
Porphyrins as Catalysts in Scalable Organic Reactions


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
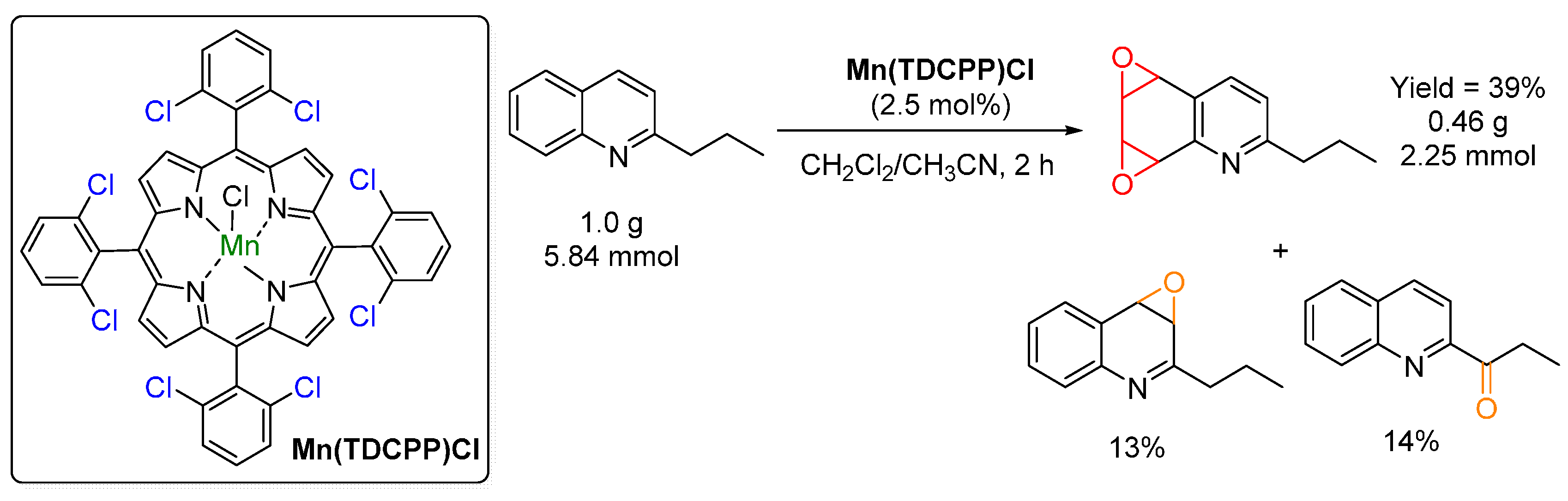{kind=link}
{kind=link}
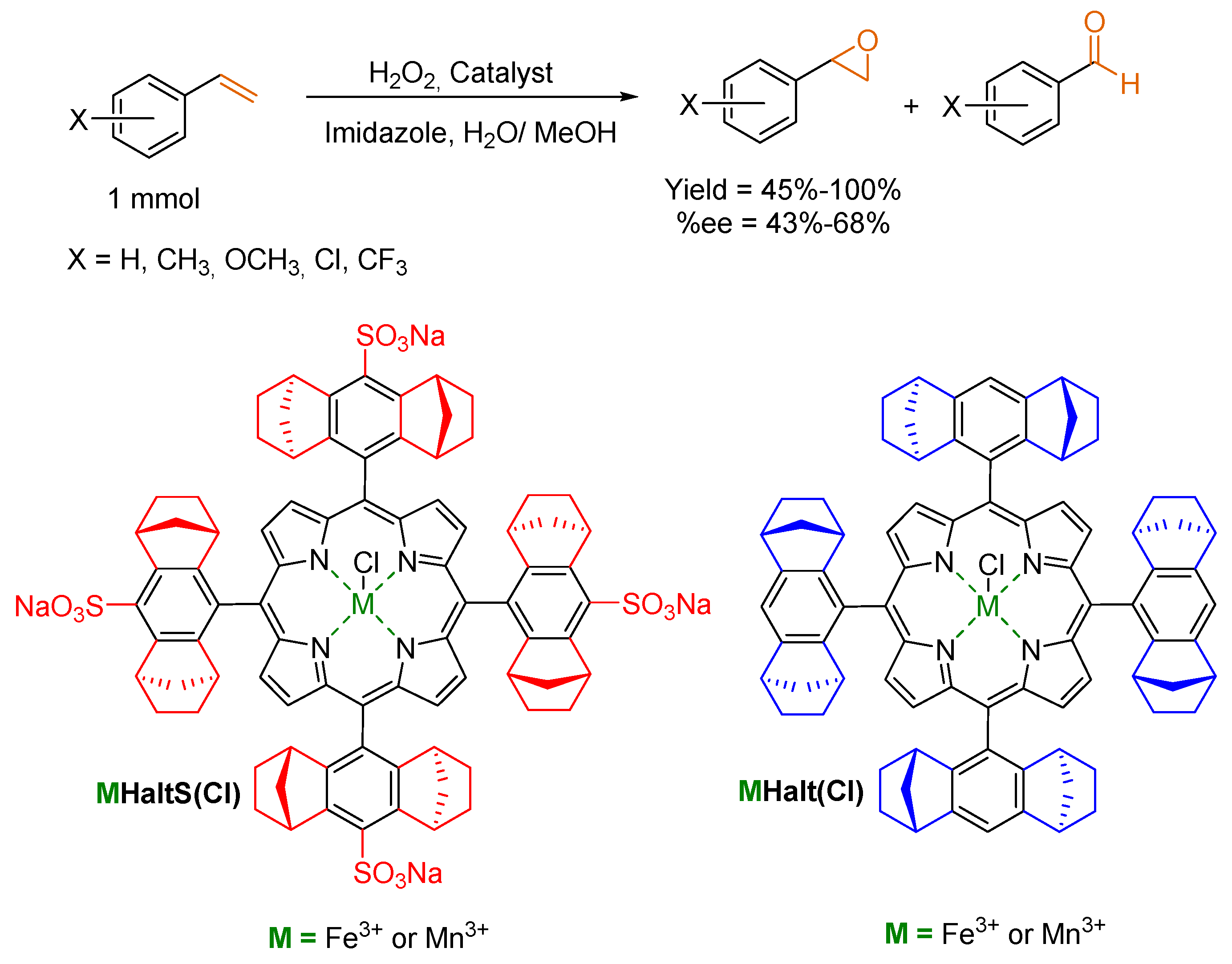{kind=link}
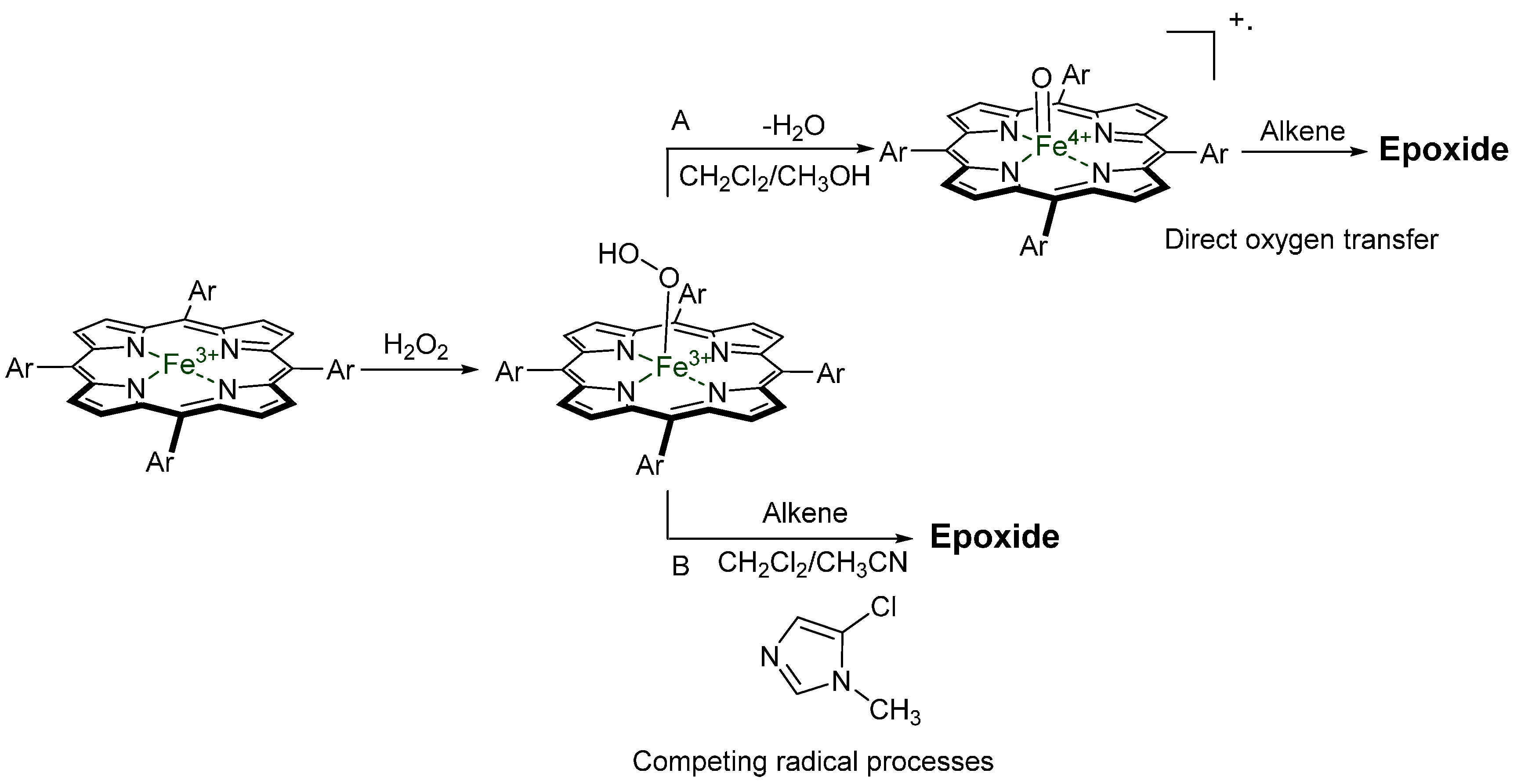{kind=link}
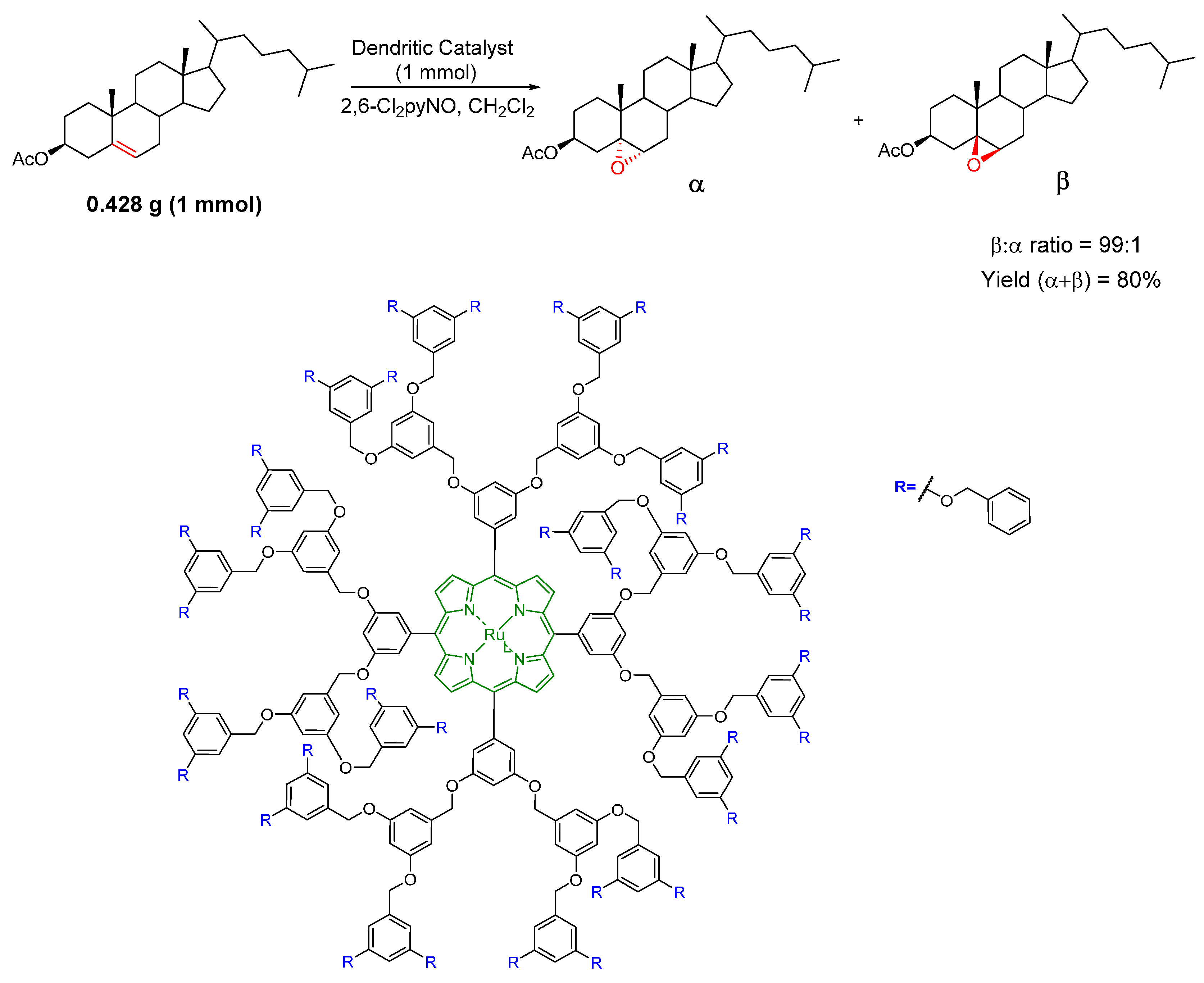{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
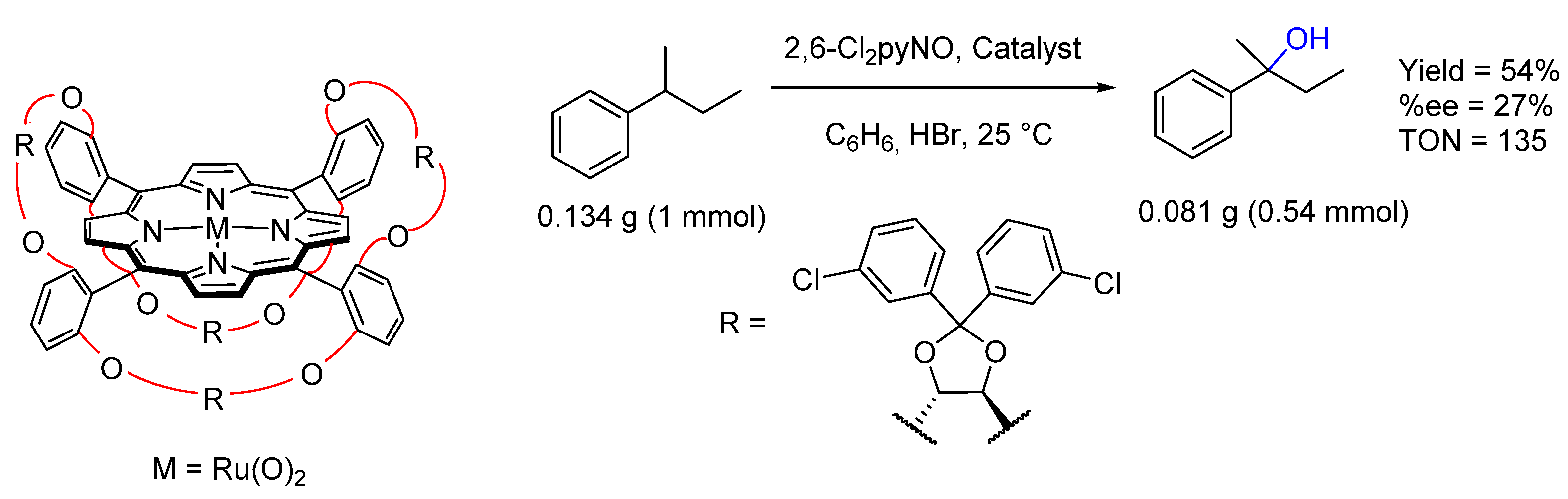{kind=link}
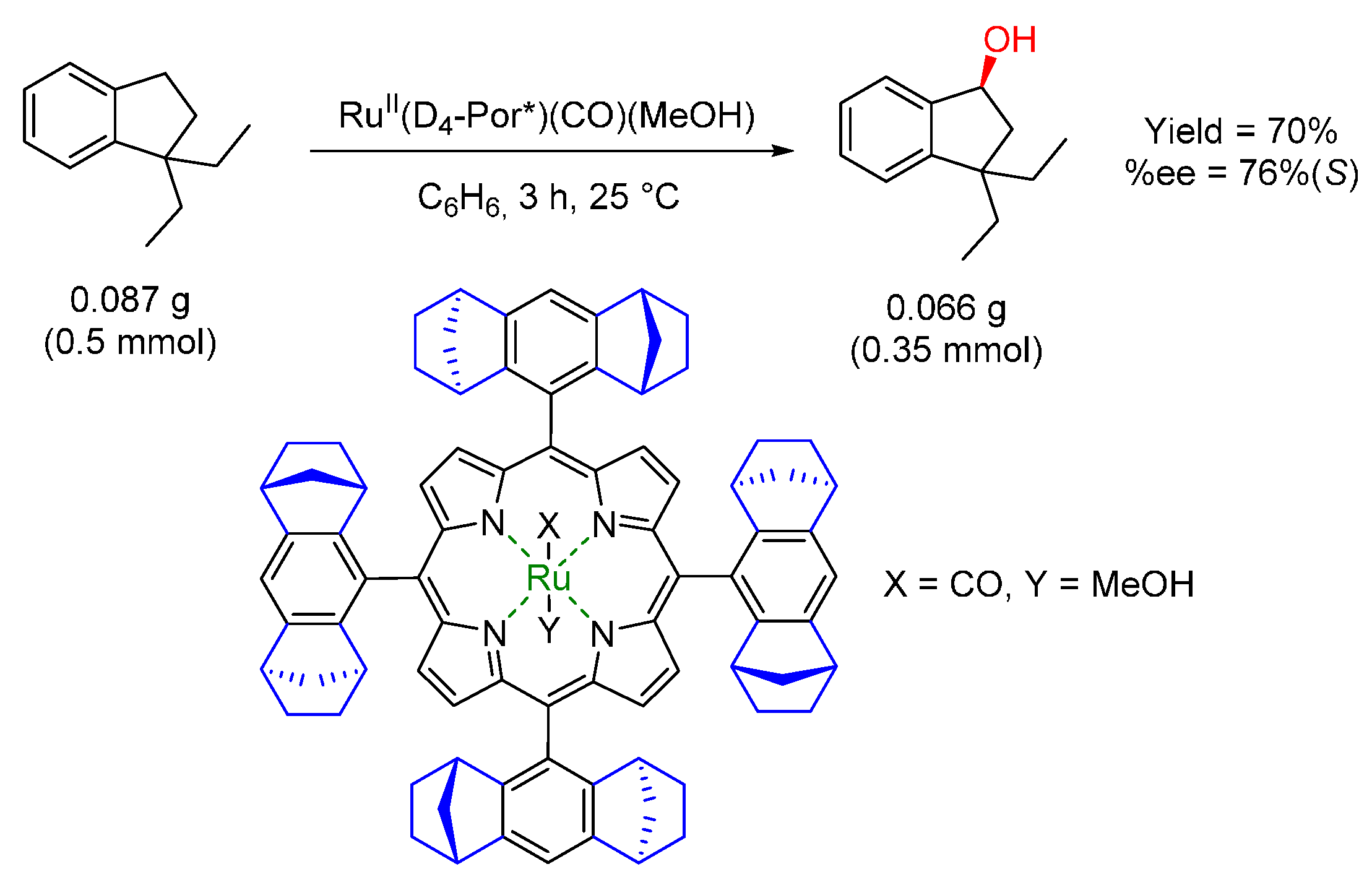{kind=link}
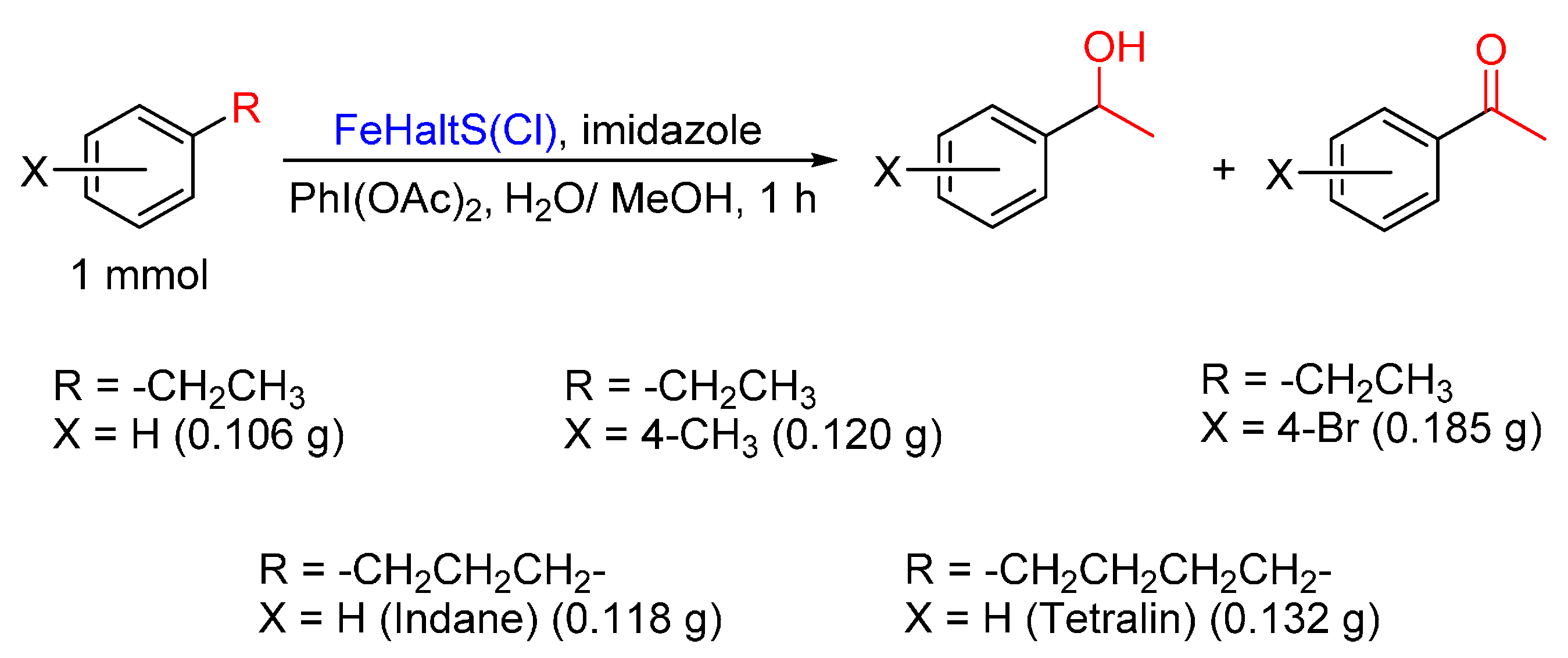{kind=link}
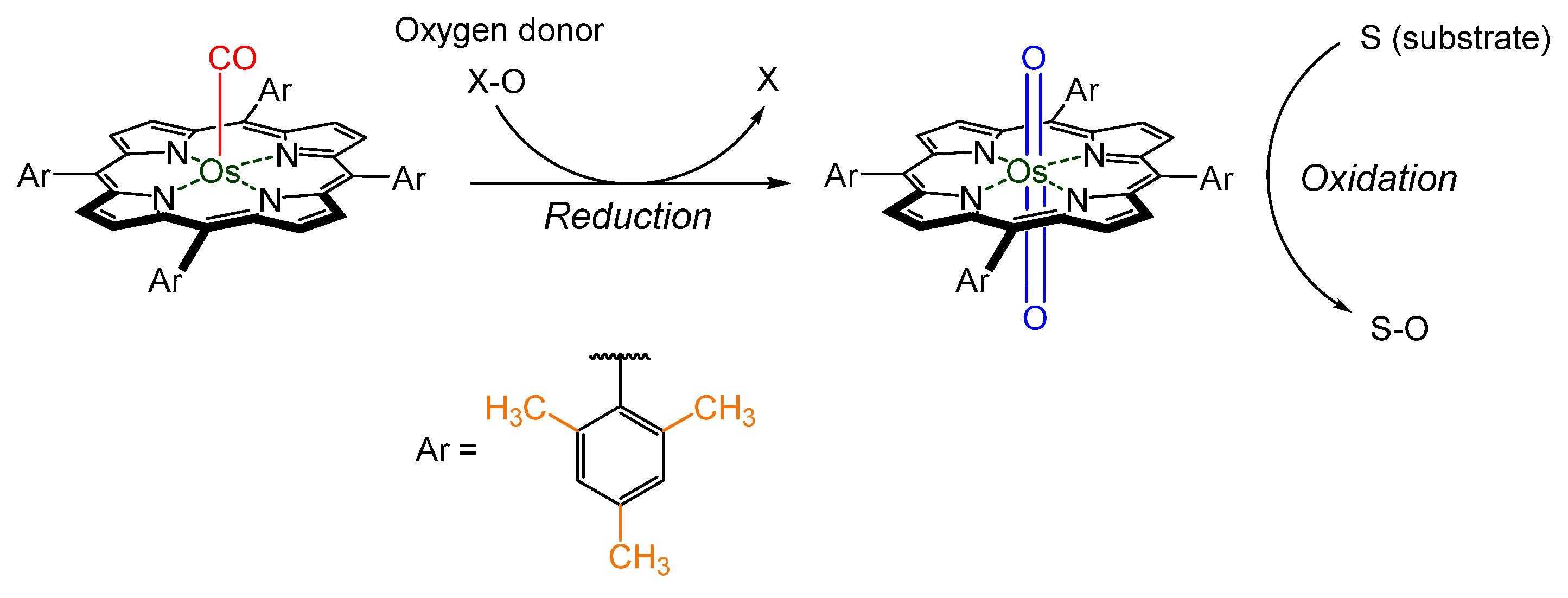{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
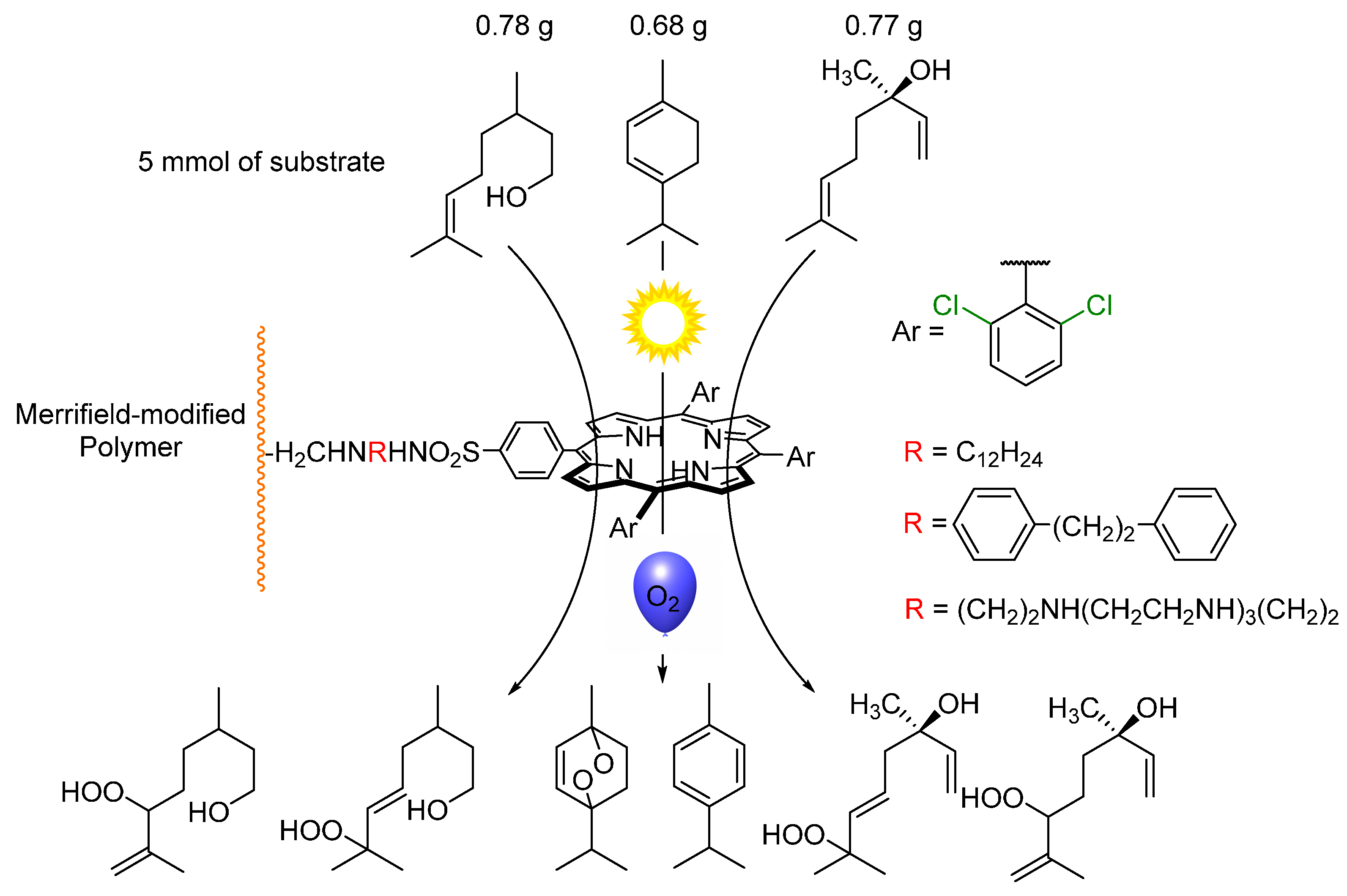{kind=link}
{kind=link}
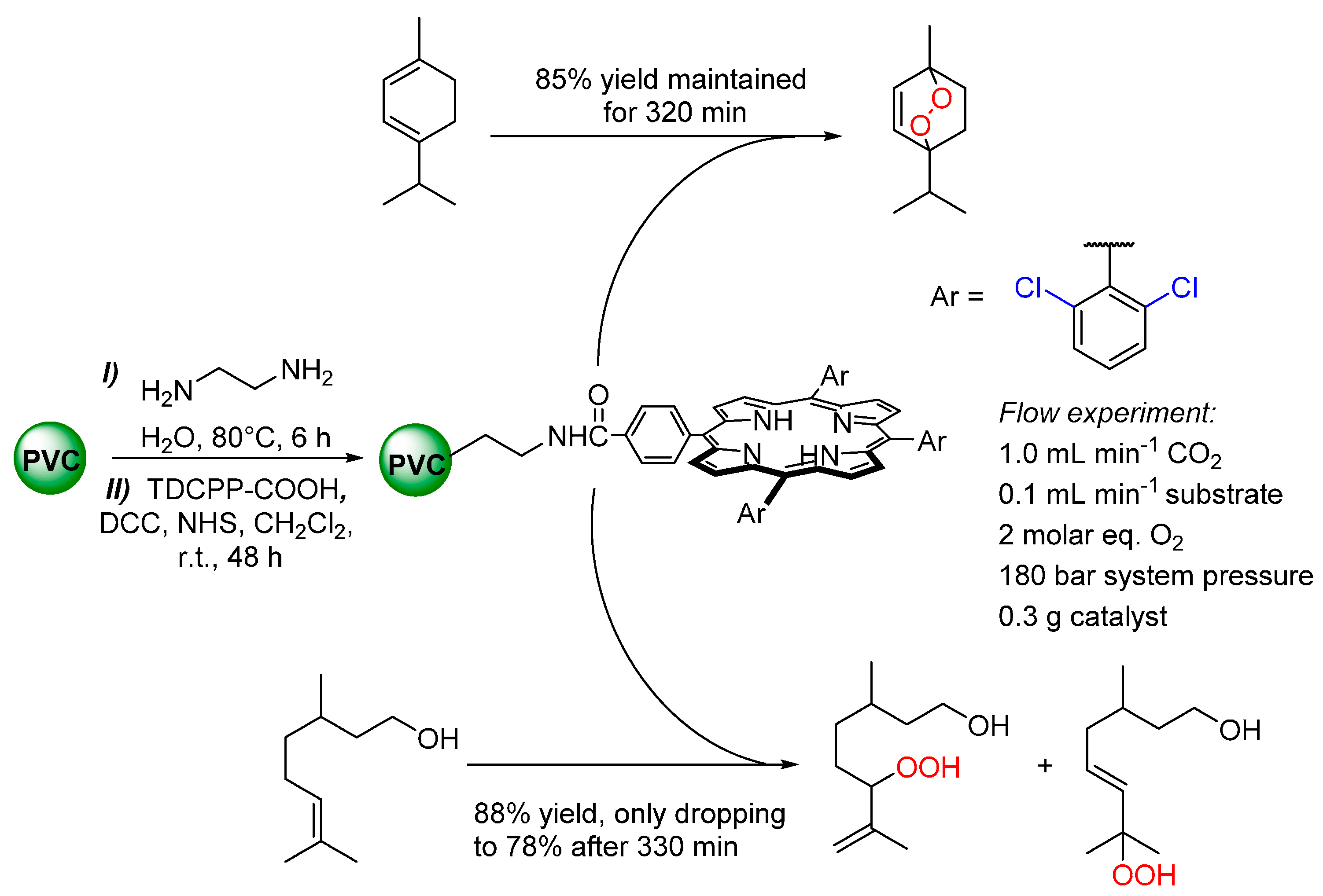{kind=link}
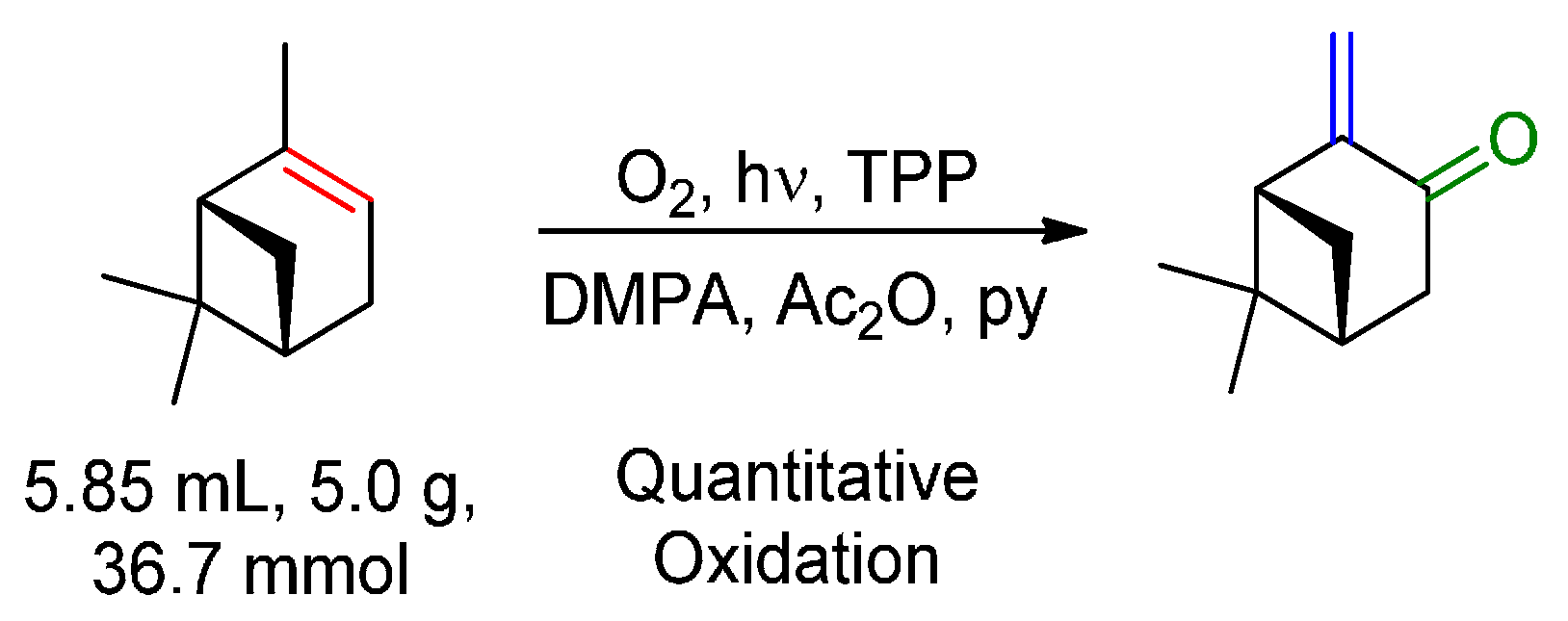{kind=link}
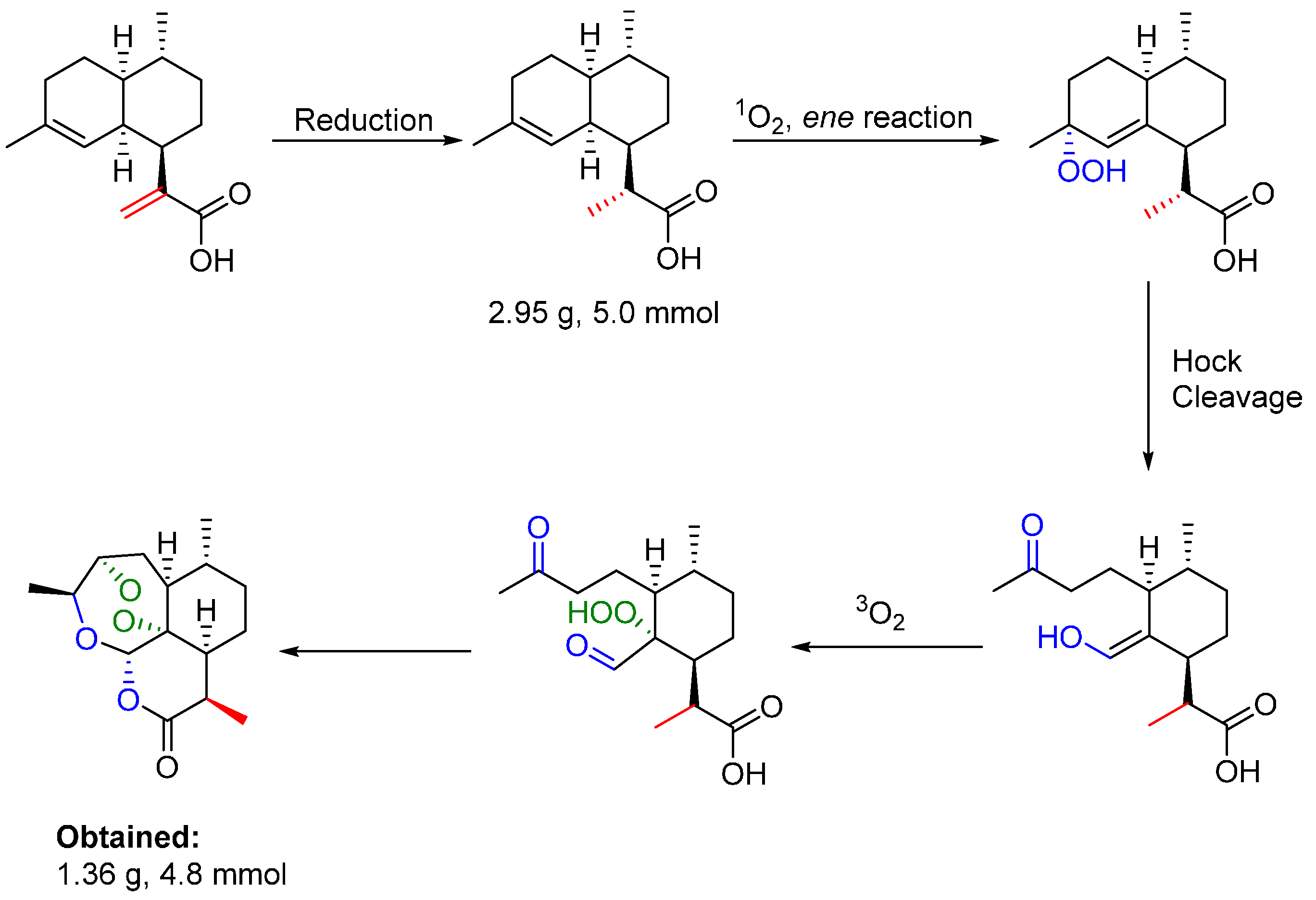{kind=link}
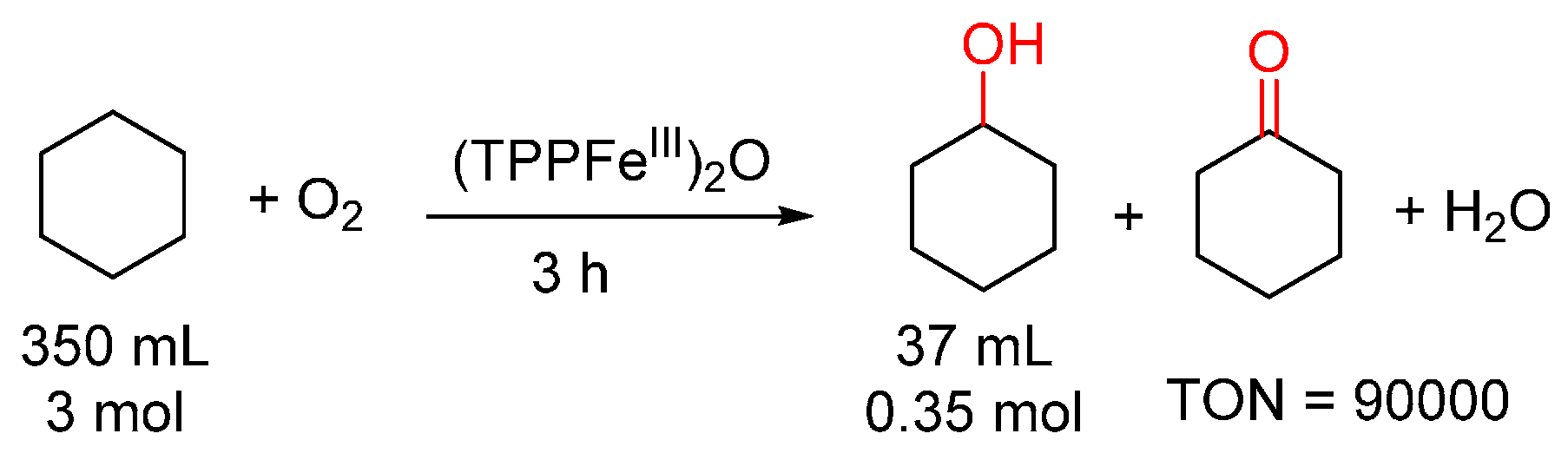
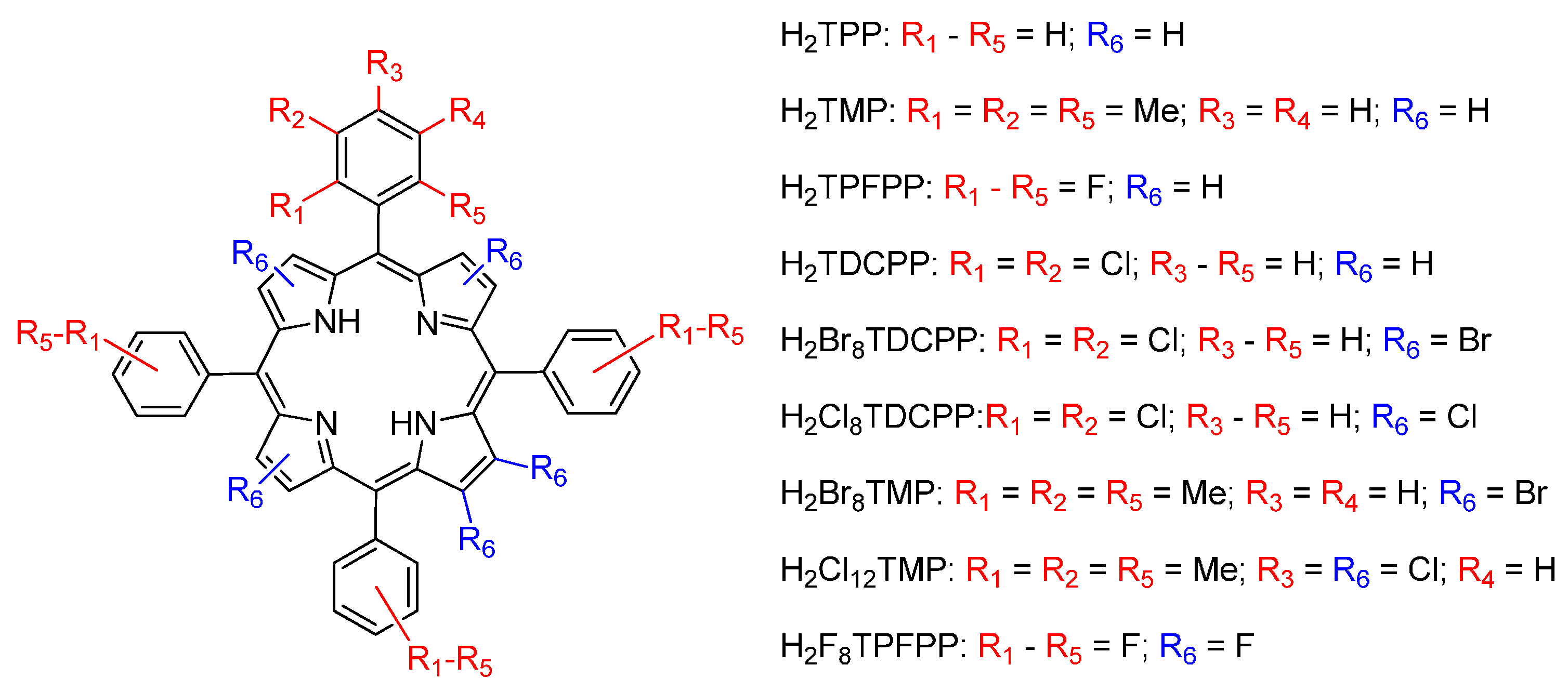
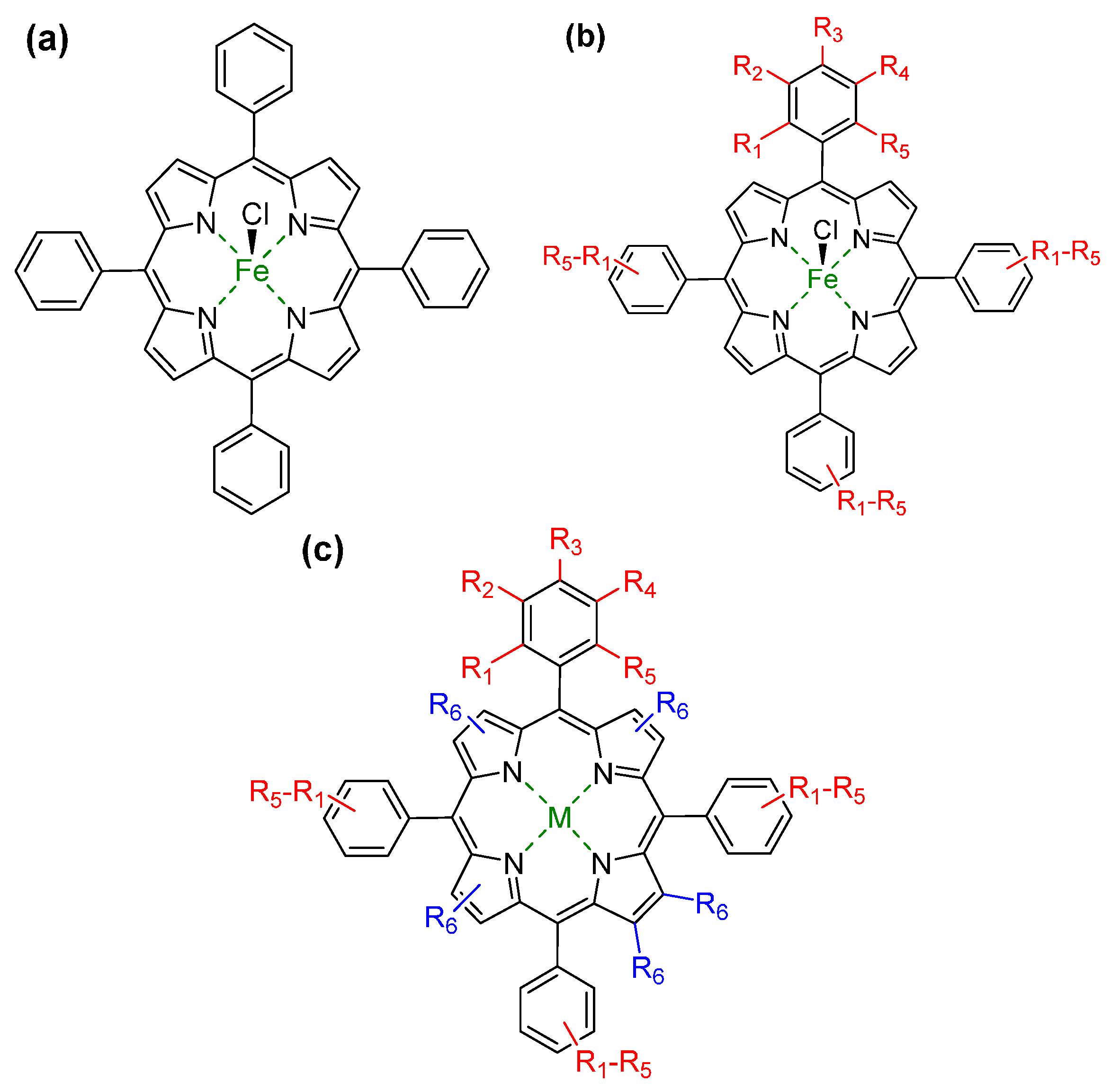
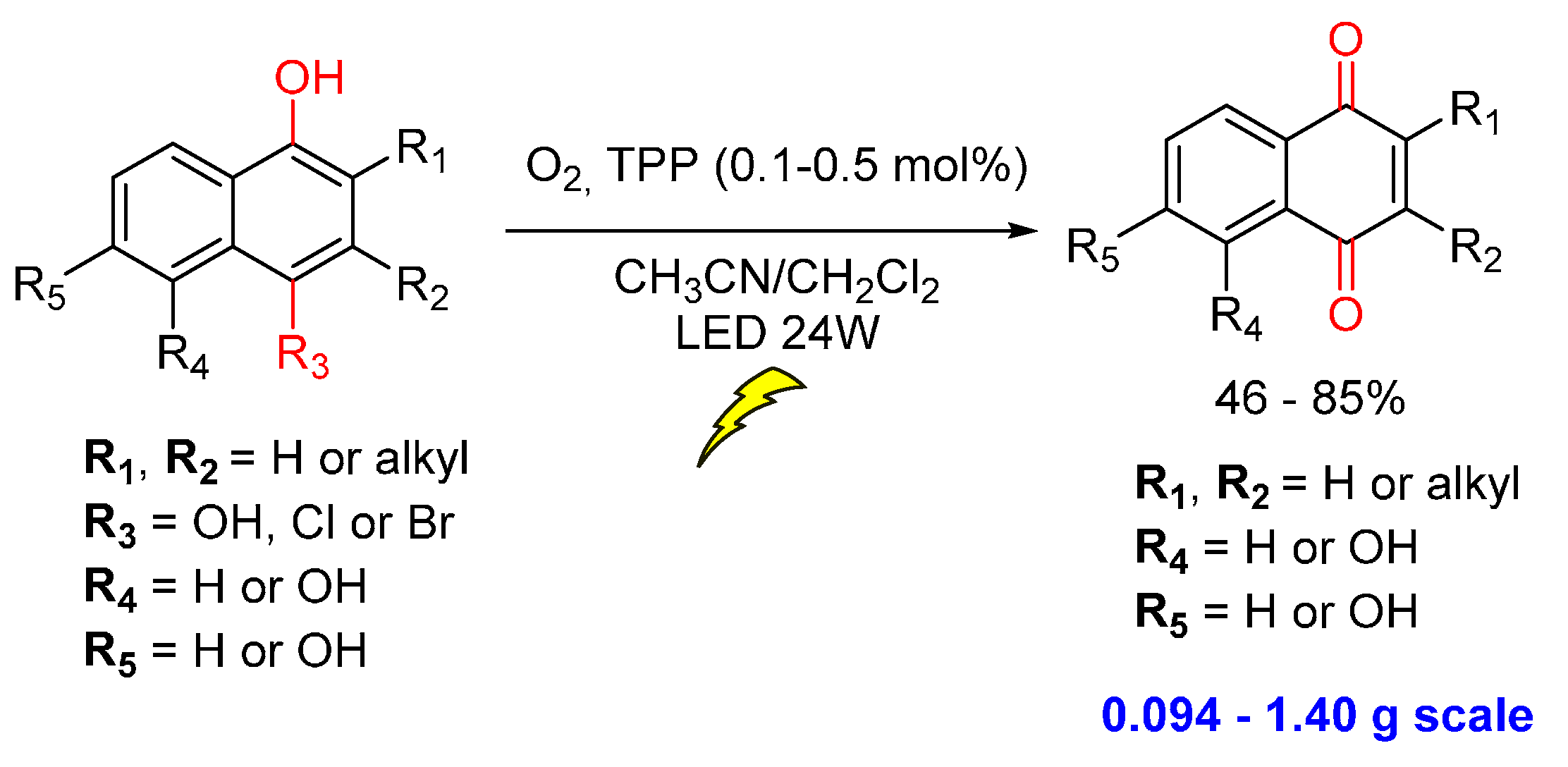{kind=link}
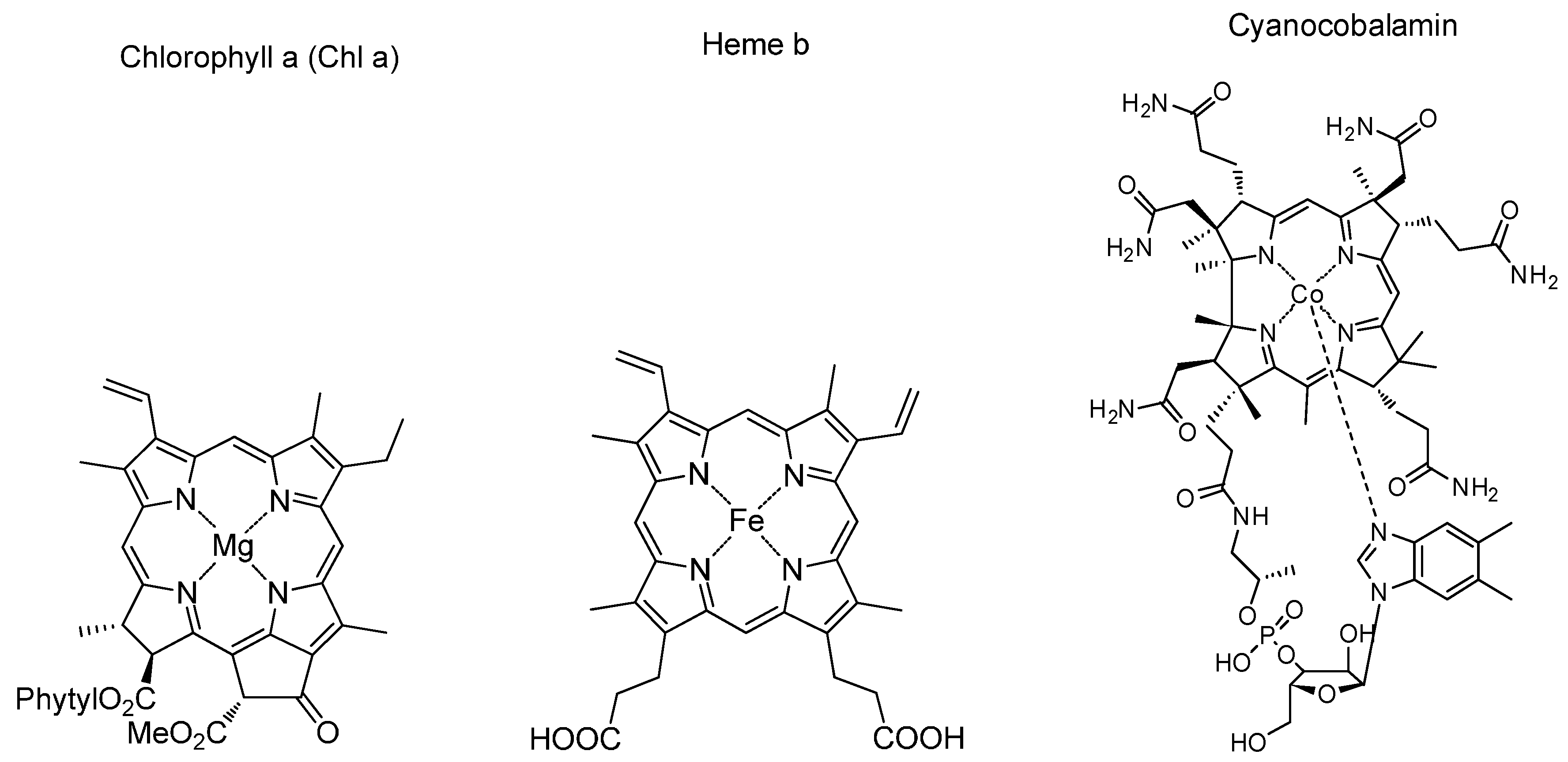
1. A Brief History of Catalysis
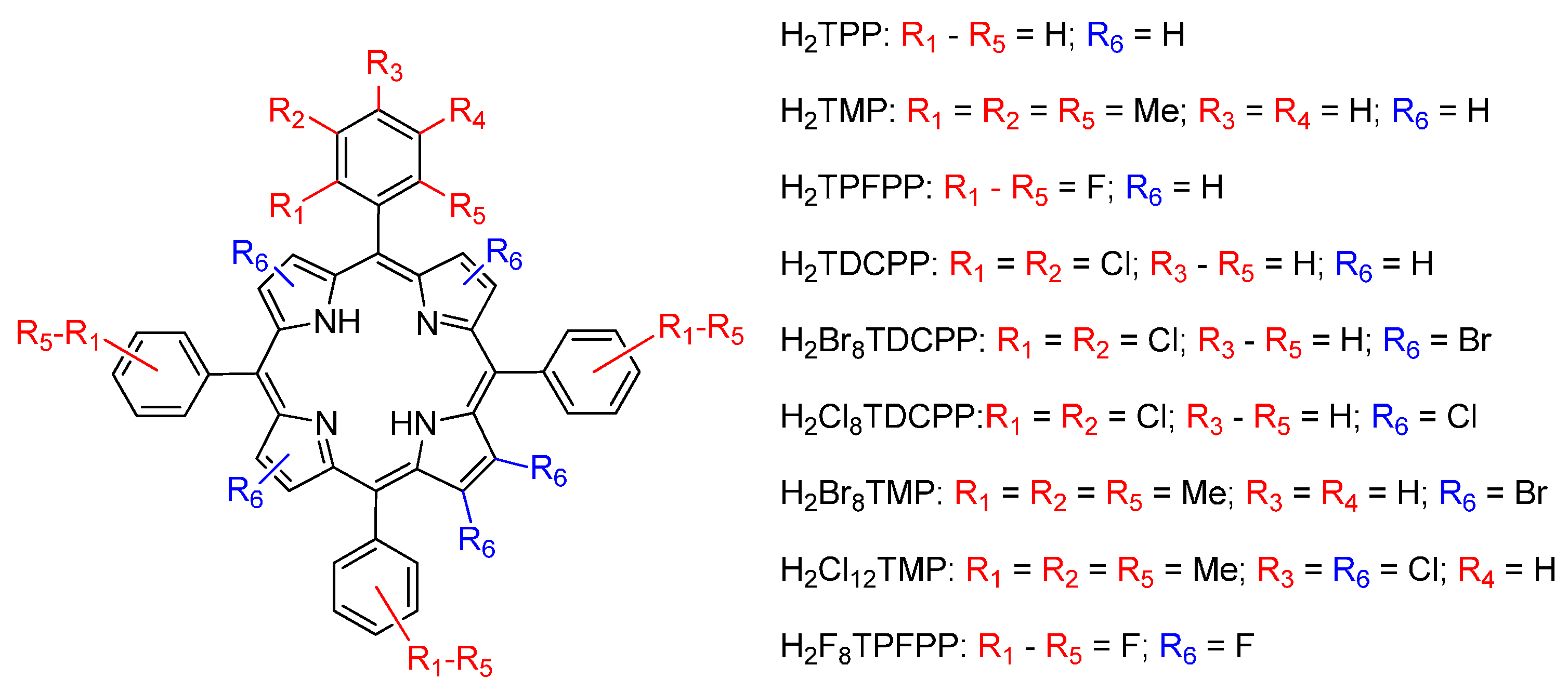
2. General Concepts in Porphyrin Catalysis
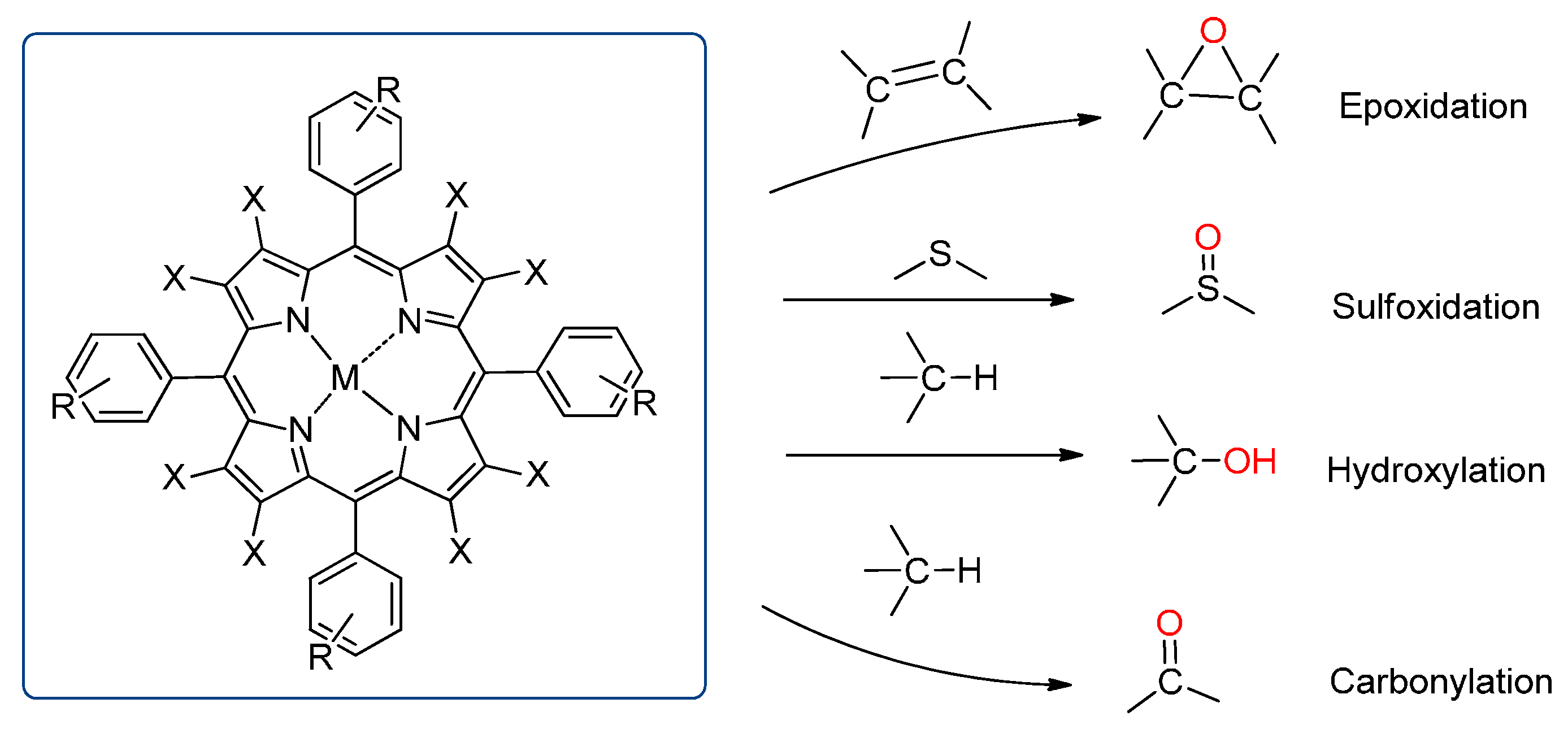
3. Oxidation Reactions with Porphyrin and Metalloporphyrin–Based Catalysts
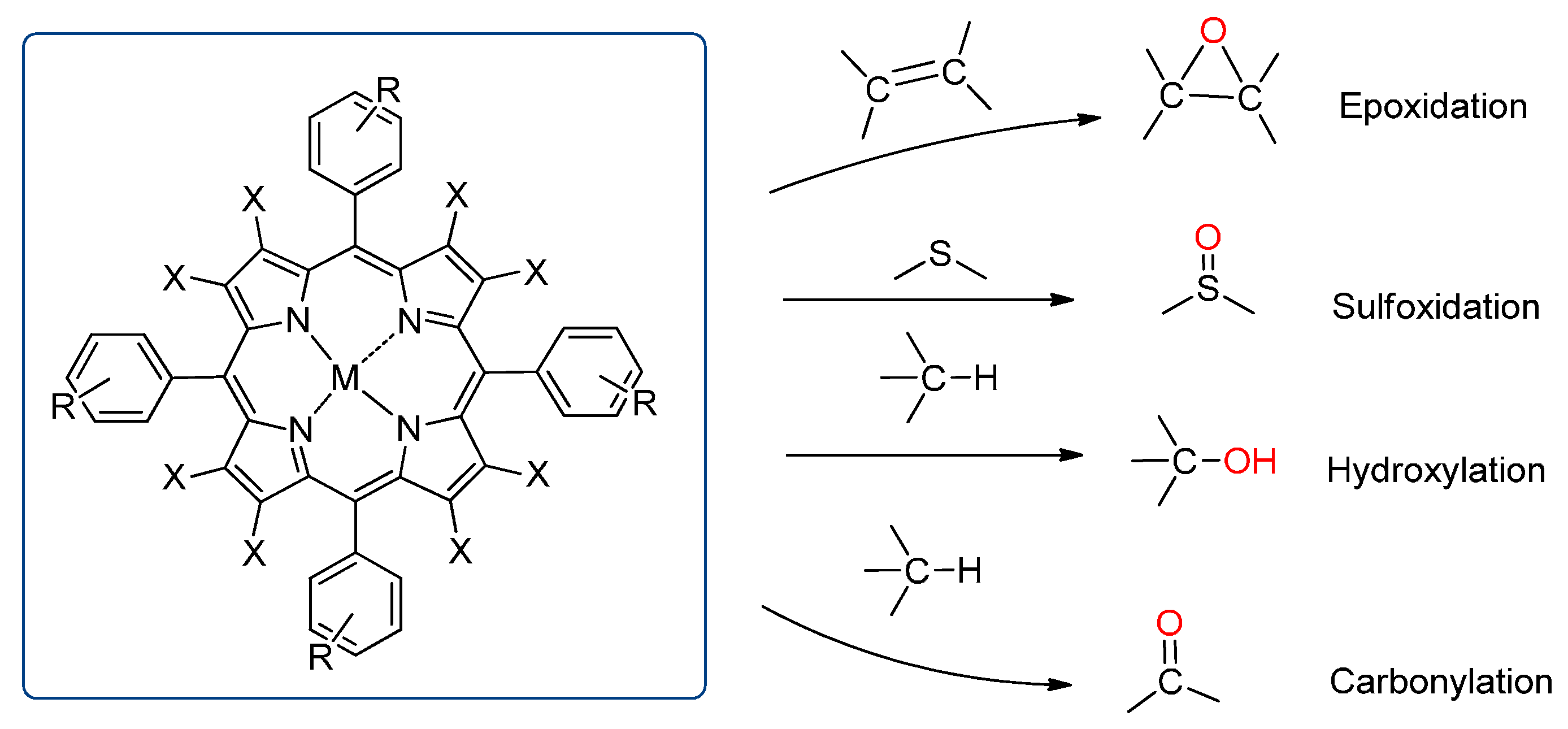
3.1. Epoxidation
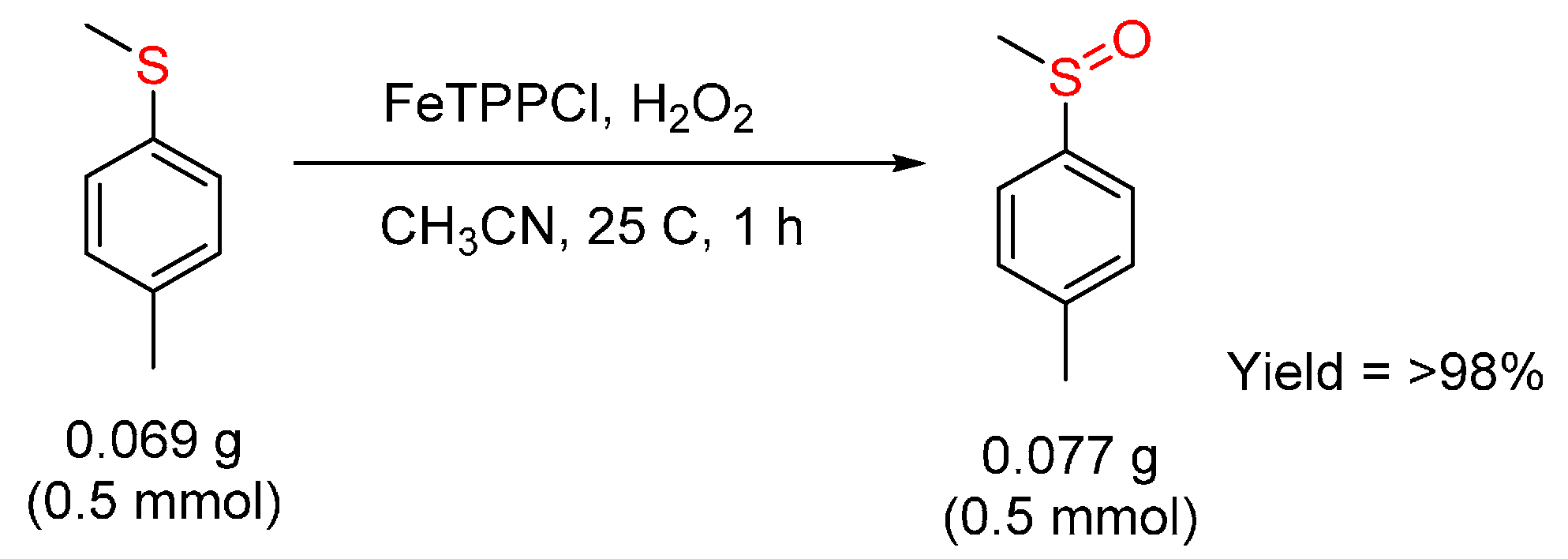
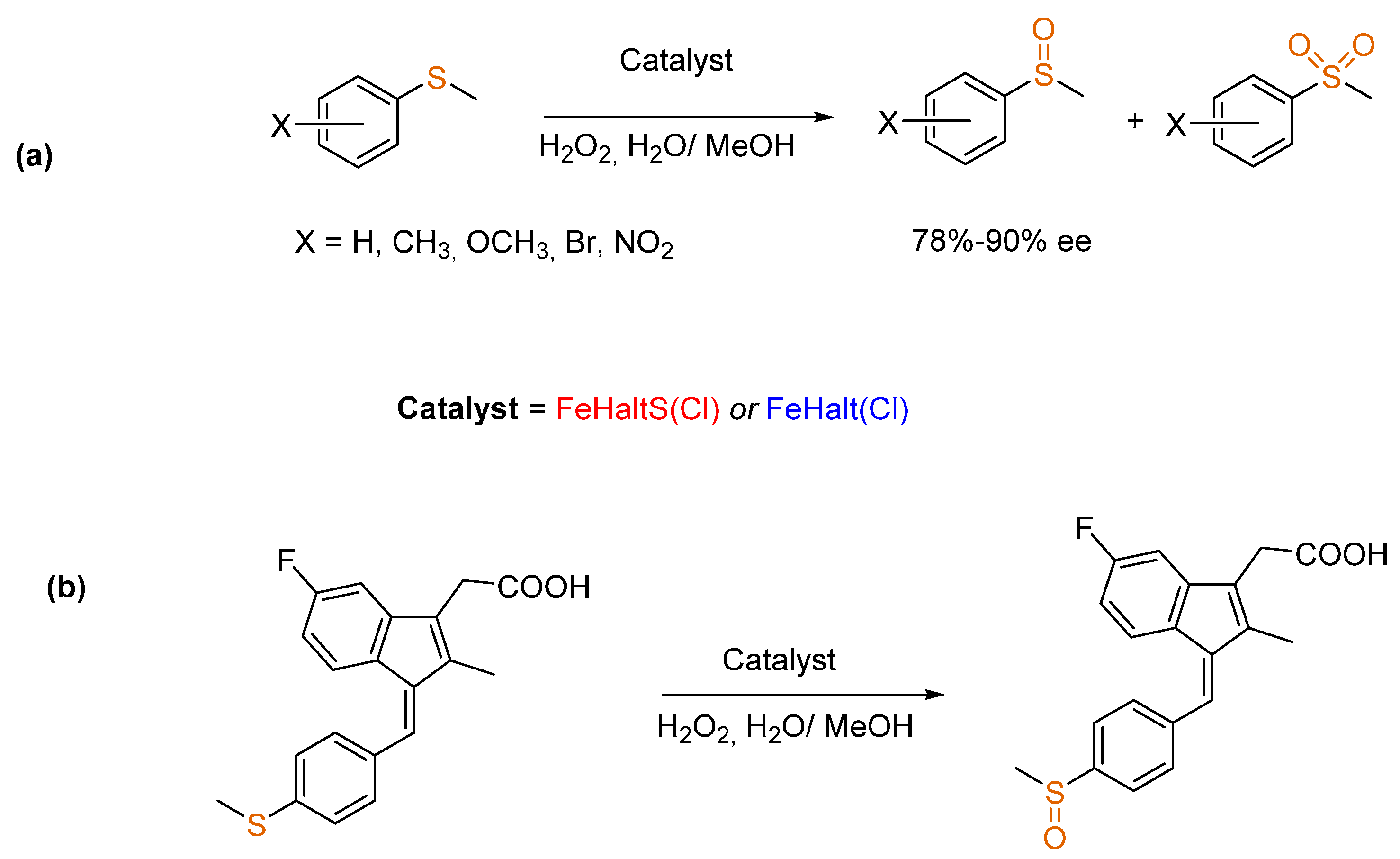
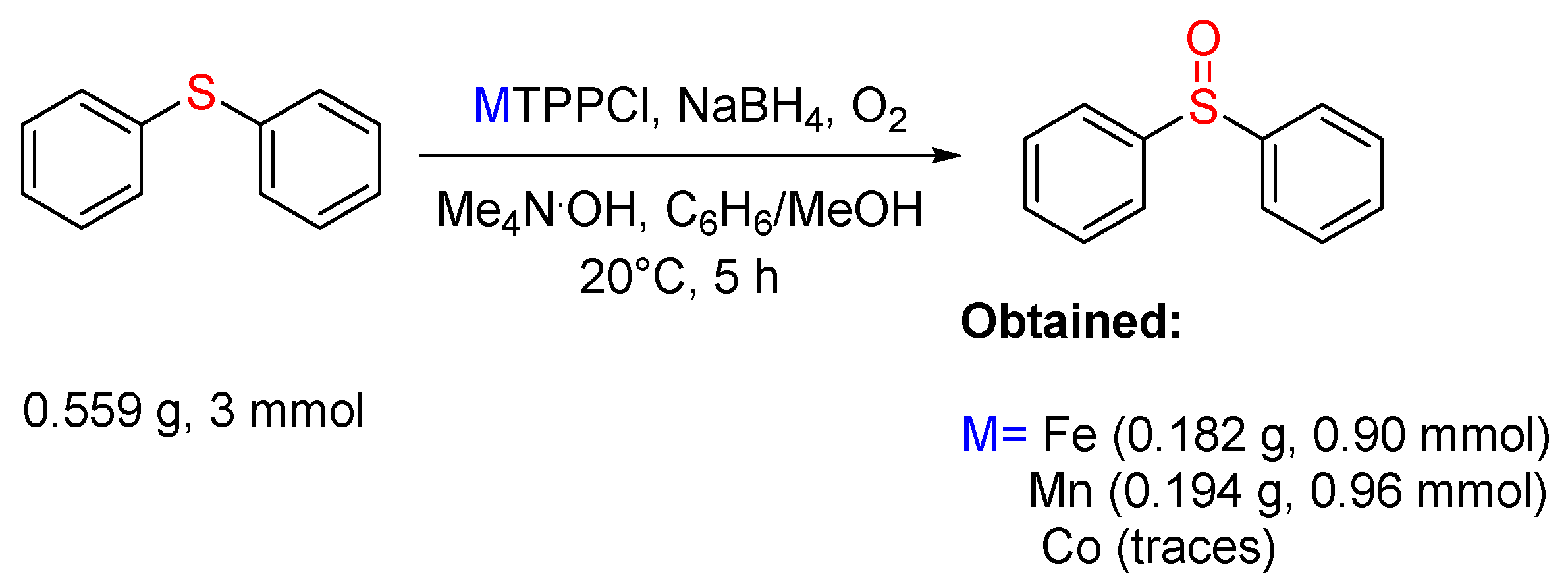
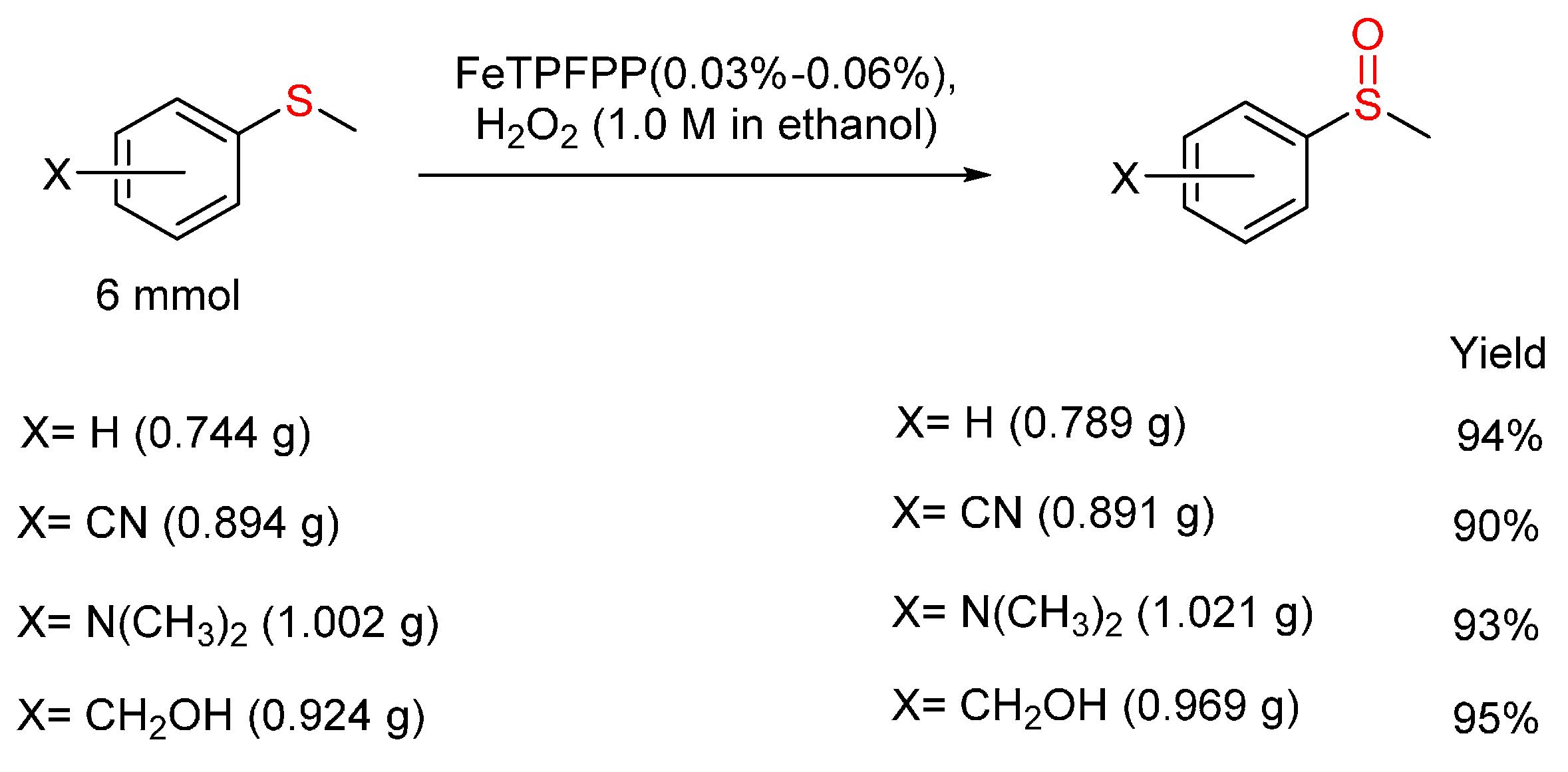
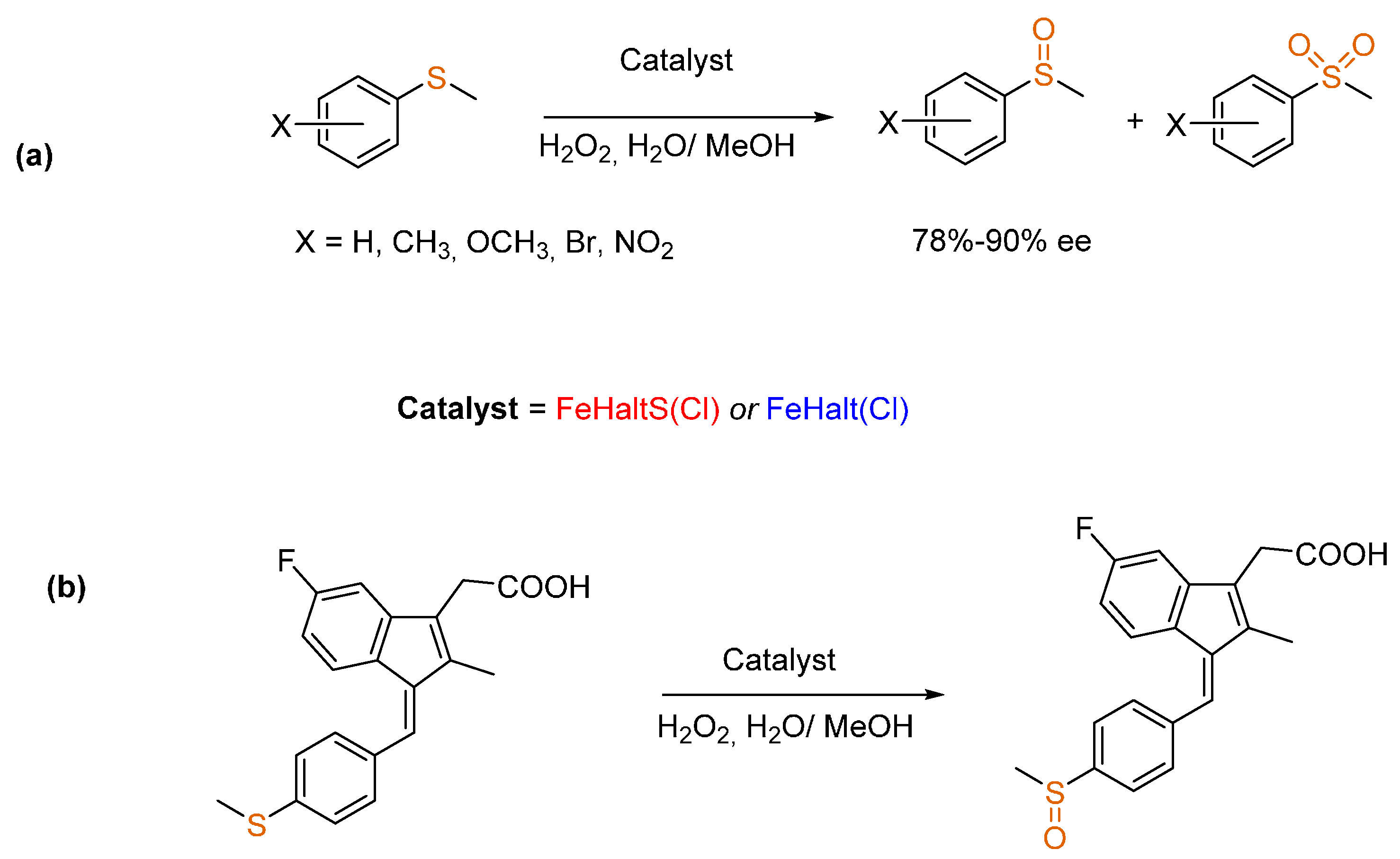
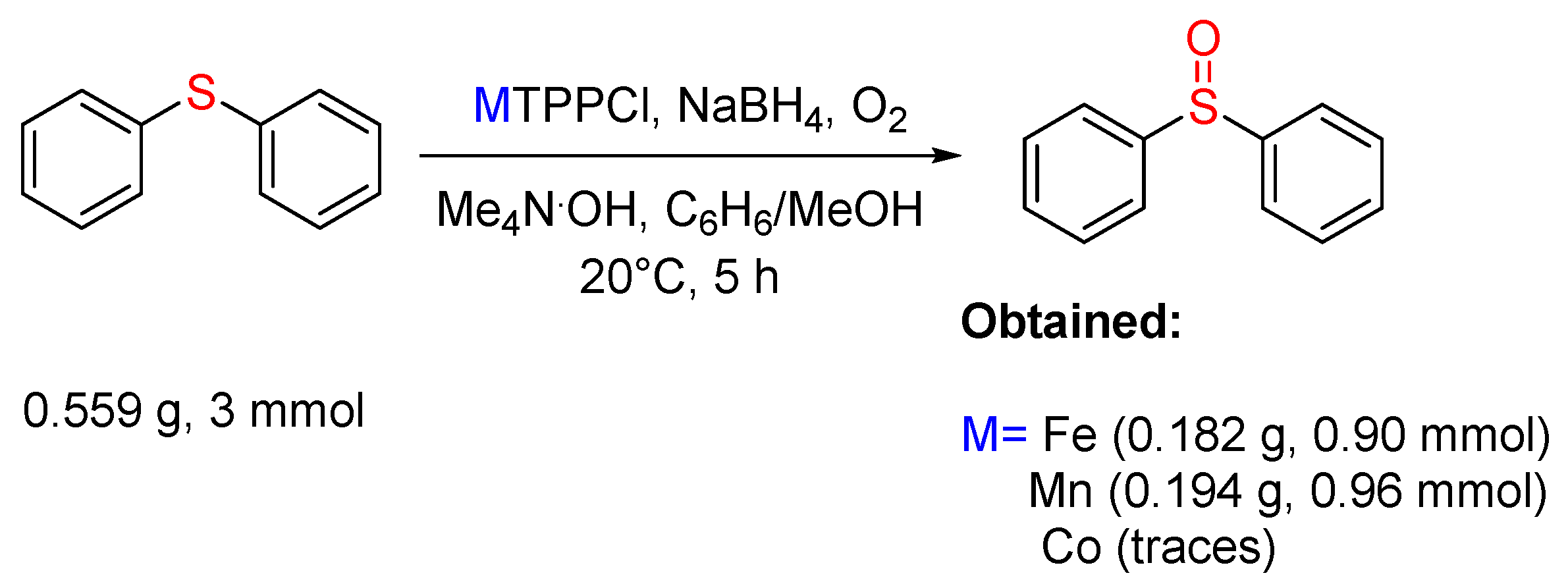
3.2. Sulfides to Sulfoxides
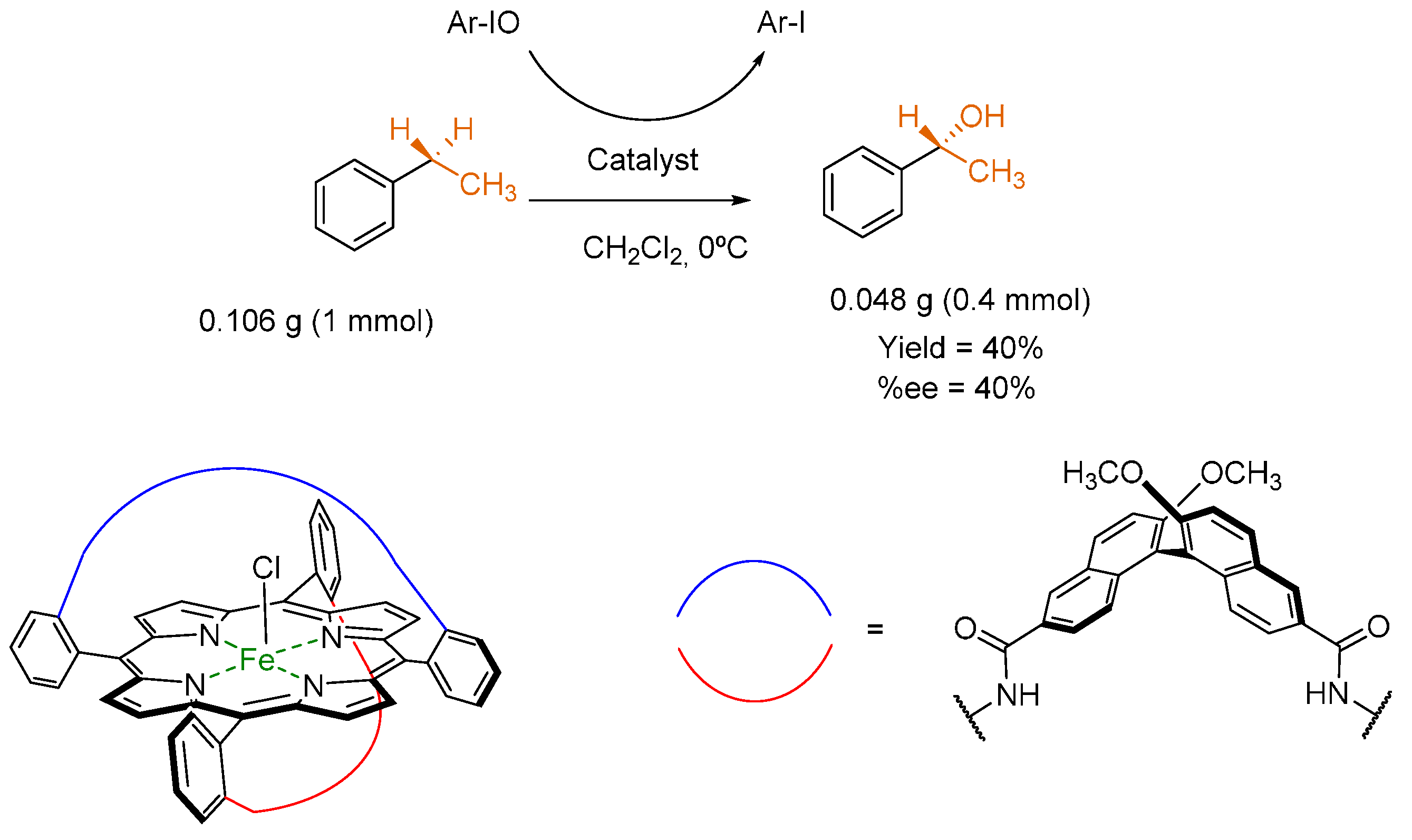
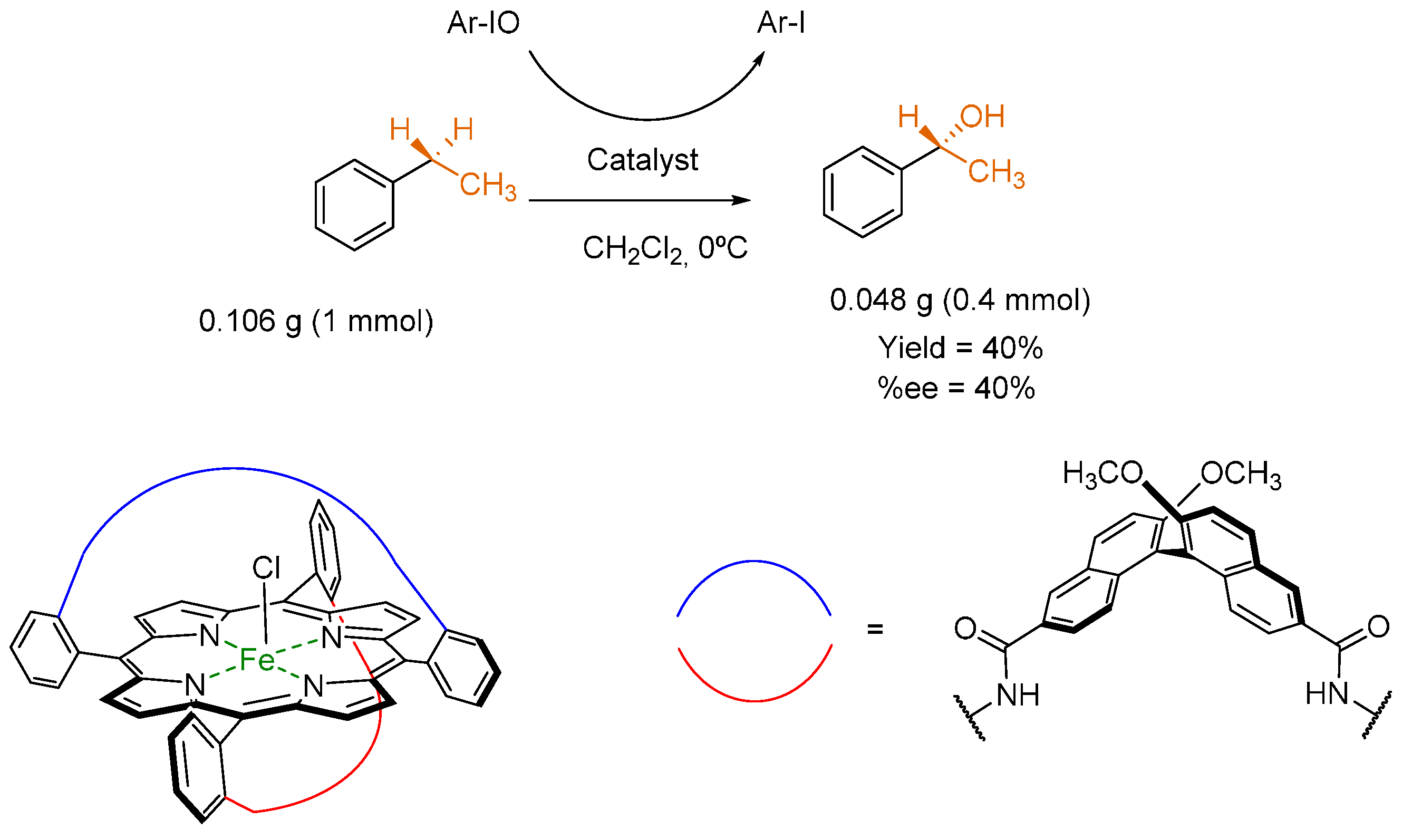
3.3. Hydroxylation
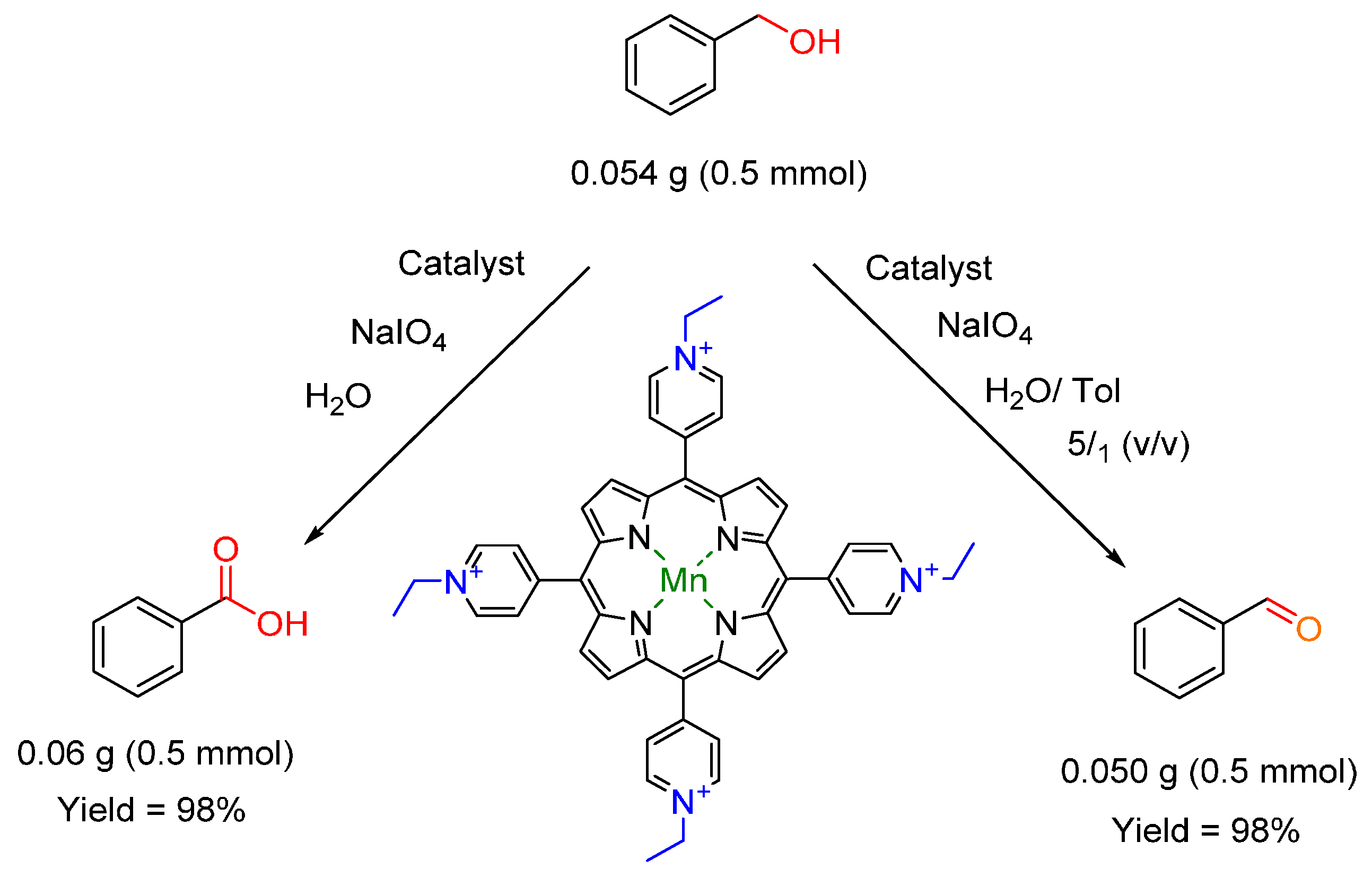
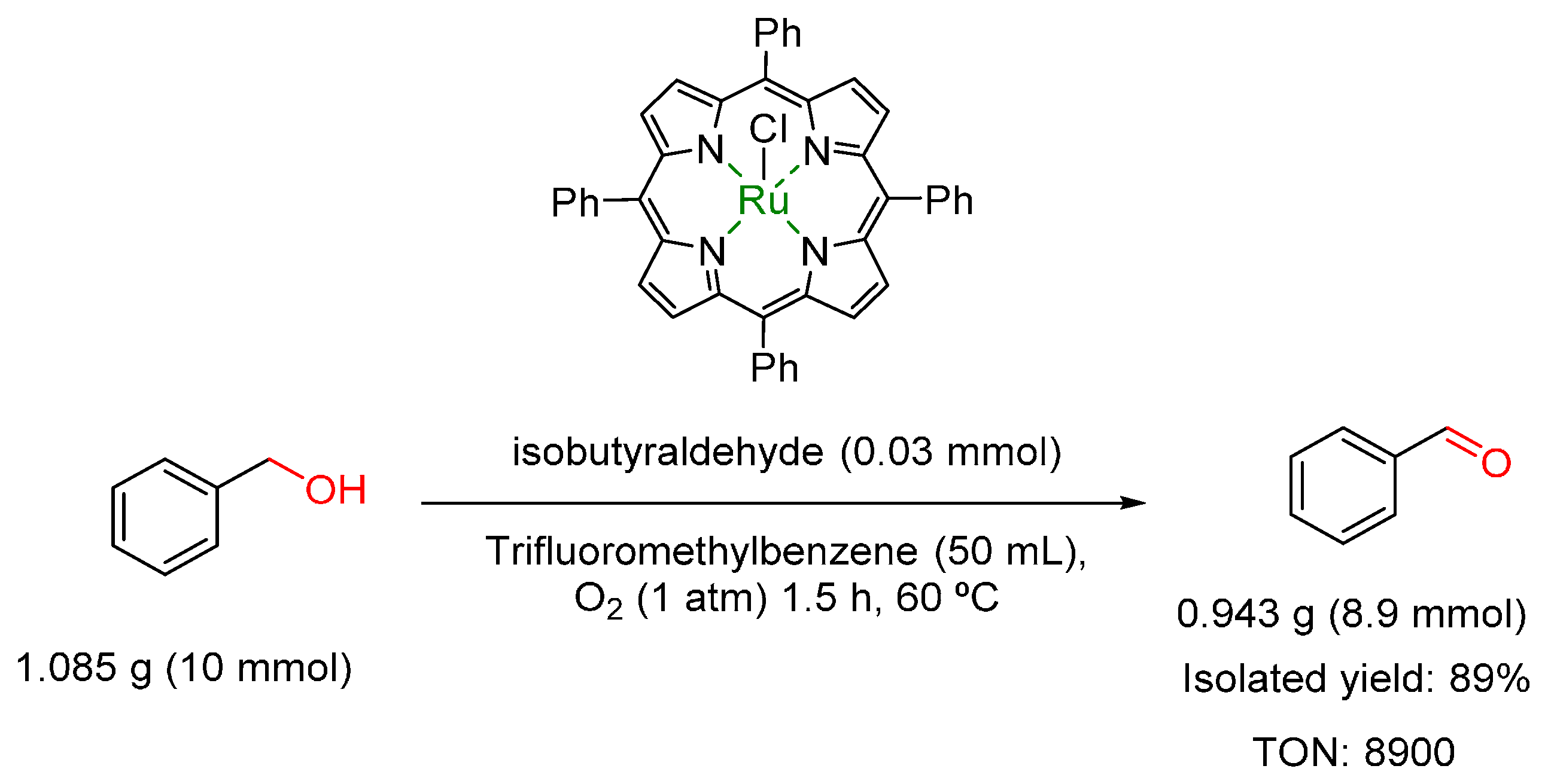
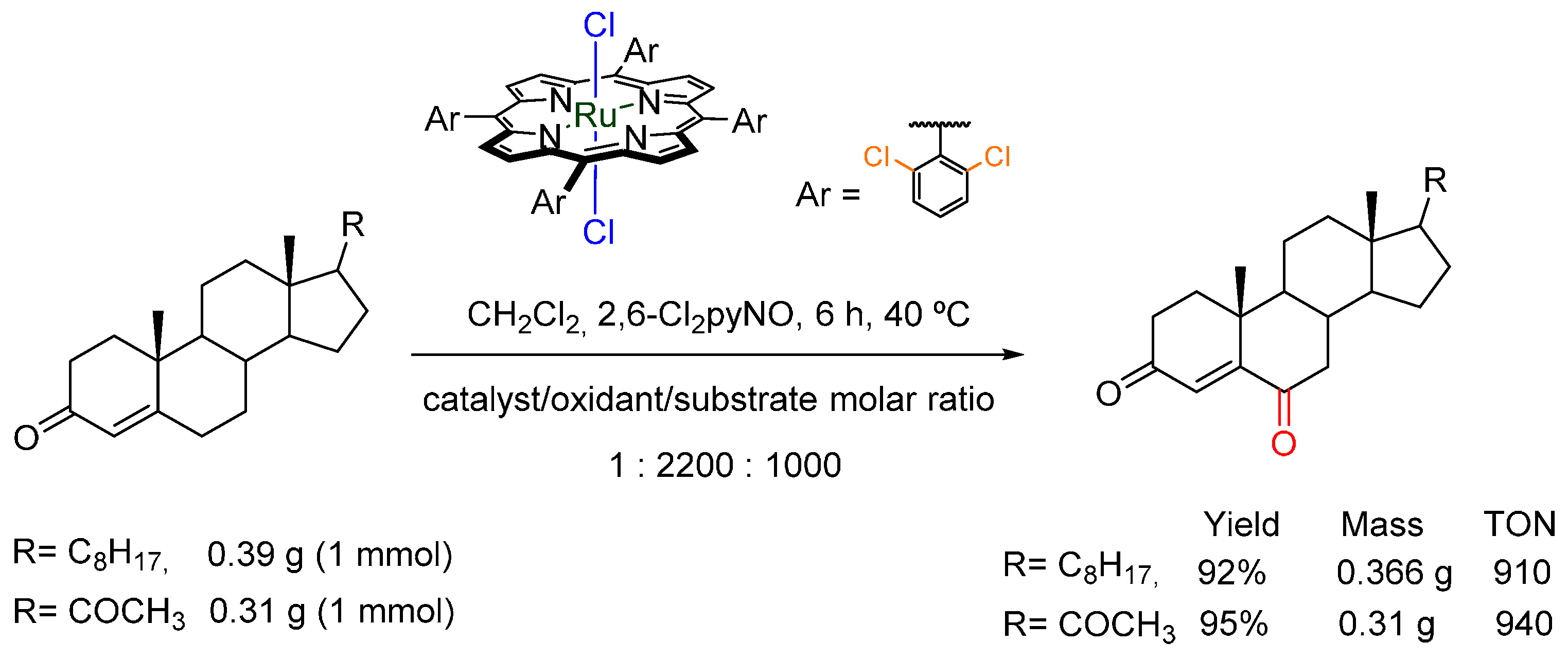
3.4. Oxidation of Alcohols to Carbonyl Compounds
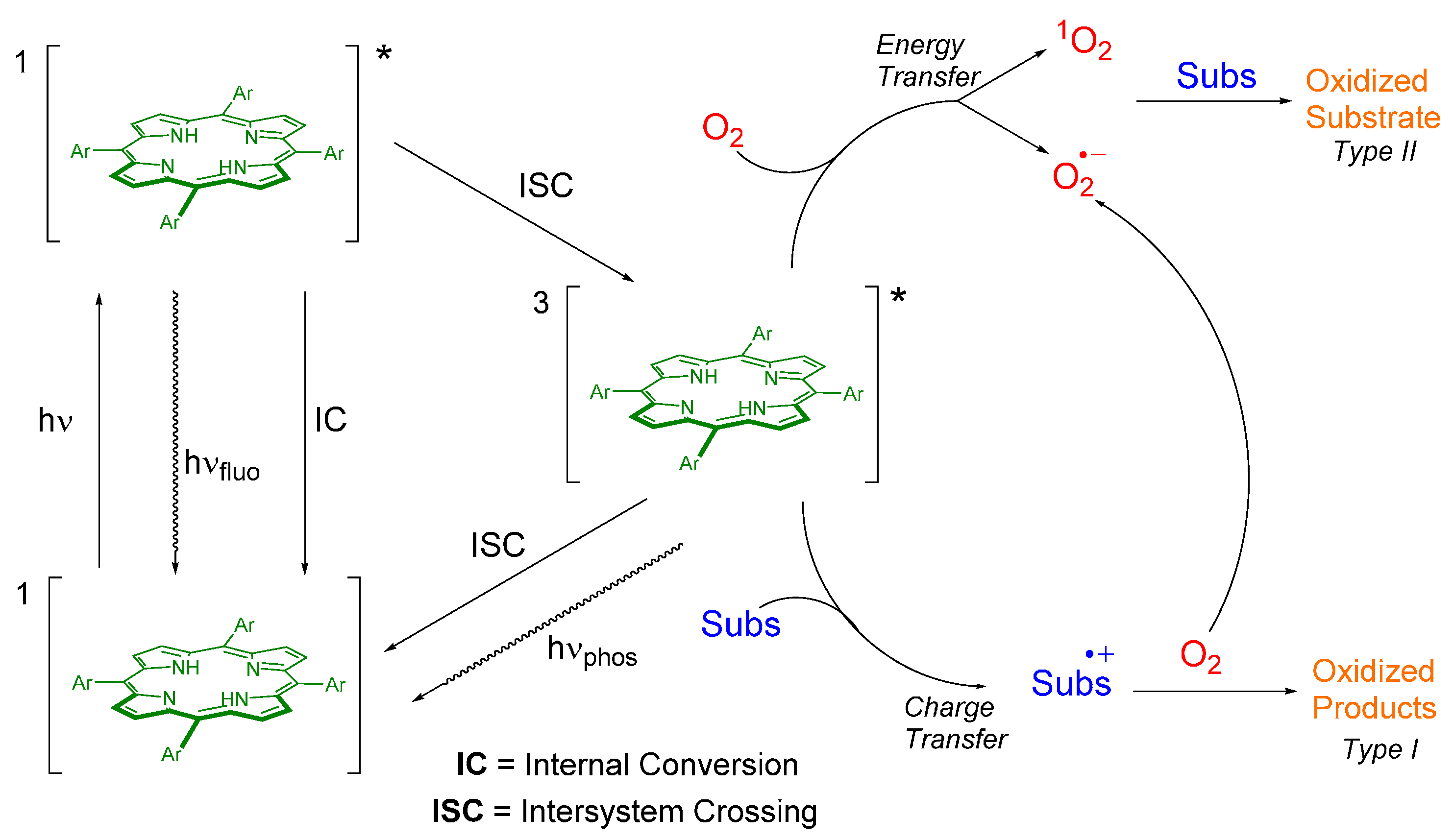
4. Porphyrin Derivatives Acting as Photocatalysts
4.1. Immobilized Porphyrins as Photocatalysts
4.2. Continuous Flow Photocatalysis
5. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wisniak, J. The history of catalysis. From the beginning to Nobel prizes. Educ. Quim. 2010, 21, 60–69. [Google Scholar]
- Roberts, M.W. Birth of the catalytic concept (1800–1900). Catal. Lett. 2000, 67, 1–4. [Google Scholar] [CrossRef]
- Van Santen, R. Catalysis in perspective: Historic review. In Catalysis: From Principles to Applications, 1st ed.; Beller, M., Renken, A., van Santen, R., Beller, M., Renken, A., Van Santen, R., Eds.; Wiley-VCH: Weinheim, Germany, 2012; pp. 3–19. [Google Scholar]
- Drain, C.M.; Hupp, J.T.; Suslick, K.S.; Wasielewski, M.R.; Chen, X. A perspective on four new porphyrin-based functional materials and devices. J. Porphyrins Phthalocyanines 2002, 06, 243–258. [Google Scholar] [CrossRef]
- Chalmers, M.; Notman, N. Living the nobel life. Chem. World 2009, 6. Available online: http://www.rsc.org/chemistryworld/Issues/2009/September/LivingtheNobellife.asp (accessed on 3 March 2016). [Google Scholar]
- Zhou, Q.L. Transition-metal catalysis and organocatalysis: Where can progress be expected? Angew. Chem. 2015, 2–4. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.B.; Brocksom, T.J.; de Oliveira, K.T. Tetrabenzoporphyrins: Synthetic developments and applications. Chem. Soc. Rev. 2013, 42, 3302–3317. [Google Scholar] [CrossRef] [PubMed]
- Betoni Momo, P.; Pavani, C.; Baptista, M.S.; Brocksom, T.J.; de Oliveira, K.T. Chemical transformations and photophysical properties of meso-tetrathienyl-substituted porphyrin derivatives. Eur. J. Org. Chem. 2014, 2014, 4536–4547. [Google Scholar] [CrossRef]
- Carvalho, C.M.B.; Fujita, M.A.; Brocksom, T.J.; de Oliveira, K.T. Synthesis and photophysical evaluations of β-fused uracil-porphyrin derivatives. Tetrahedron 2013, 69, 9986–9993. [Google Scholar] [CrossRef]
- Perutz, M.F. Stereochemical mechanism of oxygen transport by haemoglobin. Proc. R. Soc. Lond. B. Biol. 1980, 208, 135–162. [Google Scholar] [CrossRef]
- Senge, M.O.; MacGowan, S.A.; O’Brien, J.M. Conformational control of cofactors in nature—The influence of protein-induced macrocycle distortion on the biological function of tetrapyrroles. Chem. Commun. 2015, 51, 17031–17063. [Google Scholar] [CrossRef] [PubMed]
- Milgrom, L.R. The Colours of Life—An Introduction to the Chemistry of Porphyrins and Related Compounds; Oxford University Press, Inc.: New York, NY, USA, 1997; Volume 22. [Google Scholar]
- Battersby, A.R. Tetrapyrroles: The pigments of life. Nat. Prod. Rep. 2000, 17, 507–526. [Google Scholar] [CrossRef] [PubMed]
- Sheftel, A.D.; Mason, A.B.; Ponka, P. The long history of iron in the Universe and in health and disease. Biochim. Biophys. Acta—Gen. Subj. 2012, 1820, 161–187. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R. Metalloporphyrins in Catalytic Oxidations; Sheldon, R., Ed.; Marcel Dekker: New York, NY, USA, 1994. [Google Scholar]
- Collman, J.; Zhang, X.; Lee, V.; Uffelman, E.; Brauman, J. Regioselective and enantioselective epoxidation catalysed by metalloporphyrins. Science 1993, 261, 1404–1411. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; Nemo, T.E.; Myers, R.S. Hydroxylation and epoxidation catalysed by iron-porphine complexes. Oxygen transfer from iodosylbenzene. J. Am. Chem. Soc. 1979, 101, 1032–1033. [Google Scholar] [CrossRef]
- Jiang, G.; Liu, Q.; Guo, C. Biomimetic oxidation of hydrocarbons with air over metalloporphyrins. In Biomimetic Based Applications; George, A., Ed.; InTech: Rijeka, Croatia, 2011; pp. 31–58. [Google Scholar]
- Meunier, B. Metalloporphyrins as versatile catalysts for oxidation reactions and oxidative DNA cleavage. Chem. Rev. 1992, 92, 1411–1456. [Google Scholar] [CrossRef]
- Chang, C.; Kuo, M. Reaction of iron(III) porphyrins and iodosoxylene. The active oxene complex of cytochrome P-450. J. Am. Chem. Soc. 1979, 101, 3413–3415. [Google Scholar] [CrossRef]
- Dolphin, D.; Traylor, T.G.; Xie, L.Y. Polyhaloporphyrins: Unusual ligands for metals and metal-catalysed oxidations. Acc. Chem. Res. 1997, 30, 251–259. [Google Scholar] [CrossRef]
- Traylor, T.G.; Tsuchiya, S. Perhalogenated tetraphenylhemins: Stable catalysts of high turnover catalytic hydroxylations. Inorg. Chem. 1987, 26, 1338–1339. [Google Scholar] [CrossRef]
- Haber, J.; Matachowski, L.; Pamin, K.; Poltowicz, J. The effect of peripheral substituents in metalloporphyrins on their catalytic activity in Lyons system. J. Mol. Catal. A Chem. 2003, 198, 215–221. [Google Scholar] [CrossRef]
- Srour, H.; le Maux, P.; Chevance, S.; Simonneaux, G. Metal-catalysed asymmetric sulfoxidation, epoxidation and hydroxylation by hydrogen peroxide. Coord. Chem. Rev. 2013, 257, 3030–3050. [Google Scholar] [CrossRef]
- Caron, S.; Dugger, R.W.; Ruggeri, S.G.; Ragan, J.A.; Brown Ripin, D.H. Large-scale oxidations in the pharmaceutical industry. Chem. Rev. 2006, 106, 2943–2989. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, D. Selective oxidation of organic compounds-sustainable catalytic reactions with oxygen and without transition metals? Angew. Chem. Int. Ed. Engl. 2006, 45, 3206–3210. [Google Scholar] [CrossRef] [PubMed]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Kar-yan lo, V.; Zhou, C.-Y.; Huang, J.-S. Selective functionalisation of saturated C–H bonds with metalloporphyrin catalysts. Chem. Soc. Rev. 2011, 40, 1950–1975. [Google Scholar] [CrossRef] [PubMed]
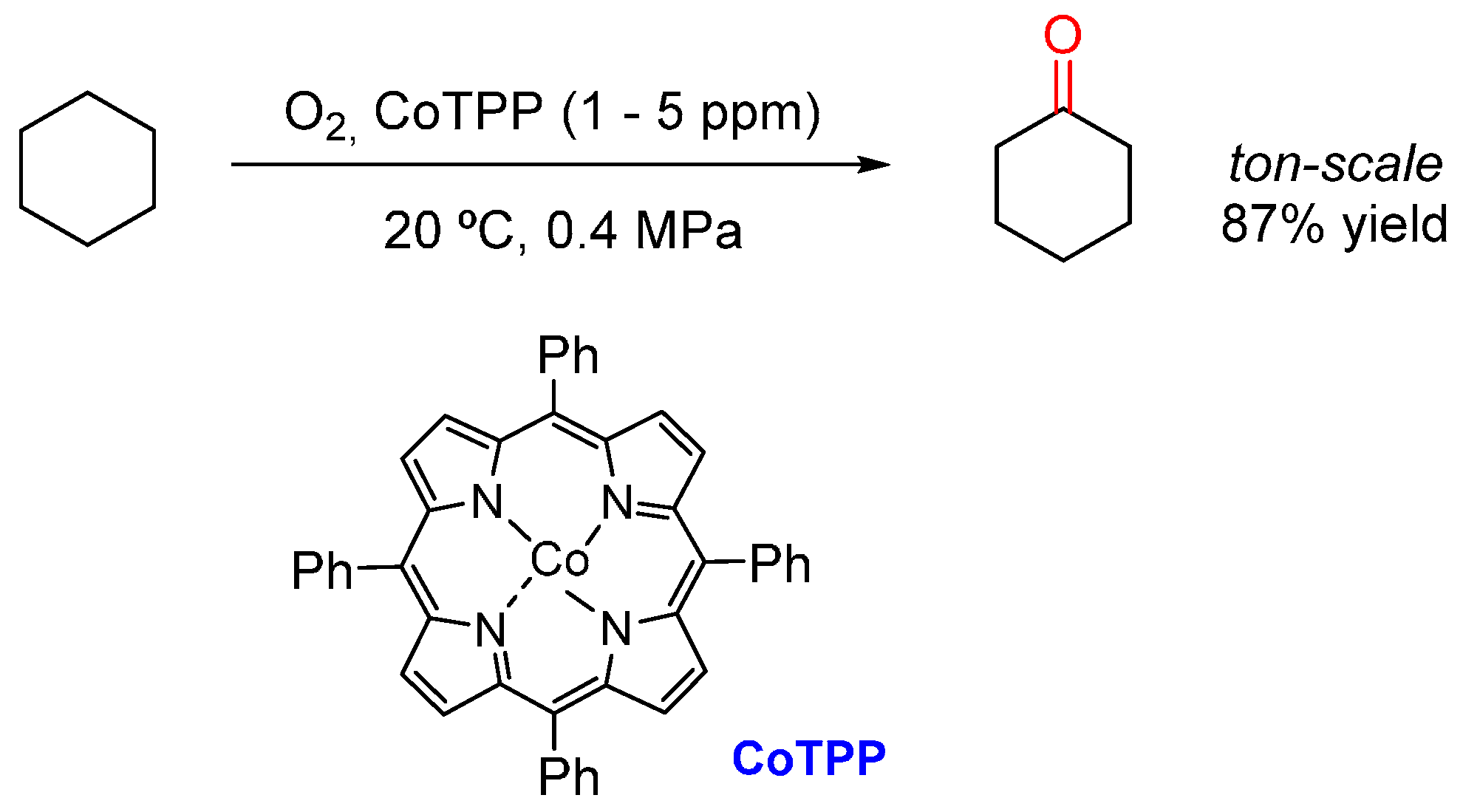
- Guo, C.-C.; Liu, X.-Q.; Liu, Q.; Liu, Y.; Chu, M.-F.; Lin, W.-Y. First industrial-scale biomimetic oxidation of hydrocarbon with air over metalloporphyrins as cytochrome P-450 monooxygenase model and its mechanistic studies. J. Porphyr. Phthalocyanines 2009, 13, 1250–1254. [Google Scholar] [CrossRef]
- Costas, M. Selective C–H oxidation catalysed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. [Google Scholar] [CrossRef]
- Nakagaki, S.; Ferreira, G.; Ucoski, G.; Dias de Freitas Castro, K. Chemical reactions catalysed by metalloporphyrin-based metal-organic frameworks. Molecules 2013, 18, 7279–7308. [Google Scholar] [CrossRef] [PubMed]
- Akagah, B.; Lormier, A.T.; Fournet, A.; Figadère, B. Oxidation of antiparasitic 2-substituted quinolines using metalloporphyrin catalysts: Scale-up of a biomimetic reaction for metabolite production of drug candidates. Org. Biomol. Chem. 2008, 6, 4494–4497. [Google Scholar] [CrossRef] [PubMed]
- Che, C.-M.; Huang, J.-S. Metalloporphyrin-based oxidation systems: From biomimetic reactions to application in organic synthesis. Chem. Commun. 2009, 27, 3996–4015. [Google Scholar] [CrossRef] [PubMed]
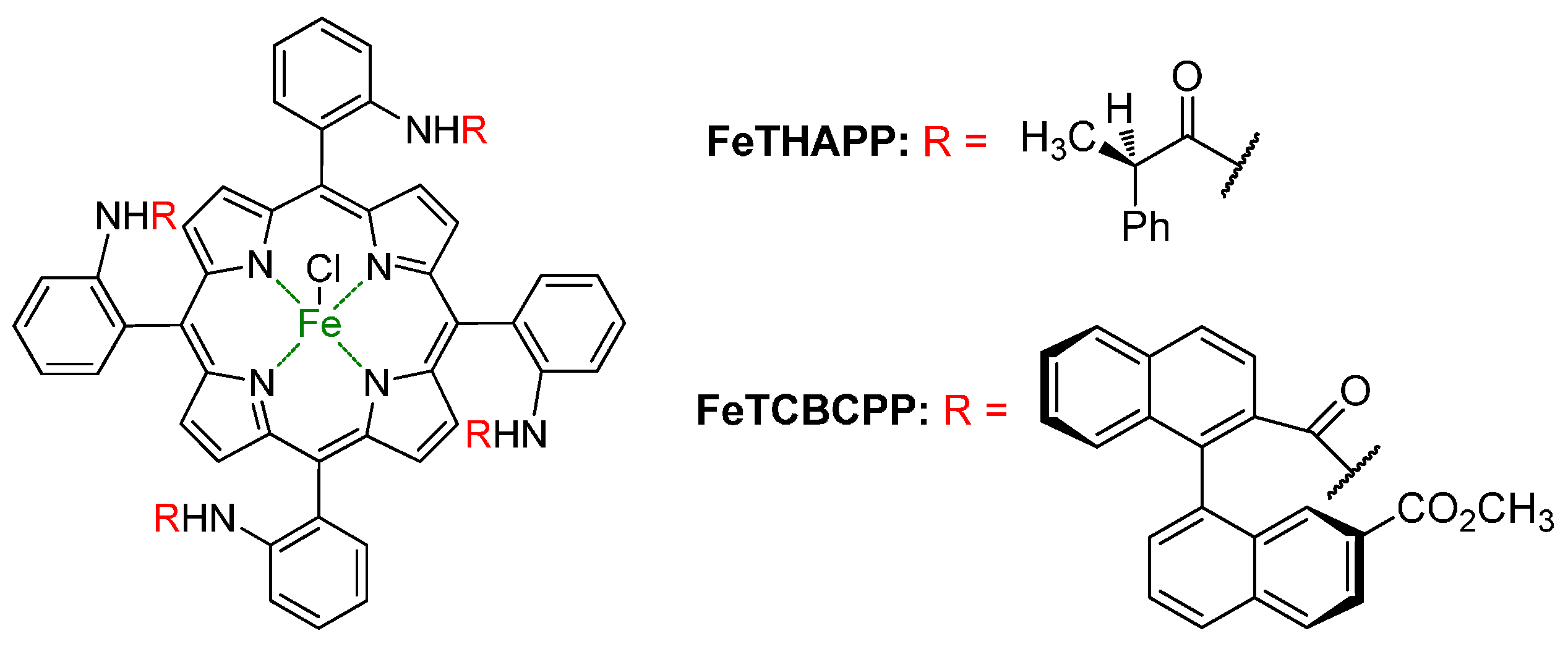
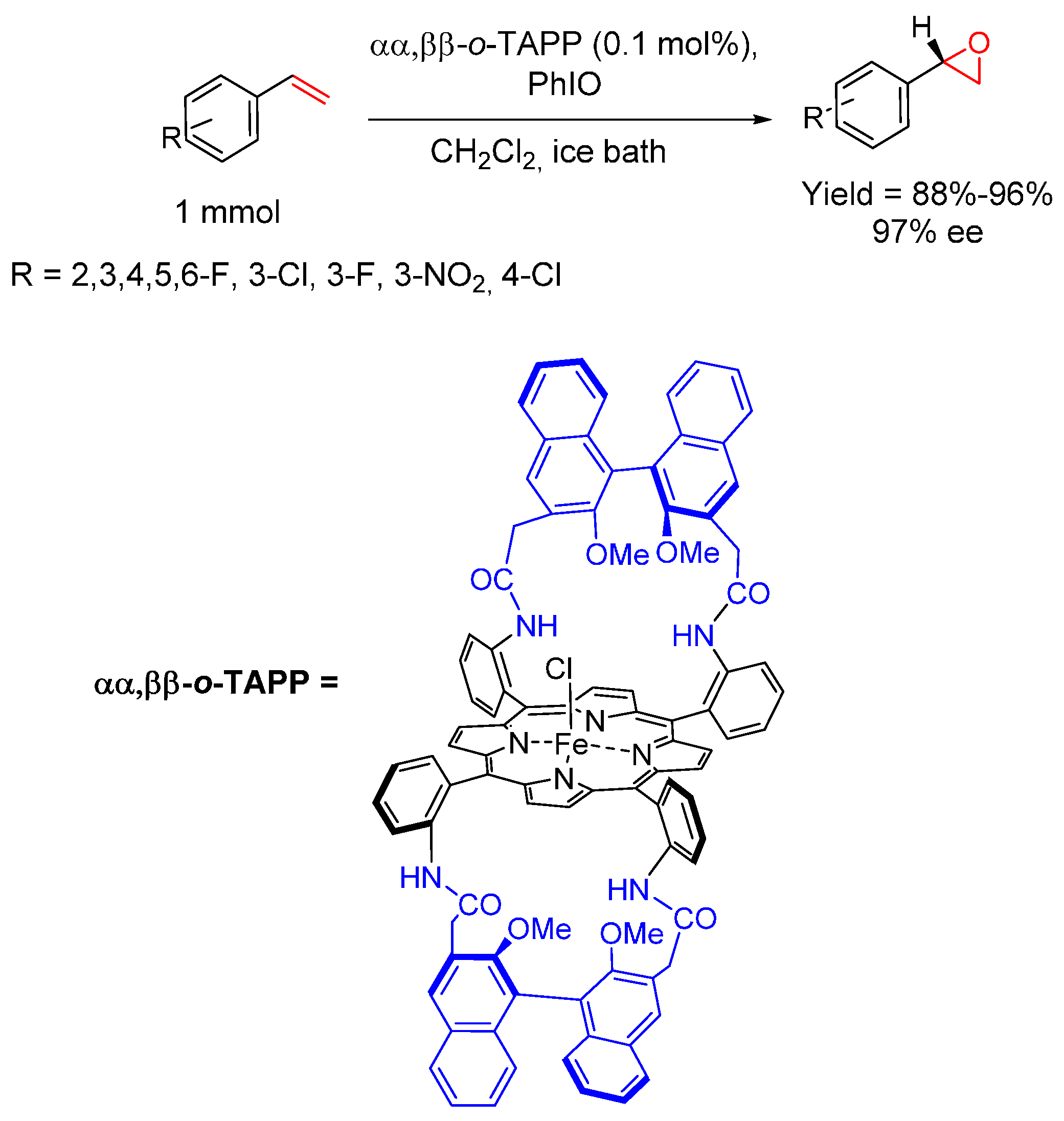
- Groves, J.T.; Myers, R.S. Catalytic asymmetric epoxidations with chiral iron porphyrins. J. Am. Chem. Soc. 1983, 105, 5791–5796. [Google Scholar] [CrossRef]
- Rose, E.; Andrioletti, B.; Zrig, S.; Quelquejeu-Ethève, M. Enantioselective epoxidation of olefins with chiral metalloporphyrin catalysts. Chem. Soc. Rev. 2005, 34, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Gopalaiah, K. Chiral iron catalysts for asymmetric synthesis. Chem. Rev. 2013, 4, 3248–3296. [Google Scholar] [CrossRef] [PubMed]
- Lane, B.S.; Burgess, K. Metal-catalysed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2473. [Google Scholar] [CrossRef] [PubMed]
- De Faveri, G.; Ilyashenko, G.; Watkinson, M. Recent advances in catalytic asymmetric epoxidation using the environmentally benign oxidant hydrogen peroxide and its derivatives. Chem. Soc. Rev. 2011, 40, 1722–1760. [Google Scholar] [CrossRef] [PubMed]
- Adam, W.; Stegmann, V.R.; Saha-mo, C.R. Regio- and diastereoselective epoxidation of chiral allylic alcohols catalysed by manganese (salen) and iron (porphyrin) complexes. J. Am. Chem. Soc. 1999, 121, 1879–1882. [Google Scholar] [CrossRef]
- Rose, E.; Ren, Q.Z.; Andrioletti, B. A Unique binaphthyl strapped iron-porphyrin catalyst for the enantioselective epoxidation of terminal olefins. Chem. Eur. J. 2004, 10, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, N.A.; Bell, A.T. Mechanistic insights into iron porphyrin-catalysed olefin epoxidation by hydrogen peroxide: Factors controlling activity and selectivity. J. Mol. Catal. A Chem. 2007, 275, 54–62. [Google Scholar] [CrossRef]
- Vilain-Deshayes, S.; Robert, A.; Maillard, P.; Meunier, B.; Momenteau, M. Enantioselective epoxidation of olefins by single-oxygen atom donors catalysed by manganese-glycoconjugated porphyrins. J. Mol. Catal. A Chem. 1996, 113, 23–34. [Google Scholar] [CrossRef]
- Le Maux, P.; Srour, H.F.; Simonneaux, G. Enantioselective water-soluble iron–porphyrin-catalysed epoxidation with aqueous hydrogen peroxide and hydroxylation with iodobenzene diacetate. Tetrahedron 2012, 68, 5824–5828. [Google Scholar] [CrossRef]
- Srour, H.; Le Maux, P.; Simonneaux, G. Enantioselective manganese-porphyrin-catalysed epoxidation and C–H hydroxylation with hydrogen peroxide in water/methanol solutions. Inorg. Chem. 2012, 51, 5850–5856. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, I.D.; Danks, T.N.; Hay, J.N.; Hamerton, I.; Gunathilagan, S. Evidence for parallel destructive, and competitive epoxidation and dismutation pathways in metalloporphyrin-catalysed alkene oxidation by hydrogen peroxide. Tetrahedron 2001, 57, 6847–6853. [Google Scholar] [CrossRef]
- Nam, W.; Jin, S.W.; Lim, M.H.; Ryu, J.Y.; Kim, C. Anionic ligand effect on the nature of epoxidizing intermediates in iron porphyrin complex-catalysed epoxidation reactions. Inorg. Chem. 2002, 41, 3647–3652. [Google Scholar] [CrossRef] [PubMed]
- Nam, W.; Oh, S.; Sun, Y.J.; Kim, J.; Kim, W.; Woo, S.K. Factors affecting the catalytic epoxidation of olefins by iron porphyrin complexes and H2O2 in protic solvents. J. Org. Chem. 2003, 68, 7903–7906. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Zhou, H.B.; Huang, J.S.; Che, C.M. Dendritic ruthenium porphyrins: A new class of highly selective catalysts for alkene epoxidation and cyclopropanation. Chem. Eur. J. 2002, 8, 1554–1562. [Google Scholar] [CrossRef]
- Oae, S.; Watanabe, Y.; Fujimori, K. Biomimetic oxidation of organic sulfides with TPPFe(III)Cl/Imidazole/Hydrogen Peroxide. Tetrahedron Lett. 1982, 23, 1189–1192. [Google Scholar] [CrossRef]
- Kowalski, P.; Mitka, K.; Ossowska, K.; Kolarska, Z. Oxidation of sulfides to sulfoxides. Part 1: Oxidation using halogen derivatives. Tetrahedron 2005, 61, 1933–1953. [Google Scholar] [CrossRef]
- Legros, J.; Dehli, J.R.; Bolm, C. Applications of catalytic asymmetric sulfide oxidations to the syntheses of biologically active sulfoxides. Adv. Synth. Catal. 2005, 347, 19–31. [Google Scholar] [CrossRef]
- Wojaczyńska, E.; Wojaczyński, J. Enantioselective synthesis of sulfoxides: 2000–2009. Chem. Rev. 2010, 110, 4303–4356. [Google Scholar] [PubMed]
- Baciocchi, E.; Gerini, M.F.; Lapi, A. Synthesis of sulfoxides by the hydrogen peroxide induced oxidation of sulfides catalysed by iron tetrakis(pentafluorophenyl)porphyrin: Scope and chemoselectivity. J. Org. Chem. 2004, 69, 3586–3589. [Google Scholar] [CrossRef] [PubMed]
- Kaczorowska, K.; Kolarska, Z.; Mitka, K.; Kowalski, P. Oxidation of sulfides to sulfoxides. Part 2: Oxidation by hydrogen peroxide. Tetrahedron 2005, 61, 8315–8327. [Google Scholar] [CrossRef]
- Carreño, M.C.; Hernández-Torres, G.; Ribagorda, M.; Urbano, A. Enantiopure sulfoxides: Recent applications in asymmetric synthesis. Chem. Commun. 2009, 7345, 6129–6144. [Google Scholar] [CrossRef] [PubMed]
- Le Maux, P.; Simonneaux, G. First enantioselective iron-porphyrin-catalysed sulfide oxidation with aqueous hydrogen peroxide. Chem. Commun. 2011, 47, 6957–6959. [Google Scholar] [CrossRef] [PubMed]
- Srour, H.; Jalkh, J.; le Maux, P.; Chevance, S.; Kobeissi, M.; Simonneaux, G. Asymmetric oxidation of sulfides by hydrogen peroxide catalysed by chiral manganese porphyrins in water/methanol solution. J. Mol. Catal. A Chem. 2013, 370, 75–79. [Google Scholar] [CrossRef]
- Mori, T.; Santa, T.; Higuchi, T.; Mashino, T.; Hirobe, M. Oxygen activation by Iron(III)-porphyrin/NaBH4/Me4N·OH system as cytochrome P-450 Model. Oxygenation of olefin, N-dealkylation of tertiary amine, oxidation of sulfide, and oxidative cleavage of ether bond. Chem. Pharm. Bull. 1993, 41, 292–295. [Google Scholar] [CrossRef]
- Ortiz De Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed]
- Vinhado, F.S.; Prado-Manso, C.M.C.; Sacco, H.C.; Iamamoto, Y. Cationic manganese(III) porphyrins bound to a novel bis-functionalised silica as catalysts for hydrocarbons oxygenation by iodosylbenzene and hydrogen peroxide. J. Mol. Catal. A Chem. 2001, 174, 279–288. [Google Scholar] [CrossRef]
- Campestrini, S. Highly efficient cascade-oxygen-transfer from H2O2 to olefins mediated by halogenated carbonyl compounds and metalloporphyrins. J. Mol. Catal. A Chem. 2001, 171, 37–42. [Google Scholar] [CrossRef]
- Shilov, A.E.; Shul, G.B. Activation of C–H bonds by metal complexes. Chem. Rev. 1997, 97, 2879–2932. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T.; Viski, P. Asymmetric hydroxylation by a chiral iron porphyrin. J. Am. Chem. Soc. 1989, 111, 8537–8538. [Google Scholar] [CrossRef]
- Groves, J.; Viski, P. Asymmetric hydroxylation, epoxidation, and sulfoxidation catalysed by vaulted binaphthyl metalloporphyrins. J. Org. Chem. 1990, 55, 3628–3634. [Google Scholar] [CrossRef]
- Gross, Z.; Ini, S. Asymmetric catalysis by a chiral ruthenium porphyrin: Epoxidation, hydroxylation, and partial kinetic resolution of hydrocarbons. Org. Lett. 1999, 1, 2077–2080. [Google Scholar] [CrossRef]
- Zhang, R.; Yu, W.-Y.; Che, C.-M. Catalytic enantioselective oxidation of aromatic hydrocarbons with D4-symmetric chiral ruthenium porphyrin catalysts. Tetrahedron Asymmetry 2005, 16, 3520–3526. [Google Scholar] [CrossRef]
- De Freitas Silva, G.; da Silva, D.C.; Guimarães, A.S.; do Nascimento, E.; Rebouças, J.S.; de Araujo, M.P.; de Carvalho, M.E.M.D.; Idemori, Y.M. Cyclohexane hydroxylation by iodosylbenzene and iodobenzene diacetate catalysed by a new β-octahalogenated Mn–porphyrin complex: The effect of meso-3-pyridyl substituents. J. Mol. Catal. A Chem. 2007, 266, 274–283. [Google Scholar] [CrossRef]
- Ogawa, S.; Hosoi, K.; Iida, T.; Wakatsuki, Y.; Makino, M.; Fujimoto, Y.; Hofmann, A.F. Osmiumporphyrin-catalysed oxyfunctionalization and isomerization of natural (5β)-bile acids with tert-butyl hydroperoxide. Eur. J. Org. Chem. 2007, 2007, 3555–3563. [Google Scholar] [CrossRef]
- Ogawa, S.; Wakatsuki, Y.; Makino, M.; Fujimoto, Y.; Yasukawa, K.; Kikuchi, T.; Ukiya, M.; Akihisa, T.; Iida, T. Oxyfunctionalization of unactivated C-H bonds in triterpenoids with tert-butylhydroperoxide catalysed by meso-5,10,15,20-tetramesitylporphyrinate osmium(II) carbonyl complex. Chem. Phys. Lipids 2010, 163, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Iida, T.; Ogawa, S.; Hosoi, K.; Makino, M.; Fujimoto, Y.; Goto, T.; Mano, N.; Goto, J.; Hofmann, A.F. Regioselective oxyfunctionalization of unactivated carbons in steroids by a model of cytochrome P-450: Osmiumporphyrin complex/tert-butyl hydroperoxide system. J. Org. Chem. 2007, 72, 823–830. [Google Scholar] [CrossRef] [PubMed]
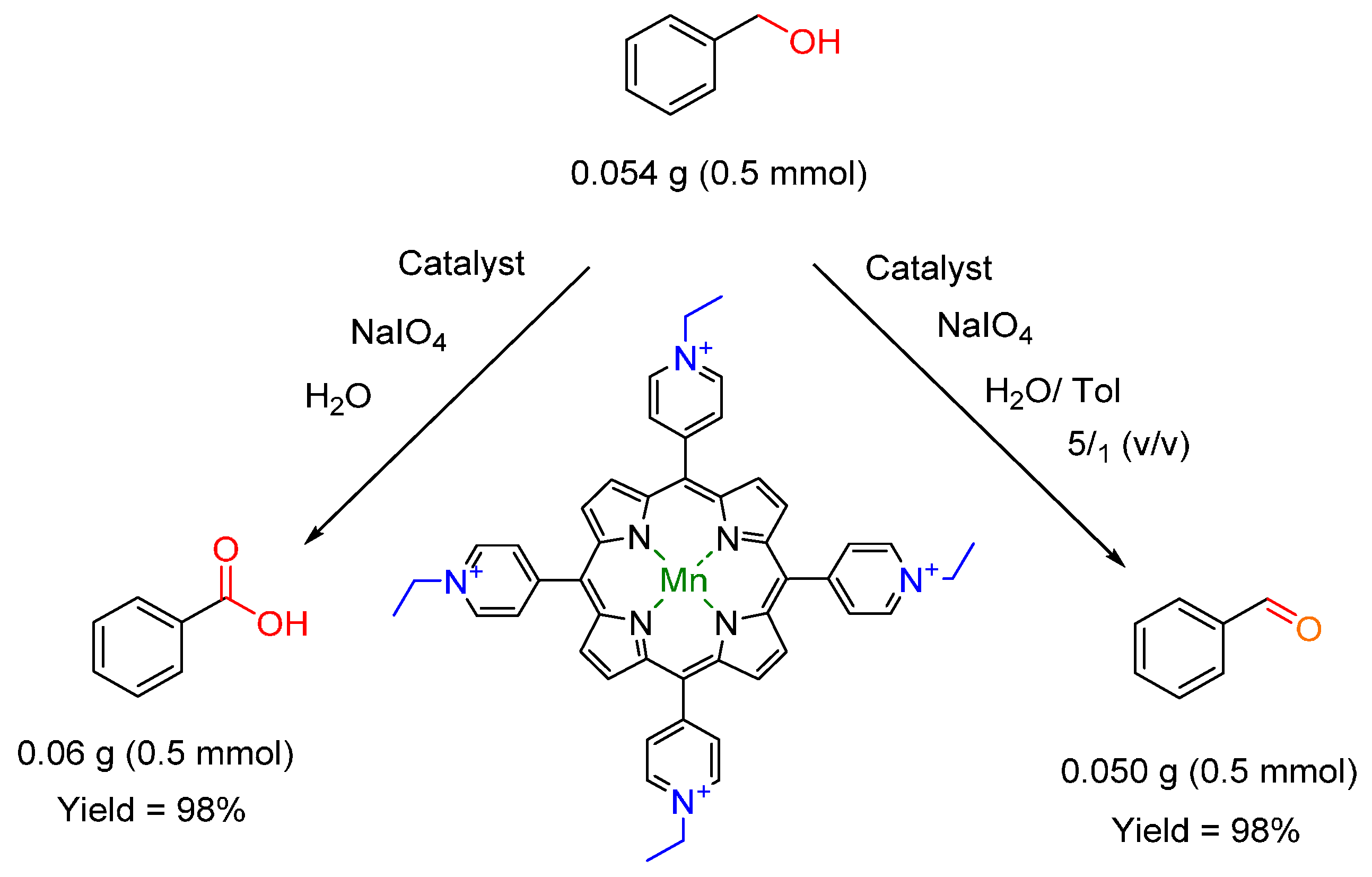
- Ren, Q.-G.; Chen, S.-Y.; Zhou, X.-T.; Ji, H.-B. Highly efficient controllable oxidation of alcohols to aldehydes and acids with sodium periodate catalysed by water-soluble metalloporphyrins as biomimetic catalyst. Bioorg. Med. Chem. 2010, 18, 8144–8149. [Google Scholar] [CrossRef] [PubMed]
- Muzart, J. Palladium-catalysed oxidation of primary and secondary alcohols. Tetrahedron 2003, 59, 5789–5816. [Google Scholar] [CrossRef]
- Ji, H.; Zhou, X. Biomimetic homogeneous oxidation catalysed by metalloporphyrins with green oxidants. In Biomimetic Learning from Nature; Mukherjee, A., Ed.; InTech: Vienna, Austria, 2010; pp. 137–166. [Google Scholar]
- Adam, W.; Prikhodovski, S.; Roschmann, K.J.; Saha-Moller, C.R. Oxidation of aryl-substituted allylic alcohols by an optically active Fe(III)(porph*) catalyst: Enantioselectivity, diastereoselectivity and chemoselectivity in the epoxide versus enone formation. Tetrahedron Asymmetry 2001, 12, 2677–2681. [Google Scholar] [CrossRef]
- Burri, E.; Öhm, M.; Daguenet, C.; Severin, K. Site-isolated porphyrin catalysts in imprinted polymers. Chem. Eur. J. 2005, 11, 5055–5061. [Google Scholar] [CrossRef] [PubMed]
- Han, J.H.; Yoo, S.; Seo, S.; Hong, J.; Kim, K.; Kim, C. Biomimetic alcohol oxidations by an iron(III) porphyrin complex: Relevance to cytochrome P-450 catalytic oxidation and involvement of the two-state radical rebound mechanism. Dalton Trans. 2005, 2, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Rezaeifard, A.; Jafarpour, M.; Moghaddam, G.K.; Amini, F. Cytochrome P-450 model reactions: Efficient and highly selective oxidation of alcohols with tetrabutylammonium peroxymonosulfate catalysed by Mn-porphyrins. Bioorg. Med. Chem. 2007, 15, 3097–3101. [Google Scholar] [CrossRef] [PubMed]
- Du, G.; Keith Woo, L. Alcohol oxidation with dioxygen mediated by oxotitanium porphyrin and related transition metal complexes. J. Porphyr. Phthalocyanines 2005, 9, 206–213. [Google Scholar] [CrossRef]
- Du, G.; Woo, L.K. Synthesis, characterization, and reactivity of group 4 metalloporphyrin diolate complexes. Organometallics 2003, 22, 450–455. [Google Scholar] [CrossRef]
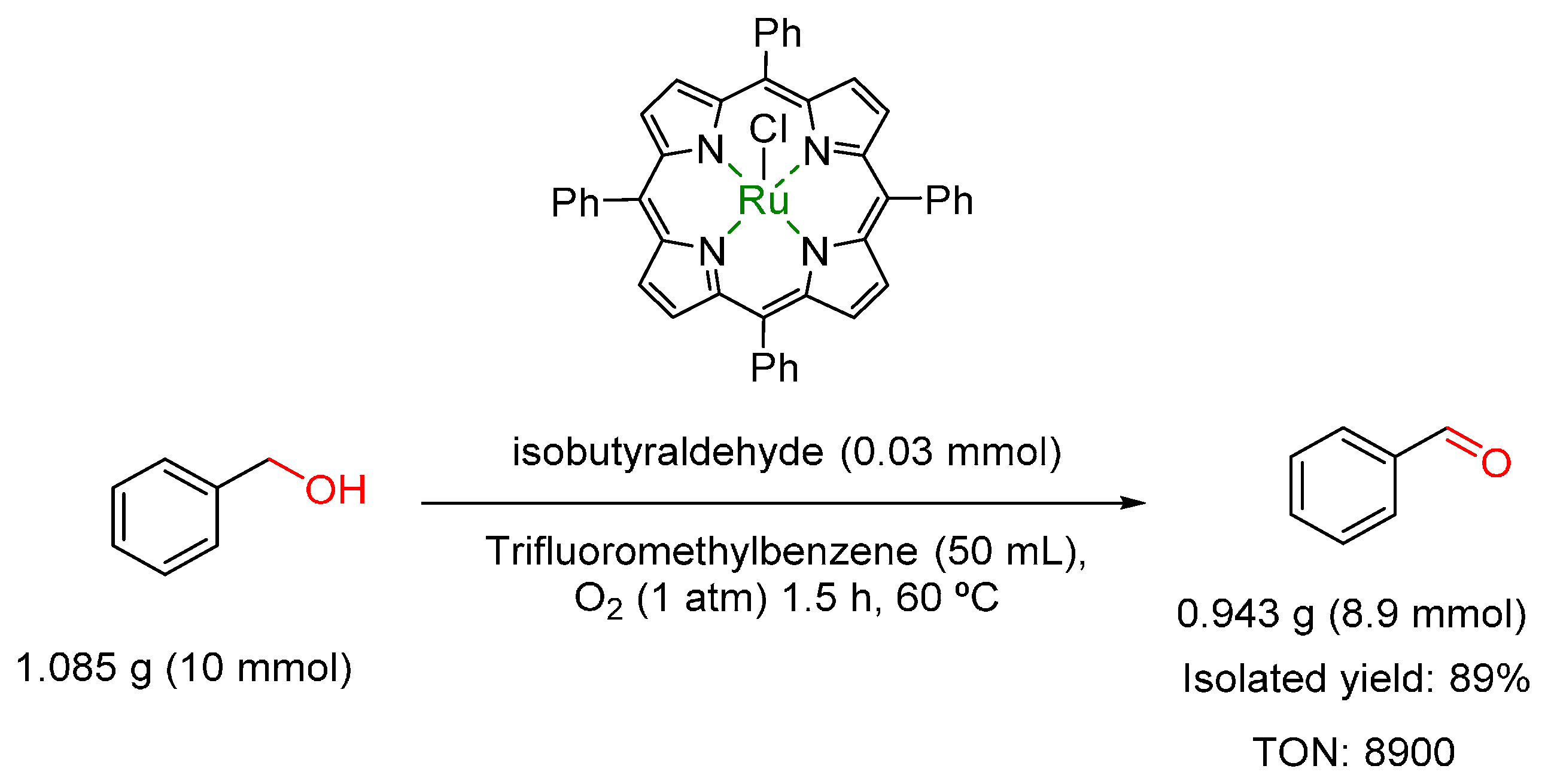
- Ji, H.-B.; Yuan, Q.-L.; Zhou, X.-T.; Pei, L.-X.; Wang, L.-F. Highly efficient selective oxidation of alcohols to carbonyl compounds catalysed by ruthenium (III) meso-tetraphenylporphyrin chloride in the presence of molecular oxygen. Bioorg. Med. Chem. Lett. 2007, 17, 6364–6368. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, S.L.; Simões, M.M.; Neves, M.G.P.M.; Cavaleiro, J.A. Oxidation of alkylaromatics with hydrogen peroxide catalysed by manganese(III) porphyrins in the presence of ammonium acetate. J. Mol. Catal. A Chem. 2003, 201, 9–22. [Google Scholar] [CrossRef]
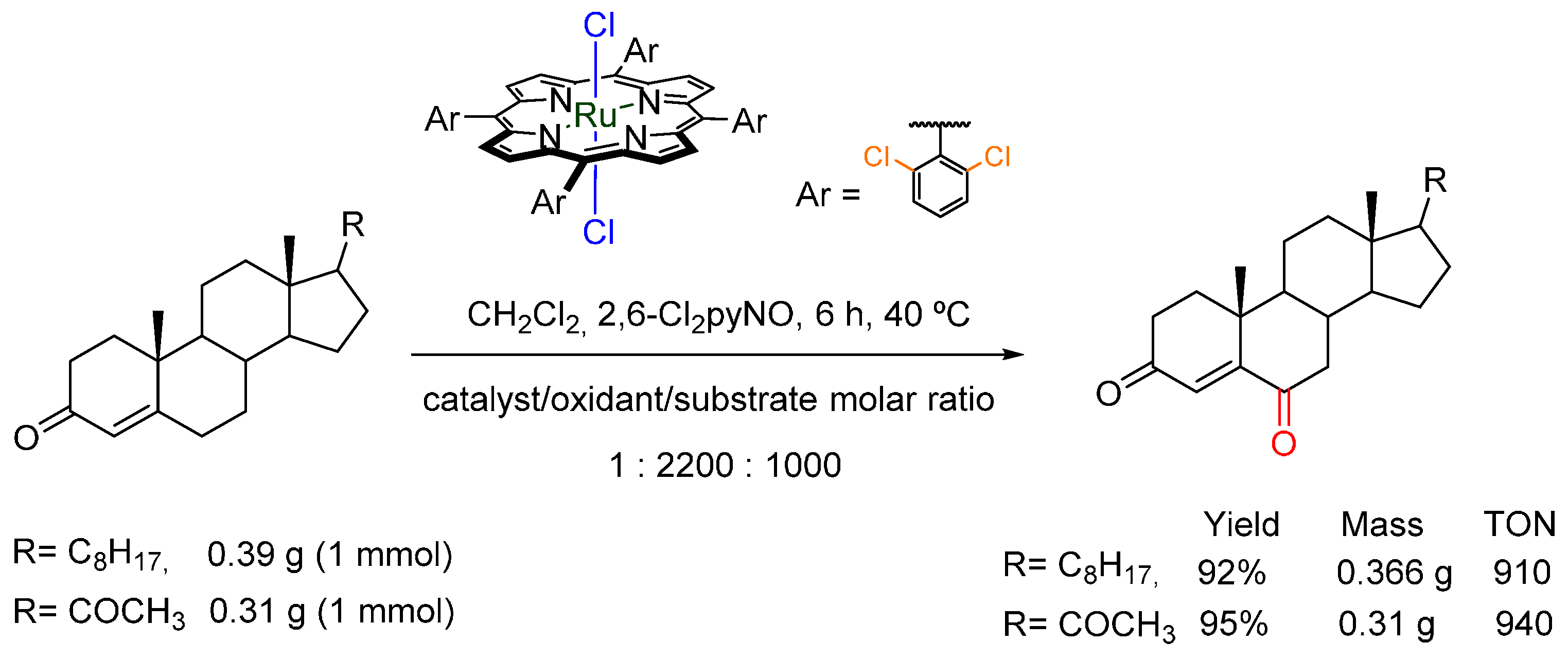
- Zhang, J.L.; Che, C.M. Dichlororuthenium(IV) complex of meso-Tetrakis(2,6-dichlorophenyl) porphyrin: Active and robust catalyst for highly selective oxidation of arenes, unsaturated steroids, and electron-deficient alkenes by using 2,6-dichloropyridine N-oxide. Chem. Eur. J. 2005, 11, 3899–3914. [Google Scholar] [CrossRef] [PubMed]
- Twarowski, A.J. Porphyrin-sensitized formation of a polymer-bound endoperoxide in the solid state. J. Phys. Chem. 1988, 92, 6580–6584. [Google Scholar] [CrossRef]
- Slavětínská, L.; Mosinger, J.; Kubát, P. Supramolecular carriers of singlet oxygen: Photosensitized formation and thermal decomposition of endoperoxides in the presence of cyclodextrins. J. Photochem. Photobiol. A 2008, 195, 1–9. [Google Scholar] [CrossRef]
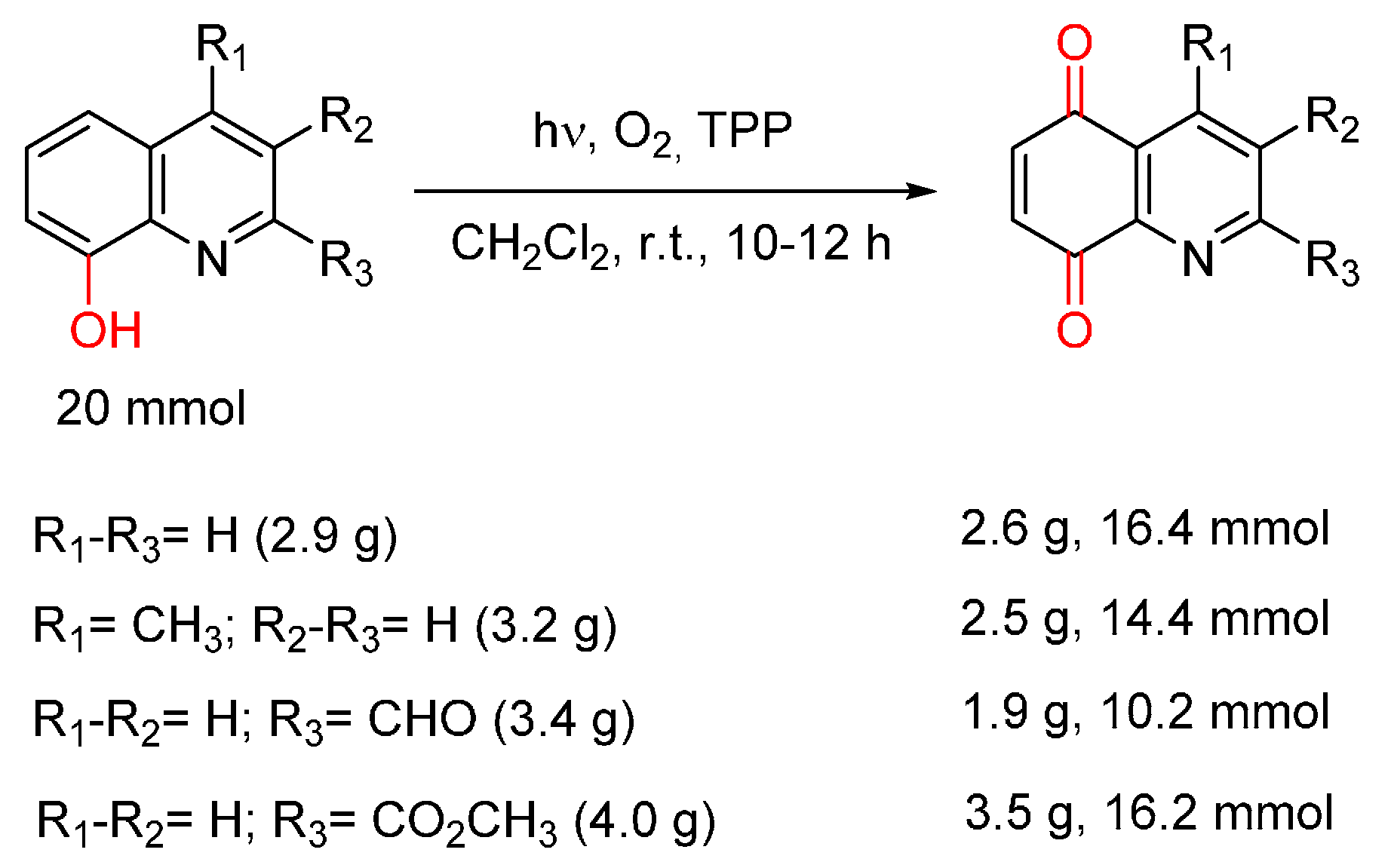
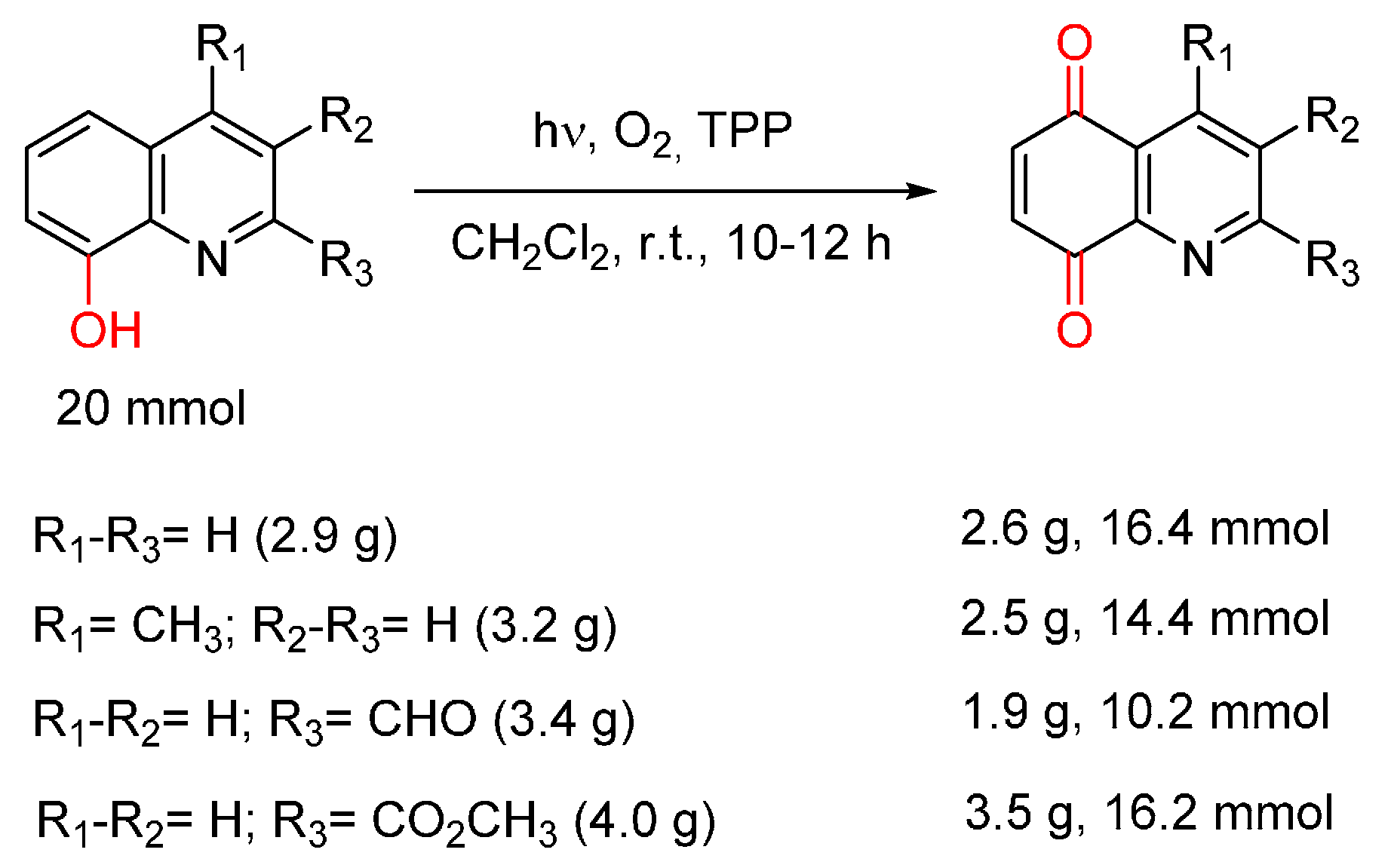
- Cossy, J.; Belotti, D. Efficient synthesis of substituted quinoline-5,8-quinones from 8-hydroxyquinolines by photooxygenation. Tetrahedron Lett. 2001, 42, 4329–4331. [Google Scholar] [CrossRef]
- Faustino, M.A.; Neves, M.G.; Cavaleiro, J.A.; Neumann, M.; Brauer, H.D.; Jori, G. meso-Tetraphenylporphyrin dimer derivatives as potential photosensitizers in photodynamic therapy. Part 2. Photochem. Photobiol. 2000, 72, 217–225. [Google Scholar] [CrossRef]
- Ogilby, P.R. Singlet oxygen: There is indeed something new under the sun. Chem. Soc. Rev. 2010, 39, 3181–3209. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, A.S. A new synthesis of quinoline-5,8-quinone. Synth. Commun. 1999, 29, 3063–3066. [Google Scholar] [CrossRef]
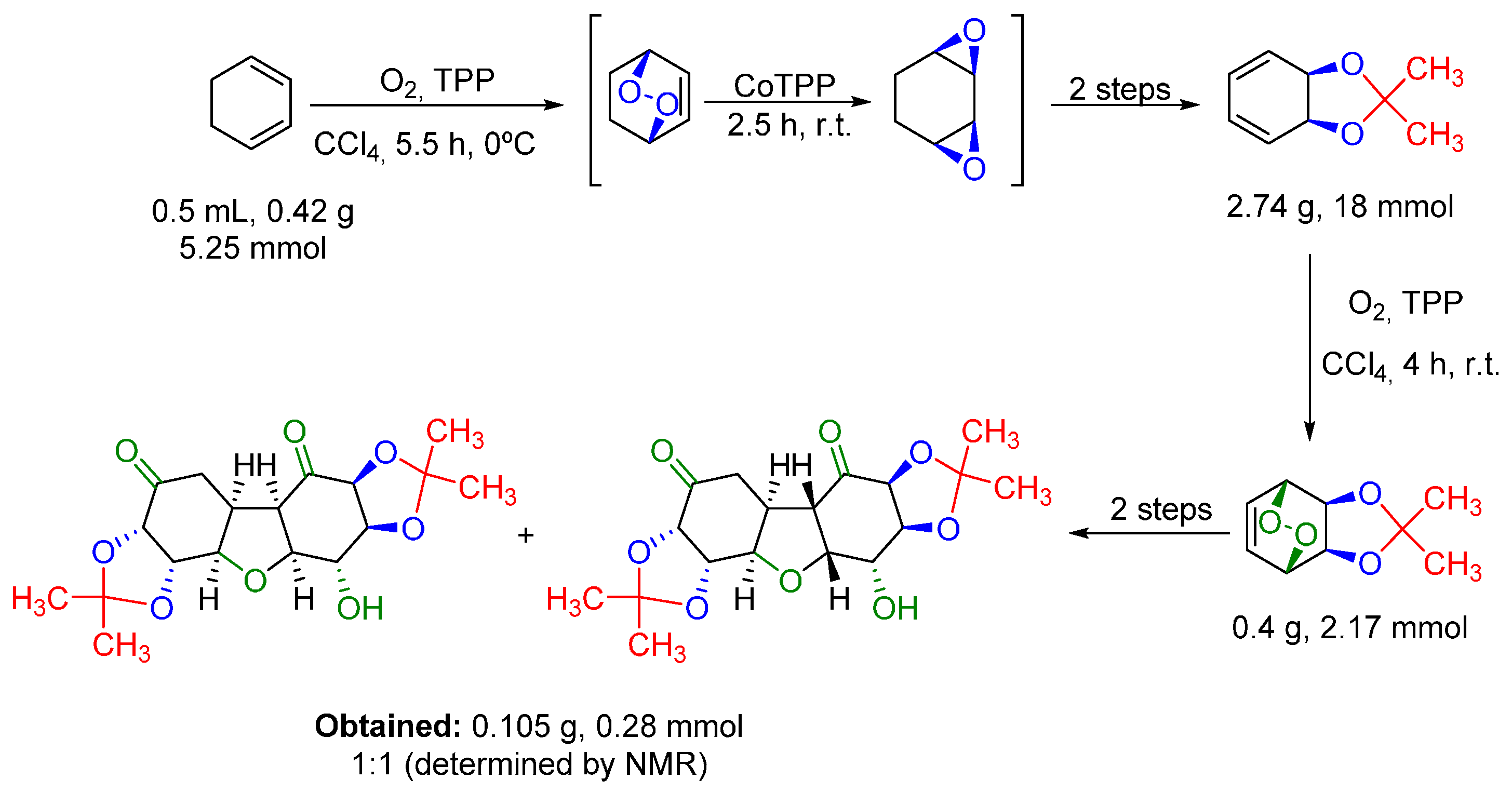
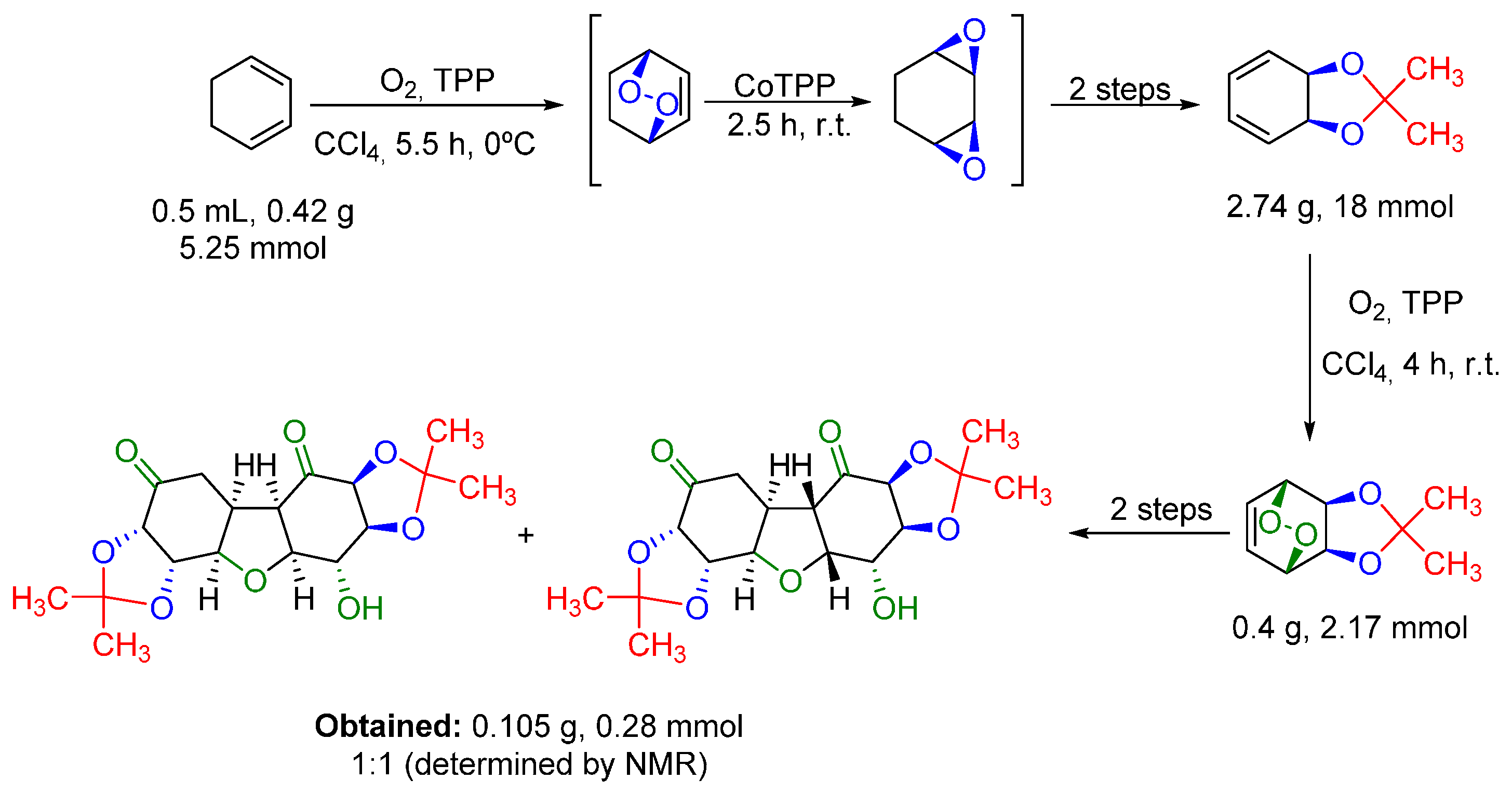
- Paddock, V.L.; Phipps, R.J.; Conde-Angulo, A.; Blanco-Martin, A.; Giró-Mañas, C.; Martin, L.J.; White, A.J.P.; Spivey, A.C. (±)-trans,cis-4-Hydroxy-5,6-di-O-isopropylidenecyclohex-2-ene-1-one: Synthesis and facile dimerization to decahydrodibenzofurans. J. Org. Chem. 2011, 76, 1483–1486. [Google Scholar] [CrossRef] [PubMed]
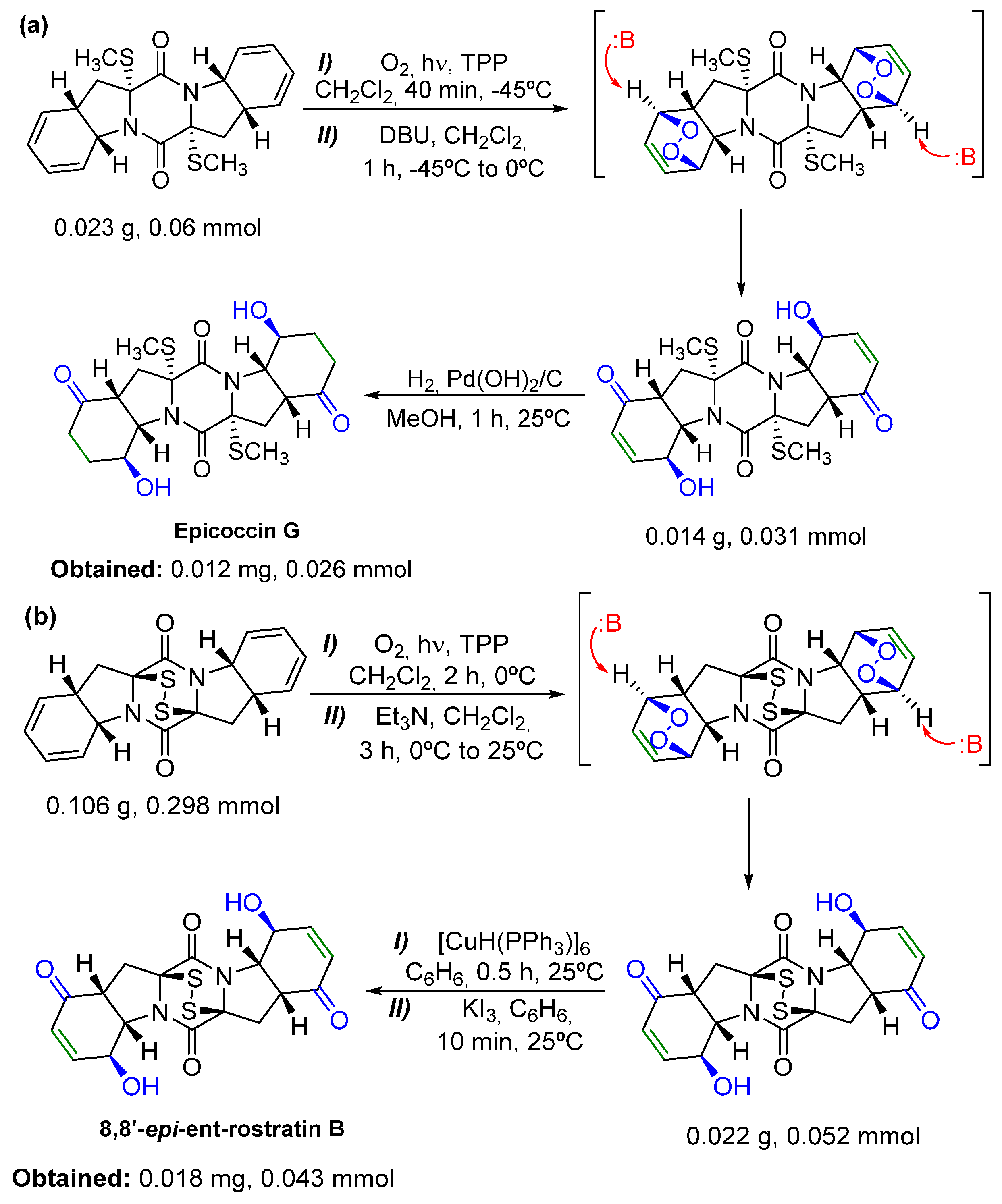
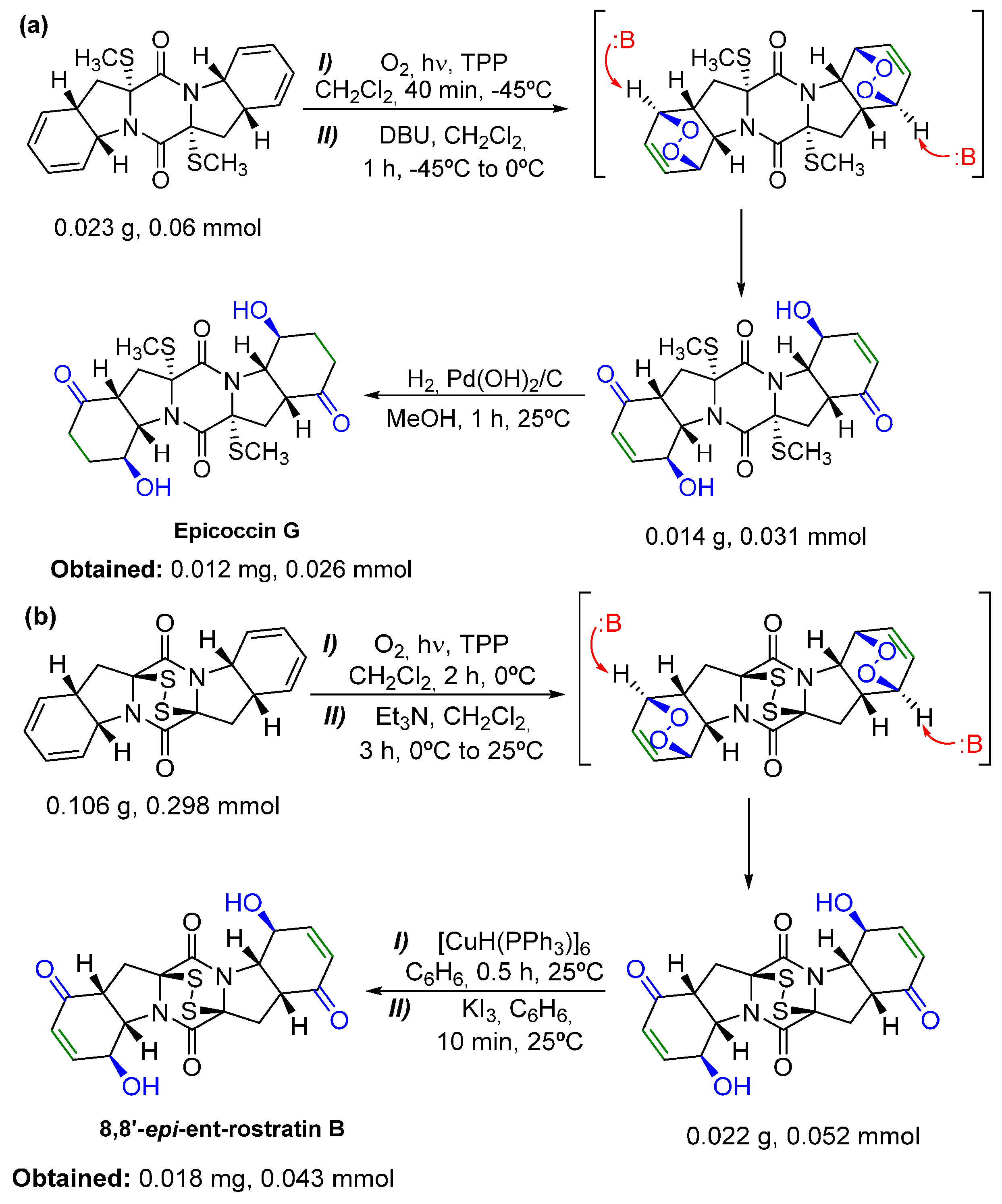
- Nicolaou, K.C.; Totokotsopoulos, S.; Giguère, D.; Sun, Y.; Sarlah, D. Total synthesis of epicoccin G. J. Am. Chem. Soc. 2011, 133, 8150–8153. [Google Scholar] [CrossRef] [PubMed]
- Murahashi, S.-I. Synthetic aspects of metal-catalysed oxidations of amines and related reactions. Angew. Chem. Int. Ed. Engl. 1995, 34, 2443–2465. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kitano, Y. Singlet Oxygenation of 1-aminomethyl-1-tert-butyl-2-methoxy-2-(3-methoxy-phenyl)ethylenes: Marked effect of allylic nitrogen on the reaction pathways and chemoselectivity. Tetrahedron Lett. 1996, 37, 8191–8194. [Google Scholar] [CrossRef]
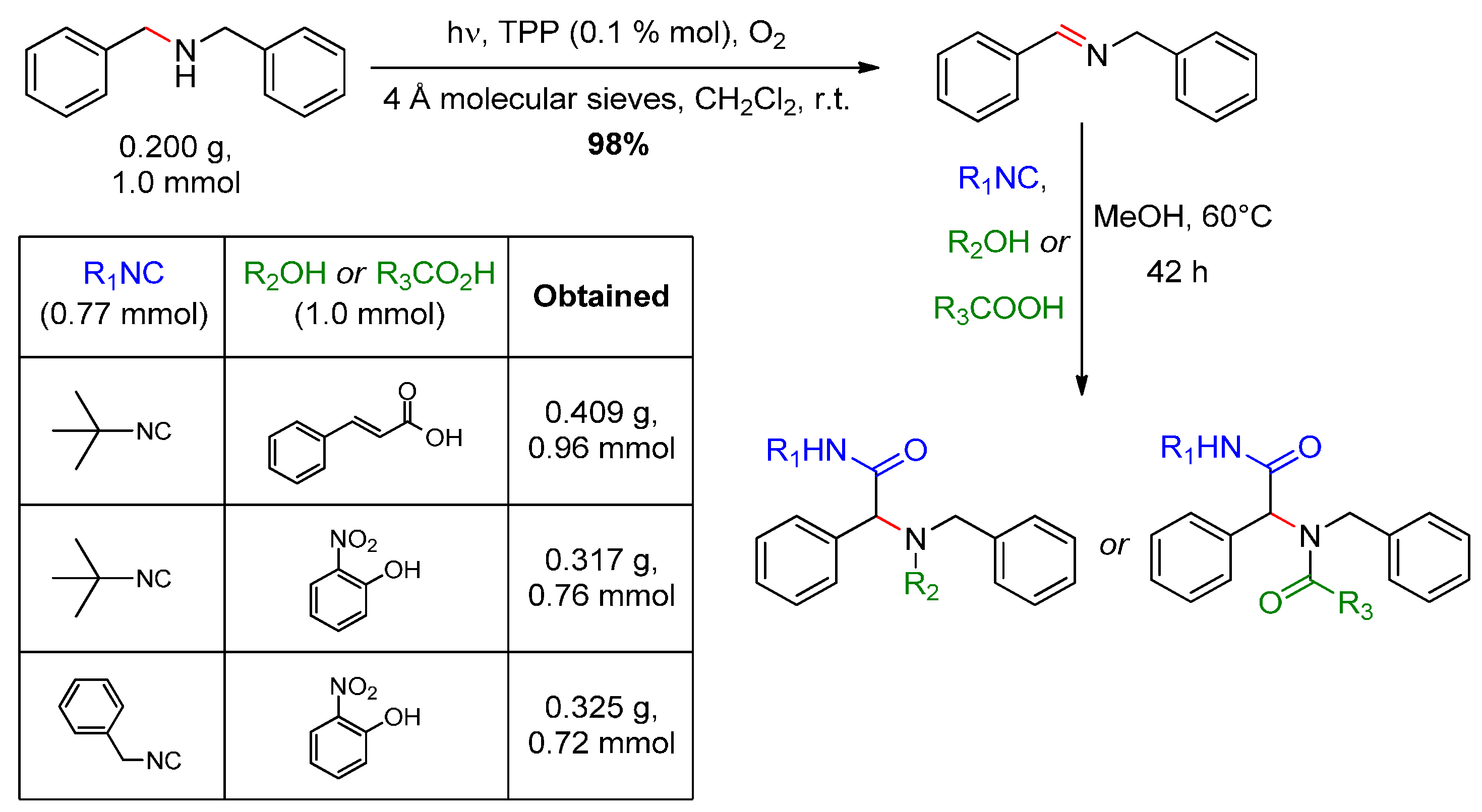
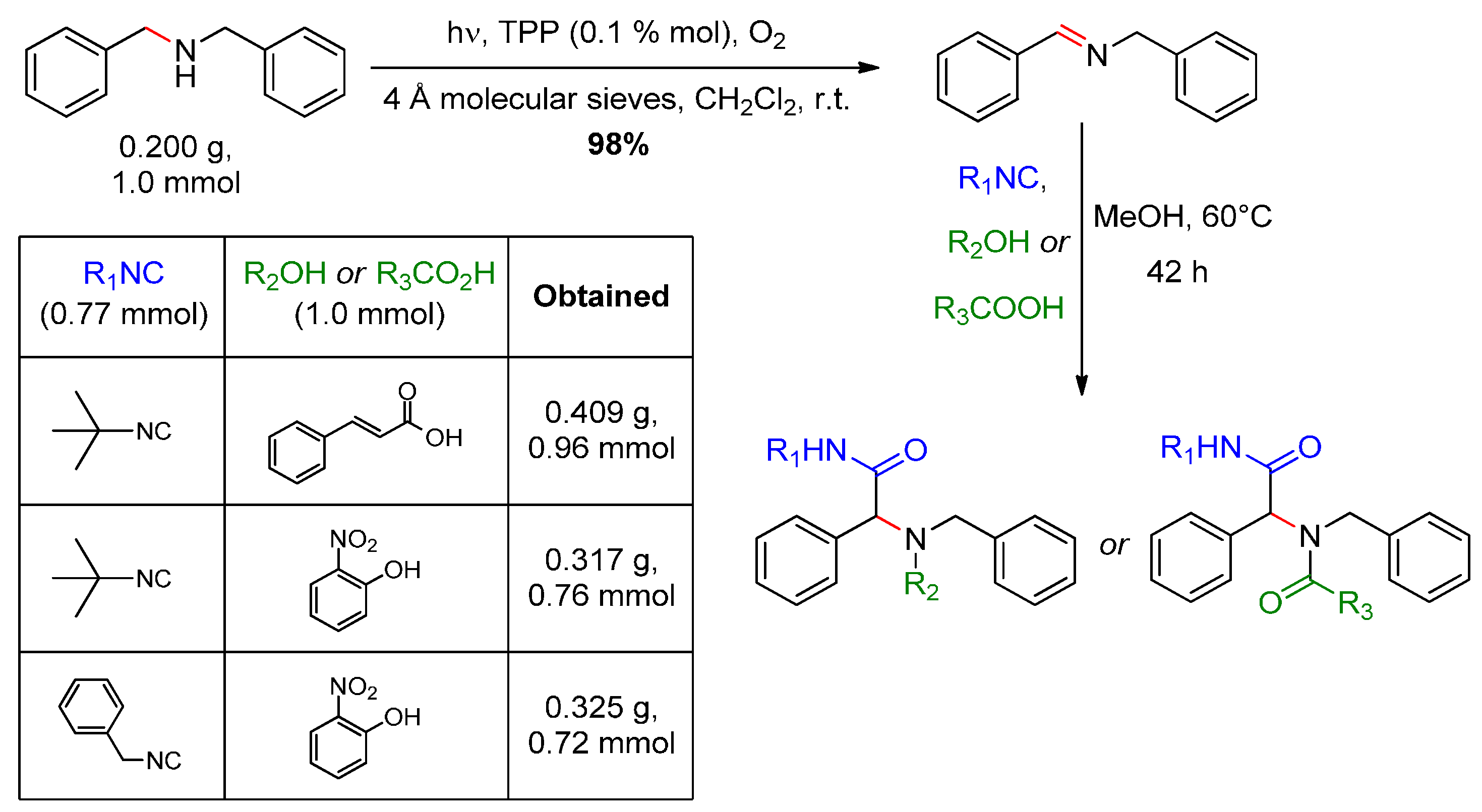
- Jiang, G.; Chen, J.; Huang, J.-S.; Che, C.-M. Highly efficient oxidation of amines to imines by singlet oxygen and its application in Ugi-type reactions. Org. Lett. 2009, 11, 4568–4571. [Google Scholar] [CrossRef] [PubMed]
- Nardello, V.; Hervé, M.; Alsters, P.L.; Aubry, J.-M. “Dark” singlet oxygenation of hydrophobic substrates in environmentally friendly microemulsions. Adv. Synth. Catal. 2002, 344, 184–191. [Google Scholar] [CrossRef]
- Coyle, E.E.; Joyce, K.; Nolan, K.; Oelgemöller, M. Green photochemistry: The use of microemulsions as green media in photooxygenation reactions. Green Chem. 2010, 12, 1544–1547. [Google Scholar] [CrossRef]
- Lu, H.; Zhang, X.P. Catalytic C–H functionalization by metalloporphyrins: Recent developments and future directions. Chem. Soc. Rev. 2011, 40, 1899–1909. [Google Scholar] [CrossRef] [PubMed]
- Song, C.E.; Lee, S.G. Supported chiral catalysts on inorganic materials. Chem. Rev. 2002, 102, 3495–3524. [Google Scholar] [CrossRef] [PubMed]
- Leadbeater, N.E.; Marco, M. Preparation of polymer-supported ligands and metal complexes for use in catalysis. Chem. Rev. 2002, 102, 3217–3274. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.M.; Serra, A.C.; Rocha Gonsalves, A.M.D. Covalently immobilized porphyrins on silica modified structures as photooxidation catalysts. J. Mol. Catal. A Chem. 2010, 326, 121–127. [Google Scholar] [CrossRef]
- Benaglia, M.; Danelli, T.; Fabris, F.; Sperandio, D.; Pozzi, G. Poly(ethylene glycol)-supported tetrahydroxyphenyl porphyrin: A convenient, recyclable catalyst for photooxidation reactions. Org. Lett. 2002, 4, 4229–4232. [Google Scholar] [CrossRef] [PubMed]
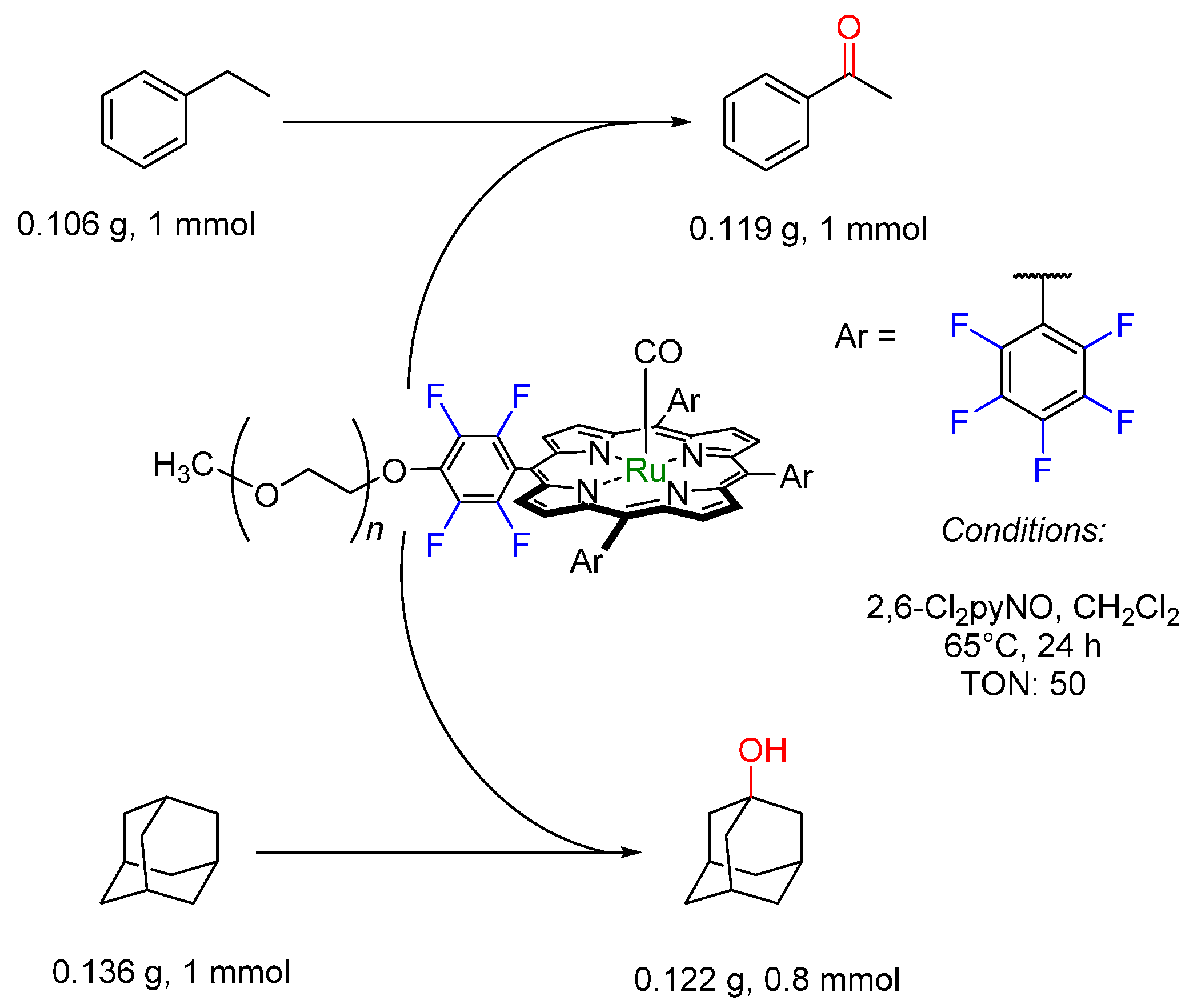
- Zhang, J.-L.; Huang, J.-S.; Che, C.-M. Oxidation chemistry of poly(ethylene glycol)-supported carbonylruthenium(II) and dioxoruthenium(VI)meso-Tetrakis(pentafluorophenyl)porphyrin. Chem. Eur. J. 2006, 12, 3020–3031. [Google Scholar] [CrossRef] [PubMed]
- Shiragami, T.; Makise, R.; Inokuchi, Y.; Matsumoto, J.; Inoue, H.; Yasuda, M. Efficient photocatalytic oxidation of cycloalkenes by dihydroxo(tetraphenylporphyrinato)antimony supported on silica gel under visible light irradiation. Chem. Lett. 2004, 33, 736–737. [Google Scholar] [CrossRef]
- Azenha, E.G.; Serra, A.C.; Pineiro, M.; Pereira, M.M.; Seixas de Melo, J.; Arnaut, L.G.; Formosinho, S.J.; Rocha Gonsalves, A.M.D. Heavy-atom effects on metalloporphyrins and polyhalogenated porphyrins. Chem. Phys. 2002, 280, 177–190. [Google Scholar] [CrossRef]
- Ribeiro, S.; Serra, A.C.; Gonsalves, A.R. Efficient solar photooxygenation with supported porphyrins as catalysts. ChemCatChem 2013, 5, 134–137. [Google Scholar] [CrossRef]
- Wegner, J.; Ceylan, S.; Kirschning, A. Flow chemistry—A key enabling technology for (multistep) organic synthesis. Adv. Synth. Catal. 2012, 354, 17–57. [Google Scholar] [CrossRef]
- Wiles, C.; Watts, P. Continuous flow reactors: A perspective. Green Chem. 2012, 14, 38–54. [Google Scholar] [CrossRef]
- Oelgemöller, M.; Shvydkiv, O. Recent advances in microflow photochemistry. Molecules 2011, 16, 7522–7550. [Google Scholar] [CrossRef] [PubMed]
- Griesbeck, A.G.; El-Idreesy, T.T. Solvent-free photooxygenation of 5-methoxyoxazoles in polystyrene nanocontainers doped with tetrastyrylporphyrine and protoporphyrine-IX. Photochem. Photobiol. Sci. 2005, 4, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Bourne, R.A.; Han, X.; Poliakoff, M.; George, M.W. Cleaner continuous photo-oxidation using singlet oxygen in supercritical carbon dioxide. Angew. Chem. Int. Ed. 2009, 48, 5322–5325. [Google Scholar] [CrossRef] [PubMed]
- Bourne, R.A.; Han, X.; Chapman, A.O.; Arrowsmith, N.J.; Kawanami, H.; Poliakoff, M.; George, M.W. Homogeneous photochemical oxidation via singlet O2 in supercritical CO2. Chem. Commun. 2008, 37, 4457–4459. [Google Scholar] [CrossRef] [PubMed]
- Amara, Z.; Bellamy, J.F.B.; Horvath, R.; Miller, S.J.; Beeby, A.; Burgard, A.; Rossen, K.; Poliakoff, M.; George, M.W. Applying green chemistry to the photochemical route to artemisinin. Nat. Chem. 2015, 7, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Bourne, R.A.; Poliakoff, M.; George, M.W. Immobilised photosensitisers for continuous flow reactions of singlet oxygen in supercritical carbon dioxide. Chem. Sci. 2011, 2, 1059–1067. [Google Scholar] [CrossRef]
- Loponov, K.N.; Lopes, J.; Barlog, M.; Astrova, E.V.; Malkov, A.V.; Lapkin, A.A. Optimization of a scalable photochemical reactor for reactions with singlet oxygen. Org. Process Res. Dev. 2014, 18, 1443–1454. [Google Scholar] [CrossRef]
- Lévesque, F.; Seeberger, P.H. Highly efficient continuous flow reactions using singlet oxygen as a “Green” reagent. Org. Lett. 2011, 13, 5008–5011. [Google Scholar] [CrossRef] [PubMed]
- Lévesque, F.; Seeberger, P.H. Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew. Chem. Int. Ed. 2012, 51, 1706–1709. [Google Scholar] [CrossRef] [PubMed]
- Turconi, J.; Griolet, F.; Guevel, R.; Oddon, G.; Villa, R.; Geatti, A.; Hvala, M.; Rossen, K.; Göller, R.; Burgard, A. Semisynthetic artemisinin, the chemical path to industrial production. Org. Process Res. Dev. 2014, 18, 417–422. [Google Scholar] [CrossRef]
- De Oliveira, K.T.; Miller, L.Z.; McQuade, D.T. Exploiting photooxygenations mediated by porphyrinoid photocatalysts under continuous flow conditions. RSC Adv. 2016, 6, 12717–12725. [Google Scholar] [CrossRef]
- Momo, P.B.; Bellete, B.S.; Brocksom, T.J.; de Souza, R.O.M.A.; de Oliveira, K.T. Exploiting novel process windows for the synthesis of meso-substituted porphyrins under continuous flow conditions. RSC Adv. 2015, 5, 84350–84355. [Google Scholar] [CrossRef]



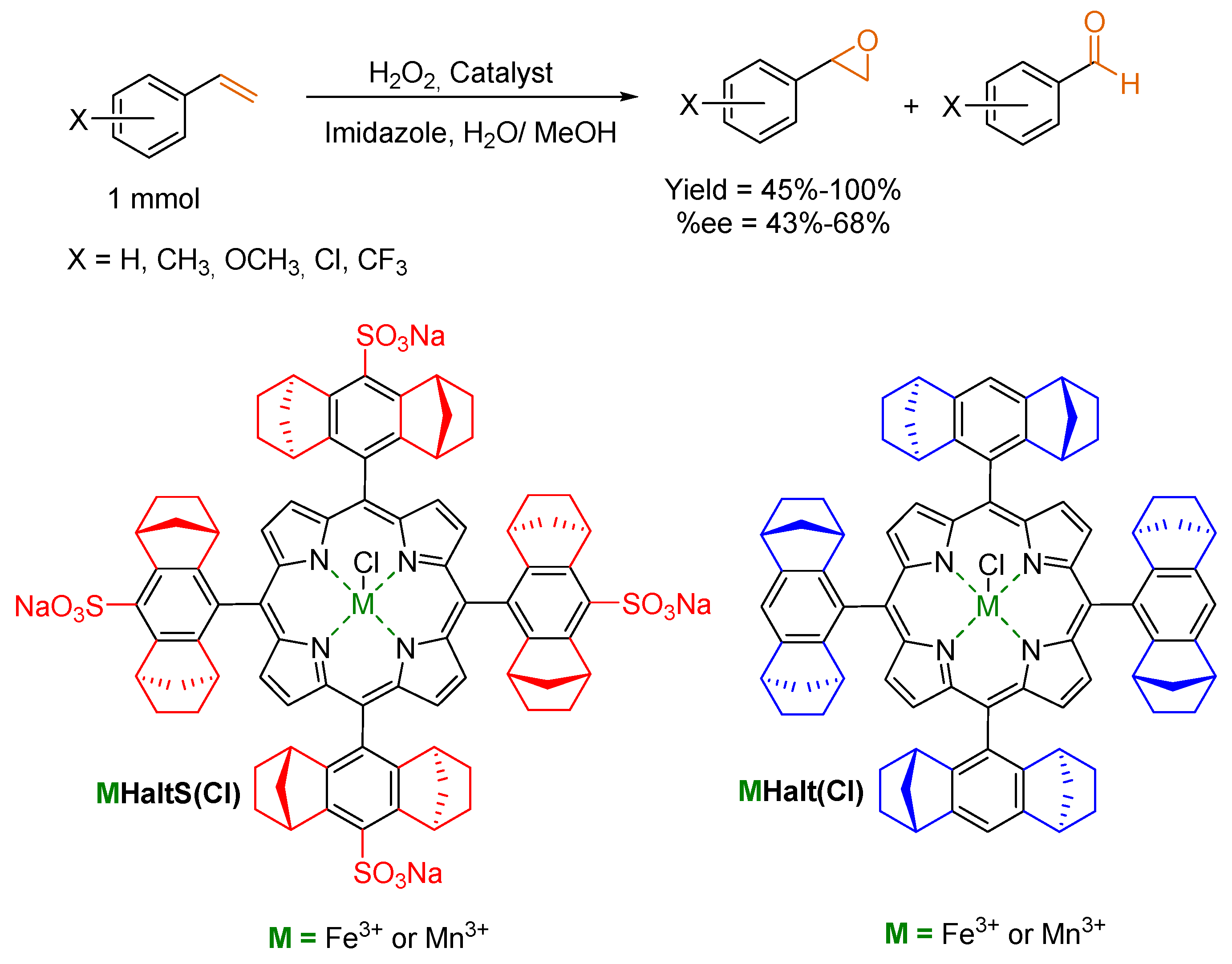
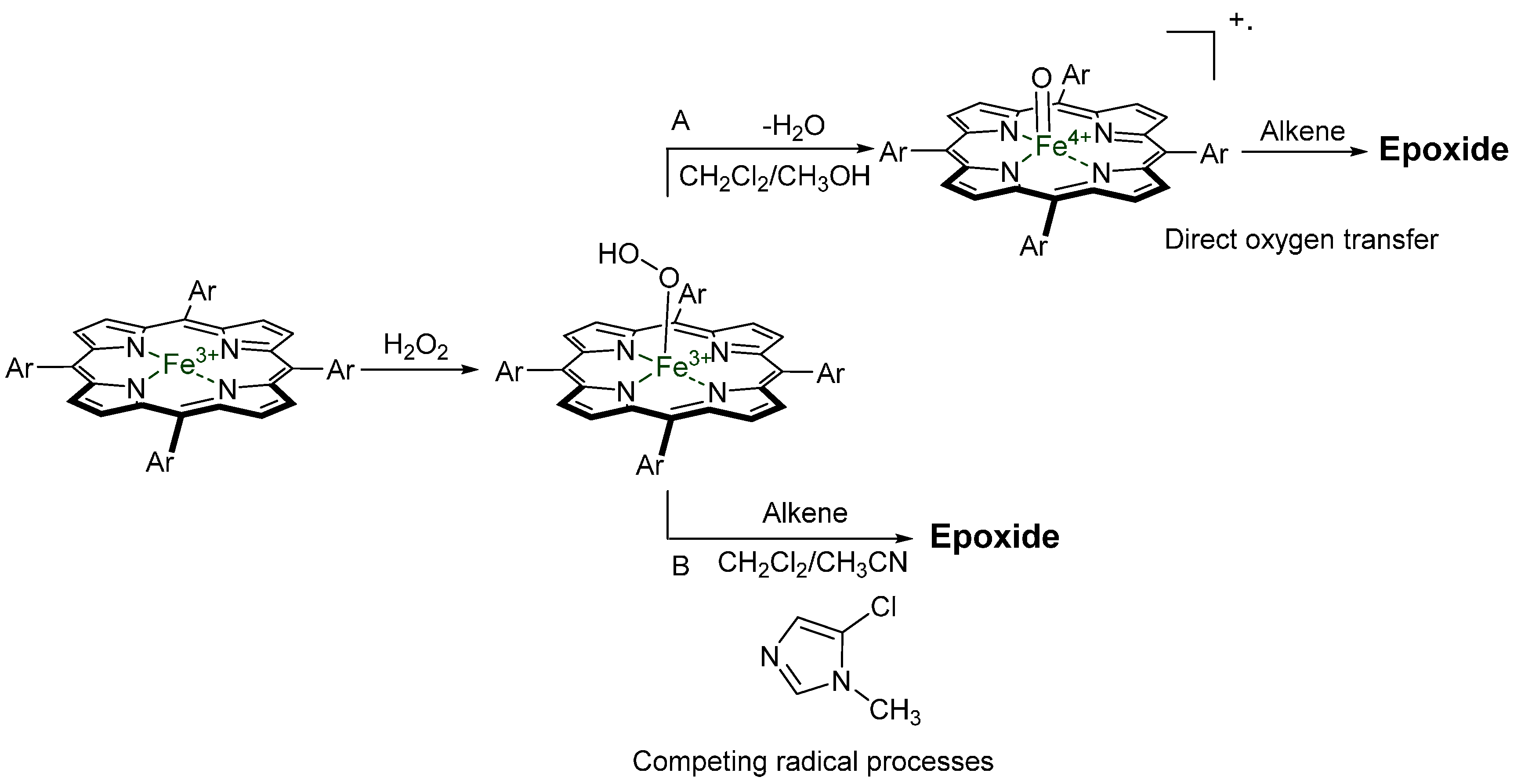
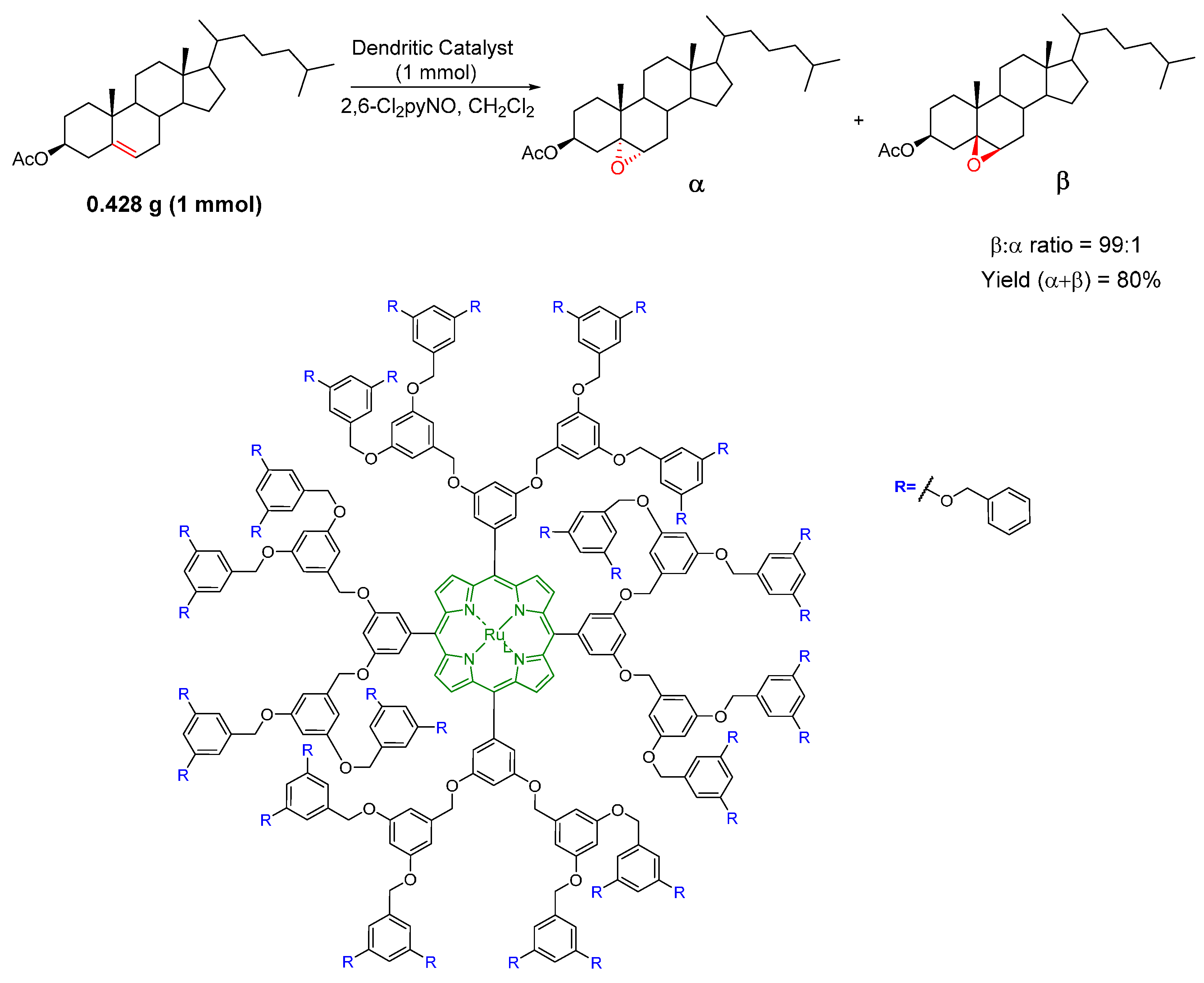
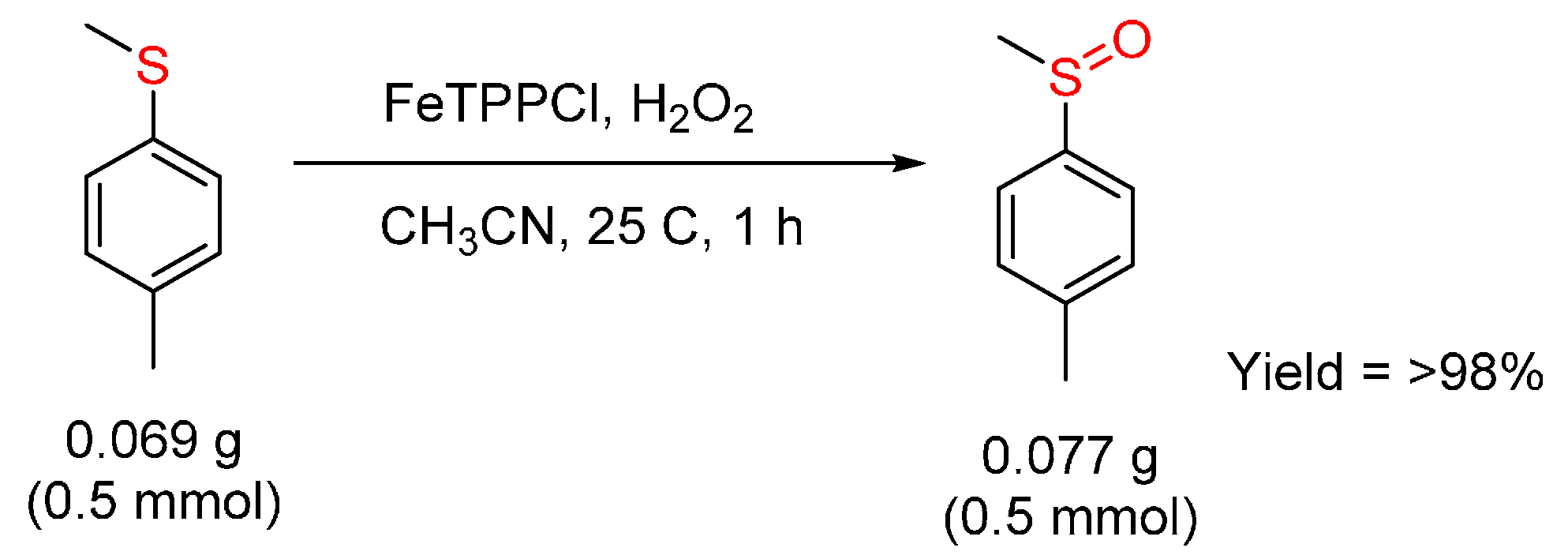
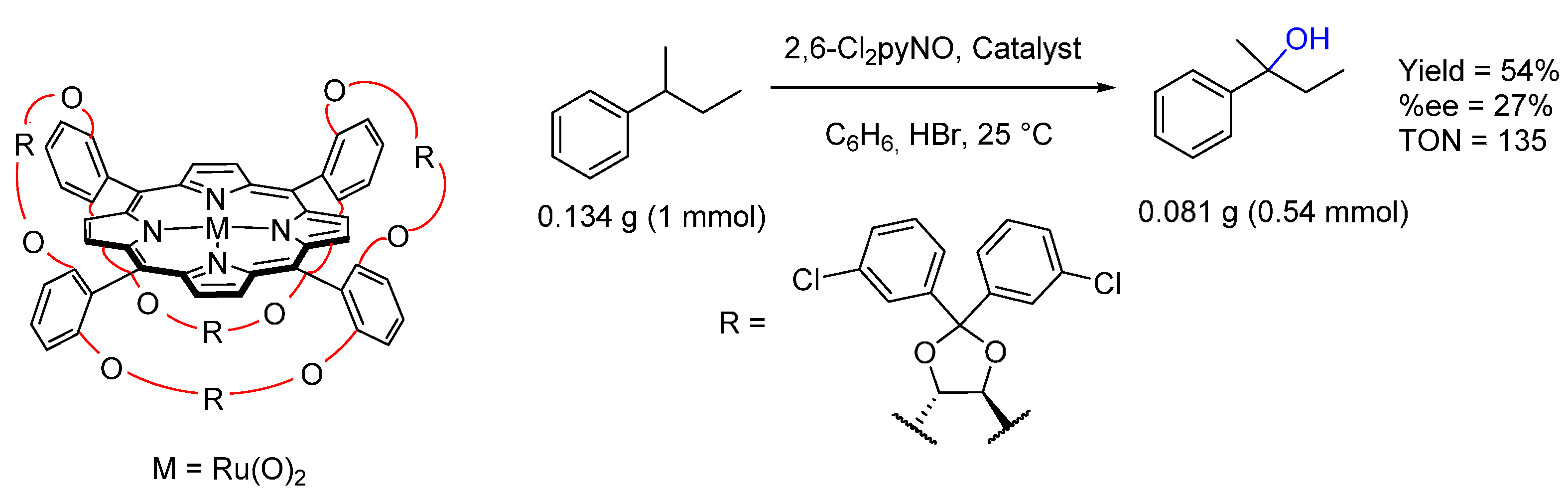
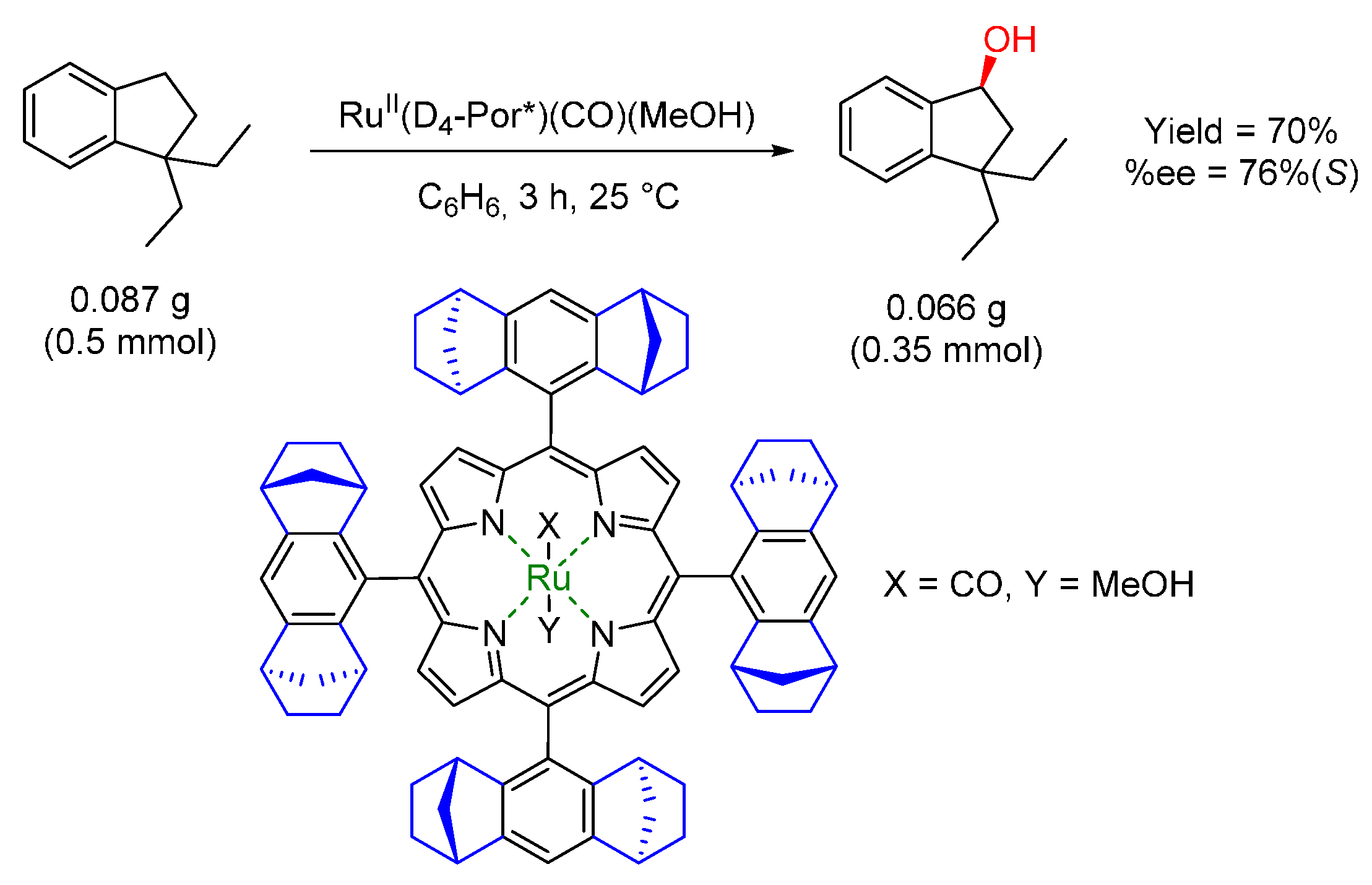

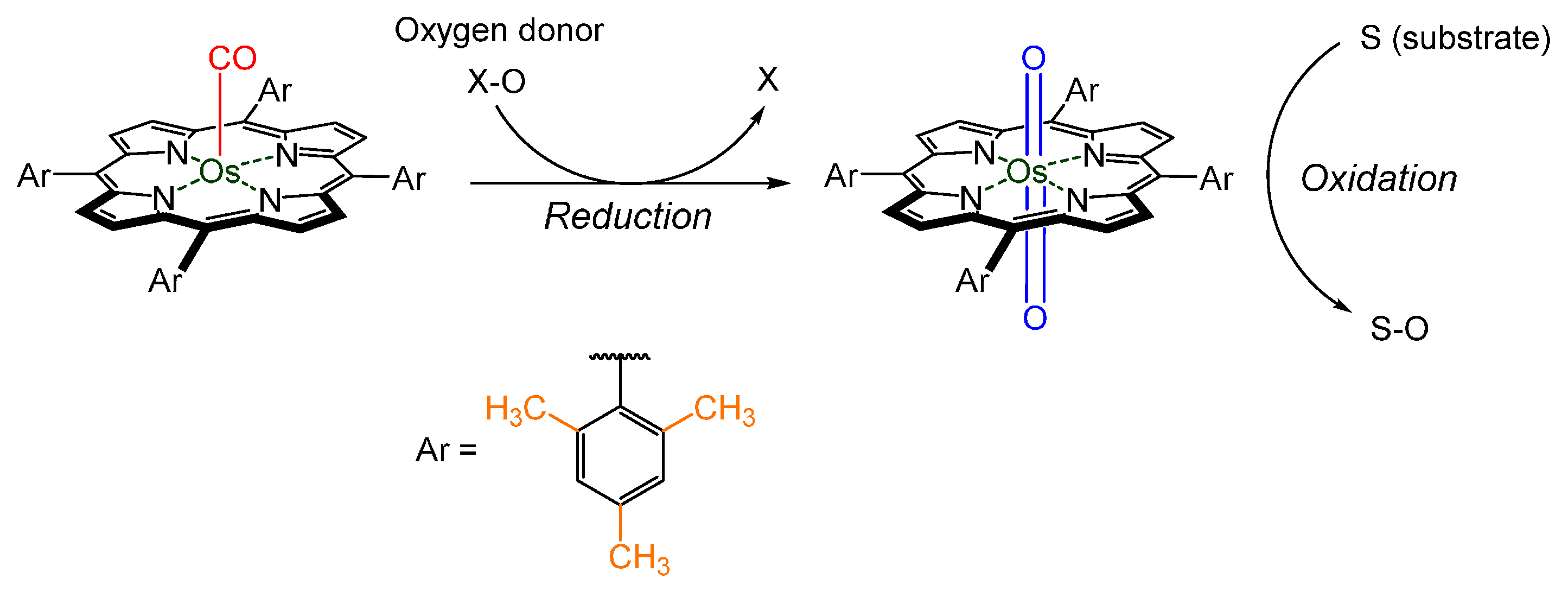
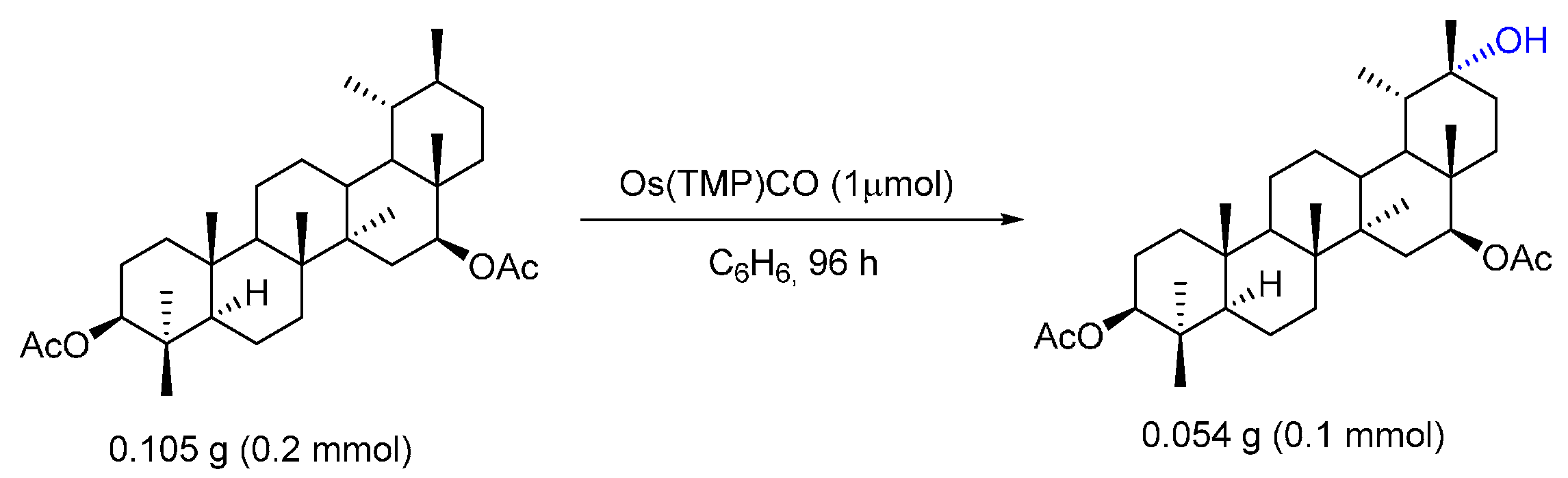
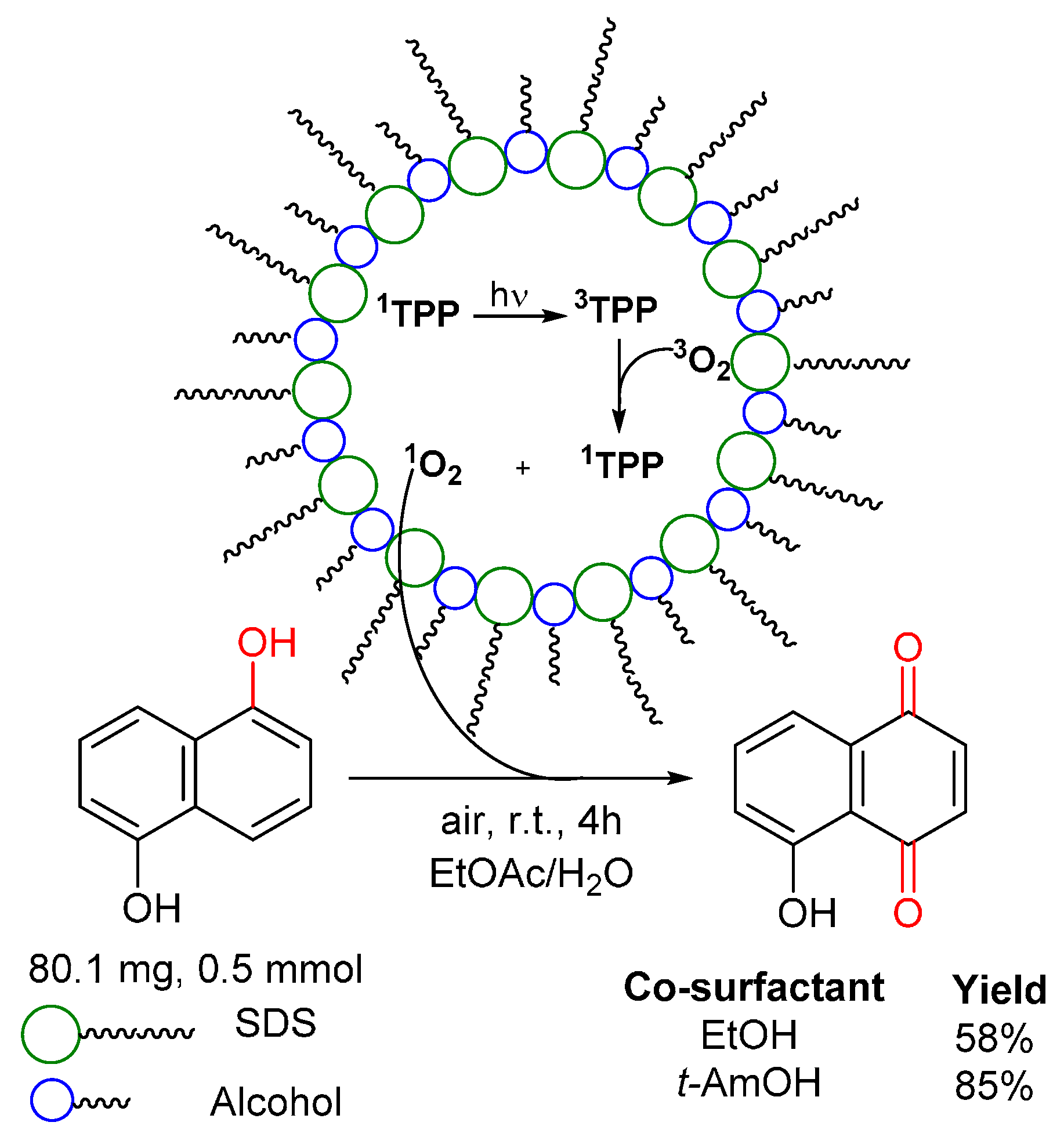
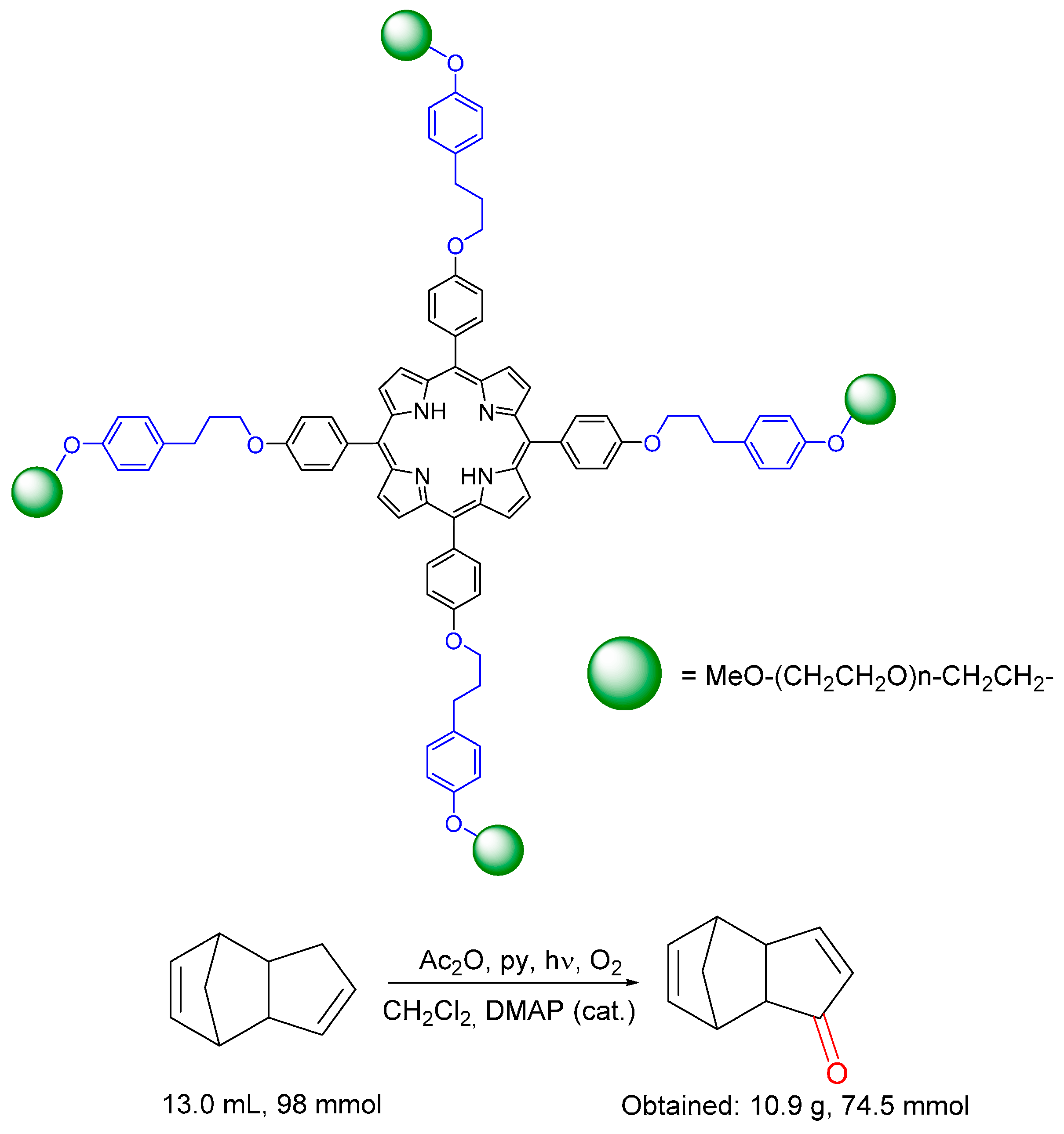
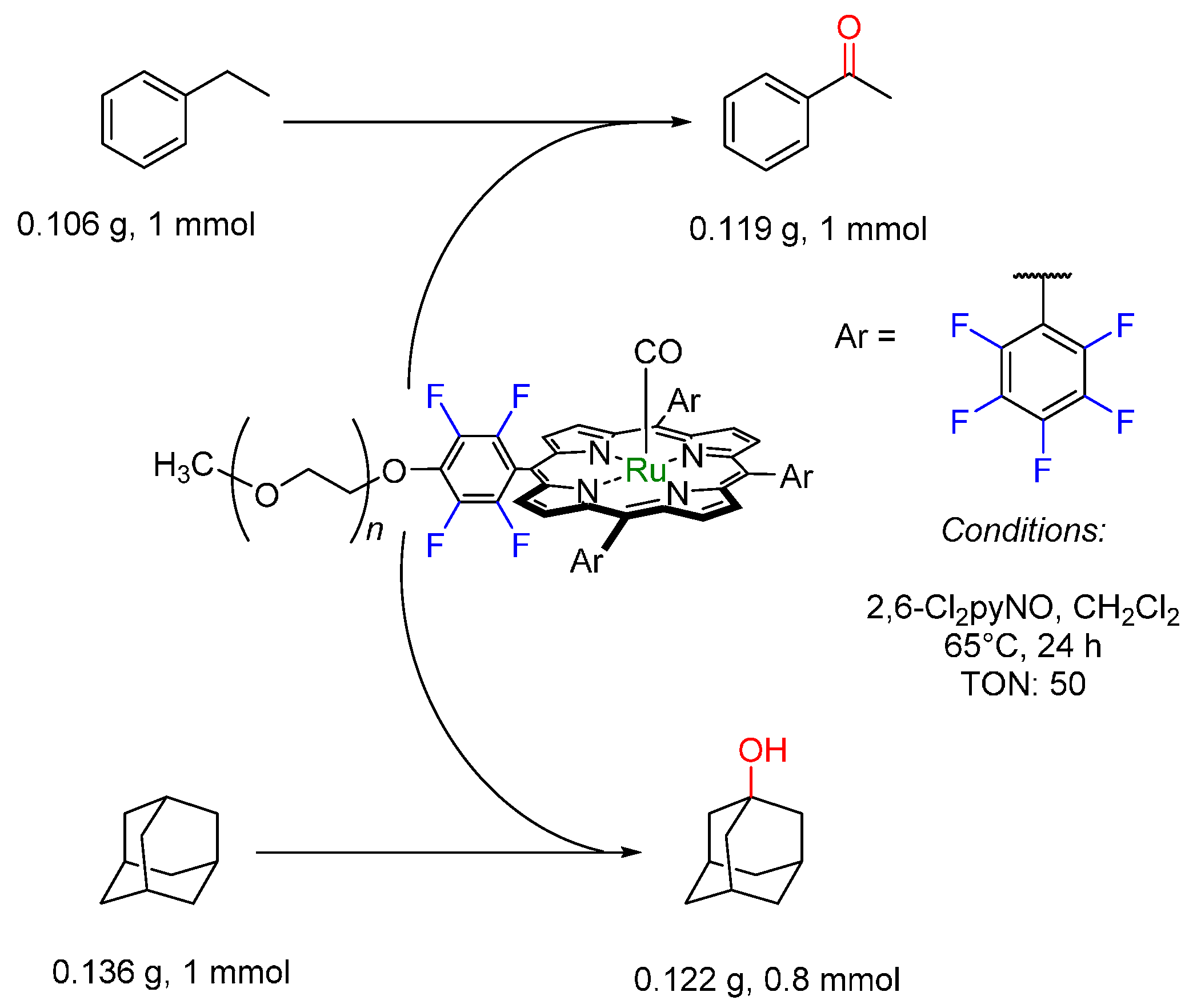
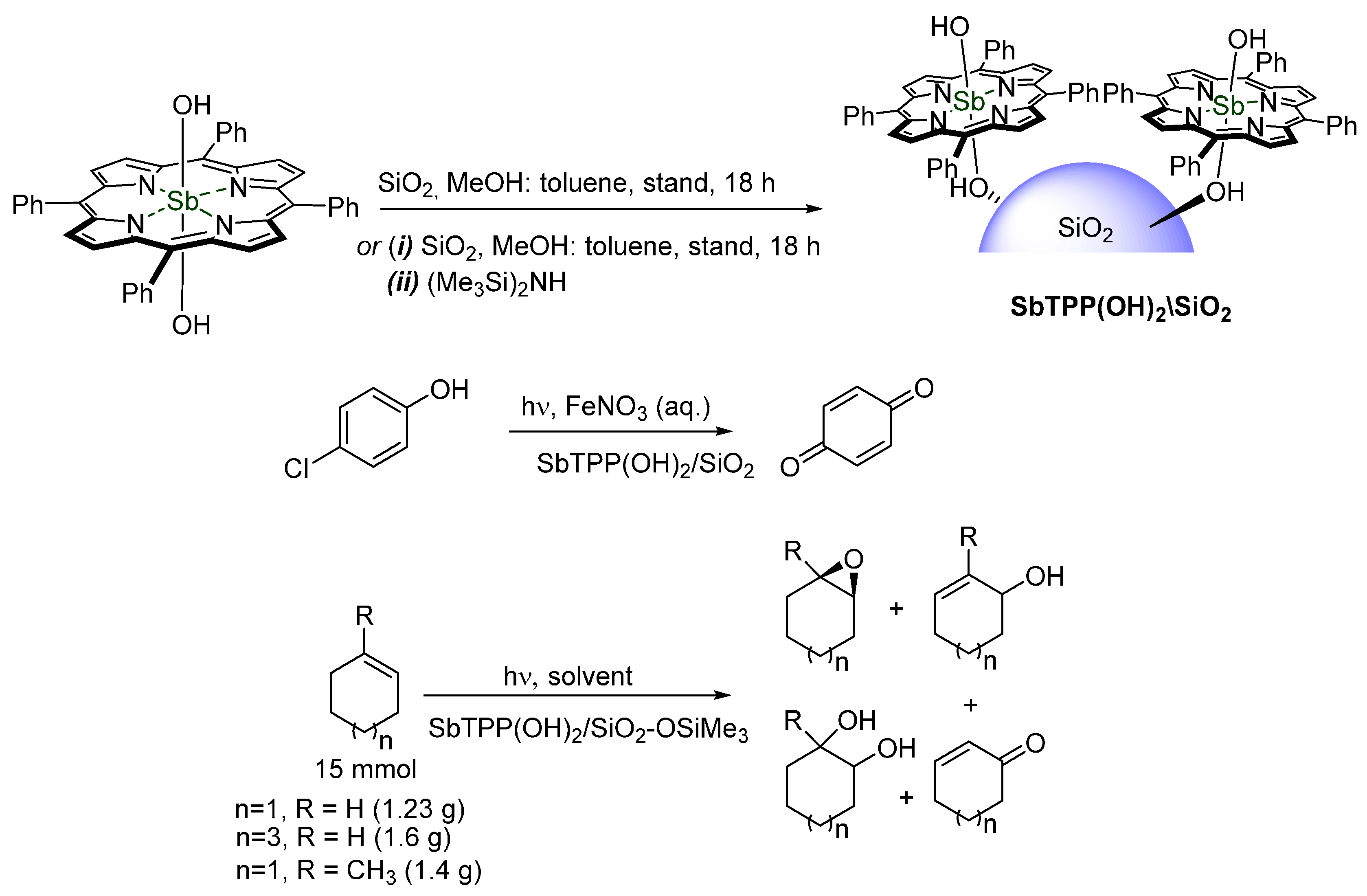

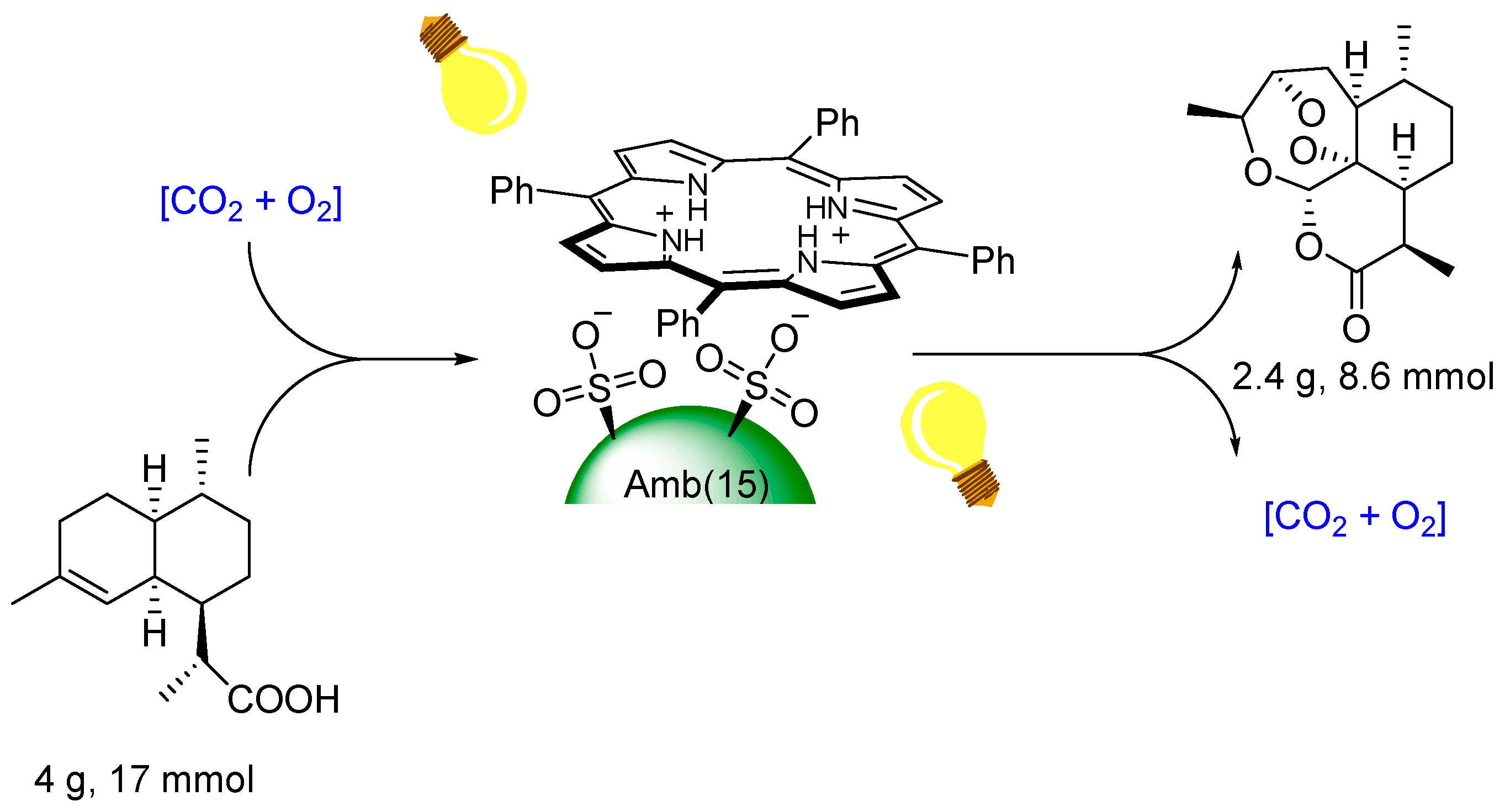
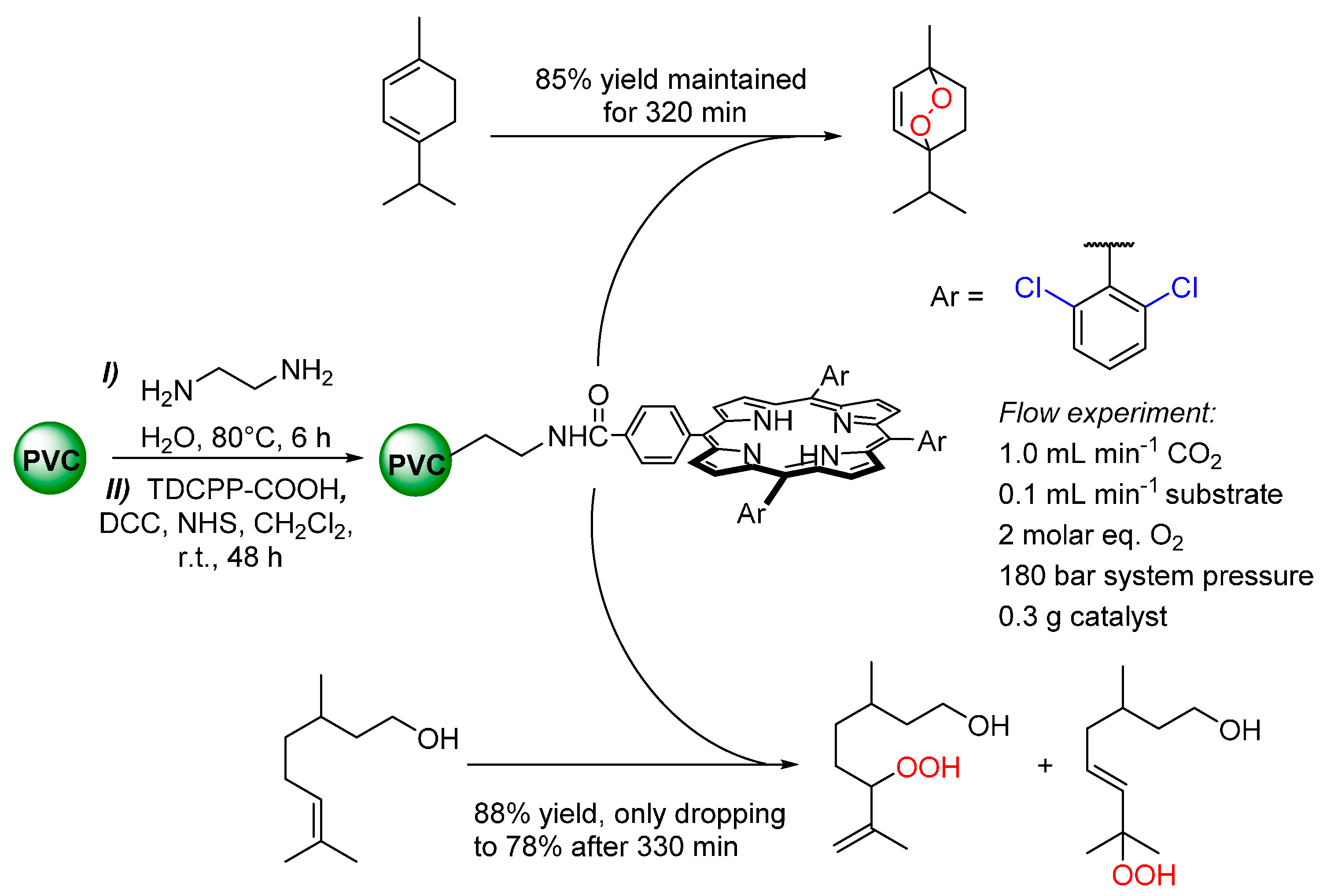
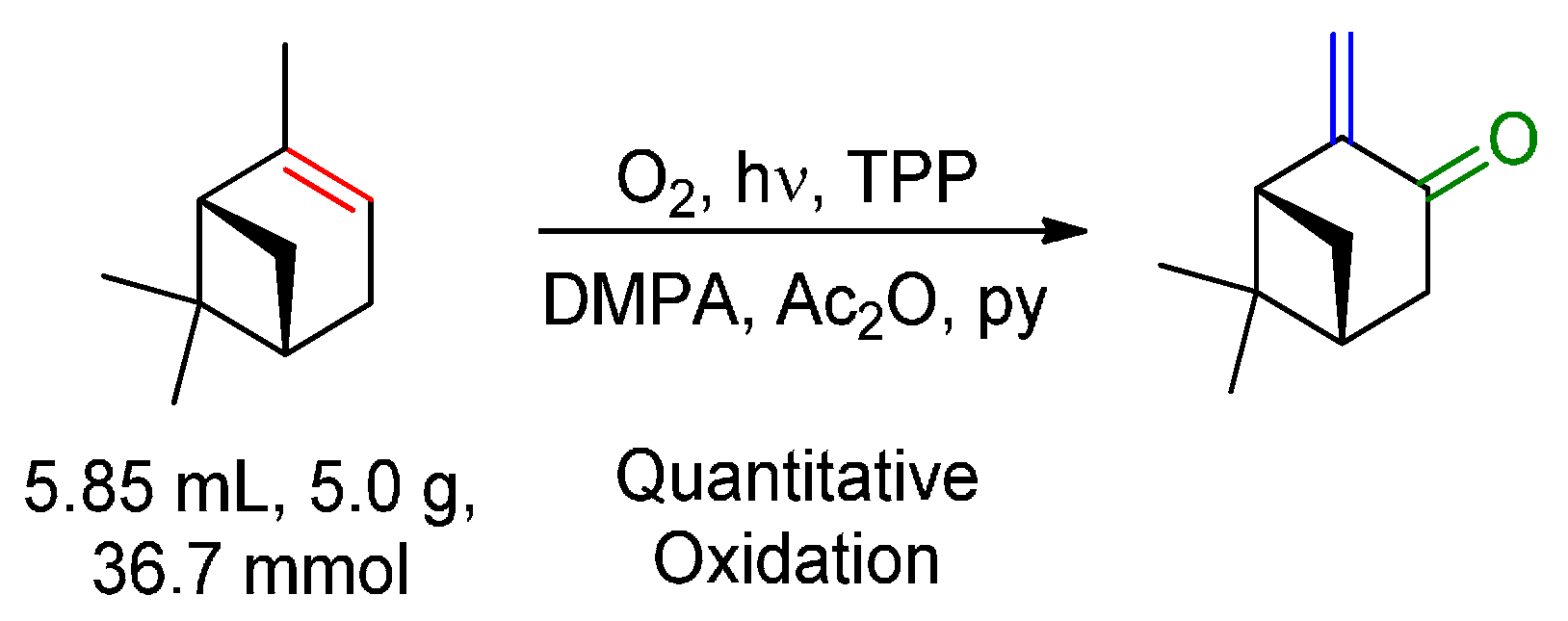

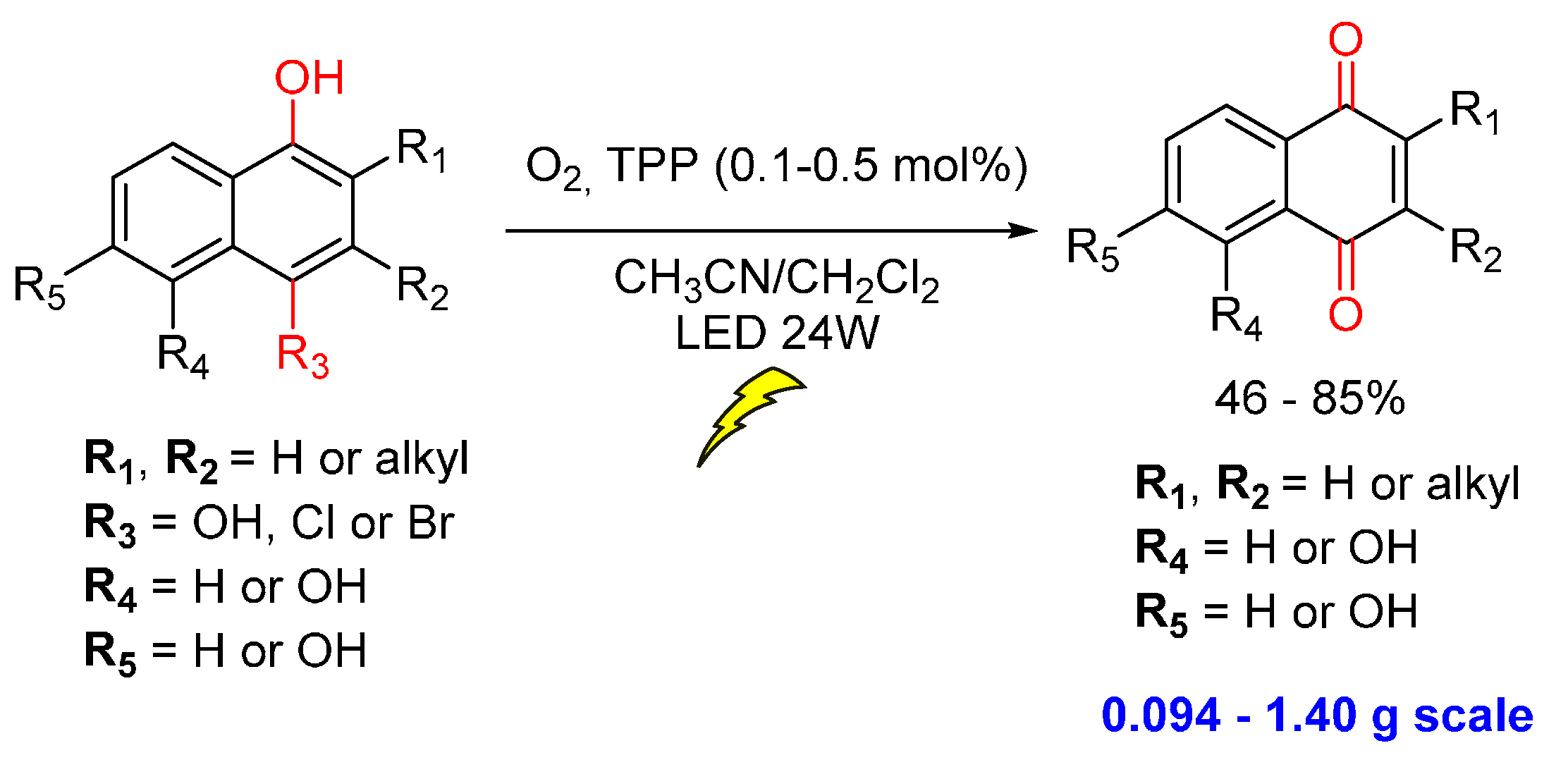
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barona-Castaño, J.C.; Carmona-Vargas, C.C.; Brocksom, T.J.; De Oliveira, K.T. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules 2016, 21, 310. https://doi.org/10.3390/molecules21030310
Barona-Castaño JC, Carmona-Vargas CC, Brocksom TJ, De Oliveira KT. Porphyrins as Catalysts in Scalable Organic Reactions. Molecules. 2016; 21(3):310. https://doi.org/10.3390/molecules21030310
Chicago/Turabian StyleBarona-Castaño, Juan C., Christian C. Carmona-Vargas, Timothy J. Brocksom, and Kleber T. De Oliveira. 2016. "Porphyrins as Catalysts in Scalable Organic Reactions" Molecules 21, no. 3: 310. https://doi.org/10.3390/molecules21030310
APA StyleBarona-Castaño, J. C., Carmona-Vargas, C. C., Brocksom, T. J., & De Oliveira, K. T. (2016). Porphyrins as Catalysts in Scalable Organic Reactions. Molecules, 21(3), 310. https://doi.org/10.3390/molecules21030310