
Capsaicin Suppresses Cell Proliferation, Induces Cell Cycle Arrest and ROS Production in Bladder Cancer Cells through FOXO3a-Mediated Pathways


Abstract
:1. Introduction
2. Results
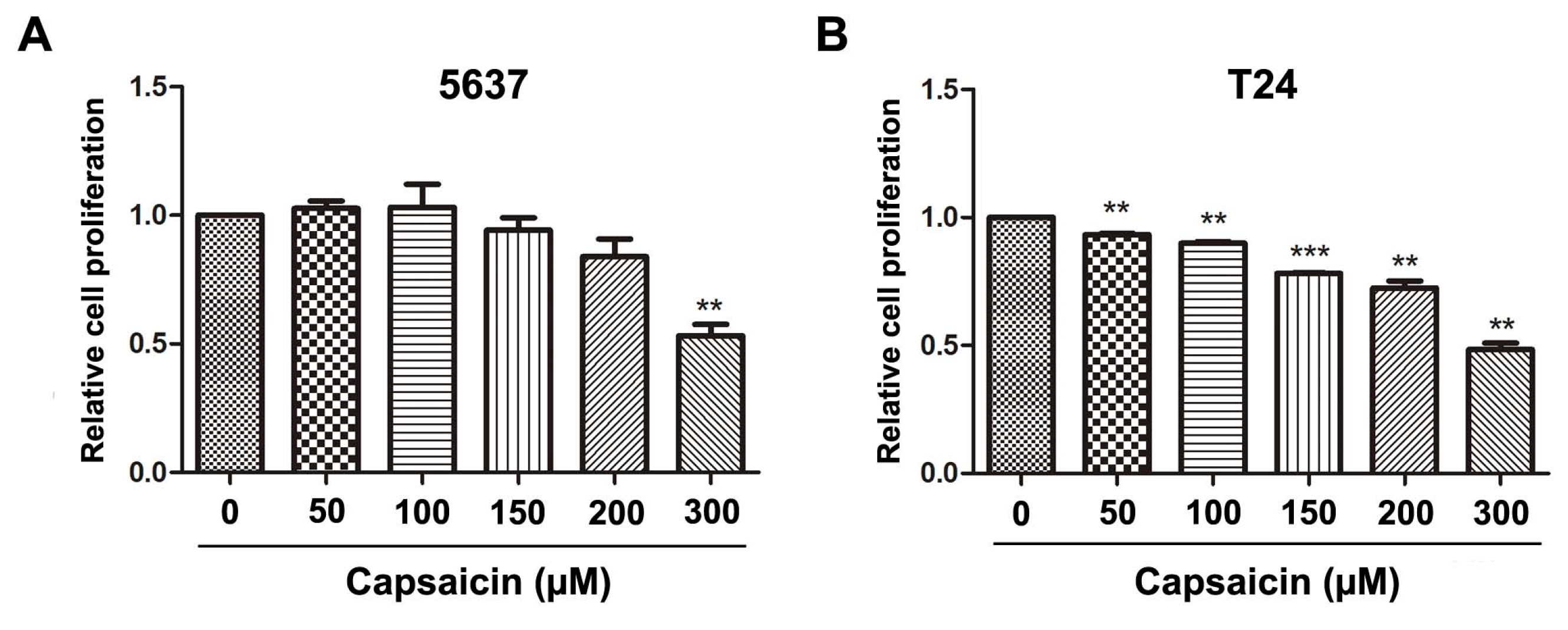
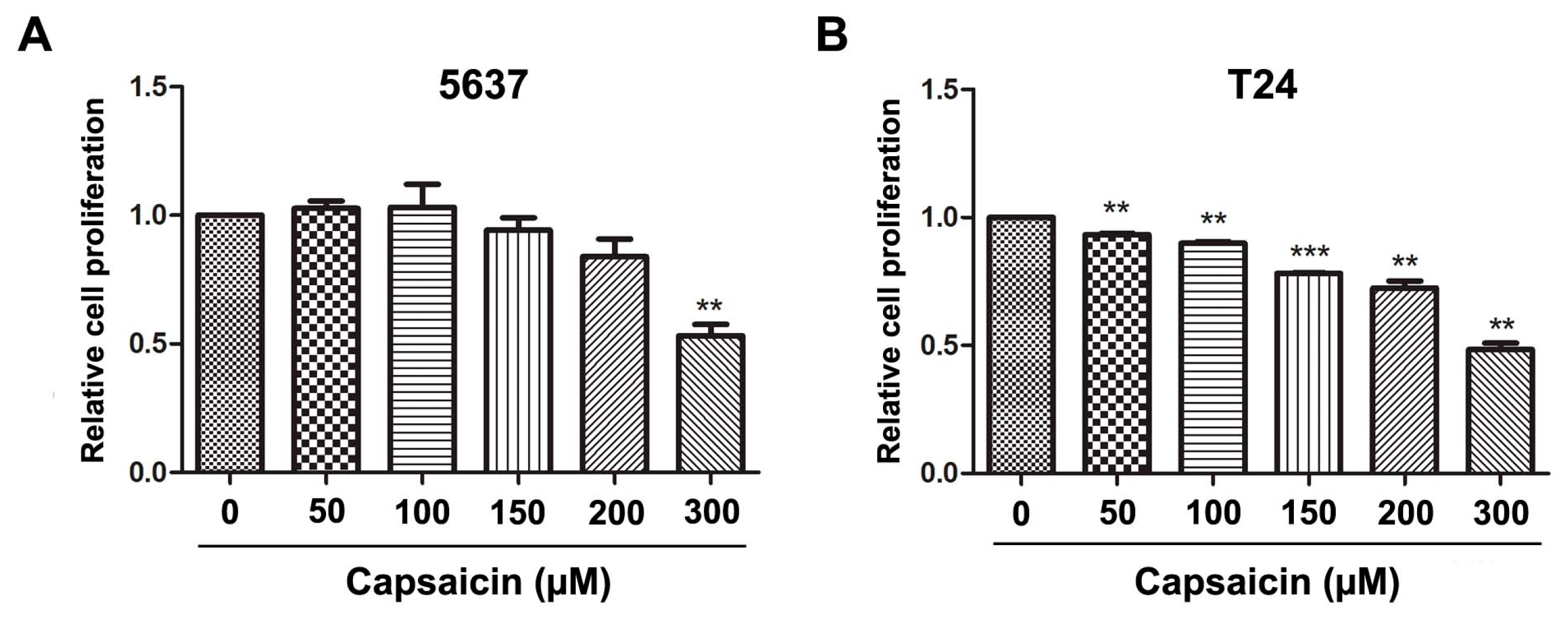
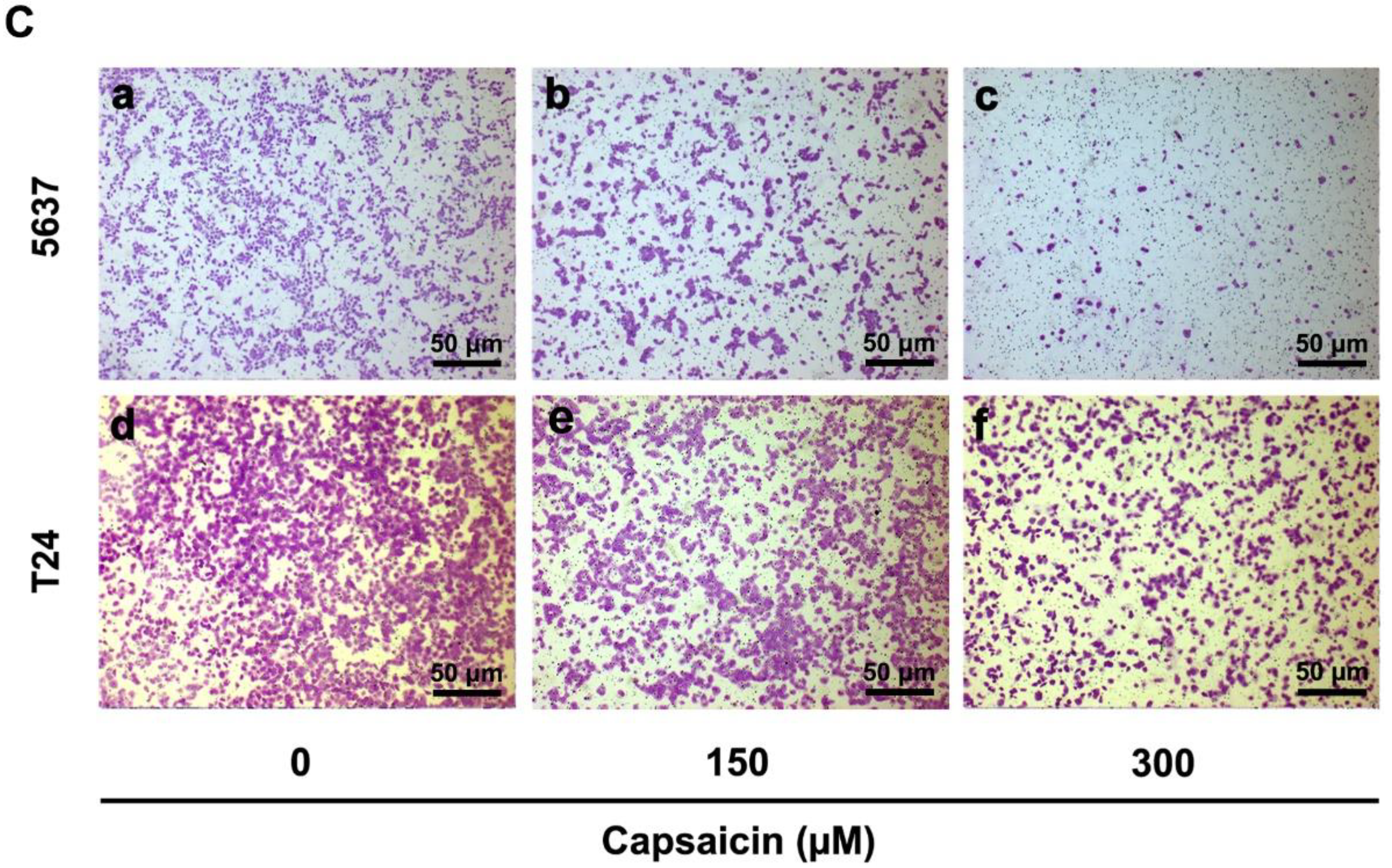
2.1. CAP Inhibited BCa Cell Proliferation and Migration
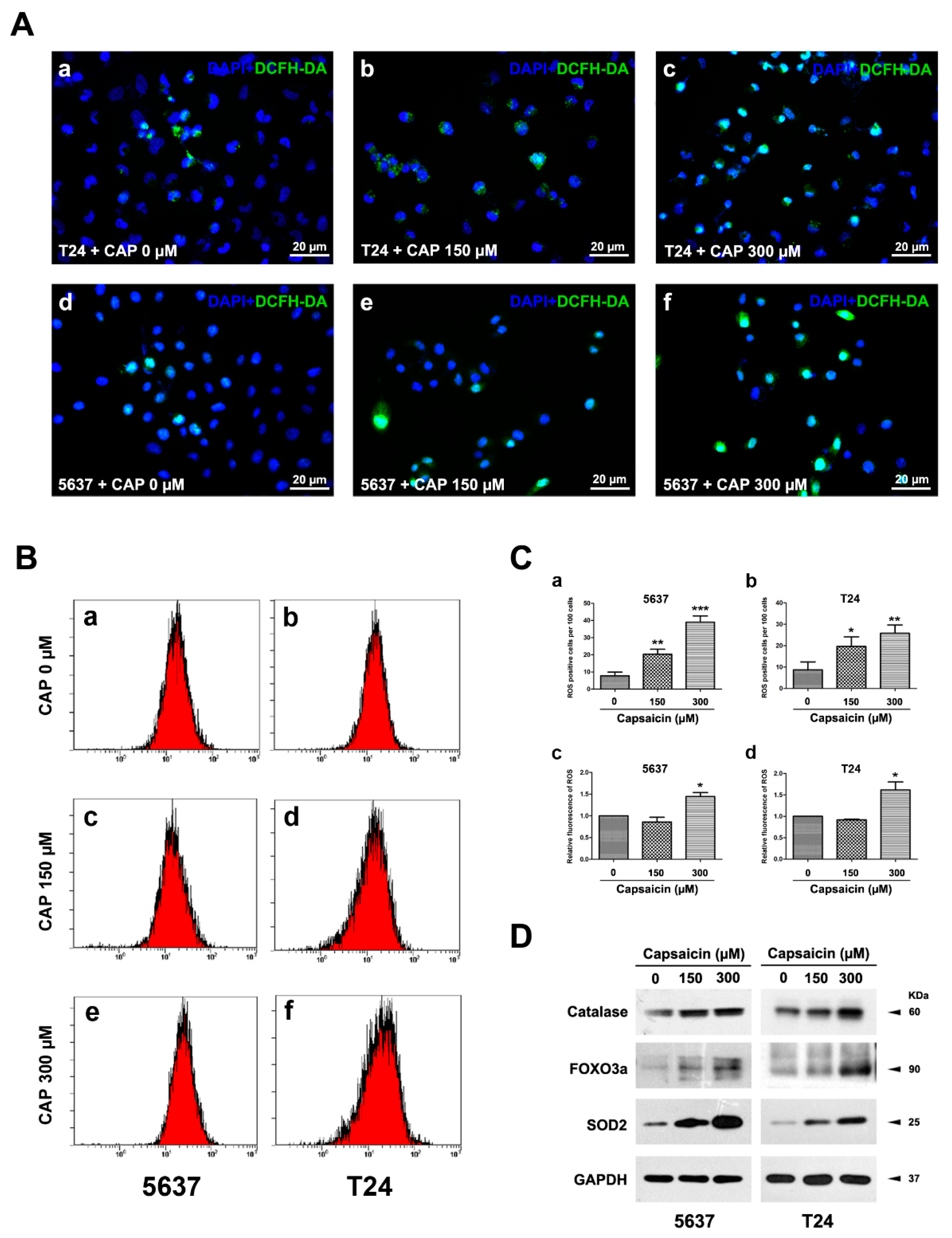
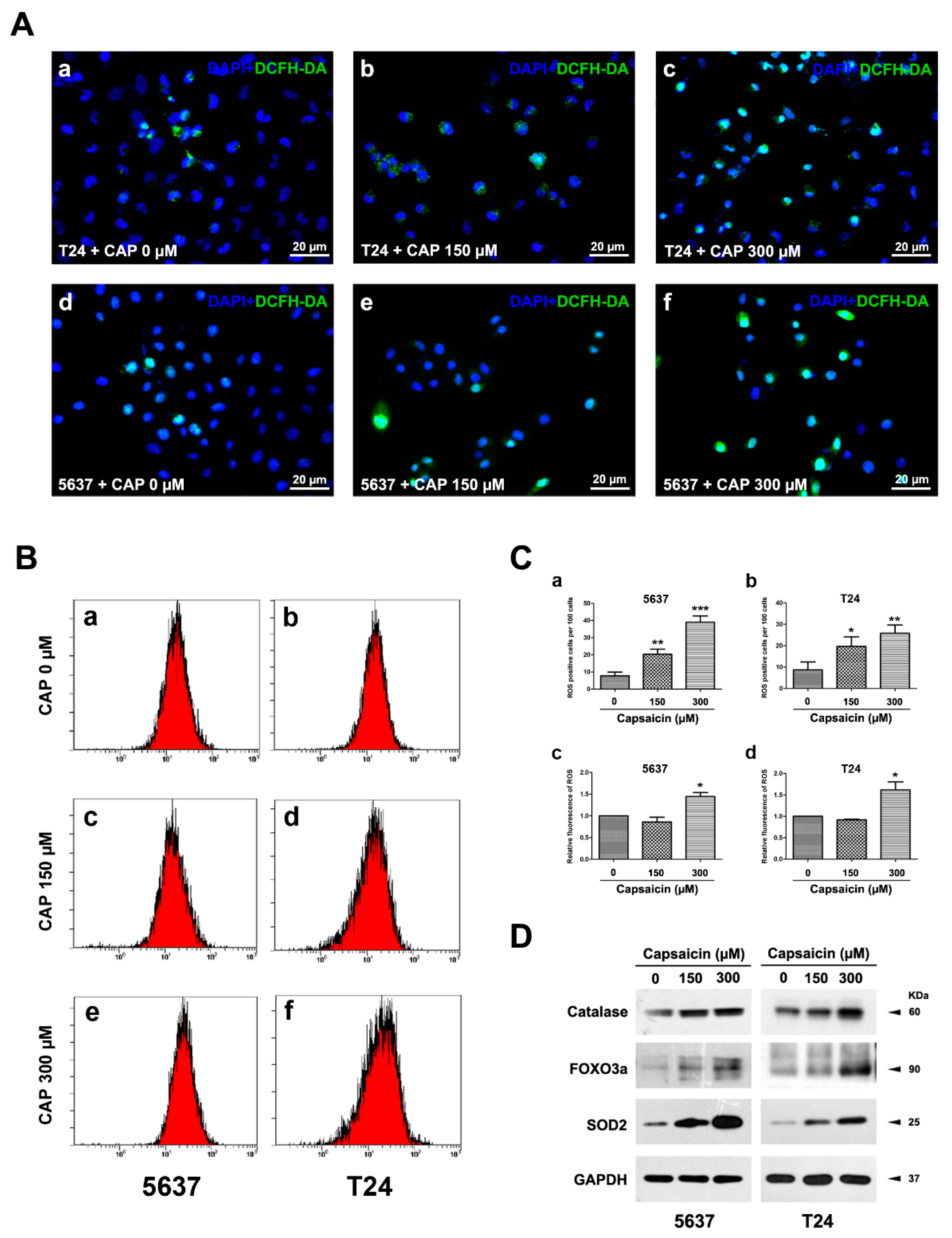
2.2. CAP Induced an Increase of ROS Production in BCa Cells
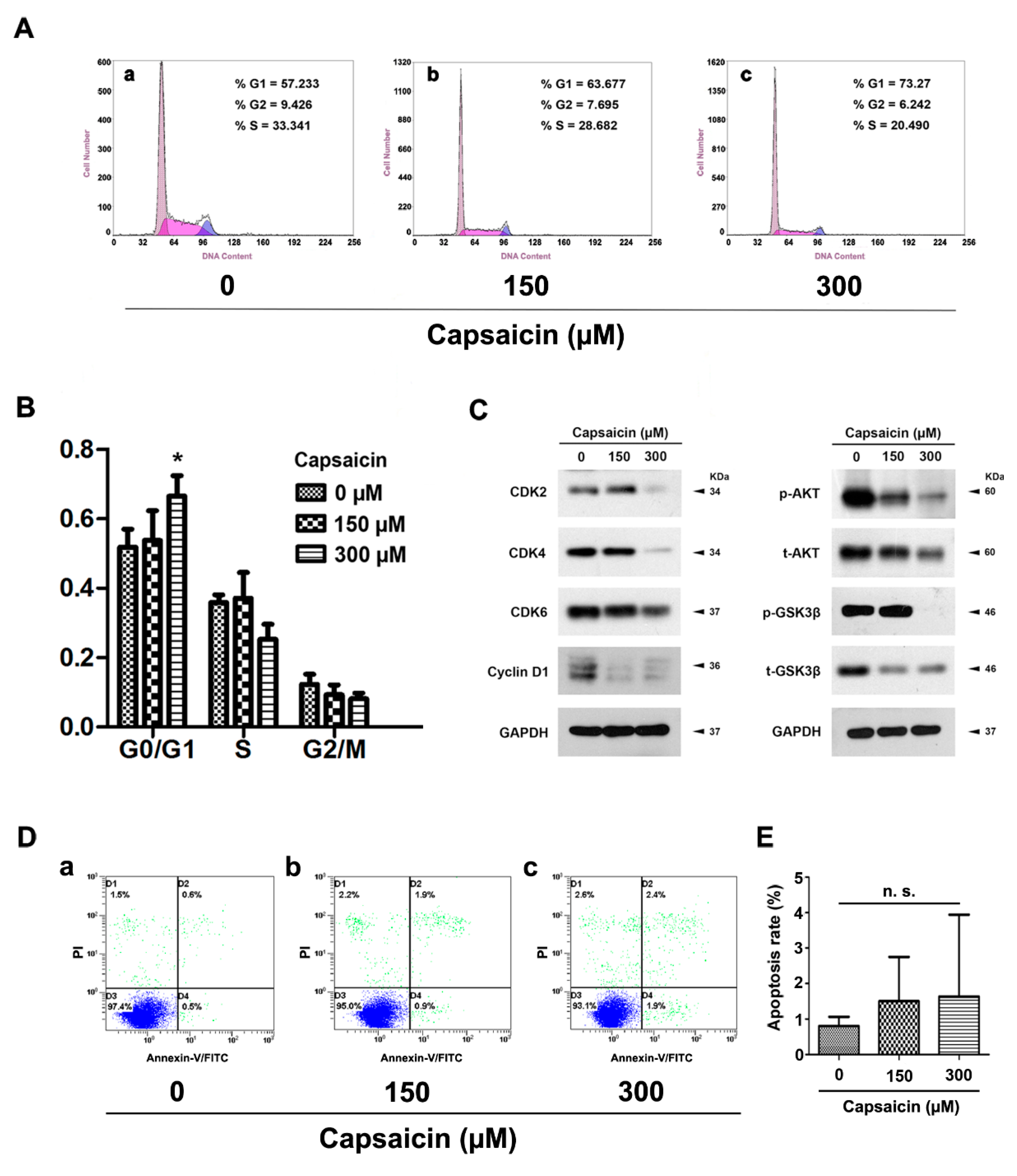
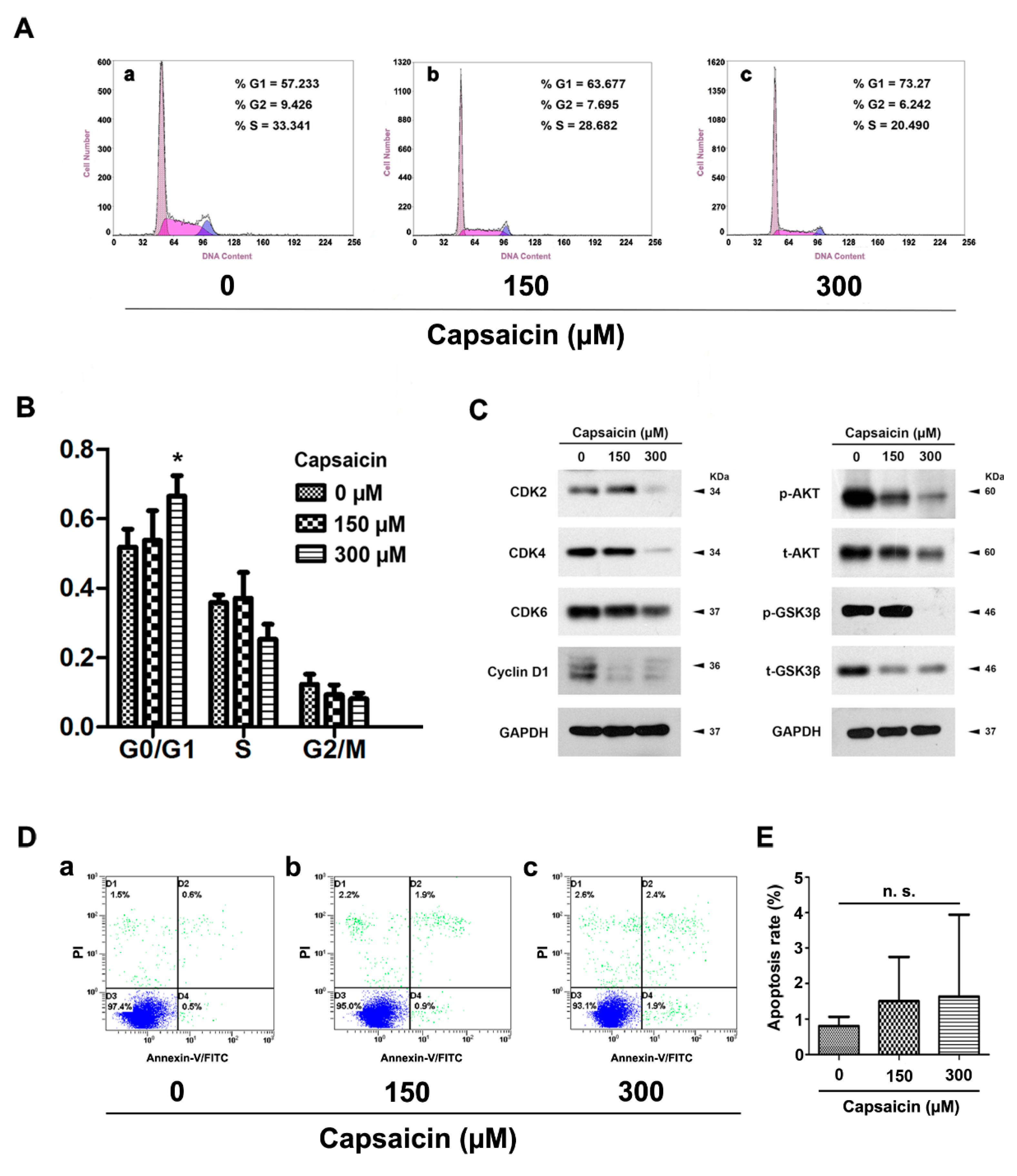
2.3. CAP Triggered Cell Cycle Arrest at G0/G1 Phase, But No Significant Effect on Apoptosis in BCa Cells
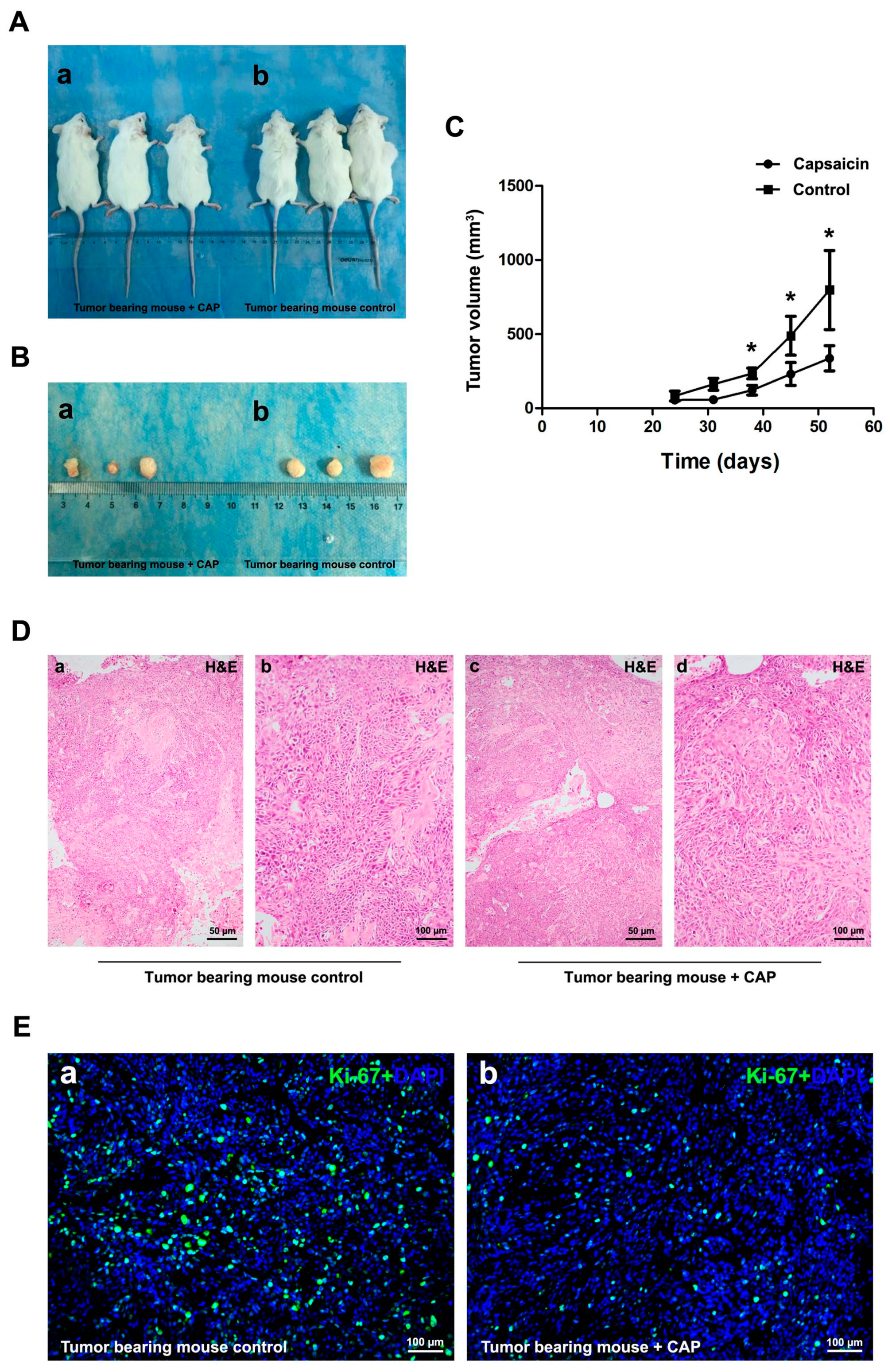
2.4. Injection of CAP Suppressed Tumor Growth In Vivo
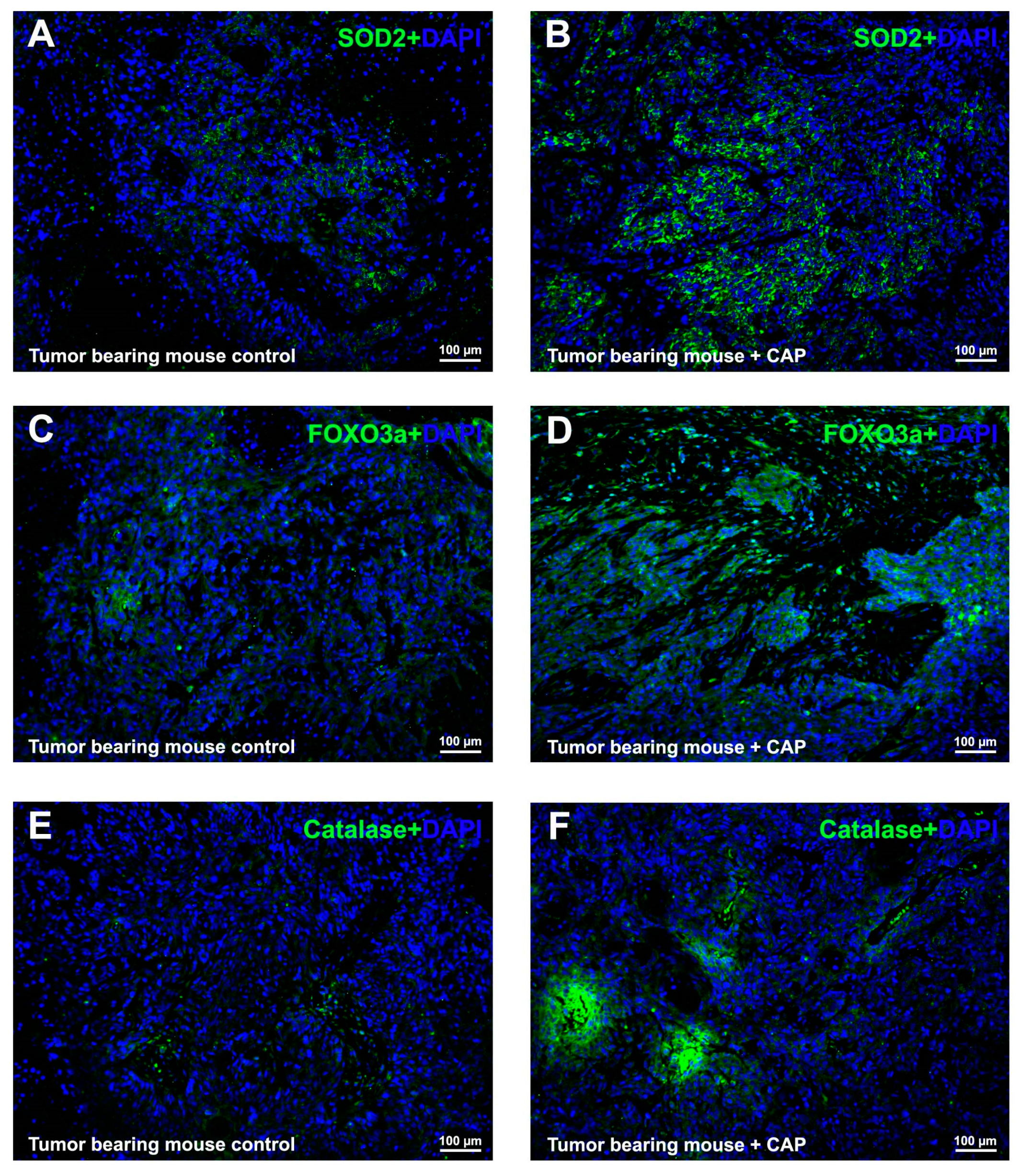
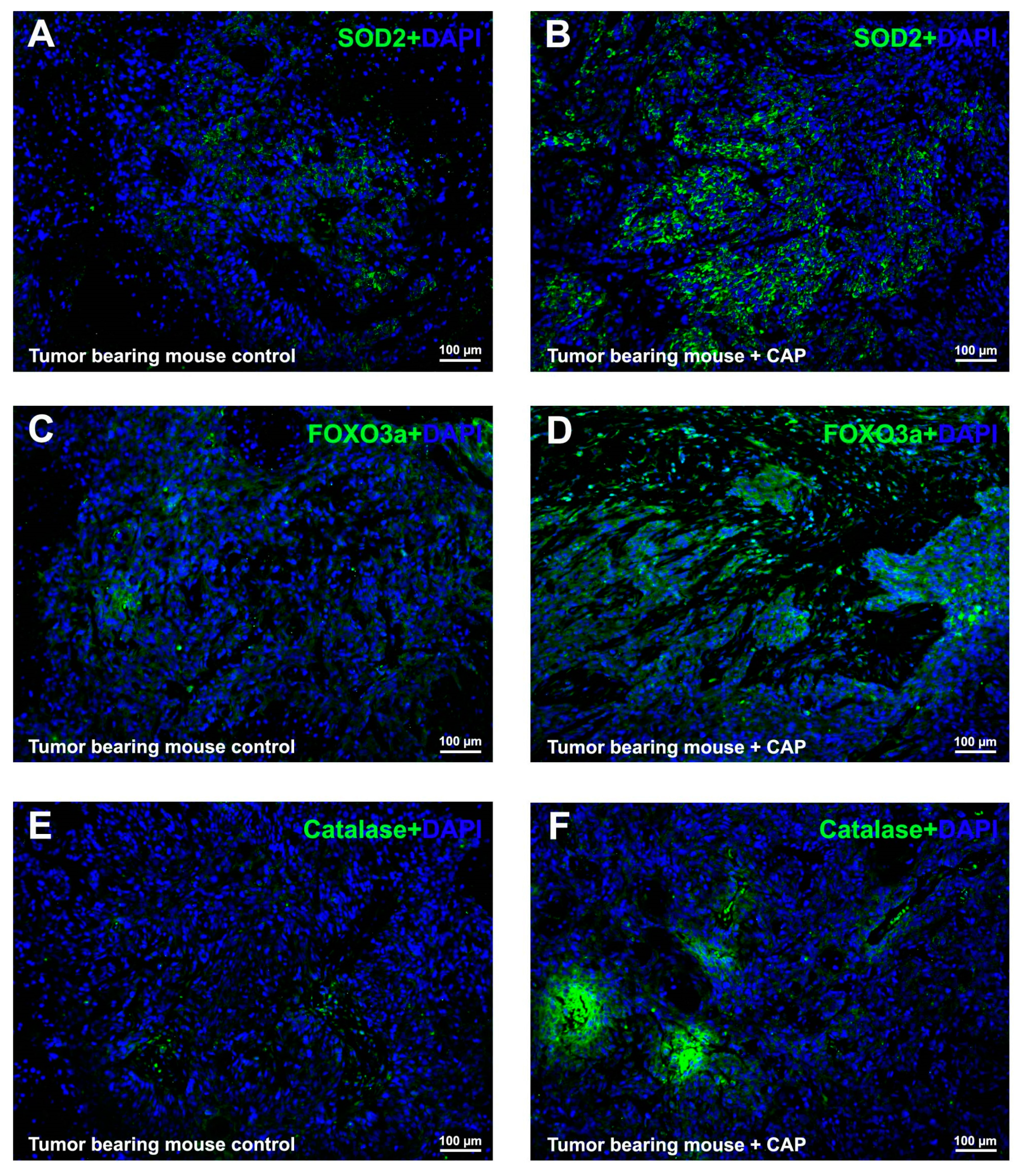
2.5. CAP Interfered ROS Metabolism In Vivo
3. Discussion
4. Experimental Section
4.1. Ethical Statement for Mice (NOD/SCID)
4.2. Human Bladder Cancer Cell Lines
4.3. Capsaicin Treatment for BCa Cells In Vitro
4.4. Xenograft Model
4.5. Cell Culture Experiments
4.5.1. MTT Test for CAP Treatment
4.5.2. Transwell Migration Assay
4.5.3. Flow Cytometry Analysis for Alterations of Cell Cycle and Apoptosis
4.5.4. ROS Detection by Staining with DCFH-DA
4.6. Protein Analyses
4.6.1. Isolation of Total Protein from BCa Cells and Western Blot Analysis
4.6.2. Immunofluorescence Staining for Xenograft Mouse Tissues
4.6.3. Hematoxylin and Eosin (H & E) Staining
4.7. Statistical Analyses
Acknowledgments
Author Contributions
Conflicts of interest
References
- Burger, M.; Catto, J.W.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Van Rhijn, B.W.; Burger, M.; Lotan, Y.; Solsona, E.; Stief, C.G.; Sylvester, R.J.; Witjes, J.A.; Zlotta, A.R. Recurrence and progression of disease in non-muscle-invasive bladder cancer: From epidemiology to treatment strategy. Eur. Urol. 2009, 56, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Karl, A.; Grimm, T.; Jokisch, F.; Gaisa, N.T.; Stief, C.G. Non-muscle invasive bladder cancer: Current aspects of diagnostics, local therapy options and the update of the 2016 WHO classification. Urol. A 2016, 55, 1247–1258. [Google Scholar] [CrossRef] [PubMed]
- Sievert, K.D.; Amend, B.; Nagele, U.; Schilling, D.; Bedke, J.; Horstmann, M.; Hennenlotter, J.; Kruck, S.; Stenzl, A. Economic aspects of bladder cancer: What are the benefits and costs? World J. Urol. 2009, 27, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Witjes, J.A. Bladder cancer in 2015: Improving indication, technique and outcome of radical cystectomy. Nat. Rev. Urol. 2016, 13, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, S.M.; Danzig, M.R.; Ghandour, R.A.; Deibert, C.M.; Decastro, G.J.; Benson, M.C.; McKiernan, J.M. Cost-effectiveness of neoadjuvant chemotherapy before radical cystectomy for muscle-invasive bladder cancer. Urol. Oncol. 2014, 32, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, M.A.; Goenka, A. Quality of Life Outcomes for Bladder Cancer Patients Undergoing Bladder Preservation with Radiotherapy. Curr. Urol. Rep. 2015, 16. [Google Scholar] [CrossRef] [PubMed]
- Schepisi, G.; Santoni, M.; Massari, F.; Gurioli, G.; Salvi, S.; Conteduca, V.; Montironi, R.; de Giorgi, U. Urothelial Cancer: Inflammatory Mediators and Implications for Immunotherapy. BioDrugs 2016, 30, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, S.; Horwich, A.; Omar, O.; Mohammed, K.; Thompson, A.; Kumar, P.; Khoo, V.; van As, N.; Eeles, R.; Dearnaley, D.; et al. Selective organ preservation with neo-adjuvant chemotherapy for the treatment of muscle invasive transitional cell carcinoma of the bladder. Br. J. Cancer 2015, 112, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Bley, K.; Boorman, G.; Mohammad, B.; McKenzie, D.; Babbar, S. A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol. Pathol. 2012, 40, 847–873. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.L.; Wease, K.N.; van Ryssen, M.P.; Paterson, S.; Agate, B.; Gallagher, K.A.; Brown, C.T.; Scott, R.H.; Conway, S.J. In vitro photo-release of a TRPV1 agonist. Bioorg. Med. Chem. Lett. 2006, 16, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, E.E.; Keller, J.M.; Kominsky, H.; Kaszas, K.; Maric, D.; Iadarola, M.J. Positive allosteric modulation of TRPV1 as a novel analgesic mechanism. Mol. Pain 2012, 8, 70. [Google Scholar] [CrossRef] [PubMed]
- Kalogris, C.; Caprodossi, S.; Amantini, C.; Lambertucci, F.; Nabissi, M.; Morelli, M.B.; Farfariello, V.; Filosa, A.; Emiliozzi, M.C.; Mammana, G.; et al. Expression of transient receptor potential vanilloid-1 (TRPV1) in urothelial cancers of human bladder: Relation to clinicopathological and molecular parameters. Histopathology 2010, 57, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Szallasi, A. Peripheral TRPV1 receptors as targets for drug development: New molecules and mechanisms. Curr. Pharm. Des. 2008, 14, 32–41. [Google Scholar] [PubMed]
- Avelino, A.; Cruz, F. TRPV1 (vanilloid receptor) in the urinary tract: Expression, function and clinical applications. Naunyn Schmiedebergs Arch. Pharmacol. 2006, 373, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.H.; Lee, Y.H.; Cheng, H.L.; Chen, H.Y.; Jhuang, F.H.; Chueh, P.J. Capsaicin Inhibits Multiple Bladder Cancer Cell Phenotypes by Inhibiting Tumor-Associated NADH Oxidase (tNOX) and Sirtuin1 (SIRT1). Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Chandiramani, V.; Parkinson, M.C.; Beckett, A.; Fowler, C.J. Treating the human bladder with capsaicin: Is it safe? Eur. Urol. 1998, 33, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Vij, A.S.; Sharma, M. Mechanisms and clinical uses of capsaicin. Eur. J. Pharmacol. 2013, 720, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wang, X.; Zhu, G.; Zhou, Z.; Wang, Y.; Chen, D.; Meng, Z. Effect of surgical castration on expression of TRPM8 in urogenital tract of male rats. Mol. Biol. Rep. 2012, 39, 4797–4802. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Meng, Z.; Liu, T.; Wang, G.; Qian, G.; Cao, T.; Guan, X.; Dan, H.; Xiao, Y.; Wang, X. Decreased TRPM7 inhibits activities and induces apoptosis of bladder cancer cells via ERK1/2 pathway. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, Y.; Cao, R.; Qian, G.; Wang, X.; Xiao, Y. Simvastatin induces cell cycle arrest and inhibites proliferation of bladder cancer cells via PPAR and ErbB signaling pathways. In Proceedings of the 41st American Society of Andrology Annual Meeting, New Orleans, LA, USA, 1–5 April 2016; p. 121.
- Cao, R.; Liu, T.; Meng, Z.; Yang, Z.; Xiao, Y.; Wang, X. Effect of TRPM7 on apoptosis and metastasis of bladder cancer via MAPK and PI3K/AKT signaling pathways. J. Urol. 2016, 195, e353. [Google Scholar] [CrossRef]
- Chen, D.; Yang, Z.; Wang, Y.; Zhu, G.; Wang, X. Capsaicin induces cycle arrest by inhibiting cyclin-dependent-kinase in bladder carcinoma cells. Int. J. Urol. 2012, 19, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chang, J.S.; Lee, J.Y.; Kim, J.A. Capsaicin-induced apoptosis and reduced release of reactive oxygen species in MBT-2 murine bladder tumor cells. Arch. Pharm. Res. 2004, 27, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.H.; Wang, X.H.; Wang, H.P.; Hu, L.Q.; Zheng, X.M.; Li, S.W. Capsaicin mediates cell death in bladder cancer T24 cells through reactive oxygen species production and mitochondrial depolarization. Urology 2010, 75, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Chelikani, P.; Fita, I.; Loewen, P.C. Diversity of structures and properties among catalases. Cell. Mol. Life Sci. 2004, 61, 192–208. [Google Scholar] [CrossRef] [PubMed]
- Scholzen, T.; Gerdes, J. The Ki-67 protein: From the known and the unknown. J. Cell. Physiol. 2000, 182, 311–322. [Google Scholar] [CrossRef]
- Arzuman, L.; Beale, P.; Yu, J.Q.; Huq, F. Synthesis of tris(quinoline)monochloroplatinum(II) Chloride and its Activity Alone and in Combination with Capsaicin and Curcumin in Human Ovarian Cancer Cell Lines. Anticancer Res. 2016, 36, 2809–2818. [Google Scholar] [PubMed]
- Ko, E.Y.; Moon, A. Natural Products for Chemoprevention of Breast Cancer. J. Cancer Prev. 2015, 20, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Anandakumar, P.; Kamaraj, S.; Jagan, S.; Ramakrishnan, G.; Asokkumar, S.; Naveenkumar, C.; Raghunandhakumar, S.; Vanitha, M.K.; Devaki, T. The Anticancer Role of Capsaicin in Experimentallyinduced Lung Carcinogenesis. J. Pharmacopunct. 2015, 18, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.P.; Chen, J.C.; Wu, C.C.; Chen, C.T.; Tang, N.Y.; Ho, Y.T.; Lo, C.; Lin, J.P.; Chung, J.G.; Lin, J.G. Capsaicin-induced apoptosis in human hepatoma HepG2 cells. Anticancer Res. 2009, 29, 165–174. [Google Scholar] [PubMed]
- Lu, H.F.; Chen, Y.L.; Yang, J.S.; Yang, Y.Y.; Liu, J.Y.; Hsu, S.C.; Lai, K.C.; Chung, J.G. Antitumor activity of capsaicin on human colon cancer cells in vitro and colo 205 tumor xenografts in vivo. J. Agric. Food Chem. 2010, 58, 12999–13005. [Google Scholar] [CrossRef] [PubMed]
- D’Eliseo, D.; Manzi, L.; Velotti, F. Capsaicin as an inducer of damage-associated molecular patterns (DAMPs) of immunogenic cell death (ICD) in human bladder cancer cells. Cell Stress Chaperones. 2013, 18, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Lewinska, A.; Jarosz, P.; Czech, J.; Rzeszutek, I.; Bielak-Zmijewska, A.; Grabowska, W.; Wnuk, M. Capsaicin-induced genotoxic stress does not promote apoptosis in A549 human lung and DU145 prostate cancer cells. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 779, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.J.; Peng, J.; Li, Y.J. Recent advances in the study on capsaicinoids and capsinoids. Eur. J. Pharmacol. 2011, 650. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.M.; Malagarie-Cazenave, S.; Olea, N.; Vara, D.; Chiloeches, A.; Diaz-Laviada, I. Apoptosis induced by capsaicin in prostate PC-3 cells involves ceramide accumulation, neutral sphingomyelinase, and JNK activation. Apoptosis 2007, 12, 2013–2024. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Chen, J.; Ma, Z.; Liu, W.; Yang, F.; Yang, Z.; Wang, K.; Wang, X.; He, D.; Li, L.; et al. Capsaicin enhances anti-proliferation efficacy of pirarubicin via activating TRPV1 and inhibiting PCNA nuclear translocation in 5637 cells. Mol. Med. Rep. 2016, 13, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Choi, C.H.; Jung, Y.K.; Oh, S.H. Autophagy induction by capsaicin in malignant human breast cells is modulated by p38 and extracellular signal-regulated mitogen-activated protein kinases and retards cell death by suppressing endoplasmic reticulum stress-mediated apoptosis. Mol. Pharmacol. 2010, 78, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and cancer: Have we moved forward? Biochem. J. 2007, 401. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.J.; Hua, X. Targeting ROS: Selective killing of cancer cells by a cruciferous vegetable derived pro-oxidant compound. Cancer Biol. Ther. 2007, 6, 646–647. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Li, Y.; Niu, B.; Wang, X.; Cheng, Y.; Zhou, Z.; You, T.; Liu, Y.; Wang, H.; Xu, J. Involvement of PI3K/Akt/FoxO3a and PKA/CREB Signaling Pathways in the Protective Effect of Fluoxetine Against Corticosterone-Induced Cytotoxicity in PC12 Cells. J. Mol. Neurosci. 2016, 59, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Ishitobi, H.; Miyaki, S.; Kawaoka, T.; Kayashima, T.; Matsubara, K. Carnosic acid protects starvation-induced SH-SY5Y cell death through Erk1/2 and Akt pathways, autophagy, and FoxO3a. Int. J. Food Sci. Nutr. 2016, 67, 977–982. [Google Scholar] [CrossRef] [PubMed]
- McClelland Descalzo, D.L.; Satoorian, T.S.; Walker, L.M.; Sparks, N.R.; Pulyanina, P.Y.; Zur Nieden, N.I. Glucose-Induced Oxidative Stress Reduces Proliferation in Embryonic Stem Cells via FOXO3A/β-Catenin-Dependent Transcription of p21(cip1). Stem Cell Rep. 2016, 7, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.M.; Tseng, H.Y.; Cheng, Y.L.; Yeh, B.W.; Wu, W.J.; Huang, H.S. TG-interacting factor transcriptionally induced by AKT/FOXO3A is a negative regulator that antagonizes arsenic trioxide-induced cancer cell apoptosis. Toxicol. Appl. Pharmacol. 2015, 285, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, A.R.; Debeb, B.G.; Lacerda, L.; Larson, R.; Bambhroliya, A.; Huang, X.; Bertucci, F.; Finetti, P.; Birnbaum, D.; van Laere, S.; et al. Simvastatin prevents triple-negative breast cancer metastasis in pre-clinical models through regulation of FOXO3a. Breast Cancer Res. Treat. 2015, 154, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, A.; Nepal, S.; Kim, M.J.; Chang, J.H.; Kim, S.H.; Jeong, G.S.; Jeong, C.H.; Park, G.H.; Jung, S.; Lim, J.; et al. Critical Role of AMPK/FoxO3A Axis in Globular Adiponectin-Induced Cell Cycle Arrest and Apoptosis in Cancer Cells. J. Cell. Physiol. 2016, 231, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; You, S.Y.; Cho, S.; Jeon, H.J.; Lee, S.; Cho, D.H.; Kim, J.S.; Oh, J.S. Eccentric localization of catalase to protect chromosomes from oxidative damages during meiotic maturation in mouse oocytes. Histochem. Cell Biol. 2016, 146, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Luo, H.; Vanek, K.N.; LaRue, A.C.; Schulte, B.A.; Wang, G.Y. Catalase inhibits ionizing radiation-induced apoptosis in hematopoietic stem and progenitor cells. Stem Cells Dev. 2015, 24, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Pias, E.K.; Ekshyyan, O.Y.; Rhoads, C.A.; Fuseler, J.; Harrison, L.; Aw, T.Y. Differential effects of superoxide dismutase isoform expression on hydroperoxide-induced apoptosis in PC-12 cells. J. Biol. Chem. 2003, 278, 13294–13301. [Google Scholar] [CrossRef] [PubMed]
- Naito, S.; von Eschenbach, A.C.; Giavazzi, R.; Fidler, I.J. Growth and metastasis of tumor cells isolated from a human renal cell carcinoma implanted into different organs of nude mice. Cancer Res. 1986, 46, 4109–4115. [Google Scholar] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigens | Species Antibodies Raised in | Dilution (IF) | Dilution (WB) | Supplier |
|---|---|---|---|---|
| Akt (pan), mouse | Rabbit, monoclonal | - | 1:2000 | Cell Signaling Technology, Danvers, MA, USA, Cat. #4691 |
| CDK2, human | Rabbit, monoclonal | - | 1:2000 | Cell Signaling Technology, Cat. #2546 |
| CDK4, human | Rabbit, monoclonal | - | 1:2000 | Cell Signaling Technology, Cat. #12790 |
| CDK6, human | Rabbit, monoclonal | - | 1:1000 | Abcam, Cat. #ab124821 |
| Cyclin D1, human | Rabbit, monoclonal | - | 1:2000 | Cell Signaling Technology, Cat. #2978 |
| E-cadherin, human | Rabbit, monoclonal | - | 1:500 | Cell Signaling Technology, Cat. #3195 |
| Glyceraldehyde 3-phosphate dehydrogenase (GAPDH), human | Mouse, monoclonal | - | 1:2000 | Santa Cruz Biotechnology Inc., Dallas, TX, USA, Cat. #sc-365062 |
| N-Cadherin, human | Rabbit, monoclonal | - | 1:1000 | Cell Signaling Technology, Cat. #13116 |
| β-Catenin, human | Rabbit, monoclonal | - | 1:1000 | Cell Signaling Technology, Cat. #8480 |
| p-AKT(Thr308), human | Rabbit, polyclonal | - | 1:1000 | Cell Signaling Technology, Cat. #9275 |
| Vimentin, human | Rabbit, monoclonal | - | 1:2000 | Cell Signaling Technology, Cat. #5741 |
| Ki-67, human | Rabbit, polyclonal | 1:200 | - | Novus Biologicals, Littleton, CO, USA, Cat. #NBP2-19012 |
| Catalase, human | Rabbit, monoclonal | 1:200 | 1:2000 | Abcam, Cat. #ab76024 |
| SOD2, human | Rabbit, monoclonal | 1:200 | 1:1000 | Abcam, Cat. #ab68155 |
| FOXO3a, human | Rabbit, monoclonal | 1:200 | 1:1000 | Abcam, Cat. #ab53287 |
| t-GSK-3β, human | Rabbit, monoclonal | - | 1:15,000 | Cell Signaling Technology, Cat. #12456 |
| p-GSK-3β, human | Rabbit, monoclonal | - | 1:15,000 | Cell Signaling Technology, Cat. #5558 |
| Secondary Detection System Used | Host | Method | Dilution | Supplier |
|---|---|---|---|---|
| Anti-Mouse-IgG (H + L)-HRP | Goat | WB | 1:10,000 | Sungene Biotech, Tianjin, China, Cat. #LK2003 |
| Anti-Rabbit-IgG (H + L)-HRP | Goat | WB | 1:10,000 | Sungene Biotech, Cat. #LK2001 |
| Anti-rabbit IgG (H + L), F(ab′)2 Fragment (Alexa Fluor® 488 Conjugate) | Goat | IF | 1:50 | Cell Signaling Technology, Cat. #4412 |
| Hoechst 33342 nucleic acid staining (DAPI) | - | IF | 1:750 | Molecular Probes/Invitrogen, Carlsbad, CA, USA, Cat. #A11007 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, K.; Wang, G.; Cao, R.; Liu, T.; Qian, G.; Guan, X.; Guo, Z.; Xiao, Y.; Wang, X. Capsaicin Suppresses Cell Proliferation, Induces Cell Cycle Arrest and ROS Production in Bladder Cancer Cells through FOXO3a-Mediated Pathways. Molecules 2016, 21, 1406. https://doi.org/10.3390/molecules21101406
Qian K, Wang G, Cao R, Liu T, Qian G, Guan X, Guo Z, Xiao Y, Wang X. Capsaicin Suppresses Cell Proliferation, Induces Cell Cycle Arrest and ROS Production in Bladder Cancer Cells through FOXO3a-Mediated Pathways. Molecules. 2016; 21(10):1406. https://doi.org/10.3390/molecules21101406
Chicago/Turabian StyleQian, Kaiyu, Gang Wang, Rui Cao, Tao Liu, Guofeng Qian, Xinyuan Guan, Zhongqiang Guo, Yu Xiao, and Xinghuan Wang. 2016. "Capsaicin Suppresses Cell Proliferation, Induces Cell Cycle Arrest and ROS Production in Bladder Cancer Cells through FOXO3a-Mediated Pathways" Molecules 21, no. 10: 1406. https://doi.org/10.3390/molecules21101406
APA StyleQian, K., Wang, G., Cao, R., Liu, T., Qian, G., Guan, X., Guo, Z., Xiao, Y., & Wang, X. (2016). Capsaicin Suppresses Cell Proliferation, Induces Cell Cycle Arrest and ROS Production in Bladder Cancer Cells through FOXO3a-Mediated Pathways. Molecules, 21(10), 1406. https://doi.org/10.3390/molecules21101406