
A Computational Study of Structure and Reactivity of N-Substitued-4-Piperidones Curcumin Analogues and Their Radical Anions


Abstract
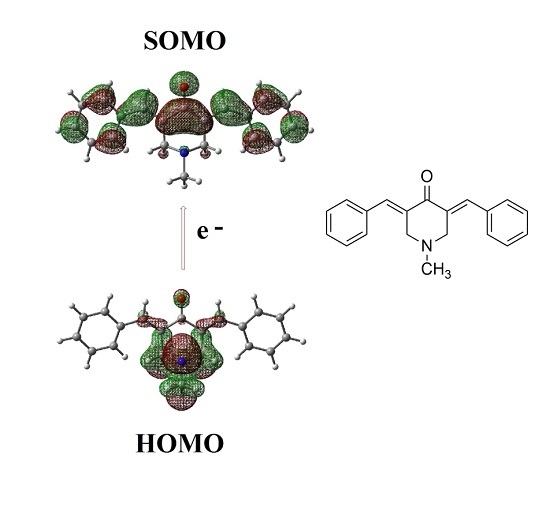
:
1. Introduction
2. Results and Discussion
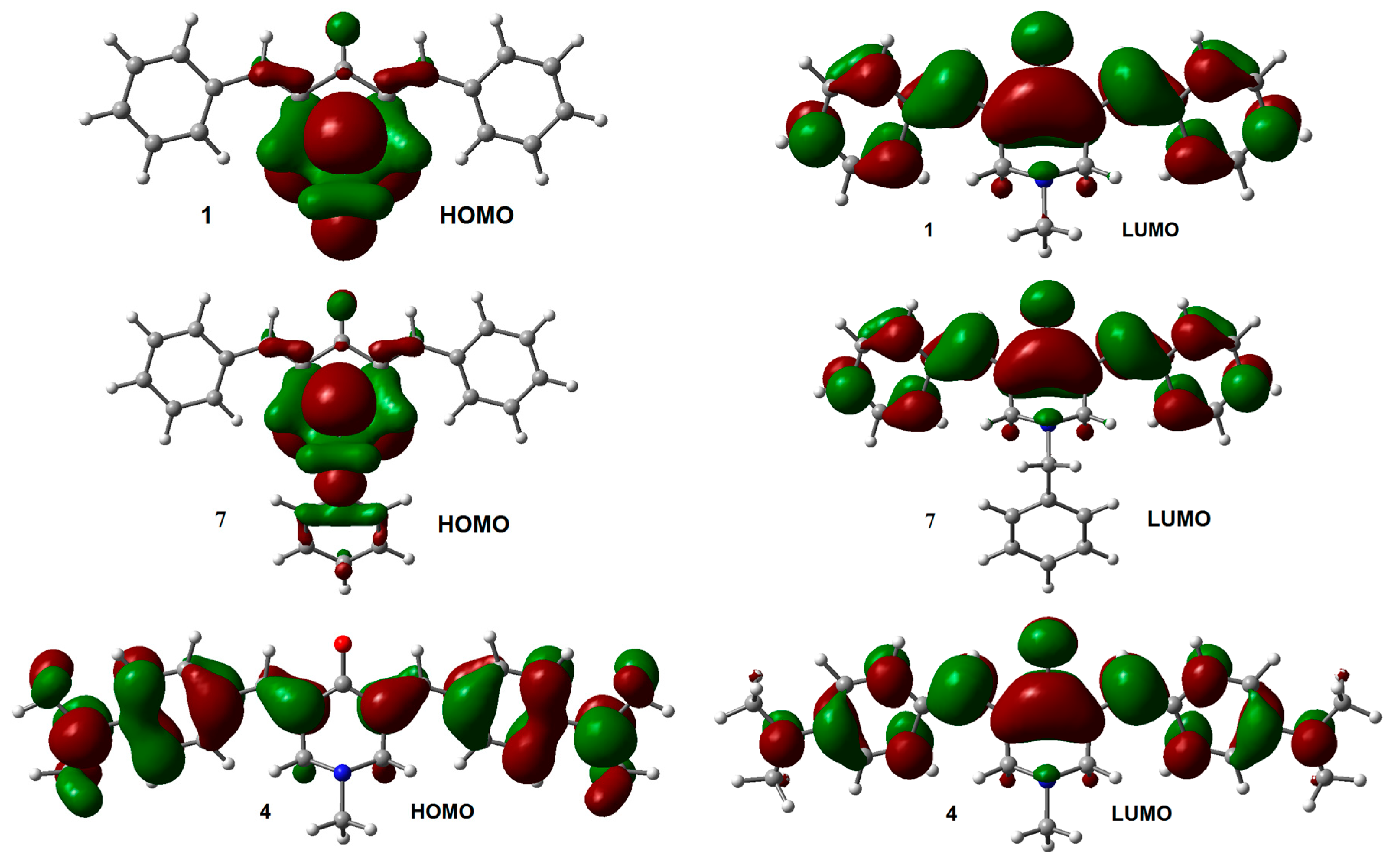
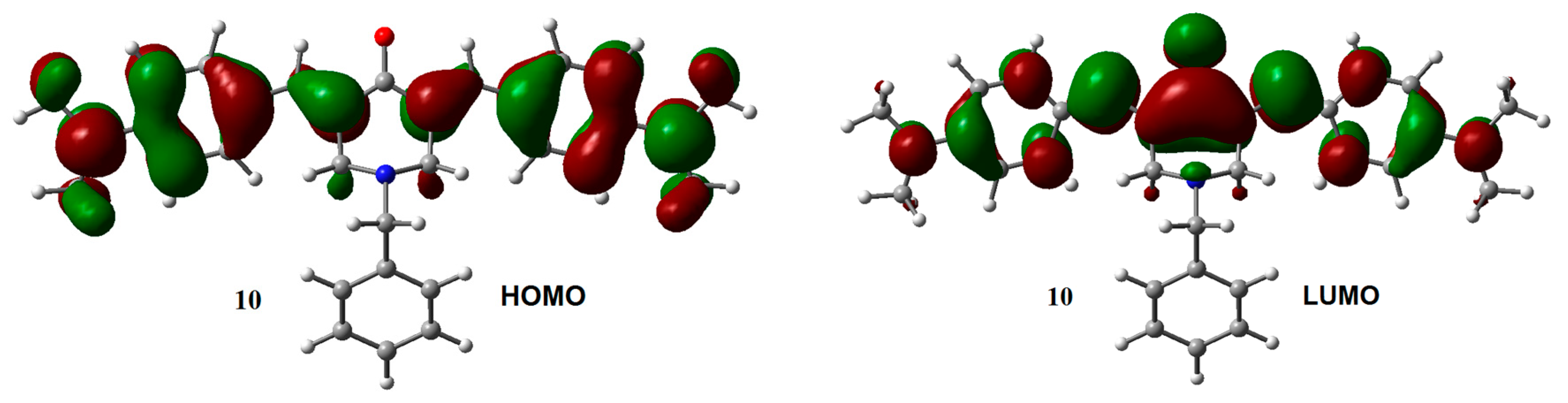
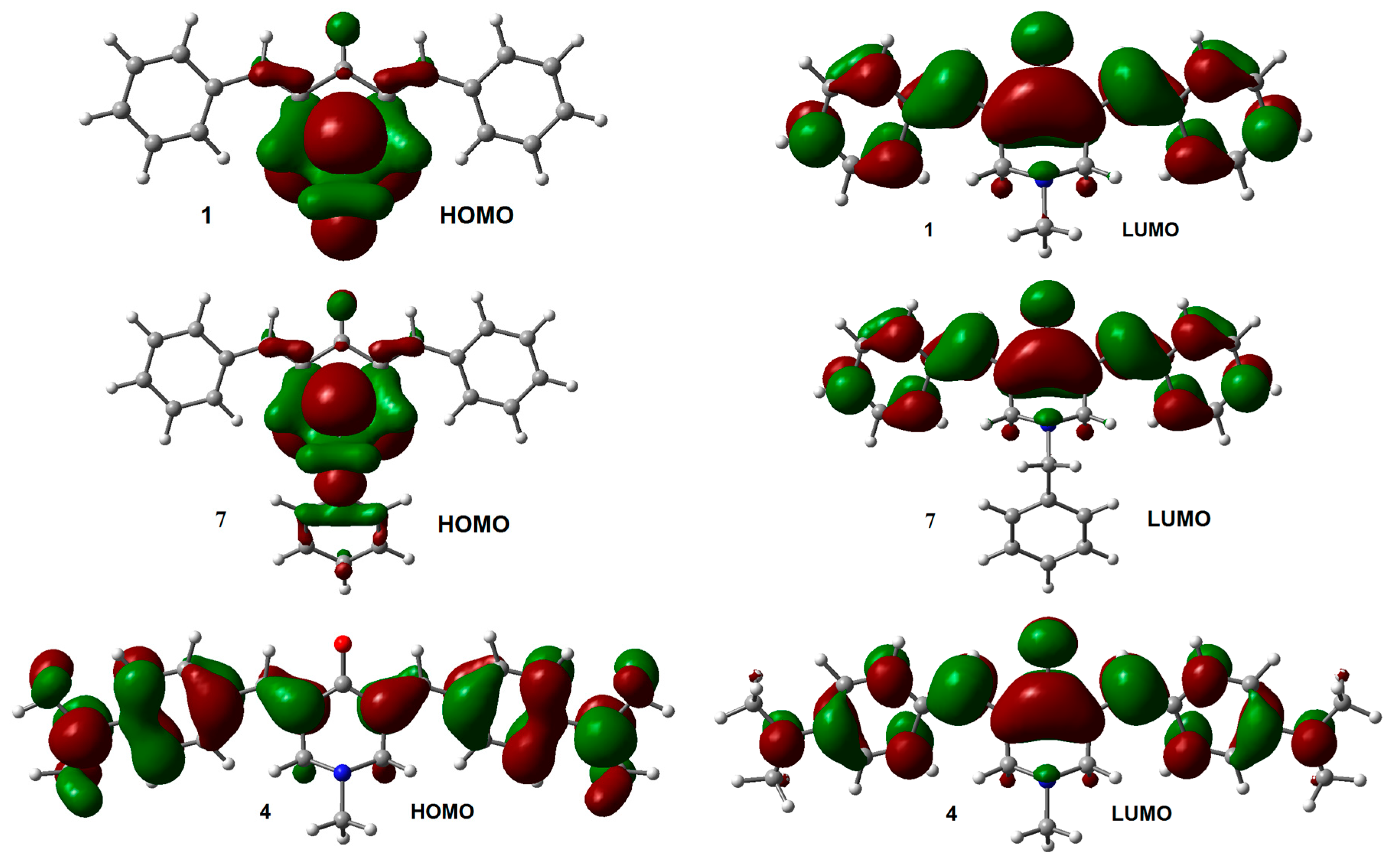
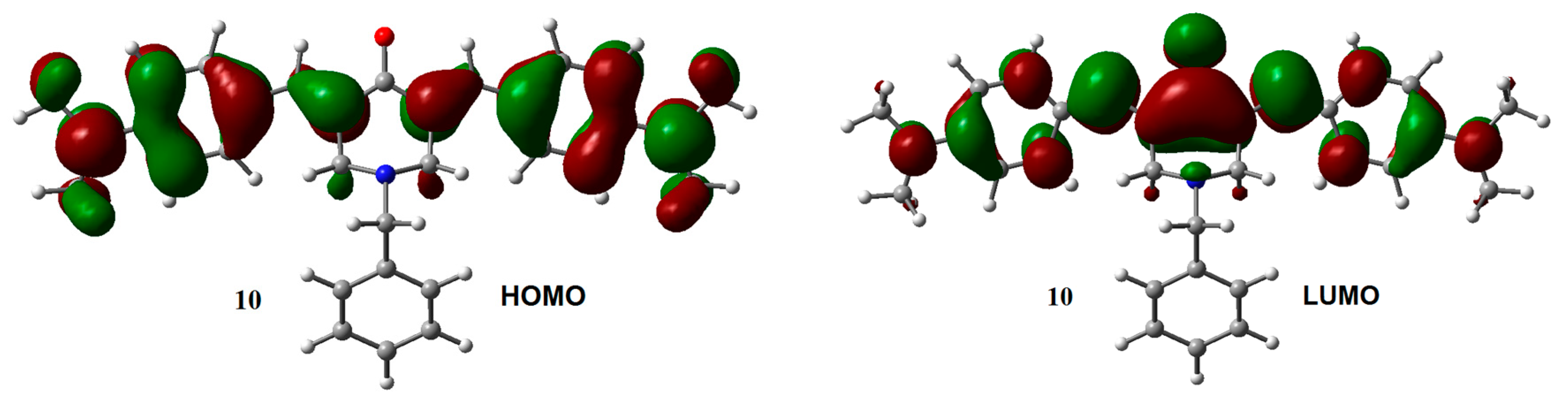
2.1. Geometry and Electronic Structure Analysis of Neutral Species
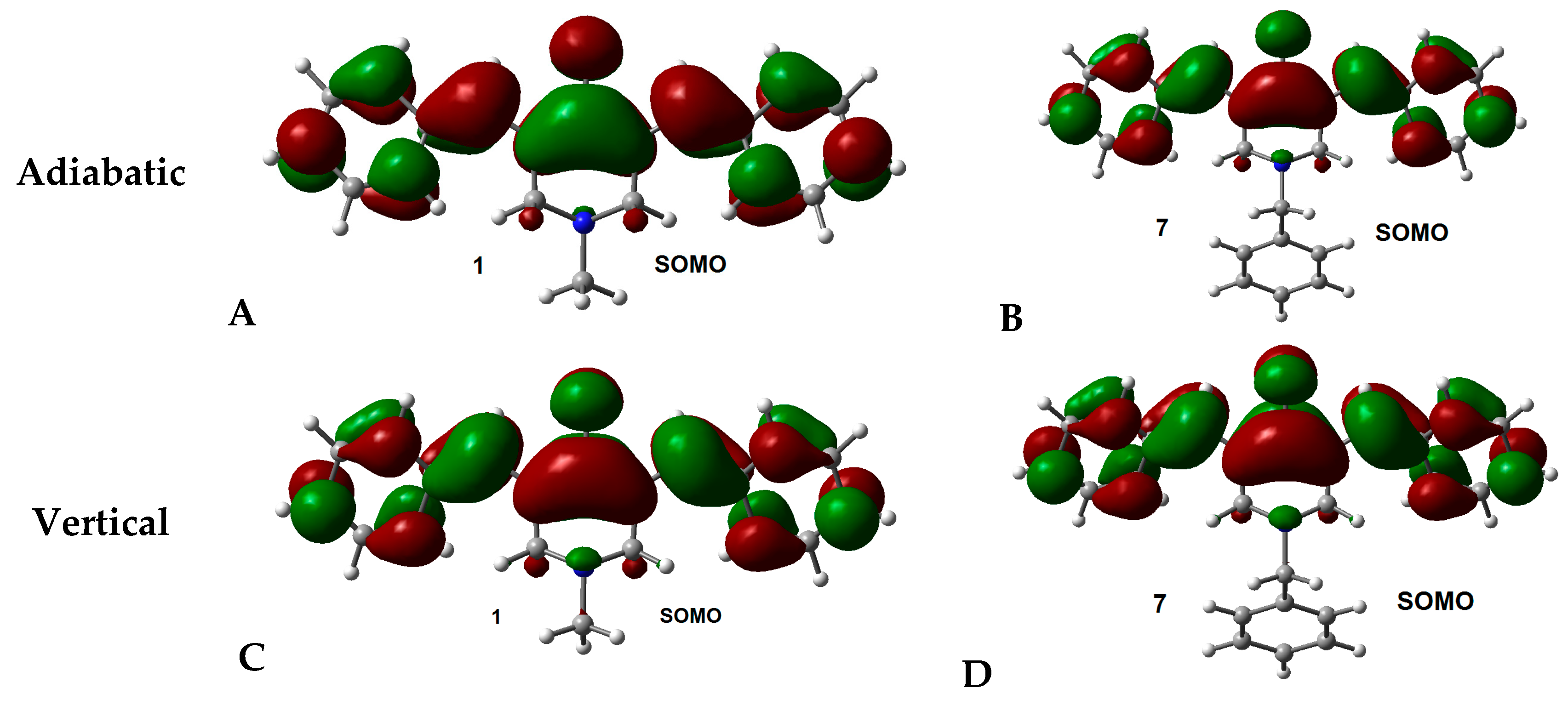
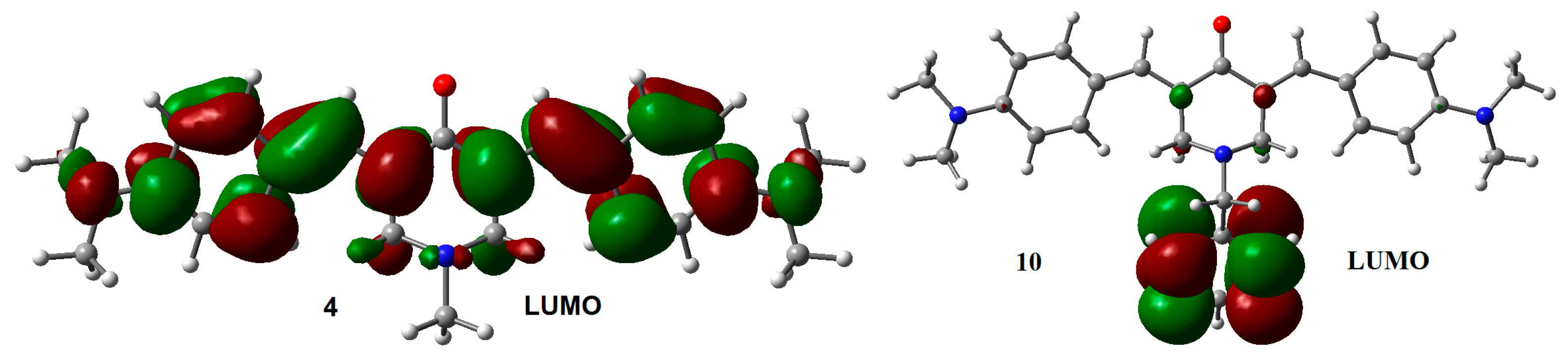
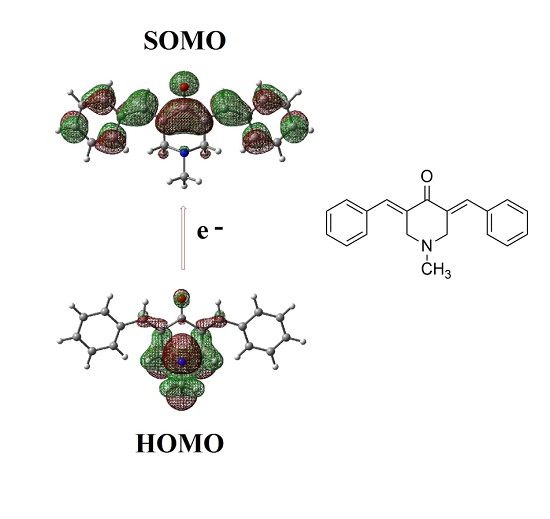
2.2. Radical Anions and Electron Affinities
2.3. Local Reactivity
3. Materials and Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Martinez-Cifuentes, M.; Weiss-Lopez, B.E.; Santos, L.S.; Araya-Maturana, R. Heterocyclic curcumin derivatives of pharmacological interest: Recent progress. Curr. Top. Med. Chem. 2015, 15, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Bairwa, K.; Grover, J.; Kania, M.; Jachak, S.M. Recent developments in chemistry and biology of curcumin analogues. Rsc Adv. 2014, 4, 13946–13978. [Google Scholar] [CrossRef]
- Zhou, H.; Beevers, C.S.; Huang, S. The targets of curcumin. Curr. Drug. Targets 2011, 12, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.; Sanderson, I.R.; MacDonald, T.T. Curcumin as a therapeutic agent: The evidence from in vitro, animal and human studies. Br. J. Nutr. 2010, 103, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Teiten, M.H.; Eifes, S.; Dicato, M.; Diederich, M. Curcumin-the paradigm of a multi-target natural compound with applications in cancer prevention and treatment. Toxins 2010, 2, 128–162. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Sung, B. Pharmacological basis for the role of curcumin in chronic diseases: An age-old spice with modern targets. Trends Phermacol. Scl. 2009, 30, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.; El-meligy, A.; Lungu, G.; Fetaih, H.; Dessouki, A.; Stoica, G.; Barhoumi, R. Curcumin induces apoptosis in a murine mammary gland adenocarcinoma cell line through the mitochondrial pathway. Eur. J. Pharmacol. 2011, 668, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm. 2007, 4, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Moron, E.; Calderon-Montano, J.M.; Salvador, J.; Robles, A.; Lopez-Lazaro, M. The dark side of curcumin. Int. J. Cancer 2010, 126, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.A.; Shao, L.L.; Wang, Y.; Zhao, C.G.; Chu, Y.H.; Xiao, J.; Zhao, Y.; Li, X.K.; Yang, S.L. Exploration and synthesis of curcumin analogues with improved structural stability both in vitro and in vivo as cytotoxic agents. Bioorgan. Med. Chem. 2009, 17, 2623–2631. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Thomas, S.G.; Kunnumakkara, A.B.; Sundaram, C.; Harikumar, K.B.; Sung, B.; Tharakan, S.T.; Misra, K.; Priyadarsini, I.K.; Rajasekharan, K.N.; et al. Biological activities of curcumin and its analogues (congeners) made by man and mother nature. Biochem. Pharmacol. 2008, 76, 1590–1611. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Surh, Y.-J.; Shishodia, S. The Molecular Targets and Therapeutic uSes of Curcumin in Health and Disease, Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2007. [Google Scholar]
- Bazzaro, M.; Anchoori, R.K.; Mudiam, M.K.R.; Issaenko, O.; Kumar, S.; Karanam, B.; Lin, Z.H.; Vogel, R.I.; Gavioli, R.; Destro, F.; et al. Alpha,beta-unsaturated carbonyl system of chalcone-based derivatives is responsible for broad inhibition of proteasomal activity and preferential killing of human papilloma virus (hpv) positive cervical cancer cells. J. Med. Chem. 2011, 54, 449–456. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, Y.; Cai, Y.; Wang, J.; Weng, B.; Tang, Q.; Chen, X.; Pan, Z.; Liang, G.; Yang, S. Discovery and evaluation of piperid-4-one-containing mono-carbonyl analogs of curcumin as anti-inflammatory agents. Bioorg. Med. Chem. 2013, 21, 3058–3065. [Google Scholar] [CrossRef] [PubMed]
- Nunes, L.M.; Hossain, M.; Varela-Ramirez, A.; Das, U.; Ayala-Marin, Y.M.; Dimmock, J.R.; Aguilera, R.J. A novel class of piperidones exhibit potent, selective and pro-apoptotic anti-leukemia properties. Oncol. Lett. 2016, 11, 3842–3848. [Google Scholar] [CrossRef] [PubMed]
- Pati, H.N.; Das, U.; Sharma, R.K.; Dimmock, J.R. Cytotoxic thiol alkylators. Mini-Rev. Med. Chem. 2007, 7, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Sharma, R.K.; Dimmock, J.R. 1,5-diaryl-3-oxo-1,4-pentadienes: A case for antineoplastics with multiple targets. Curr. Med. Chem. 2009, 16, 2001–2020. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Das, U.; Varela-Ramirez, A.; Lema, C.; Aguilera, R.J.; Balzarini, J.; de Clercq, E.; Dimmock, S.G.; Gorecki, D.K.J.; Dimmock, J.R. Bis 3,5-bis(benzylidene)-4-oxo-1-piperidinyl amides: A novel class of potent cytotoxins. Chemmedchem 2011, 6, 1892–1899. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Rios-Gutierrez, M.; Perez, P. Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 2016, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Ao, G.Z.; Chu, X.J.; Ji, Y.Y.; Wang, J.W. Antioxidant properties and pc12 cell protective effects of a novel curcumin analogue (2e,6e)-2,6-bis(3,5-dimethoxybenzylidene)cyclohexanone (mch). Int. J. Mol. Sci. 2014, 15, 3970–3988. [Google Scholar] [CrossRef] [PubMed]
- Leon-Gonzalez, A.J.; Auger, C.; Schini-Kerth, V.B. Pro-oxidant activity of polyphenols and its implication on cancer chemoprevention and chemotherapy. Biochem. Pharmacol. 2015, 98, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Nimiya, Y.; Wang, W.C.; Du, Z.Y.; Sukamtoh, E.; Zhu, J.L.; Decker, E.; Zhang, G.D. Redox modulation of curcumin stability: Redox active antioxidants increase chemical stability of curcumin. Mol. Nutr. Food Res. 2016, 60, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorg. Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Modzelewska, A.; Pettit, C.; Achanta, G.; Davidson, N.E.; Huang, P.; Khan, S.R. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg. Med. Chem. 2006, 14, 3491–3495. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cifuentes, M.; Salazar, R.; Escobar, C.A.; Weiss-Lopez, B.E.; Santos, L.S.; Araya-Maturana, R. Correlating experimental electrochemistry and theoretical calculations in 2′-hydroxy chalcones: The role of the intramolecular hydrogen bond. RSC Adv. 2015, 5, 50929–50937. [Google Scholar] [CrossRef]
- Pandith, A.H.; Islam, N. Electron transport and nonlinear optical properties of substituted aryldimesityl boranes: A dft study. PLoS ONE 2014, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.M.; Pluth, M.D. Linear free energy relationships reveal structural changes in hydrogen-bonded host-guest interactions. J. Org. Chem. 2014, 79, 11797–11801. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Al-Sehemi, A.G.; Al-Assiri, M.S. Push-pull effect on the electronic, optical and charge transport properties of the benzo 2,3-b thiophene derivatives as efficient multifunctional materials. Comp. Theor. Chem. 2014, 1031, 76–82. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Domingo, L.R.; Perez, P.; Saez, J.A. Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic parr functions. RSC Adv. 2013, 3, 1486–1494. [Google Scholar] [CrossRef]
- Moore, T.W.; Zhu, S.; Randolph, R.; Shoji, M.; Snyder, J.P. Liver s9 fraction-derived metabolites of curcumin analogue ubs109. ACS Med. Chem. Lett. 2014, 5, 288–292. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T.; DeCaprio, A.; Barber, D.S. Application of the hard and soft, acids and bases (hsab) theory to toxicant-target interactions. Chem. Res. Toxicol. 2012, 25, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Revision a.01, Gaussian 09, Gaussian Inc.: Wallingford, CT, USA, 2009.
- Liu, H.N.; Ding, Y.Q.; Walker, L.A.; Doerksen, R.J. Computational study on the effect of exocyclic substituents on the ionization potential of primaquine: Insights into the design of primaquine-based antimalarial drugs with less methemoglobin generation. Chem. Res. Toxicol. 2015, 28, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.


| Compounds | R1 | R2 |
|---|---|---|
| 1 | -H | CH3 |
| 2 | 4-Cl | CH3 |
| 3 | 4-Br | CH3 |
| 4 | 4-NMe2 | CH3 |
| 5 | 4-CN | CH3 |
| 6 | 4-CF3 | CH3 |
| 7 | -H | CH2Ph |
| 8 | 4-Cl | CH2Ph |
| 9 | 4-Br | CH2Ph |
| 10 | 4-NMe2 | CH2Ph |
| 11 | 4-CN | CH2Ph |
| 12 | 4-CF3 | CH2Ph |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
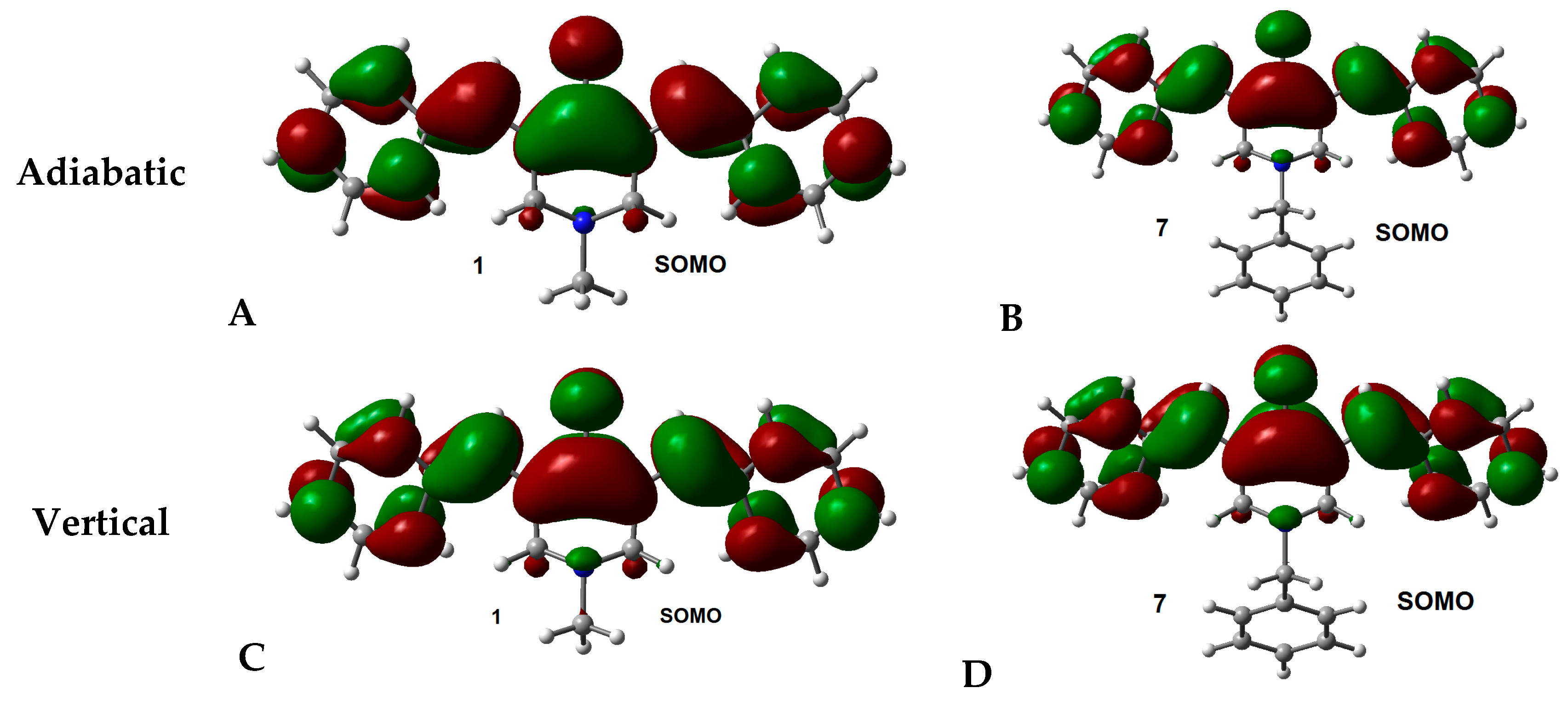{kind=link}
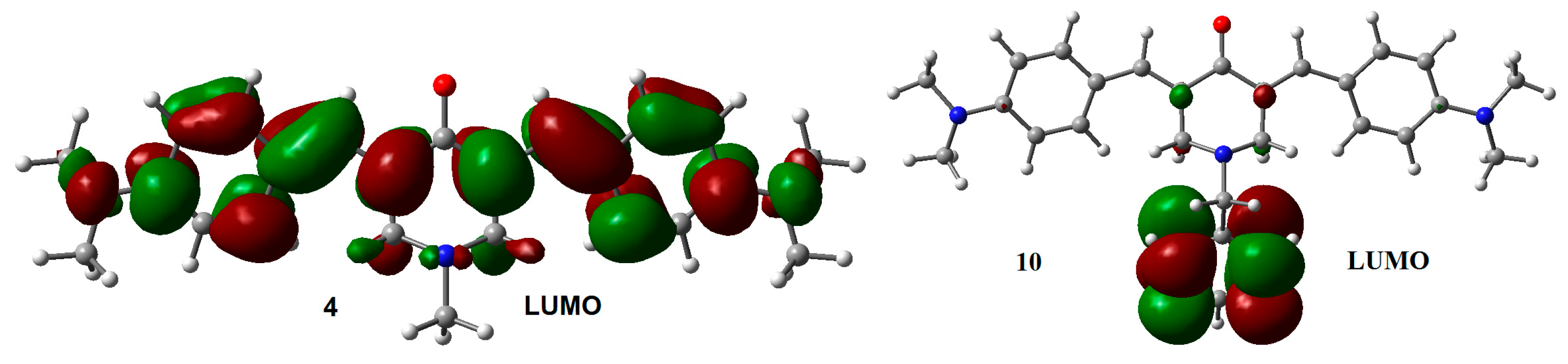{kind=link}
| Compounds | HOMO | LUMO | GAPH-L | μ | η | ώ |
|---|---|---|---|---|---|---|
| 1 | −6.01 | −2.09 | 3.92 | −4.05 | 1.96 | 4.18 |
| 2 | −6.22 | −2.37 | 3.85 | −4.30 | 1.92 | 4.81 |
| 3 | −6.21 | −2.38 | 3.83 | −4.30 | 1.91 | 4.81 |
| 4 | −4.87 | −1.51 | 3.36 | −3.19 | 1.68 | 3.03 |
| 5 | −6.54 | −2.92 | 3.62 | −4.73 | 1.81 | 6.18 |
| 6 | −6.36 | −2.59 | 3.84 | −4.48 | 1.89 | 5.31 |
| 7 | −5.91 | −2.06 | 3.85 | −3.99 | 1.92 | 4.14 |
| 8 | −6.11 | −2.34 | 3.77 | −4.22 | 1.88 | 4.74 |
| 9 | −6.11 | −2.34 | 3.77 | −4.22 | 1.88 | 4.74 |
| 10 | −4.87 | −1.50 | 3.37 | −3.18 | 1.68 | 3.01 |
| 11 | −6.41 | −2.88 | 3.53 | −4.65 | 1.77 | 6.11 |
| 12 | −6.22 | −2.55 | 3.67 | −4.38 | 1.84 | 5.21 |
| Compounds | AEA | VEA | VDE | |||
|---|---|---|---|---|---|---|
| Basis 1 a | Basis 2 b | Basis 1 a | Basis 2 b | Basis 1 a | Basis 2 b | |
| 1 | 0.932 | 1.398 | 0.791 | 1.295 | 1.059 | 1.556 |
| 2 | 1.263 | 1.669 | 1.124 | 1.571 | 1.391 | 1.829 |
| 3 | 1.280 | 1.707 | 1.144 | 1.606 | 1.406 | 1.859 |
| 4 | 0.578 | 0.977 | 0.361 | 0.839 | 0.851 | 1.319 |
| 5 | 1.856 | 1.902 | 1.718 | 2.181 | 1.998 | 2.454 |
| 6 | 1.512 | 2.002 | 1.332 | 1.856 | 1.685 | 2.202 |
| 7 | 0.939 | 1.407 | 0.798 | 1.303 | 1.062 | 1.560 |
| 8 | 1.264 | 1.672 | 1.122 | 1.571 | 1.386 | 1.826 |
| 9 | 1.282 | 1.709 | 1.142 | 1.606 | 1.400 | 1.855 |
| 10 | 0.531 | 0.970 | 0.379 | 0.858 | 0.690 | 1.167 |
| 11 | 1.848 | 2.271 | 1.706 | 2.170 | 1.985 | 2.443 |
| 12 | 1.510 | 1.999 | 1.324 | 1.849 | 1.676 | 2.193 |
| Molecule | Fukui Function for Nucleophilic Attack | Parr Function for Nucleophilic Attack | ||
|---|---|---|---|---|
| C7 | C9 | C7 | C9 | |
| 1 | 0.062 | 0.057 | 0.199 | 0.121 |
| 2 | 0.059 | 0.054 | 0.185 | 0.113 |
| 3 | 0.059 | 0.054 | 0.182 | 0.111 |
| 4 | 0.063 | 0.054 | 0.205 | 0.124 |
| 5 | 0.049 | 0.047 | 0.129 | 0.082 |
| 6 | 0.056 | 0.053 | 0.160 | 0.099 |
| 7 | 0.061 | 0.056 | 0.198 | 0.121 |
| 8 | 0.058 | 0.054 | 0.183 | 0.113 |
| 9 | 0.058 | 0.053 | 0.180 | 0.112 |
| 10 | 0.061 | 0.053 | 0.213 | 0.130 |
| 11 | 0.047 | 0.046 | 0.127 | 0.083 |
| 12 | 0.055 | 0.053 | 0.158 | 0.099 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Cifuentes, M.; Weiss-López, B.; Araya-Maturana, R. A Computational Study of Structure and Reactivity of N-Substitued-4-Piperidones Curcumin Analogues and Their Radical Anions. Molecules 2016, 21, 1658. https://doi.org/10.3390/molecules21121658
Martínez-Cifuentes M, Weiss-López B, Araya-Maturana R. A Computational Study of Structure and Reactivity of N-Substitued-4-Piperidones Curcumin Analogues and Their Radical Anions. Molecules. 2016; 21(12):1658. https://doi.org/10.3390/molecules21121658
Chicago/Turabian StyleMartínez-Cifuentes, Maximiliano, Boris Weiss-López, and Ramiro Araya-Maturana. 2016. "A Computational Study of Structure and Reactivity of N-Substitued-4-Piperidones Curcumin Analogues and Their Radical Anions" Molecules 21, no. 12: 1658. https://doi.org/10.3390/molecules21121658
APA StyleMartínez-Cifuentes, M., Weiss-López, B., & Araya-Maturana, R. (2016). A Computational Study of Structure and Reactivity of N-Substitued-4-Piperidones Curcumin Analogues and Their Radical Anions. Molecules, 21(12), 1658. https://doi.org/10.3390/molecules21121658