
Methods to Increase the Metabolic Stability of 18F-Radiotracers



Abstract
:1. Introduction

| Nuclide | t½ (min) | Production Route | Average Range in H2O (mm) | Eav. (β+) (keV) |
|---|---|---|---|---|
| 11C | 20.4 | 14N(p,α)11C | 1 | 385 |
| 13N | 10 | 16O(p,α)13N | 1.5 | 491 |
| 15O | 2 | 15N(d,n)15O | 2.7 | 735 |
| 18F | 109.8 | 20Ne(d,α)18F 18O(p,n)18F | 0.3 | 242 |
| 68Ga | 67.6 | 68Ge-68Ga generator | 3.7 | 740 |
| 124I | 250.6 | 124Te(p,n)124I | 3 | 188 |
1.1. Nature of the C-F Bond
| Element X | Van der Waals Radius (pm) | Electronegativity (Pauling Scale) | Bond Length of C-X (pm) |
|---|---|---|---|
| H | 120 | 2.1 | 109 |
| C | 170 | 2.5 | 154 |
| O | 152 | 3.5 | 143 |
| F | 147 | 4.0 | 135 |
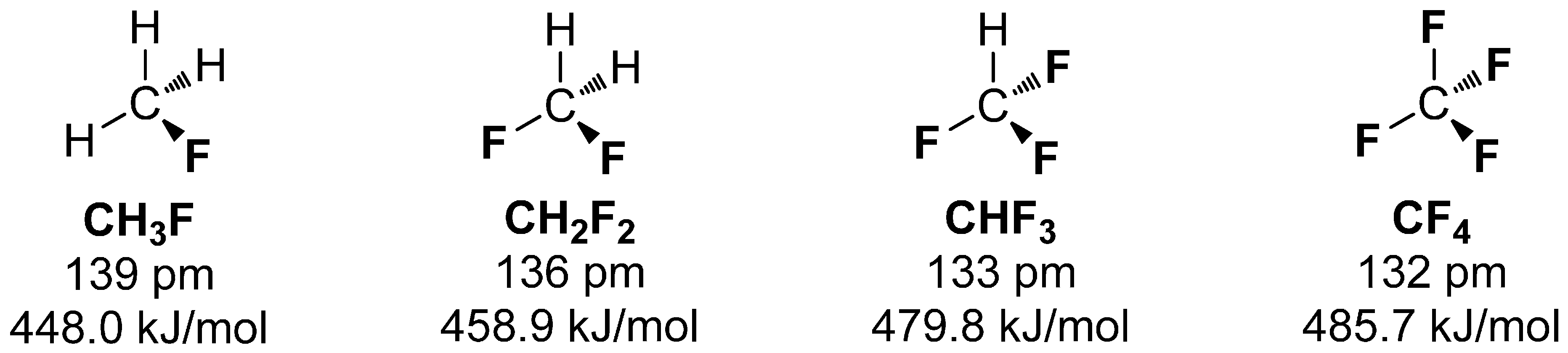
1.2. Possibilities to Introduce Fluorine-18—Short Overview
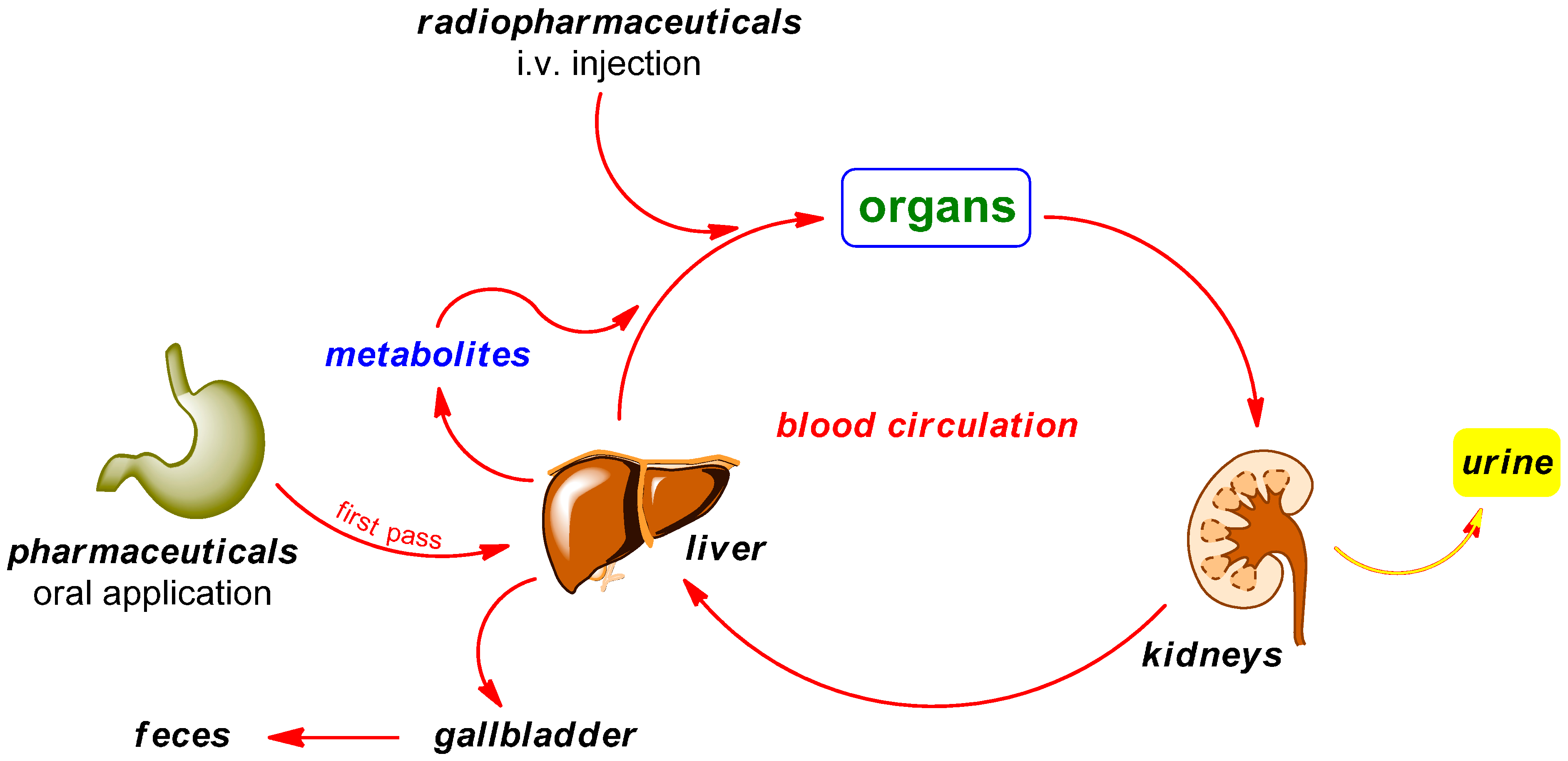
2. Radiodefluorination
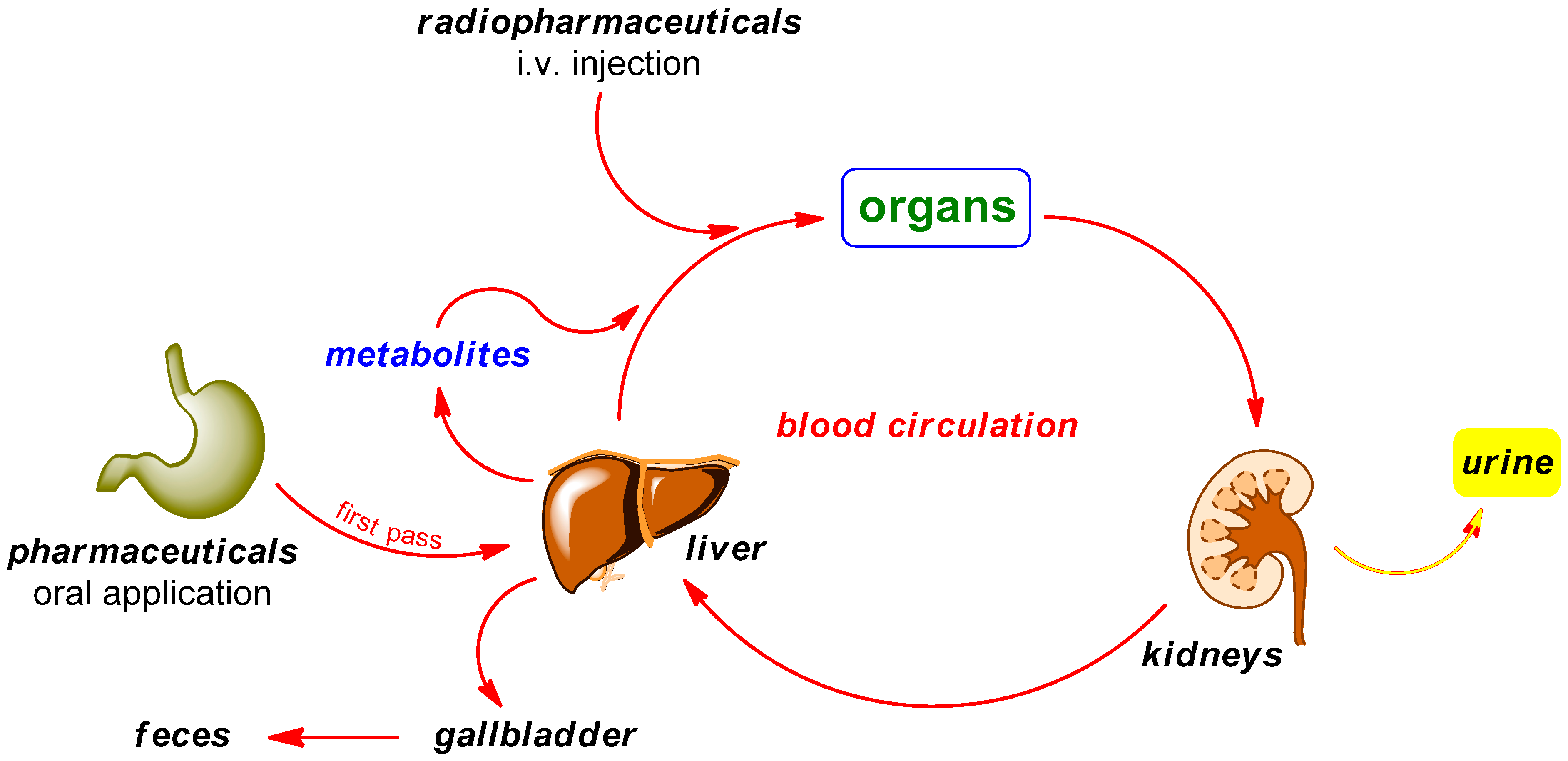
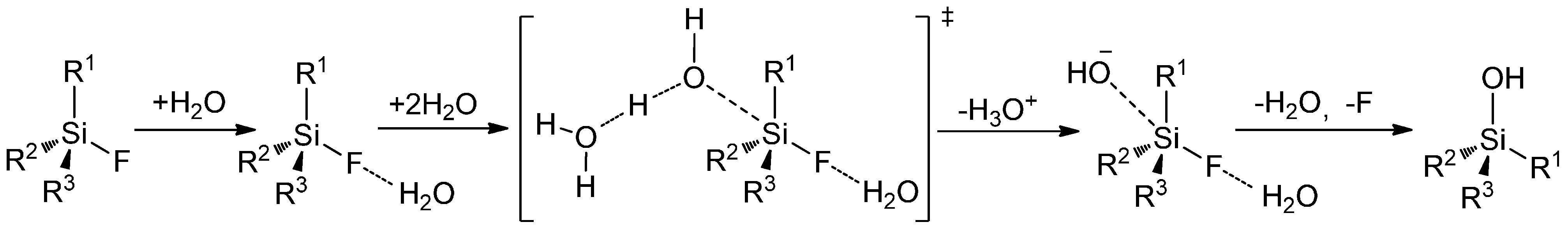
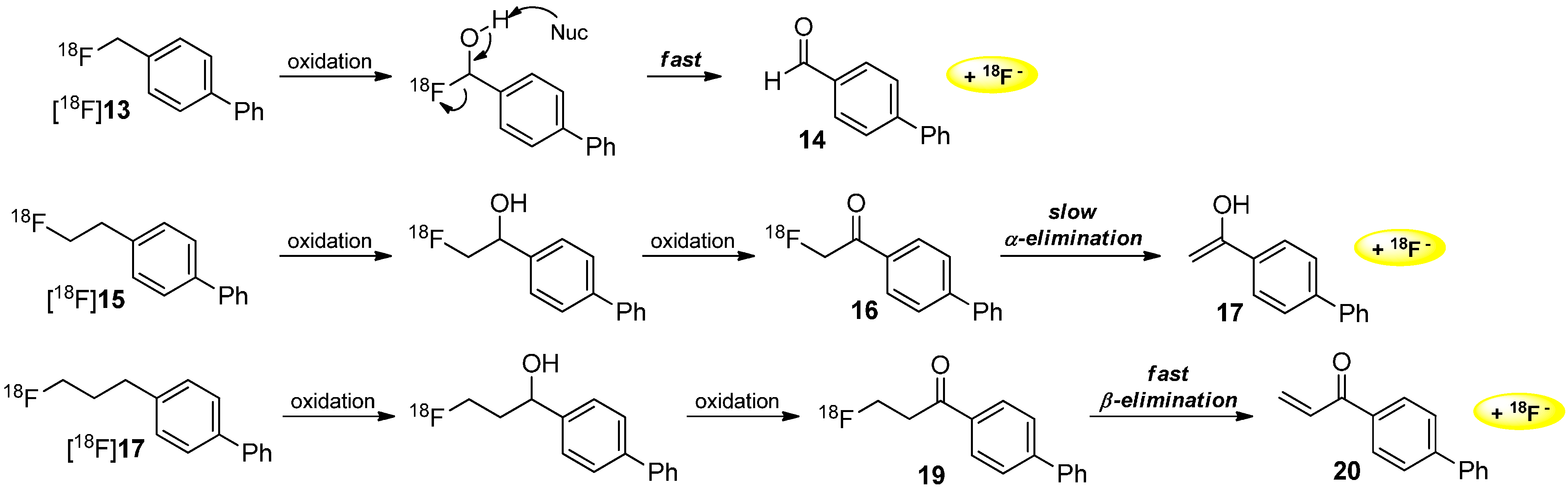
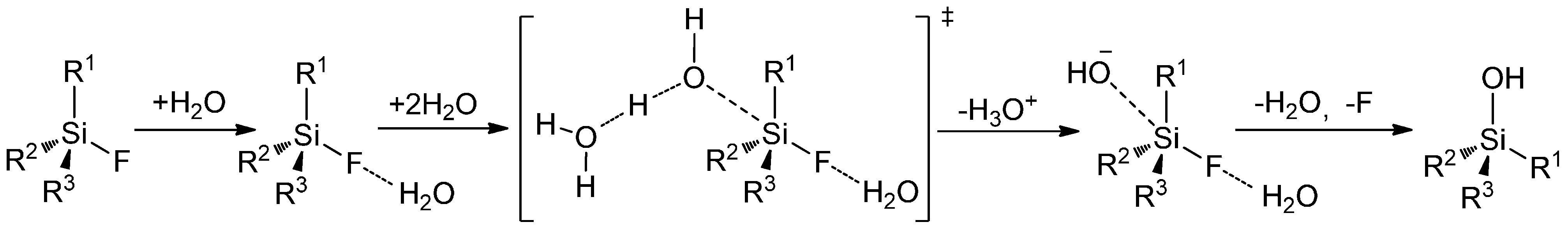
Mechanisms of Radiodefluorination
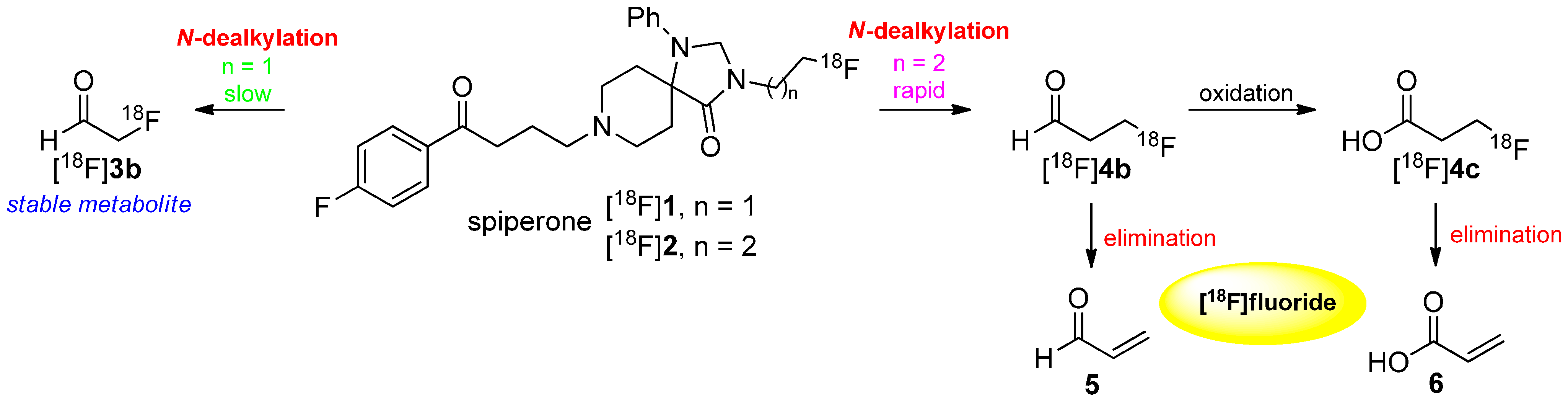
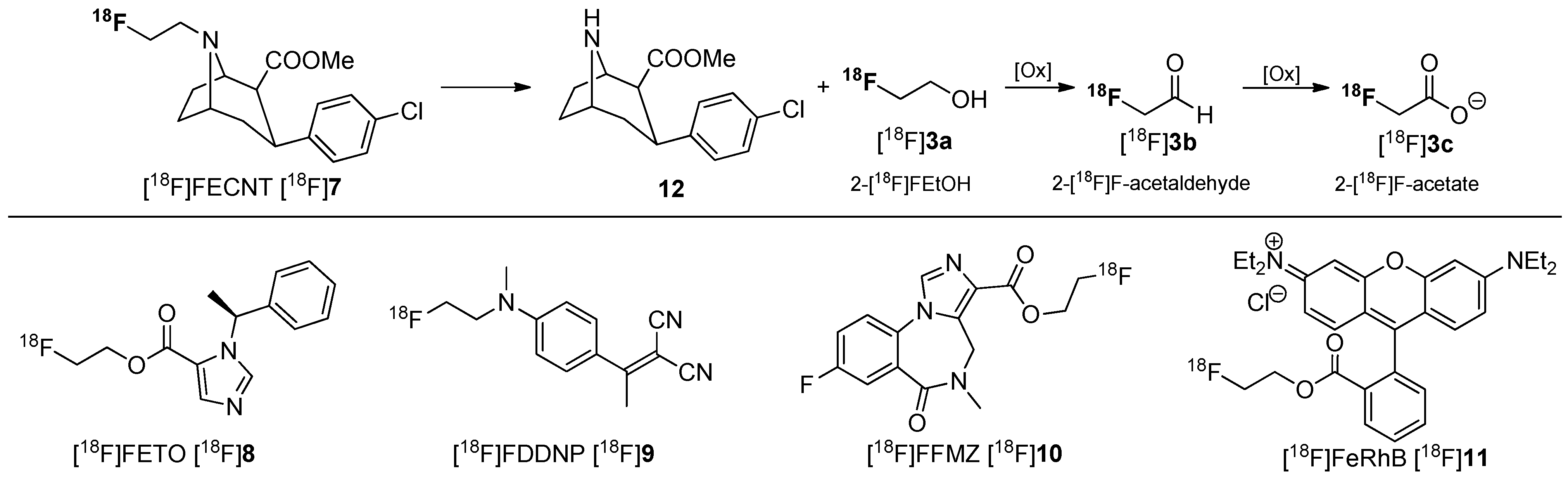
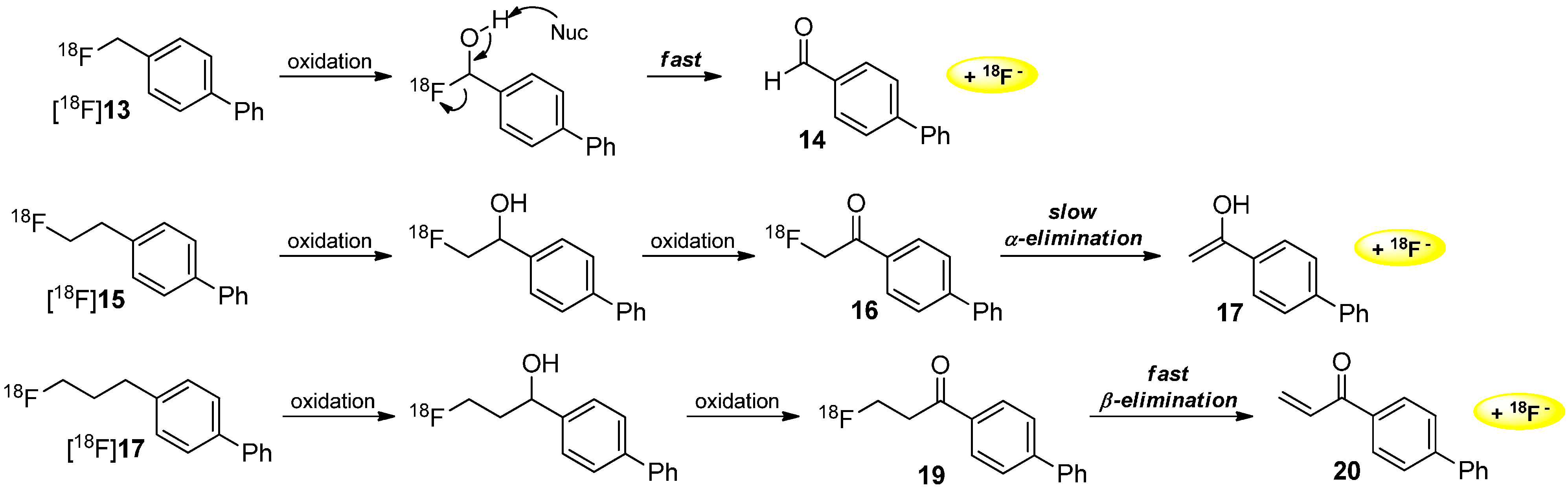
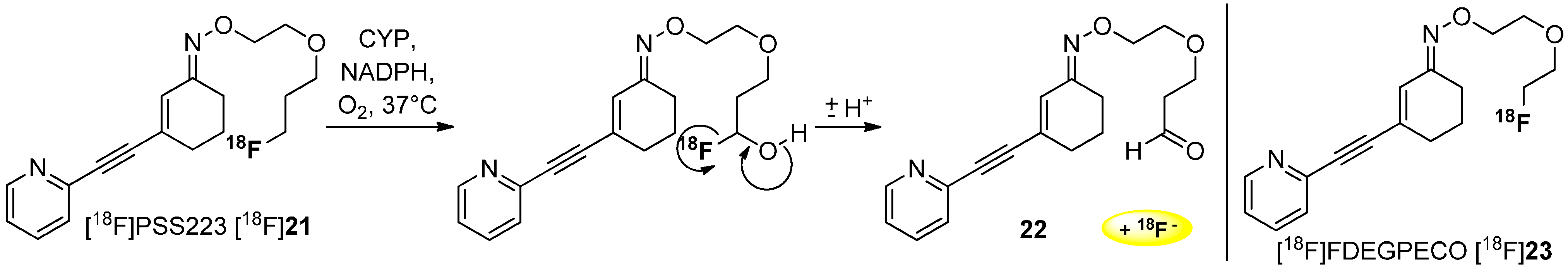
3. Methods to Avoid Radiodefluorination
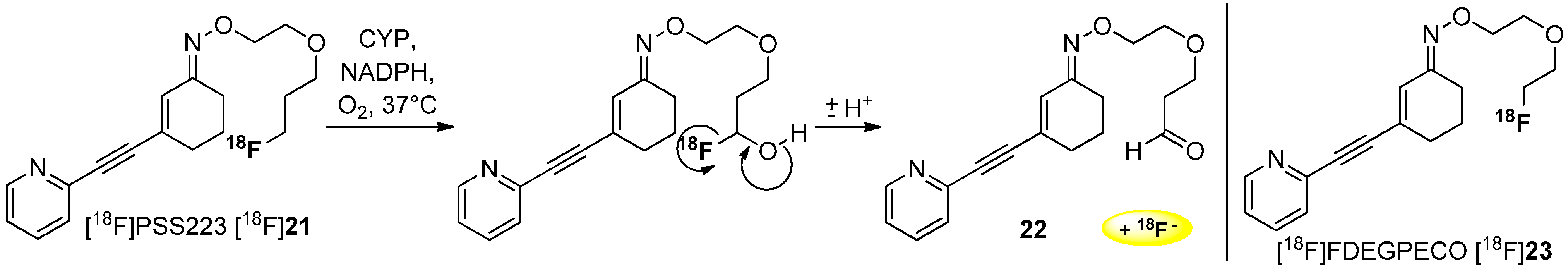
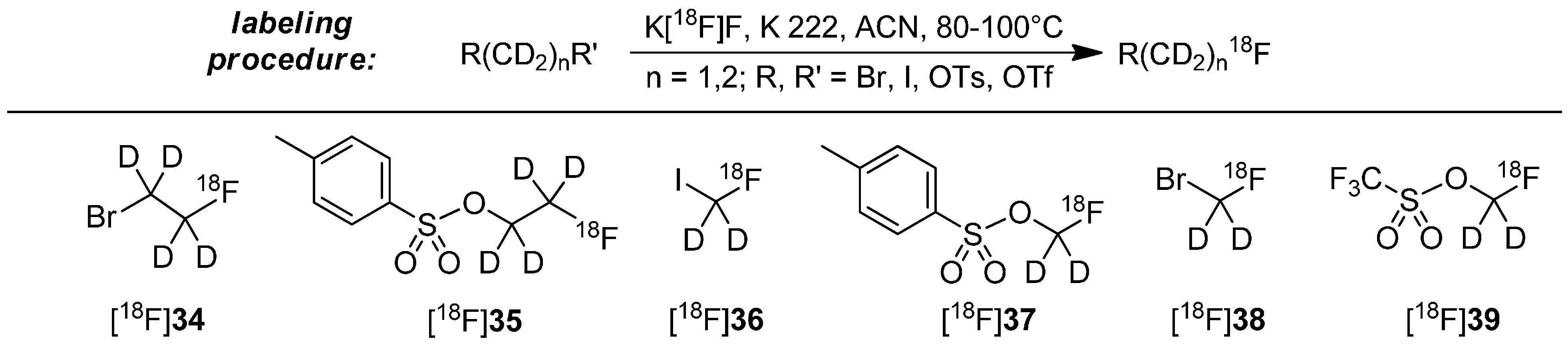
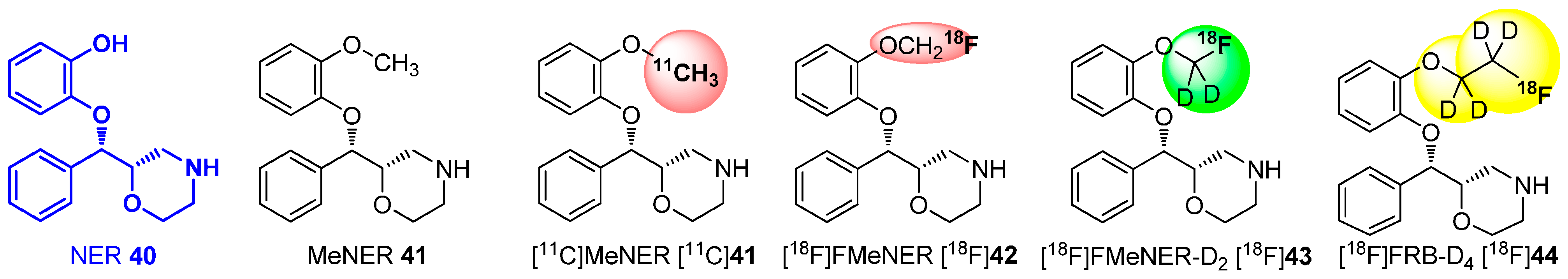
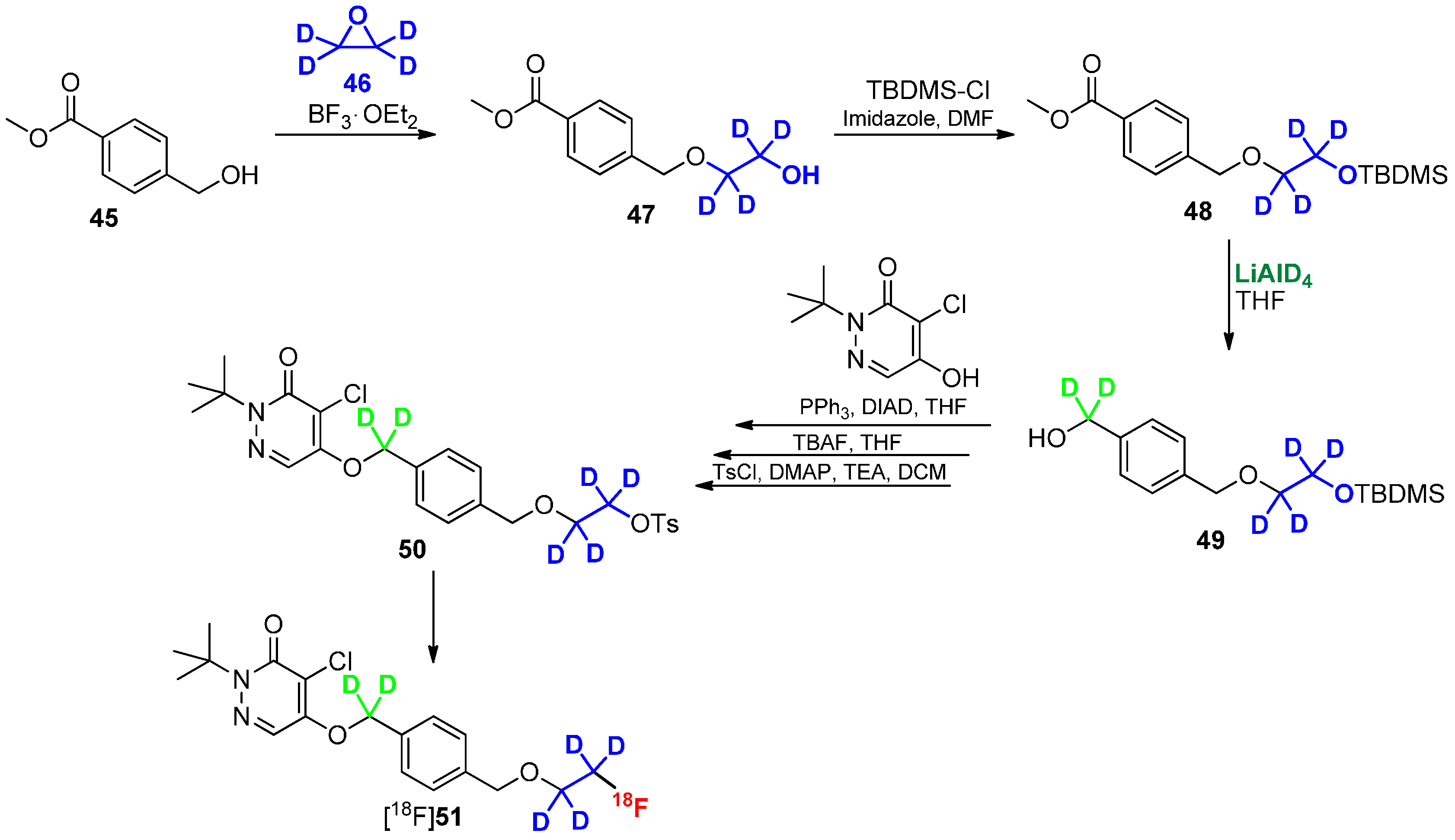
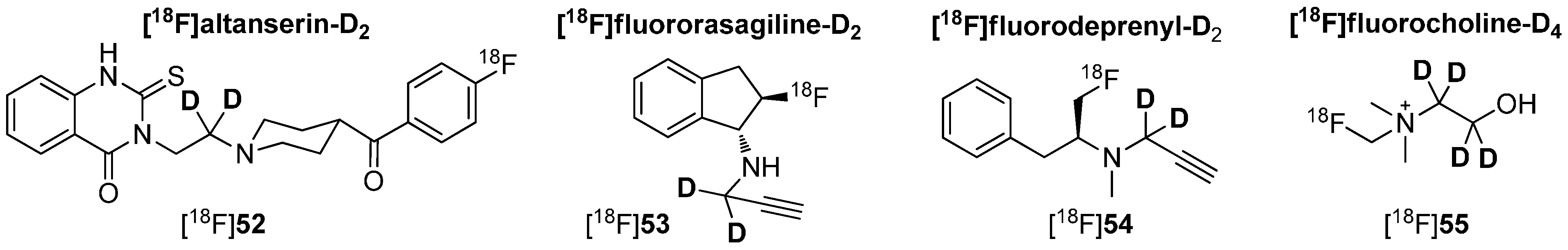
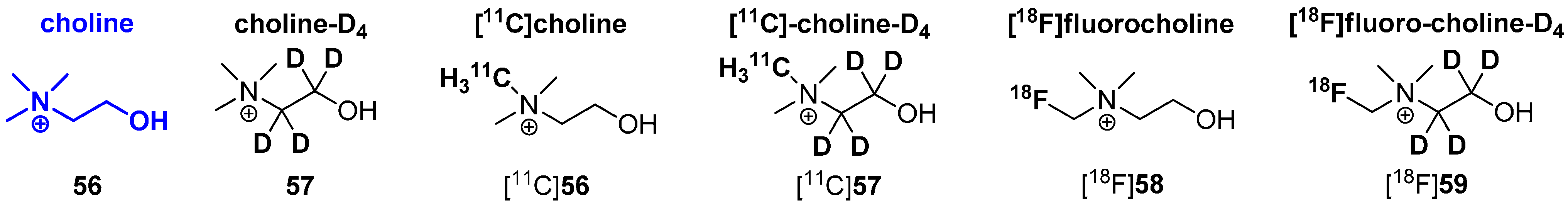
Deuteration in Direct Neighborhood of Fluorine-18
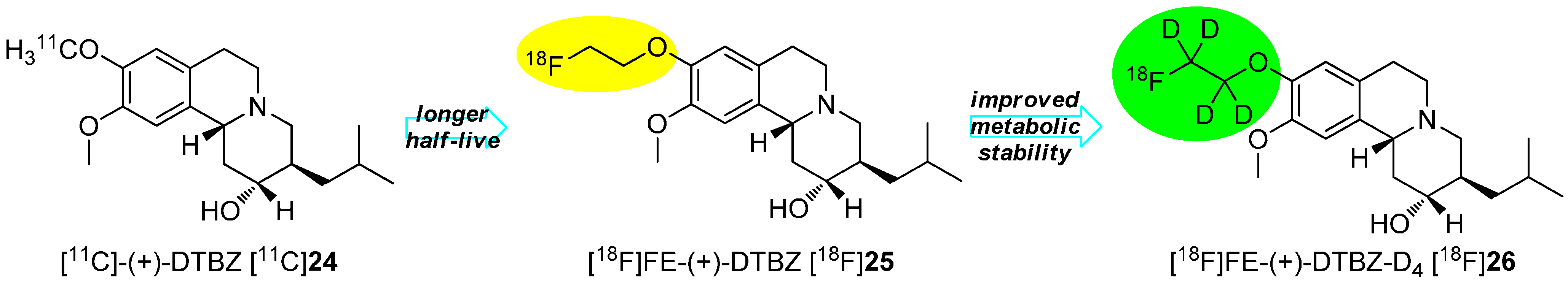
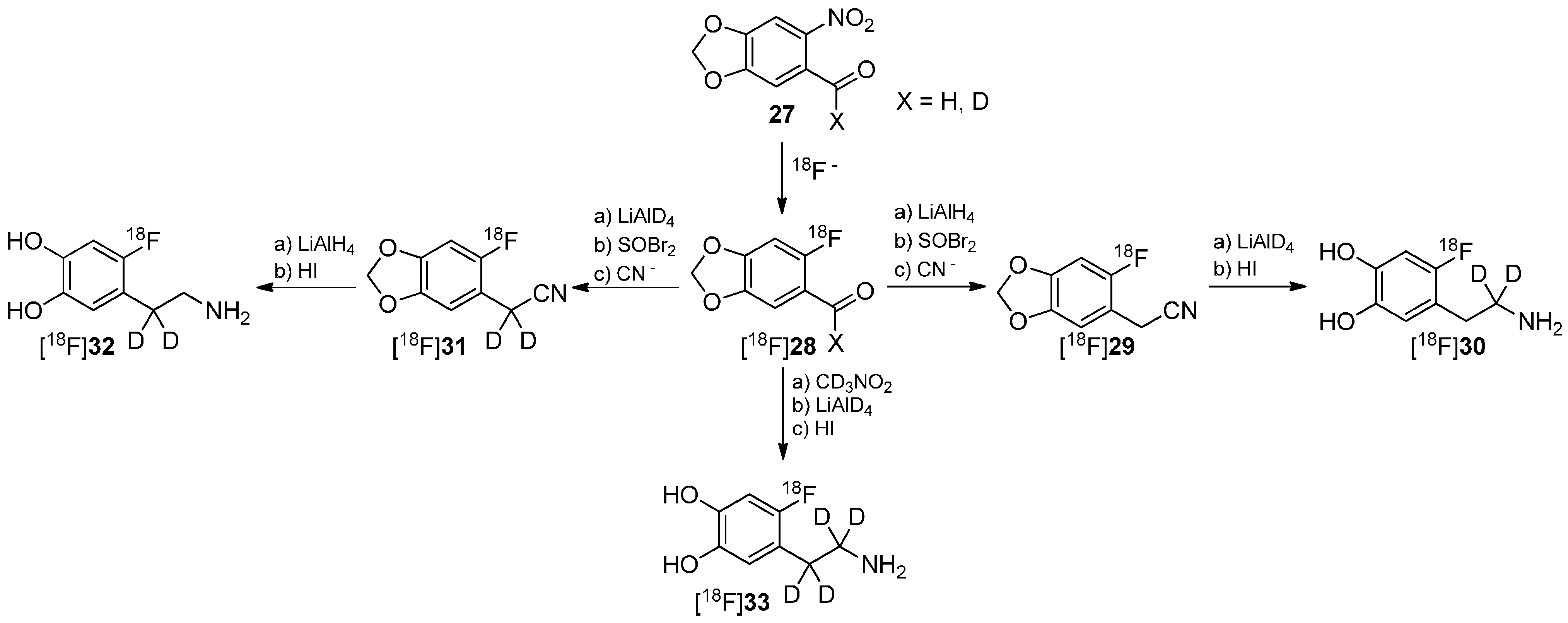
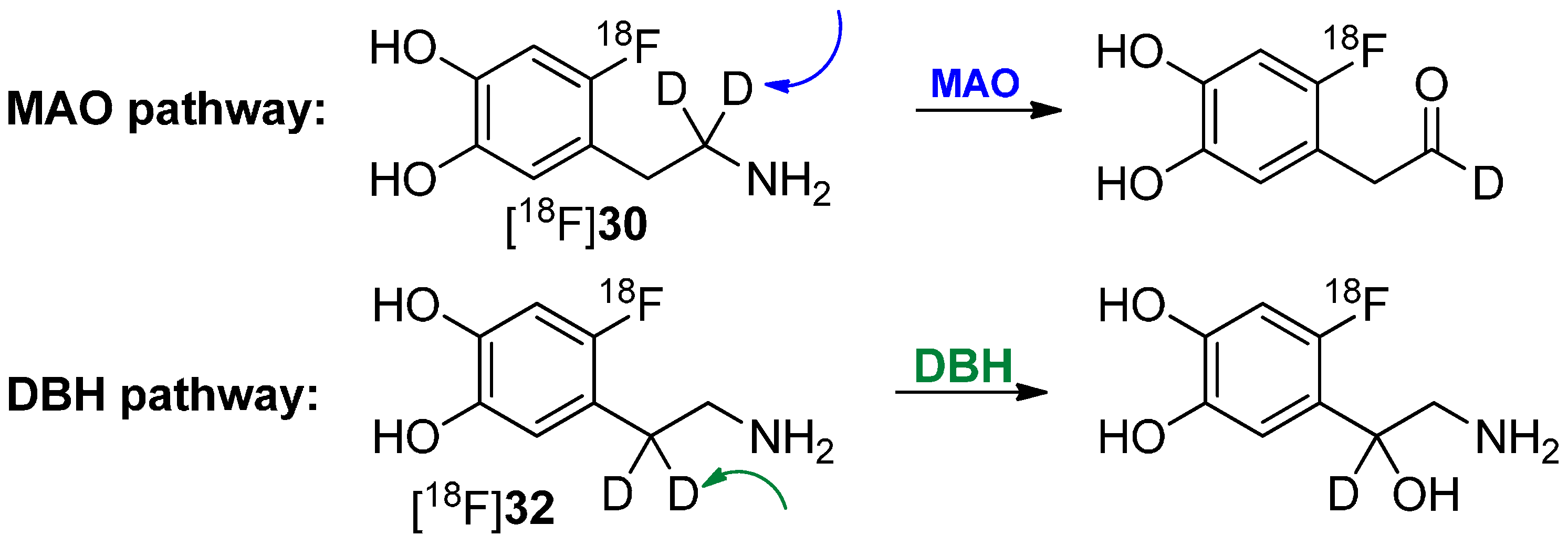
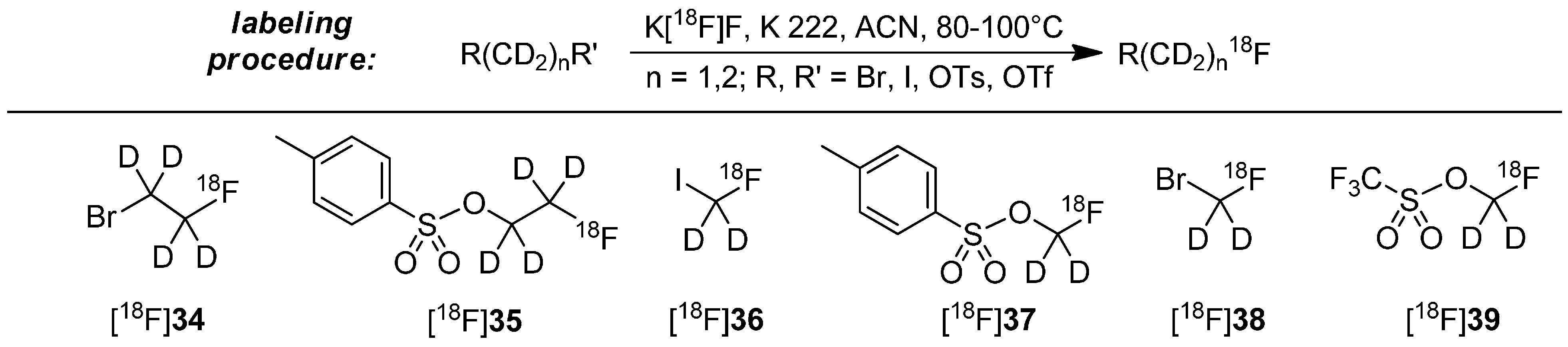
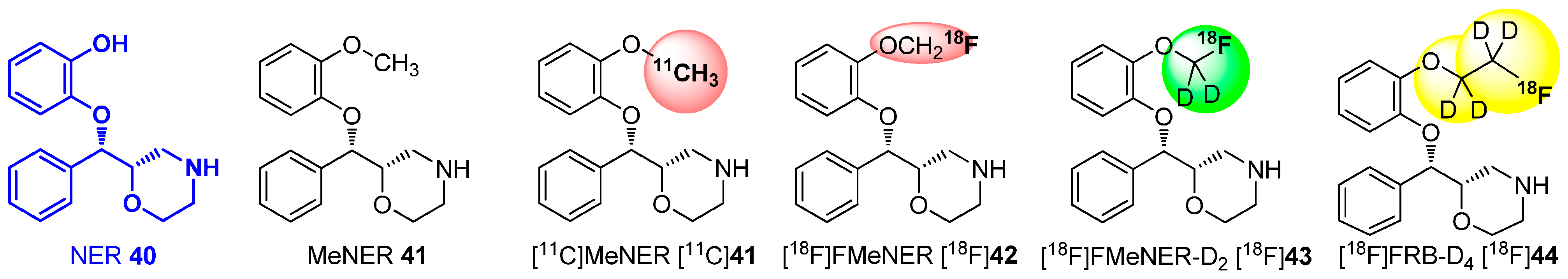
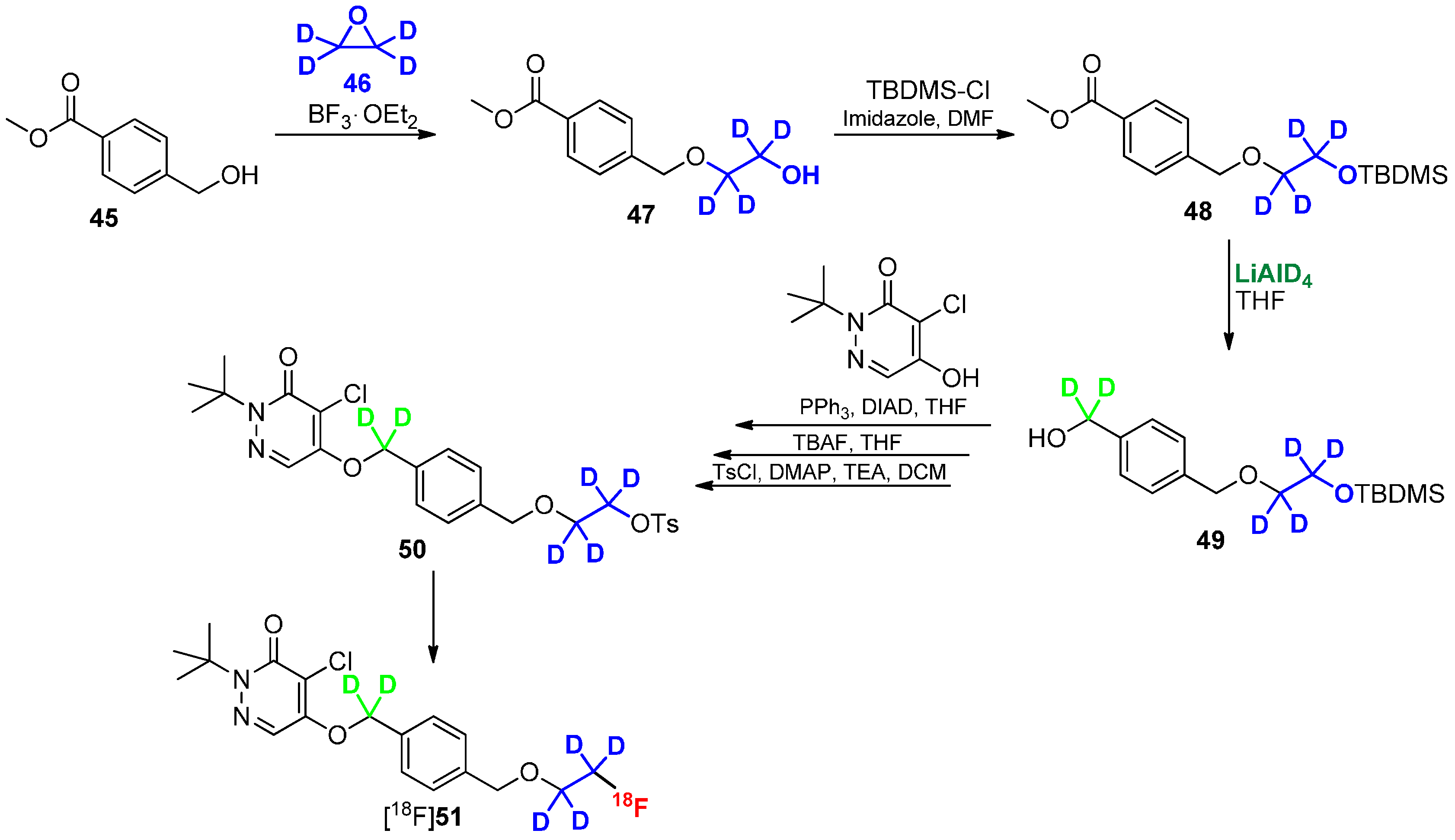
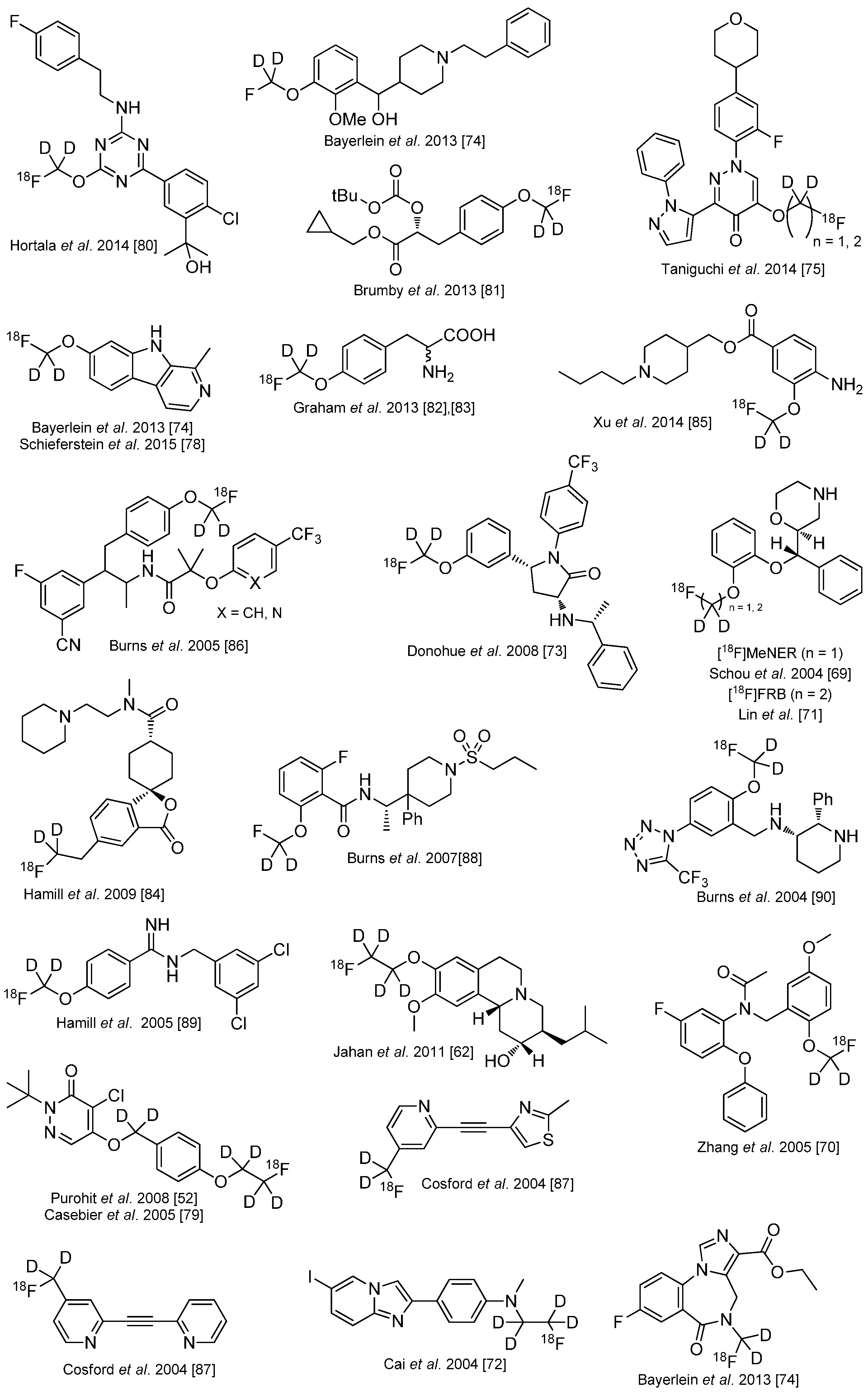
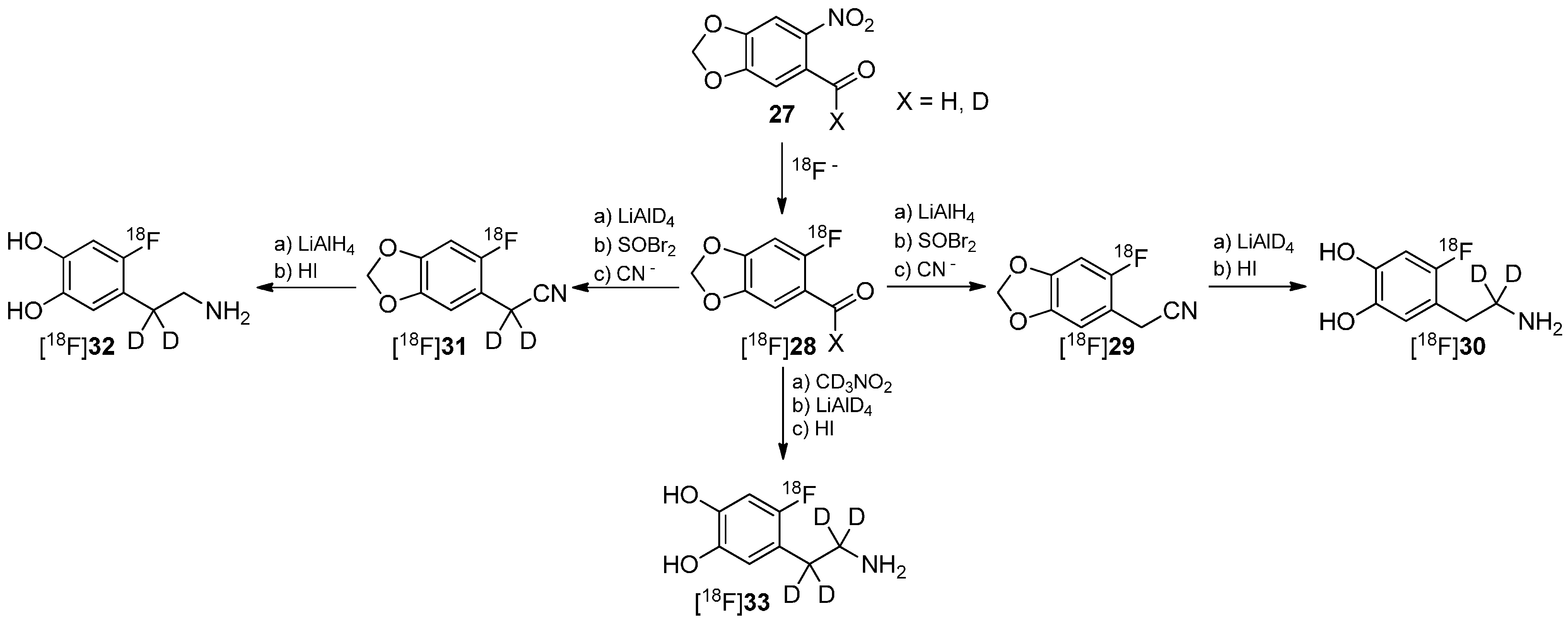
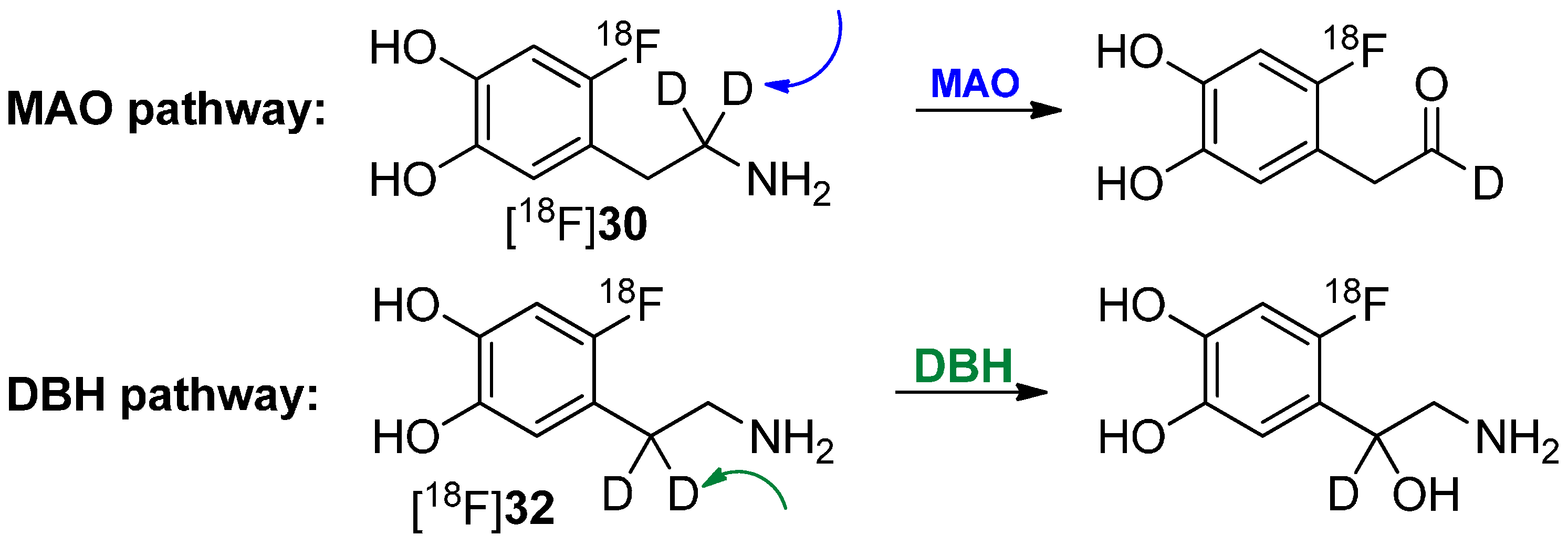
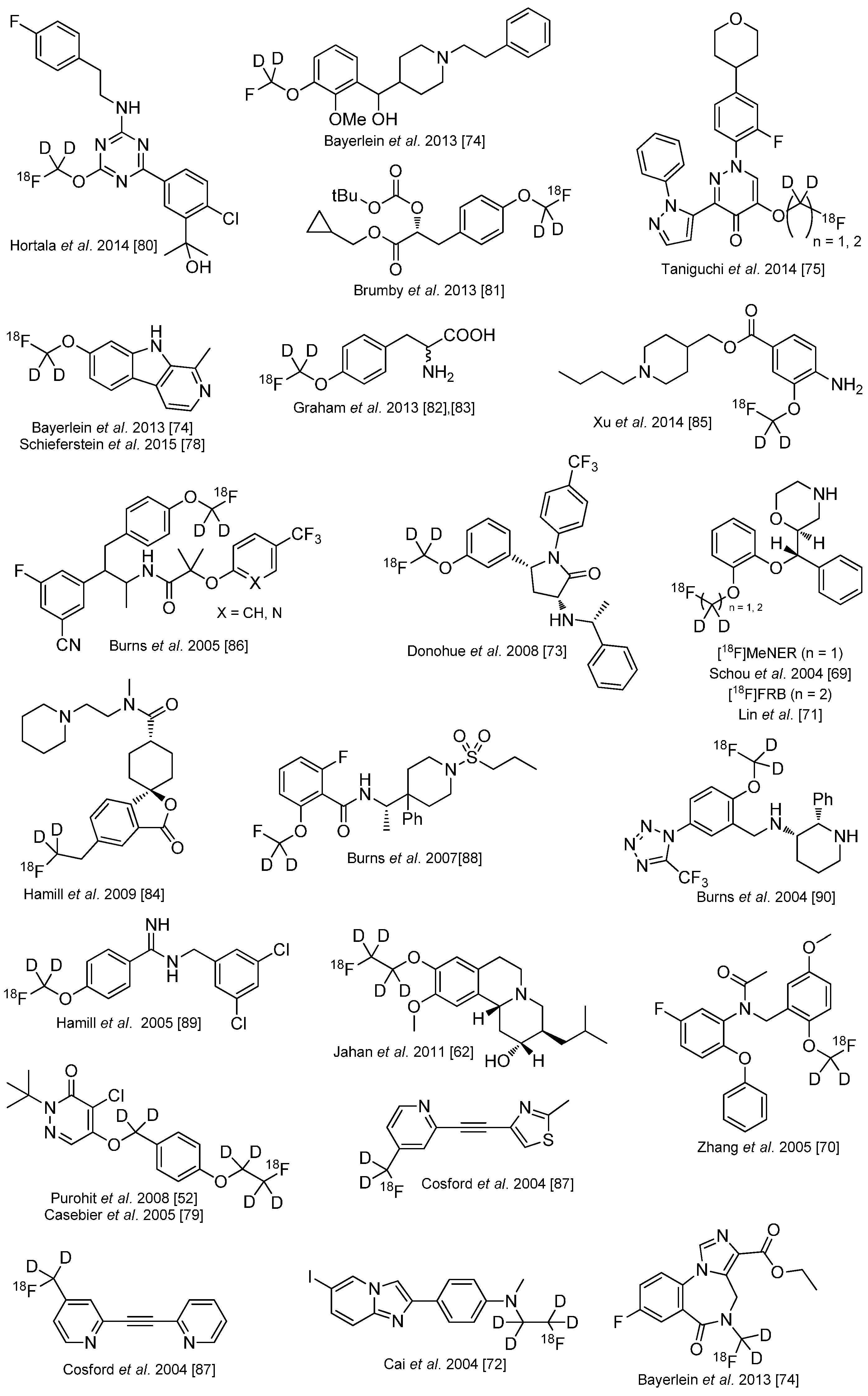
4. Deuteration on other Parts of the Molecule to Avoid Degradation

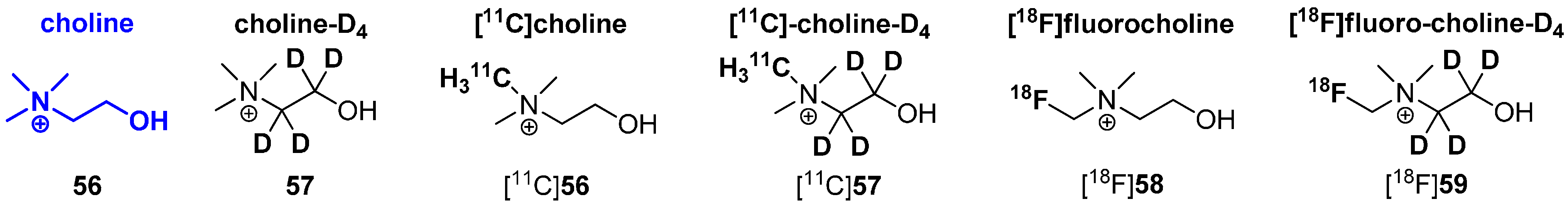
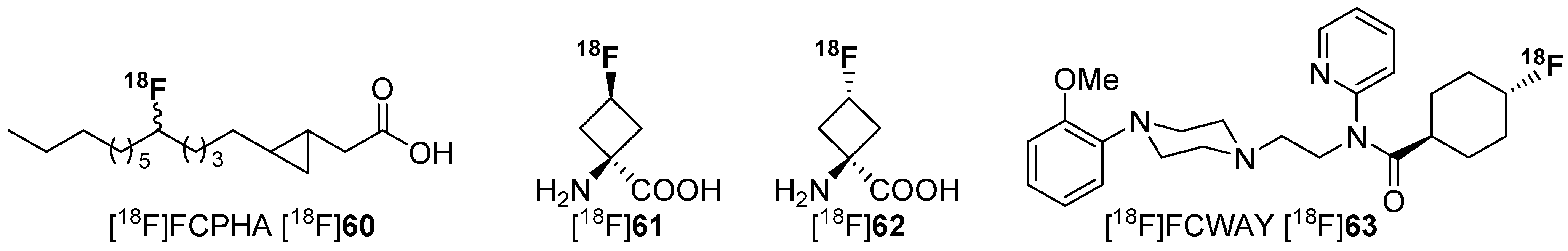
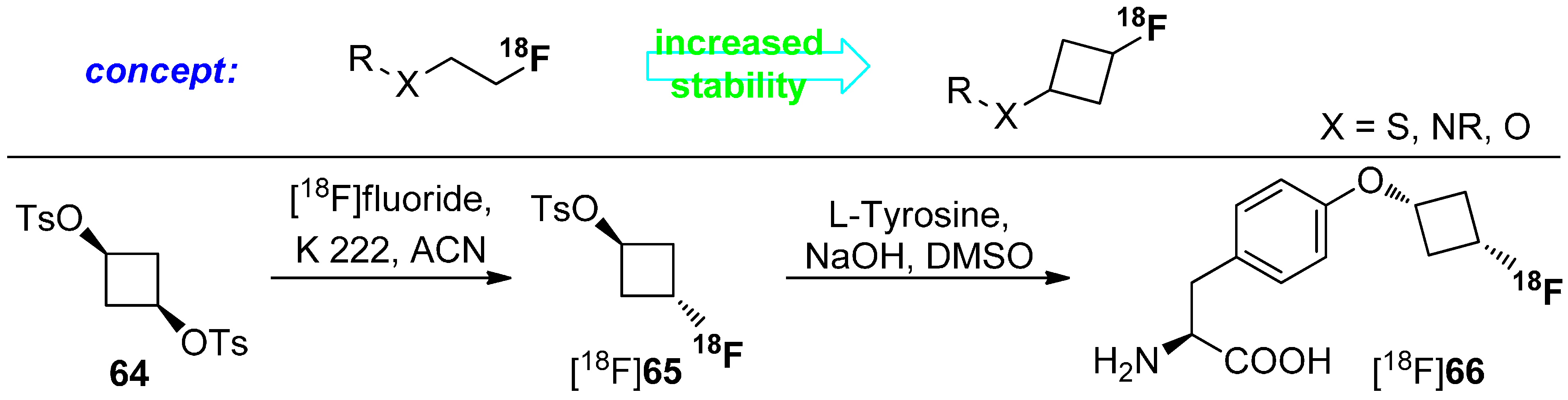
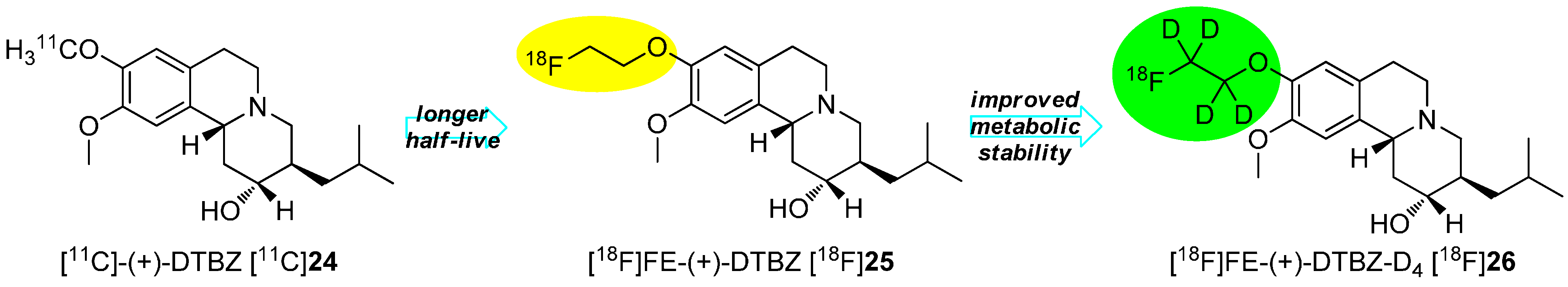
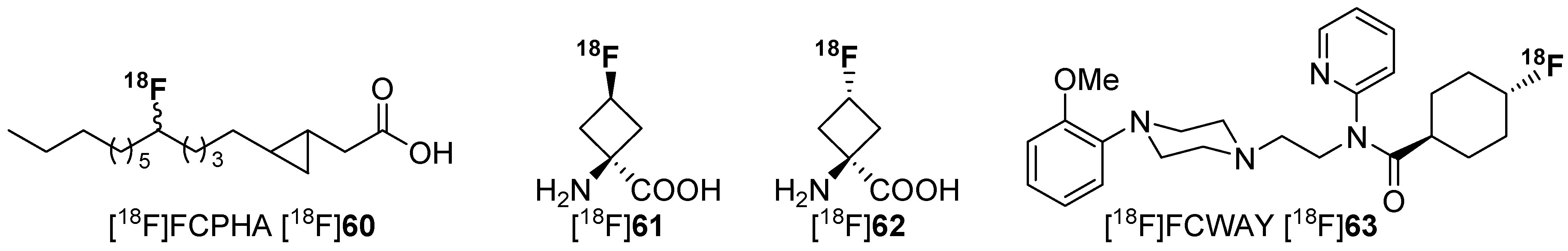
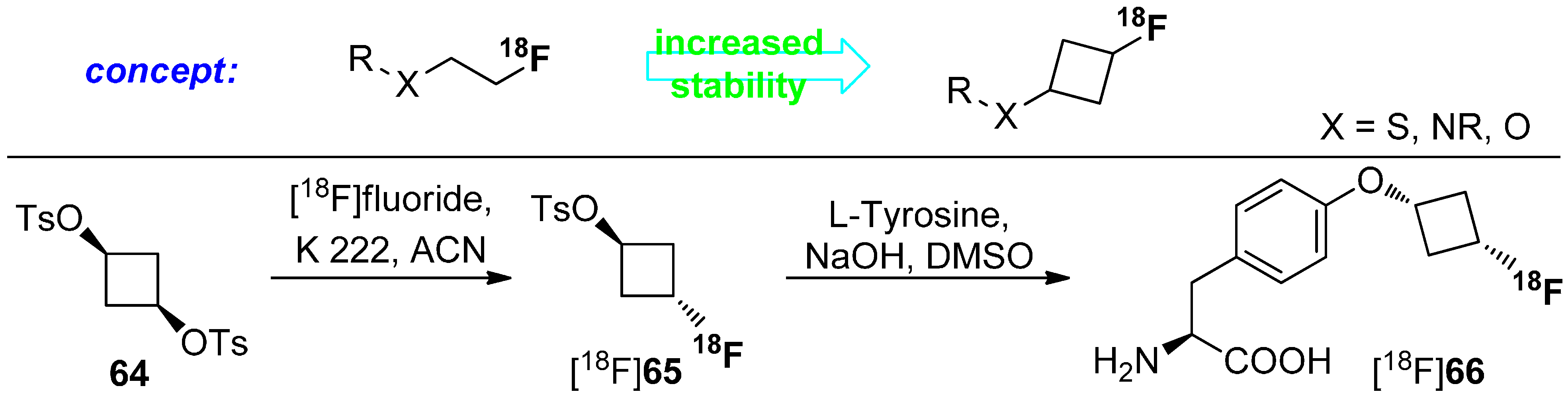
4.1. Cycloalkyl Derivatives and Fluorine Connected to a Secondary Carbon Atom
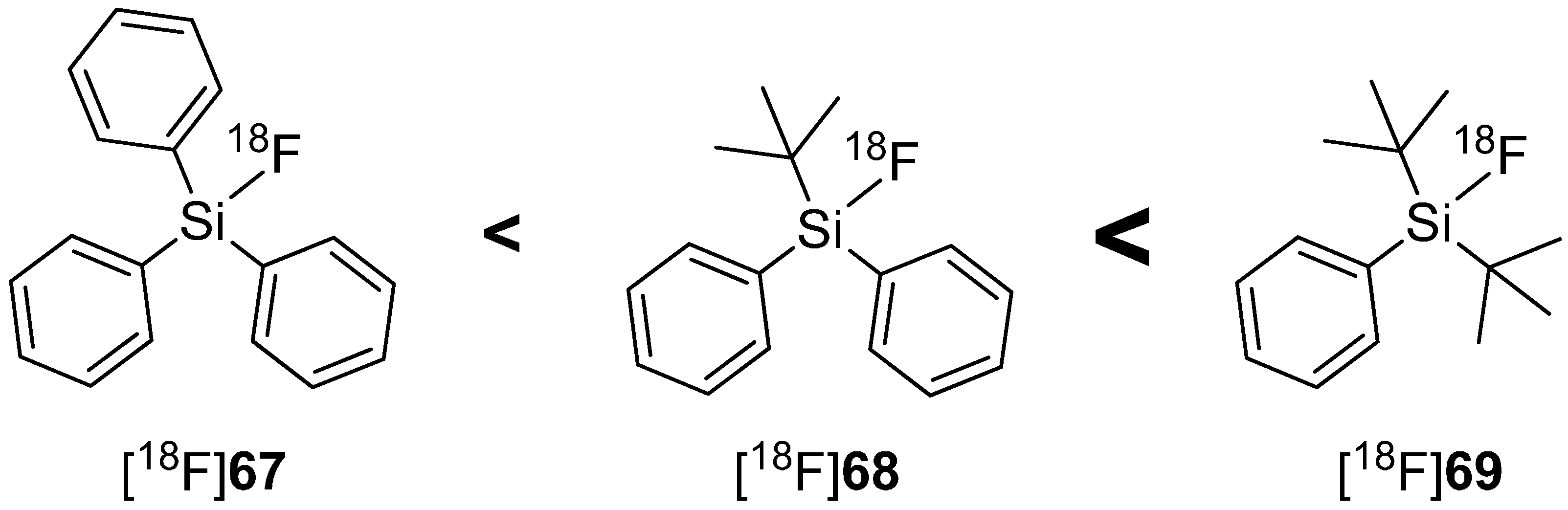
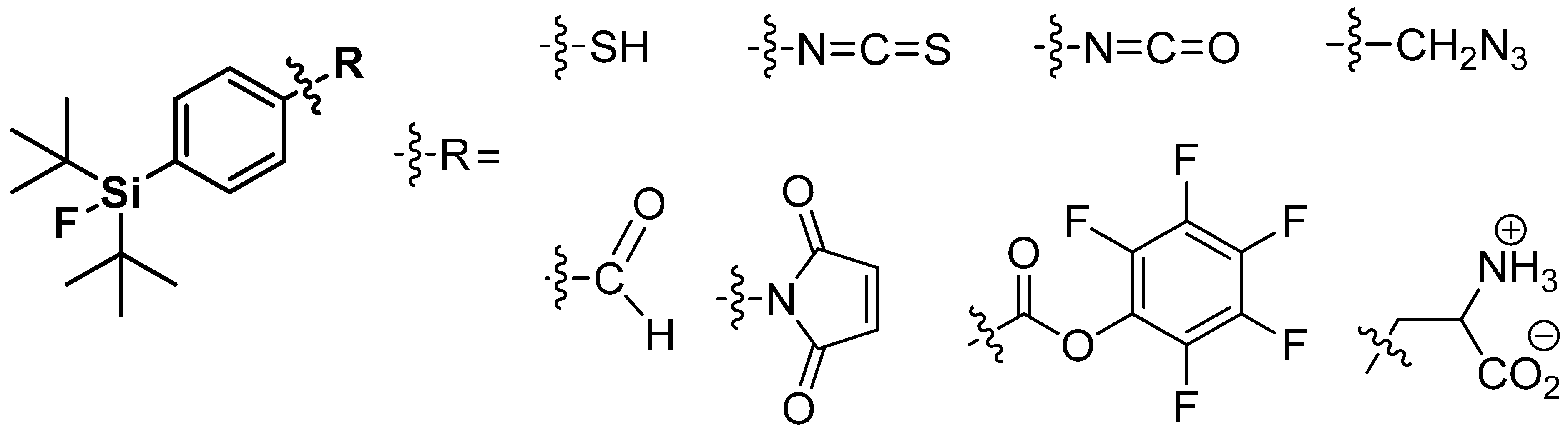
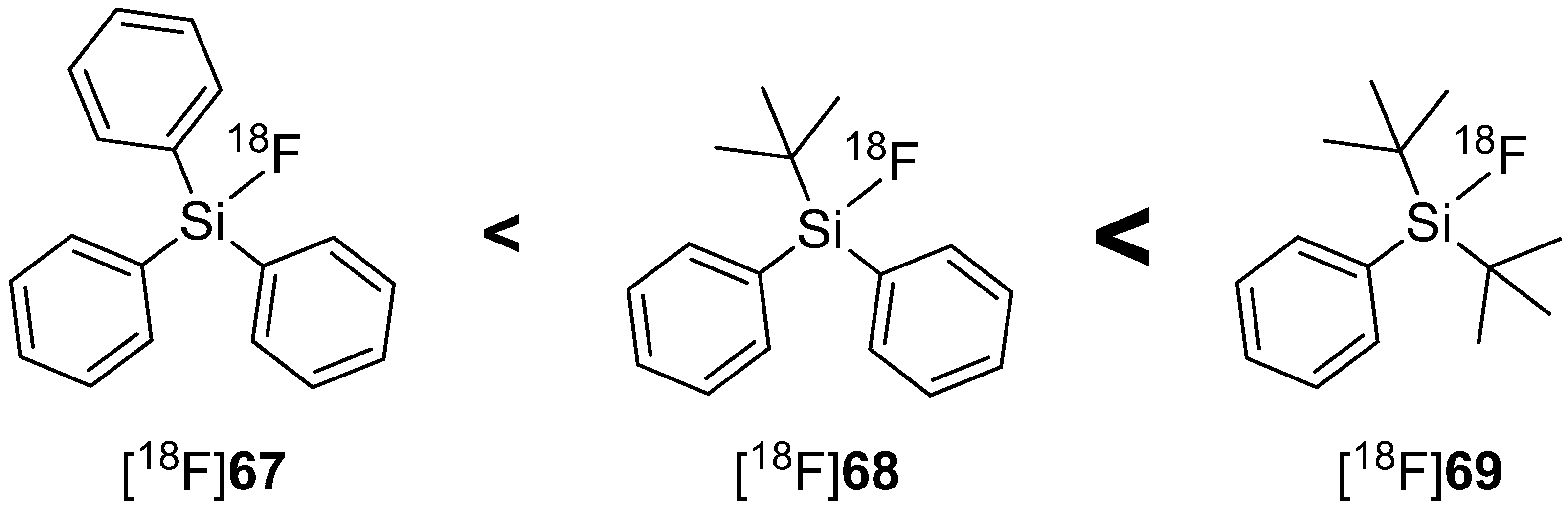
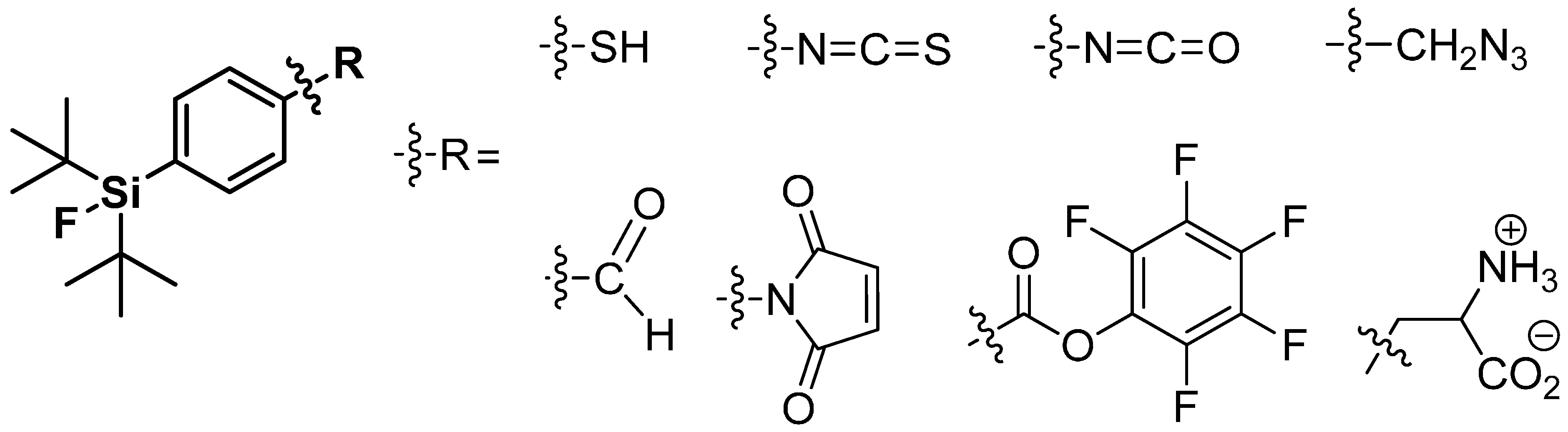
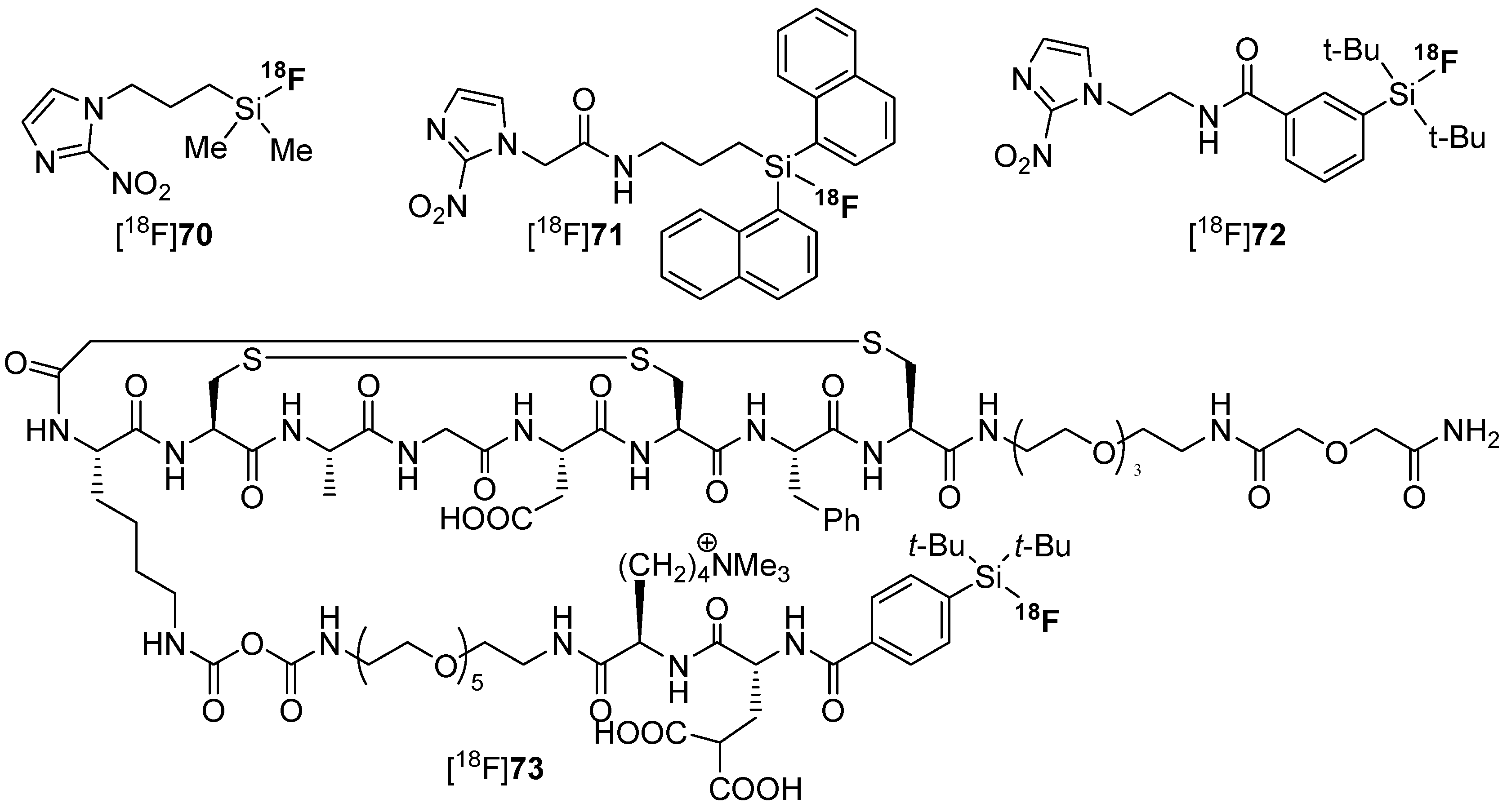
4.2. SiFA-Techniology

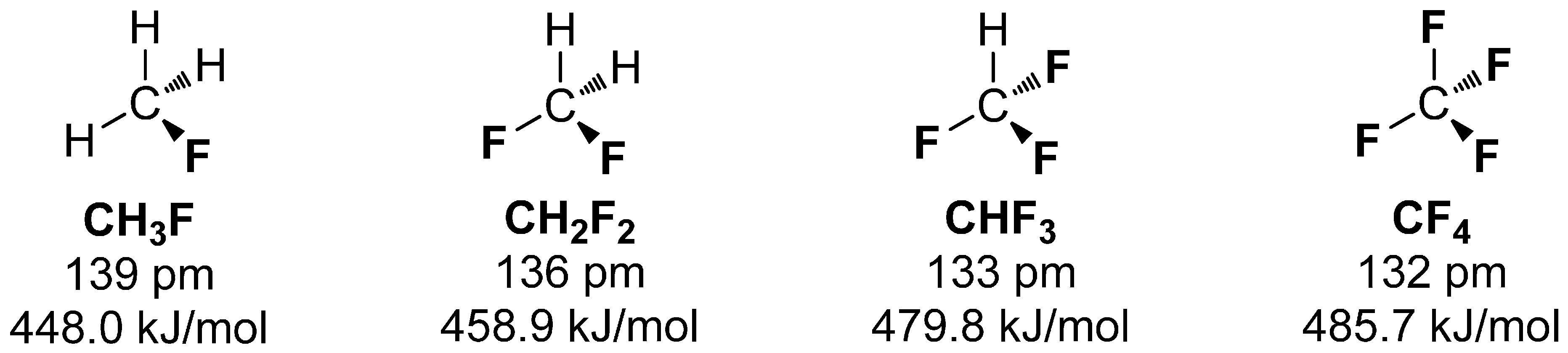{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
5. Miscellaneous
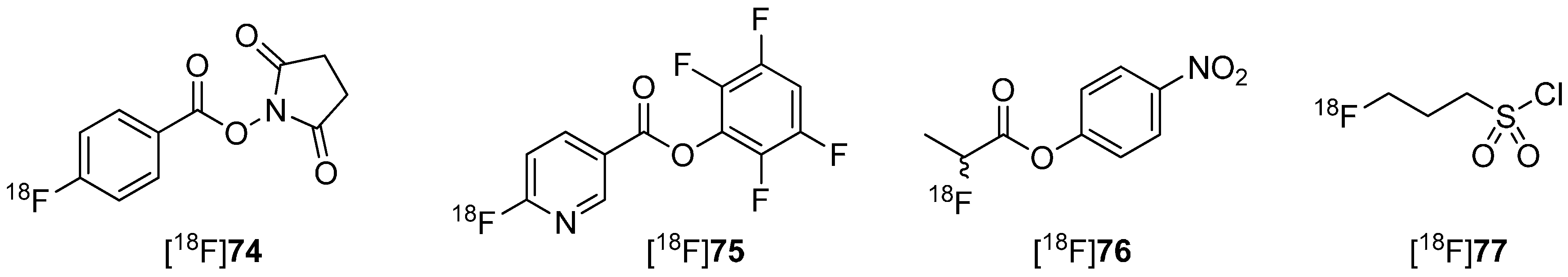
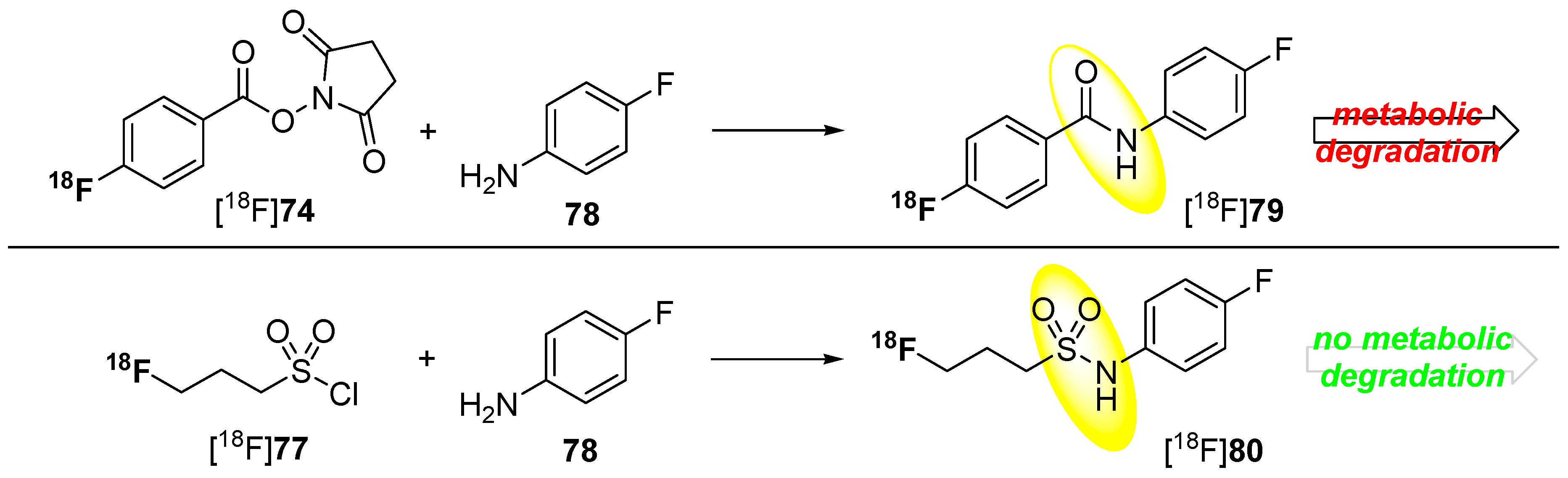
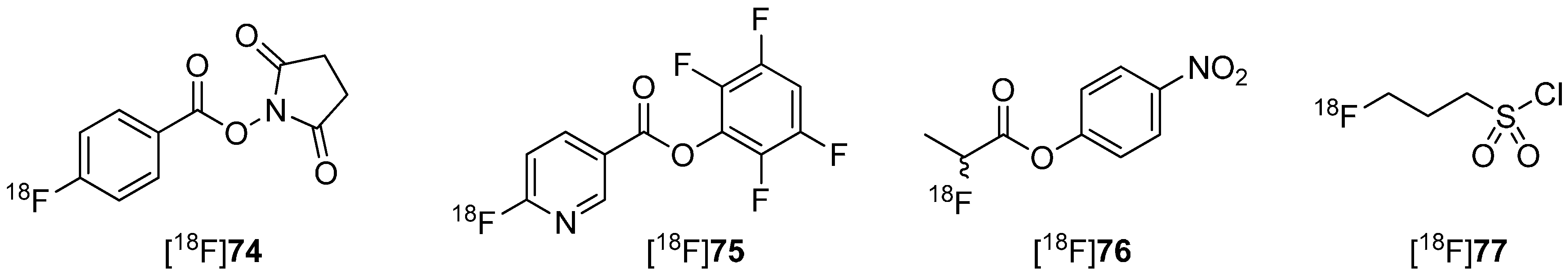
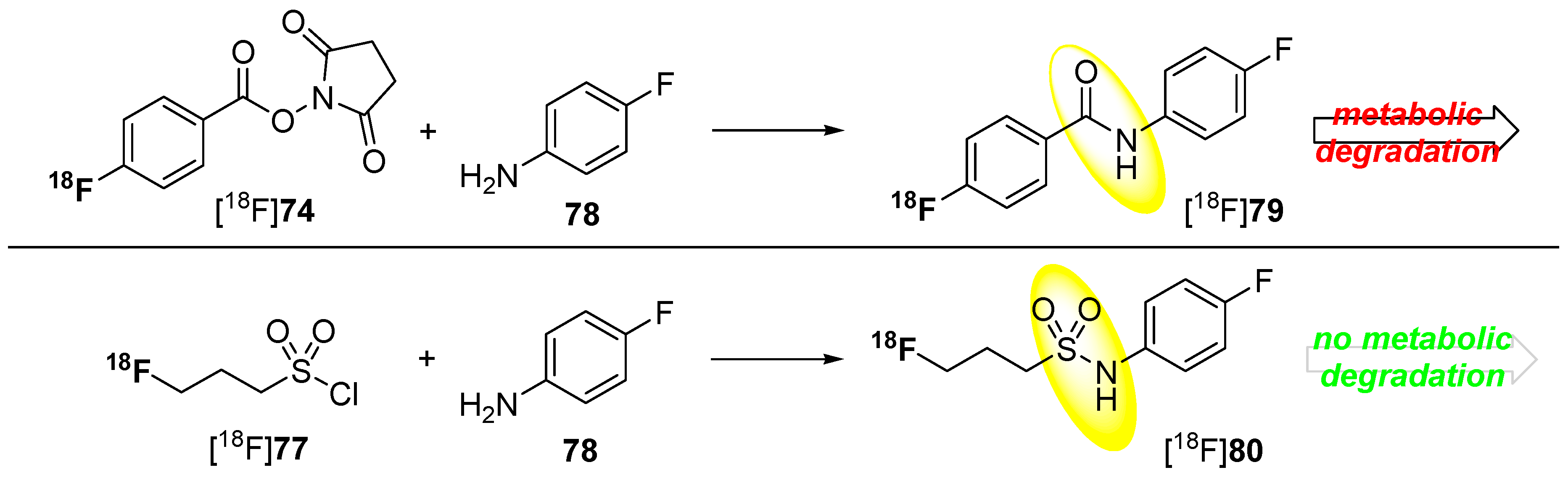
5.1. Fluorosulfonamides
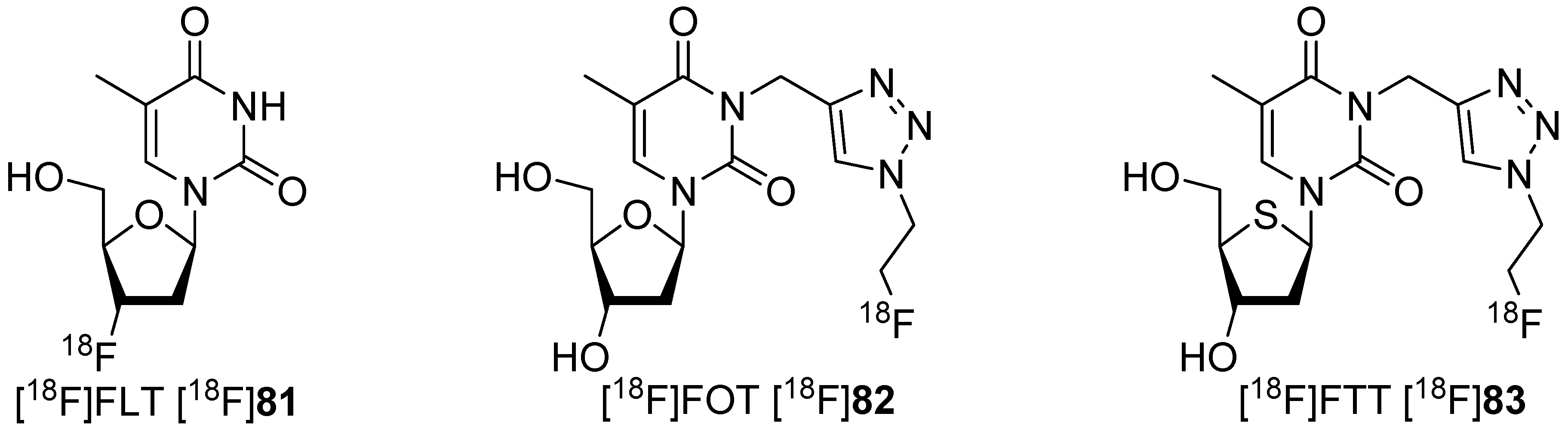
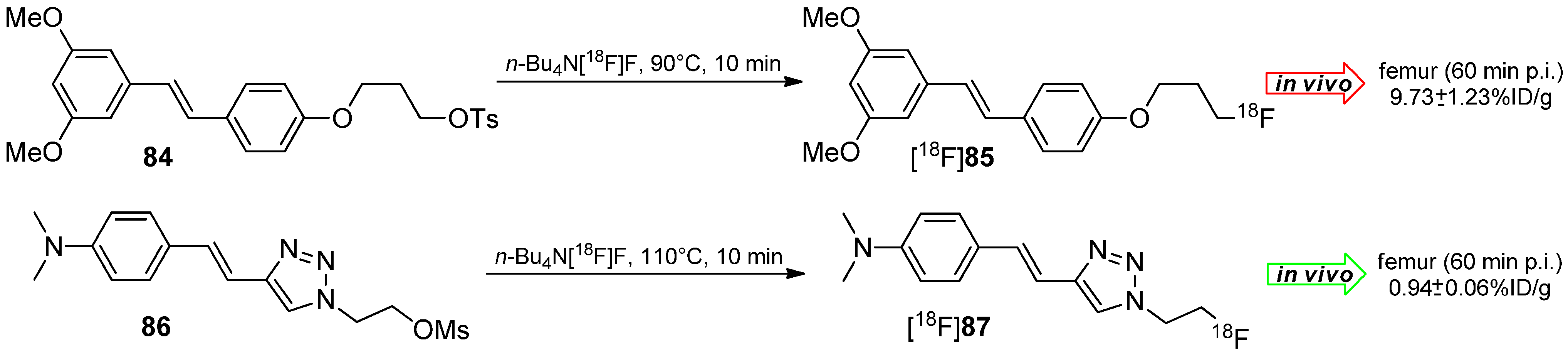
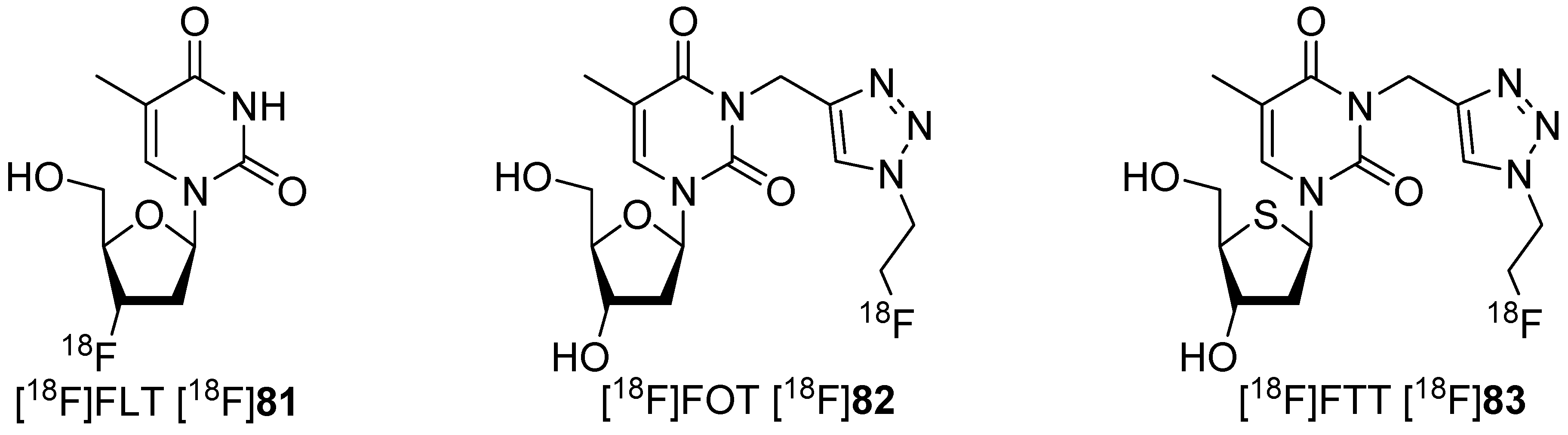
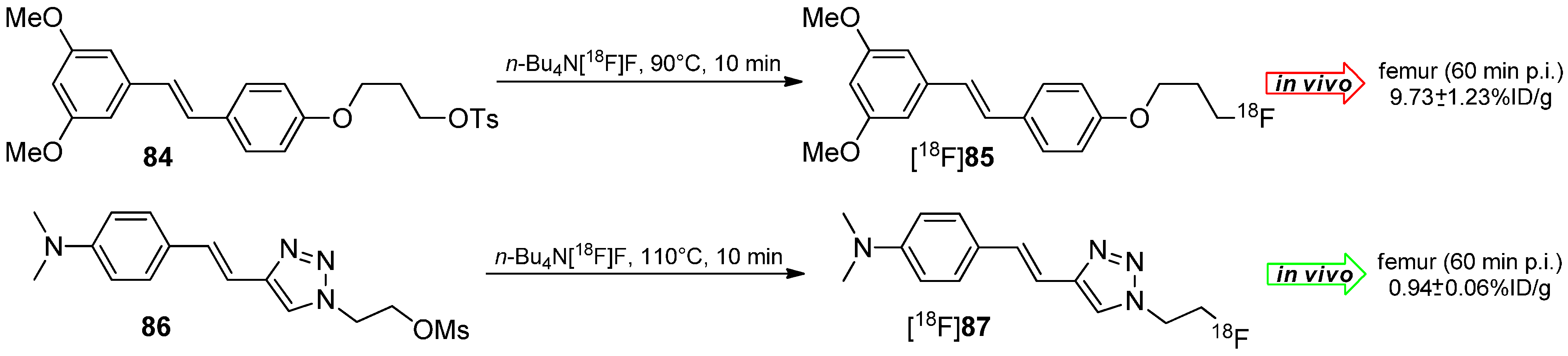
5.2. Click-Chemistry
5.3. CF3-Derivatives

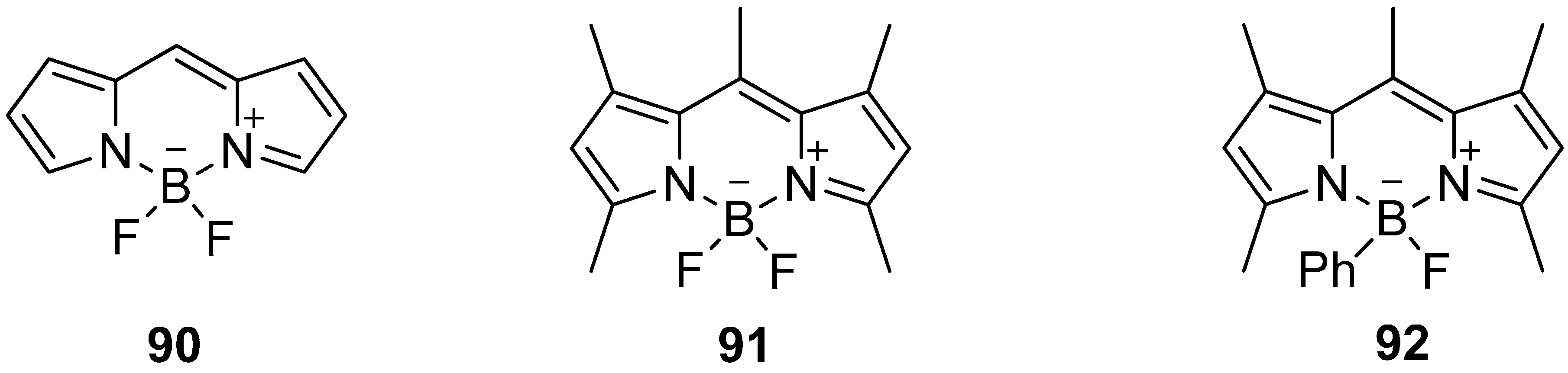
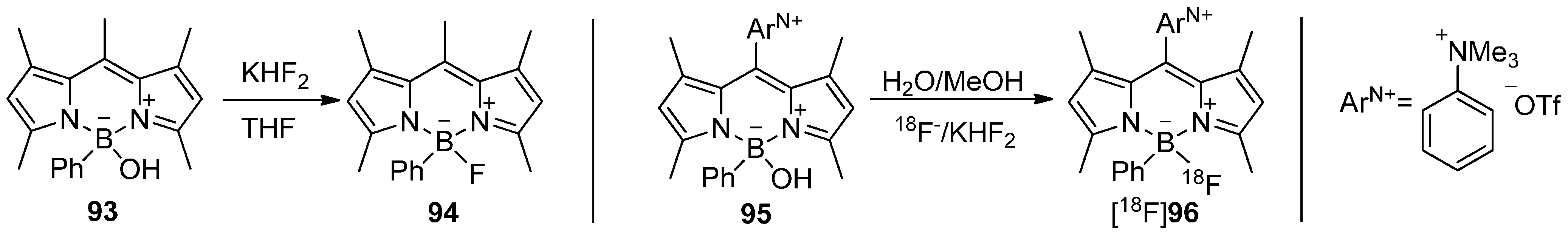
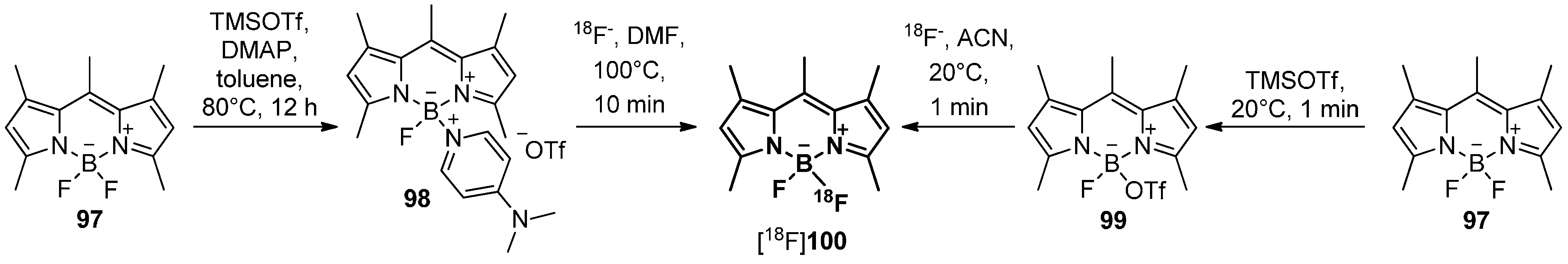
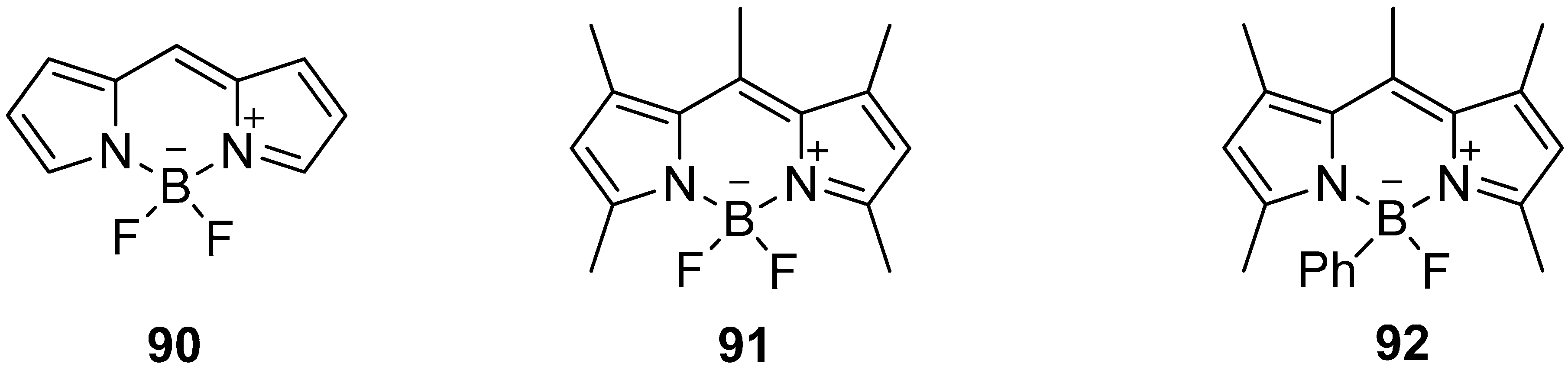
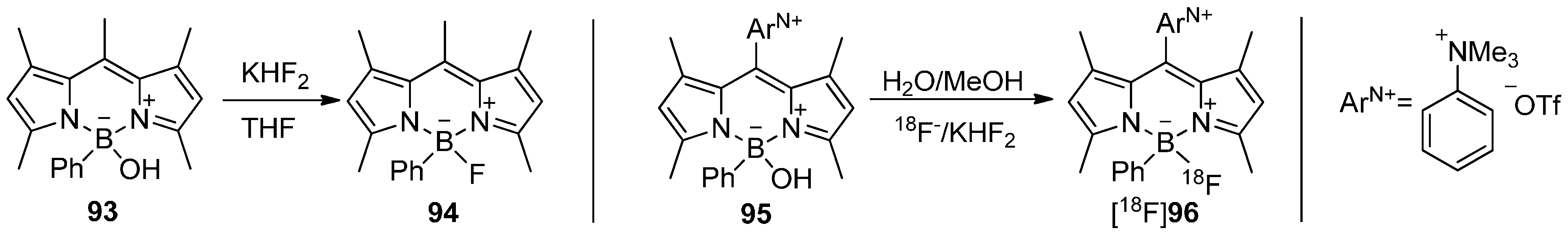
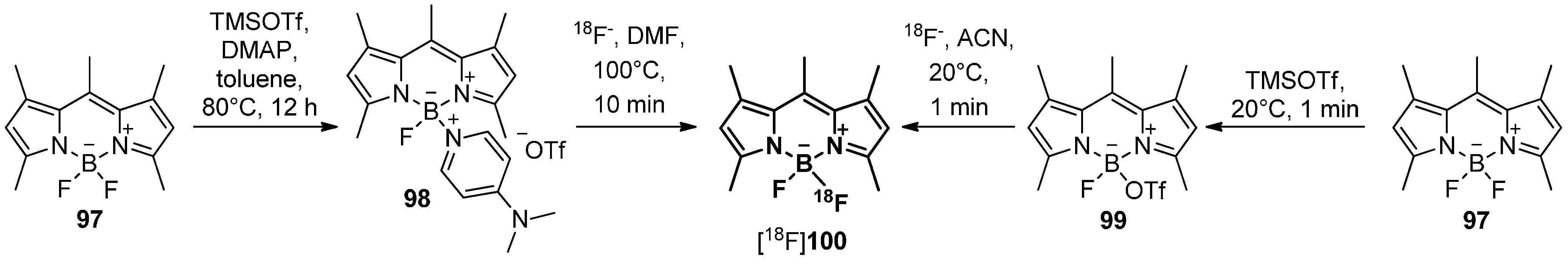
5.4. 18F-Fluoroborates
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Valk, P.E.; Baily, D.L.; Townsend, D.W.; Maisey, M.N. Positron Emission Tomography: Basic Science and Clinical Practice; Springer: London, UK, 2003. [Google Scholar]
- Fowler, J.S.; Ding, Y.-S. Radiotracer Chemistry. In Principles and Practice of PET and PET/CT, 2nd ed.; Wahl, R.L., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009. [Google Scholar]
- Ross, T.L.; Wester, H.J. 18F: Labeling Chemistry and Labeled Compounds. In Handbook of Nuclear Chemistry, 2nd ed.; Vértes, A., Nagy, S., Klencsár, Z., Lovas, R.G., Rösch, F., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 4, pp. 2023–2025. [Google Scholar]
- Miller, P.W.; Long, N.J.; Vilar, R.; Gee, A.D. Synthese von 11C-, 18F-, 15O- und 13N-Radiotracern für die Positronenemissionstomographie. Angew. Chem. 2008, 120, 9136–9172. [Google Scholar] [CrossRef]
- Pretze, M.; Große-Gehling, P.; Mamat, C. Cross-Coupling Reactions as Valuable Tool for the Preparation of PET Radiotracers. Molecules 2011, 16, 1129–1165. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.; Harper, D.B. Fluorine-containing natural products. J. Fluor. Chem. 1999, 100, 127–133. [Google Scholar] [CrossRef]
- Reddy, V.P. Organofluorine Compounds in Biology and Medicine, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1–23. [Google Scholar]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Wuest, F. 18F-Labeled Peptides: The Future Is Bright. Molecules 2014, 19, 20536–20556. [Google Scholar] [CrossRef] [PubMed]
- Pimlott, S.L.; Sutherland, A. Molecular tracers for the PET and SPECT imaging of disease. Chem. Soc. Rev. 2011, 40, 149–162. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 48, 441–451. [Google Scholar] [CrossRef]
- Sharpe, A.G. The physical properties of the carbon-fluorine bond. In Ciba Foundation Symposium 2—Carbon-Fluorine Compounds: Chemistry, Biochemistry and Biological Activities; Elliot, K., Birch, J., Eds.; Associated Scientific Publishers: Amsterdam, The Netherlands, 1972; pp. 33–54. [Google Scholar]
- Peters, D. Problem of the Lengths and Strengths of Carbon-Fluorine Bonds. J. Chem. Phys. 1963, 38, 561–563. [Google Scholar] [CrossRef]
- Bent, H.A. An Appraisal of Valence-bond Structures and Hybridization in Compounds of the First-row elements. Chem. Rev. 1961, 61, 275–311. [Google Scholar] [CrossRef]
- Lemal, D.M. Perspective on Fluorocarbon Chemistry. J. Org. Chem. 2004, 69, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tozer, D.J. The conformation and internal rotational barrier of benzyl fluoride. Chem. Phys. Lett. 1999, 308, 160–164. [Google Scholar] [CrossRef]
- Kochi, J.K.; Hammond, G.S. Benzyl Tosylates. II. The Application of the Hammett Equation to the Rates of their Solvolysis. J. Am. Chem. Soc. 1953, 75, 3445–3451. [Google Scholar] [CrossRef]
- Wüst, F.; Müller, M.; Bergmann, R. Synthesis of 4-([18F]fluoromethyl)-2-chlorophenylisothiocyanate: A novel bifunctional 18F-labelling agent. Radiochim. Acta 2004, 92, 349–353. [Google Scholar] [CrossRef]
- Zavitsas, A.A.; Rogers, D.W.; Matsunag, N. Remote Substituent Effects on Allylic and Benzylic Bond Dissociation Energies. Effects on Stabilization of Parent Molecules and Radicals. J. Org. Chem. 2007, 72, 7091–7101. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, T. Organofluorine Compounds: Chemistry and Applications; Springer: Berlin, Germany, 2000; p. 126. [Google Scholar]
- Frank, H.; Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of Bond Lengths determined by X-ray and Neutron Diffraction. Part 1. Bond Lengths in Organic Compounds. J. Chem. Soc. Perkin Trans. 1987, 2, S1–S19. [Google Scholar]
- Wiberg, K.B.; Rablen, P.R. Substituent Effects. 7. Phenyl Derivatives. When Is a Fluorine a π-Donor? J. Org. Chem. 1998, 63, 3722–3730. [Google Scholar] [CrossRef]
- Carroll, T.X.; Thomas, T.D.; Bergersen, H.; Børve, K.J.; Sæthre, L.J. Fluorine as a π Donor. Carbon 1s Photoelectron Spectroscopy and Proton Affinities of Fluorobenzenes. J. Org. Chem. 2006, 71, 1961–1968. [Google Scholar] [CrossRef] [PubMed]
- Liotta, C.L.; Harris, H.P. Chemistry of naked anions. I. Reactions of the 18-crown-6 complex of potassium fluoride with organic substrates in aprotic organic solvents. J. Am. Chem. Soc. 1974, 96, 2250–2252. [Google Scholar] [CrossRef]
- Tressaud, A.; Haufe, G. Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2008; pp. 35–42. [Google Scholar]
- Ermert, J. 18F-Labelled Intermediates for Radiosynthesis by Modular Build-Up Reactions: Newer Developments. BioMed. Res. Int. 2014, 15. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Fischer, C.R.; Holland, J.P.; Becaud, J.; Schubiger, P.A.; Schibli, R.; Ametamey, S.M.; Graham, K.; Stellfeld, T.; Dinkelborg, L.M.; et al. 18F-Radiolabeling of Aromatic Compounds Using Triarylsulfonium Salts. Eur. J. Org. Chem. 2012, 2012, 889–892. [Google Scholar] [CrossRef]
- Sander, K.; Gendron, T.; Yiannaki, E.; Cybulska, K.; Kalber, T.L.; Lythgoe, M.F.; Årstad, E. Sulfonium Salts as Leaving Groups for Aromatic Labelling of Drug-like Small Molecules with Fluorine-18. Sci. Rep. 2015, 5, 9941. [Google Scholar] [CrossRef] [PubMed]
- Brandt, J.R.; Lee, E.; Boursalian, G.B.; Ritter, T. Mechanism of electrophilic fluorination with Pd(IV): Fluoride capture and subsequent oxidative fluoride transfer. Chem. Sci. 2014, 5, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-stage [18F]fluorination: New solutions to old problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.L.; Wester, H.J. 18F: Labeling Chemistry and Labeled Compounds. In Handbook of Nuclear Chemistry, 2nd ed.; Vértes, A., Nagy, S., Klencsár, Z., Lovas, R.G., Rösch, F., Eds.; Springer: Dordrecht, The Netherlands, 2011; Volume 4, pp. 2026–2032. [Google Scholar]
- Vallabhajosula, S. Molecular Imaging: Radiopharmaceuticals for PET and SPECT; Springer: Dordrecht, The Netherlands, 2009; pp. 142–144. [Google Scholar]
- Tressaud, A.; Haufe, G. Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2008; p. 7. [Google Scholar]
- Scott, P.J.H.; Hockley, B.G.; Kung, H.F.; Manchanda, R.; Zhang, W.; Kilbourn, M.R. Studies into radiolytic decomposition of fluorine-18 labeled radiopharmaceuticals for positron emission tomography. Appl. Radiat. Isot. 2009, 67, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Langguth, P.; Seydel, J. Überarbeitetes Glossar zu Begriffen der Pharmazeutik. Angew. Chem. 2011, 123, 3635–3651. [Google Scholar] [CrossRef]
- Middleton, R.K. Drug Interactions. In Textbook of Therapeutics: Drug and Disease Management, 8th ed.; Helms, R.A., Herfindal, E.T., Quan, D.J., Eds.; Lipincott Williams & Wilkins: Philadelphia, PA, USA, 2006; p. 50. [Google Scholar]
- Kharasch, E.D.; Thummel, K.E. Identification of cytochrome P450 2E1 as the predominant enzyme catalyzing human liver microsomal defluorination of sevoflurane, isoflurane, and methoxyflurane. Anesthesiology 1993, 79, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Anders, M.W.; Jones, J.P. Metabolism of 1,2-dichloro-1-fluoroethane and 1-fluoro-1,2,2-trichloroethane: Electronic factors govern the regioselectivity of cytochrome P450-dependent oxidation. Chem. Res. Toxicol. 1996, 9, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Bier, D.; Holschbach, M.H.; Wutz, W.; Olsson, R.A.; Coenen, H.H. Metabolism of the a1 adenosine receptor positron emission tomography ligand [18F]8-cyclopentyl-3-(3-fluoropropyl)-1-propylxanthine ([18F]cpfpx) in rodents and humans. Drug Metabol. Dispos. 2006, 34, 570–576. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: Crossing the blood–brain barrier and surviving metabolism. Trends Pharm. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Park, B.K.; Kitteringham, N.R. Effects of fluorine substitution on drug metabolism: Pharmacological and toxicological implications. Drug Metab. Rev. 1994, 26, 605–643. [Google Scholar] [CrossRef] [PubMed]
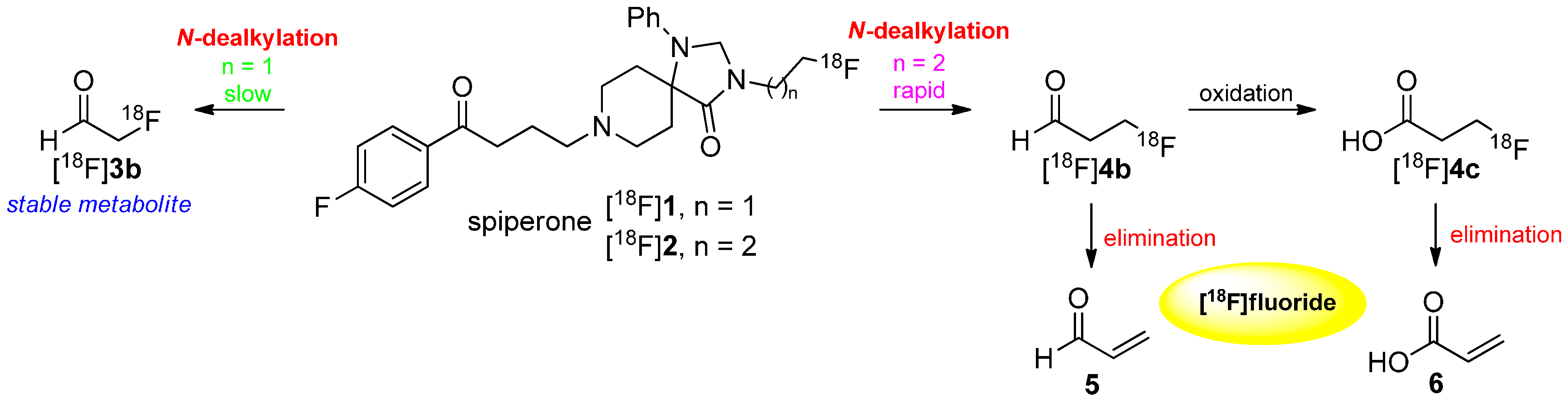
- Welch, M.J.; Katzenellenbogen, J.A.; Mathias, C.J.; Brodack, J.W.; Carlson, K.E.; Chi, D.Y.; Dence, C.S.; Kilbourn, M.R.; Perlmutter, J.S.; Raichle, M.E.; et al. N-(3-[18F]fluoropropyl)-spiperone: The preferred 18F labeled spiperone analog for positron emission tomographic studies of the dopamine receptor. Nucl. Med. Biol. 1988, 15, 83–97. [Google Scholar] [CrossRef]
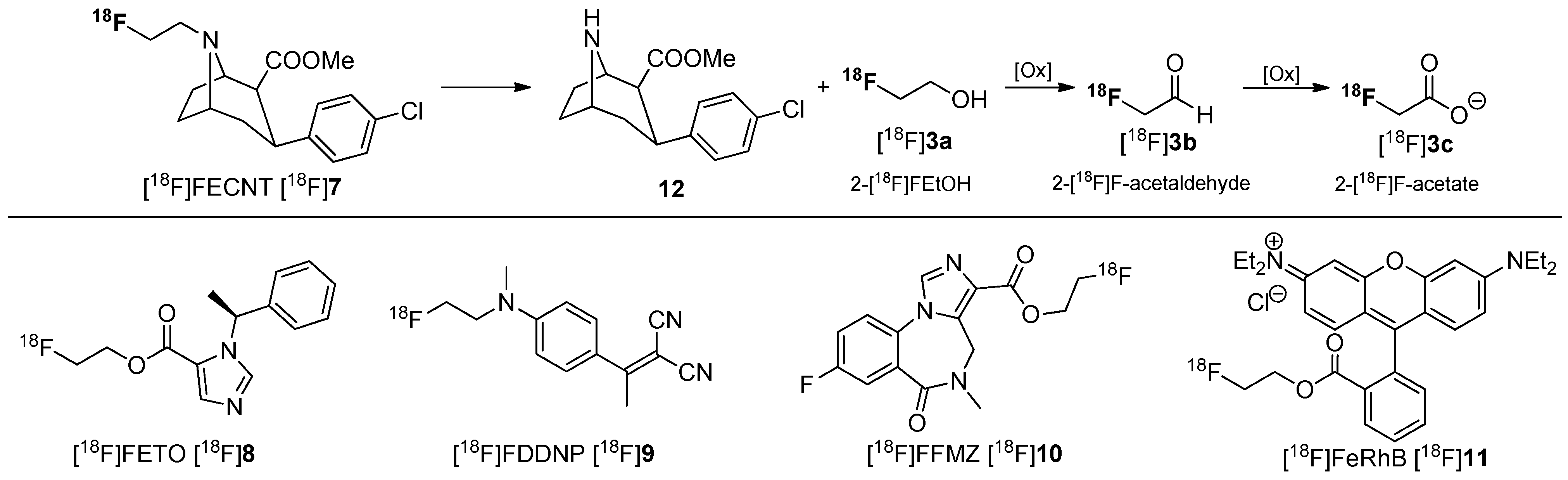
- Zoghbi, S.S.; Shetty, H.U.; Ichise, M.; Fujita, M.; Imaizumi, M.; Liow, J.S.; Shah, J.; Musachio, J.L.; Pike, V.W.; Innis, R.B. PET Imaging of the Dopamine Transporter with 18F-FECNT: A Polar Radiometabolite Confounds Brain Radioligand Measurements. J. Nucl. Med. 2006, 47, 520–527. [Google Scholar] [PubMed]
- Ettlinger, D.E.; Wadsak, W.; Mien, L.-K.; Machek, M.; Wabnegger, L.; Rendl, G.; Karanikas, G.; Viernstein, H.; Kletter, K.; Dudczak, R.; et al. [18F]FETO: Metabolic considerations. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Agdeppa, E.D.; Kepe, V.; Liu, J.; Flores-Torres, S.; Satyamurthy, N.; Petric, A.; Cole, G.M.; Small, G.W.; Huang, S.-C.; Barrio, J.R. Binding Characteristics of Radiofluorinated 6-Dialkylamino-2-Naphthylethylidene Derivatives as Positron Emission Tomography Imaging Probes for β-Amyloid Plaques in Alzheimer’s Disease. J. Neurosci. 2001, 21, RC189. [Google Scholar] [PubMed]
- Mitterhauser, M.; Wadsak, W.; Wabnegger, L.; Mien, L.K.; Tögel, S.; Langer, O.; Sieghart, W.; Viernstein, H.; Kletter, K.; Dudczak, R. Biological evaluation of 2ʹ-[18F]fluoroflumazenil ([18F]FFMZ), a potential GABA receptor ligand for PET. Nucl. Med. Biol. 2004, 31, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Gottumukkala, V.; Heinrich, T.K.; Baker, A.; Dunning, P.; Fahey, F.H.; Treves, S.T.; Packard, A.B. Biodistribution and Stability Studies of [18F]Fluoroethylrhodamine B, a Potential PET Myocardial Perfusion Agent. Nucl. Med. Biol. 2010, 37, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Pourghiasian, M.; Hundal, N.; Lau, J.; Bénard, F.; Dedhar, S.; Lin, K.-S. 2-[18F]Fluoroethanol and 3-[18F]fluoropropanol: Facile preparation, biodistribution in mice, and their application as nucleophiles in the synthesis of [18F]fluoroalkyl aryl ester and ether PET tracers. Nucl. Med. Biol. 2013, 40, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Tewson, T.J.; Welch, M.J. Preparation and preliminary biodistribution of “no carrier added” fluorine F-18 fluoroethanol. J. Nucl. Med. 1980, 21, 559–564. [Google Scholar] [PubMed]
- Lee, K.C.; Lee, S.-Y.; Choe, Y.S.; Chi, D.Y. Metabolic Stability of [18F]Fluoroalkylbiphenyls. Bull. Korean Chem. Soc. 2004, 25, 1225–1230. [Google Scholar]
- Milicevic Sephton, S.; Dennler, P.; Leutwiler, D.S.; Mu, L.; Wanger-Baumann, C.A.; Schibli, R.; Krämer, S.D.; Ametamey, S.M. Synthesis, radiolabelling and in vitro and in vivo evaluation of a novel fluorinated ABP688 derivative for the PET imaging of metabotropic glutamate receptor subtype 5. Am. J. Med. Mol. Imaging 2012, 2, 14–18. [Google Scholar]
- French, A.N.; Napolitano, E.; van Brocklin, H.F.; Brodack, J.W.; Hanson, R.N.; Welch, M.J.; Katzenellenbogen, J.A. The β-heteroatom effect in metabolic defluorination: The interaction of resonance and inductive effects may be a fundamental determinant in the metabolic liability of fluorine-substituted compounds. J. Label. Compd. Radiopharm. 1991, 30, 431–433. [Google Scholar]
- Purohit, A.; Radeke, H.; Azure, M.; Hanson, K.; Benetti, R.; Su, F.; Yalamanshili, P.; Yu, M.; Hayes, M.; Guaraldi, M.; et al. Synthesis and biological evaluation of pyridazinone analogues as potential cardiac positron emission tomography tracers. J. Med. Chem. 2008, 51, 2954–2970. [Google Scholar] [CrossRef] [PubMed]
- Dollé, F. Fluorine-18-labelled fluoropyridines: Advances in radiopharmaceutical design. Curr. Pharm. Des. 2005, 11, 3221–3235. [Google Scholar] [CrossRef] [PubMed]
- Gant, T.G. Using Deuterium in Drug Discovery: Leaving the Label in the Drug. J. Med. Chem. 2014, 57, 3595–3611. [Google Scholar] [CrossRef] [PubMed]
- Kohen, A.; Limbach, H.-H. Isotope Effects in Chemistry and Biology; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Roston, D.; Islam, Z.; Kohen, A. Isotope Effects as Probes for Enzyme Catalyzed Hydrogen-Transfer Reactions. Molecules 2013, 18, 5543–5567. [Google Scholar] [CrossRef] [PubMed]
- Fagerholm, V.; Mikkola, K.K.; Ishizu, T.; Arponen, E.; Kauhanen, S.; Nagren, K.; Solin, O.; Nuutila, P.; Haaparanta, M. Assessment of islet specificity of dihydrotetrabenazine radiotracer binding in rat pancreas and human pancreas. J. Nucl. Med. 2010, 51, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.R.; Souza, F.; Witkowski, P.; Maffei, A.; Raffo, A.; Herron, A.; Kilbourn, M.; Jurewicz, A.; Herold, K.; Liu, E.; et al. Visualizing pancreatic-cell mass with [11C]DTBZ. Nucl. Med. Biol. 2006, 33, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Goland, R.; Freeby, M.; Parsey, R.; Saisho, Y.; Kumar, D.; Simpson, N.; Hirsch, J.; Prince, M.; Maffei, A.; Mann, J.J.; et al. 11C-Dihydrotetrabenazine PET of the pancreas in subjects with long-standing type 1 diabetes and in healthy controls. J. Nucl. Med. 2009, 50, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.; Jahan, M.; Johnström, P.; Korsgren, O.; Sundin, A.; Halldin, C.; Johansson, L. In vivo and in vitro characterization of [18F]-FE-(+)-DTBZ as a tracer for beta-cell mass. Nucl. Med. Biol. 2010, 37, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.J.; Weng, Y.H.; Wey, S.P.; Hsiao, I.T.; Lu, C.S.; Skovronsky, D.; Chang, H.P.; Kung, M.P.; Yen, T.C. Whole-body biodistribution and radiation dosimetry of 18F-FP-(+)-DTBZ (18F-AV-133): A novel vesicular monoamine transporter 2 imaging agent. J. Nucl. Med. 2010, 51, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Jahan, M.; Eriksson, O.; Johnström, P.; Korsgren, O.; Sundin, A.; Johansson, L.; Halldin, C. Decreased defluorination using the novel betacell imaging agent [18F]FE-DTBZ-d4 in pigs examined by PET. EJNMMI Res. 2011, 1, 33. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.-S.; Fowler, J.S.; Wolf, A.P. Rapid, regiospecific syntheses of deuterium substituted 6-[18F]fluorodopamine (α,α-D2; β,β-D2 and α,α,β,β-D4) for mechanistic studies with positron emission tomography. J. Labelled Compd. Radiopharm. 1993, 33, 645–654. [Google Scholar] [CrossRef]
- DeWolf, W.E.J., Jr.; Carr, S.A.; Varrichio, A.; Goodhart, P.J.; Mentzer, M.A.; Roberts, G.D.; Southan, C.; Dolle, R.E.; Kurse, L.I. Inactivation of dopamine β-hydroxylase by p-cresol: Isolation and characterization of covalently modified active site peptides. Biochemistry 1988, 27, 9093–9101. [Google Scholar] [CrossRef] [PubMed]
- Coleman, A.A.; Hindsgaul, O.; Palcic, M.M. Stereochemistry of copper amine oxidase reactions. J. Biol. Chem. 1989, 264, 19500–19505. [Google Scholar] [PubMed]
- Yu, P.H. Three types of stereospecificity and the kinetic deuterium isotope effect in the oxidative deamination of dopamine as catalyzed by different amine oxidases. Biochem. Cell Biol. 1988, 66, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.H.; Bailey, B.A.; Durden, D.A.; Boulton, A.A. Stereospecific deuterium substitution at the alpha-carbon position of dopamine and its effect on oxidative deamination catalyzed by MAO-A and MAO-B from different tissues. Biochem. Pharmacol. 1986, 35, 1027–1036. [Google Scholar] [CrossRef]
- Ding, Y.-S.; Fowler, J.S.; Gatley, S.J.; Logan, J.; Volkow, N.D.; Shea, C. Mechanistic Positron Emission Tomography Studies of 6-[18F]Fluorodopamine in Living Baboon Heart: Selective Imaging and Control of Radiotracer Metabolism Using the Deuterium Isotope Effect. J. Neurochem. 1995, 65, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Schou, M.; Halldin, C.; Sóvágó, J.; Pike, V.W.; Hall, H.; Gulyás, B.; Mozley, P.D.; Dobson, D.; Shchukin, E.; Innis, R.B.; et al. PET evaluation of novel radiofluorinated reboxetine analogs as norepinephrine transporter probes in the monkey brain. Synapse 2004, 53, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-R.; Maeda, J.; Ito, T.; Okauchi, T.; Ogawa, M.; Noguchi, J.; Suhara, T.; Halldin, C.; Suzuki, K. Synthesis and evaluation of N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoromethoxy-d2–5-methoxybenzyl)acetamide: A deuterium substituted radioligand for peripheral benzodiazepine receptor. Bioorg. Med. Chem. 2005, 13, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-S.; Ding, Y.-S.; Kim, S.-W.; Kil, K.-E. Synthesis, enantiomeric resolution, F-18 labeling and biodistribution of reboxetine analogs: Promising radioligands for imaging the norepinephrine transporter with positron emission tomography. Nucl. Med. Biol. 2005, 32, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Chin, F.T.; Pike, V.W.; Toyama, H.; Liow, J.-S.; Zoghbi, S.S.; Modell, K.; Briard, E.; Shetty, H.U.; Sinclair, K.; et al. Synthesis and Evaluation of Two 18F-Labeled 6-Iodo-2-(4-N,N-dimethylamino)phenylimidazo[1,2-a]pyridine Derivatives as Prospective Radioligands for β-Amyloid in Alzheimer’s Disease. J. Med. Chem. 2004, 47, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Donohue, S.R.; Krushinski, J.H.; Pike, V.W.; Chernet, E.; Phebus, L.; Chesterfield, A.K.; Felder, C.C.; Halldin, C.; Schaus, J.M. Synthesis, Ex Vivo Evaluation, and Radiolabeling of Potent 1,5-Diphenylpyrrolidin-2-one Cannabinoid Subtype-1 Receptor Ligands as Candidates for in Vivo Imaging. J. Med. Chem. 2008, 51, 5833–5842. [Google Scholar] [CrossRef] [PubMed]
- Beyerlein, F.; Piel, M.; Hoehnemann, S.; Roesch, F. Automated synthesis and purification of [18F]fluoro-[di-deutero]methyl tosylate. J. Label. Compd. Radiopharm. 2013, 56, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Miura, S.; Hasui, T.; Halldin, C.; Stepanov, V.; Takano, A. Radiolabeled Compounds and Their Use as Radiotracers for Quantitative Imaging of Phosphodiesterase (PDE10A) in Mammals. WO 2013027845 A1, 28 February 2013. [Google Scholar]
- Schieferstein, H.; Piel, M.; Beyerlein, F.; Lueddens, H.; Bausbacher, N.; Buchholz, H.-G.; Ross, T.L.; Roesch, F. Selective binding to monoamine oxidase A: In vitro and in vivo evaluation of 18F-labeled β-carboline derivatives. Bioorg. Med. Chem. 2015, 23, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Rami-Mark, C.; Zhang, M.-R.; Mitterhauser, M.; Lanzenberger, R.; Hacker, M.; Wadsak, W. [18F]FMeNER-D2: Reliable fully-automated synthesis for visualization of the norepinephrine transporter. Nucl. Med. Biol. 2013, 40, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Schou, M.; Halldin, C.; Sóvágó, J.; Pike, V.W.; Gulyás, B.; Mozley, P.D.; Johnson, D.P.; Hall, H.; Innis, R.B.; Farde, L. Specific in vivo binding to the norepinephrine transporter demonstrated with the PET radioligand, (S,S)-[11C]MeNER. Nucl. Med. Biol. 2003, 30, 707–714. [Google Scholar] [CrossRef]
- Casebier, D.S.; Robinson, S.P.; Purohit, A.; Radeke, H.S.; Azure, M.T.; Dischino, D.D. Contrast Agents for Myocardial Perfusion Imaging. WO 2005079391 A2, 1 September 2005. [Google Scholar]
- Hortala, L.; Arnaud, J.; Roux, P.; Oustric, D.; Boulu, L.; Oury-Donat, F.; Avenet, P.; Rooney, T.; Alagille, D.; Barret, O.; et al. Synthesis and preliminary evaluation of a new fluorine-18 labelled triazine derivative for PET imaging of cannabinoid CB2 receptor. Bioorg. Med. Chem. Lett. 2014, 24, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Brumby, T.; Graham, K.; Krueger, M. Direct Synthesis of [18F]fluoromethoxy Compounds for PET Imaging and New Precursors for Direct Radiosynthesis of Protected Derivatives of O-([18F]fluoromethyl)tyrosine. WO 2013001088 A1, 3 January 2013. [Google Scholar]
- Graham, K.; Ede, S. Simplified Radiosynthesis of O-[18F]fluoromethyl Tyrosine Derivatives. WO 2013026940 A1, 28 February 2013. [Google Scholar]
- Graham, K.; Zitzmann-Kolbe, S.; Brumby, T. Preparation of Fluorodeuteriomethyl Tyrosine Derivatives. WO 2012025464 A1, 1 March 2012. [Google Scholar]
- Hamill, T.G.; Sato, N.; Jitsuoka, M.; Tokita, S.; Sanabria, S.; Eng, W.; Ryan, C.; Krause, S.; Takenaga, N.; Patel, S.; et al. Inverse agonist histamine H3 receptor PET tracers labeled with carbon-11 or fluorine-18. Synapse 2009, 63, 1122–1132. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Hong, J.; Morse, C.L.; Pike, V.W. Synthesis, Structure-Affinity Relationships, and Radiolabeling of Selective High-Affinity 5-HT4 Receptor Ligands as Prospective Imaging Probes for Positron Emission Tomography. J. Med. Chem. 2010, 53, 7035–7047. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.H.; Chen, A.M.; Gibson, R.E.; Goulet, M.T.; Hagmann, W.K.; Hamill, T.G.; Jewell, J.P.; Lin, L.S.; Liu, P.; Peresypkin, A.V. Heterocyclic Radiolabeled Cannabinoid-1 Receptor Modulators. WO 2005009479 A1, 3 February 2005. [Google Scholar]
- Cosford, N.D.P.; Govek, S.P.; Hamill, T.G.; Kamenecka, T.; Roppe, J.R.; Seiders, T.J. Alkyne Derivatives as Tracers for Metabotropic Glutamate Receptor Binding. WO 2004038374 A2, 6 May 2004. [Google Scholar]
- Burns, H.D.; Hamill, T.G.; Lindsley, C.W. Radiolabeled Glycine Transporter Inhibitors. WO 2007041025 A2, 12 April 2007. [Google Scholar]
- Hamill, T.G.; McCauley, J.A.; Burns, H.D. The Synthesis of A Benzamidine-containing NR2B-selective NMDA Receptor Ligand Labelled with Tritium or Fluorine-18. J. Label. Compd. Radiopharm. 2005, 48, 1–10. [Google Scholar] [CrossRef]
- Burns, H.D.; sEng, W.-S.; Gibson, R.E.; Hamill, T.G. Radiolabeled Neurokinin-1 Receptor Antagonists. WO 2004029024 A2, 8 April 2004. [Google Scholar]
- Leyton, J.; Smith, G.; Zhao, Y.; Perumal, M.; Nguyen, Q.-D.; Robins, E.; Årstad, E.; Aboagye, E.O. [18F]Fluoromethyl-[1,2-2H4]-Choline: A Novel Radiotracer for Imaging Choline Metabolism in Tumors by Positron Emission Tomography. Cancer Res. 2009, 69, 7721–7728. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Zhao, Y.; Leyton, J.; Shan, B.; de Nguyen, Q.; Perumal, M. Radiosynthesis and pre-clinical evaluation of [18F]fluoro-[1,2-2H4]choline. Nucl. Med. Biol. 2011, 38, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Lehmann, L.; Kettschau, G.; Toth, M.; Heinrich, T.; Thiele, A.; Varrone, A.; Halldin, C. Development of a novel fluorine-18 labeled deuterated fluororasagiline ([18F]fluororasagiline-D2) radioligand for PET studies of monoamino oxidase B (MAO-B). Bioorg. Med. Chem. 2013, 21, 6634–6641. [Google Scholar] [CrossRef] [PubMed]
- Abu-Raya, S.; Tabakman, R.; Blaugrund, E.; Trembovler, V.; Lazarovici, P. Neuroprotective and neurotoxic effects of monoamine oxidase-B inhibitors and derived metabolites under ischemia in PC12 cells. Eur. J. Pharmacol. 2002, 434, 109–116. [Google Scholar] [CrossRef]
- Nag, S. Development of Novel Fluorine-18 Labeled PET Radioligands for Monoamine Oxidase B (MAO-B). Ph.D. Thesis, Karolinska Institutet, Stockholm, Sweden, 2013. [Google Scholar]
- Ramirez de Molina, A.; Gallego-Ortega, D.; Sarmentero-Estrada, J.; Lagares, D.; Bandrés, E.; Gomez del Pulgar, T.; García-Foncillas, J.; Lacal, J.C. Choline kinase as a link connecting phospholipid metabolism and cell cycle regulation: Implications in cancer therapy. Int. J. Biochem. Cell Biol. 2008, 40, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, S.; Imamura, S.; Misaki, H.; Horiuti, Y. Purification and Characterization of Choline oxidase from Arthrobacter globiformis. J. Biochem. 1977, 82, 1741–1749. [Google Scholar] [PubMed]
- Gadda, G. pH and deuterium kinetic isotope effects studies on the oxidation of choline to betaine-aldehyde catalyzed by choline oxidase. Biochim. Biophys. Acta 2003, 1650, 4–9. [Google Scholar] [CrossRef]
- Witney, T.H.; Alam, I.S.; Turton, D.R.; Smith, G.; Carroll, L.; Brickute, D.; Twyman, F.J.; Nguyen, Q.-D.; Tomasi, G.; Awais, R.O.; et al. Evaluation of Deuterated 18F- and 11C-Labeled Choline Analogs for Cancer Detection by Positron Emission Tomography. Clin. Cancer Res. 2012, 18, 1063–1072. [Google Scholar] [CrossRef] [PubMed]
- Challapalli, A.; Sharma, R.; Hallett, W.A.; Kozlowski, K.; Carroll, L.; Brickute, D.; Twyman, F.; Al-Nahhas, A.; Aboagye, E.O. Biodistribution and Radiation Dosimetry of Deuterium-Substituted 18F-Fluoromethyl-[1, 2–2H4]Choline in Healthy Volunteers. J. Nucl. Med. 2014, 55, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Dounay, A.; Barta, N.; Bikker, J.; Borosky, S.; Campbell, B.; Crawford, T.; Denny, L.; Evans, L.; Gray, D.; Lee, P. Synthesis and pharmacological evaluation of aminopyrimidine series of 5-HT1A partial agonists. Bioorg. Med. Chem. Lett. 2009, 19, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Manoury, P.M.; Binet, J.; Rousseau, J.; Lefevre-Borg, F.; Cavero, I. Synthesis of a series of compounds related to betaxolol, a new β1-adrenoceptor antagonist with a pharmacological and pharmacokinetic profile optimized for the treatment of chronic cardiovascular diseases. J. Med. Chem. 1987, 30, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, B.; Rohde, J.; Wang, J.; Fung, S.; Monzon, K.; Chiou, W.; Pan, L.; Deng, X.; Stolarik, D.A.; Frevert, E.U. Adamantane 11-β-HSD-1 inhibitors: Application of an isocyanide multicomponent reaction. Bioorg. Med. Chem. Lett. 2006, 16, 5958–5962. [Google Scholar] [CrossRef] [PubMed]
- Wrobleski, M.; Reichard, G.; Paliwal, S.; Shah, S.; Tsui, H.; Duffy, R.; Lachowicz, J.; Morgan, C.; Varty, G.; Shih, N. Cyclobutane derivatives as potent NK1 selective antagonists. Bioorg. Med. Chem. Lett. 2006, 16, 3859–3863. [Google Scholar] [CrossRef] [PubMed]
- Shoup, T.; Elmaleh, D.; Bonab, A.; Fischman, A. Evaluation of trans-9–18F-fluoro-3,4-methyleneheptadecanoic acid as a PET tracer for myocardial fatty acid imaging. J. Nucl. Med. 2005, 46, 297–304. [Google Scholar] [PubMed]
- Martel, F.; Berlinguet, L. Impairment of tumor growth by unnatural amino acids. Can. J. Biochem. Physiol. 1959, 37, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Connors, T.; Elson, L.; Haddow, A.; Ross, W. The pharmacology and tumour growth inhibitory activity of 1-aminocyclopentane-1-carboxylic acid and related compounds. Biochem. Pharmacol. 1960, 5, 108–129. [Google Scholar] [CrossRef]
- Wooten, D.W.; Moraino, J.D.; Hillmer, A.T.; Engle, J.W.; DeJesus, O.J.; Murali, D.; Barnhart, T.E.; Nickles, R.J.; Davidson, R.J.; Schneider, M.L.; et al. In Vivo Kinetics of [F-18]MEFWAY: A comparison with [C-11]WAY100635 and [F-18]MPPF in the nonhuman primate. Synapse 2011, 65, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Williams, L.; Camp, V.; Malveaux, E.; Olson, J.; Goodman, M. Stereoselective synthesis and biological evaluation of syn-1-amino-3-[18F]fluorocyclobutyl-1-carboxylic acid as a potential positron emission tomography brain tumor imaging agent. Bioorg. Med. Chem. 2009, 17, 1982–1990. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, T.; Oku, T.; Uehara, H.; Desai, R.; Beattie, B.; Tjuvajev, J.; Blasberg, R. Facilitated amino acid transport is upregulated in brain tumors. J. Celeb. Blood Flow. Metab. 1998, 18, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Franck, D.; Kniess, T.; Steinbach, J.; Zitzmann-Kolbe, S.; Friebe, M.; Dinkelborg, L.M.; Graham, K. Investigations into the synthesis, radiofluorination and conjugation of a new [18F]fluorocyclobutyl prosthetic group and its in vitro stability using a tyrosine model system. Bioorg. Med. Chem. 2013, 21, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Franck, D. Radiofluorinated Cyclobutyl Group for Increased Metabolic Stability Using Tyrosine Derivatives as Model System. Ph.D. Thesis, TU Dresden, Dresden, Germany, 2012. [Google Scholar]
- Tipre, D.N.; Zoghbi, S.S.; Liow, J.S.; Green, M.V.; Seidel, J.; Ichise, M.; Innis, R.B.; Pike, V.W. PET Imaging of Brain 5-HT1A Receptors in Rat in Vivo with 18F-FCWAY and Improvement by Successful Inhibition of Radioligand Defluorination with Miconazole. J. Nucl. Med. 2006, 47, 345–353. [Google Scholar] [PubMed]
- Holleman, A.; Wiberg, N.; Wiberg, E. Lehrbuch der Anorganischen Chemie, 102nd ed.; De Gruyter Verlag: Berlin, Germany, 2008; pp. 1097–1103. [Google Scholar]
- Rosenthal, M.S.; Bosch, A.L.; Nickles, R.J.; Gatley, S.J. Synthesis and some characteristics of no-carrier added [18F]fluorotrimethylsilane. Int. J. Appl. Radiat. Isot. 1985, 36, 318–319. [Google Scholar] [CrossRef]
- Gatley, S.J. Rapid production and trapping of [F-18]fluorotrimethylsilane, and its use in nucleophilic F-18 labeling without an aqueous evaporation step. Appl. Radiat. Isot. 1989, 40, 541–544. [Google Scholar] [CrossRef]
- Mulholland, G.K. Recovery and purification of no-carrier-added [18F]fluoride with bistrimethylsilylsulfate (BTMSS). Int. J. Radiat. Appl. Instr. 1991, 42, 1003–1008. [Google Scholar] [CrossRef]
- Walsh, J.C.; Fleming, L.M.; Satyamurthy, N.; Barrio, J.R.; Phelps, M.E.; Gambhir, S.S.; Toyokuni, T. Application of silicon-fluoride chemistry for the development of amine-reactive F-18-labeling agents for biomolecules. J. Nucl. Med. 2000, 41, 249. [Google Scholar]
- Choudhry, U.; Martin, K.E.; Biagini, S.; Blower, P.J. Alkoxysilane groups for instant labeling of biomolecules with 18F. Nucl. Med. Commun. 2006, 27, 293. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Bradtmöller, G.; Schirrmacher, E.; Thews, O.; Tillmanns, J.; Siessmeier, T.; Buchholz, H.G.; Bartenstein, P.; Wängler, B.; Niemeyer, C.M.; et al. 18F-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew. Chem. Int. Ed. 2006, 45, 6047–6050. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, R.; Kostikov, A.; Wängler, C.; Jurkschat, K.; Bernard-Gauthier, V.; Schirrmacher, E.; Wängler, B. Silicon Fluoride Acceptors (SIFAs) for Peptide and Protein Labeling with 18F. In Radiochemical Syntheses, 1st ed.; Scott, P.J.H., Ed.; J. Wiley & Sons: Hoboken, NJ, USA, 2015; Volume 2, pp. 149–162. [Google Scholar]
- Fleischer, H. Molecular “Floppyness” and the Lewis Acidity of Silanes: A Density Functional Theory Study. Eur. J. Inorg. Chem. 2001, 393–404. [Google Scholar] [CrossRef]
- Höhne, A.; Yu, L.; Mu, L.; Reiher, M.; Voigtmann, U.; Klar, U.; Graham, K.; Schubiger, P.A.; Ametamey, S.M. Organofluorosilanes as model compounds for 18F-labeled silicon-based PET tracers and their hydrolytic stability: Experimental data and theoretical calculations (PET = Positron Emission Tomography). Chemistry 2009, 15, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.P.; Bickelhaupt, F.M. Nucleophilic Substitution at Silicon (SN2@Si) via a Central Reaction Barrier. J. Org. Chem. 2007, 72, 2201–2207. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Gauthier, V.; Wängler, C.; Schirrmacher, E.; Kostikov, A.; Jurkschat, K.; Wängler, B.; Schirrmacher, R. 18F-Labeled Silicon-Based Fluoride Acceptors: Potential Opportunities for Novel Positron Emitting Radiopharmaceuticals. BioMed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Neto, P.; Wängler, B.; Iovkova, L.; Boening, G.; Reader, A.; Jurkschat, K.; Schirrmacher, E. [18F]SiFA-isothiocyanate: A new highly effective radioactive labeling agent for lysine-containing proteins. ChemBioChem 2009, 10, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Wängler, C.; Niedermoser, S.; Chin, J.; Orchowski, K.; Schirrmacher, E.; Jurkschat, K.; Kostikov, A.P.; Iovkova-Berends, L.; Schirrmacher, R.; Wängler, B. One-step 18F-labeling of peptides for positron emission tomography imaging using the SiFA methodology. Nat. Prot. 2012, 7, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Lindner, S.; Michler, C.; Leidner, S.; Rensch, C.; Wängler, C.; Schirrmacher, R.; Bartenstein, P.; Wängler, B. Synthesis and in Vitro and in Vivo Evaluation of SiFA-Tagged Bombesin and RGD Peptides as Tumor Imaging Probes for Positron Emission Tomography. Bioconjugate Chem. 2014, 25, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Balentova, E.; Collet, C.; Lamandé-Langle, S.; Chrétien, F.; Thonon, D.; Aerts, J.; Lemaire, C.; Luxen, A.; Chapleuret, Y. Synthesis and hydrolytic stability of novel 3-[18F]fluoroethoxybis(1-methylethyl)silyl]propanamine-based prosthetic groups. J. Fluor. Chem. 2011, 132, 250–257. [Google Scholar] [CrossRef]
- Ting, R.; Adam, M.J.; Ruth, T.J.; Perrin, D.M. Arylfluoroborates and alkylfluorosilicates as potential PET imaging agents: High-yielding aqueous biomolecular18F-labeling. J. Am. Chem. Soc. 2005, 127, 13094–13095. [Google Scholar] [CrossRef] [PubMed]
- Mu, L.; Höhne, A.; Schubiger, P.A.; Ametamey, S.M.; Graham, K.; Cyr, J.E.; Dinkelborg, L.; Stellfeld, T.; Srinivasan, A.; Voigtmann, U.; et al. Silicon-Based Building Blocks for One-Step 18F-Radiolabeling of Peptides for PET Imaging. Angew. Chem. Int. Ed. 2008, 47, 4922–4925. [Google Scholar] [CrossRef] [PubMed]
- Bohn, P.; Deyine, A.; Azzouz, R.; Bailly, L.; Fiol-Petit, C.; Bischoff, L.; Fruit, C.; Marsais, F.; Vera, P. Design of siliconbased misonidazole analogues and 18F-radiolabelling. Nucl. Med. Biol. 2009, 36, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Briard, E.; Zoghbi, S.S.; Siméon, F.G.; Imaizumi, M.; Gourley, J.P.; Shetty, H.U.; Lu, S.; Fujita, M.; Innis, R.B.; Pike, V.W. Single-Step High-Yield Radiosynthesis and Evaluation of a Sensitive 18F-Labeled Ligand for Imaging Brain Peripheral Benzodiazepine Receptors with PET. J. Med. Chem. 2009, 52, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Mayer, J.M. Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry and Enzymology; Verlag Helvetica Chimica Acta: Zürich, Switzerland, 2003. [Google Scholar]
- Fukami, T.; Yokoi, T. The Emerging Role of Human Esterases. Drug Metabol. Pharmacokin. 2012, 27, 466–477. [Google Scholar] [CrossRef]
- Li, Z.; Lang, L.; Ma, Y.; Kiesewetter, D.O. [18F]Fluoropropylsulfonyl chloride: A new reagent for radiolabeling primary and secondary amines for PET imaging. J. Label. Compd. Radiopharm. 2008, 51, 23–27. [Google Scholar] [CrossRef]
- Löser, R.; Fischer, S.; Hiller, A.; Köckerling, M.; Funke, U.; Maisonial, A.; Brust, P.; Steinbach, J. Use of 3-[18F]fluoropropanesulfonyl chloride as a prosthetic agent for the radiolabelling of amines: Investigation of precursor molecules, labelling conditions and enzymatic stability of the corresponding sulfonamides. Beilstein J. Org. Chem. 2013, 9, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.; Sala, R.; Carroll, L.; Behan, K.; Glaser, M.; Robins, E.; Nguyen, Q.-D.; Aboagye, E.O. Synthesis and evaluation of nucleoside radiotracers for imaging proliferation. Nucl. Med. Biol. 2012, 39, 652–665. [Google Scholar] [CrossRef] [PubMed]
- Mamat, C.; Ramenda, T.; Wuest, F.R. Recent applications of click chemistry for the synthesis of radiotracers for molecular imaging. Mini-Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Pretze, M.; Pietzsch, D.; Mamat, C. Recent Trends in Bioorthogonal Click-Radiolabeling Reactions Using Fluorine-18. Molecules 2013, 18, 8618–8665. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, J.; Hayashi, A.; Gogami, A.; Hamada, M.; Hamashima, Y.; Katoh, T.; Node, M.; Fujibayashi, Y. Alkyl- fluorinated thymidine derivatives for imaging cell proliferation I. The in vitro evaluation of some alkyl-fluorinated thymidine derivatives. Nucl. Med. Biol. 2006, 33, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, U.; Soghomonyan, S.; Yeh, H.H.; Flores, L.G.; Shavrin, A.; Volgin, A.Y.; Gelovani, J.G.; Alauddin, M.M. Synthesis and preliminary PET imaging of N3-[18F]fluoroethyl thymidine and N3-[18F]fluoropropyl thymidine. Nucl. Med. Biol. 2008, 35, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Fluorine in Medicinal Chemistry and Chemical Biology; Ojima, I. (Ed.) Wiley-Blackwell: Sussex, UK, 2009.
- Lee, I.; Choe, Y.S.; Choi, J.Y.; Lee, K.-H.; Kim, B.-T. Synthesis and Evaluation of 18F-Labeled Styryltriazole and Resveratrol Derivatives for β-Amyloid Plaque Imaging. J. Med. Chem. 2012, 55, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Lien, V.T.; Riss, P.J. Radiosynthesis of [18F]Trifluoroalkyl Groups: Scope and Limitations. BioMed. Res. Int. 2014, 10. [Google Scholar] [CrossRef] [PubMed]
- Ido, T.; Irie, T.; Kasida, Y. Isotope exchange with 18F on superconjugate system. J. Label. Compd. Radiopharm. 1979, 16, 153–154. [Google Scholar]
- Satter, M.R.; Martin, C.C.; Oakes, T.R.; Christian, B.; Nickles, R.J. Synthesis of the fluorine-18 labeled inhalation Anesthetics. Appl. Radiat. Isot. 1994, 45, 1093–1100. [Google Scholar] [CrossRef]
- Suehiro, M.; Yang, G.; Torchon, G.; Ackerstaff, E.; Humm, J.; Koutcher, J.; Ouerfelliet, O. Radiosynthesis of the tumor hypoxia marker [18F]TFMISO via O-[18F]trifluoroethylation reveals a striking difference between trifluoroethyl tosylate and iodide in regiochemical reactivity toward oxygen nucleophiles. Bioorg. Med. Chem. 2011, 19, 2287–2297. [Google Scholar] [CrossRef] [PubMed]
- Angelini, G.; Speranza, M.; Shiue, C.-Y.; Wolf, A.P. H18F + Sb2O3: A new selective radiofluorinating agent. Chem. Commun. 1986, 12, 924–925. [Google Scholar] [CrossRef]
- Angelini, G.; Speranza, M.; Wolf, A.P.; Shiue, C.-Y. Synthesis of N-(α,α,α-tri[18F]fluoro-m-tolyl)piperazine. A potent serotonin agonist. J. Label. Compd. Radiopharm. 1990, 28, 1441–1448. [Google Scholar] [CrossRef]
- Kilbourn, M.R.; Pavia, M.R.; Gregor, V.E. Synthesis of fluorine-18 labeled GABA uptake inhibitors. Appl. Radiat. Isot. 1990, 41, 823–828. [Google Scholar] [CrossRef]
- Das, M.K.; Mukherjee, J. Radiosynthesis of [F-18]fluoxetine as a potential radiotracer for serotonin reuptake sites. Appl. Radiat. Isot. 1993, 44, 835–842. [Google Scholar] [CrossRef]
- Johnstrom, P.; Stone-Elander, S. The 18F-labelled alkylating agent 2,2,2-trifluoroethyl triflate: Synthesis and specific activity. J. Label. Compd. Radiopharm. 1995, 36, 537–547. [Google Scholar] [CrossRef]
- Riss, P.J.; Aigbirhio, F.I. A simple, rapid procedure for nucleophilic radiosynthesis of aliphatic [18F]trifluoromethyl groups. Chem. Commun. 2011, 47, 11873–11875. [Google Scholar] [CrossRef] [PubMed]
- Dolbier, W.R., Jr.; Li, A.-R.; Koch, C.J.; Shiue, C.-Y.; Kachur, A.V. [18F]-EF5, a marker for PET detection of hypoxia: Synthesis of precursor and a new fluorination procedure. Appl. Radiat. Isot. 2001, 54, 73–80. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Alauddin, M.M.; Hu, J.; Conti, P.S.; Olah, G.A. Expedient synthesis of [18F]-labeled α-trifluoromethyl ketones. J. Label. Compd. Radiopharm. 2003, 46, 1087–1092. [Google Scholar] [CrossRef]
- Prabhakaran, J.; Underwood, M.D.; Parsey, R.V.; Arango, V.; Majo, V.J.; Simpson, N.R.; van Heertum, R.; Mann, J.J.; Kumar, J.S.D. Synthesis and in vivo evaluation of [18F]-4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benzenesulfonamide as a PET imaging probe for COX-2 expression. Bioorg. Med. Chem. 2007, 15, 1802–1807. [Google Scholar] [CrossRef] [PubMed]
- Schilling, B.; Kaufmann, D.E. Organometallics: Boron Compounds. In Science of Synthesis, Houben-Weyl Methods of Molecular Transformations; Kaufmann, D.E., Matteson, D.S., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 2004; Volume 6, pp. 247–256. [Google Scholar]
- Hartman, J.S.; Miller, J.M. Adducts of Mixed Trihalides of Boron. In Advances in Inorganic Chemistry and Radiochemistry; Emeléus, H.J., Sharpe, A.G., Eds.; Academic Press: New York, NY, USA, 1978; Volume 21, pp. 147–177. [Google Scholar]
- Wade, C.R.; Broomsgrove, E.J.; Aldridge, S.; Gabbaï, F.P. Fluoride Ion Complexation and Sensing Using Organoboron Compounds. Chem. Rev. 2010, 110, 3958–3984. [Google Scholar] [CrossRef] [PubMed]
- Treibs, A.; Kreuzer, F.-H. Difluorboryl-Komplexe von Di- und Tripyrrylmethenen. Liebigs Ann. Chem. 1968, 718, 208–223. [Google Scholar] [CrossRef]
- Schmitt, A.; Hinkeldey, B.; Wild, M.; Jung, G. Synthesis of the Core Compound of the BODIPY Dye Class: 4,4-Difluoro-4-bora-(3a,4a)-diaza-s-indacene. J. Fluoresc. 2009, 19, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Tram, K.; Yan, H.; Jenkins, H.A.; Vassiliev, S.; Bruce, D. The synthesis and crystal structure of unsubstituted 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY). Dyes Pigments 2009, 82, 392–395. [Google Scholar] [CrossRef]
- Arroyo, I.J.; Hu, R.; Merino, G.; Tang, B.Z.; Pena-Cabrera, E. The smallest and one of the brightest. Efficient preparation and optical description of the parent borondipyrromethene system. J. Org. Chem. 2009, 74, 5719–5722. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lin, T.-P.; Liu, S.; Huang, C.-W.; Hudnall, T.W.; Gabbaï, F.P.; Conti, P.S. Rapid aqueous [18F]-labeling of a bodipy dye for positron emission tomography/fluorescence dual modality imaging. Chem. Commun. 2011, 47, 9324–9326. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, J.A.; Keliher, E.J.; Wan, D.; Hilderbrand, S.A.; Weissleder, R.; Mazitschek, R. Synthesis of [18F]BODIPY: Bifunctional Reporter for Hybrid Optical/Positron Emission Tomography Imaging. Angew. Chem. 2012, 124, 4681–4684. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuchar, M.; Mamat, C. Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules 2015, 20, 16186-16220. https://doi.org/10.3390/molecules200916186
Kuchar M, Mamat C. Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules. 2015; 20(9):16186-16220. https://doi.org/10.3390/molecules200916186
Chicago/Turabian StyleKuchar, Manuela, and Constantin Mamat. 2015. "Methods to Increase the Metabolic Stability of 18F-Radiotracers" Molecules 20, no. 9: 16186-16220. https://doi.org/10.3390/molecules200916186
APA StyleKuchar, M., & Mamat, C. (2015). Methods to Increase the Metabolic Stability of 18F-Radiotracers. Molecules, 20(9), 16186-16220. https://doi.org/10.3390/molecules200916186