
The Prodrug Approach: A Successful Tool for Improving Drug Solubility

 ,
, 
Abstract
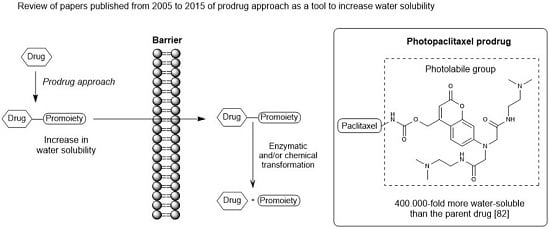
:
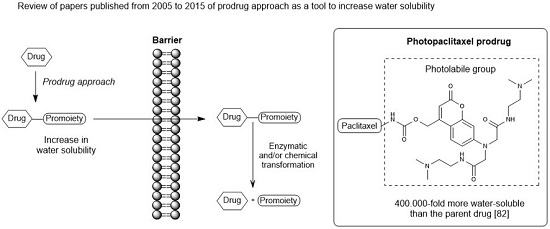
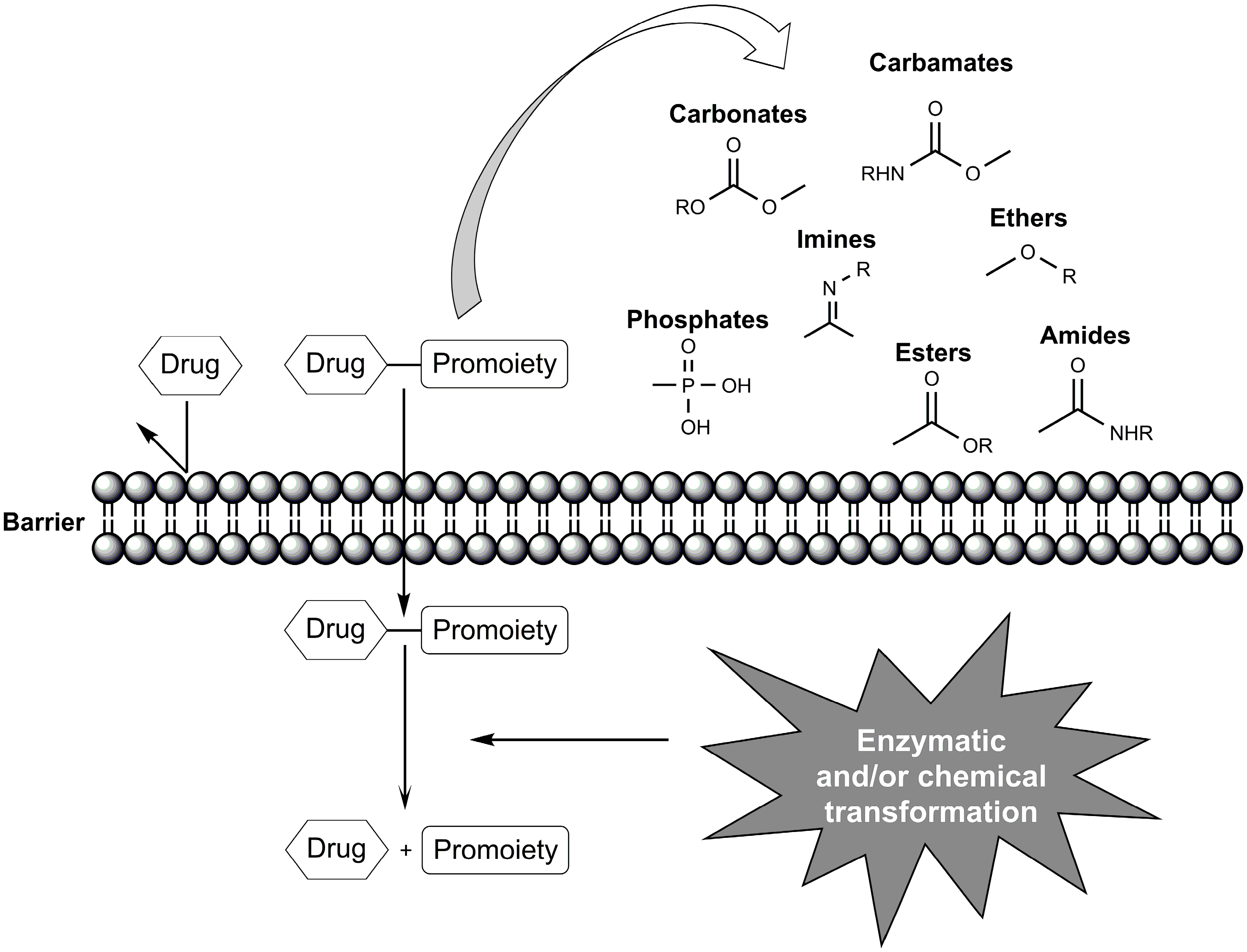
1. Introduction
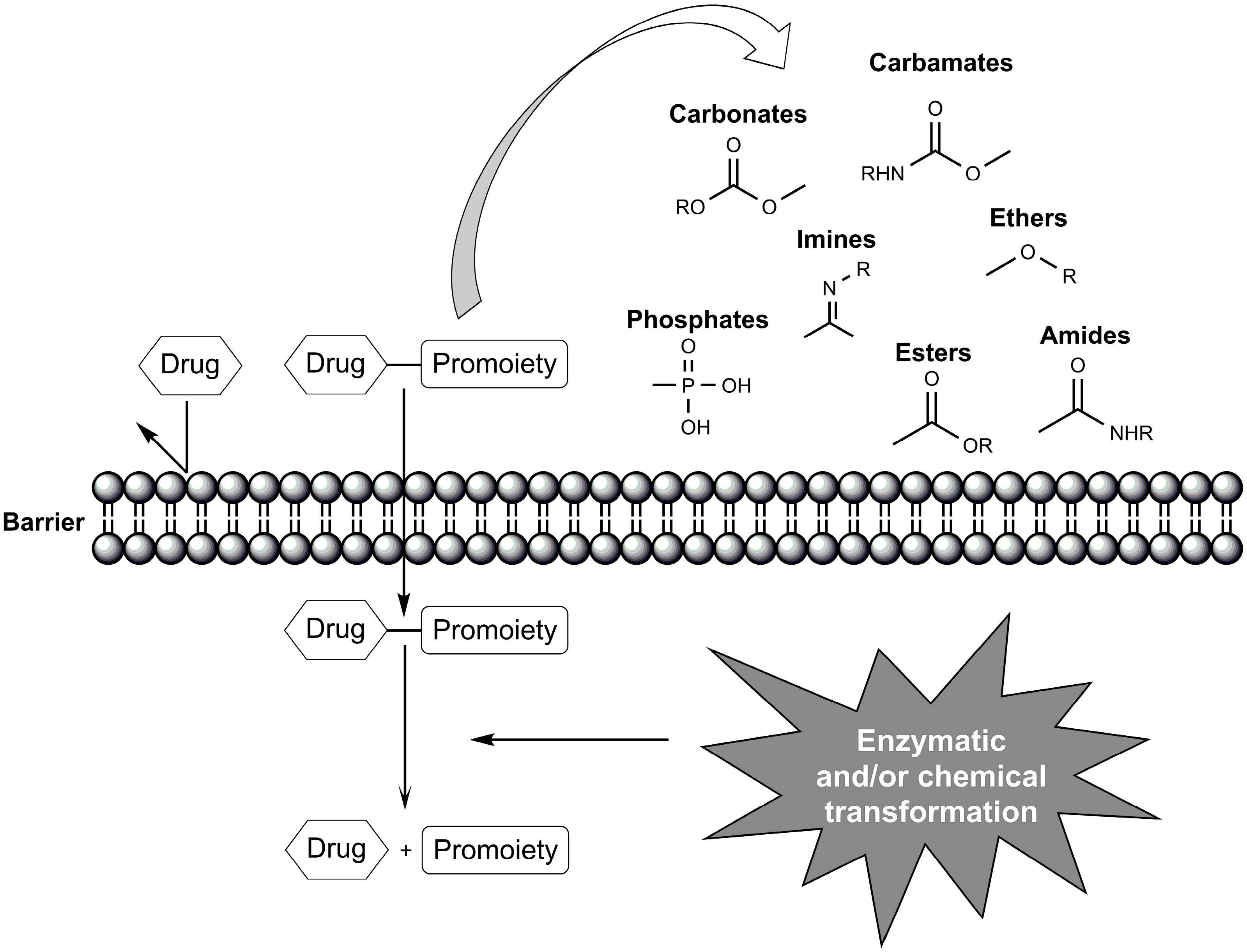
2. Ester Prodrugs
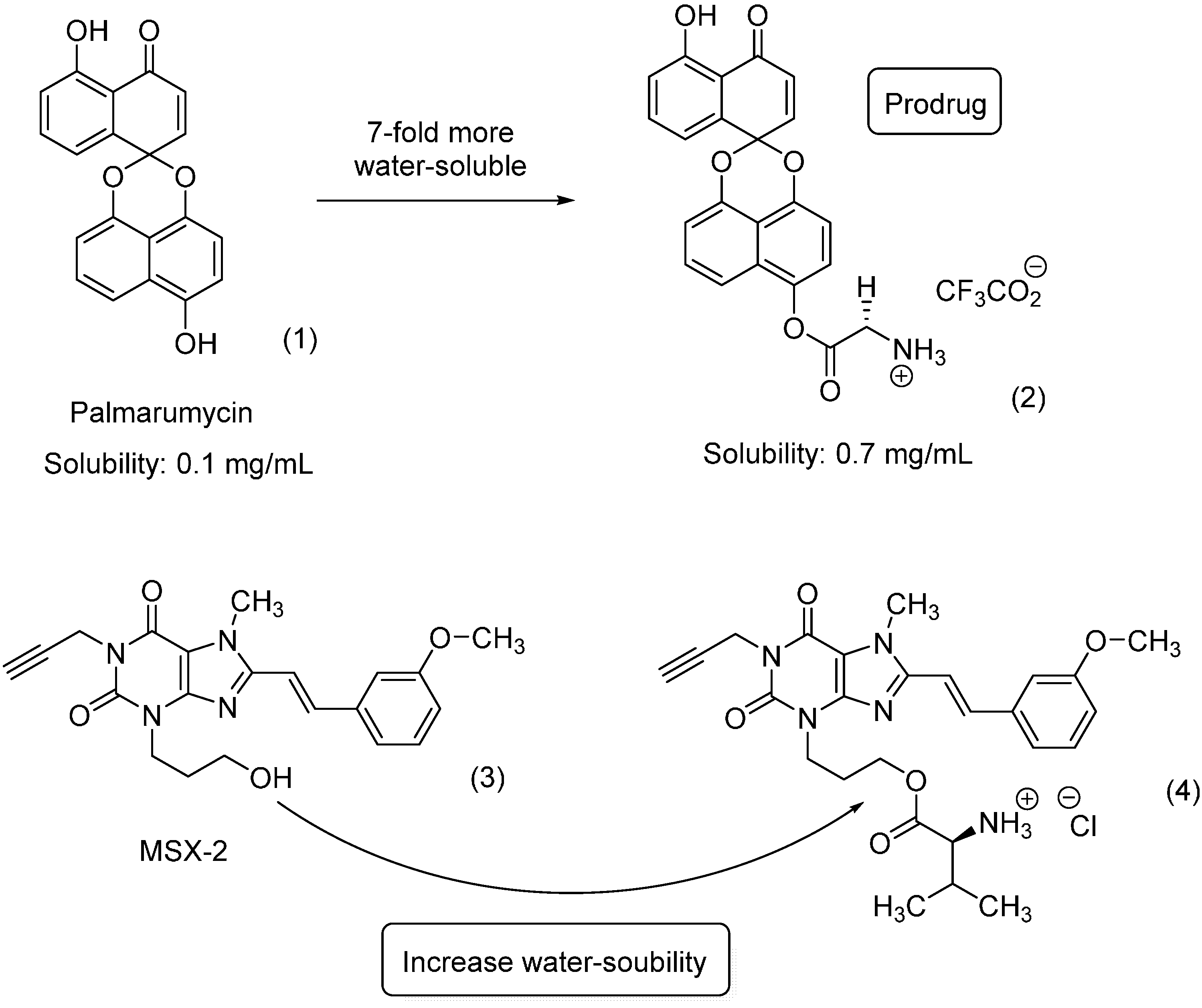

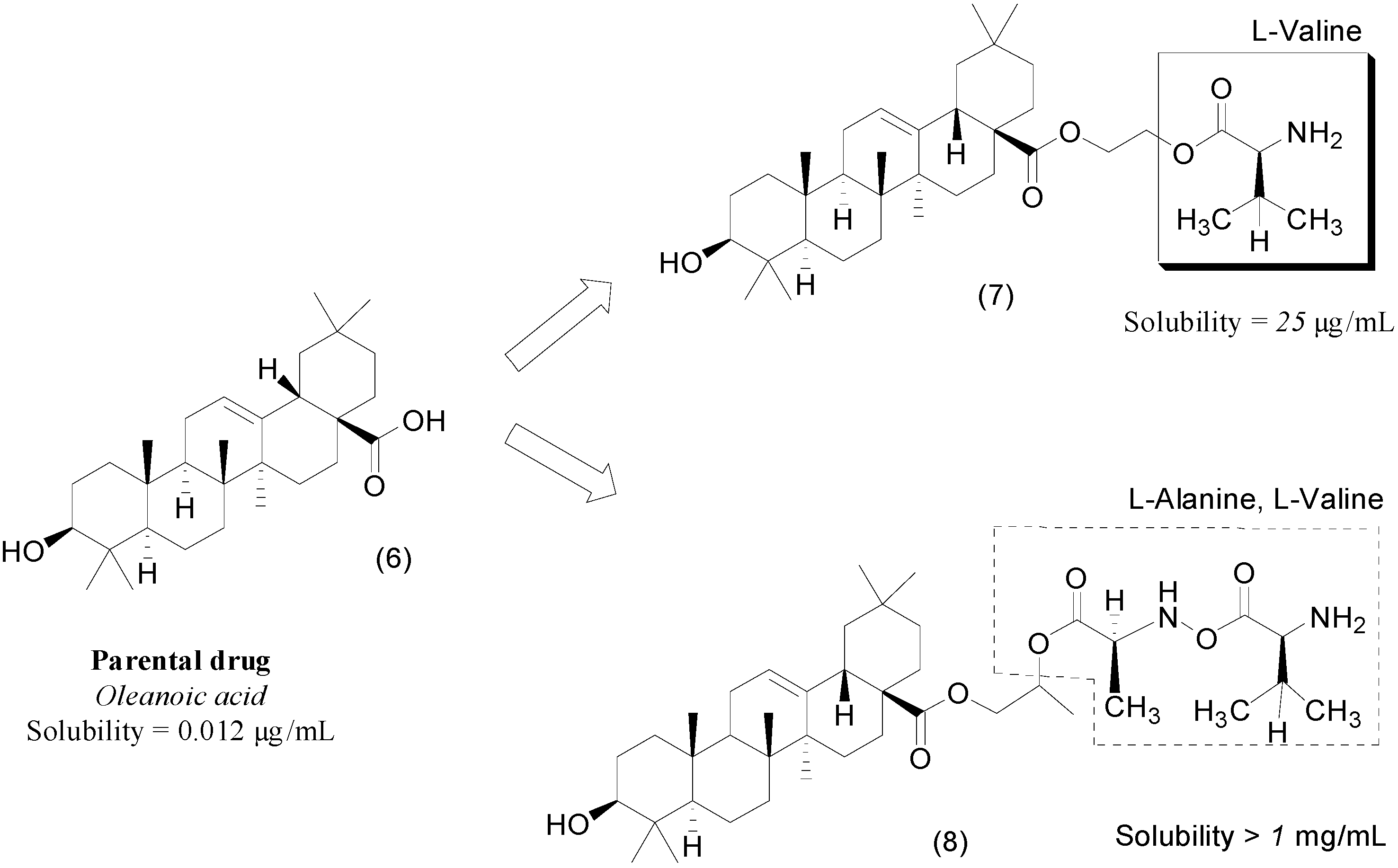
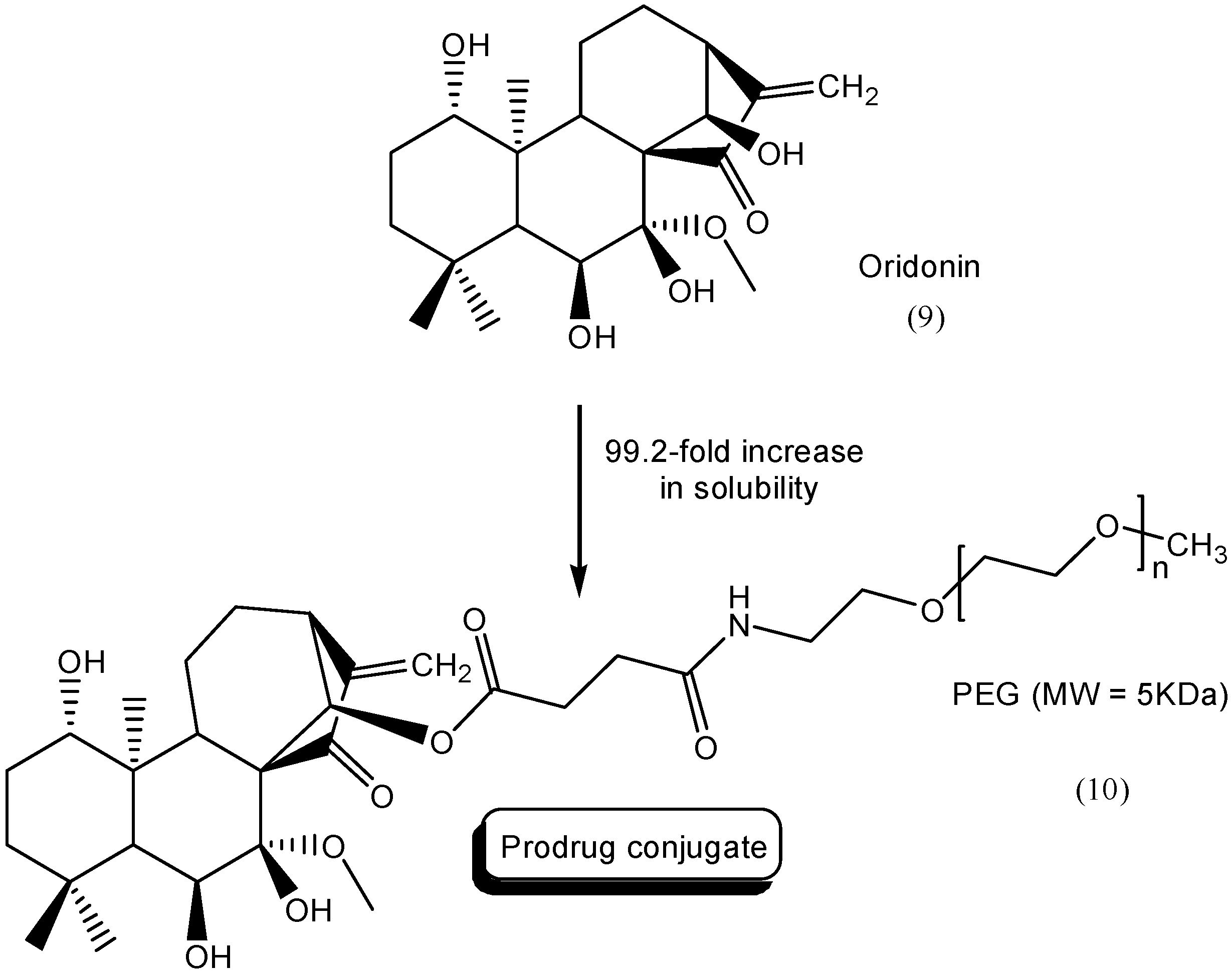
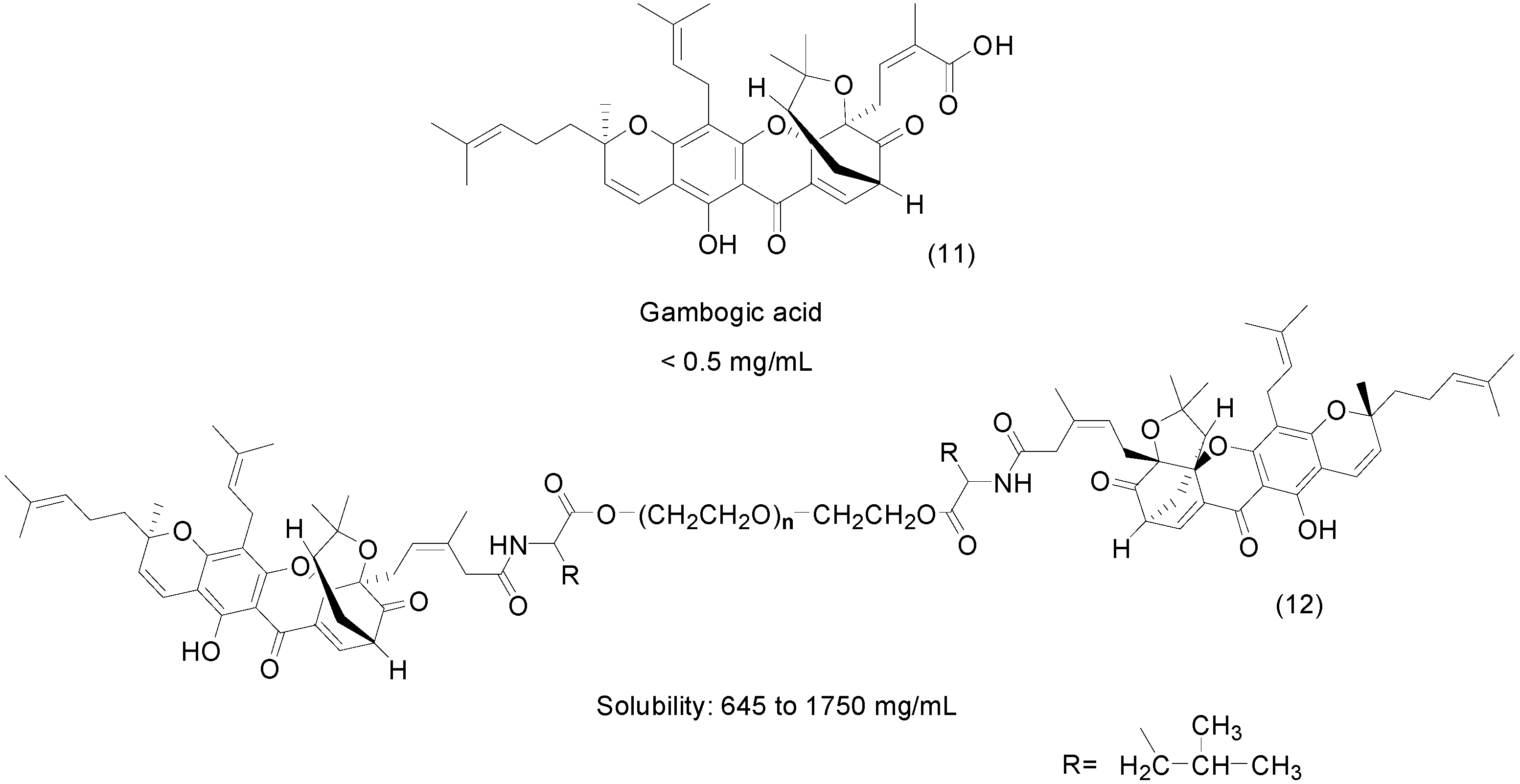
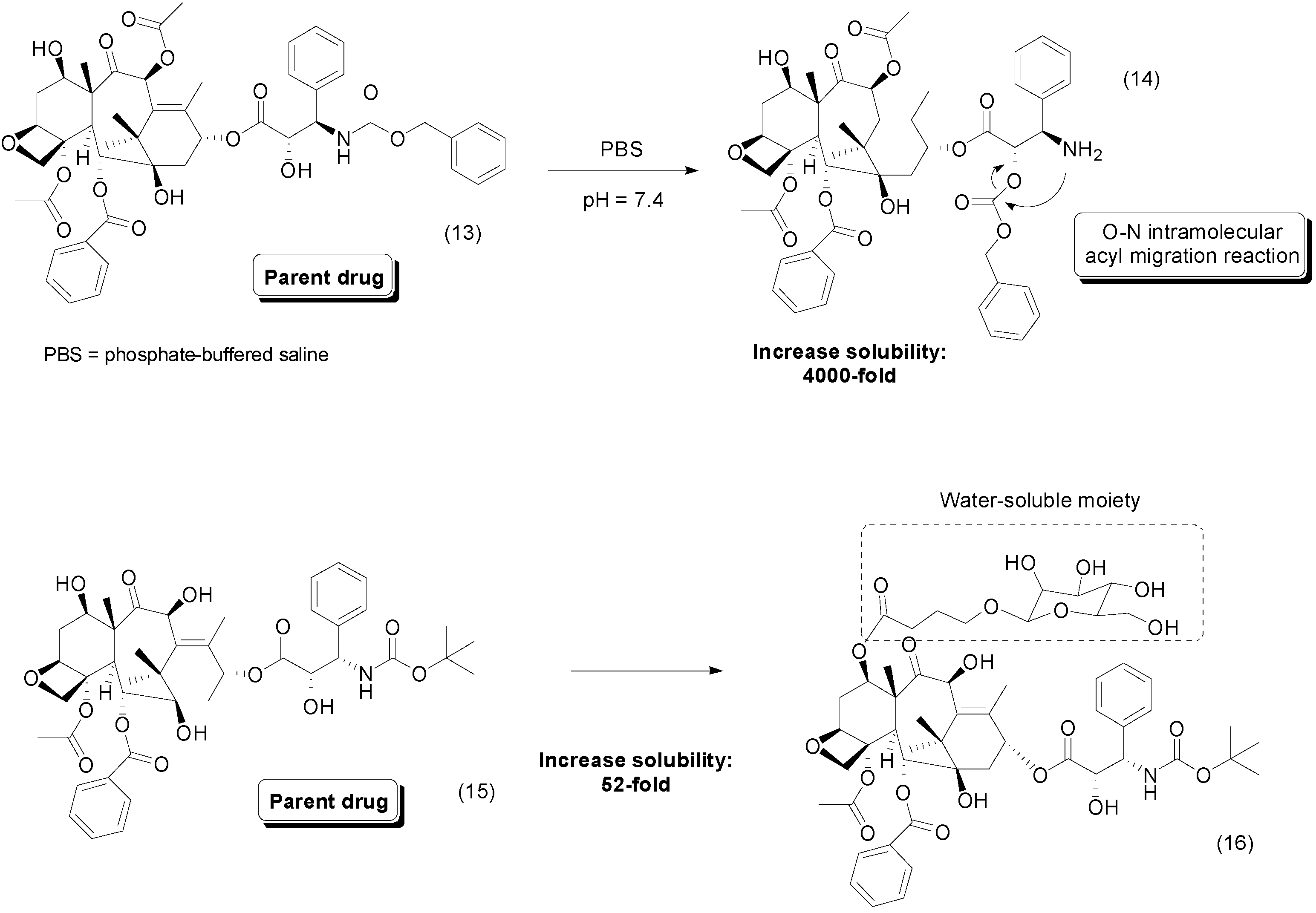
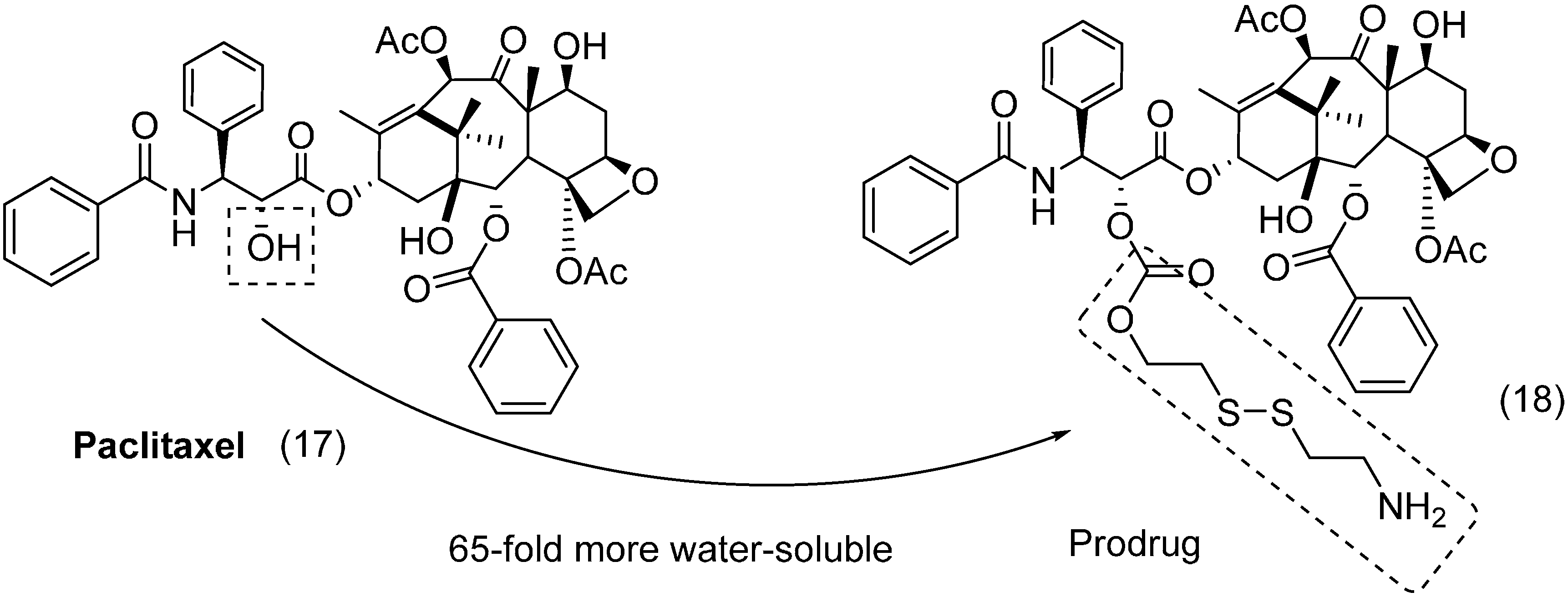
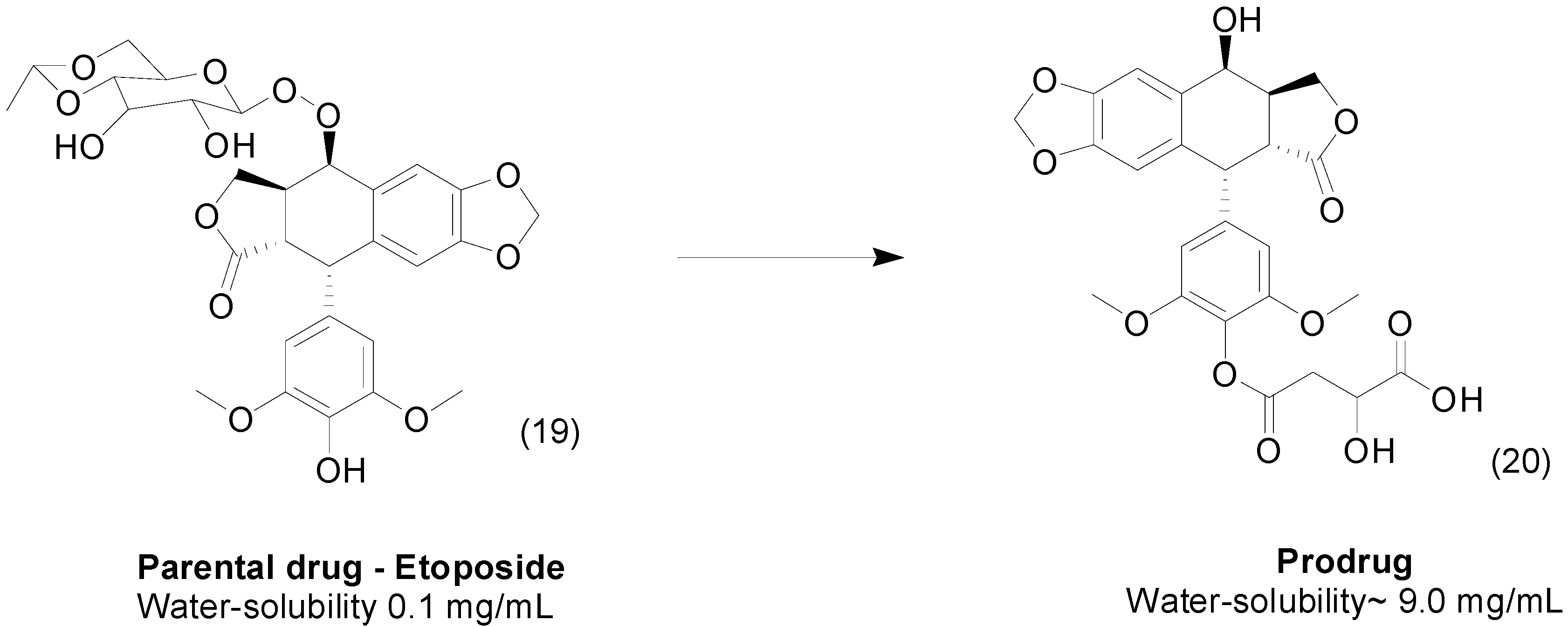
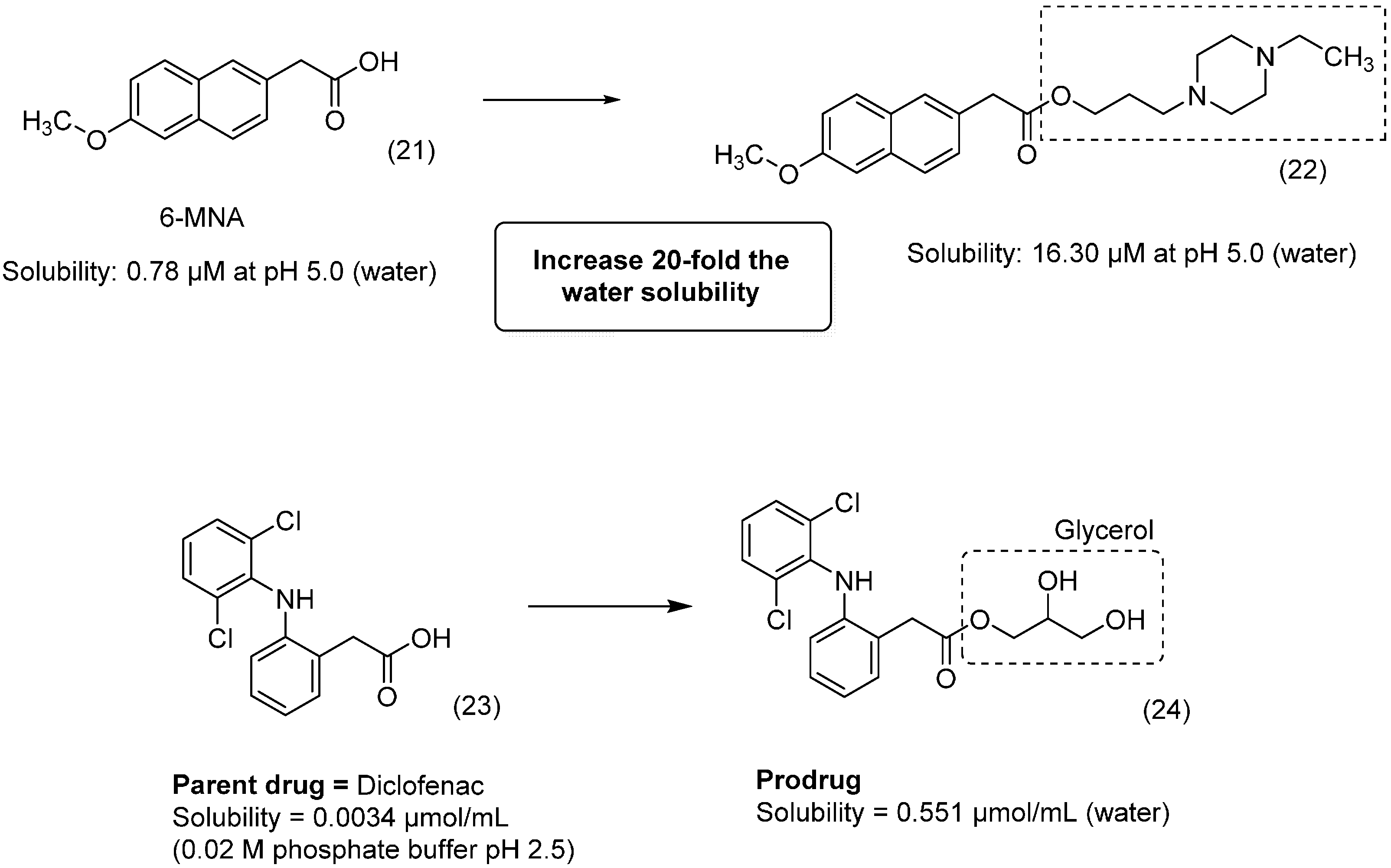
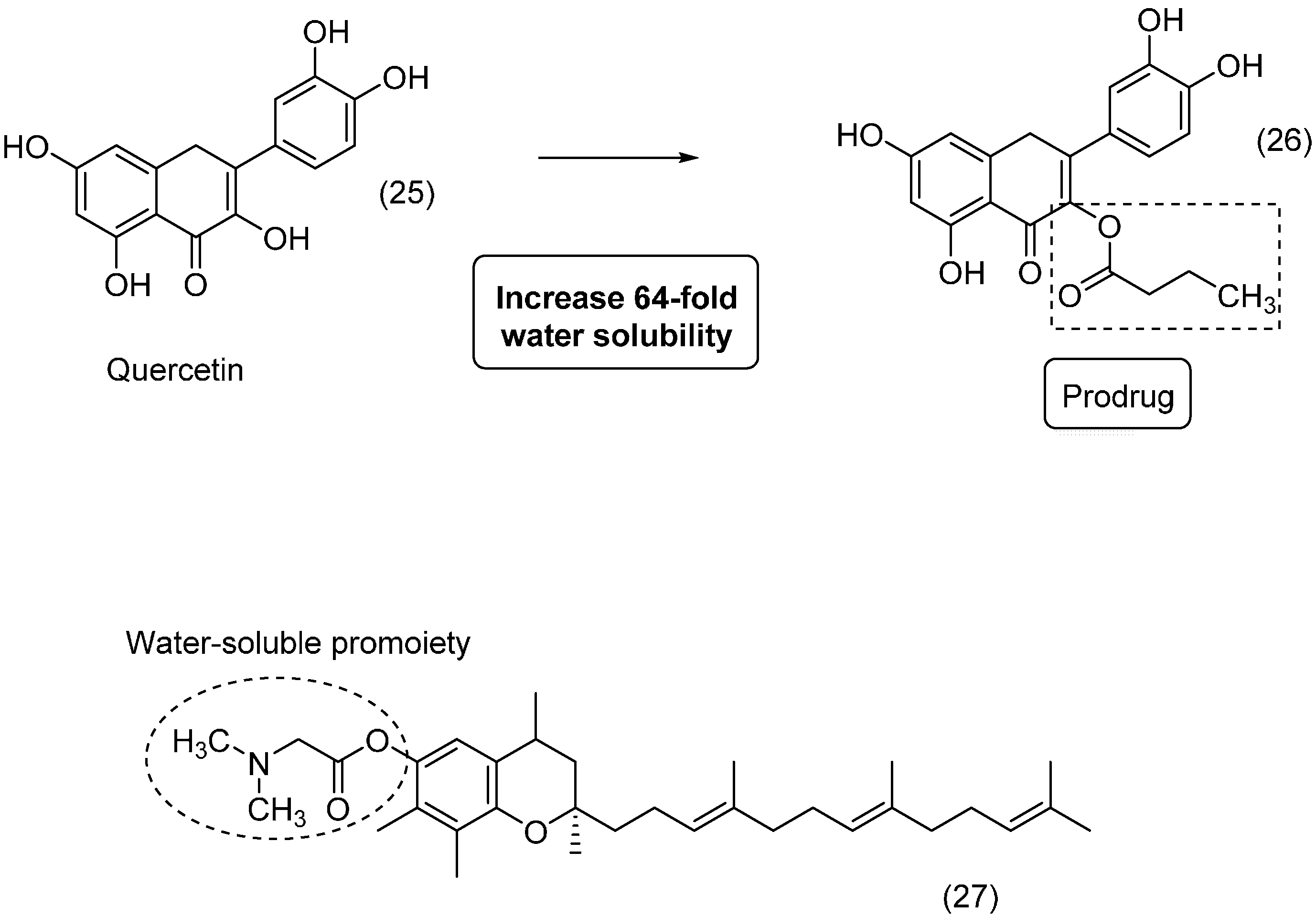
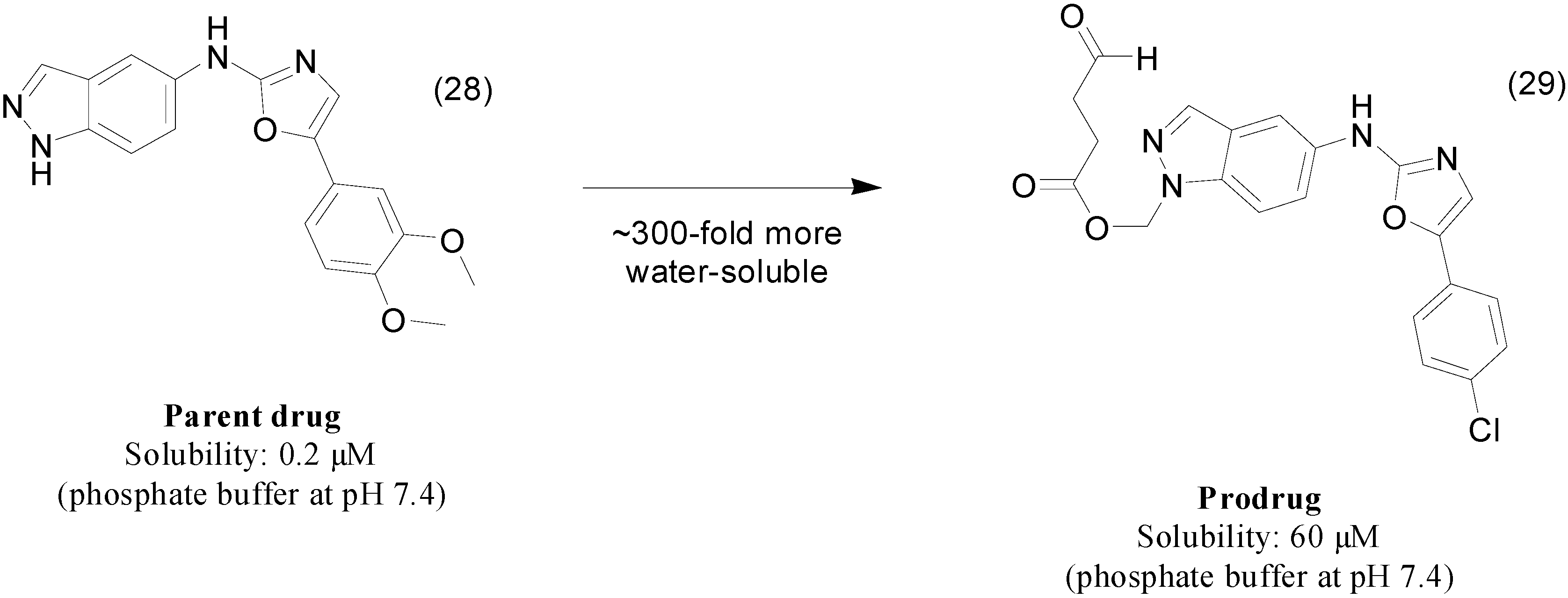
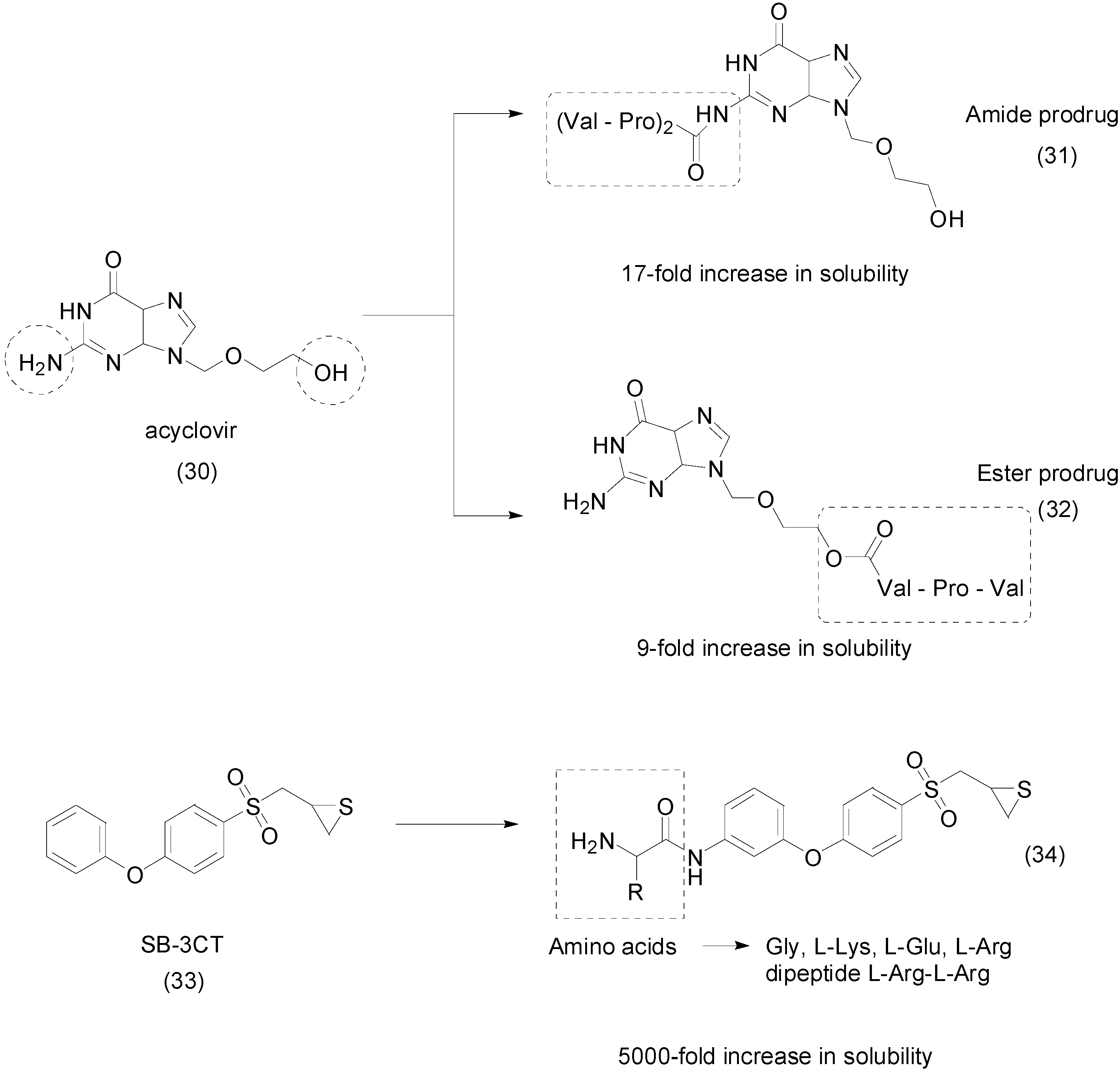
3. Amide Prodrugs
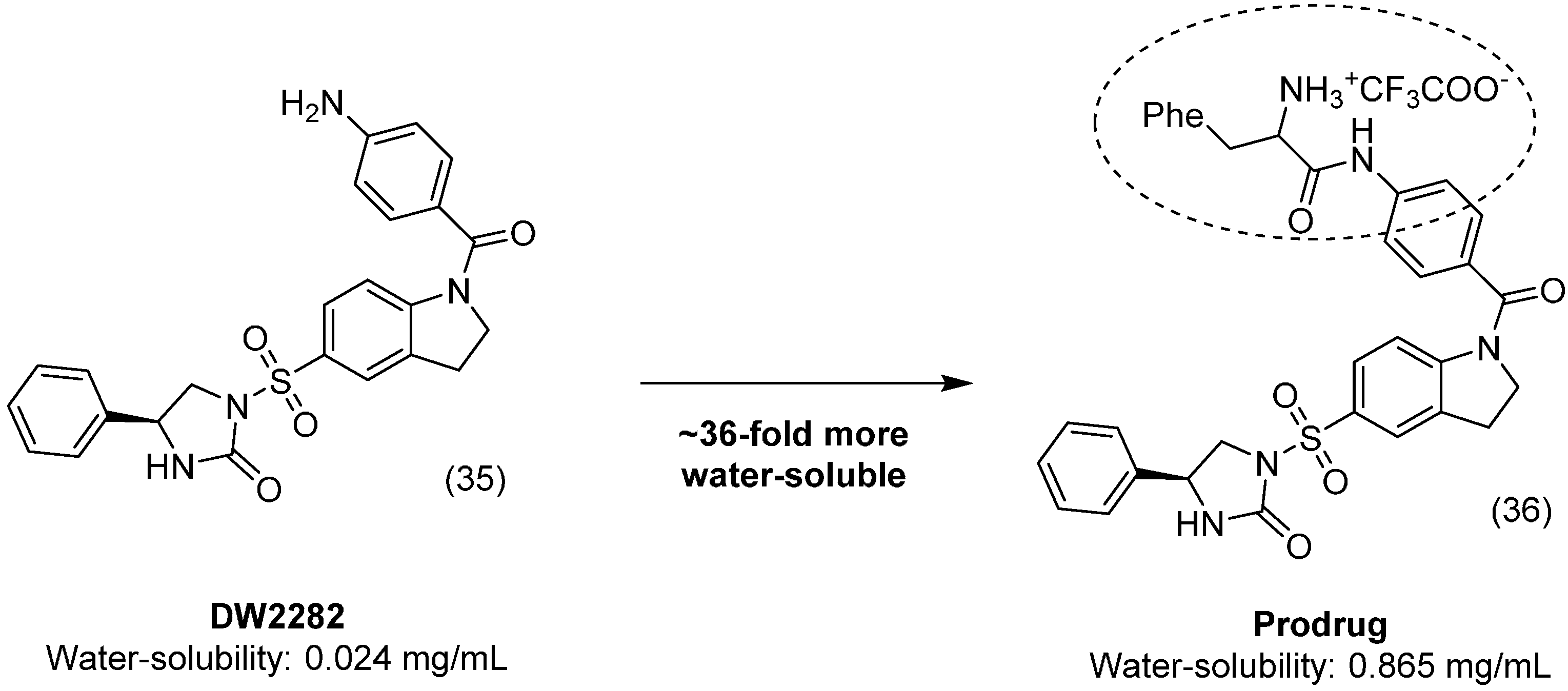

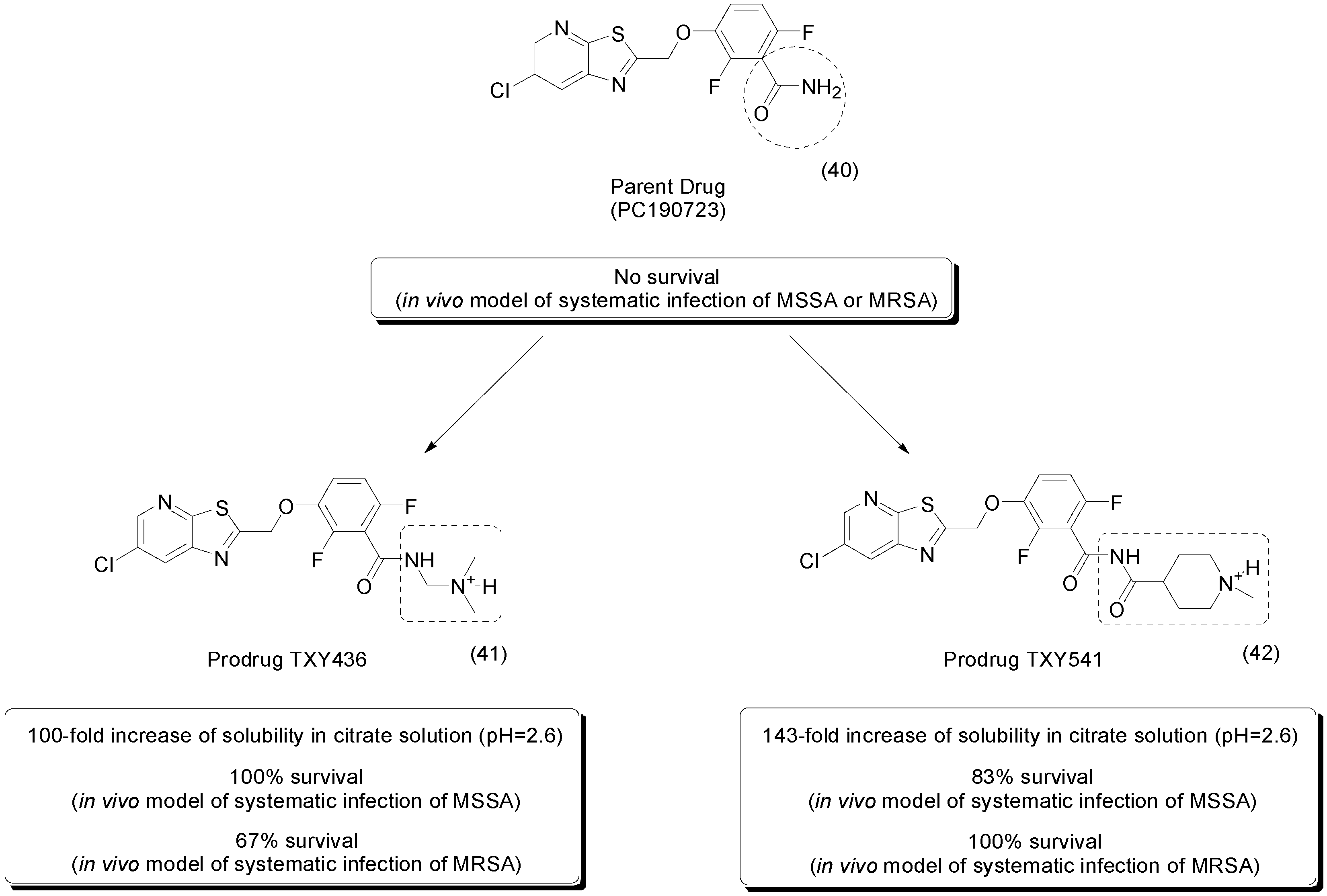
4. Carbamate Prodrugs
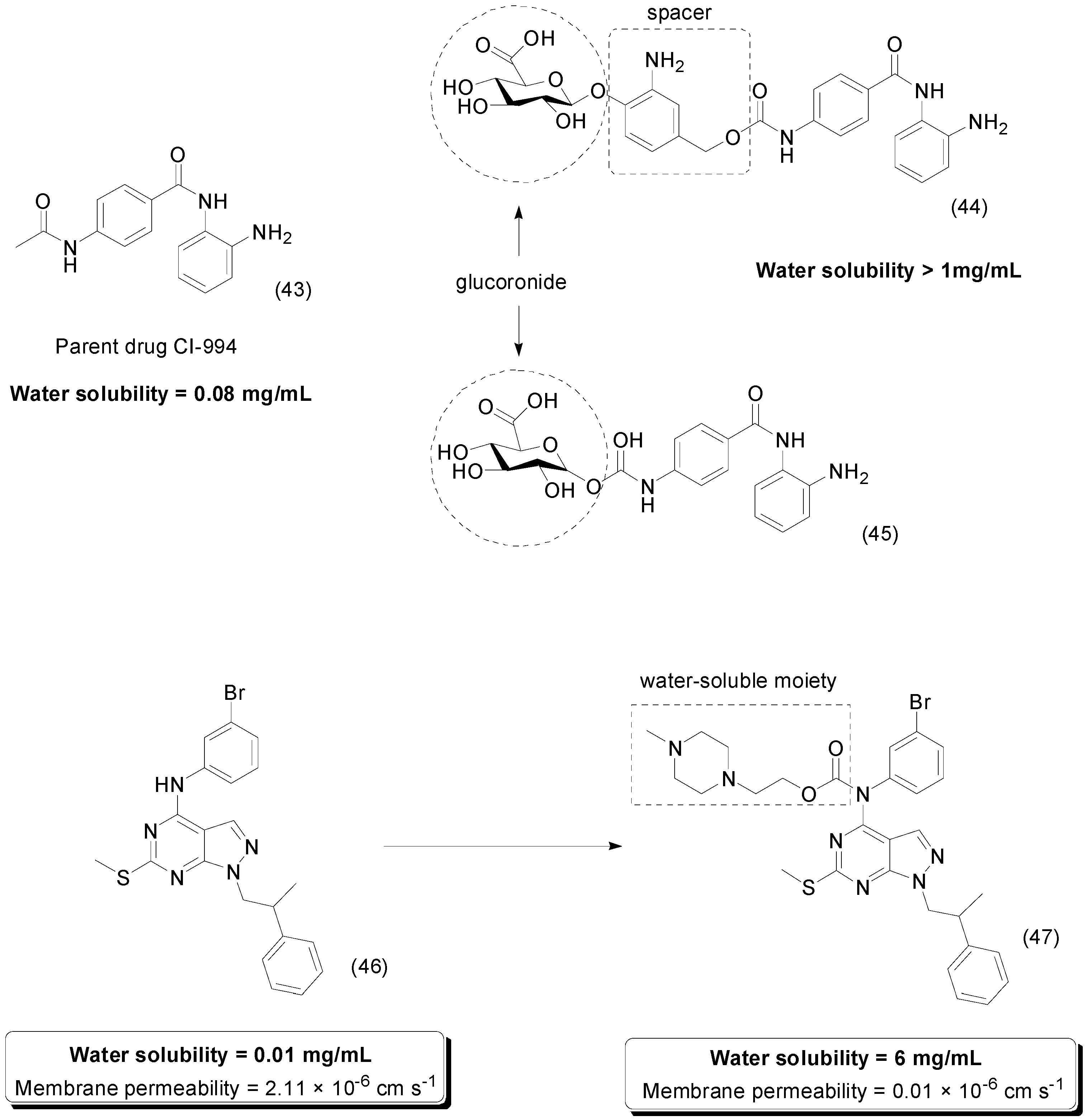
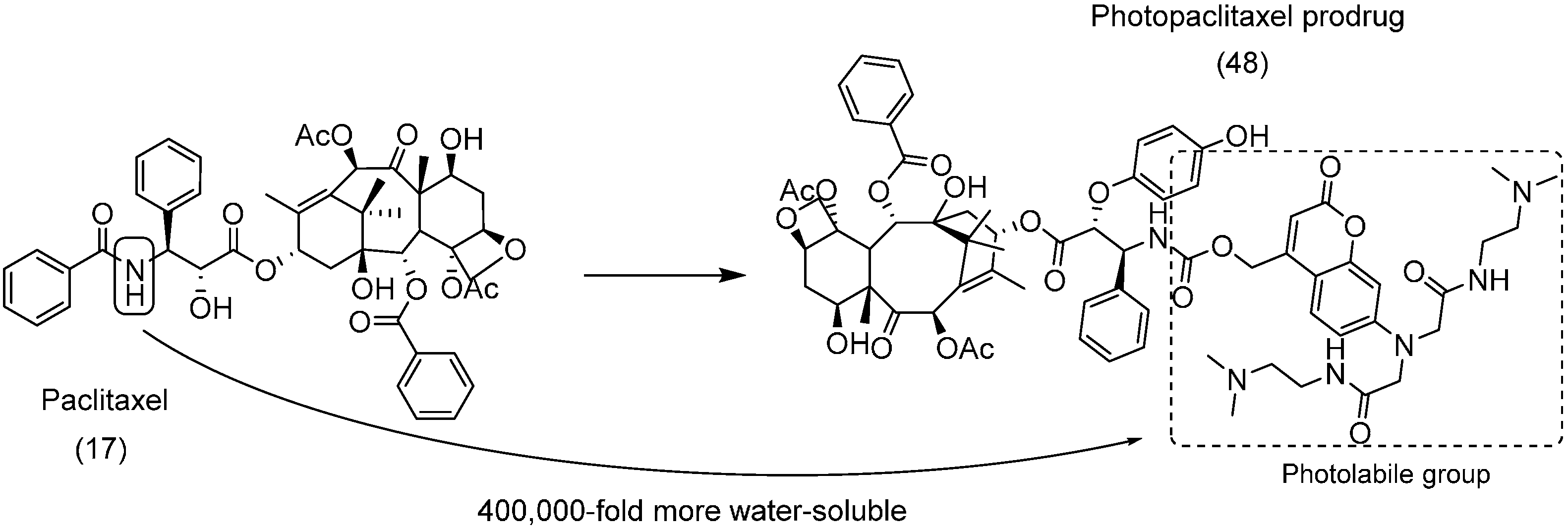
5. Carbonate Prodrugs
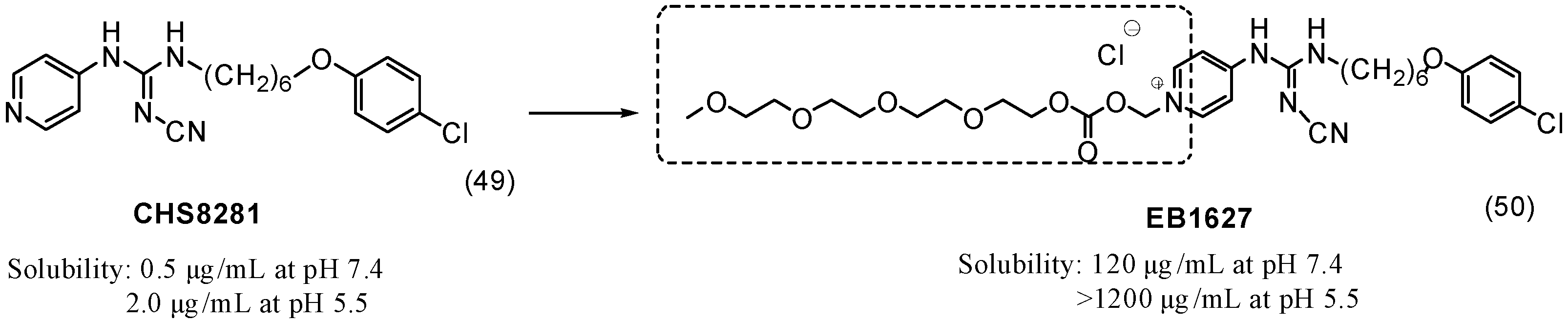
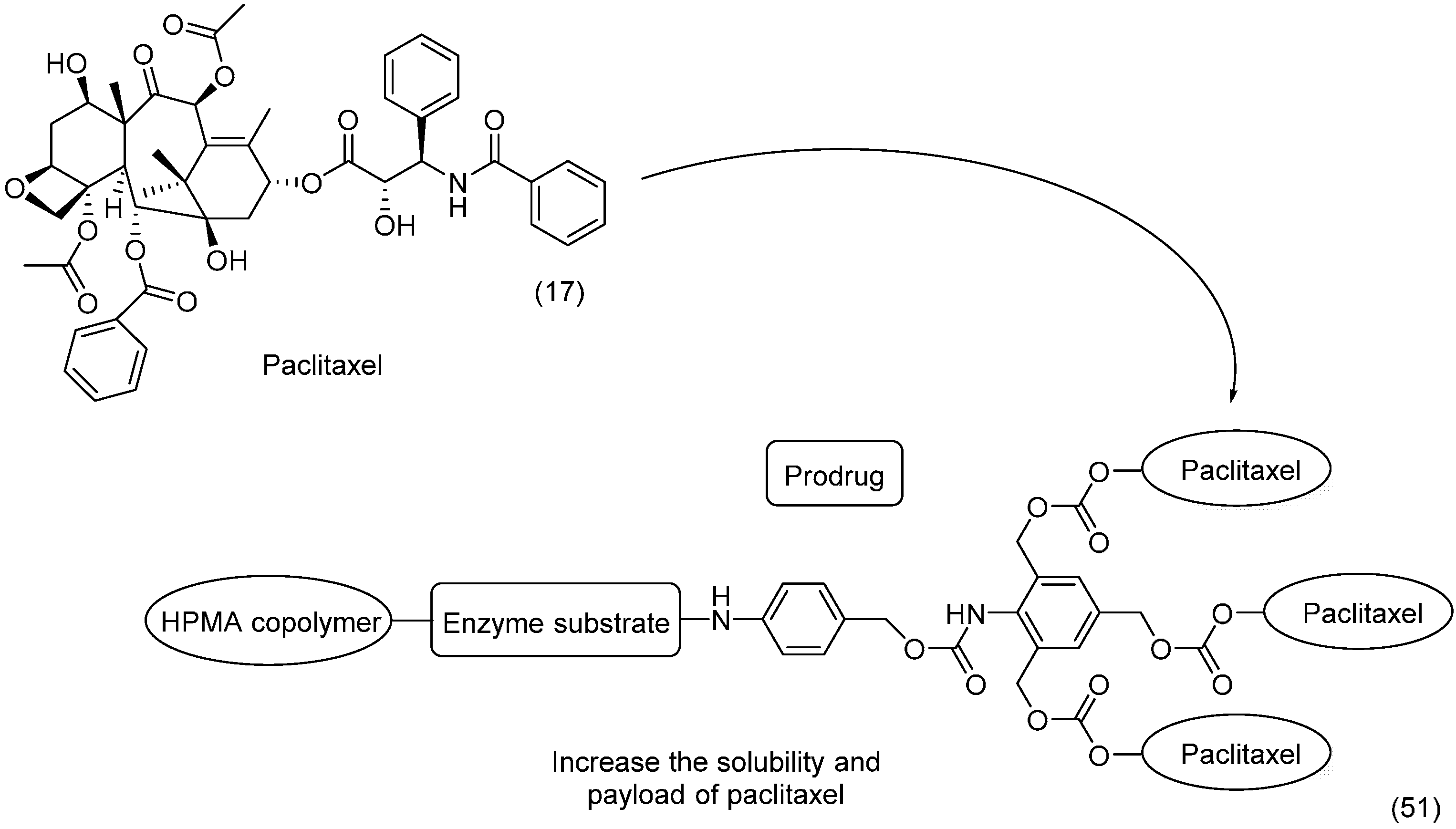
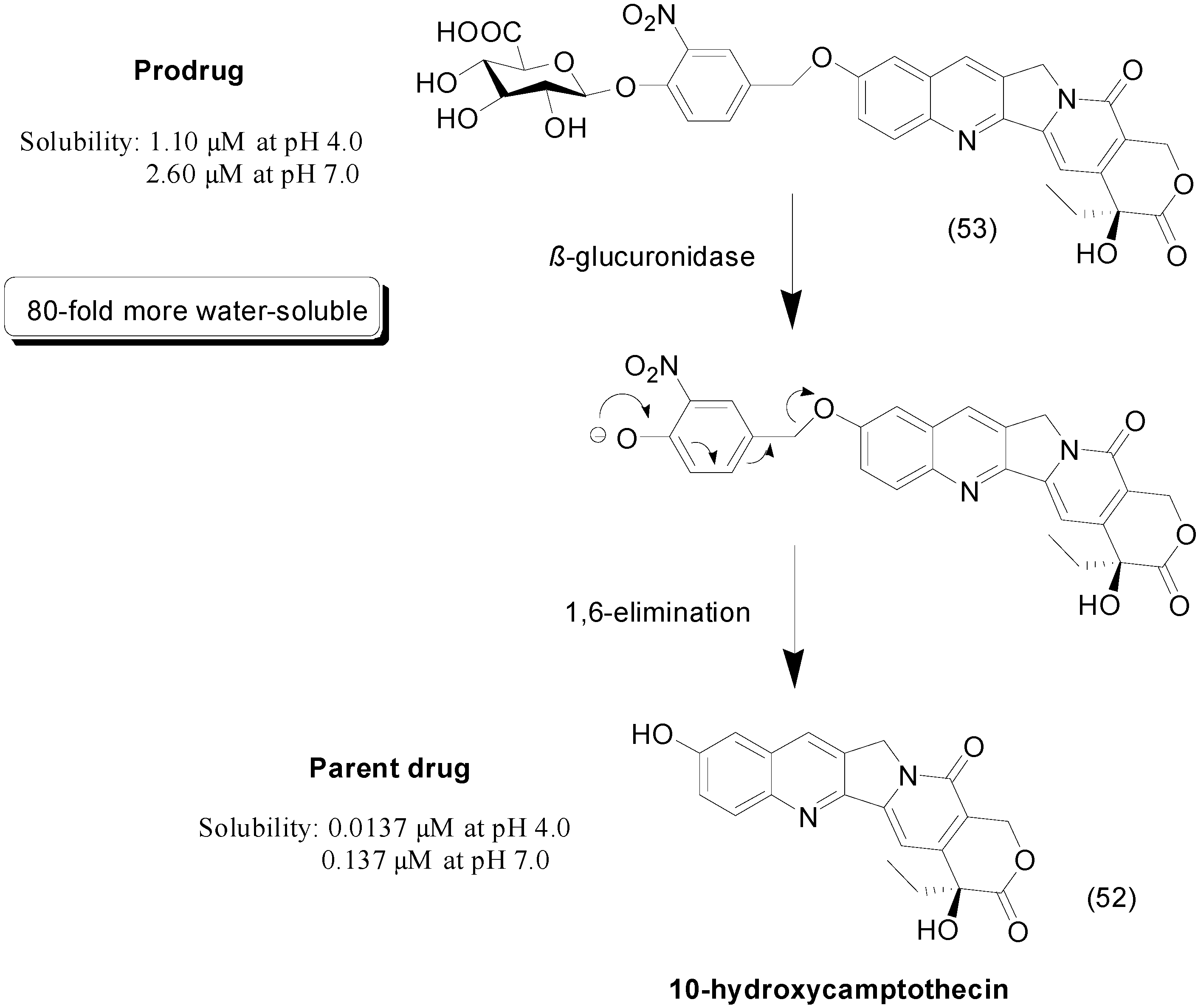
6. Ether Prodrugs
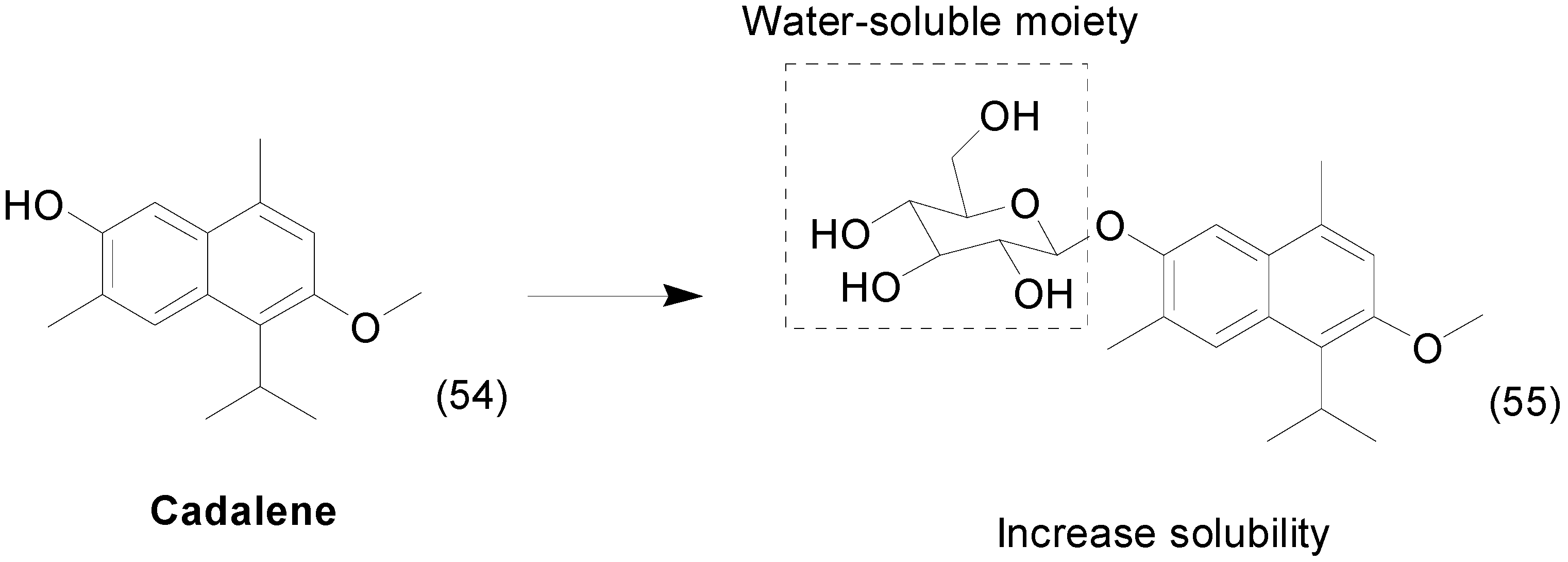
7. Imine Prodrugs
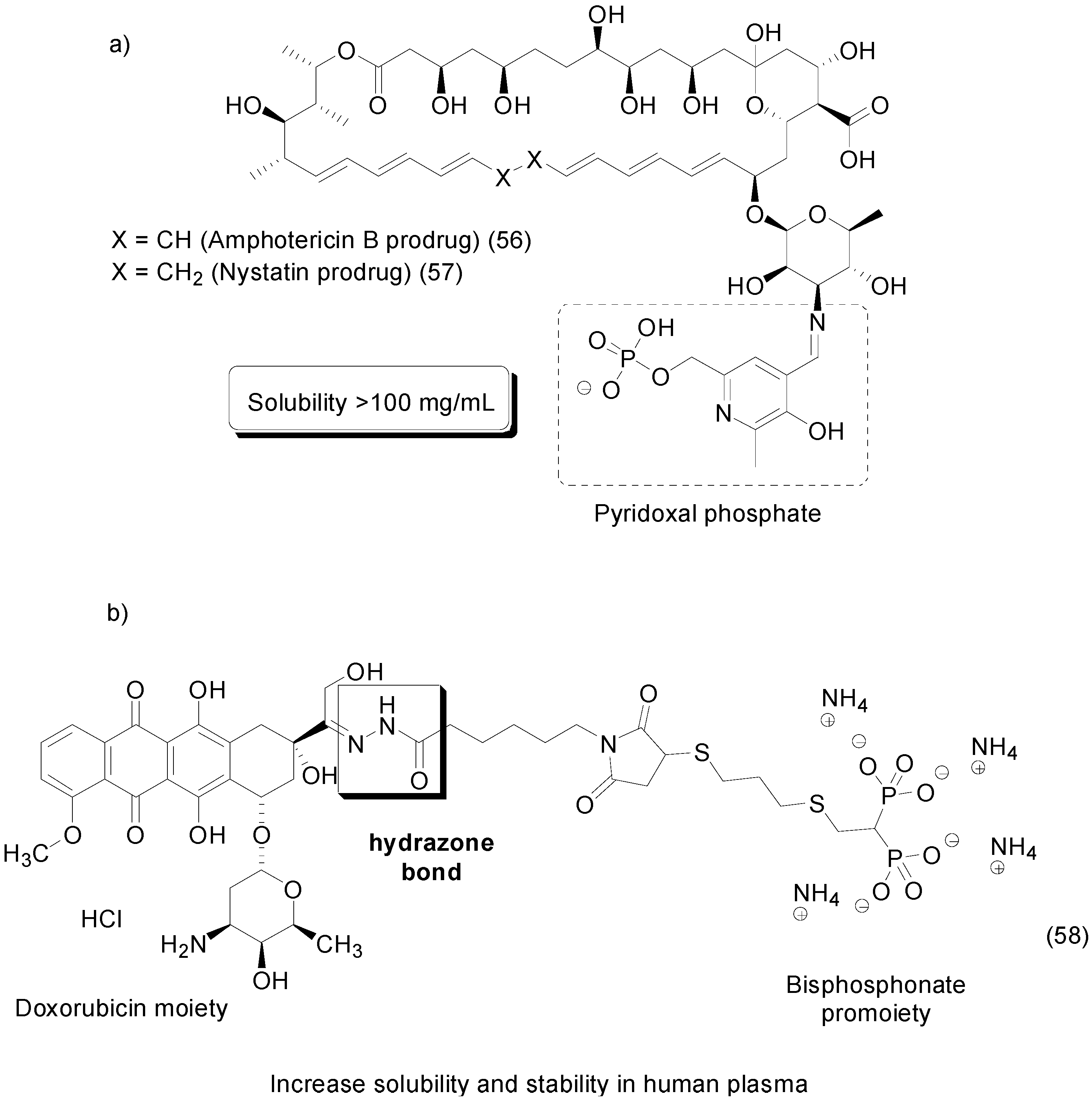
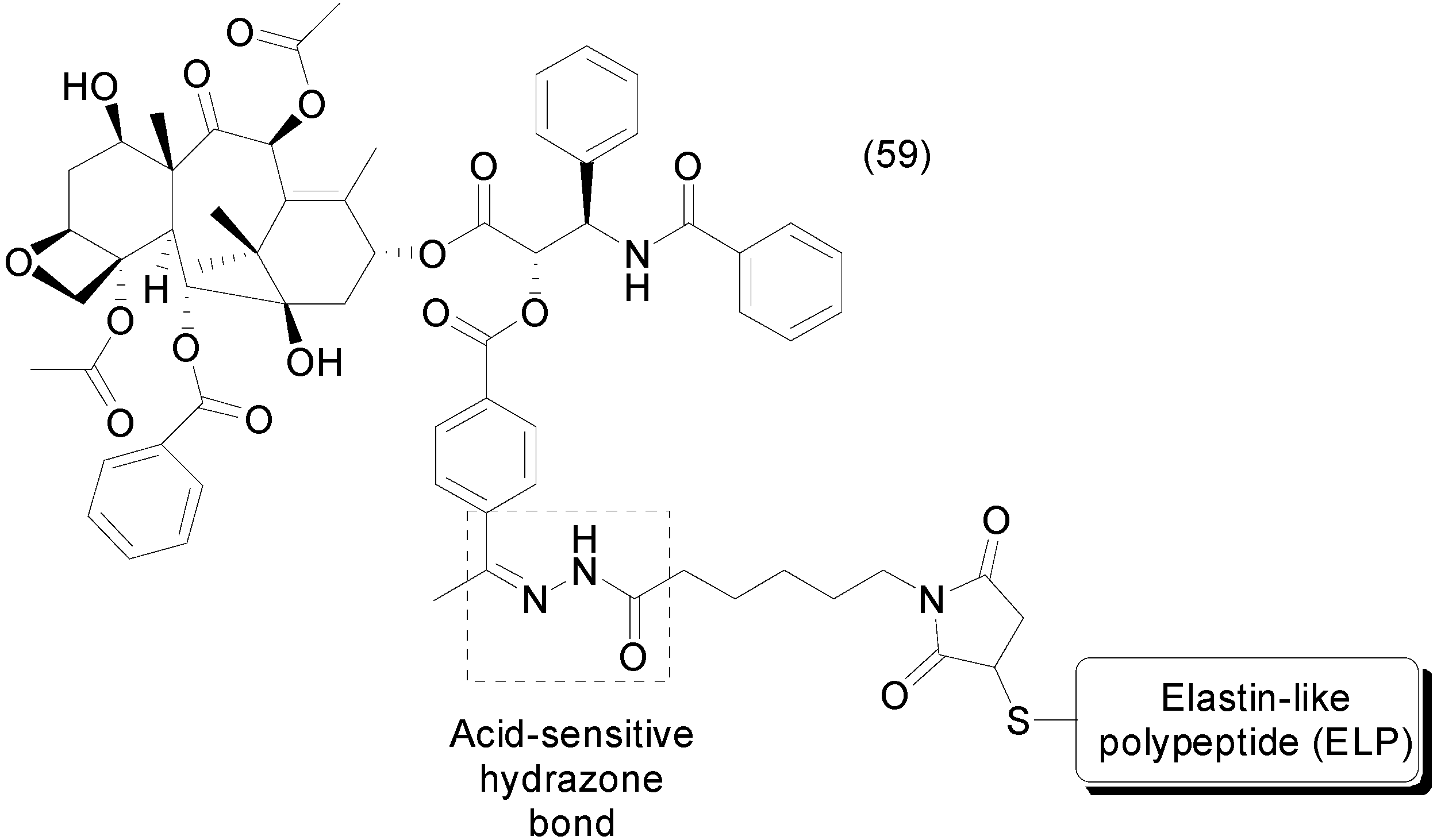
8. Phosphate Prodrugs
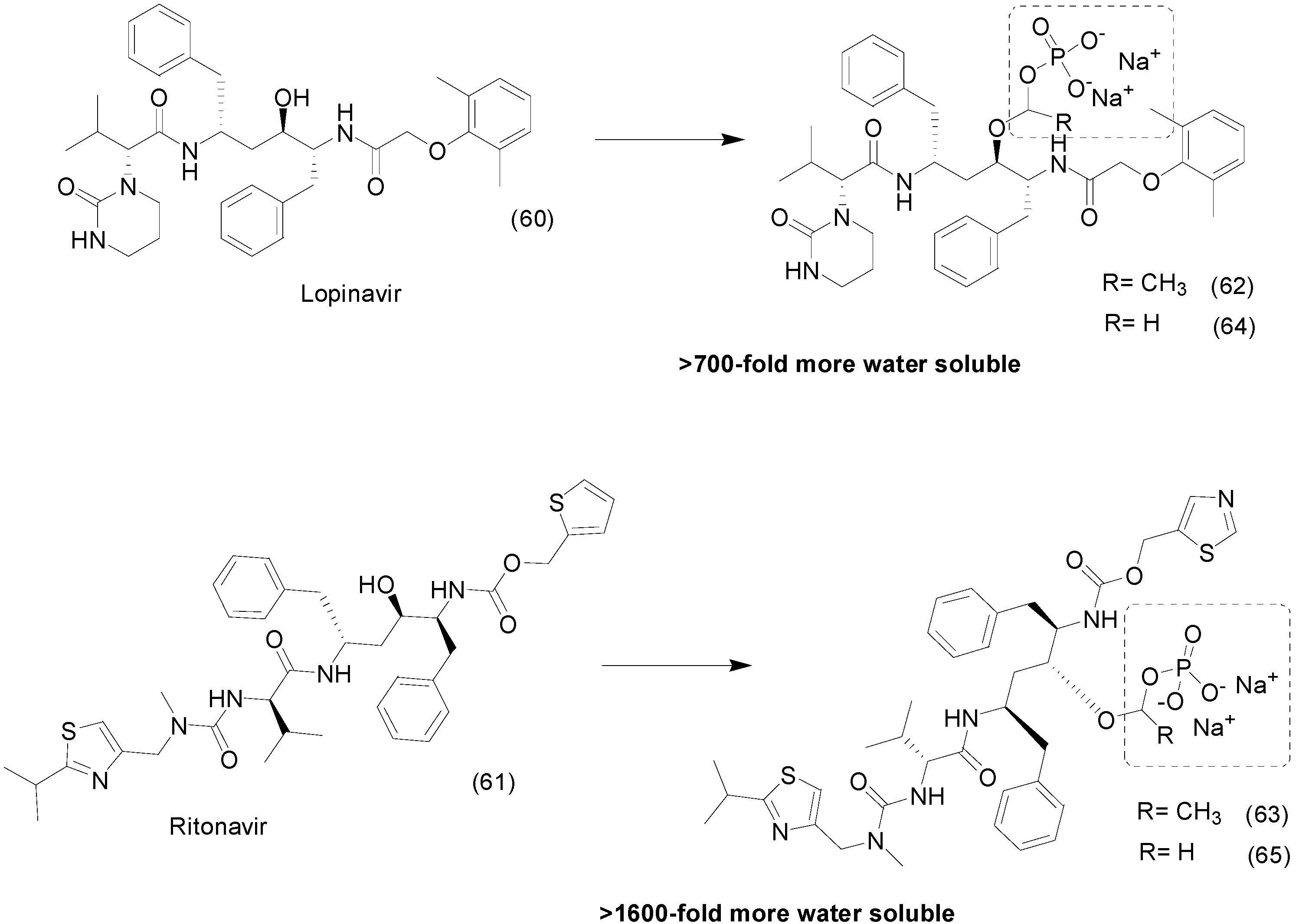
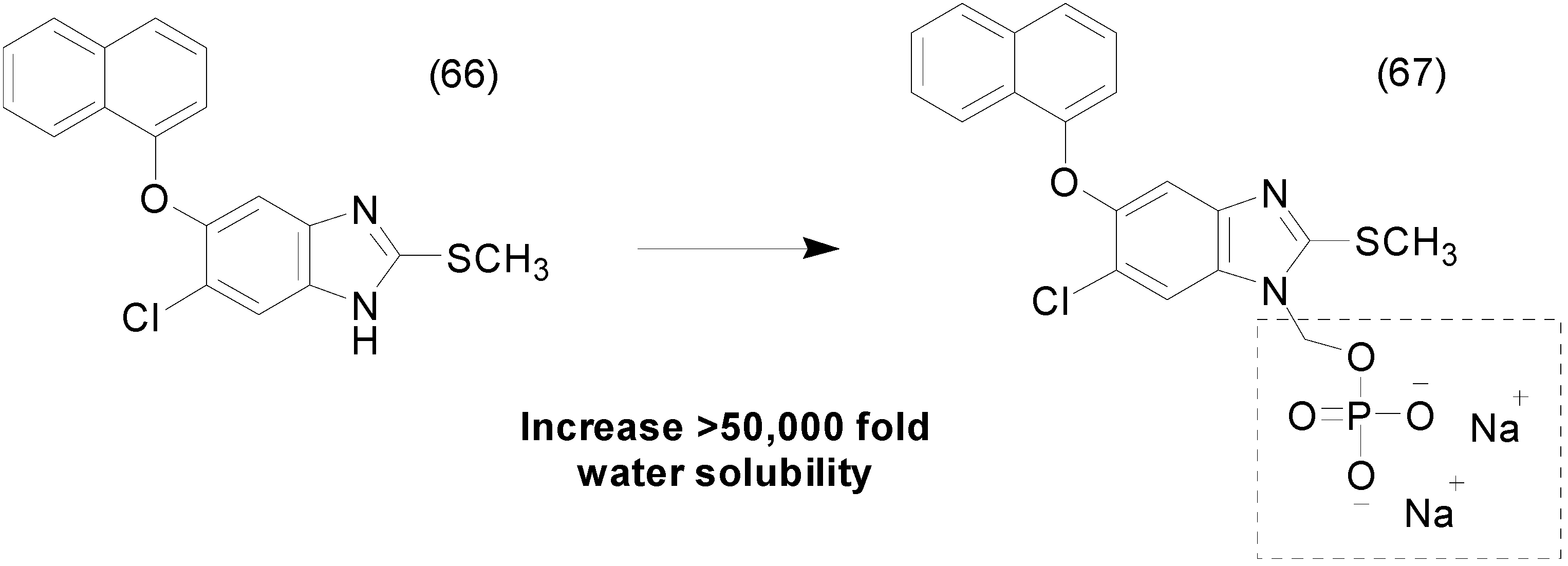
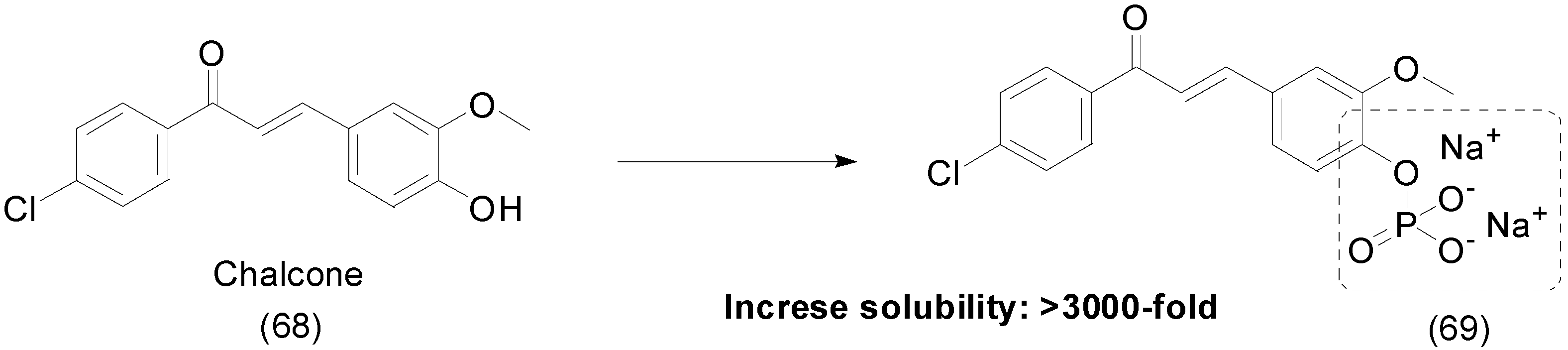
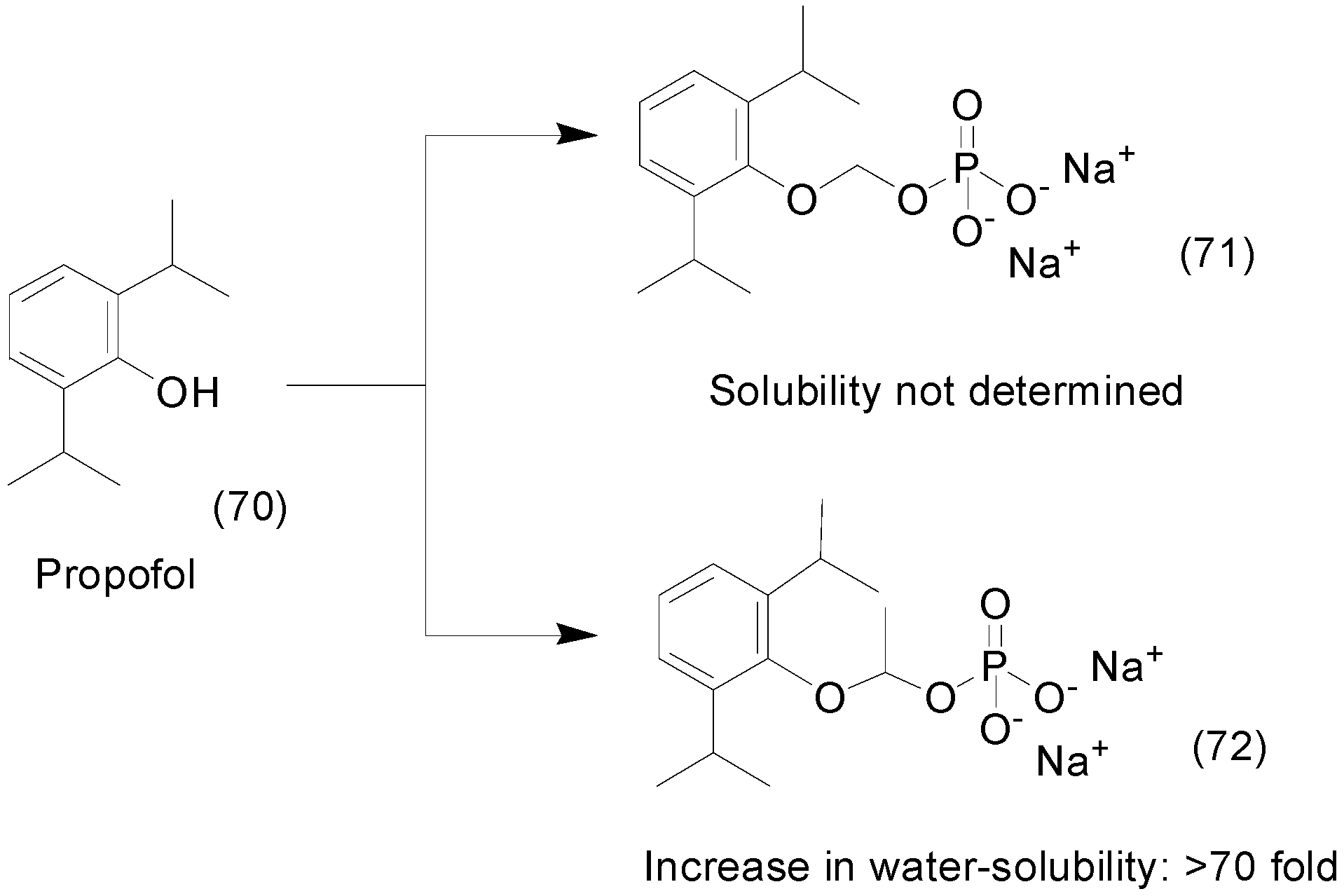
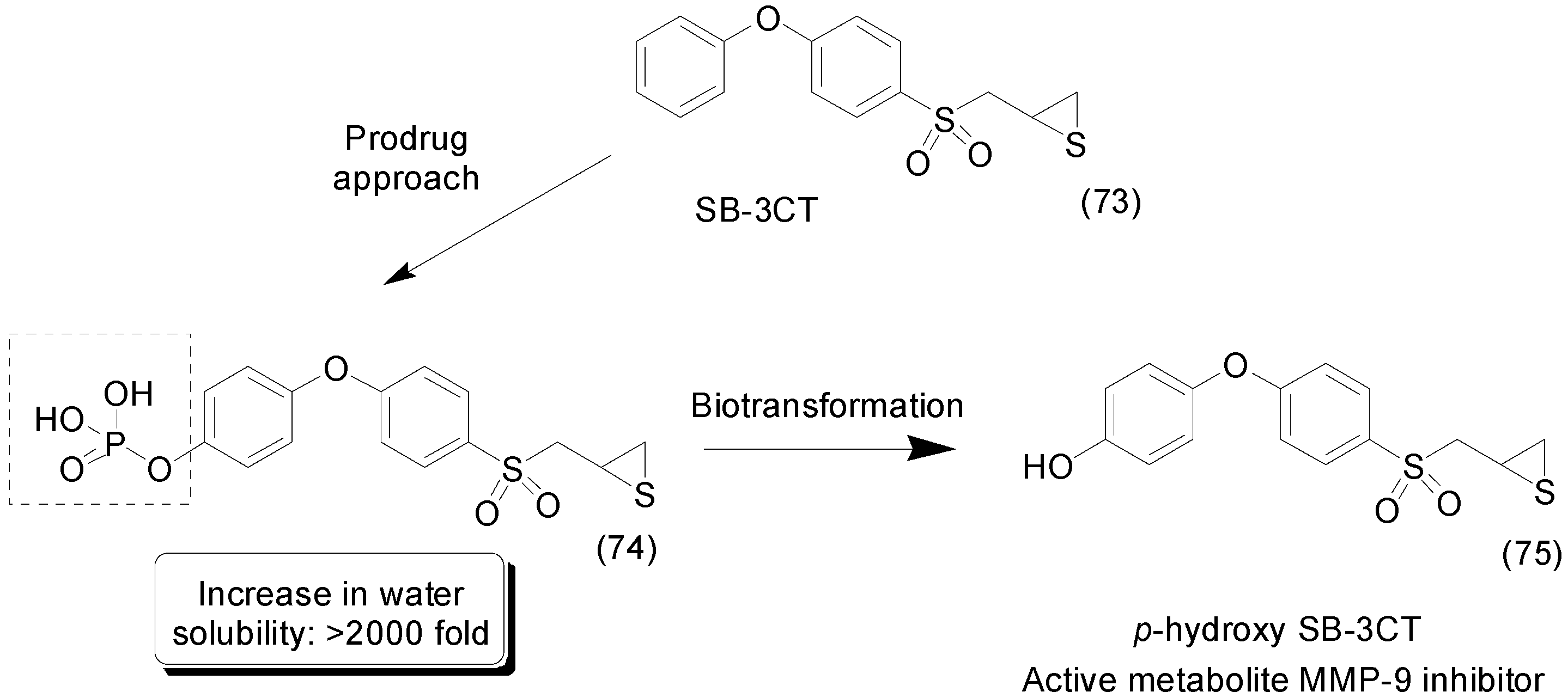
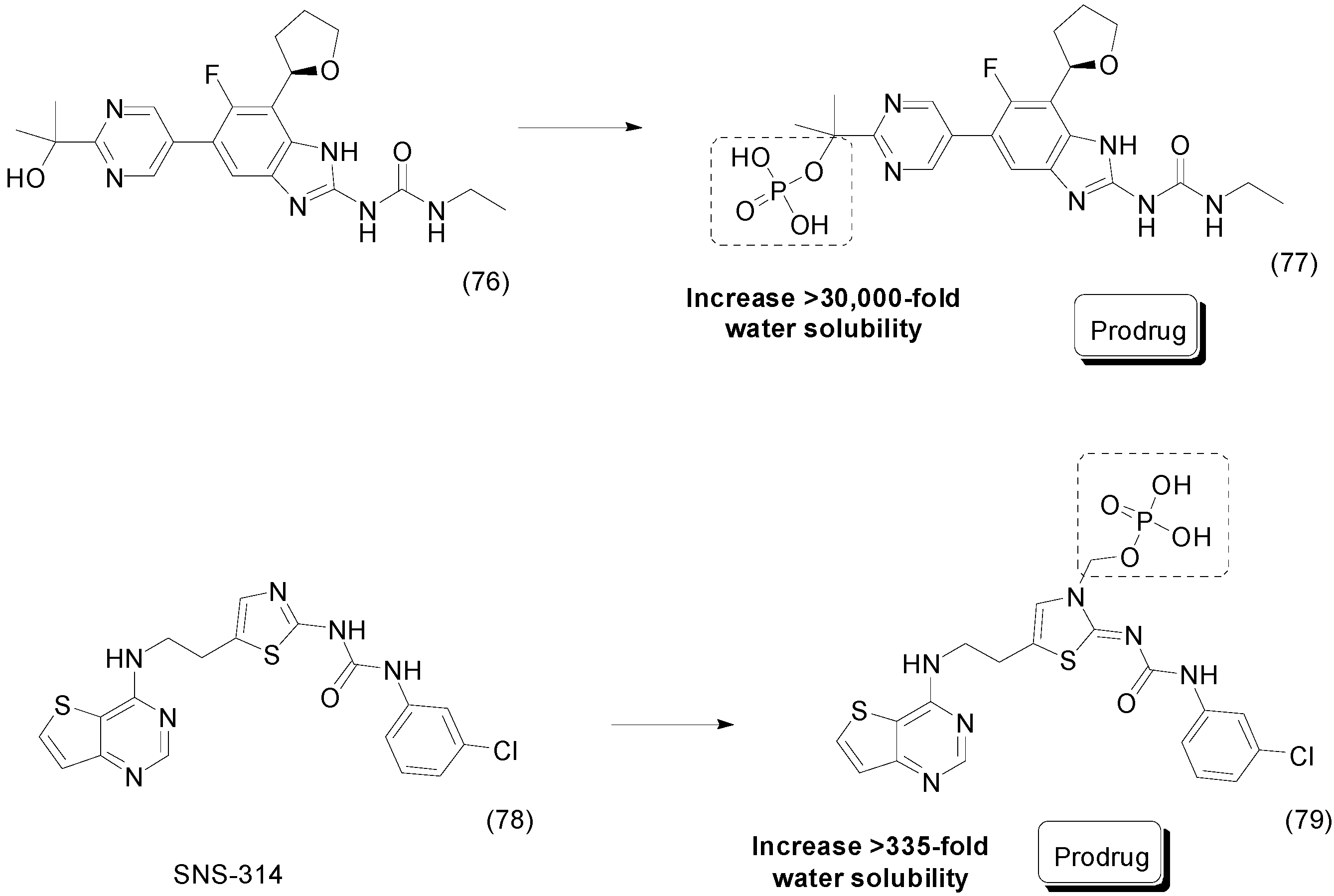
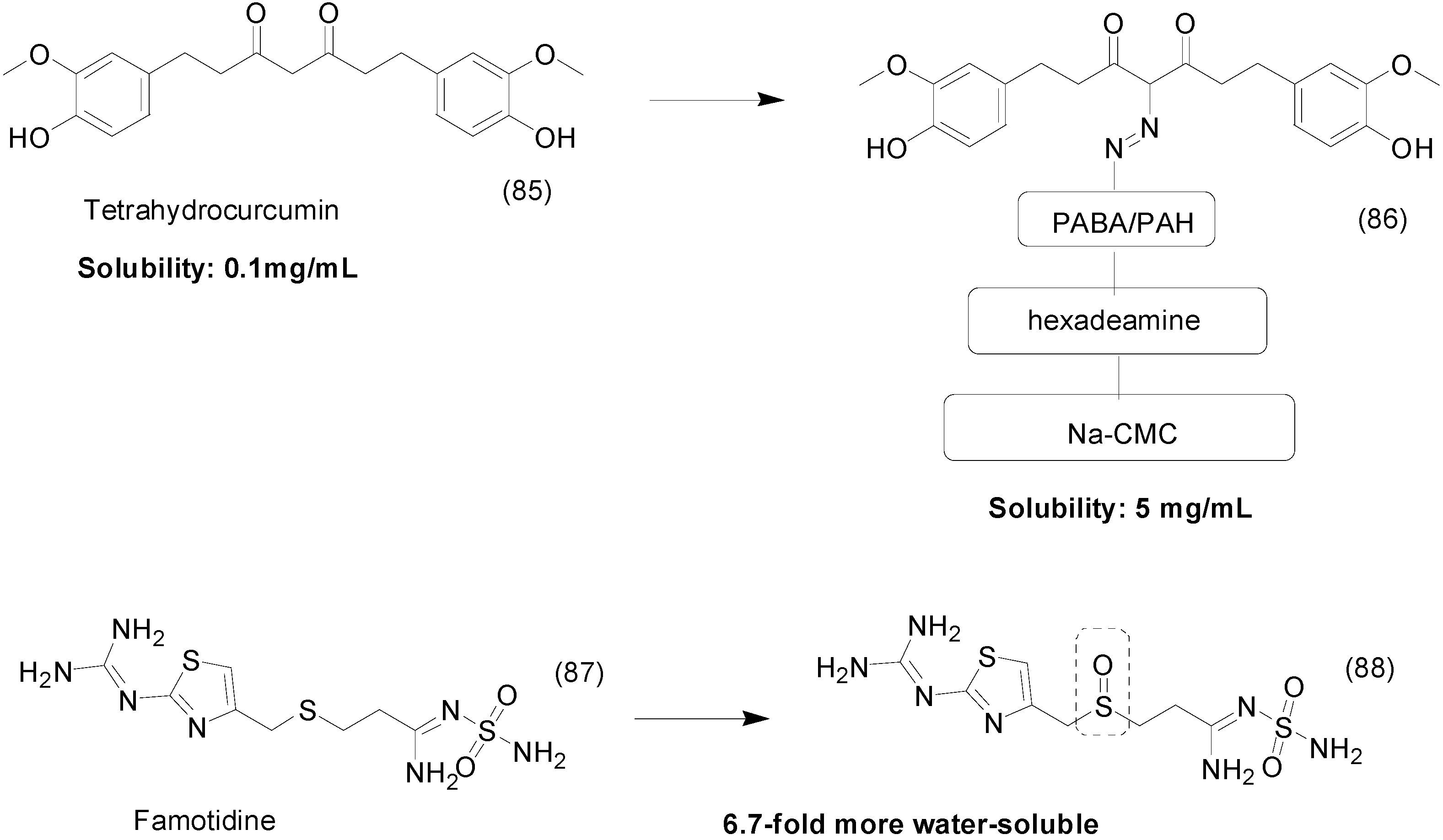
9. Other Chemical Prodrugs

10. Marketed Prodrugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
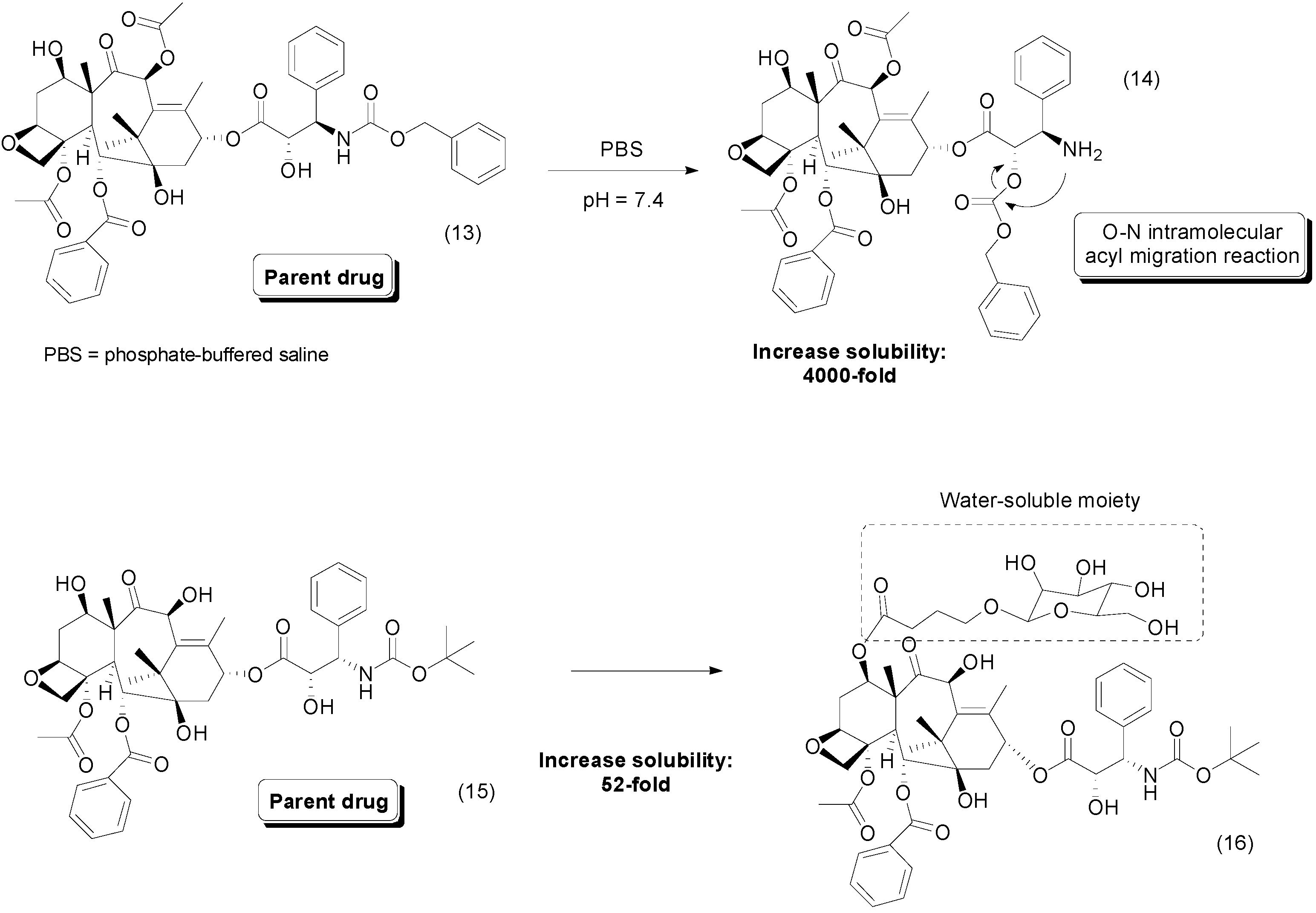{kind=link}
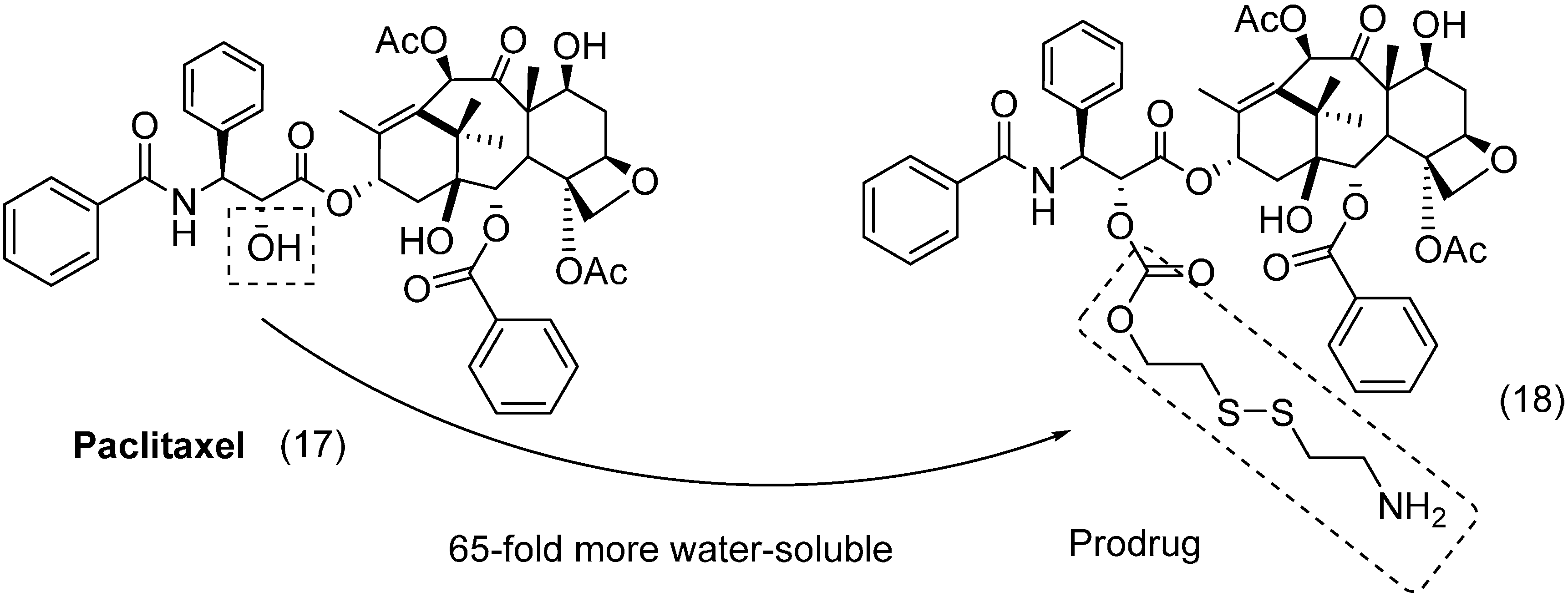{kind=link}
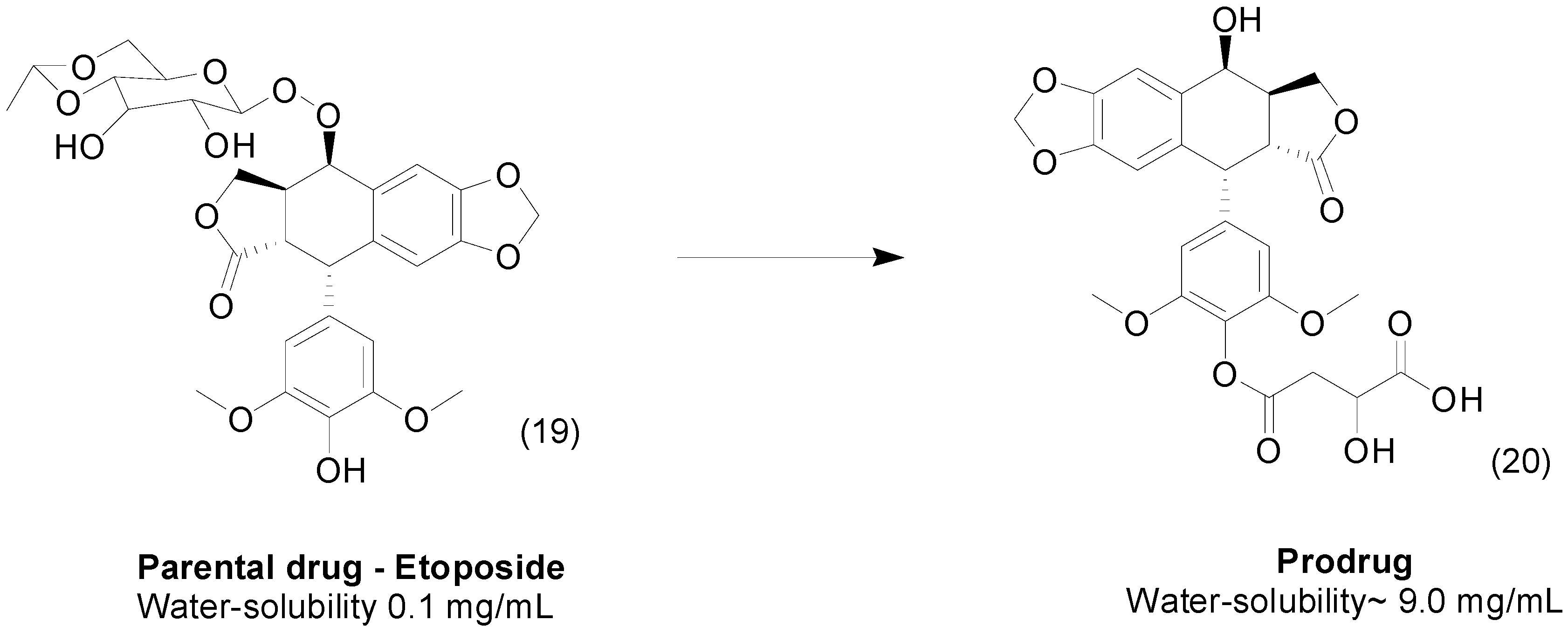{kind=link}
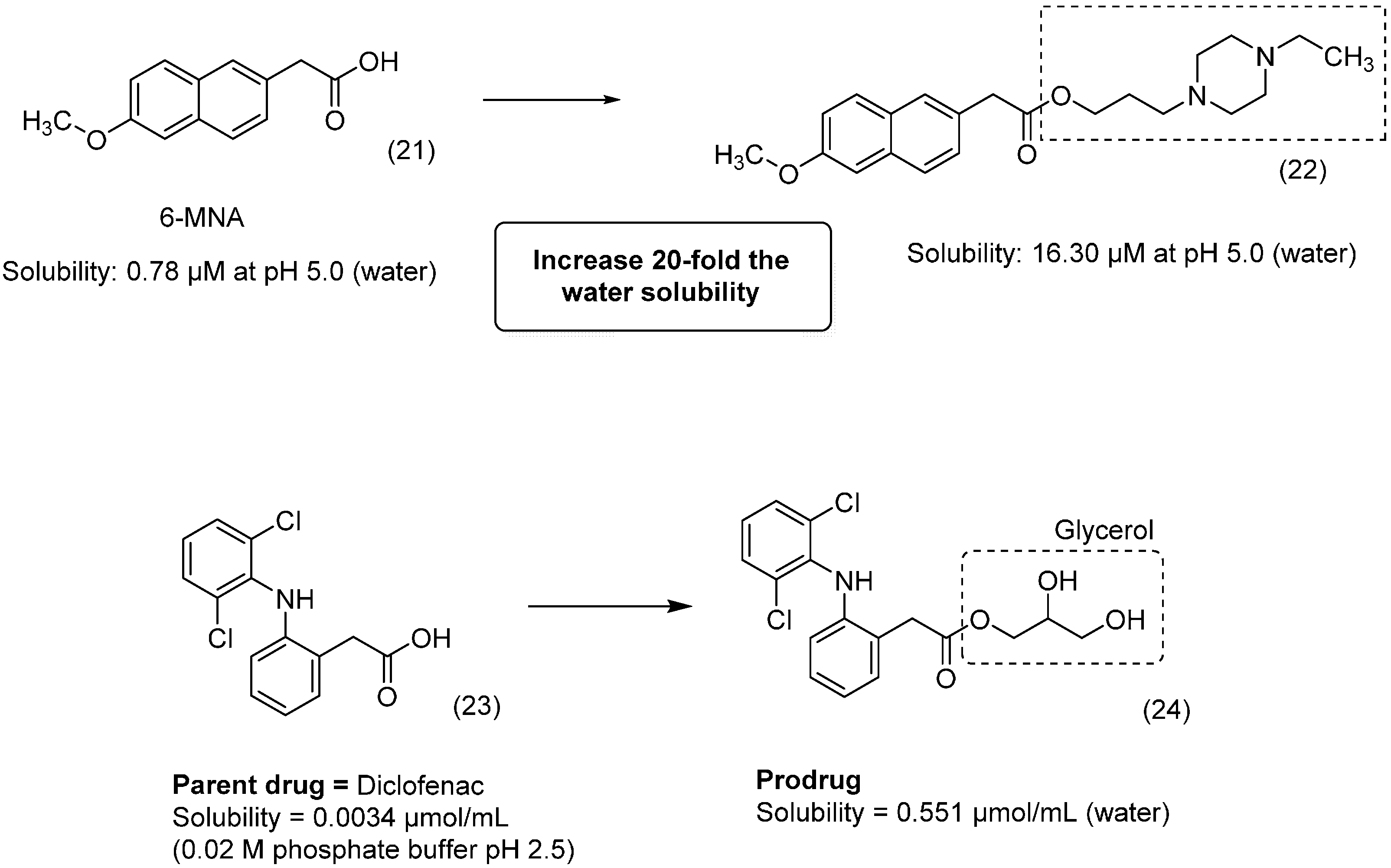{kind=link}
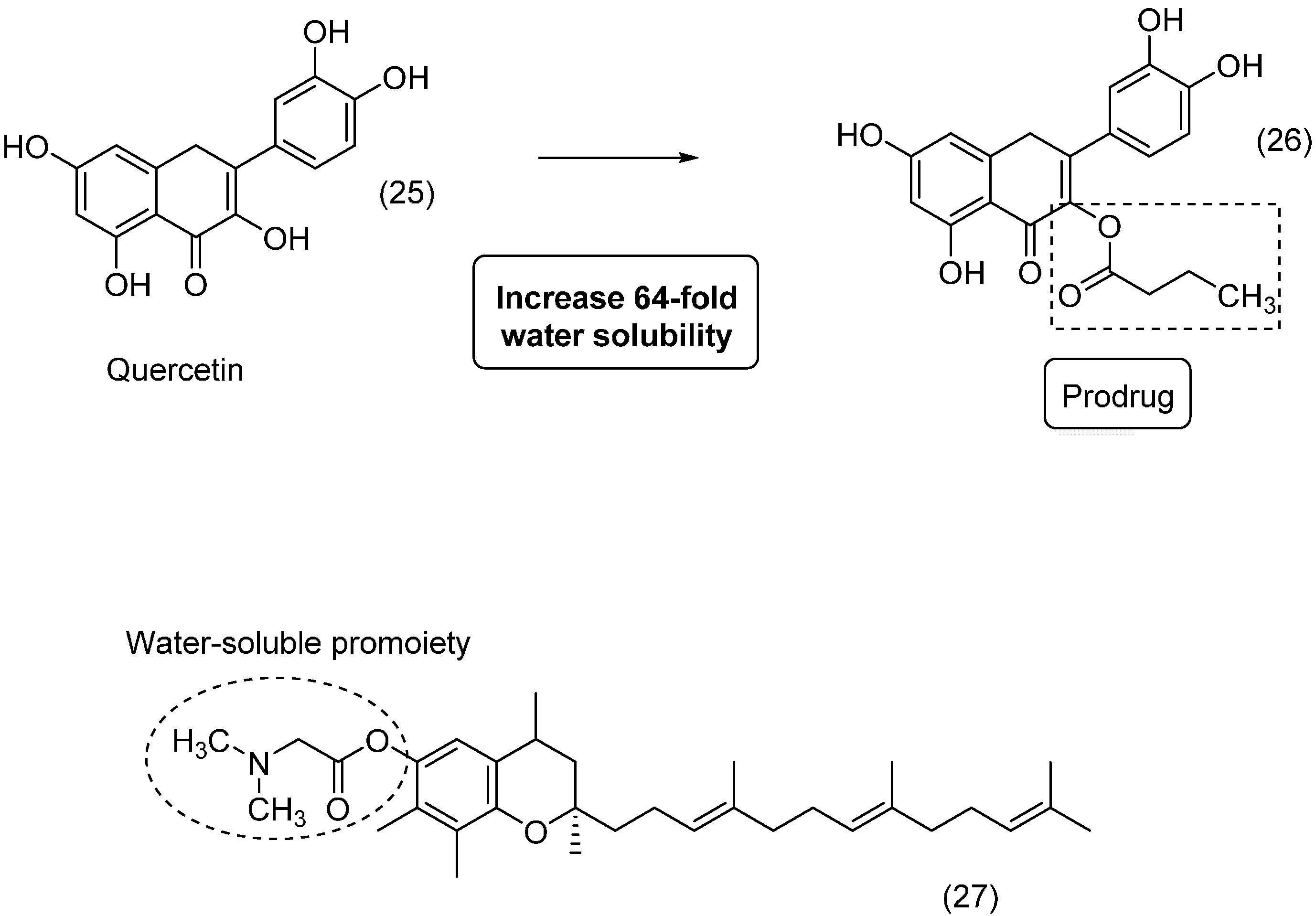{kind=link}
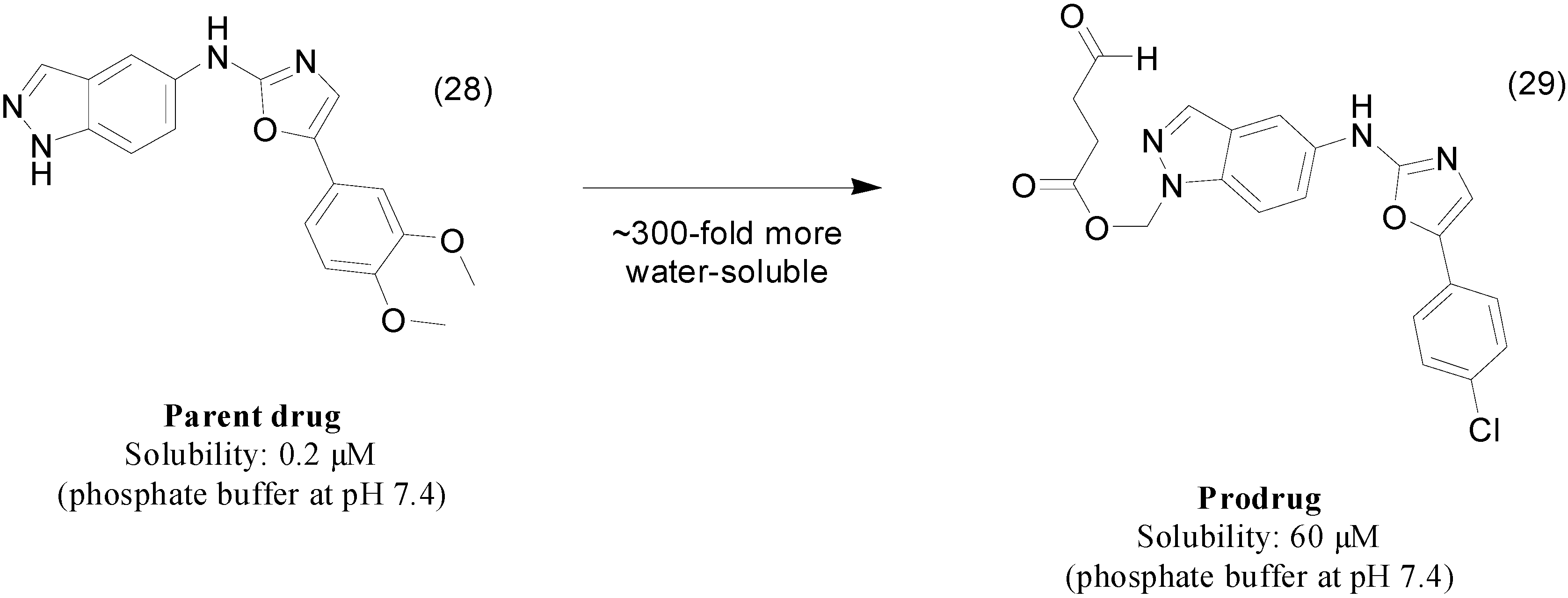{kind=link}
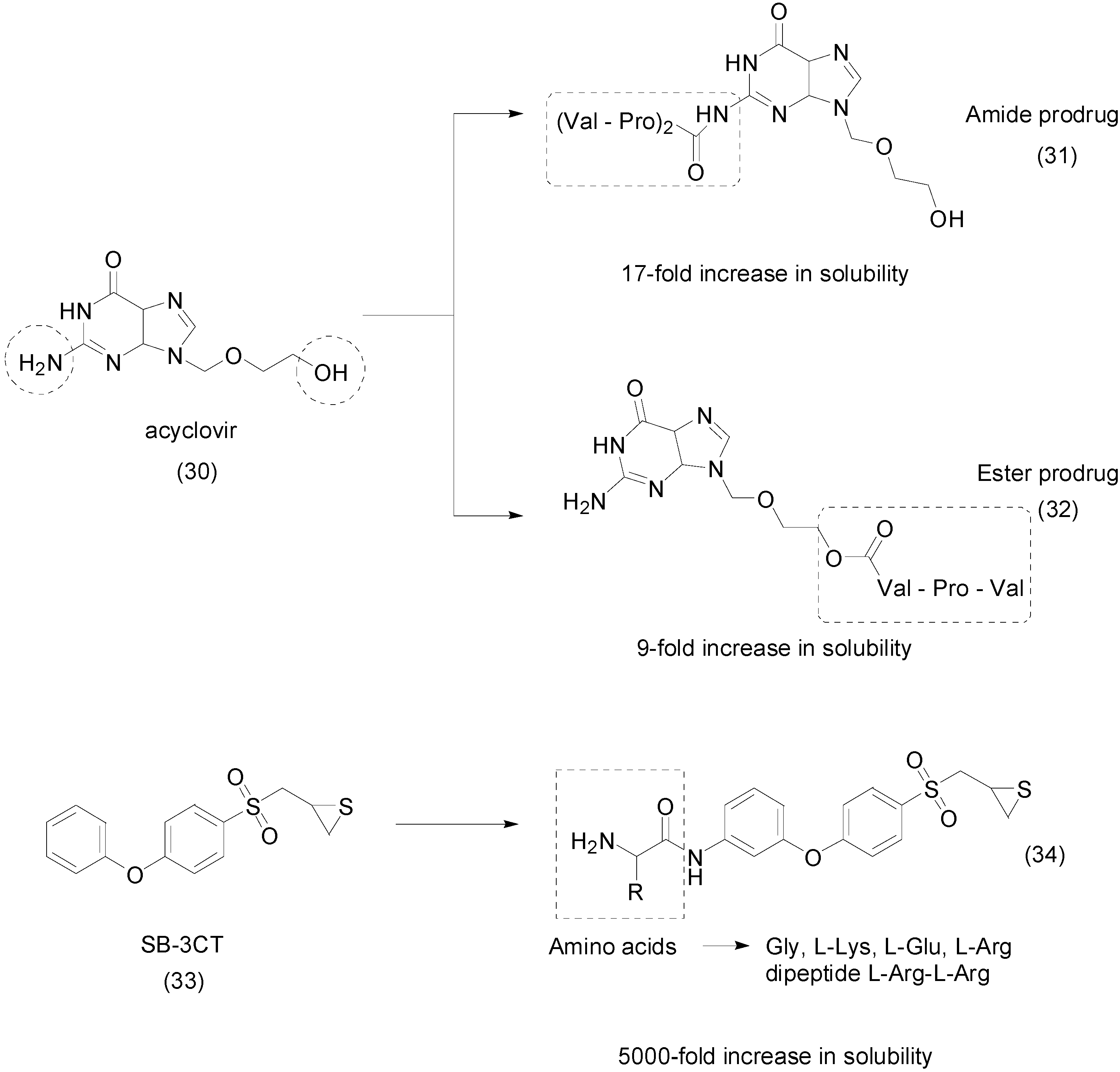{kind=link}
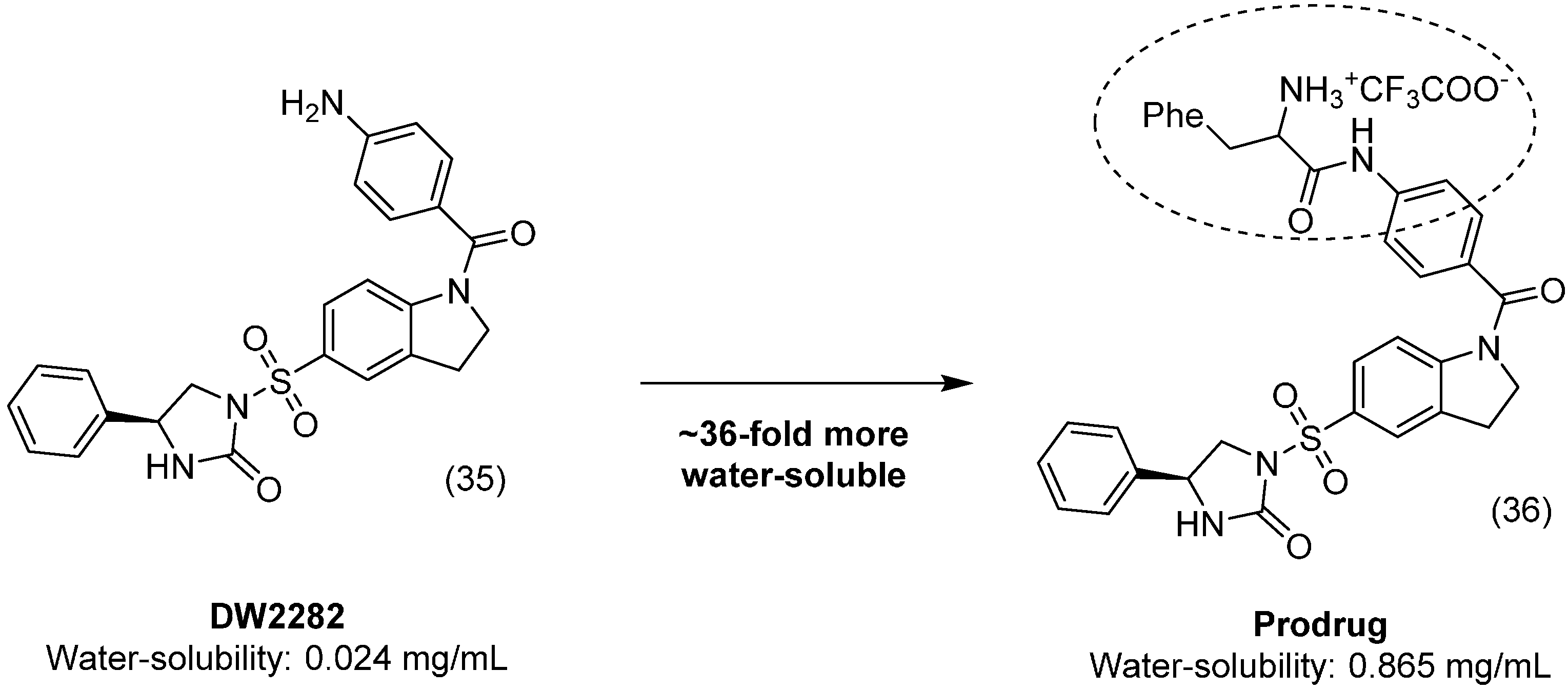{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Prodrug Name | Chemical Structure | Indication |
|---|---|---|---|
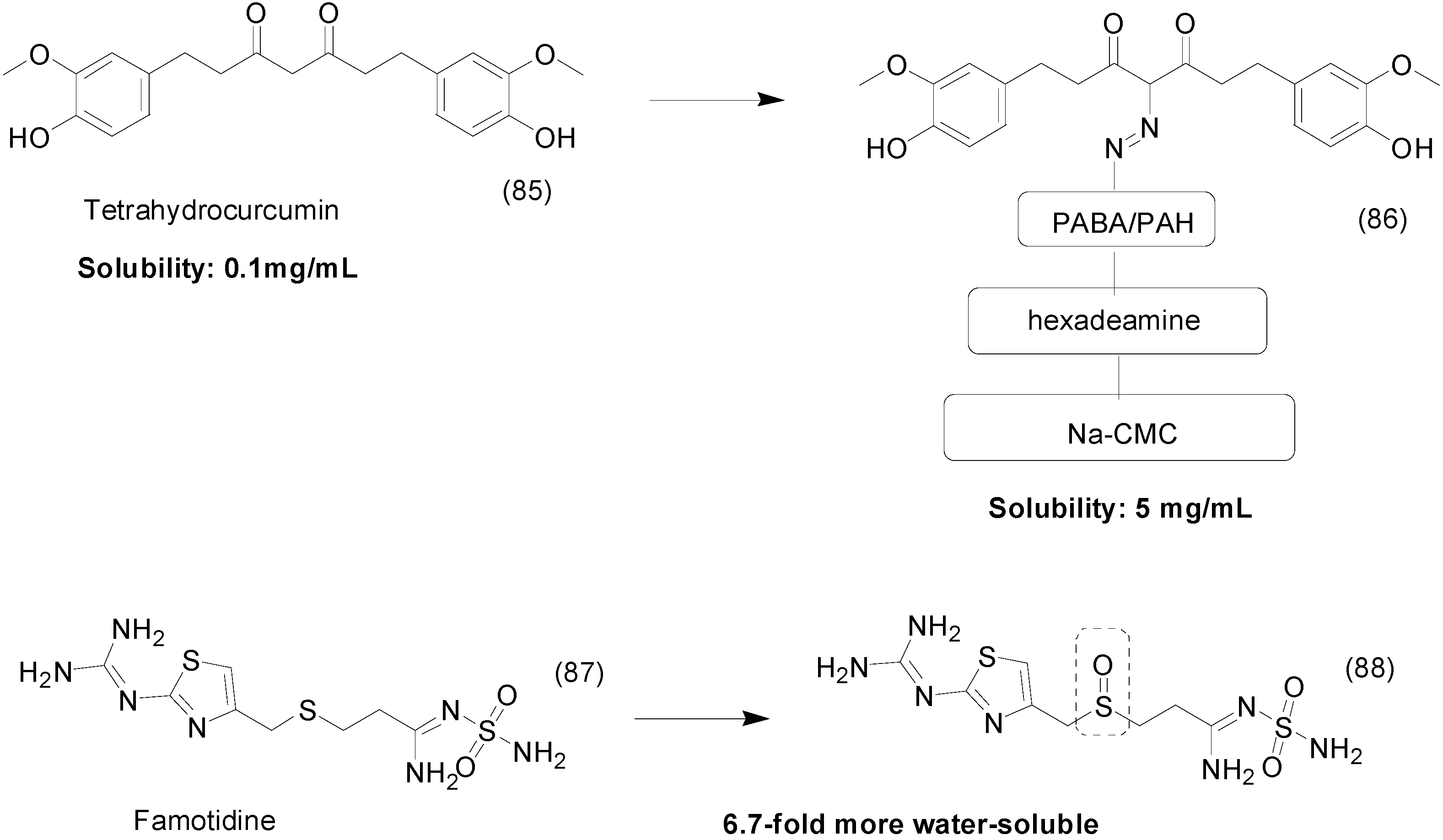
| 2005 | Nelarabine Arranon® GlaxoSmithKline plc | 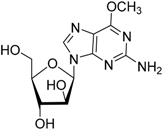 | Lymphoblastic leukemia |
| 2006 | - | - | - |
| 2007 | Lisdexamfetamine dimesylate Vyvanse® New River, Inc. | 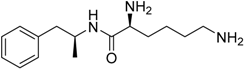 | Attention–deficit/hyperactivity disorder |
| Temsirolimus Torisel® Wyeth Pharm., Inc. | 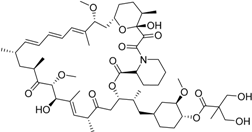 | Advanced renal cell carcinoma | |
| 2008 | Fesoterodinefumarate Toviaz® Pfizer, Inc. | 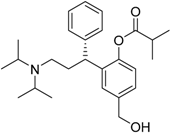 | Overactive bladder disorder |
| Fospropofoldisodium Lusedra® ElanPharm. plc |  | Monitored anesthesia care sedation. # discontinued | |
| 2009 | Prasugrel Effient® Eli Lilly (developed with Daiichi Sankyo) |  | Prevention of thrombotic cardiovascular complications in acute coronary syndromes |
| Romidepsin Istodax® Gloucester Pharm. |  | Cutaneous T cell lymphoma | |
| 2010 | Dabigatranetexilate mesylate Pradaxa® Boehringer Ingelheim GmbH | 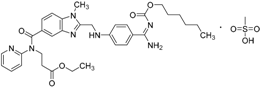 | Thromboembolism acute coronary syndrome |
| Fingolimod Gilenya® Novartis International AG | 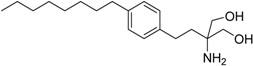 | Multiple sclerosis [sphingosin-1-phosphate (S1P) agonist with cannabinoid antagonist] | |
| Ceftarolinefosamil Teflaro® Forest Laboratories, Inc. |  | Bacterial skin infection | |
| 2011 | Abiraterone acetate Zytiga® Janssen Biotech, Inc. | 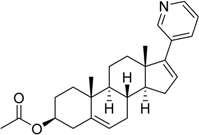 | Hormone-refractory prostate cancer (17-α-hydrolase/C17,20lyase) |
| Azilsartanmedoxomil Edarbi® Takeda Pharm. | 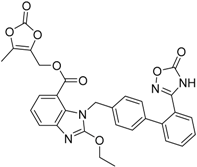 | Hypertension (angiotensin II antagonist) | |
| Gabapentin encarbil Horizant® GlaxoSmithKline plc | 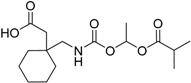 | Restless leg syndrome (GABA and Ca channel modulator) new indication | |
| 2012 | Tafluprost Zioptan® Merck & Co., Inc. | 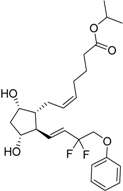 | Elevated intraocular pressure (prostaglandin analog) |
| 2013 | Dimethyl fumarate Tecfidera® Biogen Idec, Inc. | 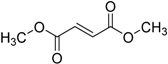 | Relapsing multiple sclerosis |
| Eslicarbazepineacetate Aptiom® Bial-Portela & Ca. S.A. | 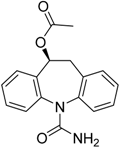 | Anticonvulsant (partial-onset seizures) | |
| Sofosbuvir Sovaldi® Gilead Sciences, Inc. | 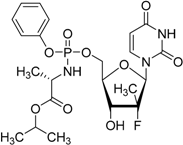 | Treatment of hepatitis C virus (HCV) infection | |
| 2014 | Droxidopa Northera® Lundbeck A/S | 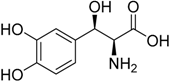 | Neurogenic orthostatic hypotension |
| Tedizolidphosphate Sivextro® Cubist Pharma., Inc. |  | Acute bacterial skin and skin structure infections | |
| 2015 | Isavuconazonium sulfate Cresemba® Astellas Pharma, Inc. | 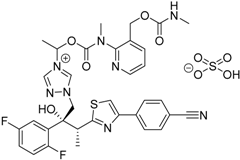 | Invasive aspergillosis and invasive mucormycosis |
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Huttunen, K.; Raunio, H.; Rautio, J. Prodrugs: Design and clinical applications. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Abu-jaish, A.; Jumaa, S.; Karaman, R. Prodrug Overview. In Prodrugs Design: A New Era; Karaman, R., Ed.; Nova Publisher: Hauppauge, NY, USA, 2014; pp. 77–102. [Google Scholar]
- Beaumont, K.; Webster, R.; Gardner, I.; Dack, K. Design of ester prodrugs to enhance oral absorption of poorly permeable compounds: Challenges to the discovery scientist. Curr. Drug Metab. 2003, 4, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Testa, B. Prodrugs: Bridging pharmacodynamic/pharmacokinetic gaps. Curr. Opin. Chem. Biol. 2009, 13, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.-C.; Silva, A.T.D.A.; Castro, L.F.; Güido, R.V.C.; Nassute, J.C.; Ferreira, E.I. Latenciação e formas avançadas de transporte de fármacos. Rev. Bras. Ciênc. Farm. 2005, 41, 155–180. [Google Scholar] [CrossRef]
- Redasani, V.K.; Bari, S.B. Prodrug Design: Perspectives, Approaches and Applications in Medicinal Chemistry, 1st ed.; Academic Press: London, UK, 2015. [Google Scholar]
- Zovko, M.; Zorc, B.; Novak, P.; Tepeš, P.; Cetina-Čižmek, B.; Horvat, M. Macromolecular prodrugs: XI. Synthesis and characterization of polymer–estradiol conjugate. Int. J. Pharm. 2004, 285, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons Learned from Marketed and Investigational Prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Han, H.K.; Amidon, G.L. Targeted prodrug design to optimize drug delivery. AAPS PharmSci 2000, 2, E6. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.T.D.A.; Chung, M.C.; Castro, L.F.; Güido, R.V.C.; Ferreira, E.I. Advances in prodrug design. Mini Rev. Med. Chem. 2005, 5, 893–914. [Google Scholar] [CrossRef] [PubMed]
- Williams, H.D.; Trevaskis, N.L.; Charman, S.A.; Shanker, R.M.; Charman, W.N.; Pouton, C.W.; Porter, C.J.H. Strategies to address low drug solubility in discovery and development. Pharmacol. Rev. 2013, 65, 315–499. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.E. Prodrug approaches for enhancing the bioavailability of drugs with low solubility. Chem. Biodivers. 2009, 6, 2071–2083. [Google Scholar] [CrossRef] [PubMed]
- Wenlock, M.C.; Austin, R.P.; Barton, P.; Davis, A.M.; Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 2003, 46, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.C.; Ferreira, E.I.; Leandro, J.; Giarolla, J.; Rando, D.G.; Almeida, A.E.; Bosquesi, P.L.; Menegon, R.F.; Blau, L. Prodrugs for the Treatment of Neglected Diseases. Molecules 2008, 616–677. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and Thioredoxin Target Proteins: From Molecular Mechanisms to Functional Significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Lynch, S.M.; Powis, G.; Birmingham, A.; Englund, E.E. Synthesis and biological activity of prodrug inhibitors of the thioredoxin-thioredoxin reductase system. Org. Biomol. Chem. 2005, 3, 3880–3882. [Google Scholar] [CrossRef] [PubMed]
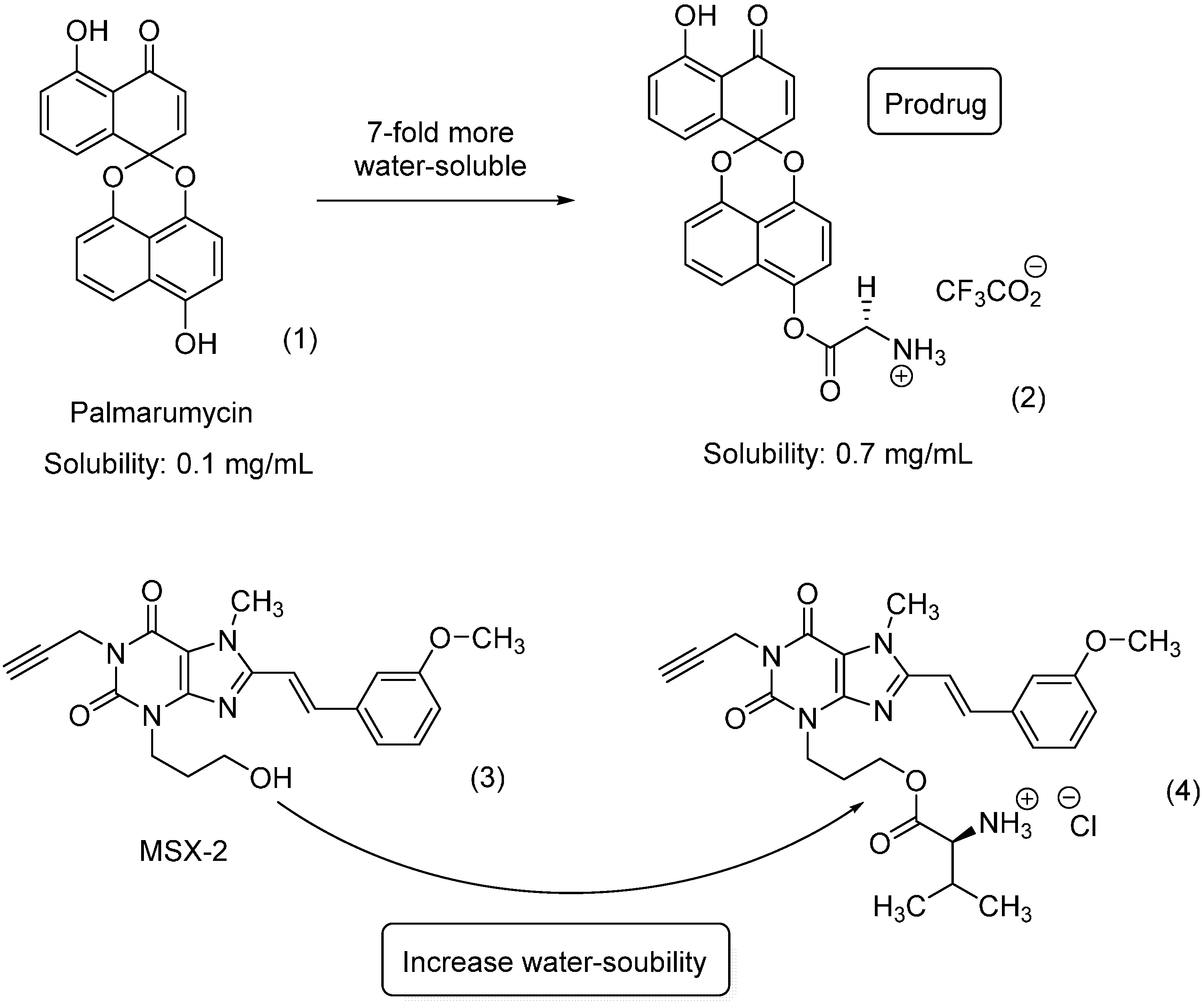
- Vollmann, K.; Qurishi, R.; Hockemeyer, J.; Müller, C.E. Synthesis and properties of a new water-soluble prodrug of the adenosine A 2A receptor antagonist MSX-2. Molecules 2008, 13, 348–359. [Google Scholar] [CrossRef] [PubMed]
- De Lera Ruiz, M.; Lim, Y.; Zheng, J. Adenosine A2A receptor as a drug discovery target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef] [PubMed]
- Burbiel, J.C.; Mu, C.E. Multigram-Scale Syntheses, Stability, and Photoreactions of A 2A Adenosine Receptor Antagonists with 8-Styrylxanthine Structure: Potential Drugs for Parkinson’s Disease. J. Org. Chem. 2004, 69, 3308–3318. [Google Scholar]
- Sauer, R.; Maurinsh, J.; Reith, U.; Fülle, F.; Klotz, K.N.; Müller, C.E. Water-soluble phosphate prodrugs of 1-propargyl-8-styrylxanthine derivatives, A2A-selective adenosine receptor antagonists. J. Med. Chem. 2000, 43, 440–448. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Balzarini, J. Aryl furano pyrimidines: The most potent and selective anti-VZV agents reported to date. Antiviral Res. 2006, 71, 149–153. [Google Scholar] [CrossRef] [PubMed]
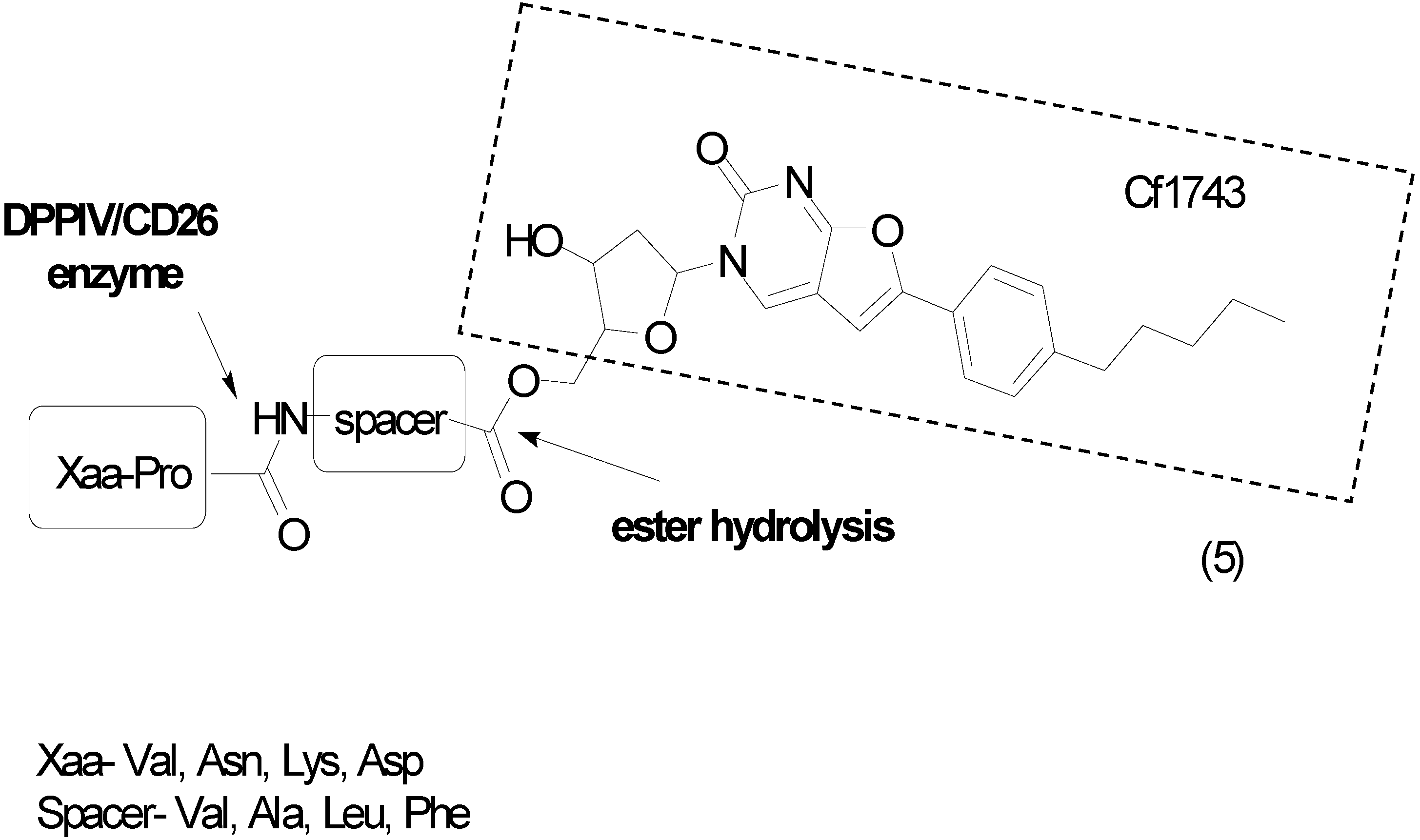
- Diez-Torrubia, A.; Balzarini, J.; Andrei, G.; Snoeck, R.; de Meester, I.; Camarasa, M.J.; Velázquez, S. Dipeptidyl peptidase IV dependent water-soluble prodrugs of highly lipophilic bicyclic nucleoside analogues. J. Med. Chem. 2011, 54, 1927–1942. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; de Vrueh, R.L.; Rhie, J.K.; Covitz, K.M.; Smith, P.L.; Lee, C.P.; Oh, D.M.; Sadée, W.; Amidon, G. 5’-Amino acid esters of antiviral nucleosides, acyclovir, and AZT are absorbed by the intestinal PEPT1 peptide transporter. Pharm. Res. 1988, 15, 1154–1159. [Google Scholar] [CrossRef]
- Ohwada, J.; Ozawa, S.; Kohchi, M.; Fukuda, H.; Murasaki, C.; Suda, H.; Murata, T.; Niizuma, S.; Tsukazaki, M.; Ori, K.; et al. Synthesis and biological activities of a pH-dependently activated water-soluble prodrug of a novel hexacyclic camptothecin analog. Bioorg. Med. Chem. Lett. 2009, 19, 2772–2776. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Huang, W.; Fei, Y.J.; Leibach, F.H.; Ganapathy, V.; Ganapathy, M.E. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J. Pharm. Sci. 2000, 89, 781–789. [Google Scholar] [CrossRef]
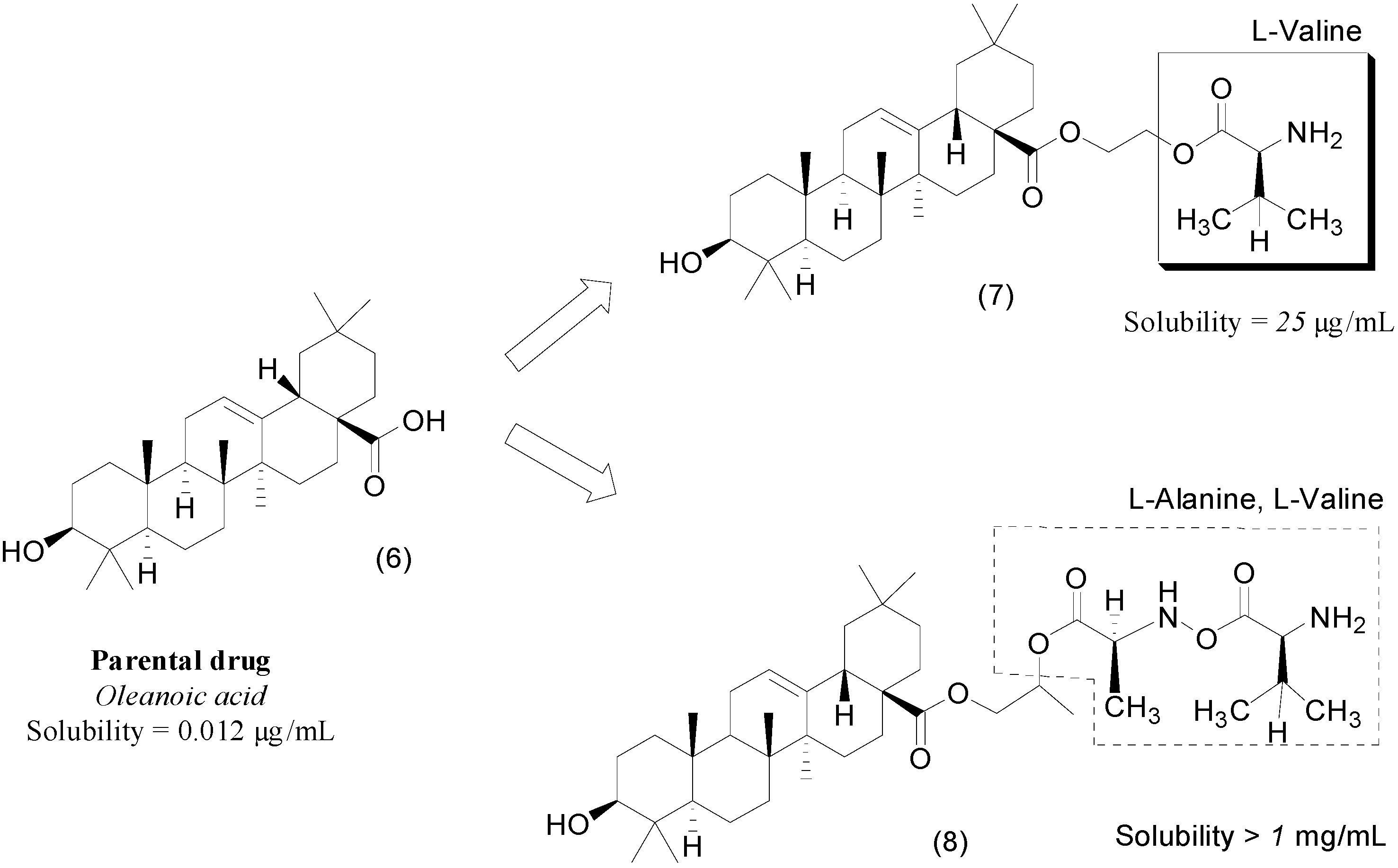
- Cao, F.; Jia, J.; Yin, Z.; Gao, Y.; Sha, L.; Lai, Y.; Ping, Q.; Zhang, Y. Ethylene Glycol-Linked Amino Acid Diester Prodrugs of Oleanolic Acid for PepT1-Mediated Transport: Synthesis, Intestinal Permeability and Pharmacokinetics. Mol. Pharm. 2012, 9, 2127–2135. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Gao, Y.; Wang, M.; Fang, L.; Ping, Q. Propylene glycol-linked amino acid/dipeptide diester prodrugs of oleanolic acid for PepT1-mediated transport: Synthesis, intestinal permeability, and pharmacokinetics. Mol. Pharm. 2013, 10, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.B.; Kang, H.; Wang, L.; Gao, L.; Liu, P.; Xie, J.; Zhang, F.X.; Weng, X.Q.; Shen, Z.X.; Chen, J.; et al. Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t(8;21) leukemia in vitro and in vivo. Blood 2007, 109, 3441–3450. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, E.; Cheng, Y.; Bao, J. Oridonin: An active diterpenoid targeting cell cycle arrest, apoptotic and autophagic pathways for cancer therapeutics. Int. J. Biochem. Cell Biol. 2011, 43, 701–704. [Google Scholar] [CrossRef] [PubMed]
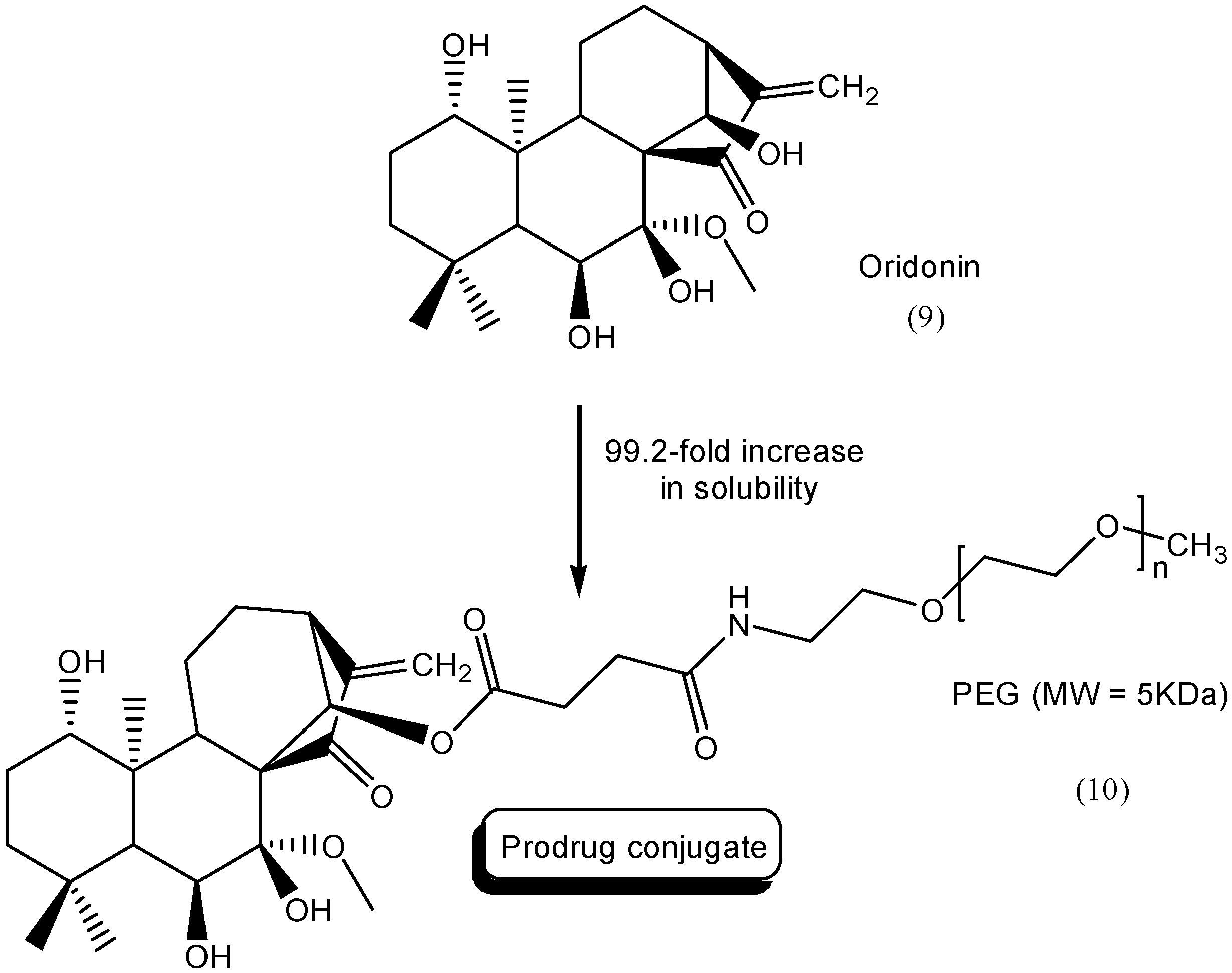
- Shen, J.; Zhang, D.; Zhao, Z.; Jia, L.; Zheng, D.; Liu, G.; Hao, L.; Zhang, Q.; Tian, X.; Li, C.; et al. Synthesis, characterization, in vitro and in vivo evaluation of PEGylated oridonin conjugates. Int. J. Pharm. 2013, 456, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.M.; Sun, Y.; Ge, X.; Wan, X.; Li, C.B. Gambogic acid induces apoptosis and inhibits colorectal tumor growth via mitochondrial pathways. World J. Gastroenterol. 2015, 21, 6194–6205. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, P.; Ye, H.; Zhang, C.; Shen, W.; Ping, Q. Water-soluble gambogic acid PEGylated prodrugs: Synthesis, characterization, physicochemical properties and in vitro hydrolysis. Pharmazie 2008, 63, 711–717. [Google Scholar] [PubMed]
- Adams, J.D.; Flora, K.; Goldspiel, B.R.; Wilson, J.W.; Arbuck, S.G.; Finley, R. Taxol: A history of pharmaceutical development and current pharmaceutical concerns. J. Natl. Cancer Inst. Monogr. 1993, 15, 141–147. [Google Scholar] [PubMed]
- Skwarczynski, M.; Sohma, Y.; Noguchi, M.; Kimura, M.; Hayashi, Y.; Hamada, Y.; Kimura, T.; Kiso, Y. No auxiliary, no byproduct strategy for water-soluble prodrugs of taxoids: Scope and limitation of O–N intramolecular acyl and acyloxy migration reactions. J. Med. Chem. 2005, 48, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, K.; Kubota, N. Chemo-enzymatic synthesis of ester-linked docetaxel-monosaccharide conjugates as water-soluble prodrugs. Molecules 2011, 16, 6769–6777. [Google Scholar] [CrossRef] [PubMed]
- Estrelaa, J.M.; Ortegaa, A.; Obradora, E. Glutathione in Cancer Biology and Therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef] [PubMed]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar]
- Gund, M.; Khanna, A.; Dubash, N.; Damre, A.; Singh, K.S.; Satyam, A. Water-soluble prodrugs of paclitaxel containing self-immolative disulfide linkers. Bioorg. Med. Chem. Lett. 2015, 25, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Du, W. Synthesis and Evaluation of Water-Soluble Etoposide Esters of Malic Acid as Prodrugs. Med. Chem. (Los. Angeles). 2013, 3, 740–747. [Google Scholar] [CrossRef]
- Ullah, N.; Huang, Z.; Sanaee, F.; Rodriguez-Dimitrescu, A.; Aldawsari, F.; Jamali, F.; Bhardwaj, A.; Islam, N.; Velázquez-Martínez, C. NSAIDs do not require the presence of a carboxylic acid to exert their anti-inflammatory effect—Why do we keep using it? J. Enzym. Inhib. Med. Chem. 2015, 24, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Halen, P.; Murumkar, P.; Giridhar, R.; Yadav, M. Prodrug designing of NSAIDs. Mini Rev. Med. Chem. 2009, 9, 124–139. [Google Scholar] [CrossRef] [PubMed]
- Pawar, V.; Thosani, R.; Kanhed, A.; Giridhar, R.; Yadav, M.R. Potential of Piperazinylalkylester Prodrugs of 6-Methoxy-2-Naphthylacetic Acid (6-MNA) for Percutaneous Drug Delivery. AAPS PharmSciTech 2015, 16, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Lobo, S.; Li, H.; Farhan, N.; Yan, G. Evaluation of diclofenac prodrugs for enhancing transdermal delivery. Drug Dev. Ind. Pharm. 2014, 40, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Formica, J.V.; Regelson, W. Review of the biology of quercetin and related bioflavonoids. Food Chem. Toxicol. 1995, 33, 1061–1080. [Google Scholar] [CrossRef]
- Montenegro, L.; Carbone, C.; Maniscalco, C.; Lambusta, D.; Nicolosi, G.; Ventura, C.A.; Puglisi, G. In vitro evaluation of quercetin-3-O-acyl esters as topical prodrugs. Int. J. Pharm. 2007, 336, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Kamat, J.P.; Devasagayam, T.P.A. Tocotrienols from palm oil as potent inhibitors of lipid peroxidation and protein oxidation in rat brain mitochondria. Neurosci. Lett. 1995, 195, 179–182. [Google Scholar] [CrossRef]
- Newaz, M.A.; Nawal, N.N.; Rohaizan, C.H.; Muslim, N.; Gapor, A. alpha-Tocopherol increased nitric oxide synthase activity in blood vessels of spontaneously hypertensive rats. Am. J. Hypertens. 1999, 12, 839–844. [Google Scholar] [CrossRef]
- Qureshi, A.A.; Dewitt, G.; Kramer, G.; Gapor, A. Lowering humans of serum cholesterol in hypercholesterolemic by tocotrienols. Am. J. Clin. Nutr. 1991, 53, 1021S–1026S. [Google Scholar] [PubMed]
- Qureshi, A.A.; Bradlow, B.A.; Brace, L.; Manganello, J.; Peterson, D.M.; Pearce, B.C.; Wright, J.J.; Gapor, A.; Elson, C. Response of hypercholesterolemic subjects to administration of tocotrienols. Lipids 1995, 30, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.A.; Bradlow, B.A.; Salser, W.A.; Brace, L.D. Novel tocotrienols of rice bran modulate cardiovascular disease risk parameters of hypercholesterolomic humans. J. Nutr. Biochem. 1997, 8, 290–298. [Google Scholar] [CrossRef]
- Parker, B.A.; Pearce, B.C.; Clark, R.W.; Gordon, D.A.; Wright, J.J.K. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme a reductase. J. Biol. Chem. 1993, 268, 11230–11238. [Google Scholar] [PubMed]
- Raederstorff, D.; Elste, V.; Aebischer, C.; Weber, P. Effect of either gamma-tocotrienol or a tocotrienol mixture on the plasma lipid profile in hamsters. Ann. Nutr. Metab. 2002, 46, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Guthrie, N.; Gapor, A.; Chambers, A.F.; Carroll, K.K. Inhibition of proliferation of estrogen receptor-negative MDA-MB-435 and -positive MCF-7 human breast cancer cells by palm oil tocotrienols and tamoxifen, alone and in combination. J. Nutr. 1997, 127, 544S–548S. [Google Scholar] [PubMed]
- Nesaretnam, K.; Ambra, R.; Selvaduray, K.R.; Radhakrishnan, A.; Reimann, K.; Razak, G.; Virgili, F. Tocotrienol-rich fraction from palm oil affects gene expression in tumors resulting from MCF-7 cell inoculation in athymic mice. Lipids 2004, 39, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.J.; Sylvester, P. Gamma-tocotrienol inhibits neoplastic mammary epithelial cell proliferation by decreasing Akt and nuclear factor kappaB activity. Exp. Biol. Med. 2005, 230, 235–241. [Google Scholar]
- Akaho, N.; Takata, J.; Fukushima, T.; Matsunaga, K.; Hattori, A.; Hidaka, R.; Fukui, K.; Yoshida, M.; Fujioka, T.; Karube, Y.; et al. Preparation and In Vivo Evaluation of a Water-Soluble Prodrug for 2R-γ-Tocotrienol and as a Two-Step Prodrug for 2,7,8-Trimethyl-2S-(β-carboxyethyl)-6-hydroxychroman (S-γ-CEHC) in Rat. Drug Metab. Dispos. 2007, 35, 1502–1510. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Markovitz, B.; Trovato, R.; Murphy, B.R.; Austin, H.; Willardsen, A.J.; Baichwal, V.; Morham, S.; Bajji, A. Discovery of a new HIV-1 inhibitor scaffold and synthesis of potential prodrugs of indazoles. Bioorg. Med. Chem. Lett. 2013, 23, 2888–2892. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, P.; Blum, M. Pharmacokinetics of acyclovir after intravenous and oral administration. J. Antimicrob. Chemother. 1983, 12, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Diez-torrubia, A.; Cabrera, S.; Castro, S.D.; García-aparicio, C.; Mulder, G.; Meester, I.D.; Camarasa, M.; Balzarini, J.; Velázquez, S. Novel water-soluble prodrugs of acyclovir cleavable by the dipeptidyl-peptidase IV (DPP IV/CD26) enzyme. Eur. J. Med. Chem. 2013, 70, 456–468. [Google Scholar] [CrossRef] [PubMed]
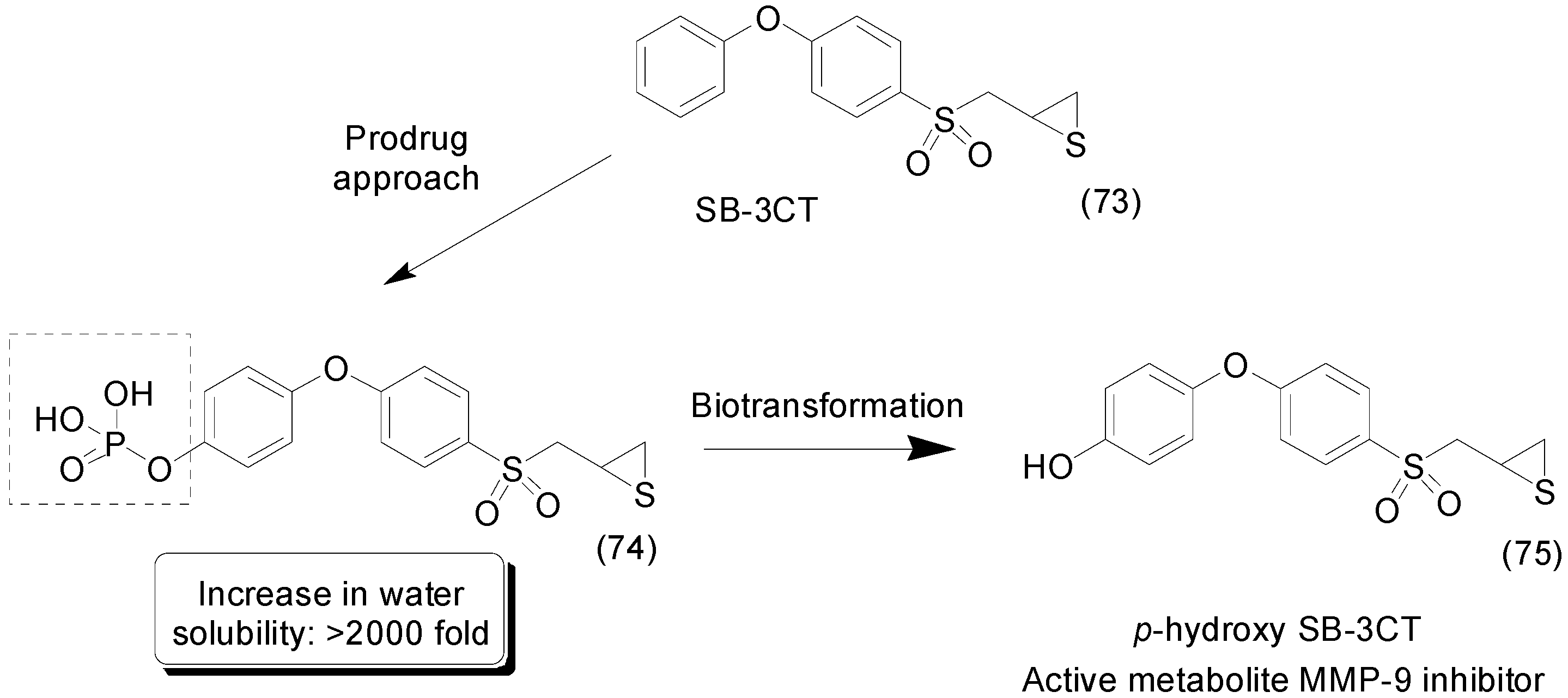
- Zhou, J.; Tao, P.; Fisher, J.F.; Shi, Q.; Mobashery, S.; Schlegel, H.B. QM/MM Studies of the Matrix Metalloproteinase 2 (MMP2) Inhibition Mechanism of (S)-SB-3CT and its Oxirane Analogue. J. Chem. Theory Comput. 2010, 6, 3580–3587. [Google Scholar] [CrossRef] [PubMed]
- Gooyit, M.; Lee, M.; Schroeder, V.A.; Ikejiri, M.; Suckow, M.A.; Mobashery, S.; Chang, M. Selective water-soluble gelatinase inhibitor prodrugs. J. Med. Chem. 2011, 54, 6676–6690. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Hong, D.H.; Han, S.B.; Jung, S.-H.; Kim, H.C.; Fine, R.L.; Lee, S.-H.; Kim, H.M. A novel stereo-selective sulfonylurea, 1-[1-(4-aminobenzoyl)-2,3-dihydro-1H-indol-6-sulfonyl]-4-phenyl-imidazolidin-2-one, has antitumor efficacy in in vitro and in vivo tumor models. Biochem. Pharmacol. 2002, 64, 473–480. [Google Scholar] [CrossRef]
- Lee, K.-C.; Venkateswararao, E.; Sharma, V.K.; Jung, S.-H. Investigation of amino acid conjugates of (S)-1-[1-(4-aminobenzoyl)-2,3-dihydro-1H-indol-6-sulfonyl]-4-phenyl-imidazolidin-2-one (DW2282) as water soluble anticancer prodrugs. Eur. J. Med. Chem. 2014, 80, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, H.; Takahashi, T.; Fujisaki, J.; Masunaga, T.; Sato, S.; Hiroi, J.; Tokunaga, Y.; Kimura, S.; Hata, T. Bone-specific delivery and sustained release of diclofenac, a non-steroidal anti-inflammatory drug, via bisphosphonic prodrug based on the Osteotropic Drug Delivery System (ODDS). J. Control. Release 2001, 70, 183–191. [Google Scholar] [CrossRef]
- Wang, D.; Miller, S.C.; Kopečková, P.; Kopeček, J. Bone-targeting macromolecular therapeutics. Adv. Drug Deliv. Rev. 2005, 57, 1049–1076. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.B.; Hartgerink, J.D.; Goepferich, A.; Mikos, A.G. Synthesis and in vitro hydroxyapatite binding of peptides conjugated to calcium-binding moieties. Biomacromolecules 2007, 8, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, J.; Waki, Y.; Takahashi-Nishioka, T.; Yokogawa, K.; Miyamoto, K.I. Selective drug delivery to bone using acidic oligopeptides. J. Bone Miner. Metab. 2009, 27, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sarig, S. Aspartic acid nucleates the apatite crystallites of bone: A hypothesis. Bone 2004, 35, 108–113. [Google Scholar] [CrossRef] [PubMed]
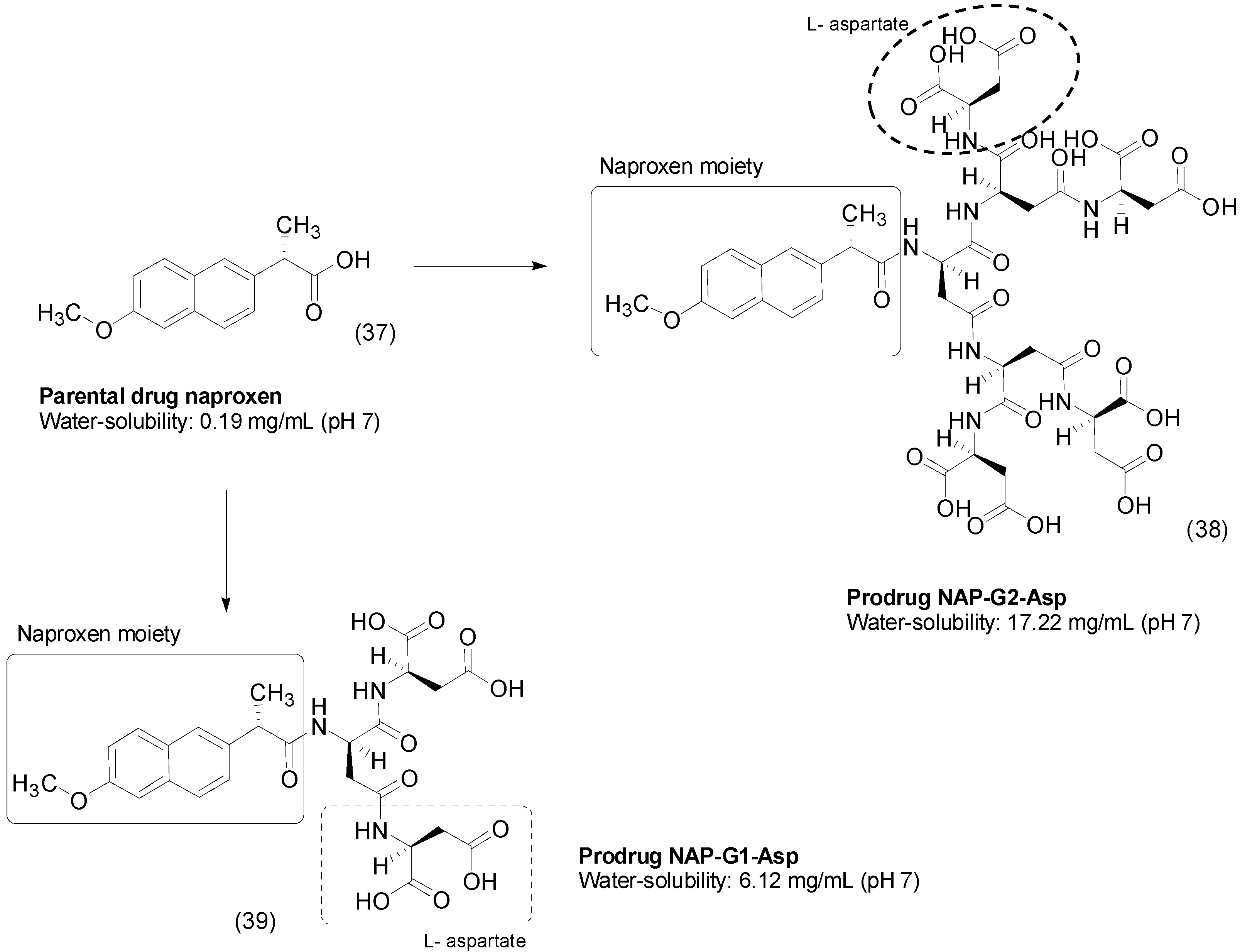
- Ouyang, L.; Zhang, J.; Pan, J.; Yan, L.; Guo, L. Synthesis and preliminary evaluation in vitro of novel naproxen-dendritic peptide conjugates. Drug Deliv. 2009, 16, 348–356. [Google Scholar] [CrossRef] [PubMed]
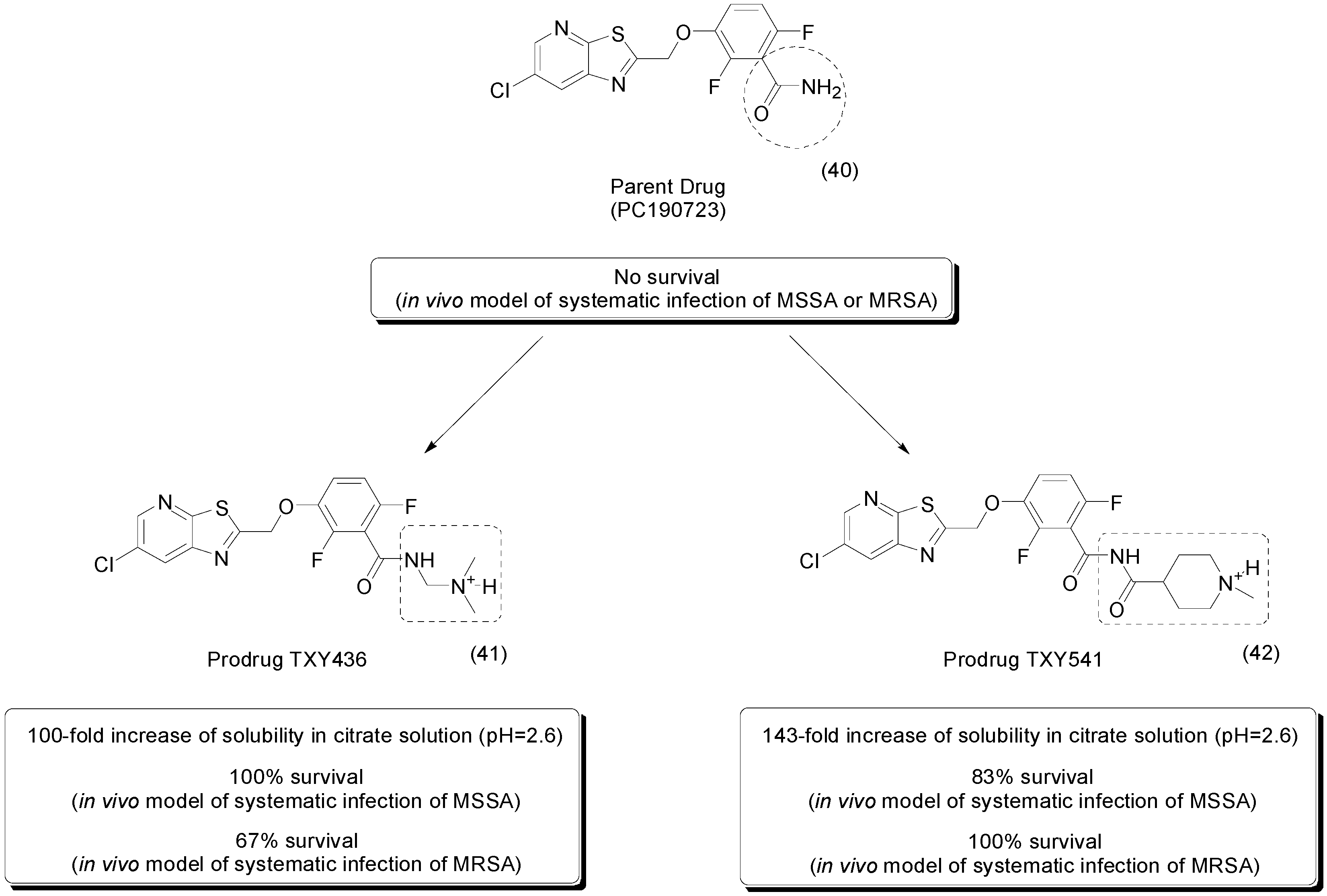
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; LaVoie, E.J.; Pilch, D.S. An FtsZ-targeting prodrug with oral antistaphylococcal efficacy in vivo. Antimicrob. Agents Chemother. 2013, 57, 5860–5869. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Mark, L.; Zhang, Y.; Parhi, A.K.; Lavoie, E.J.; Pilch, D.S. Pharmacokinetics and in vivo antistaphylococcal efficacy of TXY541, a 1-methylpiperidine-4-carboxamide prodrug of PC190723. Biochem. Pharmacol. 2013, 86, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Monneret, C. Histone deacetylase inhibitors. Eur. J. Med. Chem. 2005, 40, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed]
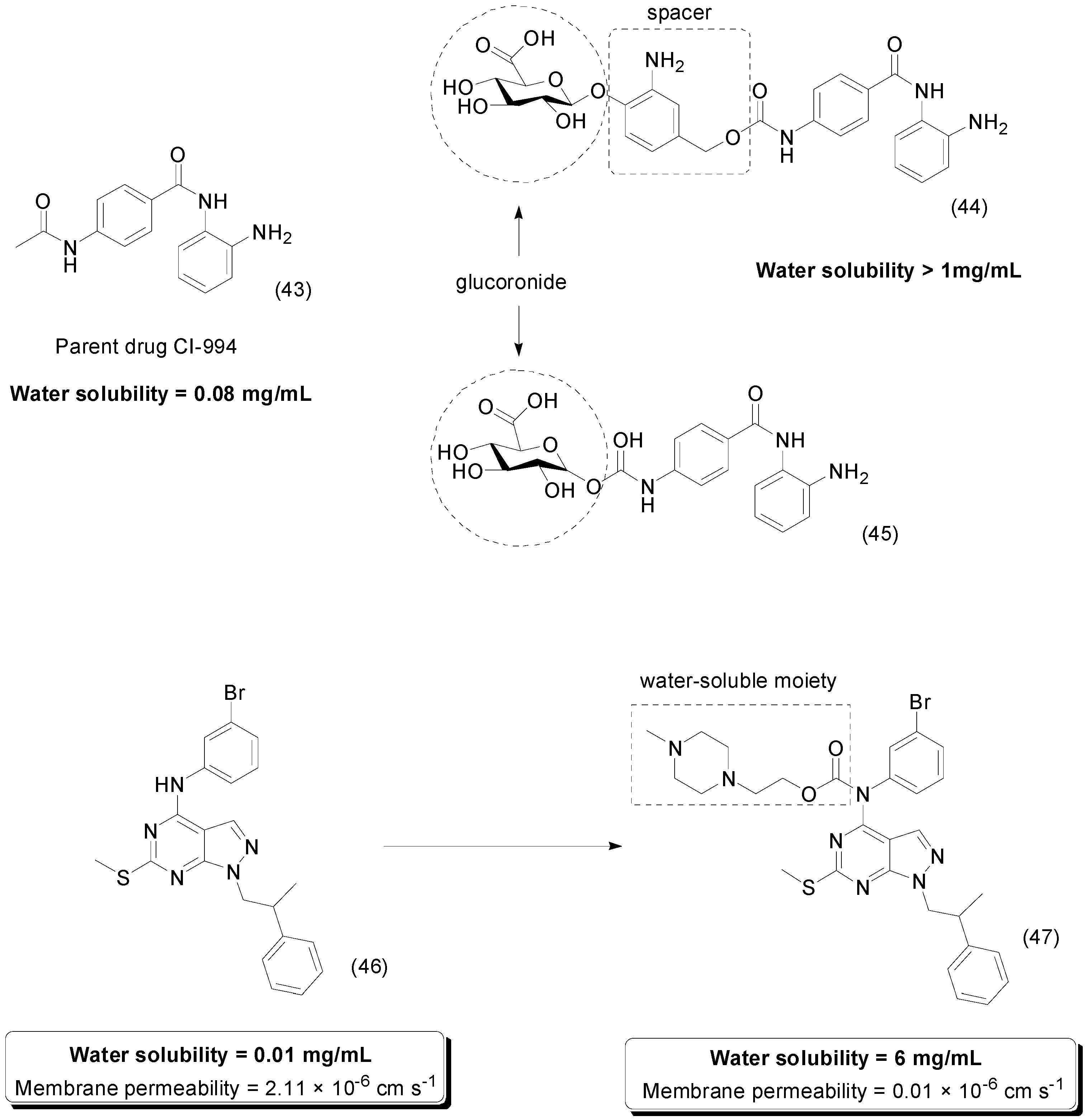
- Thomas, M.; Clarhaut, J.; Tranoy-Opalinski, I.; Gesson, J.P.; Roche, J.; Papot, S. Synthesis and biological evaluation of glucuronide prodrugs of the histone deacetylase inhibitor CI-994 for application in selective cancer chemotherapy. Bioorg. Med. Chem. 2008, 16, 8109–8116. [Google Scholar] [CrossRef] [PubMed]
- Vignaroli, G.; Zamperini, C.; Dreassi, E.; Radi, M.; Angelucci, A.; Sanità, P.; Crespan, E.; Kissova, M.; Maga, G.; Schenone, S.; et al. Pyrazolo[3,4-d]pyrimidine prodrugs: Strategic optimization of the aqueous solubility of dual Src/Abl inhibitors. ACS Med. Chem. Lett. 2013, 4, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Scholar, E.M. Role of tyrosine kinase inhibitors in cancer therapy. J. Pharmacol. Exp. Ther. 2005, 315, 971–979. [Google Scholar]
- Debele, T.; Peng, S.; Tsai, H.-C. Drug Carrier for Photodynamic Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 22094–22136. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, I.J.; Dougherty, T.J. Basic principles of photodynamic therapy. J. Porphyr. Phthalocyanines 2001, 5, 105–129. [Google Scholar] [CrossRef]
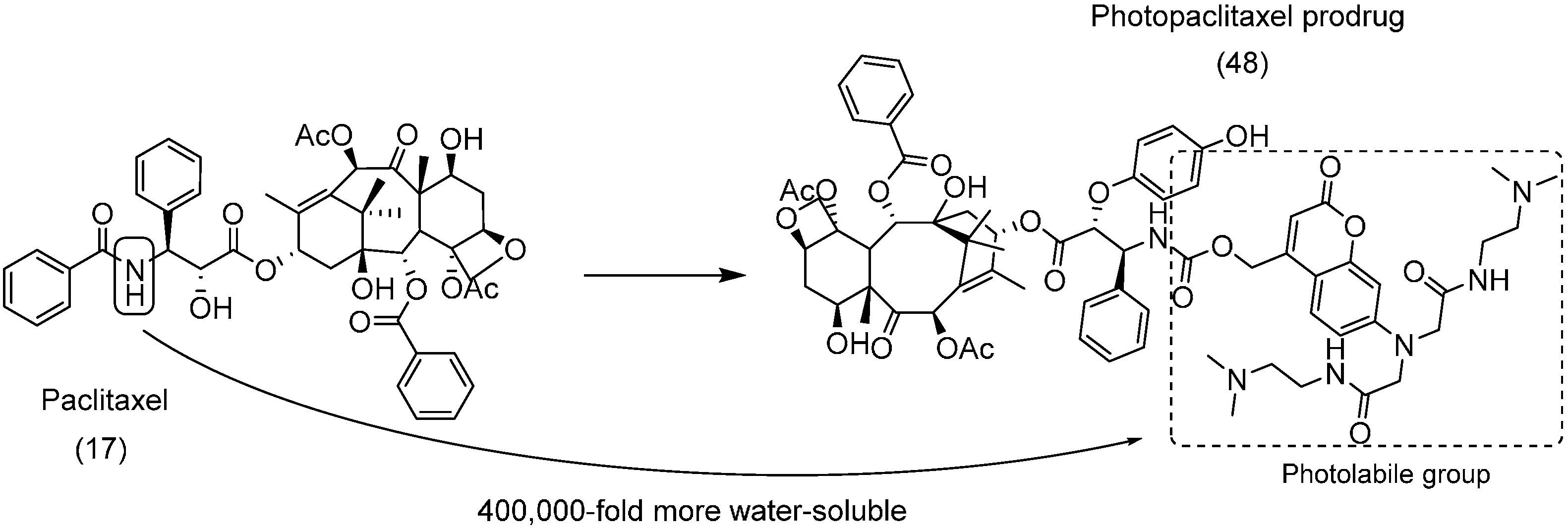
- Noguchi, M.; Skwarczynski, M.; Prakash, H.; Hirota, S.; Kimura, T.; Hayashi, Y.; Kiso, Y. Development of novel water-soluble photocleavable protective group and its application for design of photoresponsive paclitaxel prodrugs. Bioorg. Med. Chem. 2008, 16, 5389–5397. [Google Scholar] [CrossRef] [PubMed]
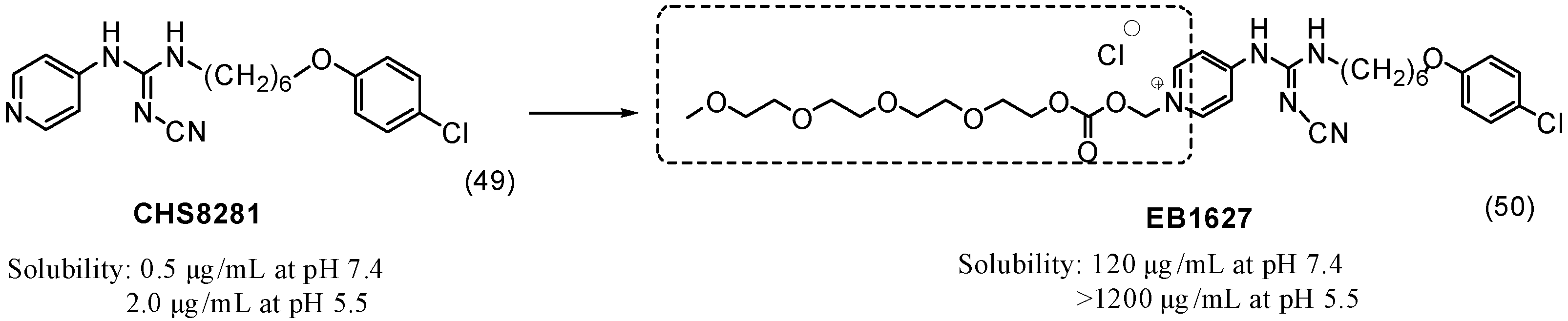
- Binderup, E.; Björkling, F.; Hjarnaa, P.V.; Latini, S.; Baltzer, B.; Carlsen, M.; Binderup, L. EB1627: A soluble prodrug of the potent anticancer cyanoguanidine CHS828. Bioorg. Med. Chem. Lett. 2005, 15, 2491–2494. [Google Scholar] [CrossRef] [PubMed]
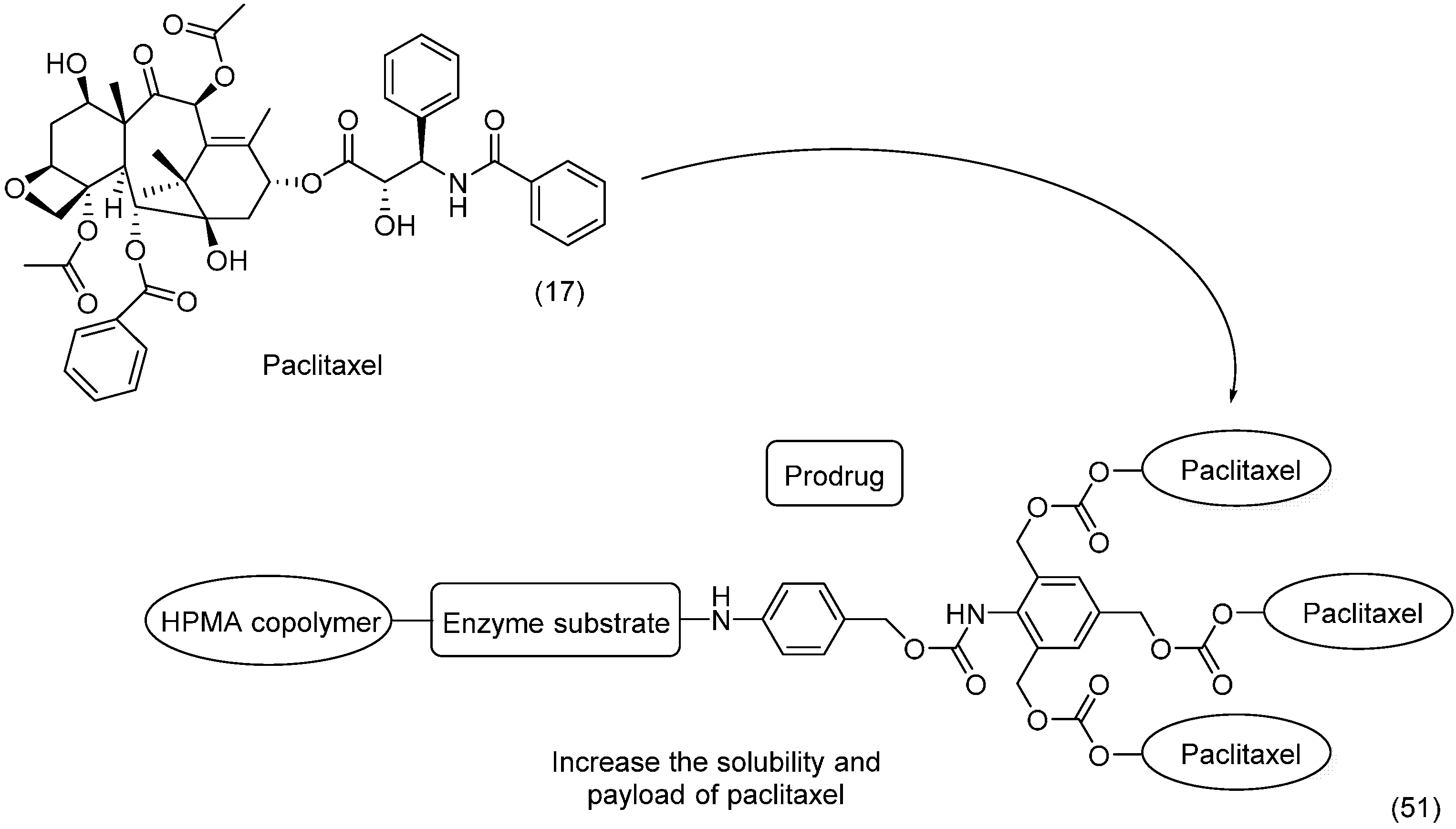
- Erez, R.; Segal, E.; Miller, K.; Satchi-Fainaro, R.; Shabat, D. Enhanced cytotoxicity of a polymer-drug conjugate with triple payload of paclitaxel. Bioorg. Med. Chem. 2009, 17, 4327–4335. [Google Scholar] [CrossRef] [PubMed]
- Satchi-Fainaro, R.; Hailu, H.; Davies, J.W.; Summerford, C.; Duncan, R. PDEPT: Polymer-Directed Enzyme Prodrug Therapy. 2. HPMA Copolymer-β-lactamase and HPMA Copolymer-C-Dox as a Model Combination. Bioconjugate Chem. 2003, 14, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Wani, M.C.; Ronman, P.E.; Lindley, J.T.; Wall, M.E. Plant antitumor agents. 18. Synthesis and biological activity of camptothecin analogs. J. Med. Chem. 1980, 23, 554–560. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.H.; Andersson, B.S.; Nelson, J.A. DNA topoisomerase I as a site of action for 10-hydroxycamptothecin in human promyelocytic leukemia cells. Cancer Biochem. Biophys. 1990, 11, 23–30. [Google Scholar] [PubMed]
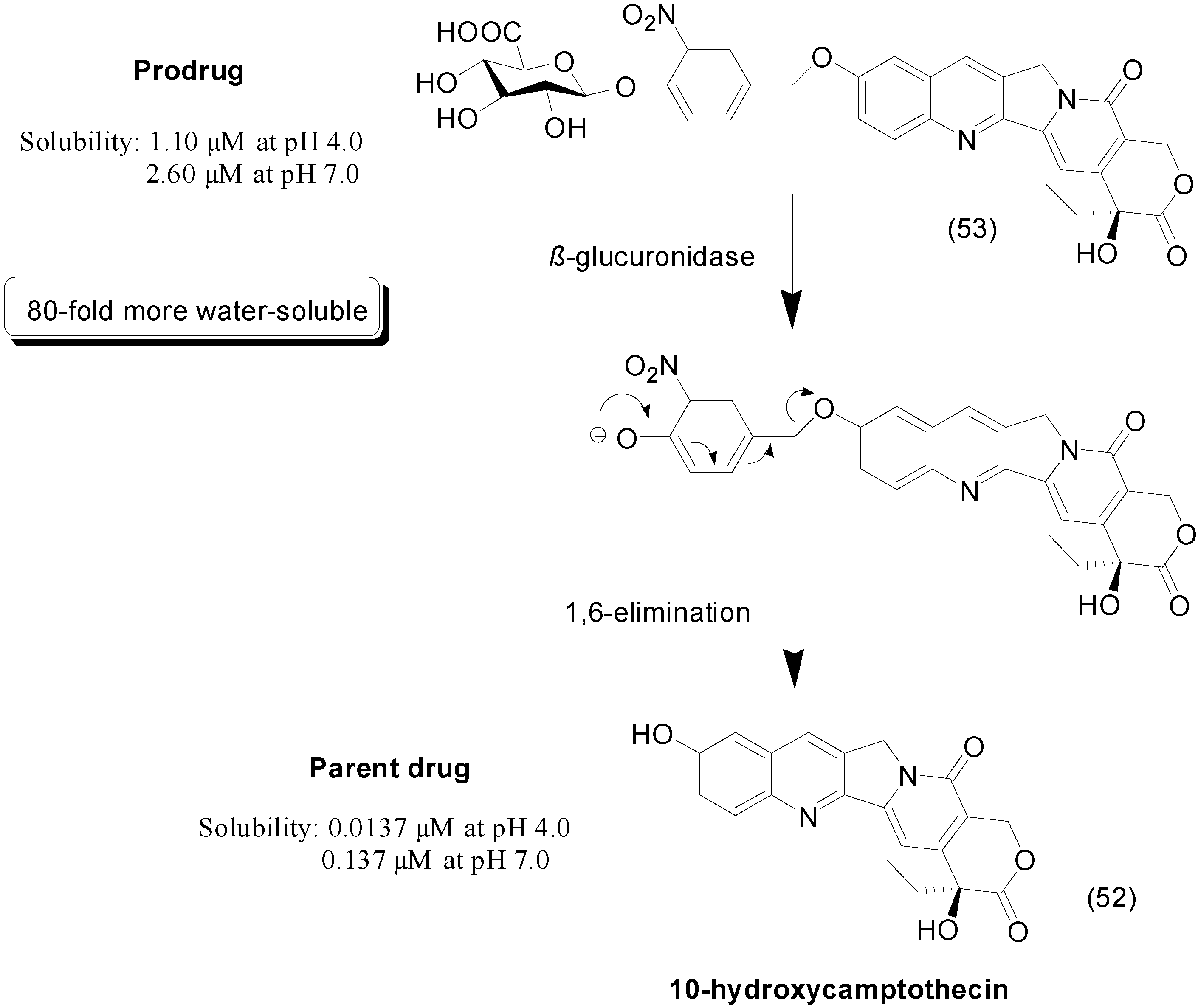
- Leu, Y.L.; Chen, C.S.; Wu, Y.J.; Chern, J.W. Benzyl ether-linked glucuronide derivative of 10-hydroxycamptothecin designed for selective camptothecin-based anticancer therapy. J. Med. Chem. 2008, 51, 1740–1746. [Google Scholar] [CrossRef] [PubMed]
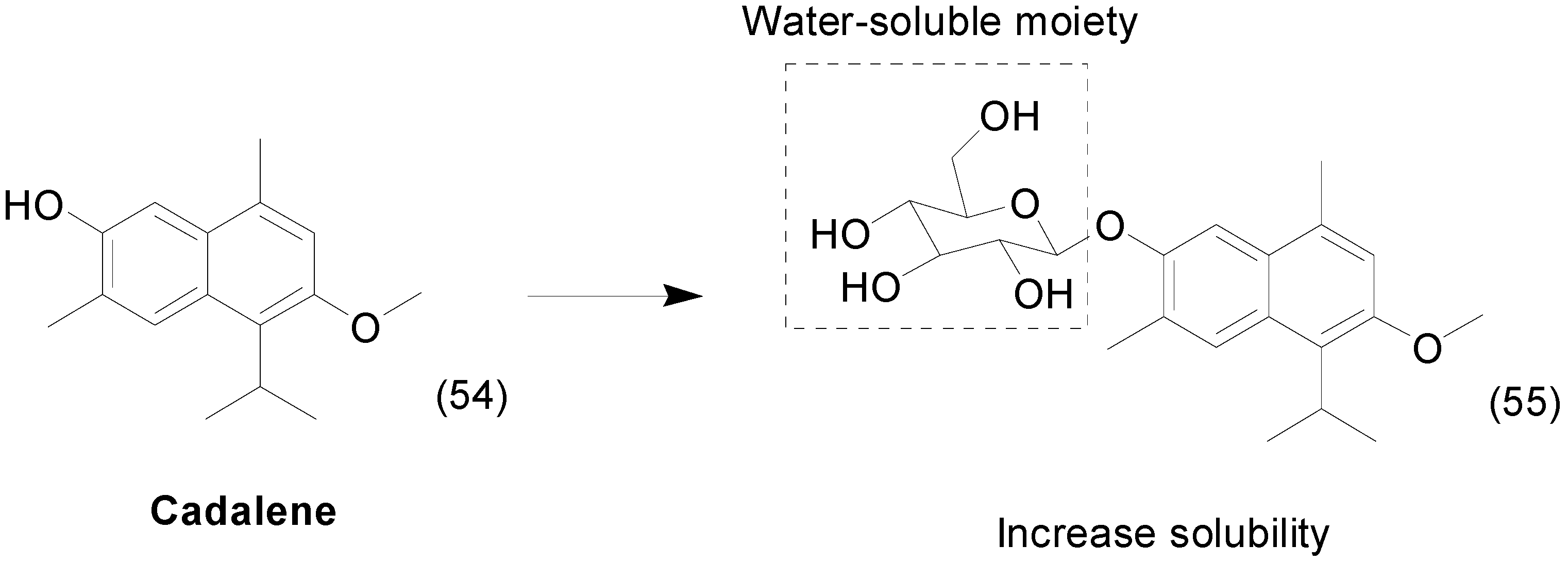
- Kim, J.H.; Lee, H.J.; Yeon, S.C.; Choi, D.H.; Lee, S.S.; Kang, J.K.; Chae, C.H.; Paik, N.W.; Lee, K.H.; Cho, M.H. Antioxidative Effects of 7-Hydroxy-3-Methoxy-Cadalene Extracted from Zelkova serrata on Butanone-induced Oxidative Stress in A/J Mice. Phyther. Res. 2004, 18, 425–427. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Kwon, J.T.; Koh, M.; Cho, M.H.; Park, S.B. Enhanced efficacy of 7-hydroxy-3-methoxycadalene via glycosylation in in vivo xenograft study. Bioorg. Med. Chem. Lett. 2007, 17, 6335–6339. [Google Scholar] [CrossRef] [PubMed]
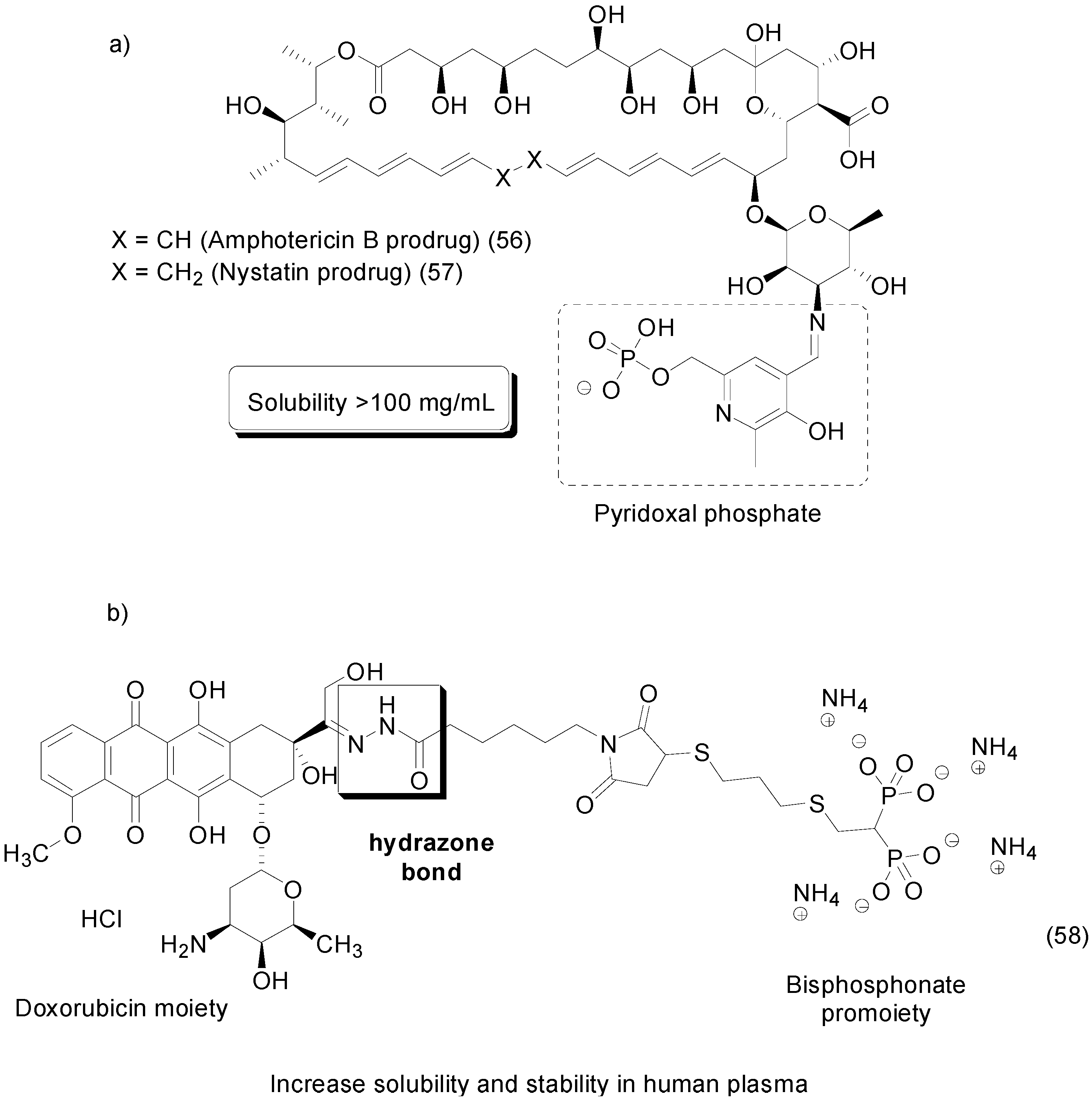
- Day, T.P.; Sil, D.; Shukla, N.M.; Anbanandam, A.; Day, V.W.; David, S.A. Imbuing aqueous solubility to amphotericin B and nystatin with a vitamin. Mol. Pharm. 2010, 8, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Hochdörffer, K.; Abu Ajaj, K.; Schäfer-Obodozie, C.; Kratz, F. Development of novel bisphosphonate prodrugs of doxorubicin for targeting bone metastases that are cleaved pH dependently or by cathepsin B: Synthesis, cleavage properties, and binding properties to hydroxyapatite as well as bone matrix. J. Med. Chem. 2012, 55, 7502–7515. [Google Scholar] [CrossRef] [PubMed]
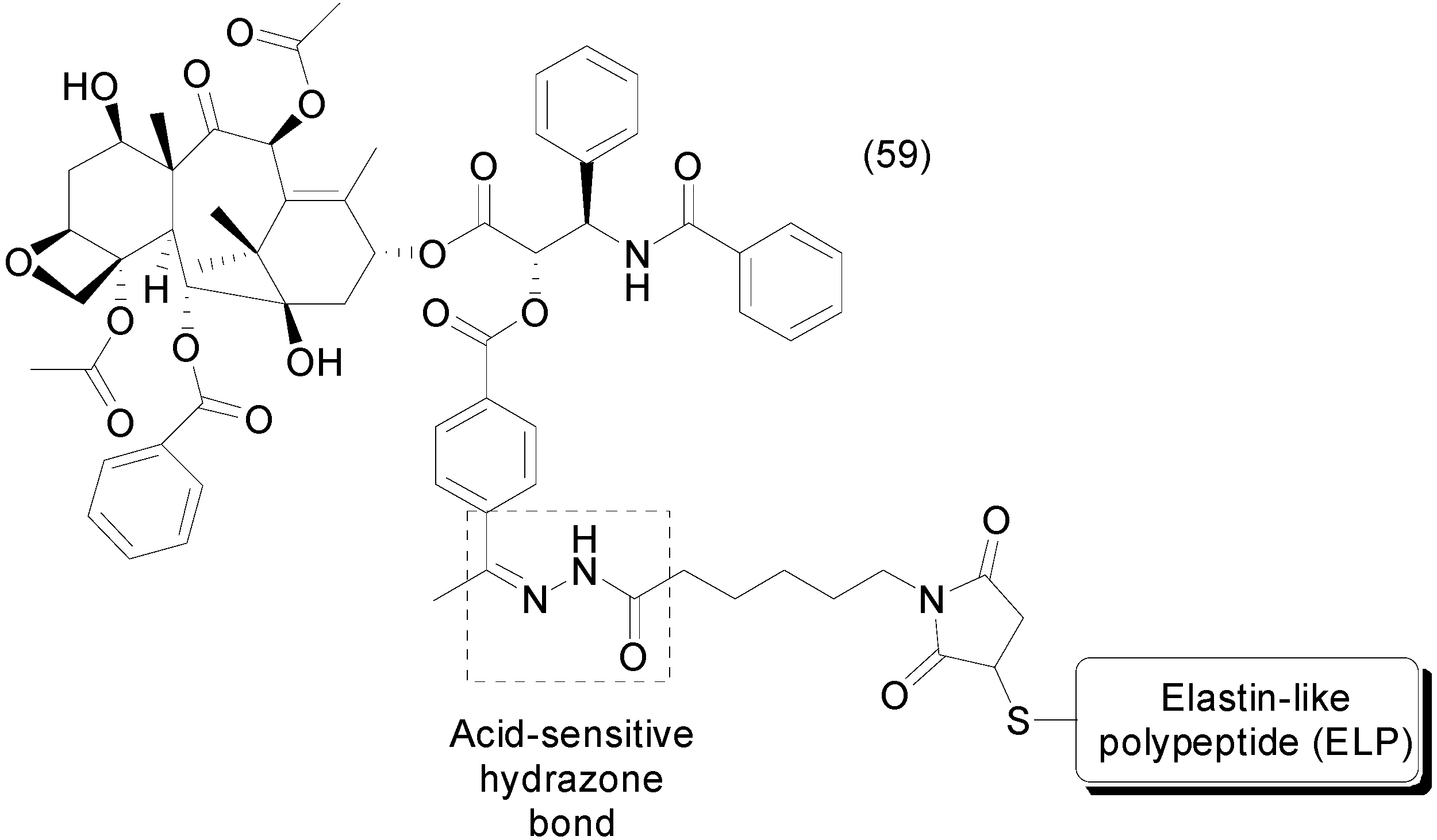
- Moktan, S.; Ryppa, C.; Kratz, F.; Raucher, D. A thermally responsive biopolymer conjugated to an acid-sensitive derivative of paclitaxel stabilizes microtubules, arrests cell cycle, and induces apoptosis. Investig. New Drugs 2012, 30, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C. Prodrugs of biologically active phosphate esters. Bioorg. Med. Chem. 2003, 11, 885–898. [Google Scholar] [CrossRef]
- Wiemer, A.; Wiemer, D. Prodrugs of phosphonates and phosphates: Crossing the membrane barrier. Top. Curr. Chem. 2014, 360, 115–160. [Google Scholar]
- Ghosh, K.; Mazumder Tagore, D.; Anumula, R.; Lakshmaiah, B.; Kumar, P.P.B.S.; Singaram, S.; Matan, T.; Kallipatti, S.; Selvam, S.; Krishnamurthy, P.; et al. Crystal structure of rat intestinal alkaline phosphatase—Role of crown domain in mammalian alkaline phosphatases. J. Struct. Biol. 2013, 184, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Hecker, S.J.; Erion, M.D. Prodrugs of Phosphates and Phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [Google Scholar] [CrossRef] [PubMed]
- Krise, J.P.; Stella, V.J. Prodrugs of Phosphates, Phosphonates, and Phosphinates. Adv. Drug Deliv. Rev. 1996, 19, 287–310. [Google Scholar] [CrossRef]
- Degoey, D.A.; Grampovnik, D.J.; Flosi, W.J.; Marsh, K.C.; Wang, X.C.; Klein, L.L.; Mcdaniel, K.F.; Liu, Y.; Long, M.A.; Kati, W.M.; Molla, A.; Kempf, D.J. Water-Soluble Prodrugs of the Human Immunodeficiency Virus Protease Inhibitors Lopinavir and Ritonavir Water-Soluble Prodrugs of the Human Immunodeficiency Virus Protease Inhibitors Lopinavir and Ritonavir. J. Med. Chem. 2009, 52, 2964–2970. [Google Scholar] [CrossRef] [PubMed]
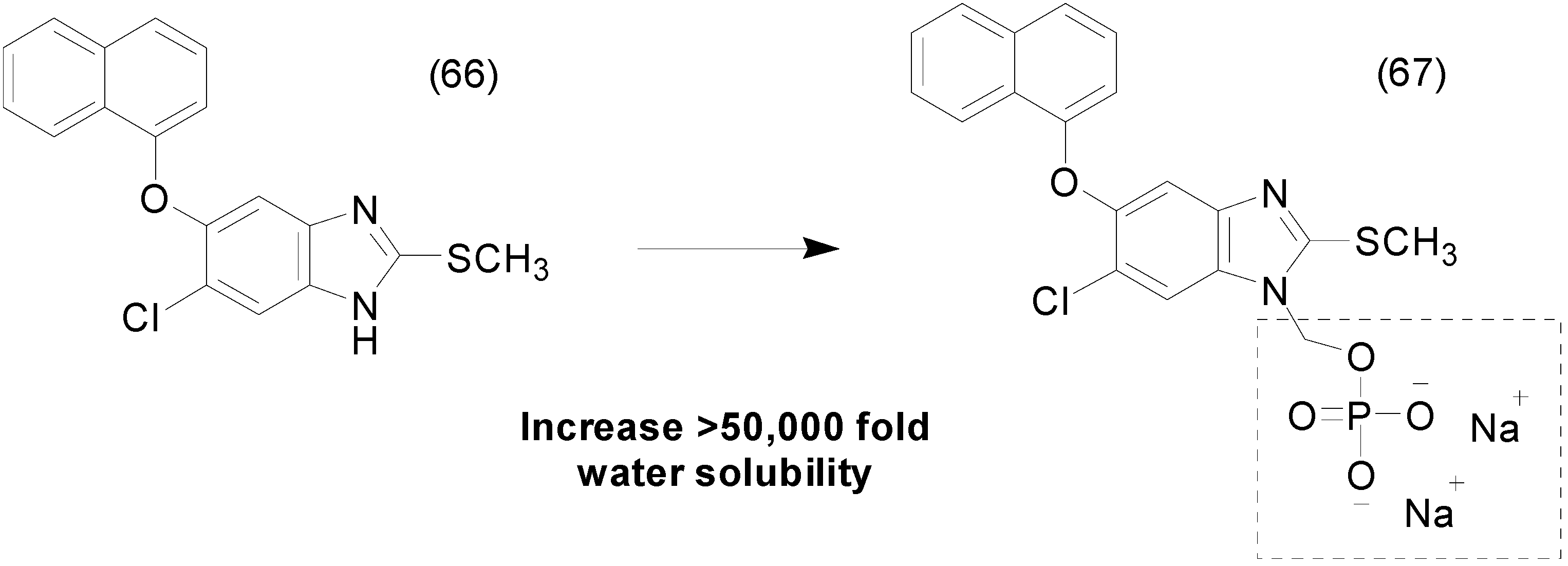
- Flores-ramos, M.; Ibarra-velarde, F.; Hernández-campos, A.; Vera-montenegro, Y.; Jung-cook, H.; Cantó-alarcón, G.J.; Misael, L.; Castillo, R. A highly water soluble benzimidazole derivative useful for the treatment of fasciolosis. Bioorg. Med. Chem. Lett. 2014, 24, 5814–5817. [Google Scholar] [CrossRef] [PubMed]
- Hachet-Haas, M.; Balabanian, K.; Rohmer, F.; Pons, F.; Franchet, C.; Lecat, S.; Chow, K.Y.C.; Dagher, R.; Gizzi, P.; Didier, B.; et al. Small Neutralizing Molecules to Inhibit Actions of the Chemokine CXCL12. J. Biol. Chem. 2008, 283, 23189–23199. [Google Scholar] [CrossRef] [PubMed]
- Gasparik, V.; Daubeuf, F.; Hachet-Haas, M.; Rohmer, F.; Gizzi, P.; Haiech, J.; Galzi, J.L.; Hibert, M.; Bonnet, D.; Frossard, N. Prodrugs of a CXC chemokine-12 (CXCL12) neutraligand prevent inflammatory reactions in an asthma model in vivo. ACS Med. Chem. Lett. 2012, 3, 10–14. [Google Scholar] [CrossRef] [PubMed]
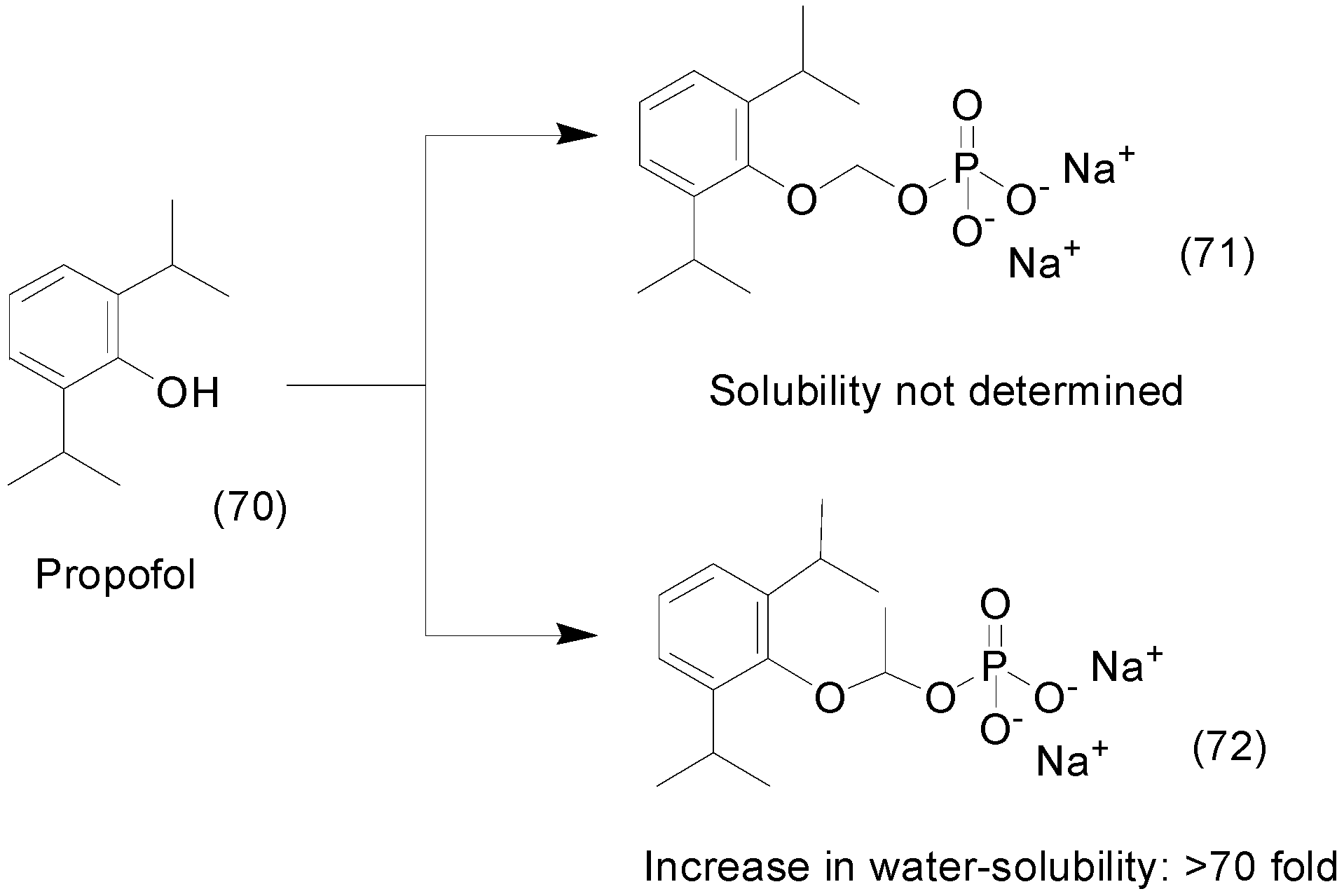
- Kumpulainen, H.; Järvinen, T.; Mannila, A.; Leppänen, J.; Nevalainen, T.; Mäntylä, A.; Vepsäläinen, J.; Rautio, J. Synthesis, in vitro and in vivo characterization of novel ethyl dioxy phosphate prodrug of propofol. Eur. J. Pharm. Sci. 2008, 34, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Chen, Z.; Tomlinson, B.N.; Gooyit, M.; Hesek, D.; Juárez, M.R.; Nizam, R.; Boggess, B.; Lastochkin, E.; Schroeder, V.A.; et al. Water-Soluble MMP-9 Inhibitor Reduces Lesion Volume after Severe Traumatic Brain Injury. ACS Chem. Neurosci. 2015, 10, 1658–1664. [Google Scholar] [CrossRef] [PubMed]
- Charifson, P.S.; Grillot, A.L.; Grossman, T.H.; Parsons, J.D.; Badia, M.; Bellon, S.; Deininger, D.D.; Drumm, J.E.; Gross, C.H.; LeTiran, A.; et al. Novel dual-targeting benzimidazole urea inhibitors of DNA gyrase and topoisomerase IV possessing potent antibacterial activity: Intelligent design and evolution through the judicious use of structure-guided design and structure-activity relationships. J. Med. Chem. 2008, 51, 5243–5263. [Google Scholar] [CrossRef] [PubMed]
- Grossman, T.H.; Bartels, D.J.; Mullin, S.; Gross, C.H.; Parsons, J.D.; Liao, Y.; Grillot, A.-L.; Stamos, D.; Olson, E.R.; Charifson, P.S.; Mani, N. Dual targeting of GyrB and ParE by a novel aminobenzimidazole class of antibacterial compounds. Antimicrob. Agents Chemother. 2007, 51, 657–666. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, H.; Shannon, D.E.; Chandupatla, K.R.; Dixit, V.; Engtrakul, J.J.; Ye, Z.; Jones, S.M.; O’Brien, C.F.; Nicolau, D.P.; Tessier, P.R.; et al. Discovery and Characterization of a Water-Soluble Prodrug of a Dual Inhibitor of Bacterial DNA Gyrase and Topoisomerase IV. ACS Med. Chem. Lett. 2015, 6, 822–826. [Google Scholar] [CrossRef] [PubMed]
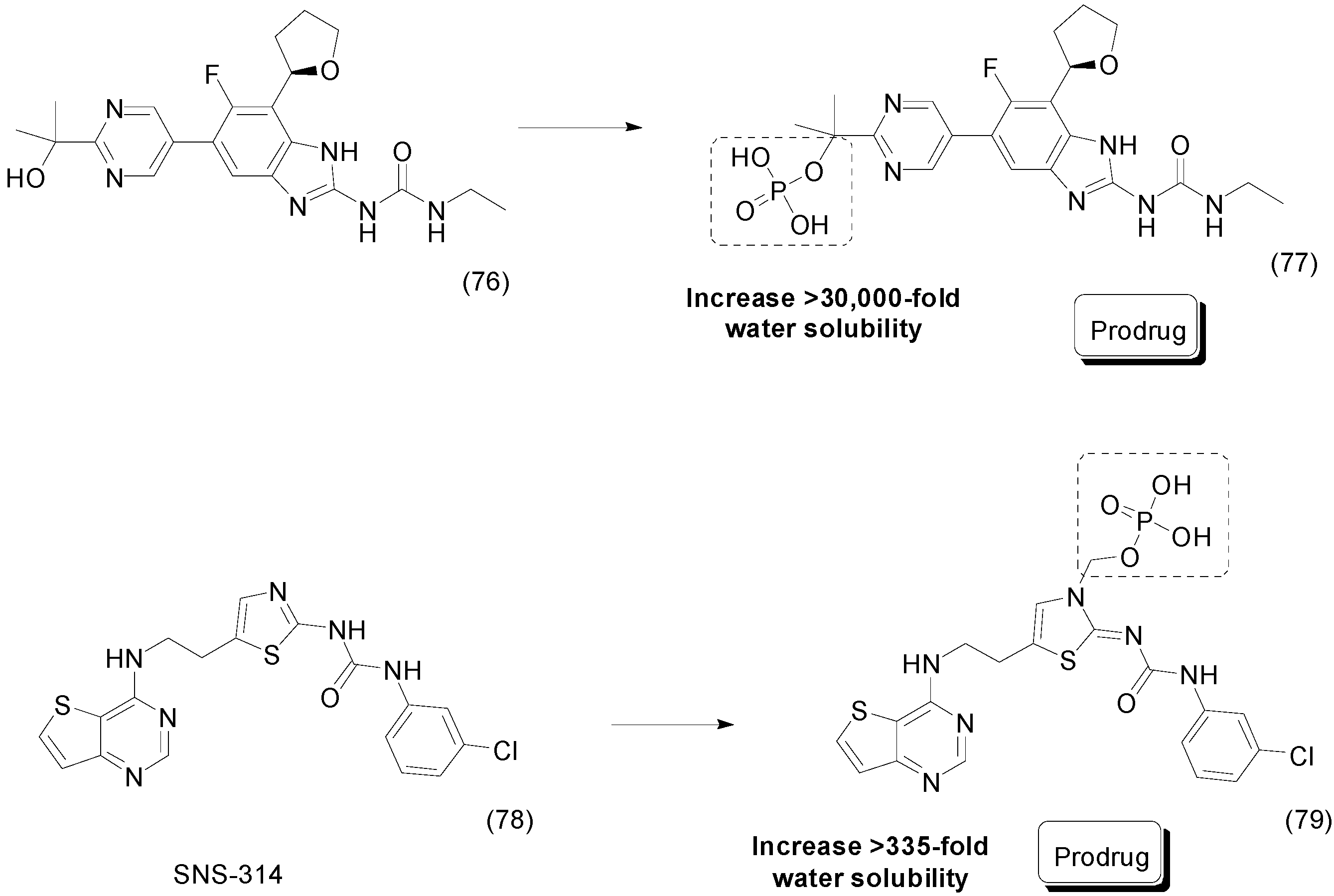
- Oslob, J.D.; Heumann, S.A.; Yu, C.H.; Allen, D.A.; Baskaran, S.; Bui, M.; Delarosa, E.; Fung, A.D.; Hashash, A.; Hau, J.; et al. Water-soluble prodrugs of an Aurora kinase inhibitor. Bioorg. Med. Chem. Lett. 2009, 19, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Mortlock, A.A.; Keen, N.J.; Jung, F.H.; Heron, N.M.; Foote, K.M.; Wilkinson, R.W.; Green, S. Progress in the development of selective inhibitors of aurora kinases. Curr. Top. Med. Chem. 2005, 5, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Venkatesh, S. Miniature device for aqueous and non-aqueous solubility measurements during drug discovery. Pharm Res 2004, 21, 1758–1761. [Google Scholar] [CrossRef] [PubMed]
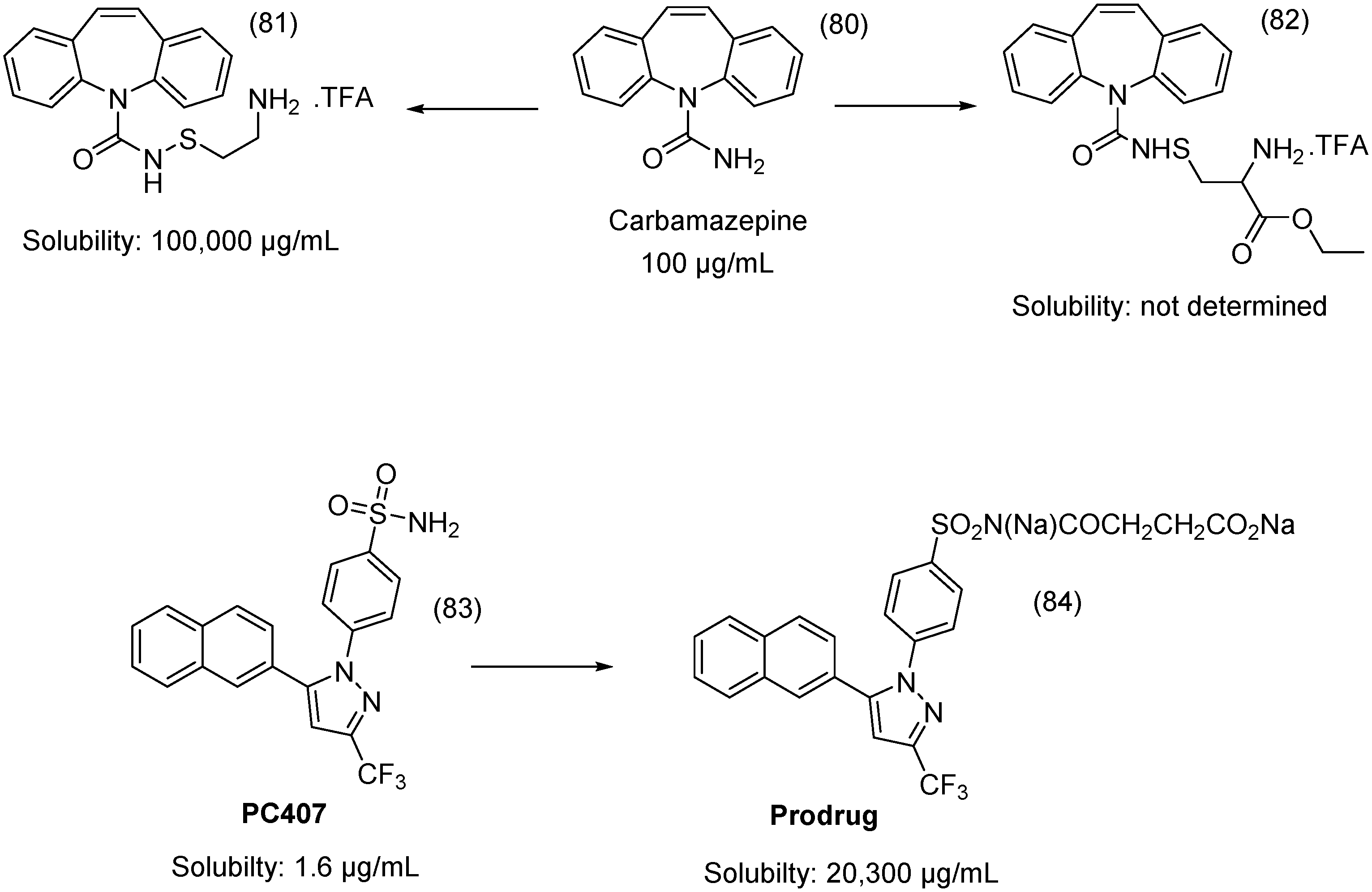
- Hemenway, J.N.; Nti-Addae, K.; Guarino, V.R.; Stella, V.J. Preparation, characterization and in vivo conversion of new water-soluble sulfenamide prodrugs of carbamazepine. Bioorg. Med. Chem. Lett. 2007, 17, 6629–6632. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-F.; Wan, N.; Huan, M.-L.; Jia, Y.-Y.; Yuan, X.-F.; Zhou, S.-Y.; Zhang, B.-L. Enhanced water-soluble derivative of PC407 as a novel potential COX-2 inhibitor injectable formulation. Bioorg. Med. Chem. Lett. 2014, 24, 4794–4797. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.K.; Pan, M.H.; Lin-Shiau, S.Y. Recent studies on the biofunctions and biotransformations of curcumin. Biofactors 2000, 13, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.H.; Huang, T.M.; Lin, J.K. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab. Dispos. 1999, 27, 486–494. [Google Scholar] [PubMed]
- Kawakishi, S. Antioxidative of the P-Diketone Moiety in the Mechanism of Tetrahydrocurcumin. Science 1996, 52, 519–525. [Google Scholar]
- Plyduang, T.; Lomlim, L.; Yuenyongsawad, S.; Wiwattanapatapee, R. Carboxymethylcellulose-tetrahydrocurcumin conjugates for colon-specific delivery of a novel anti-cancer agent, 4-amino tetrahydrocurcumin. Eur. J. Pharm. Biopharm. 2014, 88, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.R.; Vavia, P.R. Preparation and Evaluation of Taste Masked Famotidine Formulation Using Drug/β-cyclodextrin/Polymer Ternary Complexation Approach. AAPS PharmSciTech 2008, 9, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraj, S.; Omshanthi, B.; Anitha, S.; Sampath Kumar, K.P. Synthesis and Characterization of Novel Sulphoxide prodrug of Famotidine. Indian J. Pharm. Educ. Res. 2014, 48, 35–44. [Google Scholar] [CrossRef]
- Rocamora, R. A review of the efficacy and safety of eslicarbazepine acetate in the management of partial-onset seizures. Ther. Adv. Neurol. Disord. 2015, 8, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Ben-Menachem, E. Eslicarbazepine Acetate: A Well-Kept Secret? Epilepsy Curr. 2010, 10, 7–8. [Google Scholar] [PubMed]
- Mueller, S.W.; Moore, G.D.; Maclaren, R. Fospropofol Disodium for Procedural Sedation: Emerging Evidence of its Value? Clin. Med. Insights Ther. 2010, 2, 513–522. [Google Scholar]
- Boules, R.; Szkiladz, A.; Nogid, A. Fospropofol disodium (lusedra) injection for anesthesia-care sedation: a clinical review. Pharm. Ther. 2012, 37, 395–422. [Google Scholar]
- Ishikawa, T.; Matsunaga, N.; Tawada, H.; Kuroda, N.; Nakayama, Y.; Ishibashi, Y.; Tomimoto, M.; Ikeda, Y.; Tagawa, Y.; Iizawa, Y.; et al. TAK-599, a novel N-phosphono type prodrug of anti-MRSA cephalosporin T-91825: Synthesis, physicochemical and pharmacological properties. Bioorg. Med. Chem. 2003, 11, 2427–2437. [Google Scholar] [CrossRef]
- Flanagan, S.; Bartizal, K.; Minassian, S.L.; Fang, E.; Prokocimer, P. In vitro, in Vivo, and clinical studies of tedizolid to assess the potential for peripheral or central monoamine oxidase interactions. Antimicrob. Agents Chemother. 2013, 57, 3060–3066. [Google Scholar] [PubMed]
- Rybak, J.M.; Roberts, K. Tedizolid Phosphate: A Next-Generation Oxazolidinone. Infect. Dis. Ther. 2015, 4, 1–14. [Google Scholar]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jornada, D.H.; Dos Santos Fernandes, G.F.; Chiba, D.E.; De Melo, T.R.F.; Dos Santos, J.L.; Chung, M.C. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2016, 21, 42. https://doi.org/10.3390/molecules21010042
Jornada DH, Dos Santos Fernandes GF, Chiba DE, De Melo TRF, Dos Santos JL, Chung MC. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules. 2016; 21(1):42. https://doi.org/10.3390/molecules21010042
Chicago/Turabian StyleJornada, Daniela Hartmann, Guilherme Felipe Dos Santos Fernandes, Diego Eidy Chiba, Thais Regina Ferreira De Melo, Jean Leandro Dos Santos, and Man Chin Chung. 2016. "The Prodrug Approach: A Successful Tool for Improving Drug Solubility" Molecules 21, no. 1: 42. https://doi.org/10.3390/molecules21010042
APA StyleJornada, D. H., Dos Santos Fernandes, G. F., Chiba, D. E., De Melo, T. R. F., Dos Santos, J. L., & Chung, M. C. (2016). The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules, 21(1), 42. https://doi.org/10.3390/molecules21010042