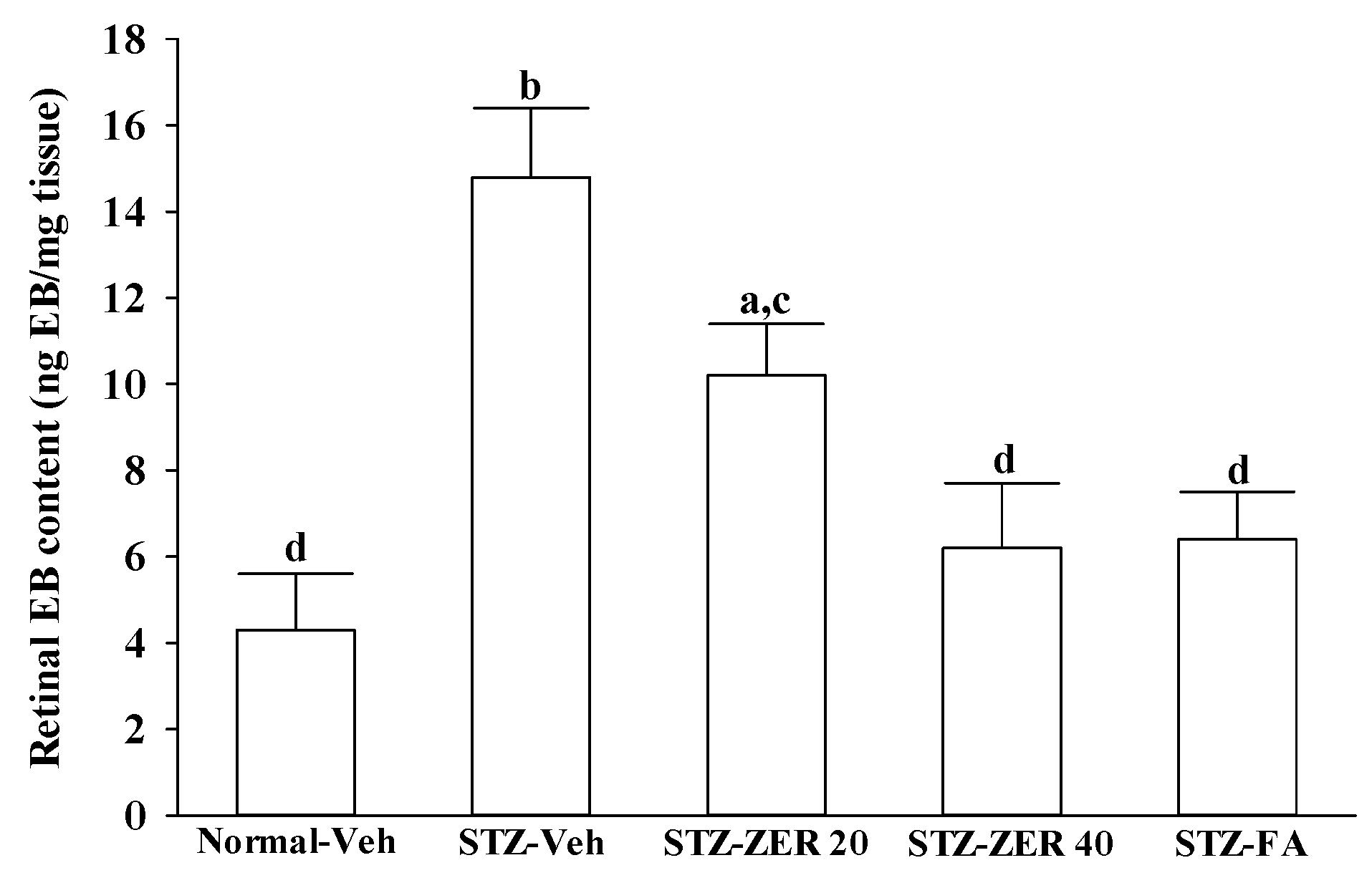1. Introduction
Diabetic retinopathy (DR) is known to be the most common microvascular complication of diabetes mellitus (DM). The relative risk of developing DR is higher for type 1 diabetes compared to type 2 [
1]. DR is characterized by capillary basement membrane thickening, pericyte and endothelial cell loss, blood-retinal barrier (BRB) breakdown and leakage, acellular capillaries, and neovascularization [
2]. Vision loss from DR is a major and leading cause of blindness in adults [
3]. There is thus a pressing need for the development of novel and effective therapeutic approaches to halt the progression of DR. The primary pathogenic factor for the progression of DM is hyperglycemia, and a reduction in glycemia has been found to inhibit the development and progression of retinopathy [
4]. Unfortunately, tight glycemic control is difficult for many patients with hyperglycemia, so effective supplemental therapies still are needed to inhibit retinopathy.
A number of biochemical pathways have been proposed as potential links between hyperglycemia and DR. There is increasing evidence that hyperglycemia-induced inflammatory processes, including activation of inducible nitric oxide synthase (iNOS) and cyclooxygenase (COX)-2 and production of tumor necrosis factor (TNF)-α, interleukin (IL)-1β and vascular endothelial growth factor (VEGF), have a considerable role in the pathogenesis of DR [
5]. These mediators in turn contribute to the upregulation of the adhesion molecules of endothelial cells and leucocytes [
5]. Further, leukostasis leads to vascular occlusion, tissue ischemia, edema, loss of neuronal functions, and neuronal cell death [
6]. In fact, the increased expression of many inflammatory proteins is regulated at the level of gene transcription through the activation of proinflammatory transcription factors, including nuclear factor-κB (NF-κB) [
7]. Due to the involvement of the inflammatory processes of low-grade chronic inflammation in the pathogenesis of DR, inhibiting the inflammatory pathway could be an appealing treatment option for DR in future practices [
8].
Zerumbone (1,(2
E,6
E,10
E)-2,6,9,9-tetramethylcycloundeca-2,6,10-trien-1-one) a southeast Asian ginger sesquiterpene, is derived from several plant species of the family Zingiberaceae [
9,
10]. It is the main component in the rhizome of the edible plant
Zingiber zerumbet Smith grown in tropical and subtropical areas [
11,
12], mainly in Southeast Asia. The plant has been shown to possess antinociceptive, anti-inflammatory, antiulcer, antihyperglycemic, and antiplatelet activities [
12]. Zerumbone is commonly used as a condiment for food flavoring and has been found to have multiple biomedical properties, such as antiproliferative, antioxidant, anti-inflammatory, and anticancer activities [
12,
13]. It has also been found that zerumbone could mitigate nutritional steatohepatitis through regulating key genes related to oxidative stress, inflammation and fibrogenesis [
14]. Furthermore, the compound is valued for its anti-diabetic properties, which involve antihyperglycemia accompanied by inhibition of hyperglycemia-affected proinflammatory factors, chemokines, or adhesion molecule expression in the kidneys of streptozotocin (STZ)-diabetic rats [
15]. In a recent work, zerumbone was found to be beneficial in the amelioration of hyperglycemia-induced retinal damage by the blockade of the advanced glycation end products (AGEs) and their receptor (RAGE) system in STZ-diabetic rats [
16]. Zerumbone might therefore be utilized as an adjuvant therapy in the control of diabetic microvascular complications.
Although uncontrolled hyperglycemia-related tissue damage is the primary cause of diabetic microvascular complications, other metabolic abnormalities, such as dyslipidemia, are involved in the progression of DR [
17]. Actually, the anti-hyperlipidemic activities induced by zerumbone have been documented in our previous study [
18]. Whether the beneficial effects of zerumbone on DR are associated with its plasma lipid lowering activity has yet to be clarified. In the present study, zerumbone was administered in different doses to assess the protective effect of this compound against DR in STZ-diabetic rats, and to evaluate the potential causal mechanisms. Fenofibrate is a common lipid-lowering drug and a potent agonist of the peroxisome proliferator-activated receptor α (PPARα), that has been shown to be effective in reducing the risk of progression of DR, although this effect does not seem to be attributable to changes in lipid profile [
19,
20]. As such, fenofibric acid, the active metabolite of fenofibrate [
20], was used as the positive control in the present study.
3. Materials and Methods
3.1. Experimental Animals
All experimental methods and animal care procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of Tajen University (approval number, IACUC 104-10; approval date: 12 October 2015), in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, as well as the guidelines of the Animal Welfare Act. Male Wistar rats weighting 200–250 g were purchased from the National Laboratory Animal Center (Taipei, Taiwan) and housed two per cage in a room under controlled temperature (20–25 °C), humidity (50% ± 5%) and lighting (12-h light/dark cycle) with food and water provided ad libitum. Rats were rendered diabetic by a single intravenous injection of 60 mg/kg streptozotocin (STZ; Sigma-Aldrich, Inc., St. Louis, MO, USA). Age-matched control rats were injected with vehicle (sterile saline 0.9%, pH 7.4). After one week, rats with non-fasting blood glucose levels >350 mg/dL, polyuria, and glucosuria were defined as diabetic and used for the experiments.
3.2. Treatment Protocols
At two weeks after the injection of STZ, a group of eight rats were dosed by oral gavage once per day for three consecutive months with zerumbone (≥98%; Sigma-Aldrich, Inc.) doses of 20 or 40 mg/kg in a volume of 1.5 mL/kg distilled water. The dosage regime was selected based on a previous report demonstrating that zerumbone at these dosages is potentially effective in improving diabetic nephropathy in STZ-diabetic rats [
15]. Another group of STZ-diabetic rats was treated orally for three months with 100 mg/kg/day fenofibric acid (purity ≥98.0%; Sigma-Aldrich, Inc.), which was based on studies which reported this can have beneficial effects on DR rats [
20]. A vehicle-treated group of normal rats and STZ-diabetic rats were treated with 1.5 mL/kg distilled water only over the same treatment period. Animals had free access to standard rat diet (Harlan Teklad, Madison, WI, USA; Cat. No. 2018) and water throughout the entire period.
At the end of the three-month treatment, the rats were weighed, fasted overnight and anesthetized using an intraperitoneal injection of sodium pentobarbital (60 mg/kg). While under anesthesia, they were painlessly sacrificed and blood was collected from the abdominal aorta of each animal into heparin sample bottles. Rat eyes from each group were removed and the retinae were isolated.
3.3. Biochemical Analysis
Blood samples were centrifuged at 2000× g for 10 min at 4 °C. The plasma was removed and placed into aliquots for analyses. Kits for determining plasma levels of glucose (Cat. No. 10009582), total cholesterol (TC; Cat. # 10007640) and triglyceride (TG; Cat. # 1001030) were purchased from Cayman Chemical Company, Inc. (Ann Arbor, MI, USA). All experimental assays were carried out according to the manufacturers’ instructions; all samples were analyzed in triplicate.
3.4. Fundus Photography and Vessel Diameter
Fundus photography was performed with a retina camera (Kowa Company Ltd., Tokyo, Japan). In order to accustom the rats to the fundus photography procedure, the animals were trained before the start of the study. Eyes were dilated with a drop of 1% tropicamide (Synpac-Kingdom Pharmaceutical Co., Ltd., Taipei, Taiwan). Moisol eye drops (0.7% HPMC) were administered periodically to avoid drying of the cornea. Fundus photography was done regularly till three months to monitor the fundus changes.
The diameter of retinal vessels was estimated by a previously described method [
21]. Before diameter estimation, the retinal photographs from all groups were randomized. The vessel diameters of the three most prominent vessels were estimated at three sites in its widest portion at an equal distance from the center. An average of three estimations was taken as the final retinal vessel diameter.
3.5. Quantification of Retinal Leukostasis
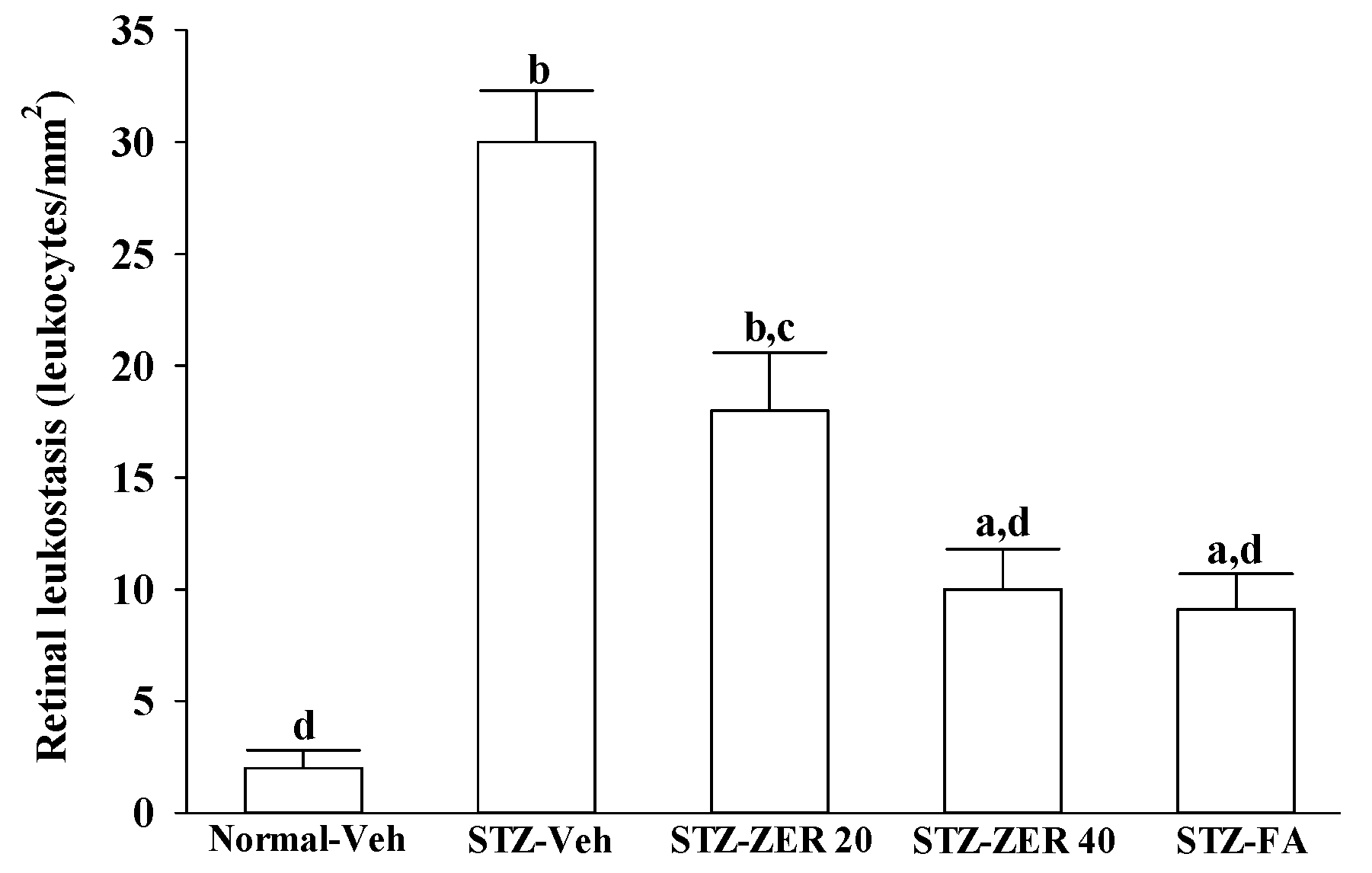
Quantification of leukostasis was performed at the end of the three-month treatment by a previously described method [
22]. Rats were anesthetized with sodium pentobarbital (40 mg/kg body weight), then a 20-gauge perfusion needle was inserted through the left ventricle into the base of the aortic arch, making sure that the needle did not obstruct the carotid arteries. The needle was clamped in place, then the right atrium was cut to allow outflow. Rats were perfused with 10 mL of phosphate buffered saline (PBS) and heparin (0.1 mg/mL) to wash out nonadherent blood cells. Fluorescein isothiocyanate-coupled concanavalin A lectin (Con A) (20 μg/mL in PBS; pH 7.4; 5 mg/kg body weight; Vector Laboratories, Burlingame, CA, USA) was then perfused to label adherent leukocytes and vascular endothelial cells. Unbound ConA was flushed by perfusion with 10 mL PBS. Eyes were removed and fixed in 4% paraformaldehyde for 1 h. Retinas were dissected and flat mounted on a microscope slide, covered with anti-fading medium and a coverslip, and imaged via fluorescence microscopy. Only whole retinas in which the entire vascular network was stained were used for analysis. The total number of adherent leukocytes within the vessels of each retina was counted.
3.6. Measurement of Retinal Vascular Permeability
Retinal vascular permeability was measured using the Evans blue (EB) dye extravasation technique at the end of the three-month treatment [
23]. EB dye (Sigma-Aldrich, Inc.) was dissolved in normal saline at 45 mg/mL and was injected through the tail vein of anesthetized rats over 10 s at a dosage of 45 mg/kg. After the dye had circulated for two hours, the rats were anesthetized with sodium pentobarbital (40 mg/kg body weight), the chest cavity was opened, and cardiac perfusion was performed via the left ventricle with 1% paraformaldehyde in citrate buffer (0.05 mol/L, pH 3.5) under a constant pressure of 120 mmHg. Immediately after perfusion, the retinas were carefully dissected under an operating microscope. After the retinas were fully dried at 4 °C, their weights were measured and the EB dye was extracted by incubating each sample in 150 µL formamide for 18 h at 70 °C. The extract was ultracentrifuged at a speed of 14,000 rpm for 60 min. Absorbance was measured using 100 µL of the supernatant at 620 nm and 740 nm. The concentration of EB in the extracts was calculated from a standard curve and normalized by the weight of the dry retina.
3.7. Retinal Inflammatory and Angiogenic Parameters Determination
Retinas were lysed in ice-cold radio-immunoprecipitation assay buffer (50 mmol/L, Tris-HCl (pH 8), 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid (EDTA), 0.1% sodium dodecyl sulfate, 1% IGEPAL
® CA-630 (Sigma-Aldrich, Inc.) and 0.5% sodium deoxicholate) containing protease inhibitor cocktails (Sigma-Aldrich, Inc.; Cat. No. P83490) and centrifuged for 15 min at 10,000×
g at 4 °C [
24]. The supernatants were collected and assayed for protein content using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Milan, Italy). Enzyme-linked immunosorbent assay (ELISA) kits for the determination of TNF-α (Cat. No. ab46070), IL-1β (Cat. No. ab100768) and intercellular adhesion molecule-1 (ICAM-1; Cat. No. ab100763) were obtained from Abcam Inc. (Cambridge, MA, USA). Vascular cell adhesion molecule 1 (VCAM-1) ELISA kit (Cat. No. NB-E30582) was obtained from Novatein Biosciences (Cambridge, MA, USA). Rat iNOS (Cat. No. E-EL-R0520), COX-2 (Cat. No. E-EL-R0792), VEGF (Cat. No. E-EL-R0020) and protein kinase C (PKC)-β1 (Cat. No. E-EL-R0808) ELISA kits were purchased from Elabscience Biotechnology Co., Ltd. (Wuhan, Hubei, China). All assays were carried out in triplicate.
3.8. Preparation of Nuclear and Cytoplasmic Fractions
Retinas were homogenized in a western lysis buffer (30 mmol/L Tris-HCl, pH 7.4, 250 mmol/L Na3VO4, 5 mmol/L EDTA, 250 mmol/L sucrose, 1% Triton X-100 with protease inhibitor and phosphatase inhibitor cocktail). The homogenate was centrifuged at 800× g for 5 min at 4 °C, and supernatant containing the cytosolic extract was stored frozen at –80 °C. The nuclear pellet was resuspended in 25 µL ice-cold nuclear extraction buffer (20 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 0.4 mmol/L NaCl, 1 mmol/L EDTA, 25% glycerol, protease inhibitors 1X). After 30 min of intermittent mixing, the extract was centrifuged at 14,000× g for 10 min at 4 °C, and supernatants containing nuclear extracts were secured.
3.9. Western Blot Analysis
Before immunoblotting, the protein concentration of each tissue was determined using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Tokyo, Japan), with bovine serum albumin as a standard to ensure equal loading among lanes. Cytosolic (70 μg total protein) and nuclear (50 μg total protein) extracts were separated on a 7.5%–15% polyacrilamide gel and transferred electophoretically to nitrocellulose membranes. Membranes were blocked with 5% non-fat dry milk in Tris-buffered saline Tween (20 mmol/L Tris, pH 7.6, 137 mmol/L NaCl, and 0.1% Tween 20) for three hours at room temperature and incubated overnight at 4 °C with primary antibodies against p38 mitogen-activated protein kinase (p38 MAPK, Cell Signaling Technology, Beverly, MA, USA; Cat. No. 9212), phospho-p38 MAPK (Thr180/Tyr182) (p-p38 MAPK, Cell Signaling Technology; Cat. No. 9211), inhibitory kappa B (IκBα, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA; Cat. No. sc-371) and NF-κB p65 (Santa Cruz Biotechnology, Inc.; Cat. No. sc-109). All antibodies were used at a dilution of 1:1000. After washing three times with Tris-buffered saline with Tween 20 (TBST), the membranes were labeled with horseradish peroxidase-conjugated secondary antibodies for one hour at room temperature. After three additional washes with TBST, the immunoreactive bands were visualized using an enhanced chemiluminescence system (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer’s instructions. Films were scanned, and band densities were quantified with densitometric analysis using ATTO Densitograph Software (ATTO Corp., Tokyo, Japan). All values were normalized by setting the density of the control samples as 1.0. Tissue sections were sampled for four independent experiments.
3.10. Real-Time Polymerase Chain Reaction (PCR)
Total RNA was extracted from rat retinas using a Trizol reagent (Invitrogen; Boston, MA, USA) according to the manufacturer’s protocol. RNA was quantified by A260 and its integrity verified by agarose gel electrophoresis using ethidium bromide for visualization. For the reverse transcriptase reaction, 1 μg of total RNA per sample and 8.5 μg/μL random hexamer primers were heated at 65 °C for 5 min and then quenched on ice. This mixture was combined with 500 μmol/L each of dATP, dTTP, dCTP, and dGTP, 10 mmol/L DTT, 20 mmol/L Tris-HCl (pH 8.4), 50 mmol/L KCl, 5 mmol/L MgCl
2, 40 units of RNaseOUT
TM recombinant ribonuclease inhibitor (Invitrogen) and 100 units SuperScript III reverse transcriptase (Invitrogen). Samples were subjected to DNase (Promega; Madison, WI, USA) treatment at 37 °C for 20 min in a GeneAmp 9700 Thermal Cycler (Applied Biosystems; Foster City, CA, USA) and then held at 4 °C. After aliquots were taken for immediate use in PCR, the remainder of the cDNA was stored at −20 °C. mRNA expression was measured by quantitative real-time PCR in a fluorescent temperature Lightcycler 480 (Roche Diagnostics; Mannheim, Germany). The following primer sequences were used: 5′-ACACCATGAGCACGGAAA GC-3′ (forward) and 5′-CCGCCACGAGCAGGAA-3′ (reverse) for TNF-𝛼; 5′-AATGGACAGAACAT AAGCCAACA-3′ (forward) and 5′-CCCAAGGCCACAGGGAT-3′ (reverse) for IL-1β; 5′-TGATCTT GTGCTGGAGGTGACCAT-3′ (forward) and 5′-TGTAGCGCTGTGTGTCACAGAAGT-3′ (reverse) for iNOS; 5′-GCATTCTTTGCCCAGCACTTCACT-3′ (forward) and 5′-TTTAAGTCCACTCCATGGCCCA GT-3′ (reverse) for COX-2; 5′-CGGGTTTGGGCTTCTCC-3′ (forward) and 5′-GCCACTGCTCGTCCAC ATAG-3′ (reverse) for ICAM-1; 5′-ATCTTCGGAGCCTCAACGG-3′ (forward) and 5′-CCAATCTGAG CGAGCGTTT-3′ (reverse) for VCAM-1; 5′-ACAGGGAAGACAATG GGATGA-3′ (forward) and 5′-GGGCCAGGGATGGGTTT-3′ (reverse) for VEGF; 5′-ACGAATTTGCTGGCTTCTCC-3′ (forward) and 5′-TGGCCTGAAGTCTTACACTCCA-3′ (reverse) for PKC-β; and 5′-TGTGATGGTGGGAATGGGTC AG-3′ (forward) and 5′-TTTGATGTCACGCACGATTTCC-3′ (reverse) for β-actin. Primers were designed using Primer Express Software version 2.0 System (Applied Biosystems; Foster City, CA, USA). The PCR reaction was performed using the following cycling protocol: 95 °C for 5 min, followed by 45 cycles of 95 °C for 5 s, 58 °C for 15 s, and 72 °C for 20 s. Dissociation curves were run after amplification to identify the specific PCR products. The mRNA expression levels were normalized to β-actin mRNA levels and calculated according to the delta-delta Ct method [
25].
3.11. Statistical Analysis
Data are expressed as the mean ± SEM. Statistical analysis and graphics were performed with the SigmaPlot 12.3 program (version 2016, Systat Software Inc., San Jose, CA, USA). Statistical analysis was performed with one-way analysis of variance (ANOVA). Dunnett range post-hoc comparisons were used to determine the source of significant differences, where appropriate. A p value of less than 0.05 was considered statistically significant.
4. Discussion
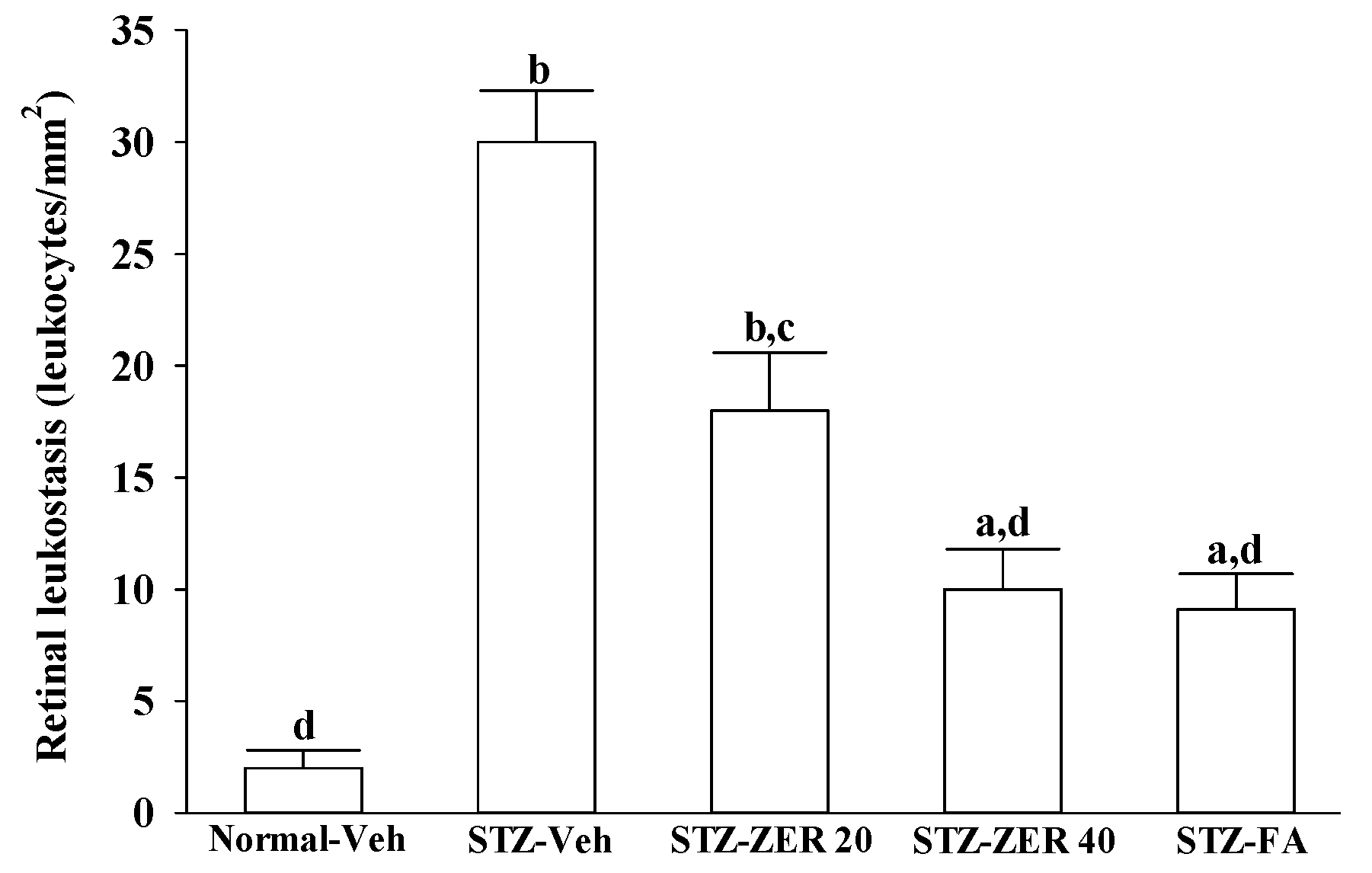
There is growing evidence that leukocyte adhesion to the retinal vasculature or retinal leukostasis results in BRB breakdown in early DR [
26,
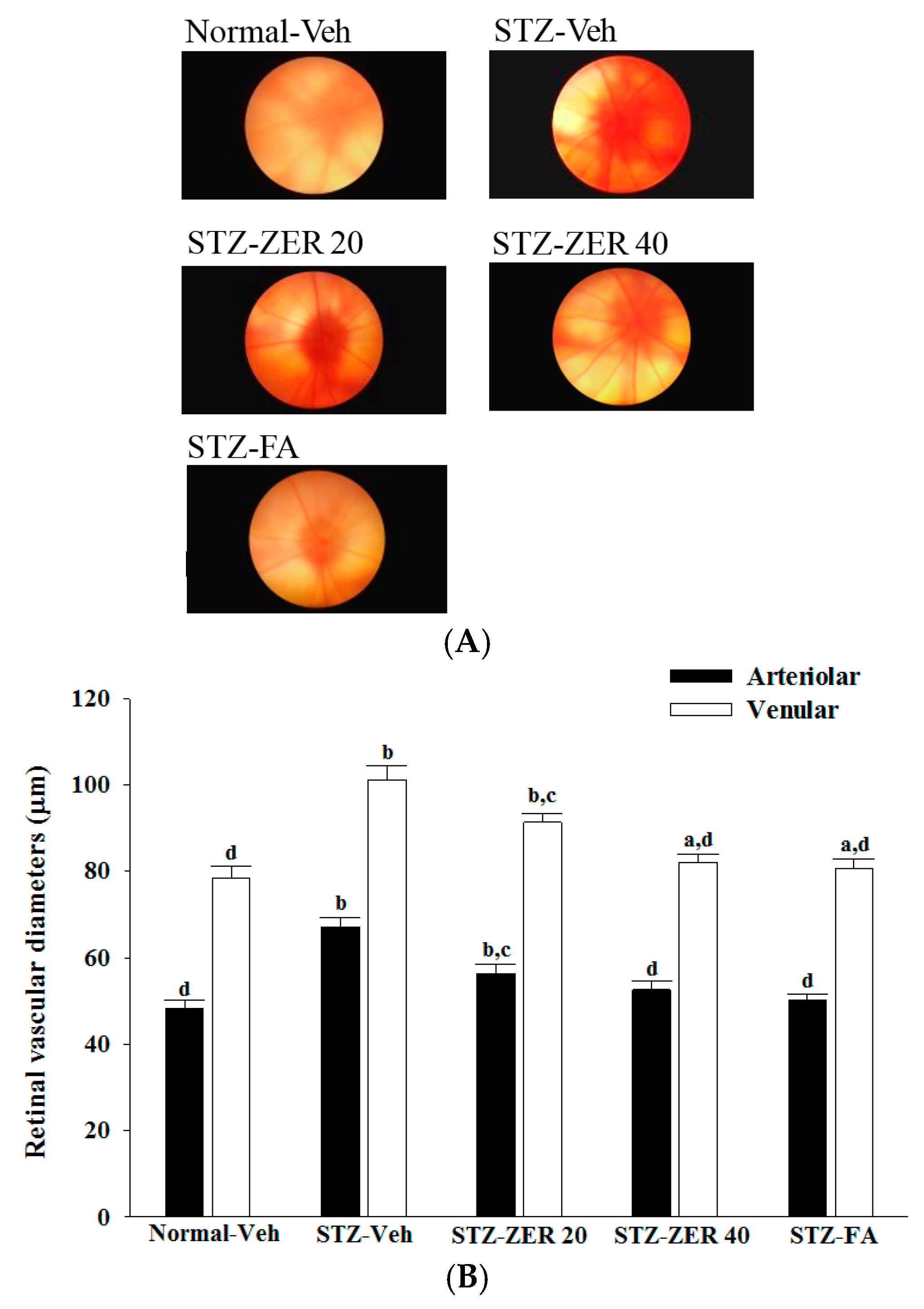
27]. We observed that the number of leukocytes adhering to the retinal vascular endothelium was increased in STZ-diabetic rats, and that vascular permeability and the thickness of retinal vessels increased dramatically. In agreement with the attenuated leukostasis, zerumbone reduced retinal permeability accompanied by decreased vascular permeability of STZ-diabetic rats. The suppressive effect of zerumbone on the BRB breakdown may be important for the prevention of the progression of DR.
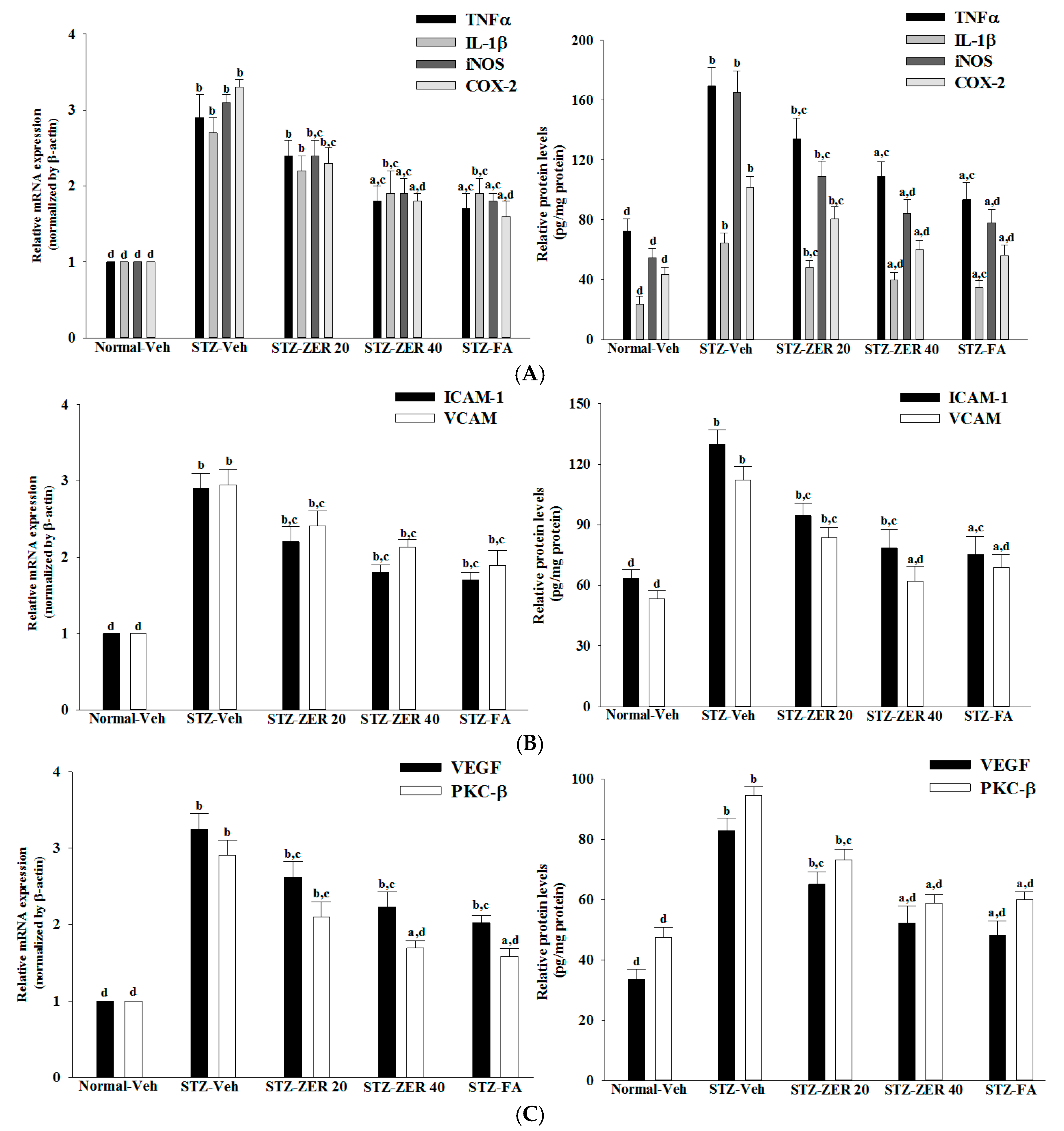
Retinal vascular hyperpermeability causes macular edema, leading to visual deterioration in retinal diseases such as DR [
28]. VEGF, an angiogenic cytokine, is known to be a key molecule leading to retinal permeability and breakdown of BRB in diabetes and other retinal diseases [
28]. VEGF-induced retinal permeability is in part through activation of PKC, specifically the β isoform [
29]. We found that the elevated contents of VEGF and PKC-β in retinas of STZ-diabetic rats were both reduced in rats receiving zerumbone treatment. It is thus suggested that zerumbone has a protective effect on diabetes induced vasculopathy via the repression of vascular hyperpermability.
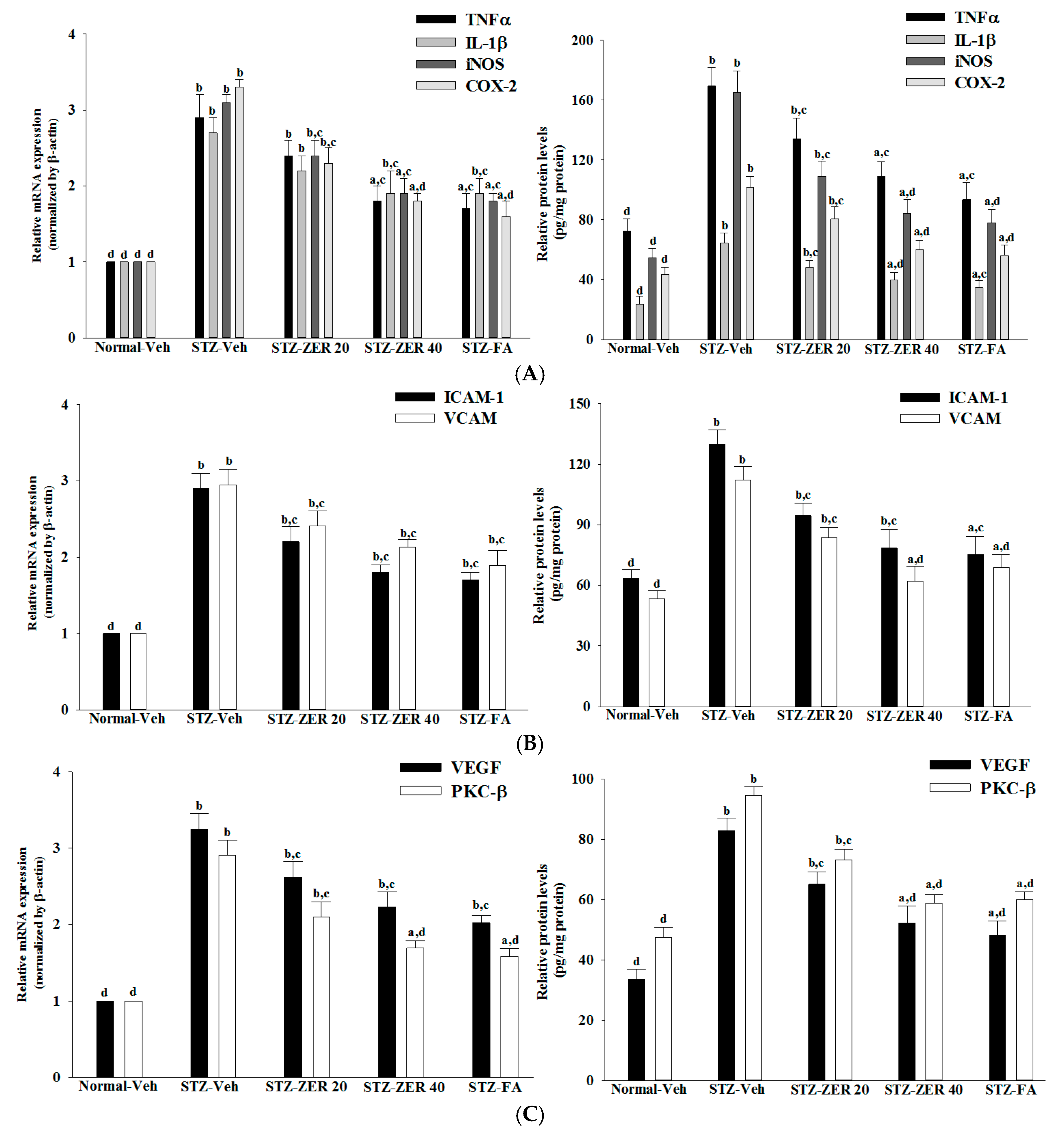
Extensive research has verified the potential role of inflammatory mediators in DR [
5]. One of these mediators is TNF-α, a proinflammatory cytokine which is known as an initiator of inflammatory reactions [
30]. Similarly, IL-1β, iNOS and COX-2 can be up-regulated in the retina with diabetes [
31,
32]. In addition to increases in the above-mentioned inflammatory mediators, ICAM-1 and VCAM-1 promote chemoattraction of leukocytes into the vascular walls and their migration into retinal tissues [
33]. Zerumbone have been demonstrated to attenuate the expression of proinflammatory factors, chemokines, or adhesion molecules in the kidneys of STZ-diabetic rats [
15]. Notably, we found that zerumbone down-regulated the gene expression of a series of proinflammatory cytokines, which may consequently act directly in the retina to reduce the expression of chemokines and adhesion molecules, finally leading to reduced leukocytosis in the retinas of STZ-diabetic rats. These results support the notion that the protective effects of zerumbone with regard to retinal damage in STZ-diabetic rats may be via blockade of inflammation and inhibition of leukostasis/monocyte adhesion to the capillary endothelium [
16].
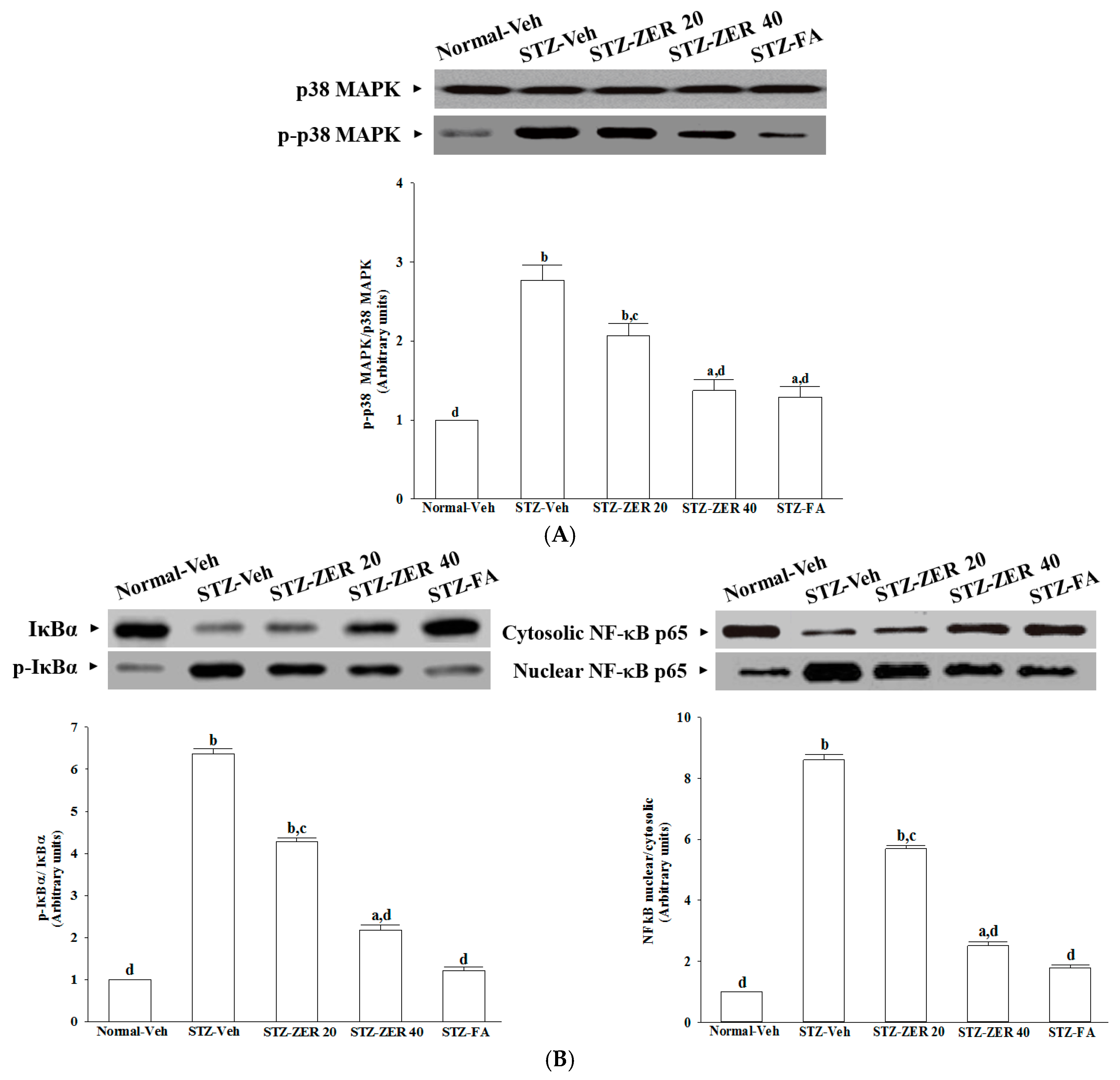
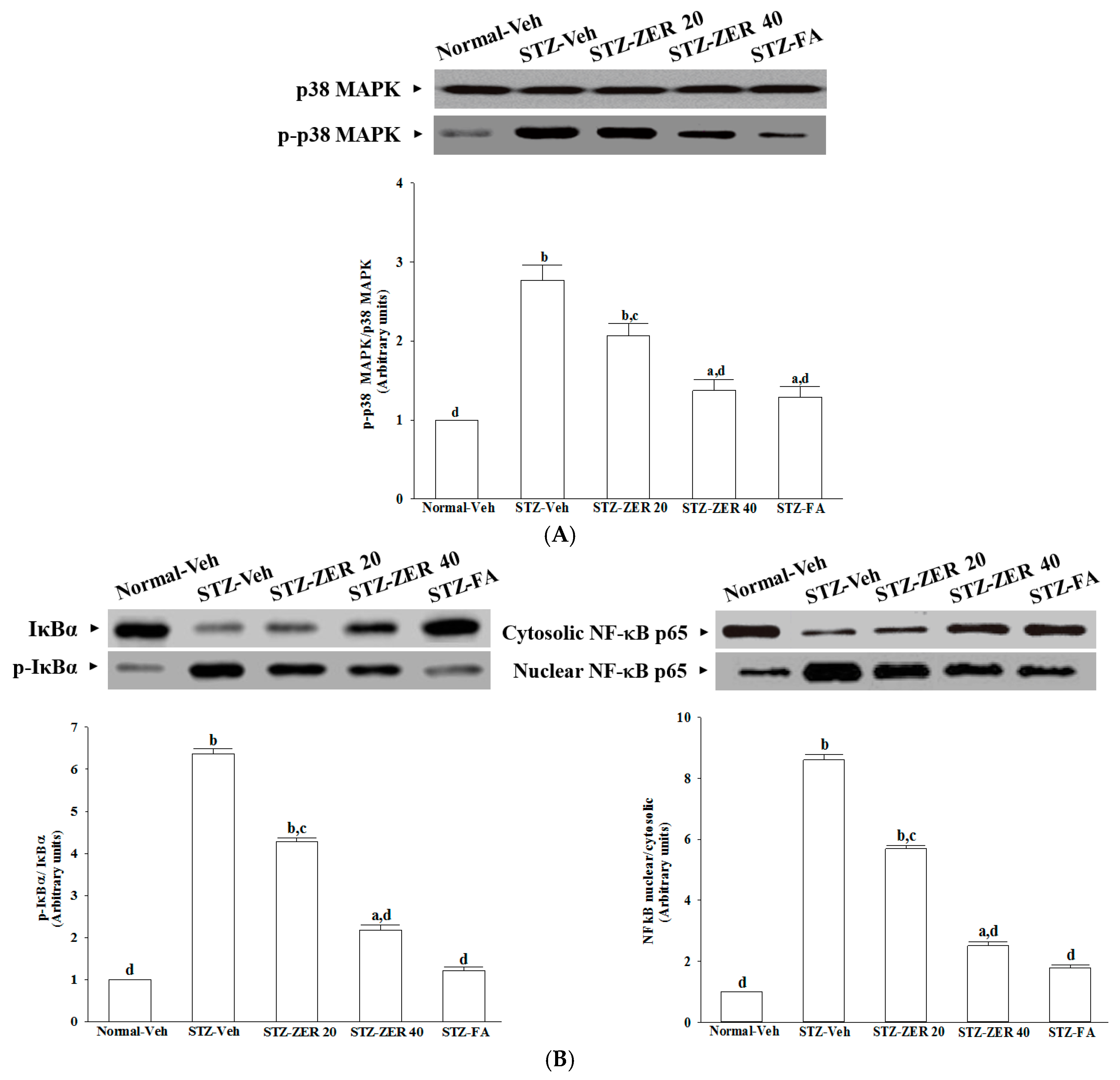
The increase in leukostasis was also associated with the activation of NF-κB, an important transcription factor involved in inflammatory responses [
7]. Once activated by inflammatory mediators, NF-κB will be dissociated from IκBα and translocate into the nucleus to modulate the transcription of its target genes [
33]. NF-κB activation is known to induce the adhesion molecules, cytokines, growth factors and iNOS expression [
34]. NF-κB inhibition has been found to reduce the levels of leukostasis and BRB breakdown in diabetic rat retinas [
31]. Zerumbone has been reported to exert anti-inflammatory activity through inhibiting NF-κB activation in mouse cornea from UVB-induced photokeratitis [
35]. The inhibition of NF-κB by zerumbone might be a critical step in preventing a cascading inflammatory response during progression of DR, as documented in our previous study [
16]. The current work provides results which suggest that zerumbone inhibits activation of the NF-κB pathway via blockade of the degradation and phosphorylation of IκB.
In addition to NF-κB, the activation of p38 MAPK has been reported in the retinas of diabetic rats, and is associated with BRB breakdown [
36]. In fact, the activation of p38 MAPK signaling contributes as an upstream regulator to stimulate the transcriptional activity of NF-κB [
37]. We found that zerumbone decreased p38 MAPK activation in diabetic retinas, and this was accompanied by the suppression of NF-κB nuclear translocation, thus suggesting that the protective effects of zerumbone on DR-related inflammatory responses appear to be due to the inhibition of p38 MAPK-NF-κB-dependent pathways. Zerumbone possesses retinal protective effects, which might be associated with the blockade of the AGEs/RAGE pathway and result in downregulation of NF-κB-mediated inflammatory signals, as documented in our previous study [
15]. It is thus likely that zerumbone has protective effects on several pharmacological targets in DR.
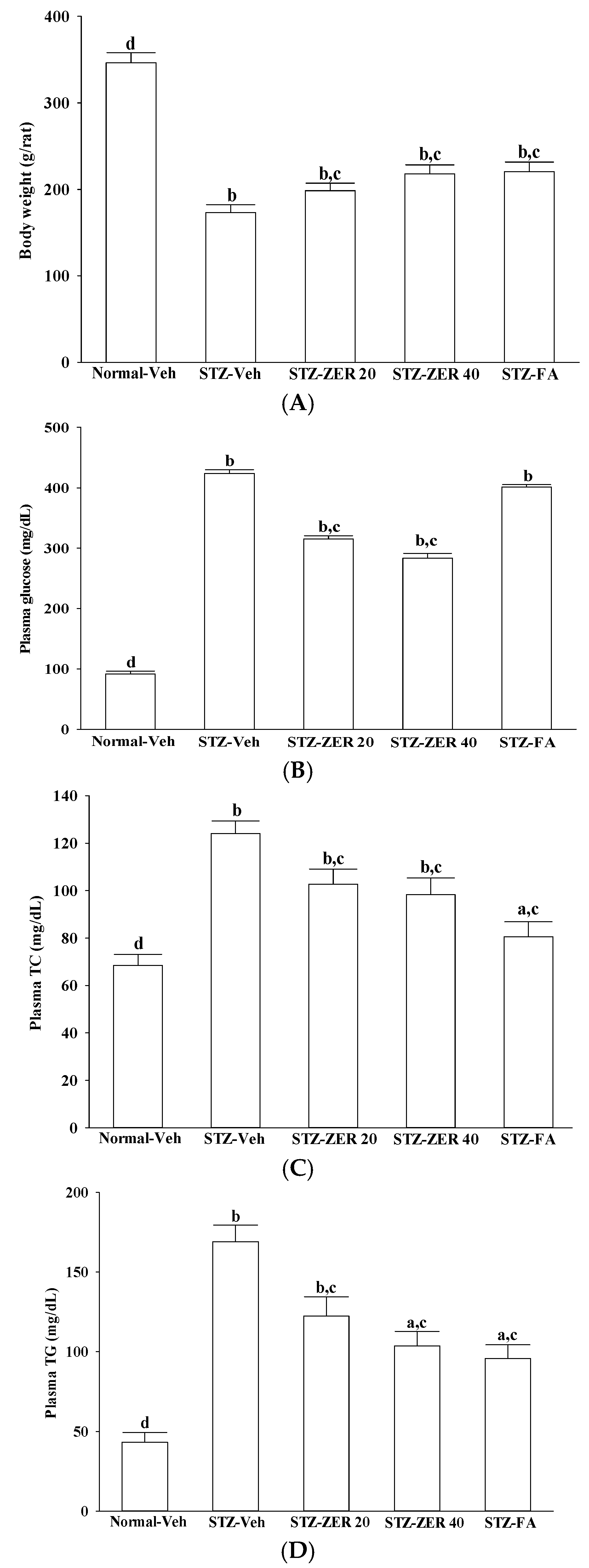
Nevertheless, as we mentioned previously, DR has a complicated etiology and involves many factors. Among these causative factors, dyslipidemia seems to contribute heavily [
17]. In fact, zerumbone has been shown to ameliorate dyslipidemia in high-fat diet-induced hyperlipidemic hamsters through the enhancement of gene expression involved in the lipid metabolism through PPARα activation [
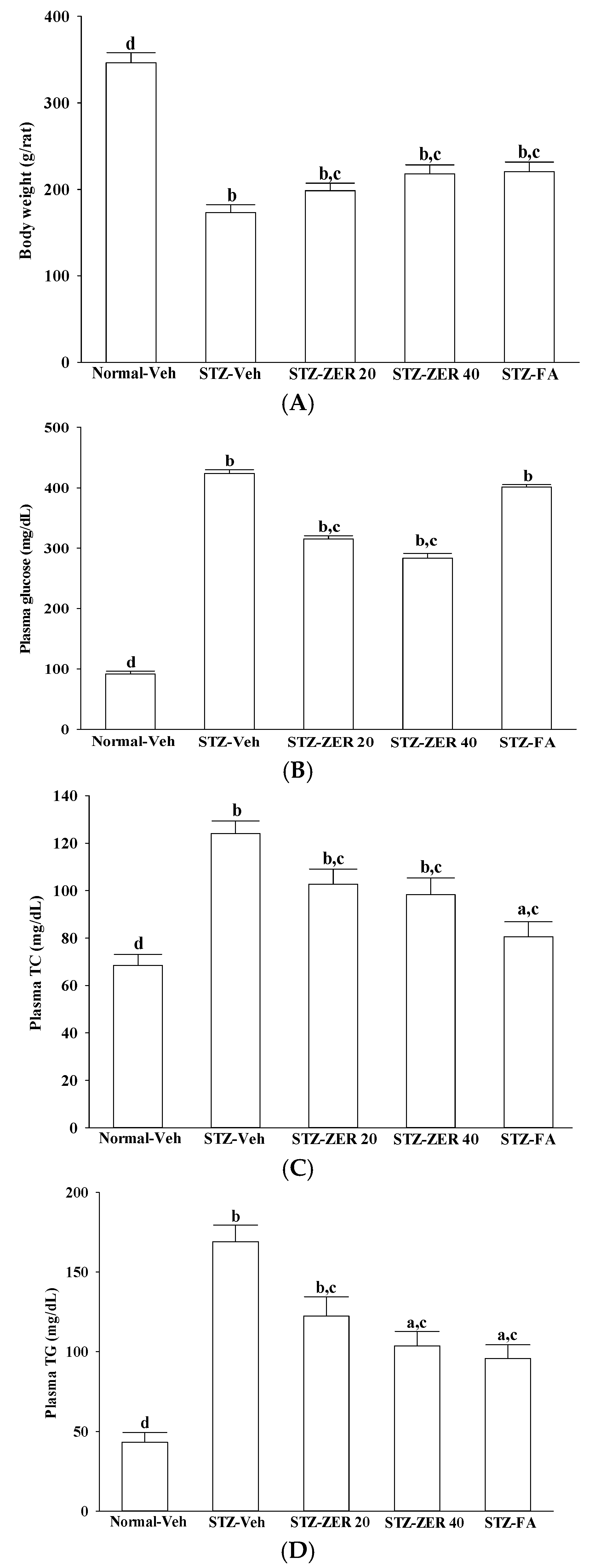
18]. Our results also show that, along with the plasma glucose lowering action, zerumbone treatment significantly decreases the higher TG or TC levels in the plasma of STZ-diabetic rats. However, fenofibric acid ameliorates retinal damage with less glycemic control, in comparison with zerumbone. We thus propose that the beneficial effects of zerumbone on DR are at least in part through its anti-hyperglycemic and anti-hyperlipidemic actions. The retinal protective impact of zerumbone is as effective as that produced by fenofibric acid. As such, the role of glucose or lipid homeostasis with regard to the action of zerumbone on the amelioration of diabetes-associated retinal dysfunction is of considerable interest, and should be further clarified in future research.


{kind=link}
{kind=link}
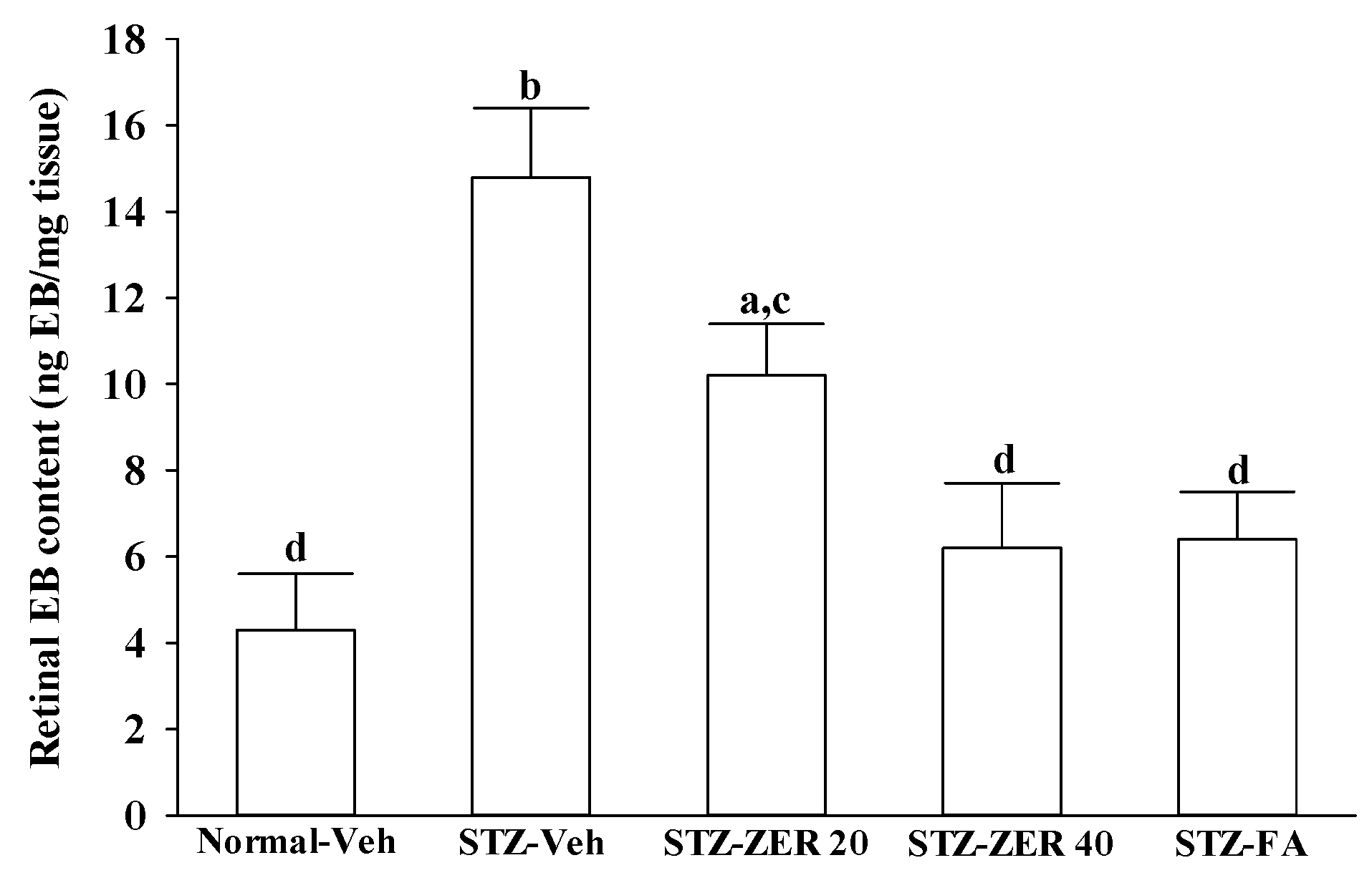{kind=link}
{kind=link}
{kind=link}
{kind=link}