m), 2.20–2.01 (10H, broad signal), 1.99–1.81 (20H, broad signal), 1.75–1.57 (5H, broad signal). MALDI-TOF-MS: C161H297N15O56S20; exact mass cald: 3976.52678; found: ~4000 (broad).
Synthesis of 16. The solution of perallylated 12 (10 mg, 0.004 mmol), thioglycerol (43 µL, 0.48 mmol, 120 equiv.) and a catalytic amount of AIBN in dioxane (2.0 mL) was stirred under reflux for 1 h. The mixture was then diluted with H2O, dialysed against water overnight, and lyophilized to yield polyol G2 (16) as yellow light oil (11 mg, 0.0024 mmol, 60%). 1H-NMR (600 MHz, D2O) δ 8.18–8.00 (5H, m), 5.09–4.81 (6H, m), 4.68 (12H, broad signal), 4.51–4.22 (11H, m), 4.22–4.02 (11H, m), 4.02–3.19 (131H, m), 3.16–2.88 (21H, m), 2.88–2.38 (61H, m), 2.21–2.00 (7H, broad signal), 2.00–1.76 (24H, broad signal), 1.74–1.51 (8H, broad signal); 13C-NMR (150 MHz, D2O) δ (ppm) 145.8, 145.4, 126.5, 103.7, 84.9, 82.7, 75.2, 73.5, 72.9, 72.7, 72.1, 71.1, 70.7, 70.3, 69.2, 68.2, 67.0, 66.3, 56.4, 55.6, 51.7, 49.7, 49.4, 35.8, 30.7, 29.9, 29.8, 24.6, 24.4, 24.3, 23.7. MALDI-TOF-MS: C181 H337N15O76S20; exact mass cald: 4576.73807; found: 4555.334.
Synthesis of 17. Perpropargylated 14 (10 mg, 0.004 mmol) and 2,2-dimethoxy-2-phenylacetophenone (DMPA), (12 mg, 0.047 mmol) in DMF (200 µL) was added in a quartz photometer cell. 2-Mercaptoethanol (200 mg, 2.56 mmol) was added into the solution. The mixture was irradiated under 365 nm light for 4 h. The mixture was then diluted with H2O, dialysed against water overnight, and lyophilized to yield polyol G2 (17) as light yellow oil (13 mg, 0.002 mmol, 56%). 1H-NMR (600 MHz, MeOD) δ (ppm) 8.25–8.11(5H, m), 5.10–4.93 (7H, m), 4.84–4.59 (19H, m), 4.50–4.27 (15H, m), 4.24–3.99 (34H, m), 3.96–3.56 (108H, m), 3.55–3.40 (14H, m), 3.28–2.63 (137H, m); 13C-NMR (150 MHz, MeOD) δ (ppm) 127.1, 126.5, 105.6, 86.5, 84.1, 79.8, 77.5, 76.6, 76.2, 76.1, 75.2, 71.8, 70.0, 63.9, 63.5, 63.3, 57.3, 56.9, 52.6, 49.1, 48.8, 48.3, 37.3, 37.2, 37.0, 36.2, 36.0. MALDI-TOF-MS: C201H377N15O76S40; exact mass: 5496.49249; found: 5590.
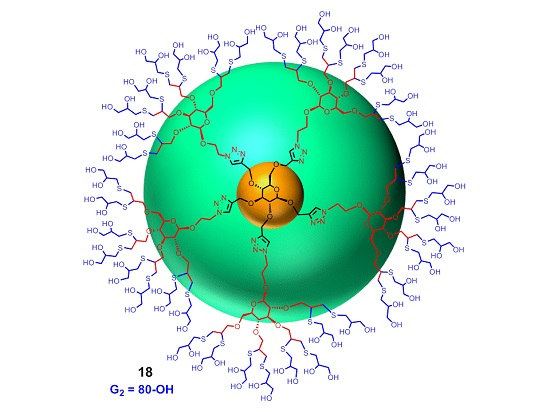
Synthesis of 18. To a solution of perpropargylated compound 14 (10 mg, 0.004 mmol) and DMPA (12 mg, 0.047 mmol) in DMF (200 µL) in a quartz photometer cell was added thiolglycerol (400 mg, 3.70 mmol). The mixture was irradiated under 365 nm light for 6 h. The mixture was then diluted with H2O, dialysed against water over night, and lyophilized yield polyol G2 (18) as light yellow solid (20 mg, 0.003 mmol, 71%).1H-NMR (600 MHz, D2O) δ (ppm) 8.23–8.04 (5H, m), 5.11–4.89 (10H, m), 4.55–4.42 (8H, m), 4.40–4.28 (7H, m), 4.27–3.98 (26H, m), 3.95–3.31(171H, m), 3.30–3.12 (23H, m), 3.10–2.59 (114H, m); 13C-NMR (150 MHz, D2O) δ (ppm) 145.1, 144.7, 126.3, 126.1, 103.2, 100.6, 100.4, 84.4, 84.0, 82.4, 78.3, 75.2, 74.5, 73.4, 71.8, 71.6, 70.1, 68.5, 67.6, 67.5, 66. 5, 65.6, 51.1, 47.0, 46.7, 46.4, 36.11, 35.4, 34.8. MALDI-TOF-MS: C241H457N15O116S40; exact mass: 6696.91507; found: 6745.
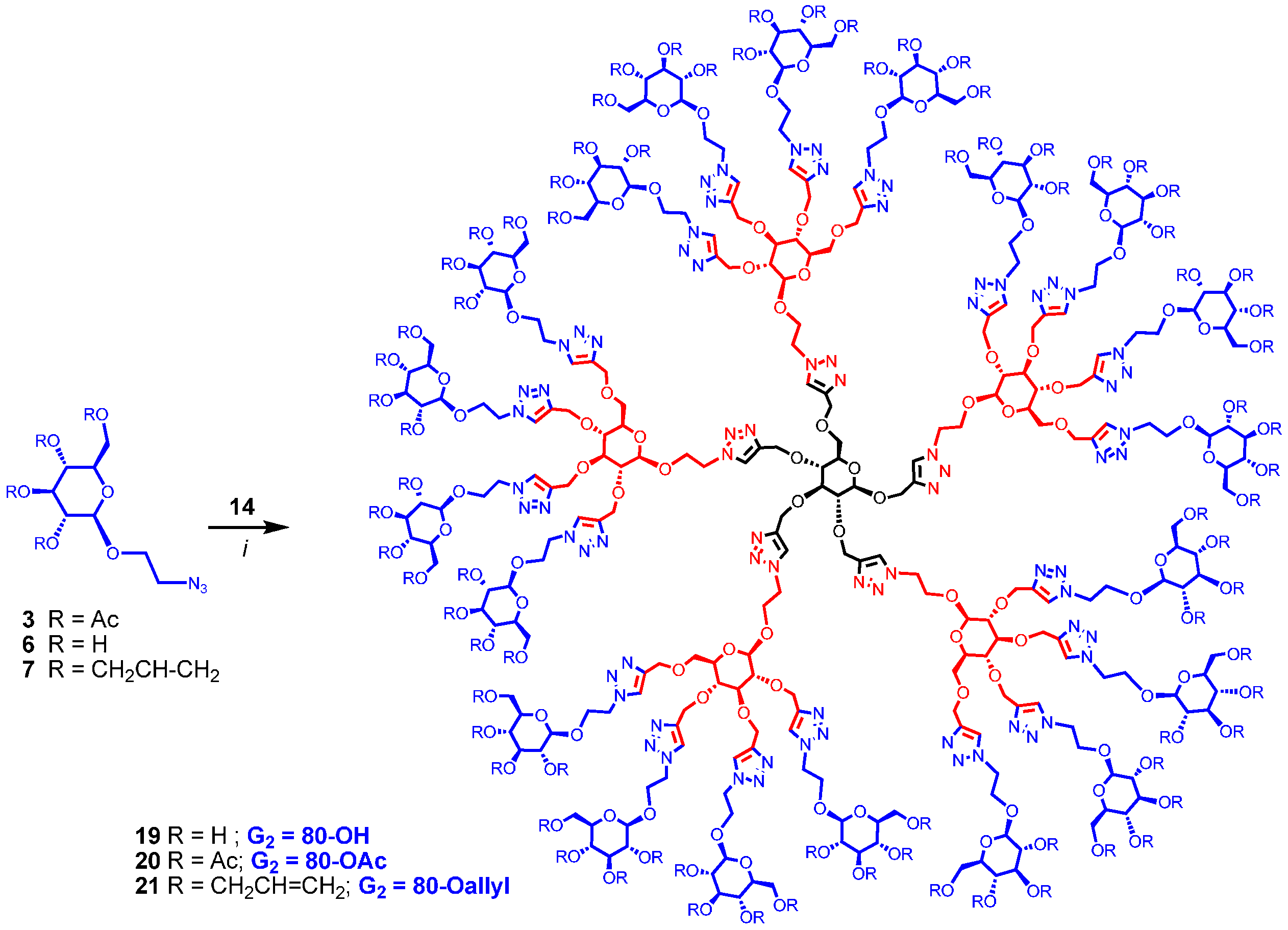
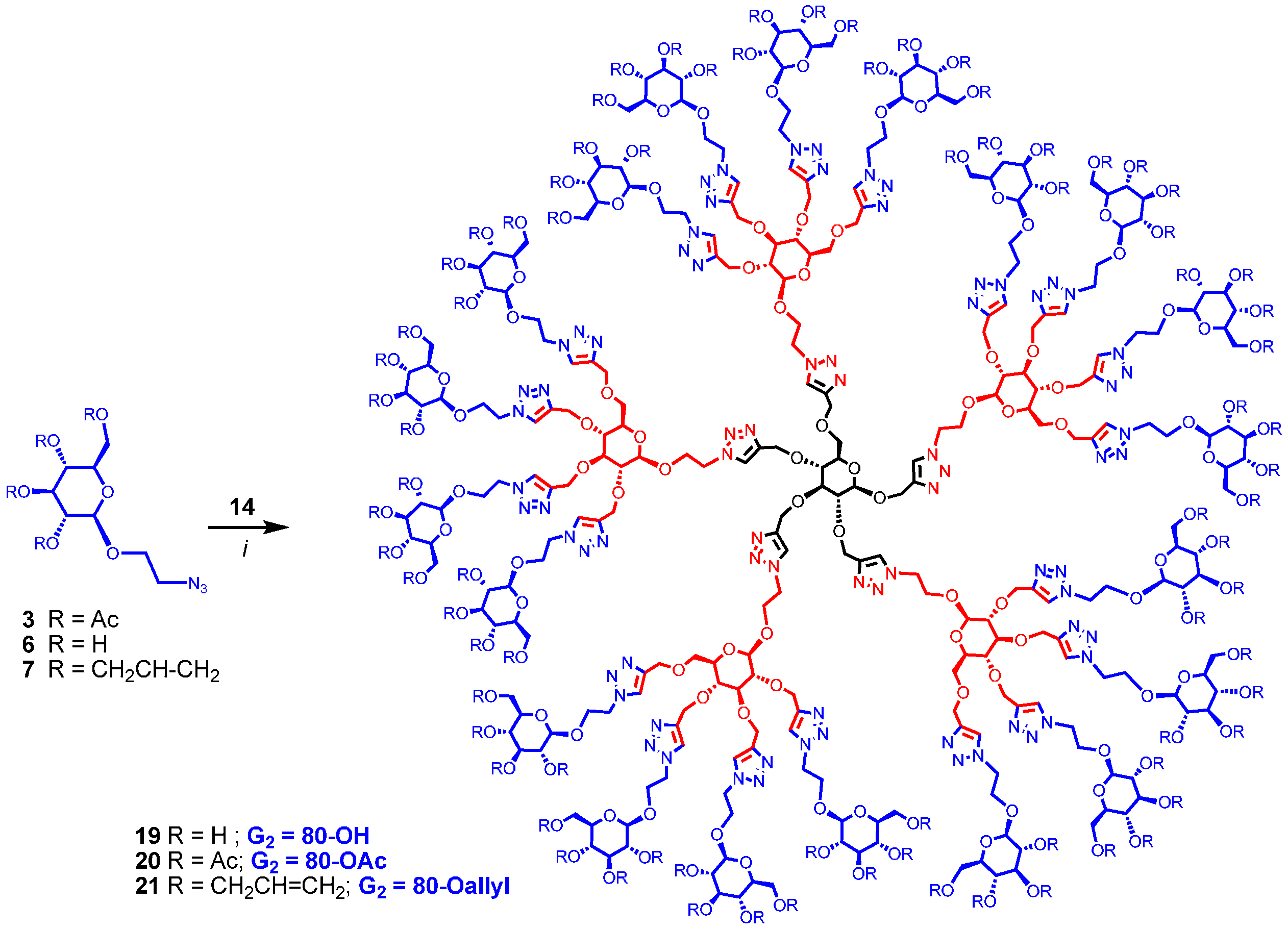
Synthesis of polyol 19. A mixture of 2-azidoethyl β-D-glucopyranoside (6) (34 mg, 0.136 mmol), 14 (8 mg, 0.003 mmol) and CuI·P(OEt)3 (3 mg, 0.008 mmol) in dry DMF (1 mL) was held at 70 °C for 21 h. It was cooled and diluted with water. The mixture was dialysed (in a dialysis bag with 1200 cut-off) against water (with frequent change of water at the start) for 72 h. The dialysed material was lyophilized to give 19 (13 mg, 0.002 mmol, 53%). 1H-NMR (600 MHz, D2O) δ (ppm) 8.07–7.95 (m, 25H), 4.69–4.51 and 4.48–4.34 (m, 116H), 4.29–4.17 (m, 28H), 4.12–3.91 (m, 27H), 3.88–3.80 (m, 22H), 3.69–3.57 (m, 32H), 3.45–3.36, 3.33–3.27, 3.22–3.16 (m, 97H); 13C-NMR (150 MHz, D2O + acetone-d6) δ (ppm) 144.2, 126.4, 103.1, 83.4, 81.3, 76.6, 76.3, 74.0, 73.62 70.3, 68.6, 66.0, 65.5, 63.9, 61.4, 51.0. MALDI-TOF-MS: C281H437N75O156; exact mass: 7357.85677; found: 7519.337.
Synthesis of 20. A mixture of
14 (13 mg, 0.006 mmol), 2-azidoethyl 2,3,4,6-tetra-
O-acetyl-β-
d-glucopyranoside (
3) (86 mg, 0.20 mmol) and CuI·P(OEt)
3 (5.5 mg, 0.02 mmol) in dry DMF (1.3 mL) was stirred at 65–70 °C for 20 h. It was cooled and evaporated. The reaction mixture was diluted with EtOAc (100 mL), washed with EDTA (2 × 5 mL), NaHCO
3 solution (2 × 5 mL) and brine, dried over MgSO
4 and evaporated. The crude product was passed through a column of silica gel with hexane-EtOAc (1:1), 3% to 5% MeOH in toluene and 5% to 8% MeOH in DCM as eluents yielding
20 as colorless oil (39 mg, 0.004 mmol, 67%).
1H-NMR (600 MHz, CDCl
3) δ (ppm) 8.13–7.61 (m, 25H), 5.20–5.14 (m, 22H), 5.09–5.04 (m, 19H), 4.99–4.82 (m, 37H), 4.80–4.47 (m, 95H), 4.39–4.33 (m, 9H), 4.31–4.17 (m, 44H), 4.17–4.08 (m, 20H), 4.07–3.97, 3.97–3.90 (m, 24 H), 3.81–3.68 (m, 30H), 3.53–3.44 (m, 10H), 3.41–3.26 (m, 15H), 2.07–1.91 (m, 240H);
13C-NMR (150 MHz, CDCl
3) δ (ppm) 170.6, 170.1, 169.4, 169.3, 169.2, 144.5, 124.6, 103.4, 100.5, 100.4, 83.7, 81.7, 74.2, 72.6, 72.5, 71.8, 70.9, 70.8, 68.3, 67.7, 65.7, 64. 5, 61.8, 49.7, 29.7, 20.7, 20.6. MALDI-TOF MS: C
441H
597N
75O
236; exact mass: 10,718.70195; found: 10,747.228. See
Supplementary Materials for GPC.
Synthesis of 21. A mixture of 2-azidoethyl 2,3,4,6-tetra-O-allyl-β-d-glucopyranoside (7) (85 mg, 0.20 mmol), 14 (13 mg, 0.006 mmol) and CuI·P(OEt)3 (5.5 mg, 0.02 mmol) in dry DMF (1.3 mL) was stirred at 65–70 °C for 20 h. The reaction mixture was evaporated and the residue was dissolved in EtOAc (100 mL). It was washed with EDTA (2 × 5 mL), saturated NaHCO3 solution (2 × 7 mL) and brine (5 mL), and dried over MgSO4. The solution was evaporated and passed through a column of silica gel with hexane-EtOAc (1:1) and 2% to 10% MeOH in DCM as eluents yielding 21 as colorless oil (33 mg, 0.003 mmol, 57%). 1H-NMR (600 MHz, CDCl3) δ (ppm) 8.14–7.70 (m, 25H), 6.09–5.65 (m, 80H), 5.42–5.01 (m, 159H), 5.01–4.49 (m, 100H), 4.37–4.18 (m, 107H), 4.15–3.86 (m, 129H), 3.77–3.53 (m, 58H), 3.46 (broad signal, 17H), 3.34 (broad signal, 69H), 3.21–3.12 (m, 22H); 13C-NMR (150 MHz, CDCl3) δ (ppm) 144.67, 144.33, 135.21, 135.16, 135.05, 134.96, 134.87, 134.78, 134.61, 134.57, 124.39, 124.00, 117.16, 116.92, 116.84, 116.63, 103.41, 103.16, 83.97, 81.23, 74.72, 74.32, 73.75, 73.43, 72.40, 68.70, 67.77, 66.42, 65.81, 64.51, 49.96, 29.66, 22.65. MALDI-TOF-MS: C521H757N75O156; exact mass: 10,560.36078; found: 10,632.893. See SI for GPC.
Computational Methods. The computational approach used for this study is the same recently adopted for the simulation of a variety of dendrimers in aqueous solution [
26,
27,
28,
29,
30]. The simulation work was conducted using the AMBER 12 software (version 12, University of California, San Francisco, CA, USA, 2012) [
31]. The molecular model for
16 has been parameterized according to the “general AMBER force field (GAFF)” [
32].
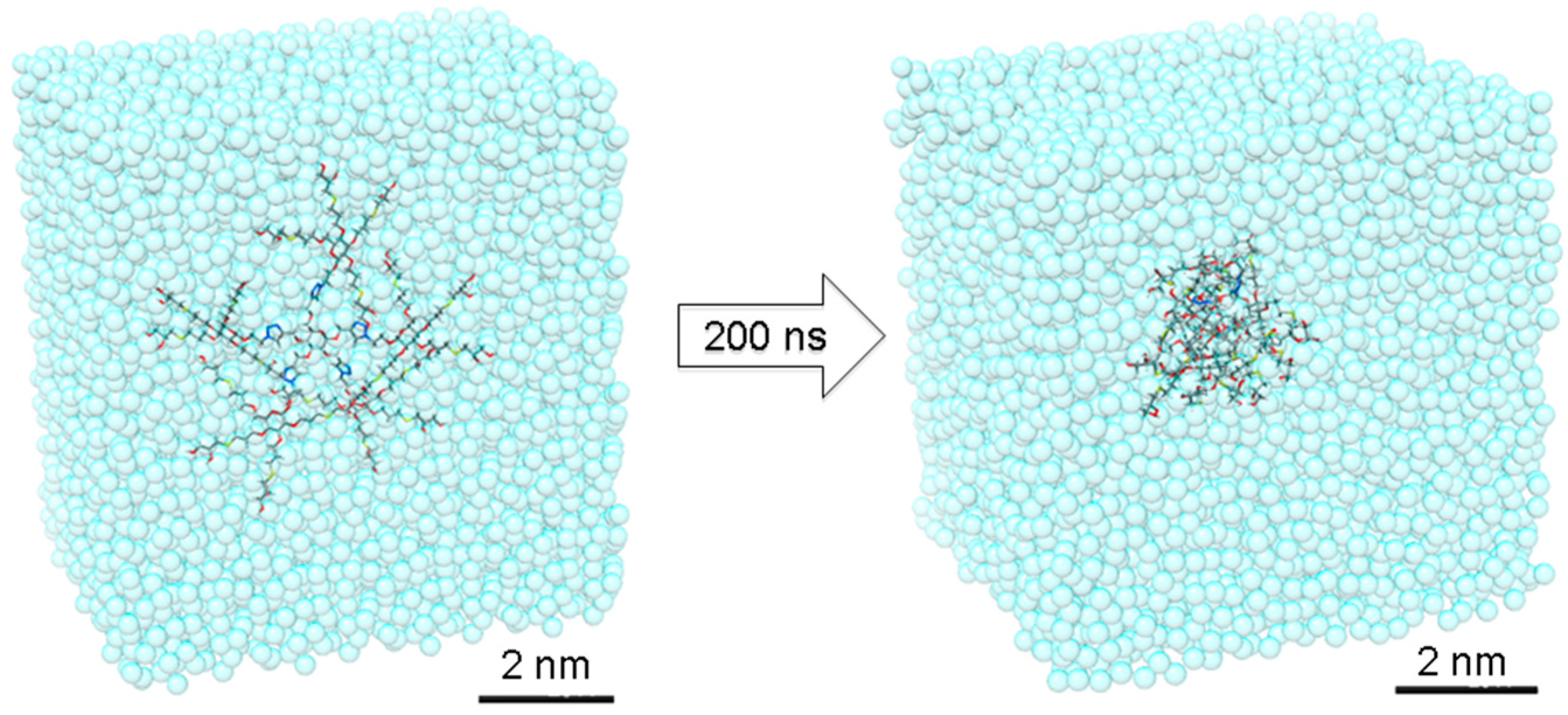
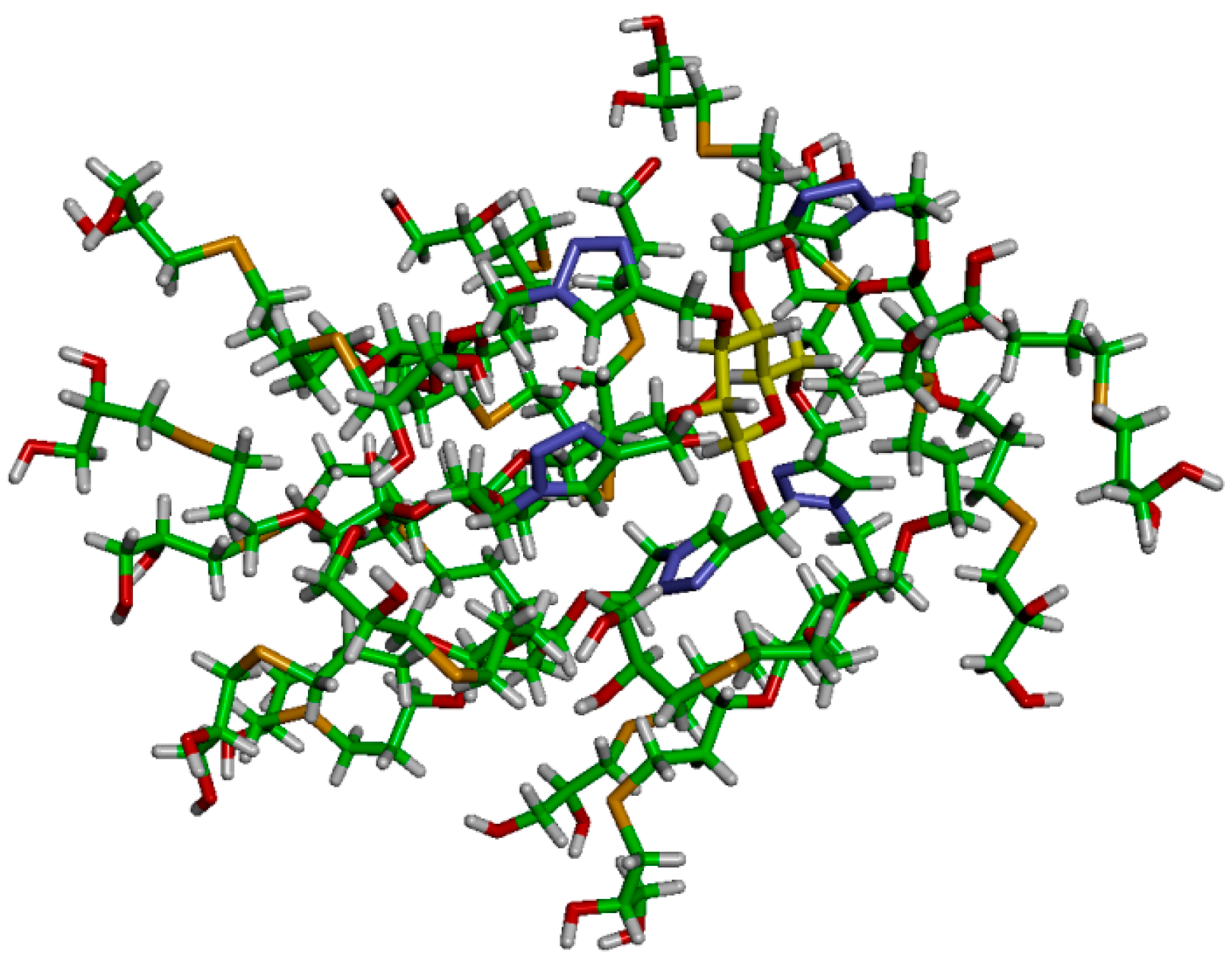
Dendrimer
16 was immersed in a simulation box filled with explicit TIP3P water molecules [
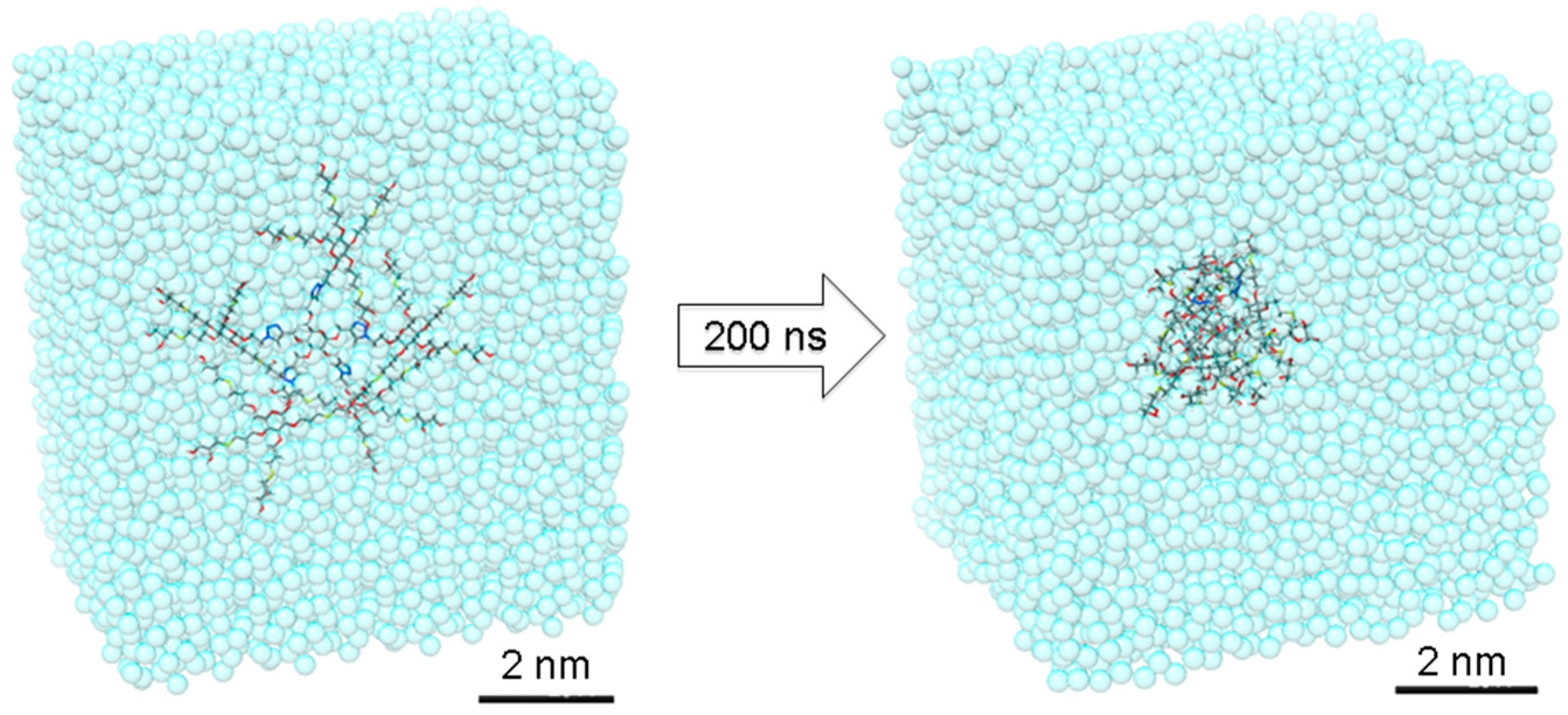
33]. After preliminary energy minimization, the system has been thermalized through a short 100 ps MD simulation in NVT (constant N: number of atoms, V: volume and T: temperature) periodic boundary conditions to reach the temperature of 37 °C (310 K). Then, dendrimer
16 has been equilibrated through 200 ns of MD simulations NPT in periodic boundary condition (constant N: number of atoms, P: pressure and T: temperature during the run) at 37 °C of temperature and 1 atm of pressure. During this time, dendrimer
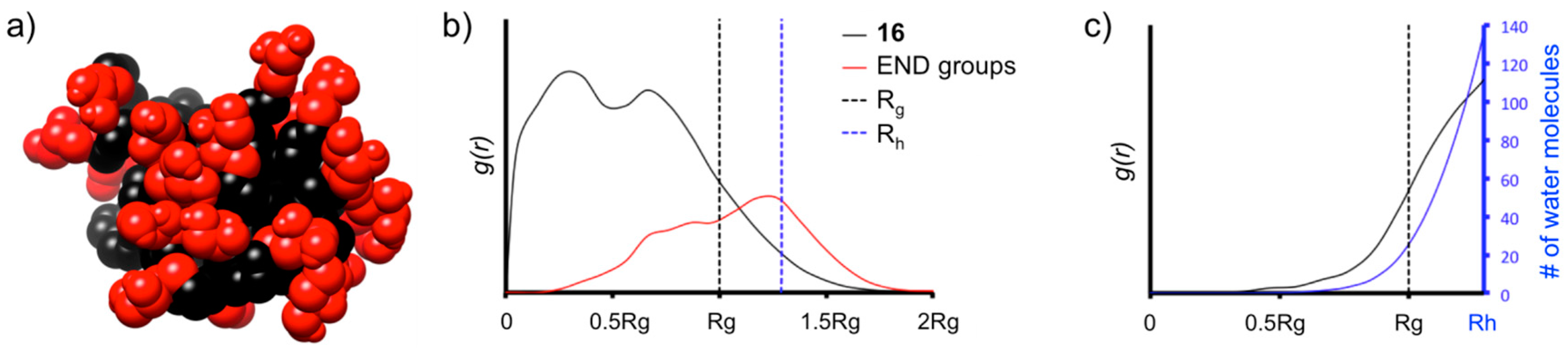
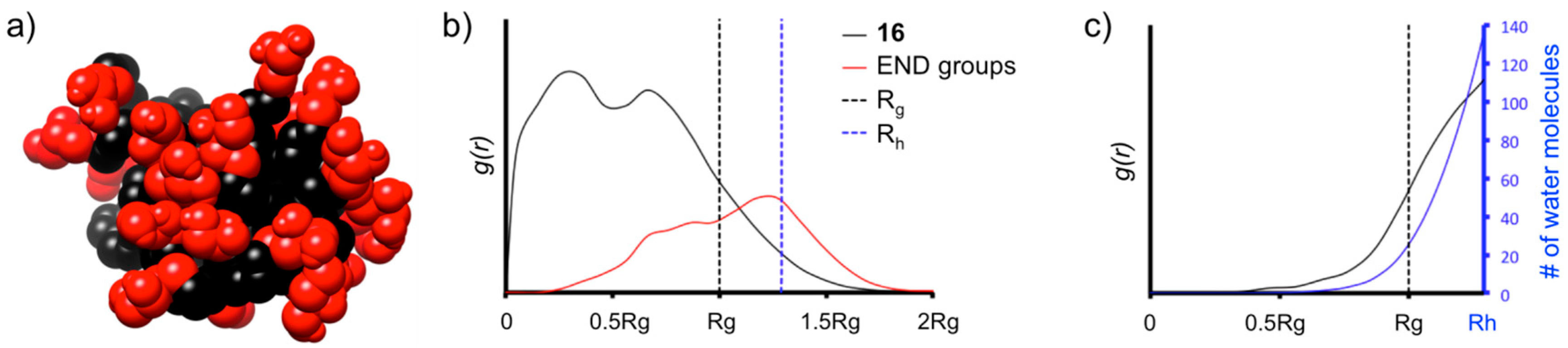
16 reached the equilibrium with good stability, as demonstrated by the root mean square displacement (RMSD), radius of gyration (R
g) and enthalpy (H) data reported in the SI. For the MD run, a 2 femtoseconds time step was used, the Langevin thermostat, a 8 Å cutoff, the particle mesh Ewald [
34] (PME) for long-range electrostatics, and the SHAKE algorithm to treat bonds involving Hydrogen atoms [
35] Enthalpy H was calculated through the MMPBSA approach [
36,
37] according to the same procedure followed previously [
26,
27,
28,
29,
30]. Namely, H is the sum of the total gas-phase in vacuo non-bonded energy (Egas) and a solvation term (Gsol = G
PB + G
NP) [
38] The polar component of G
PB was evaluated using the Poisson-Boltzmann [
39] (PB) term while the non-polar contribution to the solvation energy was calculated as G
NP = a (SASA) + b, in which a = 0.00542 kcal/Å
2, b = 0.92 kcal/mol, and the SASA (solvent accessible surface area) was estimated with the MOLSURF program included in AMBER 12 [
40]. The last 100 ns of the MD trajectories were considered as representative of the equilibrium condition for dendrimer
16 and used for the analysis of the structural parameters reported in
Table S1.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}