
Enzyme Engineering for In Situ Immobilization

Abstract
:1. Introduction
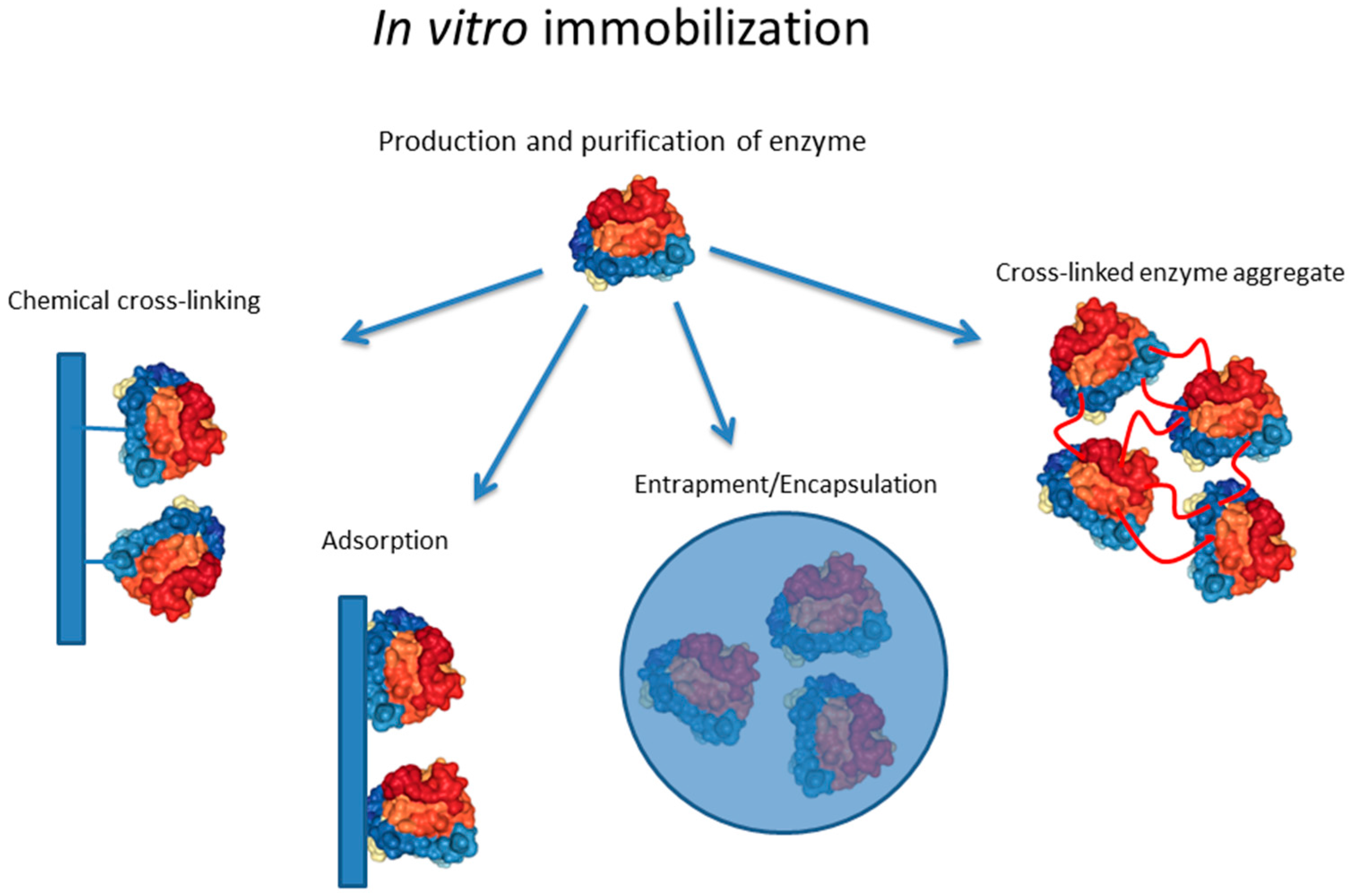
1.1. In Vitro Enzyme Immobilization Strategies
1.1.1. Materials for Immobilization
1.1.2. Chemical and Enzymatic Cross-Linking
1.1.3. Adsorption
1.1.4. Encapsulation/Entrapment
1.1.5. Enzyme Engineering for Immobilization
1.2. In Situ Enzyme Immobilization Strategies
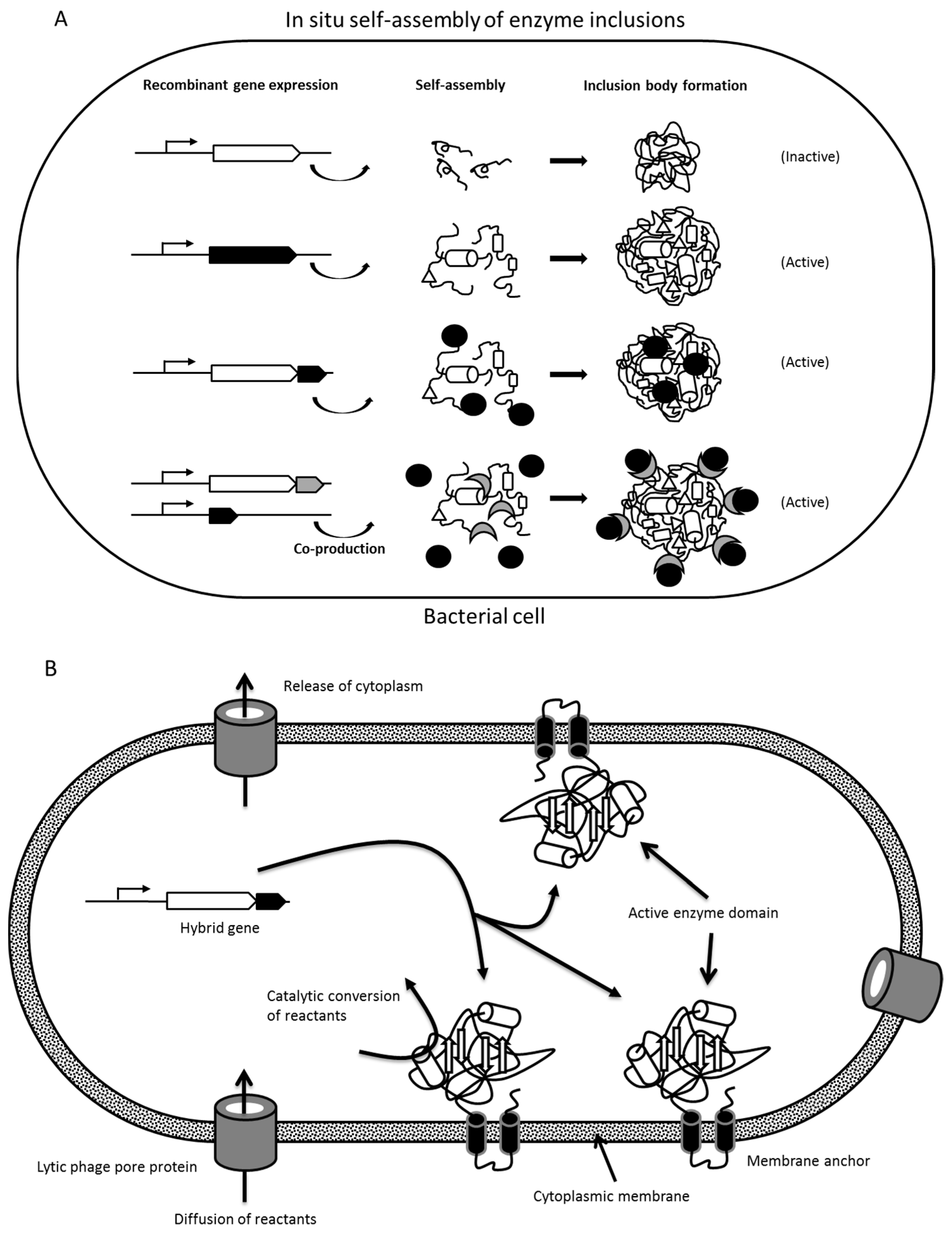
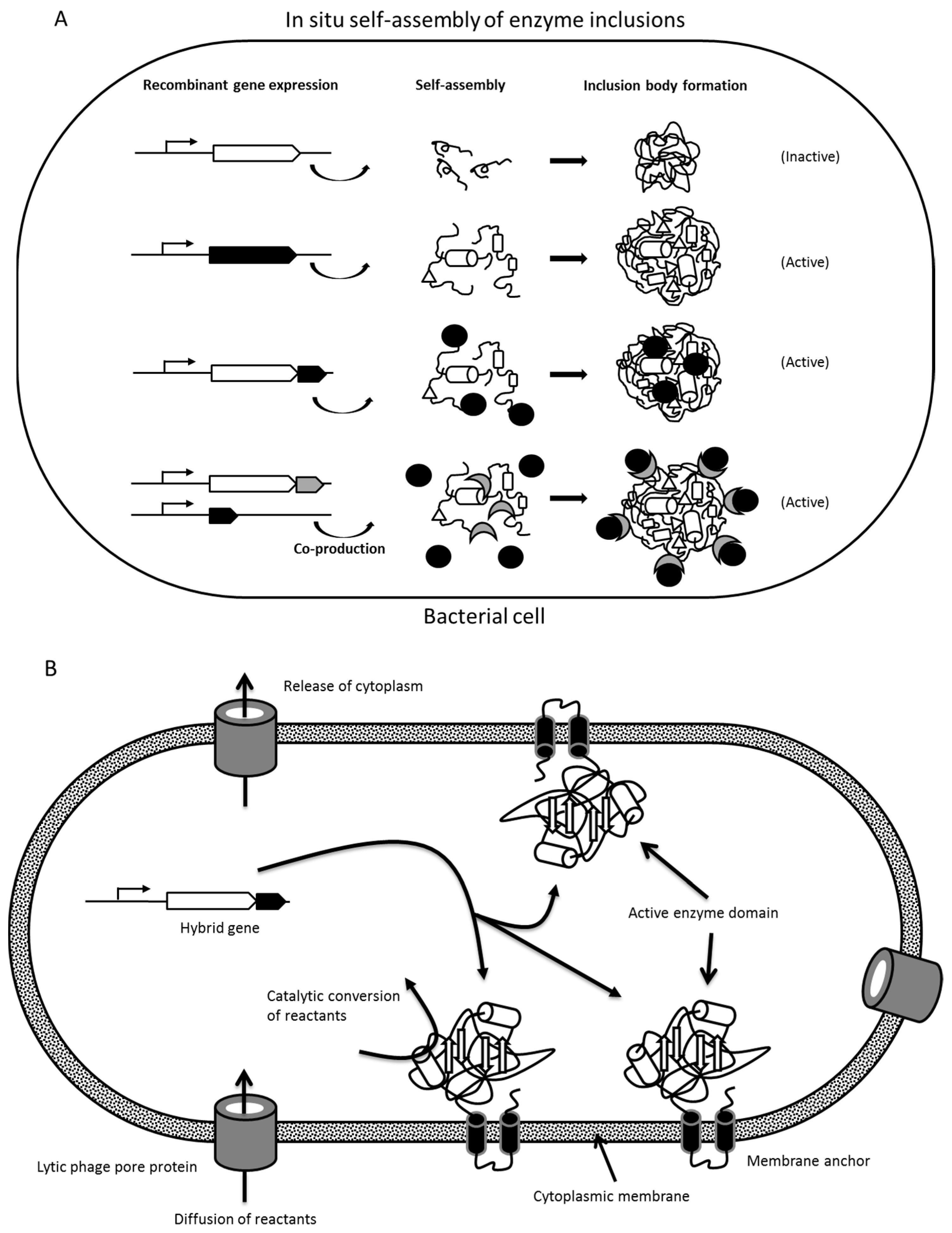
1.2.1. Formation of Protein Inclusion Bodies
1.2.2. Insolubility Tags—Self-Assembly of Engineered Enzymes
1.2.3. Polymer/Lipid Inclusions, Magnetosomes, and Membrane Vesicles
2. Intracellular Self-Assembly towards In Situ Enzyme Immobilization
2.1. Enzyme Inclusion Bodies
2.2. Protein Engineering of Enzymes for In Vivo Self-Assembly
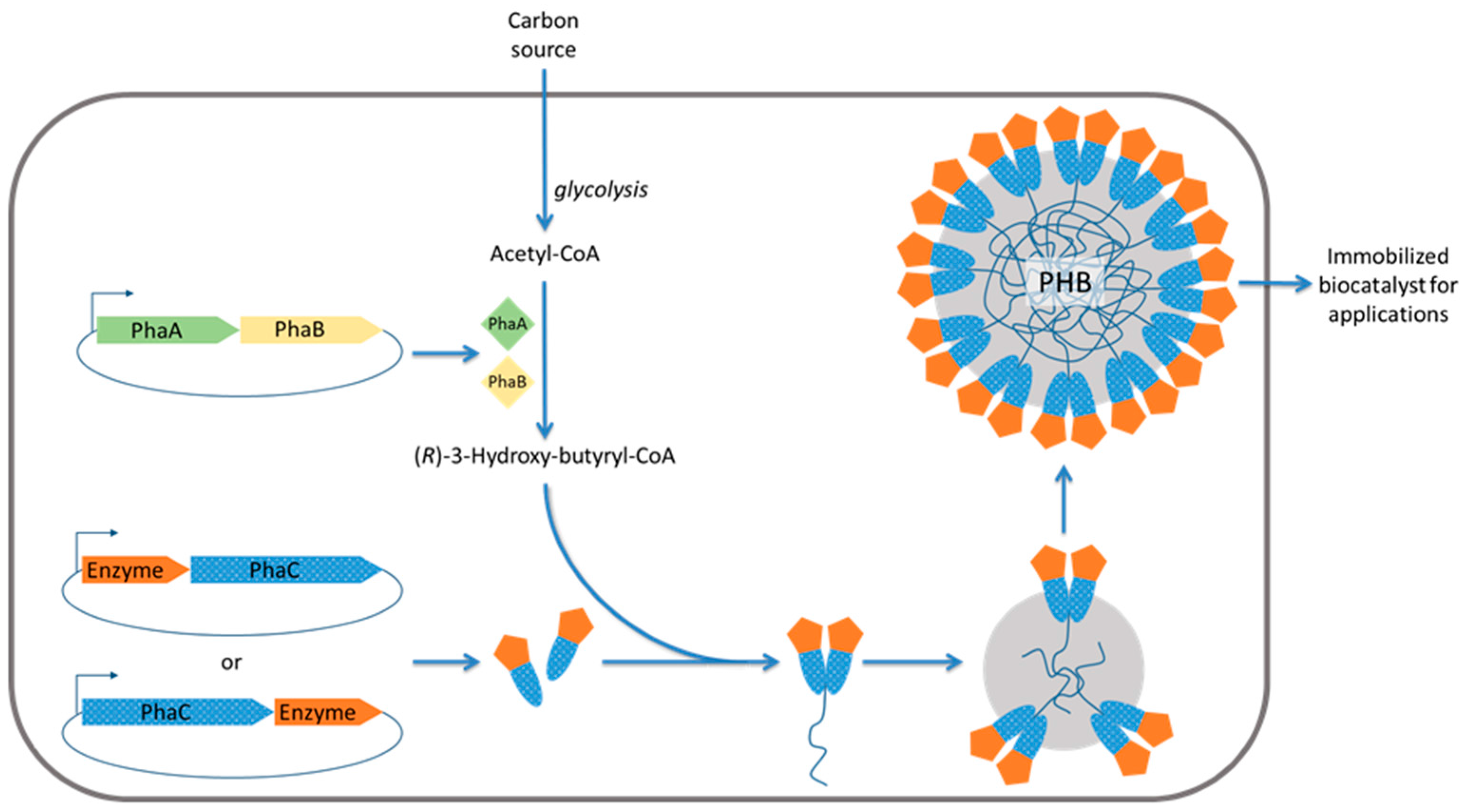
2.3. Engineered Polyhydroxyalkanoate Inclusions for Enzyme Display
2.4. Uses of In Situ Immobilized Enzymes
3. Conclusions and Prospects
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.A.; van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223–6235. [Google Scholar] [CrossRef] [PubMed]
- Bornscheuer, U.T.; Huisman, G.W.; Kazlauskas, R.J.; Lutz, S.; Moore, J.C.; Robins, K. Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Puri, M.; Barrow, C.J.; Verma, M.L. Enzyme immobilization on nanomaterials for biofuel production. Trends Biotechnol. 2013, 31, 215–216. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ramirez, J.; Martinez-Hernandez, J.L.; Segura-Ceniceros, P.; Lopez, G.; Saade, H.; Medina-Morales, M.A.; Ramos-Gonzalez, R.; Aguilar, C.N.; Ilyina, A. Cellulases immobilization on chitosan-coated magnetic nanoparticles: Application for Agave Atrovirens lignocellulosic biomass hydrolysis. Bioprocess. Biosyst. Eng. 2016. [Google Scholar] [CrossRef] [PubMed]
- Uygun, M.; Akduman, B.; Ergonul, B.; Aktas Uygun, D.; Akgol, S.; Denizli, A. Immobilization of amyloglucosidase onto macroporous cryogels for continuous glucose production from starch. J. Biomater. Sci. Polym. Ed. 2015, 26, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Stolarow, J.; Heinzelmann, M.; Yeremchuk, W.; Syldatk, C.; Hausmann, R. Immobilization of trypsin in organic and aqueous media for enzymatic peptide synthesis and hydrolysis reactions. BMC Biotechnol. 2015, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Hooks, D.O.; Blatchford, P.A.; Rehm, B.H. Bioengineering of bacterial polymer inclusions catalyzing the synthesis of N-acetylneuraminic acid. Appl. Environ. Microbiol. 2013, 79, 3116–3121. [Google Scholar] [CrossRef] [PubMed]
- Arduini, F.; Amine, A. Biosensors based on enzyme inhibition. Adv. Biochem. Eng. Biotechnol. 2014, 140, 299–326. [Google Scholar] [PubMed]
- Britton, J.; Raston, C.L.; Weiss, G.A. Rapid protein immobilization for thin film continuous flow biocatalysis. Chem. Commun. 2016, 52, 10159–10162. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kim, J.Y.; Wang, Y.Y.; Qi, L.; Wang, F.Y.; Moon, M.H. Trypsin immobilization in ordered porous polymer membranes for effective protein digestion. Anal. Chim. Acta 2016, 906, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Rastian, Z.; Khodadadi, A.A.; Guo, Z.; Vahabzadeh, F.; Mortazavi, Y. Plasma functionalized multiwalled carbon nanotubes for immobilization of Candida antarctica lipase B: Production of biodiesel from methanolysis of rapeseed oil. Appl. Biochem. Biotechnol. 2016, 178, 974–989. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.C.S.D.; Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Rodrigues, R.C.; Fernandez-Lafuente, R. Importance of the support properties for immobilization or purification of enzymes. ChemCatChem 2015, 7, 2413–2432. [Google Scholar] [CrossRef]
- Arecchi, A.; Scampicchio, M.; Brenna, O.V.; Mannino, S. Biocatalytic nylon nanofibrous membranes. Anal. Bioanal. Chem. 2010, 398, 3097–3103. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.A.; Al-Ghamdi, S.S.; El-Shishtawy, R.M. Immobilization of horseradish peroxidase on amidoximated acrylic polymer activated by cyanuric chloride. Int. J. Biol. Macromol. 2016, 91, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Brown, J.Q.; Zhu, H.; McShane, M.J. Stabilization of glucose oxidase in alginate microspheres with photoreactive diazoresin nanofilm coatings. Biotechnol. Bioeng. 2005, 91, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Hooks, D.O.; Venning-Slater, M.; Du, J.; Rehm, B.H. Polyhydroyxalkanoate synthase fusions as a strategy for oriented enzyme immobilisation. Molecules 2014, 19, 8629–8643. [Google Scholar] [CrossRef] [PubMed]
- Pagan, M.; Suazo, D.; del Toro, N.; Griebenow, K. A comparative study of different protein immobilization methods for the construction of an efficient nano-structured lactate oxidase-SWCNT-biosensor. Biosens. Bioelectron. 2015, 64, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Babadi, A.A.; Bagheri, S.; Hamid, S.B. Progress on implantable biofuel cell: Nano-carbon functionalization for enzyme immobilization enhancement. Biosens. Bioelectron. 2016, 79, 850–860. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T. Practical protocols for lipase immobilization via sol-gel techniques. Methods Mol. Biol. 2013, 1051, 241–254. [Google Scholar] [PubMed]
- Aggarwal, V.; Pundir, C.S. Rational design of nanoparticle platforms for “cutting-the-fat”: Covalent immobilization of lipase, glycerol kinase, and glycerol-3-phosphate oxidase on metal nanoparticles. Methods Enzymol. 2016, 571, 197–223. [Google Scholar] [PubMed]
- Atacan, K.; Ozacar, M. Characterization and immobilization of trypsin on tannic acid modified Fe3O4 nanoparticles. Colloids Surf. B Biointerfaces 2015, 128, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Beladiya, C.; Tripathy, R.K.; Bajaj, P.; Aggarwal, G.; Pande, A.H. Expression, purification and immobilization of recombinant AiiA enzyme onto magnetic nanoparticles. Protein Expr. Purif. 2015, 113, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Hola, K.; Markova, Z.; Zoppellaro, G.; Tucek, J.; Zboril, R. Tailored functionalization of iron oxide nanoparticles for MRI, drug delivery, magnetic separation and immobilization of biosubstances. Biotechnol. Adv. 2015, 33, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Akhond, M.; Pashangeh, K.; Karbalaei-Heidari, H.R.; Absalan, G. Efficient immobilization of porcine pancreatic alpha-amylase on amino-functionalized magnetite nanoparticles: Characterization and stability evaluation of the immobilized enzyme. Appl. Biochem. Biotechnol. 2016, 1–15. [Google Scholar]
- Mohamad, N.R.; Marzuki, N.H.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Strategies for the one-step immobilization-purification of enzymes as industrial biocatalysts. Biotechnol. Adv. 2015, 33, 435–456. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.; Guler, S. Characterization and immobilization of Trametes versicolor laccase on magnetic chitosan-clay composite beads for phenol removal. Artif. Cells Nanomed. Biotechnol. 2015, 43, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Qi, Z.; Zhu, H. Preparation of core-shell magnetic polydopamine/alginate biocomposite for Candida rugosa lipase immobilization. Colloids Surf. B Biointerfaces 2015, 128, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Zhou, L.; Zhu, H.; Wang, X.; Hu, N.; Zeng, F.; Wang, L.; Yin, H. Mussel-inspired surface modification of magnetic@graphite nanosheets composite for efficient Candida rugosa lipase immobilization. J. Ind. Microbiol. Biotechnol. 2015, 42, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Shi, J.; Nie, T.; Zhang, S.; Wang, X.; Yang, P.; Wu, H.; Jiang, Z. Conferring natural-derived porous microspheres with surface multifunctionality through facile coordination-enabled self-assembly process. ACS Appl. Mater. Interfaces 2016, 8, 8076–8085. [Google Scholar] [CrossRef] [PubMed]
- Homaei, A.A.; Sariri, R.; Vianello, F.; Stevanato, R. Enzyme immobilization: An update. J. Chem. Biol. 2013, 6, 185–205. [Google Scholar] [CrossRef] [PubMed]
- Jian, H.; Wang, Y.; Bai, Y.; Li, R.; Gao, R. Site-specific, covalent immobilization of dehalogenase ST2570 catalyzed by formylglycine-generating enzymes and its application in batch and semi-continuous flow reactors. Molecules 2016, 21, 895. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. Glutaraldehyde in bio-catalysts design: A useful crosslinker and a versatile tool in enzyme immobilization. RSC Adv. 2014, 4, 1583–1600. [Google Scholar] [CrossRef]
- Heck, T.; Faccio, G.; Richter, M.; Thony-Meyer, L. Enzyme-catalyzed protein crosslinking. Appl. Microbiol. Biotechnol. 2013, 97, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, Y.; Sheng, J.; Sun, M. Immobilization of Yarrowia lipolytica lipase on macroporous resin using different methods: Characterization of the biocatalysts in hydrolysis reaction. BioMed Res. Int. 2015, 2015, 139179. [Google Scholar] [CrossRef] [PubMed]
- Younus, H.; Rajcani, J.; Ulbrich-Hofmann, R.; Saleemuddin, M. Behaviour of a recombinant cabbage (Brassica oleracea) phospholipase D immobilized on CNBr-activated and antibody supports. Biotechnol. Appl. Biochem. 2004, 40, 95–99. [Google Scholar] [PubMed]
- Ungurean, M.; Paul, C.; Peter, F. Cellulase immobilized by sol-gel entrapment for efficient hydrolysis of cellulose. Bioprocess. Biosyst. Eng. 2013, 36, 1327–1338. [Google Scholar] [CrossRef] [PubMed]
- Buthe, A. Entrapment of enzymes in nanoporous sol-gels. Methods Mol. Biol. 2011, 743, 223–237. [Google Scholar] [PubMed]
- Sassolas, A.; Hayat, A.; Marty, J.L. Enzyme immobilization by entrapment within a gel network. Methods Mol. Biol. 2013, 1051, 229–239. [Google Scholar] [PubMed]
- Steen Redeker, E.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein engineering for directed immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Shanbhag, B.K.; Liu, B.; Fu, J.; Haritos, V.S.; He, L. Self-assembled enzyme nanoparticles for carbon dioxide capture. Nano Lett. 2016, 16, 3379–3384. [Google Scholar] [CrossRef] [PubMed]
- Peternel, S.; Komel, R. Active protein aggregates produced in Escherichia coli. Int. J. Mol. Sci. 2011, 12, 8275–8287. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, F.; Rinas, U. Physiological Stress Responses in Bioprocesses; Springer: New York, NY, USA, 2004; pp. 73–92. [Google Scholar]
- Rokney, A.; Shagan, M.; Kessel, M.; Smith, Y.; Rosenshine, I.; Oppenheim, A.B. E. Coli transports aggregated proteins to the poles by a specific and energy-dependent process. J. Mol. Biol. 2009, 392, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Schrodel, A.; de Marco, A. Characterization of the aggregates formed during recombinant protein expression in bacteria. BMC Biochem. 2005, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fruitos, E.; Sabate, R.; de Groot, N.S.; Villaverde, A.; Ventura, S. Biological role of bacterial inclusion bodies: A model for amyloid aggregation. FEBS J. 2011, 278, 2419–2427. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, B.; Christmann, A.; Heiseler, T.; Fritz, J.; Kolmar, H. In vivo enzyme immobilization by inclusion body display. Appl. Environ. Microbiol. 2010, 76, 5563–5569. [Google Scholar] [CrossRef] [PubMed]
- Peternel, S.; Bele, M.; Gaberc-Porekar, V.; Komel, R. Inclusion bodies contraction with implications in biotechnology. Acta Chim. Slov. 2008, 55, 608–612. [Google Scholar]
- Garcia-Fruitos, E.; Vazquez, E.; Diez-Gil, C.; Luis Corchero, J.; Seras-Franzoso, J.; Ratera, I.; Veciana, J.; Villaverde, A. Bacterial inclusion bodies: Making gold from waste. Trends Biotechnol. 2012, 30, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, A.K.; Murmu, A.; Singh, A.; Panda, A.K. Kinetics of inclusion body formation and its correlation with the characteristics of protein aggregates in Escherichia coli. PLoS ONE 2012, 7, e33951. [Google Scholar] [CrossRef] [PubMed]
- Peternel, S.; Jevsevar, S.; Bele, M.; Gaberc-Porekar, V.; Menart, V. New properties of inclusion bodies with implications for biotechnology. Biotechnol. Appl. Biochem. 2008, 49, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fruitos, E.; Rodriguez-Carmona, E.; Diez-Gil, C.; Ferraz, R.M.; Vazquez, E.; Luis Corchero, J.; Cano-Sarabia, M.; Ratera, I.; Ventosa, N.; Veciana, J.; et al. Surface cell growth engineering assisted by a novel bacterial nanomaterial. Adv. Mater. 2009, 21, 4249–4253. [Google Scholar] [CrossRef]
- Peternel, S.; Komel, R. Isolation of biologically active nanomaterial (inclusion bodies) from bacterial cells. Microb. Cell. Fact. 2010, 9, 66. [Google Scholar] [CrossRef] [PubMed]
- Jahns, A.C.; Maspolim, Y.; Chen, S.; Guthrie, J.; Blackwell, L.; Rehm, B. In vivo self-assembly of fluorescent protein microparticles displaying specific binding domains. Bioconjugate Chem. 2013, 24, 1314–1323. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H. Enhanced enzyme activities of inclusion bodies of recombinant beta-galactosidase via the addition of inducer analog after l-arabinose induction in the arabad promoter system of Escherichia coli. J. Microbiol. Biotechnol. 2008, 18, 434–442. [Google Scholar] [PubMed]
- Park, S.Y.; Park, S.H.; Choi, S.K. Active inclusion body formation using Paenibacillus polymyxa PoxB as a fusion partner in Escherichia coli. Anal. Biochem. 2012, 426, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Arnau, J.; Lauritzen, C.; Petersen, G.E.; Pedersen, J. Current strategies for the use of affinity tags and tag removal for the purification of recombinant proteins. Protein Express. Purif. 2006, 48, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Waugh, D.S. Making the most of affinity tags. Trends Biotechnol. 2005, 23, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biot. 2003, 60, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Venning-Slater, M.; Hooks, D.O.; Rehm, B.H. In vivo self-assembly of stable green fluorescent protein fusion particles and their uses in enzyme immobilization. Appl. Environ Microbiol 2014, 80, 3062–3071. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, H.M.; Steinbuchel, A. Triacylglycerols in prokaryotic microorganisms. Appl. Microbiol. Biotechnol. 2002, 60, 367–376. [Google Scholar] [PubMed]
- Anderson, A.J.; Haywood, G.W.; Dawes, E.A. Biosynthesis and composition of bacterial poly(hydroxyalkanoates). Int. J. Biol. Macromol. 1990, 12, 102–105. [Google Scholar] [CrossRef]
- Jendrossek, D. Polyhydroxyalkanoate granules are complex subcellular organelles (carbonosomes). J. Bacteriol. 2009, 191, 3195–3202. [Google Scholar] [CrossRef] [PubMed]
- Grage, K.; Jahns, A.C.; Parlane, N.; Palanisamy, R.; Rasiah, I.A.; Atwood, J.A.; Rehm, B.H. Bacterial polyhydroxyalkanoate granules: Biogenesis, structure, and potential use as nano-/micro-beads in biotechnological and biomedical applications. Biomacromolecules 2009, 10, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Parlane, N.; Gupta, S.; Rubio Reyes, P.; Chen, S.; Gonzalez Miro, M.; Wedlock, D.N.; Rehm, B.H.A. Self-assembled protein-coated polyhydroxyalkanoate beads: Properties and biomedical applications. ACS Biomater. Sci. Eng. 2016, in press. [Google Scholar] [CrossRef]
- Draper, J.L.; Rehm, B.H. Engineering bacteria to manufacture functionalized polyester beads. Bioengineered 2012, 3, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Peters, V.; Rehm, B.H. In vivo enzyme immobilization by use of engineered polyhydroxyalkanoate synthase. Appl. Environ. Microbiol. 2006, 72, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Rasiah, I.A.; Rehm, B.H.A. One-step production of immobilized alpha-amylase in recombinant Escherichia coli. Appl. Environ. Microbiol. 2009, 75, 2012–2016. [Google Scholar] [CrossRef] [PubMed]
- Jahns, A.C.; Rehm, B.H. Immobilization of active lipase B from Candida antarctica on the surface of polyhydroxyalkanoate inclusions. Biotechnol. Lett. 2015, 37, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, J.; Waltermann, M.; Robenek, H.; Steinbuchel, A. The Ralstonia eutropha H16 phasin PhaP1 is targeted to intracellular triacylglycerol inclusions in Rhodococcus opacus PD630 and Mycobacterium smegmatis mc2155, and provides an anchor to target other proteins. Microbiology 2006, 152, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Tanaka, T.; Yoshino, T. Stoichiometrically controlled immobilization of multiple enzymes on magnetic nanoparticles by the magnetosome display system for efficient cellulose hydrolysis. Biomacromolecules 2015, 16, 3863–3868. [Google Scholar] [CrossRef] [PubMed]
- Ginet, N.; Pardoux, R.; Adryanczyk, G.; Garcia, D.; Brutesco, C.; Pignol, D. Single-step production of a recyclable nanobiocatalyst for organophosphate pesticides biodegradation using functionalized bacterial magnetosomes. PLoS ONE 2011, 6, e21442. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, S.; Schuler, D. In vivo display of a multisubunit enzyme complex on biogenic magnetic nanoparticles. Appl. Environ. Microbiol. 2009, 75, 7734–7738. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.; Schuler, D.; Faivre, D. Synthesis of magnetite nanoparticles for bio- and nanotechnology: Genetic engineering and biomimetics of bacterial magnetosomes. Macromol. Biosci. 2007, 7, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Suhrer, I.; Langemann, T.; Lubitz, W.; Weuster-Botz, D.; Castiglione, K. A novel one-step expression and immobilization method for the production of biocatalytic preparations. Microb. Cell. Fact. 2015, 14, 180. [Google Scholar] [CrossRef] [PubMed]
- Hay, I.D.; Du, J.; Burr, N.; Rehm, B.H. Bioengineering of bacteria to assemble custom-made polyester affinity resins. Appl. Environ. Microbiol. 2015, 81, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.T.; Quyen, D.T.; Vu, H.D. Highly effective renaturation of a streptokinase from Streptococcus pyogenes DT7 as inclusion bodies overexpressed in Escherichia coli. BioMed Res. Int. 2014, 2014, 324705. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, G.S.; Poloznikov, A.A.; Chubar, T.A.; Gazaryan, I.G.; Tishkov, V.I. High-yield reactivation of anionic tobacco peroxidase overexpressed in Escherichia coli. Protein Expres. Purif. 2015, 113, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Dutta, A.K.; Ramnath; Dkhar, B.; Tandon, V.; Das, B. Cloning and expression of phosphoenolpyruvate carboxykinase from a cestode parasite and its solubilization from inclusion bodies using l-arginine. Protein Express. Purif. 2016, 125, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.; Guo, P.-C.; Jiang, W.-L.; Fan, X.-M.; Luo, X.-Y.; Li, H.-H. Expression of nattokinase in Escherichia coli and renaturation of its inclusion body. J. Biotechnol. 2016, 231, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Fruitos, E.; Gonzalez-Montalban, N.; Morell, M.; Vera, A.; Ferraz, R.M.; Aris, A.; Ventura, S.; Villaverde, A. Aggregation as bacterial inclusion bodies does not imply inactivation of enzymes and fluorescent proteins. Microb. Cell Fact. 2005, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Spieler, V.; Valldorf, B.; Maass, F.; Kleinschek, A.; Huettenhain, S.H.; Kolmar, H. Coupled reactions on bioparticles: Stereoselective reduction with cofactor regeneration on PhaC inclusion bodies. Biotechnol. J. 2016, 11, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Steinbuchel, A.; Aerts, K.; Babel, W.; Follner, C.; Liebergesell, M.; Madkour, M.H.; Mayer, F.; Pieper-Furst, U.; Pries, A.; Valentin, H.E.; et al. Considerations on the structure and biochemistry of bacterial polyhydroxyalkanoic acid inclusions. Can. J. Microbiol. 1995, 41 (Suppl. 1), 94–105. [Google Scholar] [CrossRef] [PubMed]
- Rehm, B.H. Polyester synthases: Natural catalysts for plastics. Biochem. J. 2003, 376, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Normi, Y.M.; Hiraishi, T.; Taguchi, S.; Abe, H.; Sudesh, K.; Najimudin, N.; Doi, Y. Characterization and properties of G4X mutants of Ralstonia eutropha PHA synthase for poly(3-hydroxybutyrate) biosynthesis in Escherichia coli. Macromol. Biosci. 2005, 5, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Satoh, Y.; Tajima, K.; Tannai, H.; Munekata, M. Enzyme-catalyzed poly(3-hydroxybutyrate) synthesis from acetate with CoA recycling and NADPH regeneration in vitro. J. Biosci. Bioeng. 2003, 95, 335–341. [Google Scholar] [CrossRef]
- Robins, K.J.; Hooks, D.O.; Rehm, B.H.; Ackerley, D.F. Escherichia coli NemA is an efficient chromate reductase that can be biologically immobilized to provide a cell free system for remediation of hexavalent chromium. PLoS ONE 2013, 8, e59200. [Google Scholar] [CrossRef] [PubMed]
- Blatchford, P.A.; Scott, C.; French, N.; Rehm, B.H. Immobilization of organophosphohydrolase OpdA from Agrobacterium radiobacter by overproduction at the surface of polyester inclusions inside engineered Escherichia coli. Biotechnol. Bioeng. 2012, 109, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Hay, I.D.; Du, J.; Reyes, P.R.; Rehm, B.H. In vivo polyester immobilized sortase for tagless protein purification. Microb. Cell Fact. 2015, 14, 190. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Wang, S.; Qi, Q. Expression of active recombinant human tissue-type plasminogen activator by using in vivo polyhydroxybutyrate granule display. Appl. Environ. Microbiol. 2010, 76, 7226–7230. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, J.A.; Rehm, B.H. Design of a single-chain multi-enzyme fusion protein establishing the polyhydroxybutyrate biosynthesis pathway. J. Biotechnol. 2010, 147, 31–36. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Enzyme Engineering | Carrier Type/Site | Mode of Interaction |
|---|---|---|
Genetic engineering of insertion or fusion:
|
|
|
Chemical modifications:
|
|
|
Enzymatic modifications:
|
|
|
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehm, F.B.H.; Chen, S.; Rehm, B.H.A. Enzyme Engineering for In Situ Immobilization. Molecules 2016, 21, 1370. https://doi.org/10.3390/molecules21101370
Rehm FBH, Chen S, Rehm BHA. Enzyme Engineering for In Situ Immobilization. Molecules. 2016; 21(10):1370. https://doi.org/10.3390/molecules21101370
Chicago/Turabian StyleRehm, Fabian B. H., Shuxiong Chen, and Bernd H. A. Rehm. 2016. "Enzyme Engineering for In Situ Immobilization" Molecules 21, no. 10: 1370. https://doi.org/10.3390/molecules21101370
APA StyleRehm, F. B. H., Chen, S., & Rehm, B. H. A. (2016). Enzyme Engineering for In Situ Immobilization. Molecules, 21(10), 1370. https://doi.org/10.3390/molecules21101370