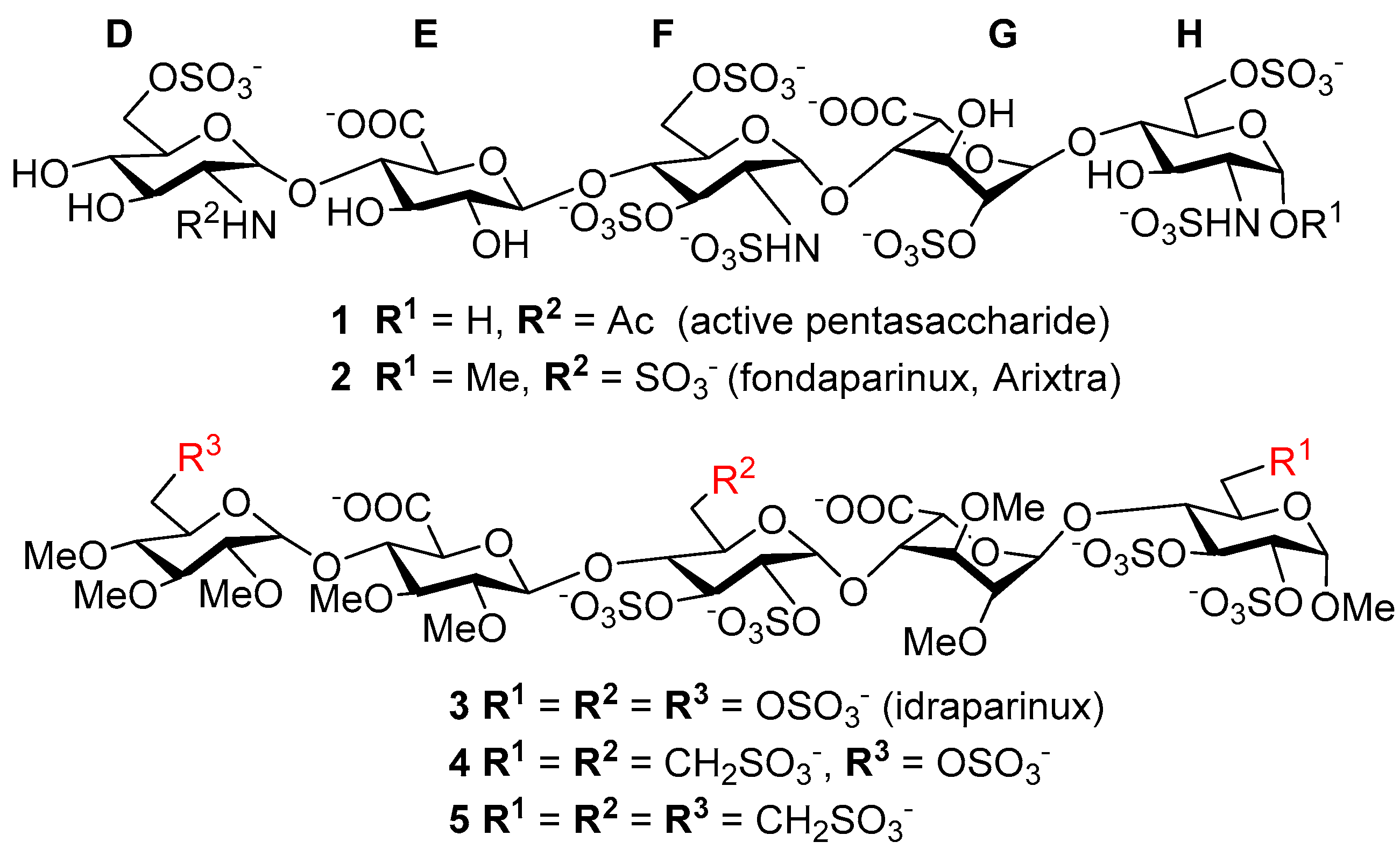
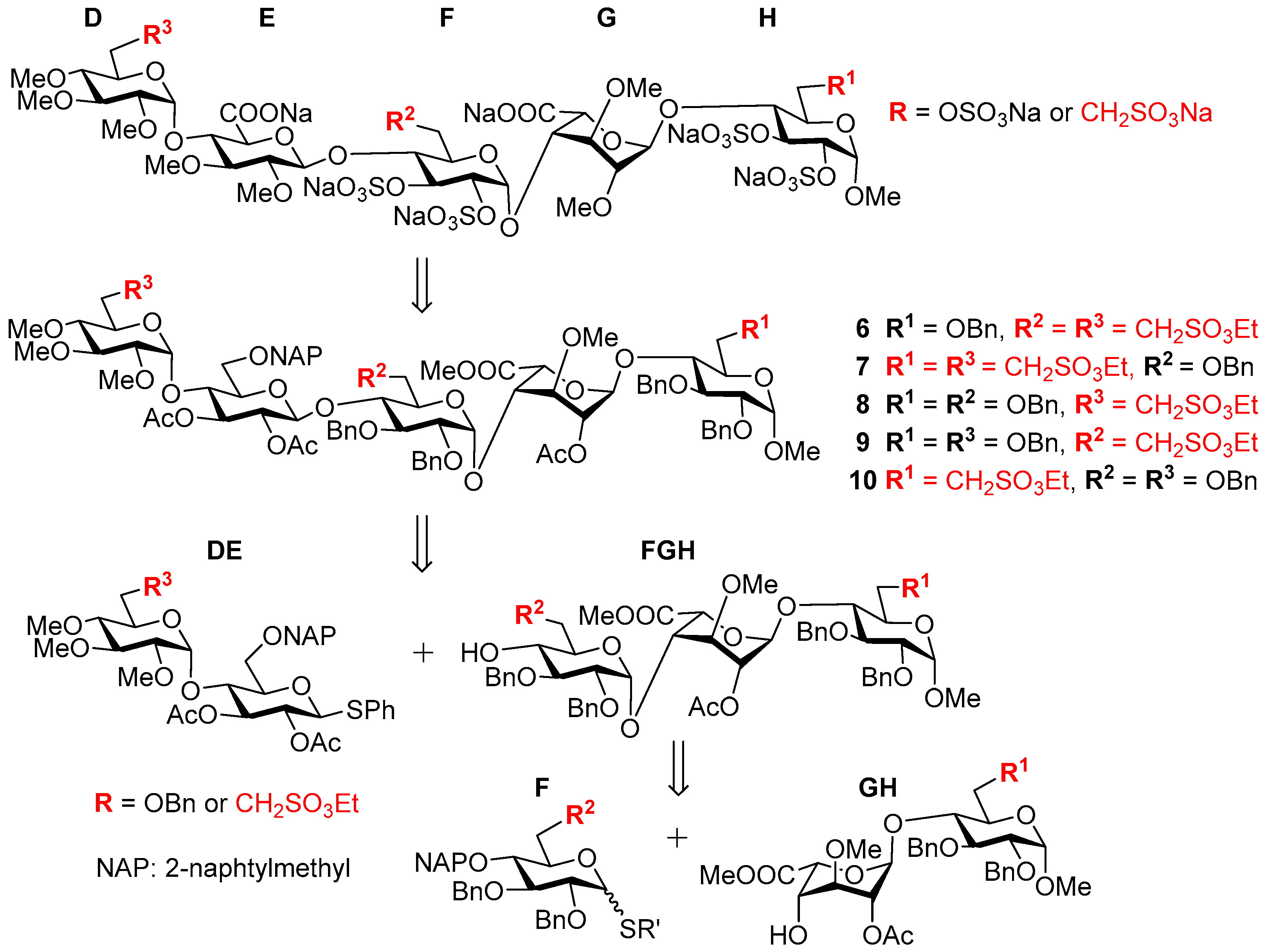
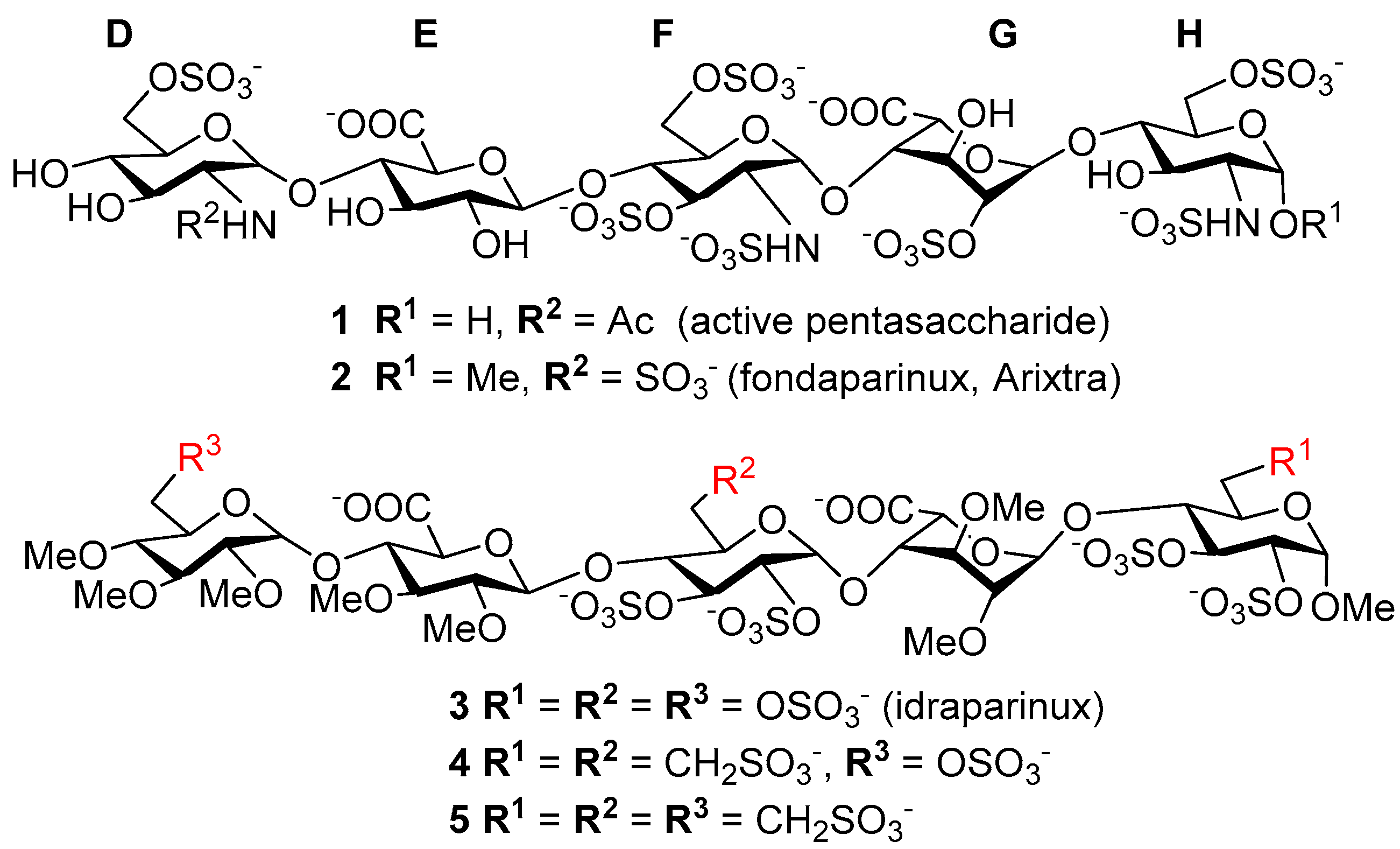
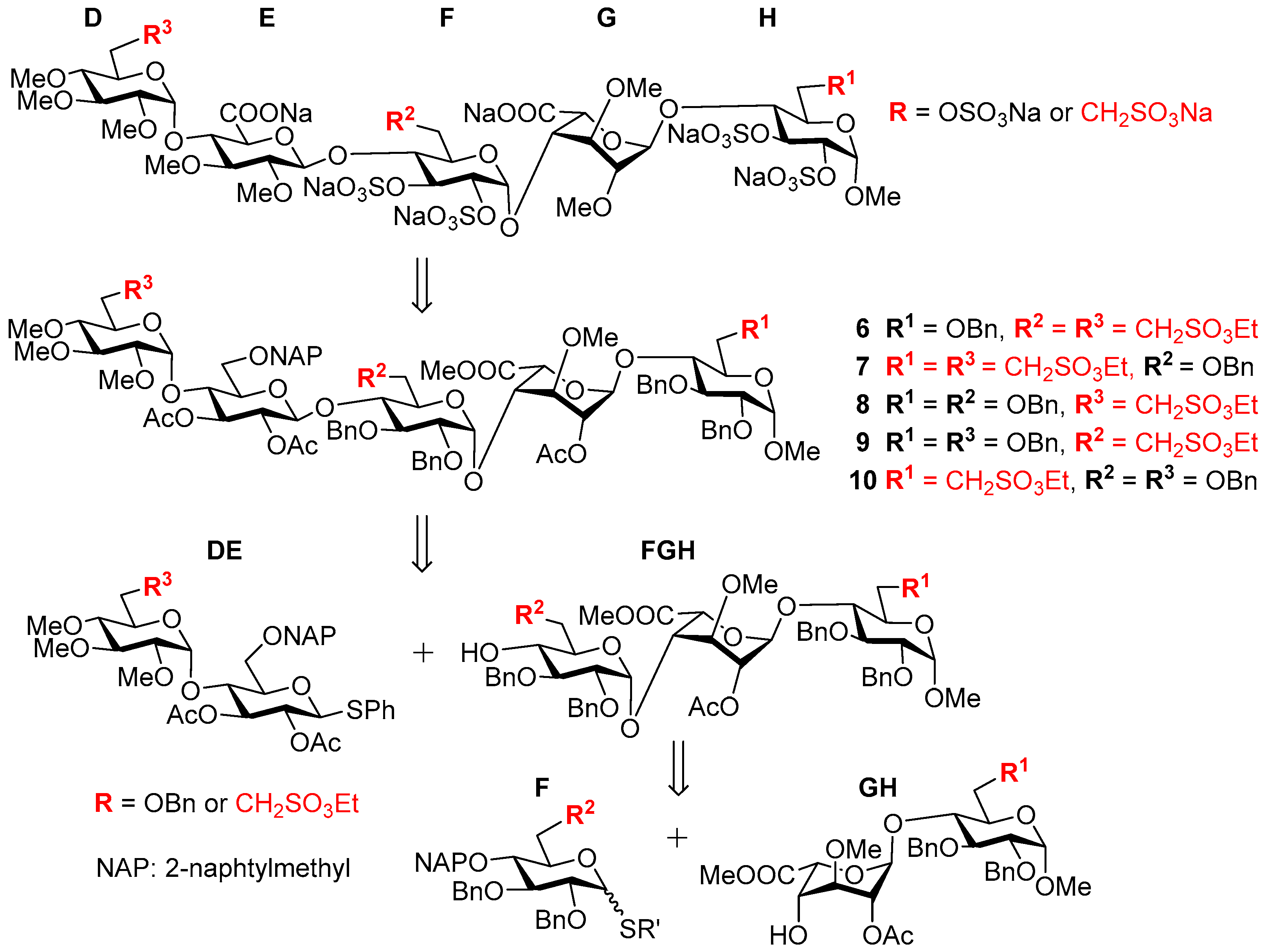
3.6. Product Characterization
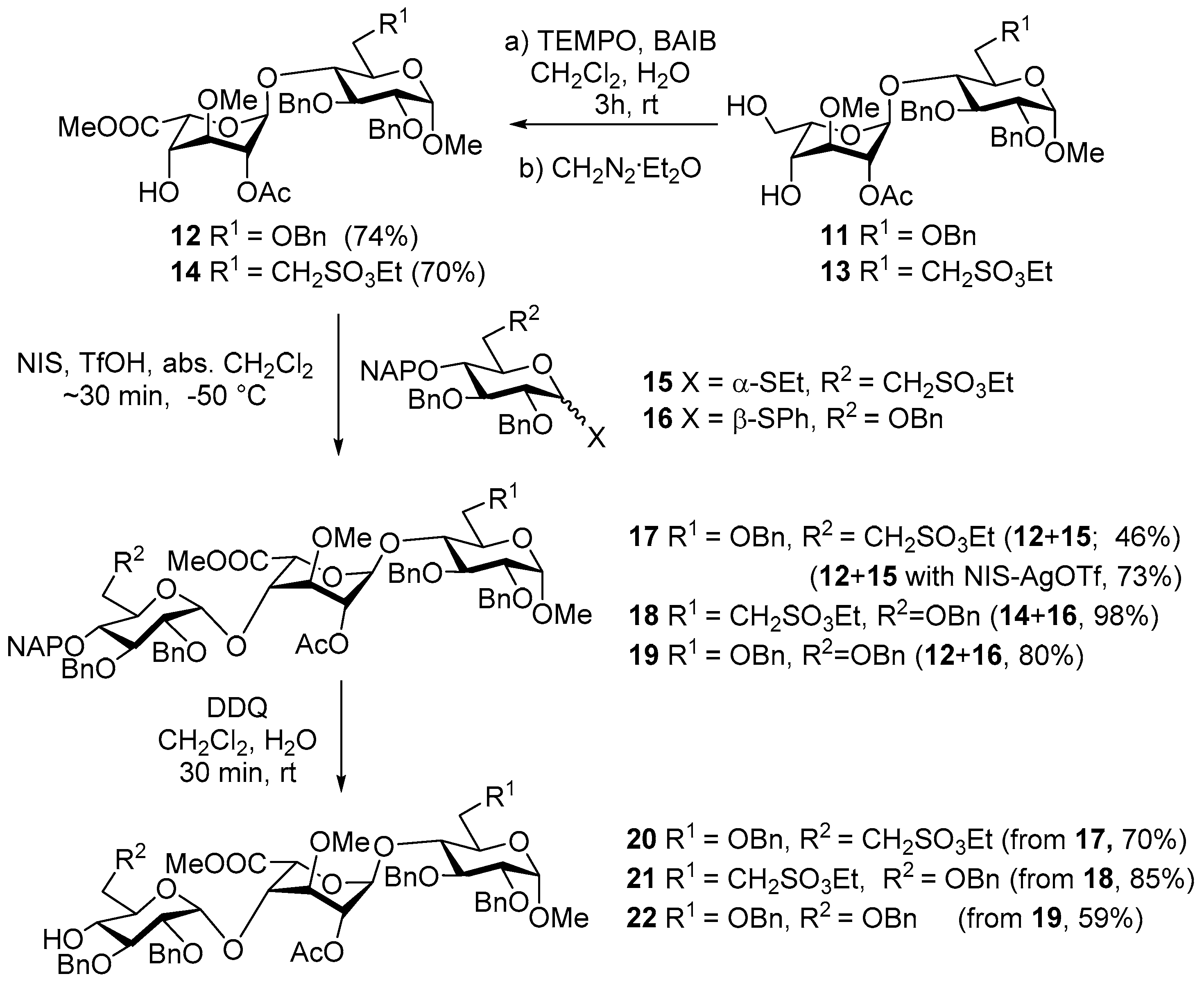
Methyl [methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate](1→4)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (
12). The starting material
11 [
18] (3.20 g, 4.7 mmol) was oxidized according to general method A. The crude uronic acid was dissolved in THF (4.5 mL) and treated with ethereal diazomethane at 0 °C. After complete disappearance of the uronic acid, the mixture was concentrated. The crude product was purified by column chromatography to give
12 (2.47 g, 74%) as a colourless syrup;
Rf = 0.36 (7:3
n-hexane/acetone); [α]
d −2.67 (
c 1.00, CHCl
3); IR ν
max (KBr): 3480, 3474, 3031, 2936, 2902, 1744, 1633, 1496, 1454, 1372, 1225, 1167, 1103, 1045, 911, 890, 854, 740, 700, 605 cm
−1;
1H-NMR (360 MHz, CDCl
3) δ 7.37–7.20 (m, 15H, arom.), 5.08 (s, 1H), 4.98 (d,
J = 11.4 Hz, 2H), 4.87 (s, 1H), 4.81 (d,
J = 11.4 Hz, 1H, PhC
H2), 4.67 (d,
J = 12.0 Hz, 1H, PhC
H2), 4.56–4.49 (m, 4H), 3.99–3.80 (m, 3H), 3.78–3.70 (m, 2H), 3.67–3.62 (m, 1H), 3.57 (dd,
J = 9.4, 3.6 Hz, 1H), 3.50–3.44 (m, 1H), 3.47, 3.40, 3.34 (3s, 9H, 3 × C
H3), 2.68 (d,
J = 11.8 Hz, 1H, OH), 2.01 (s, 3H, COC
H3) ppm;
13C-NMR (91 MHz, CDCl
3) δ 169.7, 169.2 (COOCH
3, COCH
3), 139.0, 138.0, 138.0 (3C,
Cq arom.), 128.4, 128.4, 128.2, 128.1, 127.9, 127.6, 127.6, 127.2, 127.0 (15C, arom.), 98.0, 97.6 (C-1-H, C-1-G), 80.3, 79.7, 76.8, 74.7, 70.1, 68.0, 67.2, 67.1 (C-2-G, C-2-H, C-3-G, C-3-H, C 4 G, C-4-H, C-5-G, C-5-H), 74.8, 73.4, 73.3 (3 × Ph
CH
2), 68.5 (C-6-H), 58.1 (O
CH
3, C-3-G), 55.2 (O
CH
3, C-1-H), 51.9 (COO
CH
3), 21.00 (CO
CH
3) ppm; MALDI-TOF MS:
m/
z 733.16 [M + Na]
+ (Calcd. 733.28); Anal. Calcd. for C
38H
46O
13 (710.29): C, 64.21; H, 6.52; O, 29.26. Found: C, 64.28; H, 6.55.
Methyl [2,3-di-O-Benzyl-4-O-(2-naphthyl)methyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (17)
I. To a solution of acceptor
12 (2.80 g, 3.94 mmol) and donor
15 [
16] (3.85 g, 5.91 mmol) in dry CH
2Cl
2 (20 mL), 4 Å molecular sieves (0.50 g) were added. The stirred mixture was cooled to −60 °C under argon and activated by method B. The reaction mixture was allowed to warm up to −50 °C in 1 h. The crude product was purified by column chromatography (7:3
n-hexane/EtOAc) to give
17 (2.35 g, 46%). Unreacted
12 (1.11 g, 28%) was recovered as a colourless syrup.
II. To a solution of acceptor 12 (1.11 g, 1.41 mmol) and donor 15 (1.37 g, 2.11 mmol) in dry CH2Cl2 (20 mL), 4 Å molecular sieves (0.50 g) were added. The stirred mixture was cooled to −60 °C under argon and activated by method C. The reaction mixture was allowed to warm up to −50 °C for 1 h. The crude product was purified by column chromatography (7:3 n-hexane/EtOAc) to give 17 (1.47 g, 73%) as a white foam; Rf = 0.34 (6:4 n-hexane/EtOAc); [α]d +11.30 (c 0.81, CHCl3); IR νmax (KBr): 3447, 3087, 3061, 3030, 2933, 1763, 1737, 1636, 1604, 1497, 1455, 1369, 1357, 1238, 1169, 1107, 1028, 1002, 917, 857 cm−1; 1H-NMR (360 MHz, CDCl3) δ 7.83–7.66 (m, 4H, arom.), 7.49–7.42 (m, 2H, arom.), 7.39–7.16 (m, 26H, arom.), 5.13 (s, 1H), 5.03 (d, J = 11.3 Hz, 1H, ArCH2), 4.93 (m, 2H), 4.85–4.72 (m, 7H), 4.66 (t, J = 12.6 Hz, 2H), 4.60–4.54 (m, 3H), 4.12 (q, J = 7.1 Hz, 2H, SO3CH2CH3), 3.96–3.61 (m, 9H), 3.54 (dd, J = 9.4, 3.5 Hz, 1H), 3.44–3.34 (m, 1H), 3.41, 3.36, 3.34 (3s, 9H, 3 × OCH3), 3.24–3.05 (m, 3H), 2.36–2.24 (m, 1H, H-7a), 2.00–1.83 (m, 4H, COCH3, H-7b), 1.25 (t, J = 7.1 Hz, 3H, SO3CH2CH3) ppm; 13C-NMR (91 MHz, CDCl3) δ 170.1, 169.4 (2 × CO), 139.1, 138.4, 138.1, 138.1, 138.1, 135.6, 133.3, 133.0 (8C, Cq arom.), 128.6, 128.5, 128.4, 128.4, 128.2, 128.2, 128.0, 127.9, 127.9, 127.7, 127.6, 127.3, 127.1, 126.5, 126.2, 126.0, 125.7 (32C, arom.), 98.7, 98.0, 97.7 (3 × C-1), 81.5, 81.2, 80.4, 80.2, 79.8, 76.5, 74.7, 74.7, 70.1, 69.5, 68.3, 67.7 (12C, skeleton carbons), 75.5, 75.1, 74.9, 73.6, 73.4, 73.4 (6 × ArCH2), 68.5 (C-6-F), 66.2 (SO3CH2CH3), 58.3, 55.2, 51.8 (3 × OCH3), 46.5 (C-7-H), 25.9 (C-6-H), 21.1 (COCH3), 15.1 (SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1321.57 [M + Na]+ (Calcd. 1321.50); Anal. Calcd. for C72H82O20S (1298.51): C, 66.55; H, 6.36; O, 24.62; S, 2.47. Found: C, 66.62; H, 6.40; S, 2.45.
Methyl [2,3,6-tri-O-benzyl-4-O-(2-naphthyl)methyl-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methy-α-l-idopyranosyl)uronate]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside 18. To a solution of acceptor (
14) [
17] (620 mg, 0.85 mmol) and donor
16 [
17] (874 mg, 1.28 mmol) in dry CH
2Cl
2 (20 mL) 4 Å molecular sieves (0.50 g) were added. The stirred mixture was cooled to −60 °C under argon and activated by method
B. The reaction mixture was allowed to warm up to −50 °C for 1 h. The crude product was purified by column chromatography (7:3
n-hexane/EtOAc) to give
18 (1.07 g, 98%) as a white foam;
Rf = 0.63 (1:1
n-hexane/ EtOAc); [α]
d +6.62 (
c 0.35, CHCl
3); IR ν
max (KBr): 3446, 3087, 3061, 3030, 2931, 2869, 1739, 1636, 1604, 1497, 1455, 1362, 1236, 1165, 1107, 1045, 1028, 1004, 917, 857, 818 cm
−1;
1H-NMR (360 MHz, CDCl
3) δ 7.85–7.67 (m, 3H, arom.), 7.55 (s, 1H, arom), 7.49–7.41 (m, 2H, arom.), 7.38–7.14 (m, 26H, arom.), 5.25 (s, 1H), 4.99–4.35 (m, 16H), 4.27 (q,
J = 7.1 Hz, 2H, SO
3C
H2CH
3), 3.97–3.63 (m, 7H), 3.62–3.24 (m, 4H), 3.47, 3.40, 3.32 (3 × OC
H3), 3.16–3.04 (m, 1H), 2.44–2.32 (m, 1H, H-7
a), 2.03 (s, 3H, COC
H3), 2.01–1.85 (m, 1H, H-7
b), 1.37 (t, 3H, SO
3CH
2C
H3) ppm;
13C-NMR (91 MHz, CDCl
3) δ 170.2, 169.6 (2 ×
CO), 139.0, 138.7, 138.3, 138.1, 138.0, 136.3, 133.3, 132.9 (8C,
Cq arom.), 128.5, 128.4, 128.2, 128.1, 128.0, 127.9, 127.7, 127.2, 126.1, 125.9, 125.8, 125.6 (32C, arom.), 99.6, 98.0, 98.0 (3 × C-1), 81.8, 80.3, 80.0, 79.5, 78.6, 77.0, 75.1, 71.4, 69.2, 68.4, 68.1, 57.9 (12C, skeleton carbons), 75.5, 75.2, 74.7, 73.5, 73.5, 73.4 (6 × Ar
CH
2), 68.1 (C-6-H), 66.2 (SO
3CH
2CH
3), 58.7, 55.6, 51.9 (3 × O
CH
3), 46.7 (C-7-F), 26.0 (C-6-F), 21.1 (CO
CH
3), 15.2 (SO
3CH
2CH
3) ppm; MALDI-TOF MS:
m/
z 1321.57 [M + Na]
+ (Calcd. 1321.50); Anal. Calcd. for C
72H
82O
20S (1298.51): C, 66.55; H, 6.36; O, 24.62; S, 2.47. Found: C, 66.50; H, 6.42; S, 2.51.
Methyl [2,3,6-tri-O-benzyl-4-O-(2-naphthyl)methyl-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (19). To a solution of acceptor 12 (1.50 g, 2.10 mmol) and donor 16 (2.16 g, 3.16 mmol) in dry CH2Cl2 (20 mL) 4 Å molecular sieves (0.50 g) were added. The stirred mixture was cooled to −60 °C under argon and activated by method B. The reaction mixture was allowed to warm up to −50 °C for 30 min. The crude product was purified by column chromatography (7:3 n-hexane/EtOAc) to give 18 (2.41 g, 80%) as a white foam. Rf = 0.47 (6:4 n-hexane/EtOAc).
Methyl [2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (20). Compound 17 (2.50 g, 1.92 mmol) was converted to 20 according to general method D. The crude product was purified by column chromatography (7:3 n-hexane/EtOAc) to give compound 20 (1.55 g, 70%) as a white foam; Rf = 0.40 (1:1 n-hexane/EtOAc); [α]d +13.46 (c 0.56, CHCl3); IR νmax (KBr): 3502, 3088, 3063, 3031, 2933, 1738, 1629, 1497, 1455, 1370, 1356, 1236, 1168, 1108, 1047, 1028, 917, 820, 740, 698, 606, 545, 466 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.38–7.19 (m, 25H, arom.), 5.15 (s, 1H, H-1-G), 4.93 (d, J = 11.4 Hz, 2H, BnCH2), 4.85–4.76 (m, 4H, H-2-G, H-5-G, H-1-F, BnCH2), 4.75–4.61 (m, 4H, BnCH2), 4.55 (m, 3H, H-1-H, BnCH2), 4.24 (q, J = 7.1 Hz, 2H, SO3CH2CH3), 3.94–3.79 (m, 3H, H-3-H, H-4-G), 3.77–3.59 (m, 6H, H-3-G, H-3-F, H-5-F, H-4-H, H-6a,b-H), 3.54 (dd, J = 9.3, 3.5 Hz, 1H, H-2-H), 3.43–3.31 (m, 2H, H-2-F, H-5-H), 3.40, 3.37, 3.34 (3s, 3 × CH3) 3.28–3.09 (m, 3H, H-4-F, H-7a,b), 2.48 (s, 1H, OH), 2.33–2.21 (m, 1H, H-6a-F), 1.93 (s, 3H, COCH3), 1.98–1.84 (m, 1H, H-6b-F), 1.35 (t, J = 7.1 Hz, 3H, SO3CH2CH3) ppm; 13C-NMR (101 MHz, CDCl3) δ 170.0, 169.3 (2 × CO), 139.1, 138.5, 138.1, 138.0, 137.9 (5C, Cq arom.), 128.6, 128.5, 128.4, 128.3, 128.1, 128.1, 128.0, 127.9, 127.8, 127.8, 127.7, 127.5, 127.3, 127.1 (25C, arom.), 98.5 (C-1-F), 98.0 (C-1-H), 97.7 (C-1-G), 80.6 (C-3-F), 80.1 (C-2-H), 79.9 (C-2-F), 79.8 (C-3-H), 79.8 (C-4-G), 76.5 (C-5-F), 74.8 (C-2-G), 74.4 (C-4-F), 75.0, 74.9, 73.4, 73.3, 73.2 (5 × PhCH2), 70.1 (C-4-H), 69.8 (C-5-H), 68.5 (C-3-F), 68.4 (C-6-H), 68.0 (C-5-G), 66.2 (SO3CH2CH3), 58.4, 55.2, 51.7 (3 × OCH3), 46.3 (C-7-F), 25.8 (C-6-F), 21.0 (COCH3), 15.1 (SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1181.59 [M + Na]+ (Calcd. 1181.44); Anal. Calcd. for C61H74O20S (1158.45): C, 63.20; H, 6.43; O, 27.60; S, 2.77. Found: C, 63.25; H, 6.37; S, 2.72.
Methyl [2,3,6-tri-O-benzyl-α-d-glucopyranosyl]-(1→4)-[methyl(2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (21). Compound 18 (1.06 g, 0.83 mmol) was converted to 21 according to general method D. The crude product was purified by column chromatography (65:35 n-hexane/acetone) to give compound 21 (807 mg, 85%) as a white foam; Rf = 0.38 (65:35 n-hexane/acetone); [α]d +14.44 (c 0.04, CHCl3); IR νmax (KBr): 3481, 3063, 3031, 2929, 1740, 1626, 1497, 1455, 1370, 1234, 1167, 1105, 1046, 1028, 926, 820, 739, 698, 606, 548, 460, 418 cm−1; 1H-NMR (400 MHz, CDCl3) δ 7.30–7.08 (m, 25H, arom.), 5.17 (d, J = 3.3 Hz, 1H, H-1-G), 4.86–4.65 (m, 3H, H-1-F, H-2-G, H-5-G, BnCH2), 4.59 (m, 3H, BnCH2), 4.50–4.34 (m, 4H, H-1-H, BnCH2), 4.18 (q, J = 7.1 Hz, 2H, SO3CH2CH3), 3.85–3.14 (m, 13H, skeleton protons), 3.36, 3.29, 3.23 (3s, 9H, OCH3), 3.05–2.95 (m, 1H, H-7b-H), 2.46 (s, 1H, OH), 2.34–2.24 (m, 1H, H-6a-H), 1.93 (s, 3H, COCH3), 1.89–1.76 (m, 1H, H-6b-H), 1.28 (t, J = 7.1 Hz, 3H, SO3CH2CH3) ppm; 13C-NMR (101 MHz, CDCl3) δ 170.2, 169.6 (2 × CO), 139.0, 138.8, 138.2, 138.1, 138.0 (5C, Cq arom.), 128.6, 128.5, 128.4, 128.2, 128.0, 127.9, 127.8, 127.8, 127.3 (25C, arom.), 99.5 (C-1-F), 98.1 (C-1-H), 98.0 (C-1-G), 81.1, 80.2, 79.5, 79.5, 78.9, 77.0, 75.0, 71.2, 70.9, 69.8 (12C, skeleton carbons), 75.3, 75.1,73.7, 73.6, 73.3 (5 × PhCH2), 69.3 (C-6-F), 69.0, 68.2, 66.3 (SO3CH2CH3), 58.9, 55.6, 51.9 (3 × OCH3), 46.8 (C-7-H), 25.9 (C-6-H), 21.1 (COCH3), 15.2 (SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1181.64 [M + Na]+ (Calcd. 1181.44); Anal. Calcd. for C61H74O20S (1158.45): C, 63.20; H, 6.43; O, 27.60; S, 2.77. Found: C, 63.34; H, 6.47; S, 2.79.
Methyl [2,3,6-tri-O-benzyl-α-d-glucopyranosyl]-(1→4)-[methyl-(2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (
22) [
15]. Compound
19 (2.4 g, 1.87 mmol) was converted to
22 according to general method
D. The crude product was purified by column chromatography (7:3
n-hexane/EtOAc) to give compound
22 (1.26 g, 59%) as a white foam;
Rf = 0.33 (6:4
n-hexane/EtOAc); [α]
d +8.41 (
c 0.62, CHCl
3) (lit. [
15] [α]
d +2.3 (
c 0.10, CHCl
3); IR ν
max (KBr): 3087, 3062, 3031, 2932, 2906, 1739, 1605, 1497, 1455, 1371, 1237, 1104, 1046, 1028, 908, 738, 697, 606, 538, 459, 419 cm
−1. The NMR spectroscopic and analytical data of
22 are consistent with those given in the literature [
15].
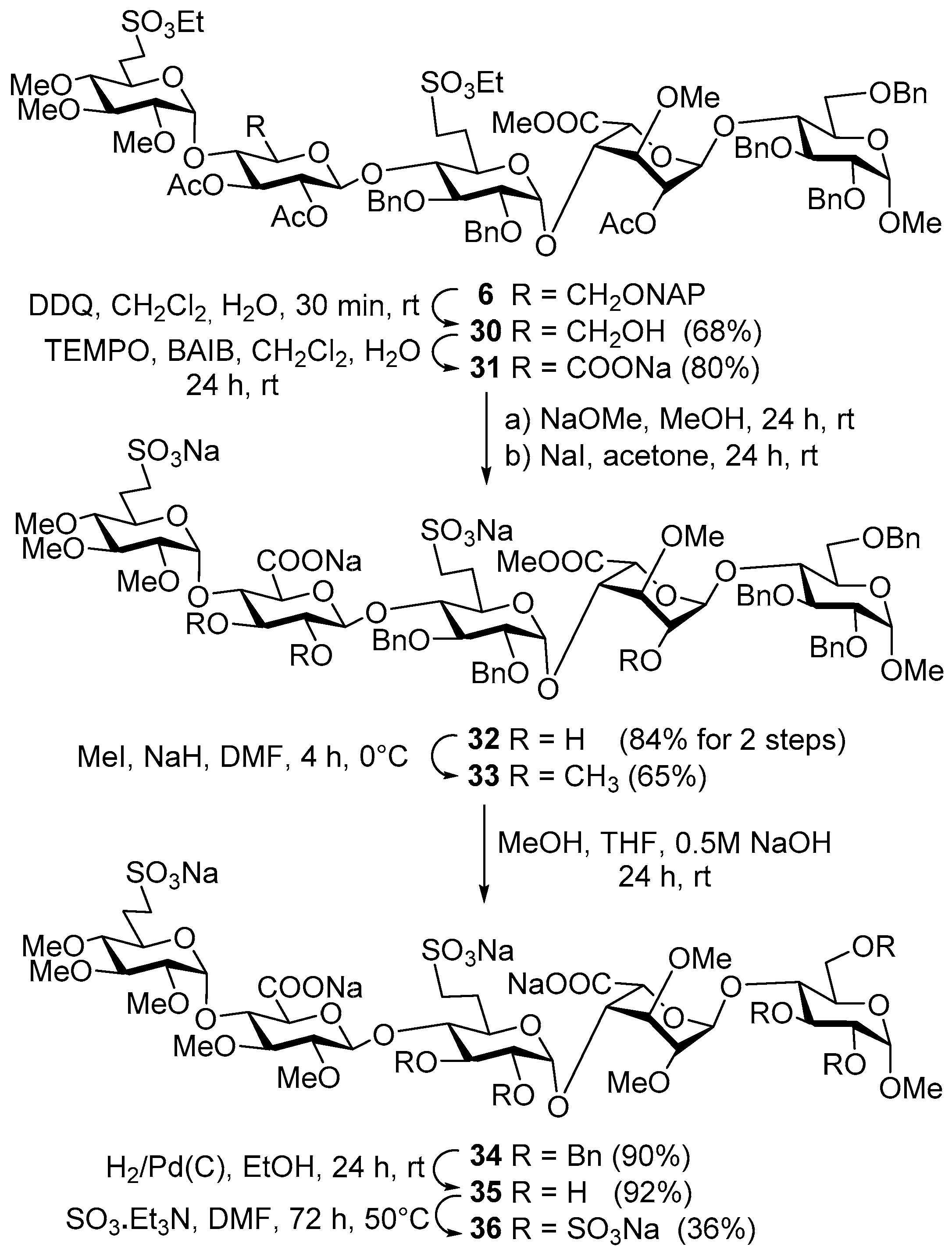
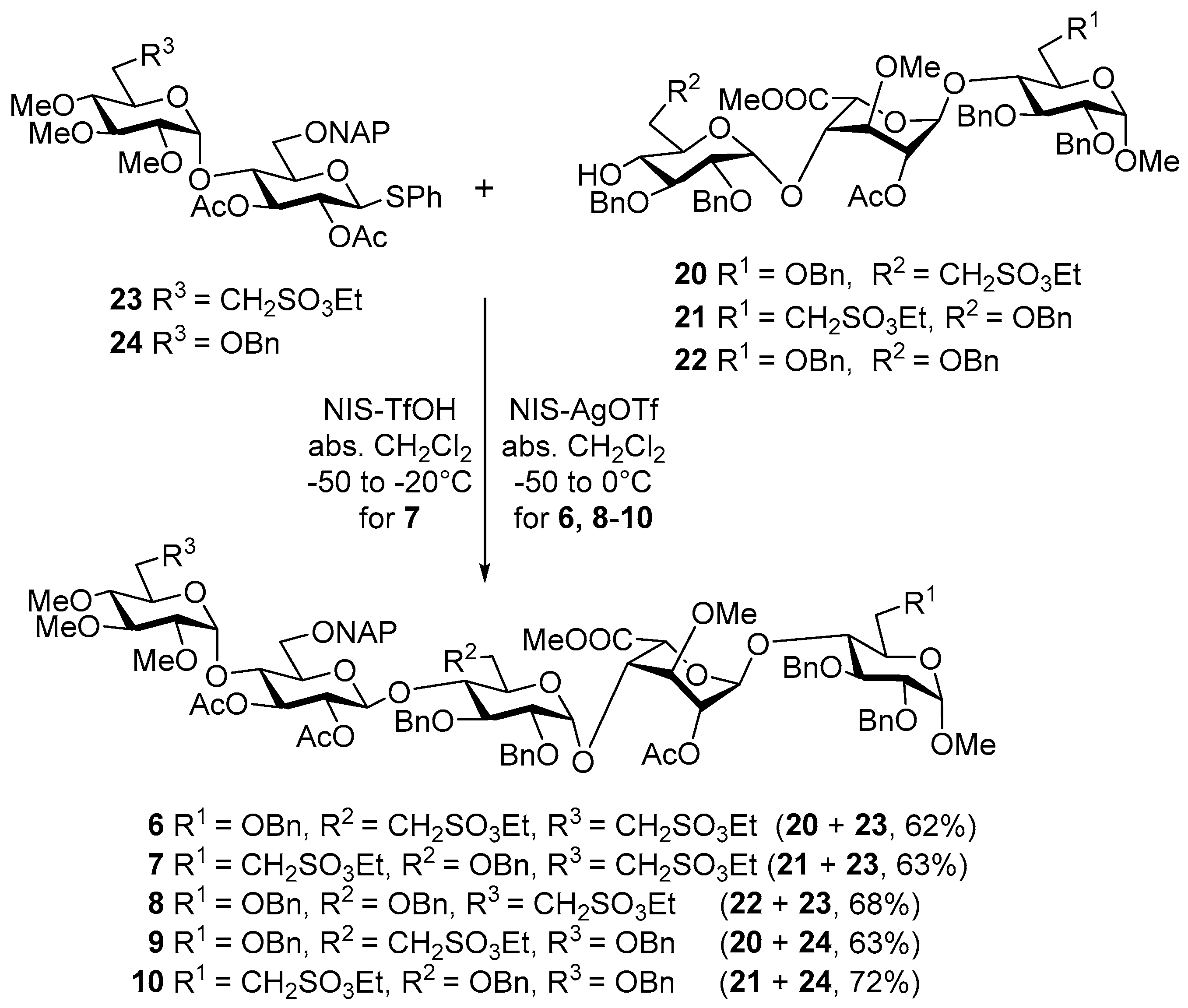
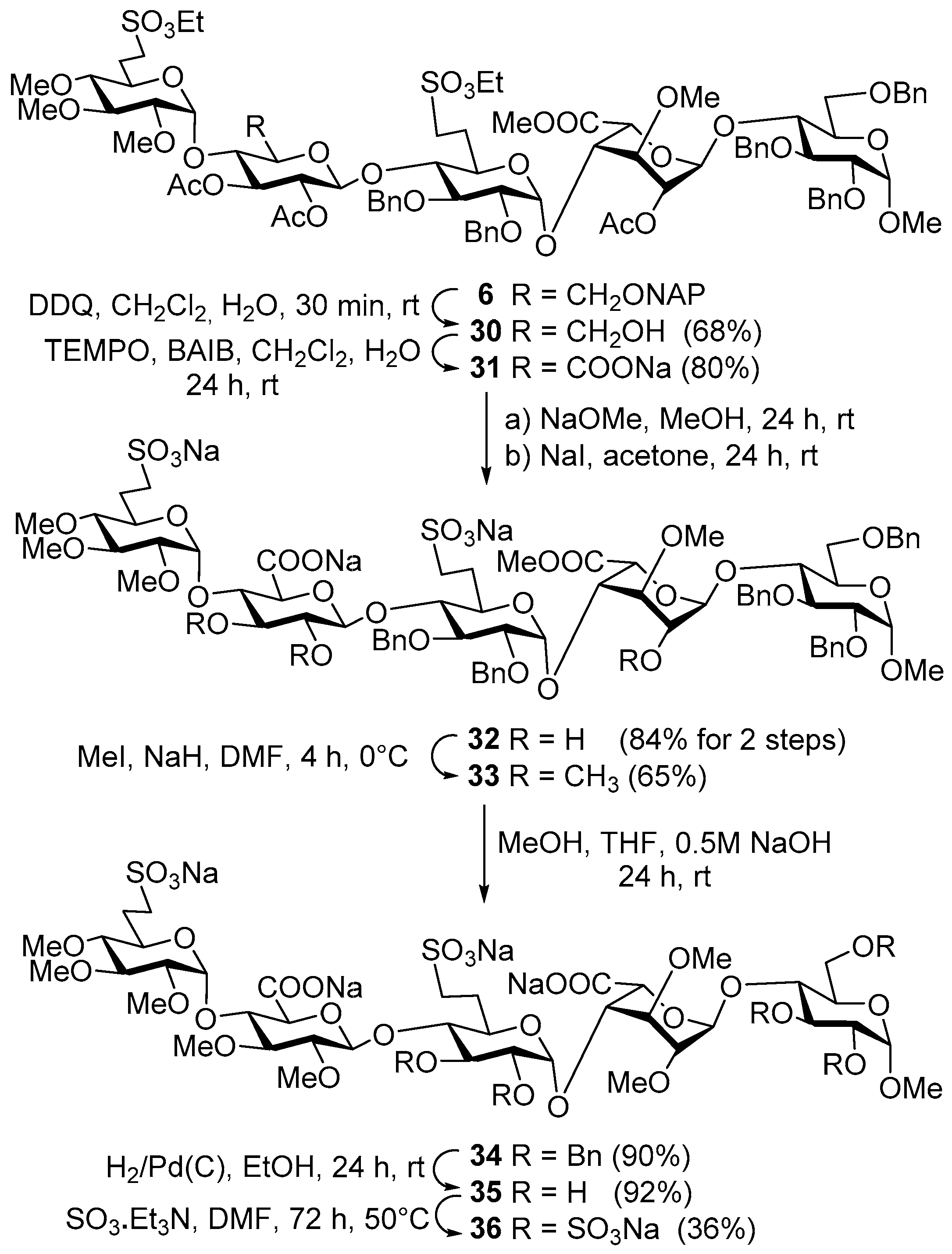
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucoopiranosyl]-(1→4)-[2,3-di-O-acetyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-2,3,6-tri-O-benzyl-α-d-gluco-pyranoside (6). To a solution of acceptor 20 (630 mg, 0.54 mmol) and donor 23 (660 mg, 0.82 mmol) in dry CH2Cl2 (20 mL), 4 Å molecular sieves (0.50 g) were added. The stirred mixture was cooled to −40 °C under argon and activated by method C. The reaction mixture was allowed to warm up to −15 °C for 4 h. The crude product was purified by column chromatography (6:4 n-hexane/EtOAc) to give 6 (626 mg, 62%) as a colourless syrup; Rf = 0.26 (1:1 n-hexane/EtOAc); [α]d +23.69 (c 0.14, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.84–7.75 (m, 3H, arom.), 7.65 (s, 1H, arom.), 7.48–7.40 (m, 2H, arom.), 7.37–7.13 (m, 26H, arom.), 5.18 (t, J = 9.2 Hz, 1H, H-3-E), 5.11 (s, 1H, H-1-G), 4.99–4.46 (m, 19H, H-1-D, H-2-E, H-5-G, H-2-G, H-1-E, H-1-H, H-1-F, 12 × ArCH2), 4.27, 4.07 (2q, 4H, SO3CH2CH3), 3.93–3.77 (m, 5H, H-4-H, H-4-E, H-3-F, H-4-G, H-3-H), 3.76–3.59 (m, 5H, H-5-H, H-5-F, H-6a,b-H, H-3-G), 3.58–3.17 (m, 9H, H-2-F, H-6a,b-E, H-5-D, H-4-F, H-2-H, H-3-D, H-5-E, H-7a-F), 3.52, 3.48, 3.40, 3.36, 3.33, 3.31 (6s, 18H, 6 × OCH3), 3.18–3.03 (m, 2H, H-7b-F, H-7a-D), 2.93–2.81 (m, 2H, H-2-D, H-7b-D), 2.65 (t, J = 9.2 Hz, 1H, H-4-D), 2.33–2.21 (m, 1H, H-6a-F), 2.21–2.08 (m, 1H, H-6a-D), 2.05, 1.99, 1.94 (3s, 9H, 3 × COCH3), 1.86–1.71 (m, 2H, H-6b-D, H-6b-F), 1.38, (t, J = 7.1 Hz, 3H, SO3CH2CH3), 1.25 (t, J = 6.8 Hz, 3H, SO3CH2CH3) ppm; 13C-NMR (101 MHz, CDCl3) δ 170.1, 169.8, 169.8, 169.3 (4 × CO), 139.1, 138.9, 138.0, 137.9, 137.7, 135.3, 133.2, 132.9 (8C, Cq arom.), 128.4, 128.3, 128.2, 128.1, 128.0, 128.0, 127.9, 127.8, 127.8, 127.6, 127.5, 127.1, 127.0, 126.4, 126.1, 125.8, 125.6 (32C, arom.), 101.1 (C-1-H), 98.0 (C-1-E), 97.8 (C-1-F), 97.4 (C-1-G), 96.8 (C-1-D), 83.2 (C-4-D), 82.3 (C-3-D), 82.1 (C-4-F), 81.9 (C-2-D), 80.0 (C-2-F), 79.7 (C-2-H), 79.6 (C-4-G), 79.4 (C-3-F), 76.0 (C-3-G), 75.1 (C-5-E), 74.7, 74.3, 73.6, 73.4, 73.2, 73.2, (6 × ArCH2), 74.5 (C-3-E), 74.5 (C-4-H), 74.4 (C-4-E), 74.0 (C-3-H), 72.5 (C-2-E), 70.0 (C-5-H), 69.2 (C-5-D), 68.9 (C-5-F), 68.3 (C-6-H), 68.2 (C-5-G), 67.7 (C-6-E), 67.3 (C-2-G), 66.2, 65.8 (2 × SO3CH2CH3), 60.5, 60.5, 58.9, 58.1, 55.1, 51.7 (6 × OCH3), 46.6 (C-7-D), 46.4 (C-7-F), 26.0 (C-6-D), 25.7 (C-6-F), 21.0, 20.8, 20.5 (3 × COCH3), 15.0, 15.0 (2 × SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1877.77 [M + Na]+ (Calcd. 1877.68); Anal. Calcd. for C94H118O34S2 (1854.69): C, 60.83; H, 6.41; O, 29.31; S, 3.46. Found: C, 60.69; H, 6.35; S, 3.41.
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-acetyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-[2,3,6-tri-O-benzyl-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (7). To a solution of acceptor 21 (720 mg, 0.62 mmol) and donor 23 (752 mg, 0.93 mmol) in dry CH2Cl2 (20 mL) 4 Å molecular sieves (0.50 g) were added. The stirred mixture was cooled to −50 °C under argon and activated by method B. The reaction mixture was allowed to warm up to −20 °C for 3 h. The crude product was purified by column chromatography (65:35 n-hexane/EtOAc) to give 7 (726 mg, 63%) as a colourless syrup; Rf = 0.21 (1:1 n-hexane/EtOAc); [α]d +32.21 (c 0.14, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.83–7.71 (m, 3H, arom.), 7.62 (s, 1H, arom.), 7.44 (m, 2H, arom.), 7.41–7.12 (m, 26H, arom.), 5.21 (d, J = 2.3 Hz, 1H, H-1-G), 5.10–4.99 (m, 1H, H-3-E, ArCH2), 4.96–4.76 (m, 6H, H-1-D, H-2-G, H-2-E, H-1-F, 2 × ArCH2), 4.75–4.63 (m, 5H, H-5-G, 4 × ArCH2), 4.60–4.38 (m, 7H, H-1-E, H-1-H, 5 × ArCH2), 4.29 (q, J = 7.1 Hz, 2H, SO3CH2CH3), 4.06 (q, J = 7.1 Hz, 2H, SO3CH2CH3), 3.95 (t, J = 9.4 Hz, 1H, H-4-H), 3.91–3.67 (m, 8H, H-4-G, H-4-E, H-6a-E, H-5-H, H-3-H, H-3-F, H-6a-F, H-5-F), 3.67–3.25 (m, 9H, H-3-G, H-6b-F, H-6b-E, H-4-F, H-5-D, H-2-H, H-2-F, H-3-D, H-7-H), 3.57, 3.53, 3.43, 3.41, 3.33 (5s, 15H, 5 × OCH3), 3.23–3.04 (m, 3H, H-5-E, H-7a-D, H-7a-H), 2.97–2.87 (m, 2H, H-2-D, H-7b-D), 2.69 (t, J = 9.2 Hz, 1H, H-4-D), 2.44–2.30 (m, 1H, H-6a-H), 2.30–2.14 (m, 1H, H-6a-D), 2.02, 1.96, 1.95 (3s, 9H, 3 × COCH3), 2.06–1.74 (m, 2H, H-6b-D, H-6b-H), 1.40 (t, J = 7.1 Hz, 3H, SO3CH2CH3), 1.23 (t, J = 7.4 Hz, 3H, SO3CH2CH3) ppm. 13C-NMR (101 MHz, CDCl3) δ 170.3, 170.0, 169.7, 169.6 (4 × CO), 139.4, 139.0, 138.3, 138.1, 137.6, 135.8, 133.4, 133.0 (8C, Cq arom.), 128.8, 128.6, 128.5, 128.4, 128.4, 128.2, 128.2, 128.1, 128.0, 127.9, 127.8, 127.6, 127.3, 126.4, 126.1, 125.9, 125.8 (32C, arom.), 99.8 (C-1-E), 99.6 (C-1-F), 98.0 (C-1-H), 97.8 (C-1-G), 96.9 (C-1-D), 83.5 (C-4-D), 82.7 (C-3-D), 82.2 (C-2-D), 80.3 (C-4-F), 79.7 (C-3-H), 79.4 (C-3-F), 79.3 (C-2-F), 78.6 (C-2-H), 76.7 (C-4-H), 76.5 (C-3-G), 75.3 (C-5-E), 74.9 (C-4-G), 74.9 (C-3-E), 74.9 (C-4-E), 72.5 (C-2-E), 71.1 (C-5-F), 69.5 (C-5-D), 69.3 (C-5-G), 68.2 (C-5-H), 68.2 (C-2-G), 75.3, 75.1, 73.9, 73.7, 73.6, 73.6 (6 × ArCH2), 68.1 (C-6-E), 67.4 (C-6-F), 66.3, 66.1 (2 × SO3CH2CH3), 60.8, 60.8, 59.2, 58.8, 55.6, 52.0 (6 × OCH3), 46.8, 46.8 (C-7-D, C-7-H), 26.2 (C-6-D), 26.0 (C-6-H), 21.1, 21.0, 20.8 (3 × COCH3), 15.2, 15.1 (2 × SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1877.77 [M + Na]+ (Calcd. 1877.68); Anal. Calcd. for C94H118O34S2 (1854.69): C, 60.83; H, 6.41; O, 29.31; S, 3.46. Found: C, 60.90; H, 6.44; S, 3.51.
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-acetyl-6-O-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-[2,3,6-tri-O-benzyl-α-d-glucopyranosyl]-(1→4)-[methyl-(2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (8). To a solution of acceptor 22 (1.18 g, 1.03 mmol) and donor 23 (1.25 g, 1.55 mmol) in dry CH2Cl2 (40 mL), 4 Å molecular sieves (1 g) were added. The stirred mixture was cooled to −50 °C under argon and activated by method C. The reaction mixture was allowed to warm up to 0 °C for 4 h. The crude product was purified by column chromatography (6:4 n-hexane/EtOAc) to give 8 (1.29 g, 68%) as a colourless syrup; Rf = 0.51 (94:6 CH2Cl2/acetone); [α]d +24.44 (c 0.05, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.86–7.71 (m, 3H, arom.), 7.62 (s, 1H, arom.), 7.44 (m, 2H, arom.), 7.40–7.11 (m, 31H, arom.), 5.15 (d, J = 2.7 Hz, 1H, H-1-G), 5.05 (m, 2H, H-3-E, ArCH2), 4.95–4.75 (m, 7H, H-1-D, H-2-E, H-1-F, H-2-G, 3 × ArCH2), 4.74–4.65 (m, 5H, H-5-G, 4 × ArCH2), 4.62–4.46 (m, 6H, H-1-H, 5 × ArCH2), 4.45–4.40 (m, 2H, H-1-E, ArCH2), 4.05 (q, J = 7.1 Hz, 2H, SO3CH2CH3), 3.95 (t, J = 9.4 Hz, 1H, H-4-F), 3.91–3.45 (m, 16H, H-4-G, H-4-H, H-4-E, H-3-H, H-5-F, H-6a,b-H, H-3-F, H-5-H, H-3-G, H-6a,b-E, H-6a,b-F, H-5-D, H-2-H), 3.57, 3.53, 3.41, 3.35, 3.34, 3.32 (6s, 18H, 6 × OCH3), 3.44–3.27 (m, 4H, H-2-F, H-3-D, H-5-E, H-7a-D), 2.97–2.85 (m, 2H, H-2-D, H-7b-D), 2.69 (t, J = 9.2 Hz, 1H, H-4-D), 2.26–2.15 (m, 1H, H-6a-D), 2.01, 1.91, 1.89 (3s, 9H, 3 × COCH3), 1.87–1.75 (m, 1H, H-6b-D), 1.23 (t, J = 7.1 Hz, 3H, SO3CH2CH3) ppm; 13C-NMR (91 MHz, CDCl3) δ 170.2, 170.0, 169.7, 169.6 (4 × CO), 139.5, 139.2, 138.3, 138.3, 138.1, 137.7, 135.8, 133.4, 133.0 (9C, Cq arom.), 128.8, 128.5, 128.4, 128.2, 128.2, 128.0, 127.9, 127.8, 127.8, 127.6, 127.5, 127.3, 127.1, 126.4, 126.1, 125.9, 125.8 (37C, arom.), 99.8 (C-1-E), 99.7 (C-1-F), 98.1 (C-1-H), 97.8 (C-1-G), 96.9 (C-1-D), 83.6 (C-4-D), 82.7 (C-3-D), 82.2 (C-2-D), 80.1 (C-2-H), 79.9 (C-3-H), 79.8 (C-3-F), 79.3 (C-2-F), 76.8 (C-3-G), 76.5 (C-4-F), 75.3 (C-5-E), 75.1 (C-3-E), 75.1 (C-4-E), 74.9 (C-4-G), 74.9 (C-4-H), 75.1, 75.1, 75.1, 73.8, 73.6, 73.5, 73.5 (7 × ArCH2), 72.5 (C-2-E), 71.0 (C-5-F), 70.2 (C-5-H), 69.5 (C-5-D), 69.2 (C-5-G), 68.5 (C-2-G), 68.3 (C-6-F), 68.3 (C-6-E), 67.4 (C-6-H), 66.0 (SO3CH2CH3), 60.8, 60.8, 59.2, 58.8, 55.3, 51.8 (6 × OCH3), 46.8 (C-7-D), 26.2 (C-6-D), 21.1, 21.1, 20.8 (3 × COCH3), 15.1 (SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1862.75 [M + Na]+ (Calcd. 1863.02); Anal. Calcd. for C98H118O32S (1840.03): C, 63.97; H, 6.46; O, 27.82; S, 1.74. Found: C, 64.03; H, 6.49; S, 1.68.
Methyl-[6-O-benzyl-2,3,4-tri-O-methyl-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-acetyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl-(2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (9). To a solution of acceptor 20 (1.20 g, 1.04 mmol) and donor 24 (1.23 g, 1.55 mmol) in dry CH2Cl2 (40 mL), 4 Å molecular sieves (1 g) were added. The stirred mixture was cooled to −35 °C under argon and activated by method C. The reaction mixture was allowed to warm up to −20 °C for 4 h. The crude product was purified twice by column chromatography (I. 6:4 n-hexane/EtOAc II. 96:4 CH2Cl2/EtOAc) to give 9 (1.20 g, 63%) as colourless; Rf = 0.33 (1:1 n-hexane/EtOAc); [α]d +26.28 (c 0.06, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.84–7.71 (m, 3H, arom.), 7.63 (s, 1H, arom.), 7.50–7.41 (m, 2H, arom.), 7.38–7.11 (m, 31H, arom), 5.22 (t, J = 9.3 Hz, 1H, H-3-E), 5.12–5.03 (m, 2H, H-1-G, H-1-D), 4.95 (t, J = 12.3 Hz, 2H, 2 × ArCH2), 4.89–4.48 (m, 15H, H-2-E, H-5-G, H-2-G, H-1-E, H-1-F, H-1-H, 9 × ArCH2), 4.40 (d, J = 10.9 Hz, 2H, 2 × ArCH2), 4.26 (m, 3H, SO3CH2CH3, ArCH2), 3.98–3.43 (m, 14H, H-4-E, H-4-H, H-4-G, H-3-F, H-3-H, H-5-F, H-5-H, H-6a,b-H, H-3-G, H-6a,b-E, H-2-H, H-5-D), 3.55, 3.39, 3.39, 3.38, 3.34, 3.30 (6s, 18H, 6 × OCH3) 3.43–2.99 (m, 9H, H-6a,b-D, H-4-F, H-5-E, H-2-F, H-3-D, H-7a,b-F, H-4-D, H-2-D), 2.31–2.20 (m, 1H, H-6a-F), 2.04, 1.99, 1.93 (3s, 9H, 3 × OCH3), 1.85–1.70 (m, 1H, H-6b-F), 1.39 (t, J = 7.1 Hz, 3H, SO3CH2CH3) ppm; 13C-NMR (91 MHz, CDCl3) δ 170.3, 170.0, 169.9, 169.5 (4 × CO), 139.2, 139.1, 138.2, 138.1, 138.1, 137.9, 136.0, 133.3, 133.0 (9C, Cq arom.), 128.5, 128.5, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 127.8, 127.7, 127.3, 127.2, 126.7, 126.2, 126.1, 125.9, 125.6 (37C, arom.), 101.1 (C-1-E), 98.3 (C-1-F), 98.1 (C-2-H), 97.9 (C-1-D), 97.6 (C-1-G), 83.2 (C-3-D), 82.1 (C-4-F), 81.8 (C-2-D), 80.2 (C-2-H), 79.9 (C-3-H), 79.8 (C-2-F), 79.5 (C-3-F), 79.3 (C-4-D), 76.1 (C-3-G), 75.1 (C-5-E), 75.0 (C-3-E), 74.8 (C-4-H), 74.5 (C-4-E), 74.2 (C-4-G), 74.9, 74.6, 73.8, 73.5, 73.4, 73.4, 73.4 (7 × ArCH2), 72.9 (C-2-E), 71.2 (C-5-D), 70.2 (C-5-F), 69.1 (C-5-H), 68.6 (C-6-E), 68.5 (C-6-H), 68.4 (C-5-G), 68.3 (C-6-D), 67.4 (C-2-G), 66.3 (SO3CH2CH3), 60.7, 60.4, 59.4, 58.2, 55.3, 51.9 (6 × OCH3), 46.6 (C-7-F), 25.9 (C-6-F), 21.2, 21.0, 20.7 (3 × COCH3), 15.2 (SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1862.74 [M + Na]+ (Calcd. 1863.02); Anal. Calcd. for C98H118O32S (1840.03): C, 63.97; H, 6.46; O, 27.82; S, 1.74. Found: C, 63.95; H, 6.51; S, 1.78.
Methyl [6-O-benzyl-2,3,4-tri-O-methyl-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-acetyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-[2,3,6-tri-O-benzyl-α-d-glucopyranosyl]-(1→4)-[methyl-(2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranoside (10). To a solution of acceptor 21. (720 mg, 0.62 mmol) and donor 24 (737 mg, 0.93 mmol) in dry CH2Cl2 (20 mL), 4 Å molecular sieves (0.50 g) were added. The stirred mixture was cooled to −40 °C under argon and activated by method C. The reaction mixture was allowed to warm up to −10 °C for 90 min. The crude product was purified twice by column chromatography (I. 9:1 CH2Cl2/EtOAc II. 6:4 n-hexane/EtOAc) to give 10 (825 mg, 72%) as a colourless syrup; Rf = 0.31 (1:1 n-hexane/EtOAc); [α]d +35.45 (c 0.05, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.82–7.69 (m, 3H, arom.), 7.62 (s, 1H, arom.), 7.48–7.09 (m, 30H, arom.), 5.21 (d, J = 5.0 Hz, 1H, H-1-G), 5.12–5.03 (m, 3H, H-3-E, H-1-D, ArCH2), 4.92–4.76 (m, 5H, H-2-E, H-1-F, H-2-G, 2 × ArCH2), 4.75–4.61 (m, 5H, H-5-G, 4 × ArCH2), 4.59–4.51 (m, 3H, 3 × ArCH2), 4.49–4.37 (m, 5H, H-1-E, H-1-H, 3 × ArCH2), 4.34–4.25 (m, 3H, SO3CH2CH3, ArCH2), 4.01–3.84 (m, 3H, H-4-H, H-4-E, H-4-G), 3.83–3.67 (m, 7H, H-3-H, H-5-F, H-6a-F, H-6a,b-E, H-5-H, H-3-F), 3.66–3.52 (m, 3H, H-3-G, H-5-D, H-6b-F), 3.50–3.26 (m, 7H, H-6a,b-D, H-2-H, H-4-F, H-3-D, H-2-F, H-7a-H), 3.59, 3.44, 3.42, 3.41, 3.33, 3.32 (6s, 18H, 6 × OCH3), 3.26–3.02 (m, 4H, H-5-E, H-4-D, H-7a-H, H-2-D), 2.42–2.32 (m, 1H, H-6a-H), 2.01, 1.96, 1.94 (3s, 9H, 3 × COCH3),1.98–1.93 (m, 1H, H-7b-H) 1.39 (t, J = 12.7 Hz, 3H, SO3CH2CH3) ppm; 13C-NMR (101 MHz, CDCl3) δ 170.3, 170.0, 169.7, 169.6 (4 × CO), 139.4, 139.0, 138.4, 138.1, 138.1, 137.7, 136.2, 133.4, 133.0 (9C, Cq arom.), 128.8, 128.6, 128.4, 128.4, 128.3, 128.2, 128.1, 128.0, 127.8, 127.7, 127.6, 127.3, 127.2, 126.0, 125.9, 125.7 (37C, arom.), 99.8 (C-1-E), 99.6 (C-1-F), 98.0 (C-1-H), 97.9 (C-1-G), 97.9 (C-1-D), 83.3 (C-3-D), 81.9 (C-2-D), 80.3 (C-4-F), 79.8 (C-5-H), 79.8 (C-3-F), 79.4 (C-4-D), 79.4 (C-3-H), 79.2 (C-2-F), 78.6 (C-2-H), 76.7 (C-3-G), 76.5 (C-4-H), 75.1 (C-4-G), 75.1 (C-4-E), 75.1 (C-5-E) 74.9 (C-3-E), 75.3, 75.1, 73.9, 73.7, 73.6, 73.4, 73.4 (7 × ArCH2), 72.7 (C-2-E), 71.4 (C-5-D), 71.1 (C-5-F), 69.3 (C-5-G), 68.9(C-6-E), 68.6 (C-6-D), 68.2 (C-2-G), 67.4 (C-6-F), 66.2 (SO3CH2CH3), 60.8, 60.5, 59.3, 58.8, 55.6, 51.9 (6 × OCH3), 46.8 (C-7-H), 26.1 (C-6-H), 21.1, 21.0, 20.8 (3 × COCH3), 15.2 (SO3CH2CH3) ppm; ESI-MS: m/z 1862.75 [M + Na]+ (Calcd. 1863.02); Anal. Calcd. for C98H118O32S (1838,73): C, 63.97; H, 6.46; O, 27.82; S, 1.74. Found: C, 64.07; H, 6.53; S, 1.82.
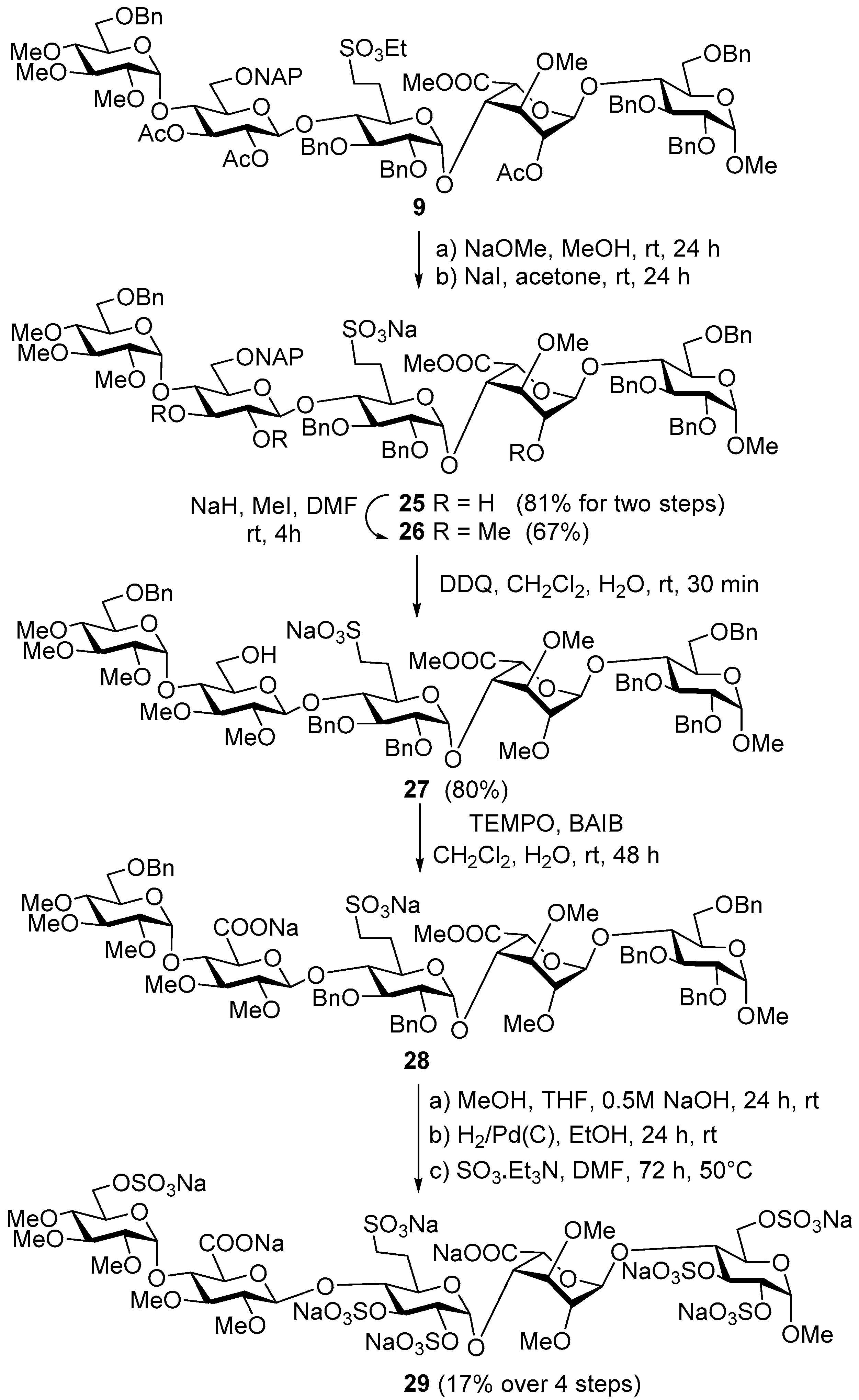
Methyl [6-O-benzyl-2,3,4-tri-O-methyl-α-d-glucopyranosyl]-(1→4)-[6-O-(2-naphthyl)-methyl-β-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-benzyl-6-deoxy-6-C-sulfonato-methyl-α-d-glucopyranosyl)]-(1→4)-[methyl (3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (25). NaOMe (36 mg) was added to the solution of compound 9 (1.22, 0.66 mmol) in MeOH (35 mL) at room temperature and stirred for 24 h. The reaction mixture was quenched by the addition of acetic acid (1–2 drops) and the solution was concentrated. Then, NaI (149 mg, 0.99 mmol) was added to the solution of crude product in acetone (40 mL) and the mixture was stirred at room temperature for 24 h. The mixture was concentrated and purified by gel chromatography (Sephadex LH-20, MeOH) to give 25 (919 mg, 81% for two steps) as a colourless syrup; Rf = 0.28 (95:5 CH2Cl2/MeOH); [α]D +33.7 (c 0.11, CHCl3); 1H-NMR (CDCl3, 400 MHz): δ (ppm) 7.79–7.07 (m, 37H, arom.), 5.34–4.21 (m, 19H, 7 × ArCH2, 5 × H-1), 3.98–2.87 (m, 49H, 20 × skeleton protons, 6 × OCH3, 3 × H-6a,b, H-7a,b, 3 × OH), 2.43–2.38 (m, 1H, H-6a-F), 2.13–2.03 (m, 1H, H-6b-F); 13C-NMR (CDCl3, 100 MHz): δ (ppm) 171.3 (CO), 140.7, 140.1, 139.4, 139.3, 138.9, 137.4, 134.6, 134.3 (9 × Cq arom.), 129.4–126.8 (37C, arom.), 104.3, 102.2, 99.5, 98.9, 96.0 (5 × C-1), 84.6, 83.5, 81.7, 81.3, 81.2, 80.7, 80.3, 79.7, 78.0, 76.3, 75.9, 75.6, 72.6, 72.3, 71.6, 70.8, 69.2, 67.9 (20C, skeleton carbons), 75.7, 74.9, 74.4, 74.2, 73.9 (7 × ArCH2), 69.8 (3 × C-6) 60.9, 60.8, 59.8, 58.5 (4 × OCH3), 55.5 (C-1-OCH3), 52.9 (COOCH3), 48.2 (C-7-F), 27.6 (C-6-F); MALDI-TOF MS: m/z 1729.72 [M + Na]+ (Calcd. 1729.64). Anal. Calcd. for C90H107NaO29S (1707.85): C, 63.29; H, 6.31; S, 1.88. Found: C, 63.37; H, 6.40; S, 1.97.
Methyl [6-O-benzyl-2,3,4-tri-O-methyl-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-methyl-6-O-(2-naphthyl)methyl-β-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-benzyl-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl)]-(1→4)-[methyl (2,3-di-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (26). An amount of 60 m/m% NaH (68 mg, 1.68 mmol) was added to the solution of compound 25 (798 mg, 0.467 mmol) in dry N,N-dimethylmethanamide (DMF) (15 mL) at 0 °C. After 30 min of stirring at room temperature, MeI (105 μL, 1.68 mmol) was added to the reaction mixture and it was stirred for 4 h. The reaction mixture was quenched by the addition of MeOH (4–5 drops). The solution was concentrated and the crude product was purified by gel chromatography (Sephadex LH-20 in MeOH) to give 26 (550 mg, 67%) as a colourless syrup; Rf = 0.58 (9:1 CH2Cl2/MeOH); [α]D +49.4 (c 0.26, CHCl3); 1H-NMR (CDCl3, 400 MHz): δ (ppm) 7.79–7.12 (m, 37H, arom.), 5.58 (d, J = 3.6 Hz, 1H), 5.21 (d, J = 5.6 Hz, 1H), 5.00–4.33 (m, 17H), 3.86–2.98 (m, 55H, 20 × skeleton protons, 9 × OCH3, 3 × H6a,b, H-7a,b), 2.48–2.39 (m, 1H, H-6a-F), 2.02–2.01 (m, 1H, H-6b-F); 13C-NMR (CDCl3, 100 MHz): δ (ppm) 169.8 (CO), 139.3, 138.9, 138.3, 138.2, 138.1, 138.0, 136.1, 133.2, 132.8 (9 × Cq arom.), 128.3–125.6 (37C, arom.), 103.0, 100.1, 99.7, 98.0, 96.0 (5 × C-1), 86.3, 84.8, 83.3, 81.6, 80.7, 80.2, 79.8, 79.6, 79.5, 79.1, 78.3, 76.4, 74.3, 74.2, 72.0, 71.5, 70.7, 70.2, 69.9, 69.6 (20C, skeleton carbons), 75.2, 75.1, 74.6, 73.5, 73.3, 73.2, 72.9 (7 × ArCH2), 69.1, 68.3, 68.0 (3 × C-6), 60.6, 60.4, 60.2, 59.7, 59.6, 59.4, 59.3 (7 × OCH3), 55.2 (C-1-OCH3), 51.7 (COOCH3), 46.8 (C-7-F), 26.4 (C-6-F); MALDI-TOF MS: m/z 1771.76 [M + Na]+ (Calcd. 1771.69). Anal. Calcd. for C93H113NaO29S (1749.93): C, 63.83; H, 6.51; S, 1.83. Found: C, 63.92; H, 7.03; S, 2.01.
Methyl [6-O-benzyl-2,3,4-tri-O-methyl-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-methyl-β-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-benzyl-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl)]-(1→4)-[methyl (2,3-di-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (27). Compound 26 (550 mg, 0.314 mmol) was converted to 27 according to general method D. The crude product was purified by column chromatography (9:1 CH2Cl2/MeOH) to give 27 (404 mg, 80%) as a colourless syrup; Rf = 0.52 (9:1 CH2Cl2/MeOH); [α]D +24.2 (c 0.06, CHCl3); 1H-NMR (CDCl3, 400 MHz): δ (ppm) 7.35–7.24 (m, 30H, arom.), 5.50 (s, 1H), 5.18 (s, 2H), 4.82–4.57 (m, 15H), 3.94–3.15 (m, 55H, 20 skeleton protons, 9 × OCH3, 3 × H-6a,b, H-7a,b), 2.52–2.41 (m, 1H, H-6a-F), 2.08–1.99 (m, 1H, H-6b-F); 13C-NMR (CDCl3, 100 MHz): δ (ppm) 171.7 (CO), 140.3–139.5 (6 × Cq arom.), 129.5–128.4 (30C, arom.), 103.5, 100.6, 99.0, 97.8, 97.0 (5 × C-1), 87.6, 86.4, 84.4, 83.0, 81.7, 81.4, 80.9, 80.6, 80.5, 80.4, 79.7, 76.8, 76.6 (20C, skeleton carbons), 76.6, 76.0, 74.9, 74.5, 74.1 (6 × ArCH2), 70.0, 69.7, 62.8 (3 × C-6), 61.0, 60.8, 60.6, 59.9, 59.7 (7 × OCH3), 55.6 (C-1-OCH3), 52.6 (COOCH3), 49.0 (C-7-F), 27.8 (C-6-F); MALDI-TOF MS: m/z 1631.77 [M + Na]+ (Calcd. 1631.63). Anal. Calcd. for C82H105NaO29S (1609.75): C, 61.18; H, 6.57; S, 1.99. Found: C, 61.24; H, 6.59; S, 2.08.
Methyl-(6-O-benzyl-2,3,4-tri-O-methyl-α-d-glucopyranosyl)-(1→4)-[sodium (2,3-di-O-methyl-β-d-glucopyranosyl) uronate]-(1→4)-(sodium (2,3-di-O-benzyl-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl)-(1→4)-[methyl (2,3-di-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (28). To a vigorously stirred solution of 27 (40 mg, 0.025 mmol) in CH2Cl2 (2.0 mL) and H2O (1.0 mL), TEMPO (0.8 mg, 0.005 mmol, 0.2 equiv.) and BAIB (32 mg, 0.099 mmol, 4 equiv.) were added. After 24 h, the TLC (9:1 CH2Cl2/MeOH) indicated moderate conversion of the starting material. Another portion of BAIB (32 mg, 0.099 mmol, 4 equiv.) were added and the stirring was continued for a further 24 h. The reaction mixture was quenched by the addition of 10% aq Na2S2O3 solution (4 mL). The mixture was then extracted twice with EtOAc (8 mL), and the combined organic layers were dried, and concentrated. The residue was purified by column chromatography (9:1 CH2Cl2/MeOH) to give a colourless syrup (31 mg). The mass spectrum contained peaks corresponding to 28 and its partially debenzylated derivatives; this mixture was used in the subsequent reaction without further purification. MALDI-TOF MS for C82H102Na2O30S (1644.60): m/z 1667.65 [M + Na]+ (Calcd. 1667.59); 1379.56 [M + Na (−3Bn)]+ (Calcd. 1379.45); 1487.56 [M + Na (−2Bn)]+ (Calcd. 1487.49).
Nona sodium [methyl (2,3,4-tri-O-methyl-6-O-sulfonato-α-d-glucopyranosyl)-(1→4)-[sodium (2,3-di-O-methyl-β-d-glucopyranosyl)uronate]-(1→4)-[sodium (2,3-di-O-sulfonato-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl)-(1→4)-[sodium (2,3-di-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-2,3,6-tri-O-sulfonato-α-d-glucopyranoside (29). An amount of 0.5 M NaOH solution (0.30 mL) was added to the solution of 28 (30 mg) in a mixture of THF (0.30 mL) and MeOH (0.30 mL) and stirred at room temperature for 24 h. The reaction was quenched by the addition of 1 N HCl solution (1 drop) and the mixture was concentrated. The crude product was converted to sodium salt by ion exchange resin (Dowex, MeOH) to give a colourless syrup (28 mg). The syrupy residue was dissolved in 96% EtOH (3.0 mL) and 10% Pd/C (10 mg) and acetic acid (100 μL) were added. The mixture was stirred at room temperature for 24 h under a 10 bar H2 atmosphere. The mixture was diluted with MeOH, the catalyst was filtered off through a pad of Celite and the filtrate was concentrated. The crude product was purified by column chromatography (7:6:1 CH2Cl2/MeOH/H2O, Rf = 0.13) and gel chromatography (Sephadex G-25, H2O) to give the corresponding pentaol (9 mg) which was characterized by MALDI MS. (MALDI-TOF MS for C39H63Na3O30S (1112.28): m/z 1091.39 [M − Na + 2H]+ (Calcd. 1091.31), 1113.39 [M + H]+ (Calcd. 1113.29), 1135.42 [M + Na]+ (Calcd. 1135.27). To the solution of the pentaol derivative (9 mg) in dry DMF (0.6 mL), SO3·Et3N complex (44 mg, 0.040 mmol) was added and the reaction mixture was stirred at 50 °C for 48 h. The reaction was quenched with satd. aq. NaHCO3 (21 mg, 0.24 mmol). The solution was concentrated. The crude product was treated with Dowex ion-exchange resin (Na+ form), and then purified by Sephadex G-25 column chromatography eluting with H2O to give 29 (7 mg, 17% from 27) as a white solid. ESI-MS for C39H57Na9O48S7 (1723.91): m/z 795.110 [M − 6Na + 4H]2− (Calcd. 795.00); 522.451 [M − 7Na + 4H]3− (Calcd.: 522.34).
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-acetyl-β-d-glucopyranosyl]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (30). Compound 6 (975 mg, 0.53 mmol) was converted to 30 according to general method D. The crude product was purified by column chromatography (94:6 CH2Cl2/acetone) to give compound 30 (612 mg, 68%) as a colourless syrup; Rf = 0.26 (93:7 CH2Cl2/acetone); [α]d +30.56 (c 0.10, CHCl3); 1H-NMR (360 MHz, CDCl3) δ 7.45–7.17 (m, 25H, arom.), 5.23 (t, J = 9.3 Hz, 1H, H-3-E), 5.11 (s, 1H, H-1-G), 5.00 (d, J = 3.6 Hz, 1H, H-1-D), 4.96–4.49 (m, 16H, H-2-E, H-5-G, H-2-G, H-1-E, H-1-H, H-1-F, 10 × PhCH2), 4.29 (q, J = 7.1, 1.2 Hz, 4H, 2 × SO3CH2CH3), 3.93–3.07 (m, 21H, skeleton protons), 3.55, 3.52, 3.41, 3.41, 3.34, 3.31 (6s, 18H, 6 × OCH3), 3.01 (dd, J = 9.8, 3.6 Hz, 1H, H-2-D), 2.72 (t, J = 9.3 Hz, 1H, H-4-D), 2.34–2.22 (m, 2H, H-6a-D, H-6a-F), 2.03, 2.01, 1.96 (3s, 9H, 3 × COCH3), 1.94–1.75 (m, 3H, H-6b-D, H-6b-F, OH), 1.41 (m, 6H, 2 × SO3CH2CH3) ppm; 13C-NMR (91 MHz, CDCl3) δ 170.2, 170.0, 169.6, 169.6 (4 × CO), 139.1, 139.0, 138.1, 138.1, 137.9 (5C, Cq arom.), 128.6, 128.6, 128.4, 128.4, 128.2, 128.1, 127.9, 127.8, 127.6, 127.3, 127.1, 126.1 (25C, arom.), 100.9, 98.4, 98.0, 97.6, 96.8 (5 × C-1), 83.8, 82.8, 82.1, 81.7, 80.2, 79.8, 79.4, 79.1, 76.1, 75.3, 74.8, 74.2, 72.6, 72.2, 70.2, 69.6, 69.2, 68.4, 67.5 (20C, skeleton carbons), 74.6, 73.9, 73.4, 73.4, 73.4 (5 × PhCH2), 68.4, 60.6 (C-6-H, C-6-E), 66.3, 66.1 (2 × SO3CH2CH3), 60.8, 60.7, 59.5, 58.2, 55.3, 51.8 (6 × OCH3), 46.8, 46.6 (C-7-D, C-7-F), 26.4, 25.5 (C-6-D, C-6-F), 21.1, 21.0, 20.6 (3 × COCH3), 15.2, 15.2 (2 × SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1737.62 [M + Na]+ (Calcd. 1737.62); Anal. Calcd. for C83H110O34S2 (1714.63): C, 58.10; H, 6.46; O, 31.70; S, 3.74. Found: C, 58.21; H, 6.41; S, 3.70.
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-acetyl-β-d-glucopyranosyl)uronate]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(ethylsulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl (2-O-acetyl-3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (31). Compound 30 (2.4 g, 1.87 mmol) was converted to 31 according to general method A. The reaction mixture was stirred for 24 h. The crude product was purified by column chromatography (98:2 CH2Cl2/MeOH) to give 31 (528 mg, 80%) as a colourless syrup; Rf = 0.50 (95:5 CH2Cl2/MeOH); [α]d +29.26 (c 0.11, CHCl3); 1H-NMR (360 MHz, CDCl3) δ 7.38–7.19 (m, 25H, arom.), 5.26–5.18 (m, 1H), 5.08 (s, 1H, H-1-G), 5.00 (d, J = 3.5 Hz, 1H, H-1-D), 4.96–4.49 (m, 16H, 10 × PhCH2, skeleton protons), 4.30, 4.28 (2q, 4H, 2 × SO3CH2CH3), 4.04 (t, J = 8.8 Hz, 1H), 3.93–3.77 (m, 4H, skeleton protons), 3.77–3.60 (m, 4H, skeleton protons), 3.59–3.18 (m, 8H skeleton protons), 3.56, 3.52, 3.42, 3.39, 3.34, 3.30 (6s, 18H, 6 × OCH3), 3.16–3.05 (m, 1H, H-7b), 3.01 (dd, J = 9.8, 3.6 Hz, 1H, H-2-D), 2.72 (t, J = 9.3 Hz, 1H, H-4-D), 2.33–2.17 (m, 2H, H-6a-D, H-6a-F), 2.03, 2.01, 1.95 (3s, 9H, 3 × COCH3), 1.91–1.77 (m, 2H, H-6b-D, H-6b-F), 1.39, 1.38 (m, 6H, 2 × SO3CH2CH3) ppm; 13C-NMR (91 MHz, CDCl3) δ 170.4, 169.9, 169.5, 169.5, 169.0 (5 × CO), 139.1, 138.7, 138.1, 138.1, 137.8 (5C, Cq arom.), 128.6, 128.5, 128.4, 128.2, 128.2, 128.1, 127.9, 127.8, 127.7, 127.6, 127.5, 127.3, 127.2 (25C, arom.), 100.8, 98.4, 98.0, 97.6, 97.6 (5 × C-1), 83.8, 82.7, 81.9, 81.6, 80.2, 80.1, 79.8, 79.0, 76.2, 75.3, 74.7, 74.4, 74.4, 74.0, 72.4, 70.2, 69.5, 69.1, 68.3, 67.4 (20C, skeleton carbons), 74.9, 74.8, 73.8, 73.4, 73.4 (5 × PhCH2), 68.5 (C-6-H), 66.6, 66.4 (2 × SO3CH2CH3), 60.7, 60.7, 59.5, 58.2, 55.3, 51.9 (6 × OCH3), 46.6, 46.5 (C-7-D, C-7-F), 26.0, 25.7 (C-6-D, C-6-F), 21.1, 20.9, 20.6 (3 × COCH3), 15.2, 15.2 (2 × SO3CH2CH3) ppm; MALDI-TOF MS: m/z 1773.53 [M + Na]+ (Calcd. 1773.58); Anal. Calcd. for C83H107O35S2 (1714.63): C, 58.10; H, 6.46; O, 31.70; S, 3.74. Found: C, 58.21; H, 6.41; S, 3.70.
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[sodium (β-d-glucopyranosyl)uronate]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl (3-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (32). NaOMe (2 mg, 0.03 mmol) was added to the solution of compound 31 (500 mg, 0.29 mmol) and stirred at room temperature for 24 h. The mixture was quenched by the addition of acetic acid (1–2 drops) and then concentrated. The crude product was dissolved in acetone (20 mL) and NaI (128 mg, 0.86 mmol) was added to the solution. The reaction mixture was stirred at room temperature for 24 h. The mixture was concentrated and the crude product was purified by gel chromatography (Sephadex LH-20, MeOH) to give 32 (386 mg, 84% for two steps) as a colourless syrup. Rf = 0.47 (7:3 CH2Cl2/MeOH); [α]d +34.32 (c 1.66, CHCl3); 1H-NMR (360 MHz, CDCl3) δ 7.44–7.15 (m, 25H, arom.), 5.51 (d, J = 3.5 Hz, 1H), 5.10 (s, 1H), 5.03 (d, J = 3.3 Hz, 1H), 5.02–4.50 (m, 14H, 5 × PhCH2, skeleton protons), 3.99–3.21 (m, 22H, skeleton protons), 3.57, 3.52, 3.52, 3.46, 3.36, 3.35 (6s, 18H, 6 × OCH3), 3.13 (dd, J = 9.8, 3.6 Hz, 1H, H-2-D), 3.08–2.95 (m, 2H, H-7a-D, H-7a-F), 2.84–2.75 (m, 1H, H-4-D), 2.43–2.31 (m, 1H, H-6a), 2.30–2.18 (m, 1H, H-6a), 2.11–1.78 (m, 2H, 2 × H-6b) ppm; 13C-NMR (91 MHz, CDCl3) δ 177.8, 171.6 (2 × CO), 140.2, 140.1, 139.5, 139.4, 139.0 (5C, Cq arom.), 129.6, 129.3, 129.2, 129.1, 128.9, 128.7, 128.4 (25C, arom.), 104.3, 102.3, 98.9, 98.2, 95.9 (5 × C-1), 85.1, 83.7, 81.8, 81.2, 80.8, 80.4, 79.3, 78.4, 78.2, 76.2, 75.9, 75.7, 75.2, 73.6, 72.5, 71.7, 70.9, 70.7, 69.1, 67.8 (20C, skeleton carbons), 76.3, 76.1, 75.0, 74.5, 74.0 (5 × PhCH2), 69.8 (C-6-H), 60.8, 60.6, 59.3, 59.2, 58.5, 55.5 (6 × OCH3), 28.3, 27.8 (C-6-D, C-6-F) ppm; MALDI-TOF MS: m/z 1635.50 [M + Na]+ (Calcd. 1635.45); Anal. Calcd. for C73H91Na3O32S2 (1612.46): C, 54.34; H, 5.68; Na, 4.27; O, 31.73; S, 3.97. Found: C, 54.23; H, 5.66; S, 4.03.
Methyl-[sodium 2,3,4-tri-O-methyl-6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-methyl-β-d-glucopyranosyl)uronate]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[methyl (2,3-di-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (33). An amount of 60 m/m% NaH (55 mg, 1.38 mmol) was added to the solution of compound 32 (370 mg, 0.23 mmol) in dry DMF (40 mL) at 0 °C. After 30 min of stirring at room temperature, MeI (64 μL, 1.04 mmol) was added to the reaction mixture and it was stirred for 4 h. The reaction mixture was quenched by the addition of MeOH and acetic acid (1–2 drops). The solution was concentrated and the crude product was purified by gel chromatography (Sephadex LH-20, MeOH) to give 33 (249 mg, 65%) as a colourless syrup; Rf = 0.53 (7:3 CH2Cl2/MeOH); [α]d +4.09 (c 0.81, CHCl3); 1H-NMR (360 MHz, CDCl3) δ 7.49–7.10 (m, 25H, arom.), 5.15–4.51 (m, 17H, 5 × H-1, H-5-E, H-5-G, 10 × PhCH2), 4.00–3.24 (m, 20H, skeleton protons), 3.57, 3.55, 3.53, 3.49, 3.46, 3.43, 3.38, 3.36, 3.35 (9s, 27H, 9 × OCH3), 3.11 (dd, J = 9.7, 3.6 Hz, 1H, H-2-D), 3.09–2.77 (m, 2H, 2 × H-7b), 2.56–2.38 (m, 1H, H-6a), 2.30–2.17 (m, 1H, H-6a), 2.07–1.85 (m, 2H, 2 × H-6b) ppm; 13C-NMR (101 MHz, MeOD) δ 170.4, 170.0 (2 × CO), 139.3, 139.3, 138.5, 138.4, 138.4 (5C, Cq arom.), 128.8, 128.7, 128.6, 128.5, 128.3, 128.3, 128.2, 128.1, 128.0, 127.9, 127.6 (25C, arom.), 100.1, 100.0, 98.4, 96.7, 96.0 (5 × C-1), 86.2, 84.2, 83.8, 83.5, 82.7, 82.1, 81.7, 81.4, 80.3, 79.5, 79.2, 78.9, 76.5, 74.7, 74.3, 71.6, 71.4, 70.7, 70.1, 68.8 (20C, skeleton carbons), 75.7, 75.5, 75.2,73.8, 73.8 (5 × PhCH2), 73.9 (C-6-H), 60.9, 60.9, 60.1, 60.0, 59.8, 59.3, 55.5, 53.0, 52.3, (9 × COCH3), 47.6, 47.5 (C-7-D, C-7-F), 27.5, 27.3 (C-6-D, C-6-F) ppm; MALDI-TOF MS: m/z 1655.53 [M + Na]+ (Calcd. 1655.52); Anal. Calcd. for C76H97Na3O32S2 (1654.51): C, 55.13; H, 5.91; Na, 4.17; O, 30.92; S, 3.87. Found: C, 55.20; H, 5.97; S, 3.79.
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-methyl-β-d-glucopyranosyl)uronate]-(1→4)-[2,3-di-O-benzyl-6-deoxy-6-C-(sulfonatomethyl)-α-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-(2,3,6-tri-O-benzyl-α-d-glucopyranoside) (34). An amount of 0.5 M NaOH solution (2 mL) was added to the solution of compound 33 (206 mg, 0.12 mmol) in a mixture of THF (2 mL) and MeOH (2 mL) and stirred at room temperature for 24 h. The reaction was quenched by the addition of 1 N HCl solution (1–2 drops) and the mixture was concentrated. The crude product was converted to sodium salt by ion exchange resin (Dowex, MeOH) to give 34 (187 mg, 90%) as a colourless syrup; Rf = 0.24 (8:2 CH2Cl2/MeOH); [α]d +42.81 (c 0.10, CHCl3); 1H-NMR (360 MHz, CDCl3) δ 7.43–7.20 (m, 25H, arom.), 5.46 (d, J = 3.6 Hz, 1H), 5.15 (d, J = 3.3 Hz, 1H), 5.10 (d, J = 3.9 Hz, 1H), 5.04–4.48 (m, 14H, 2 × H-1, H-5-E, H-5-G, 10 × PhCH2), 4.09–3.25 (m, 17H, skeleton protons), 3.58, 3.54, 3.53, 3.52, 3.50, 3.43, 3.35, 3.34 (8s, 24H, 8 × OCH3), 3.23–3.16 (m, 1H), 3.09 (dd, 1H, H-2-D), 3.07–3.86 (m, 2H, H-7b-D, H-7b-F), 2.81 (t, J = 9.8 Hz, 1H, H-4-D), 2.60–2.48 (m, 1H, H-6a), 2.33–2.19 (m, 1H, H-6a), 1.98–1.82 (m, 2H, H-6b-D, H-6b-F) ppm; 13C-NMR (101 MHz, MeOD) δ 171.1, 170.7 (2 × CO), 140.4, 140.2, 139.6, 139.5, 139.5 (5C, Cq arom.), 129.4, 129.3, 129.2, 129.0, 129.0, 128.8, 128.7, 128.6, 128.4, 128.2, 128.0 (25C, arom.), 104.6, 100.5, 98.9, 96.8, 96.7 (5 × C-1), 87.0, 85.4, 84.5, 84.1, 82.9, 82.8, 81.3, 81.2, 80.8, 80.6, 80.4, 76.6, 75.2, 74.9, 74.1, 71.6, 71.5, 71.0, 70.7 (20C, skeleton carbons), 76.0, 75.8, 74.5, 73.9, 73.8 (5 × PhCH2), 69.6 (C-6-H), 61.2, 60.9, 60.6, 59.9, 59.5, 55.9, 55.9, 55.6 (8 × OCH3), 30.7, 28.3 (C-6-D, C-6-F) ppm; MALDI-TOF MS: m/z 1685.49 [M + Na]+ (Calcd. 1685.47); Anal. Calcd. for C75H94Na4O32S2 (1662.48): C, 54.15; H, 5.70; Na, 5.53; O, 30.78; S, 3.85. Found: C, 54.08; H, 5.67; S, 3.87.
Methyl [2,3,4-tri-O-methyl-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-methyl-β-d-glucopyranosyl)uronate]-(1→4)-[6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl]-(1→4)-[sodium (2,3-di-O-methyl-α-l-idopyranosyl)uronate]-(1→4)-α-d-glucopyranoside (35). An amount of 10% Pd/C (110 mg) and acetic acid (350 μL) were added to the solution of compound 34 (180 mg, 0.11 mmol) in 96 v/v % EtOH (10 mL). The mixture was stirred at room temperature for 24 h under a 10 bar H2 atmosphere. The mixture was diluted with MeOH and the catalyst was filtered off on Celite-pad. The filtrate was concentrated. The crude product was purified by column chromatography (7:6:1 CH2Cl2/MeOH/H2O) and gel chromatography (Sephadex G-25, H2O) to give 35 (123 mg, 92%) as a colourless syrup; Rf = 0.25 (7:6:1 CH2Cl2/MeOH/H2O); [α]d +21.82 (c 0.21, CHCl3); 1H-NMR (360 MHz, CDCl3) δ 5.54 (s, 1H), 5.14 (s, 1H), 5.09 (s, 1H), 4.83 (s, 2H), 4.62 (s, 1H), 4.18 (s, 1H), 3.97–3.25 (m, 46H, skeleton protons, 8 × OCH3), 3.18–2.96 (m, 5H), 2.90 (d, J = 2.7 Hz, 1H), 2.49–2.35 (m, 1H, H-6a), 2.29–2.16 (m, 1H, H-6a), 1.99–1.85 (m, 2H, H-6b, H-6b) ppm; 13C-NMR (91 MHz, CDCl3) δ 193.3, 193.2 (2 × CO), 103.5, 103.4, 100.1, 100.0, 96.2 (5 × C-1), 86.8, 84.4, 83.9, 83.5, 82.3, 81.8, 80.4, 78.6, 78.1, 76.7, 73.9, 72.8, 72.6, 72.5, 72.4, 71.9, 71.6, 71.4, 70.3, 69.9 (20C, skeleton carbons), 61.2 (C-6-H), 61.5, 61.1, 60.4, 60.4, 59.9, 59.6, 58.9, 55.9 (8 × OCH3), 48.3, 48.2 (C-7-D, C-7-F), 27.3, 27.0 (C-6-D, C-6-F) ppm; ESI-MS: m/z 561.46 [M + 2H]2− (Calcd. 561.15); Anal. Calcd. for C40H64Na4O32S2 (1212.24): C, 39.61; H, 5.32; Na, 7.58; O, 42.21; S, 5.29. Found: C, 39.56; H, 5.29; S, 5.24.
Nona sodium [methyl (2,3,4-tri-O-methyl-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl)]-(1→4)-[2,3-di-O-methyl-β-d-glucopyranosyluronate]-(1→4)-[2,3-di-O-sulfonato-6-deoxy-6-C-sulfonatomethyl-α-d-glucopyranosyl]-(1→4)-[2,3-di-O-methyl-α-l-idopyranosyluronate]-(1→4)-2,3,6-tri-O-sulfonato-α-d-glucopyranoside (36). To the solution of compound 35 (58 mg, 0.047 mmol) in dry DMF (4 mL) sulfur trioxide–triethylamine complex (215 mg, 1.187 mmol) was added and the reaction mixture was stirred at 50 °C for 72 h. The reaction was quenched with satd. aq. NaHCO3 (262 mg, 3.12 mmol). The solution was concentrated. The crude product was treated with Dowex ion-exchange resin (Na+ form), and then purified by Sephadex G-25 column chromatography eluting with H2O to give 36 (28 mg, 36%) as a white powder. Rf = 0.53 (7:4:1 CH2Cl2/MeOH/H2O); ESI-MS: m/z for C40H59Na9O47S7 (1721.94): 837.771 [M − 2Na]2− (Calcd. 837.979); 794.136 [M − 6Na + 4H]2− (Calcd. 794.015).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}