
A Promising PET Tracer for Imaging of α7 Nicotinic Acetylcholine Receptors in the Brain: Design, Synthesis, and in Vivo Evaluation of a Dibenzothiophene-Based Radioligand


 ,
,  , , and
, , and
Abstract
:1. Introduction
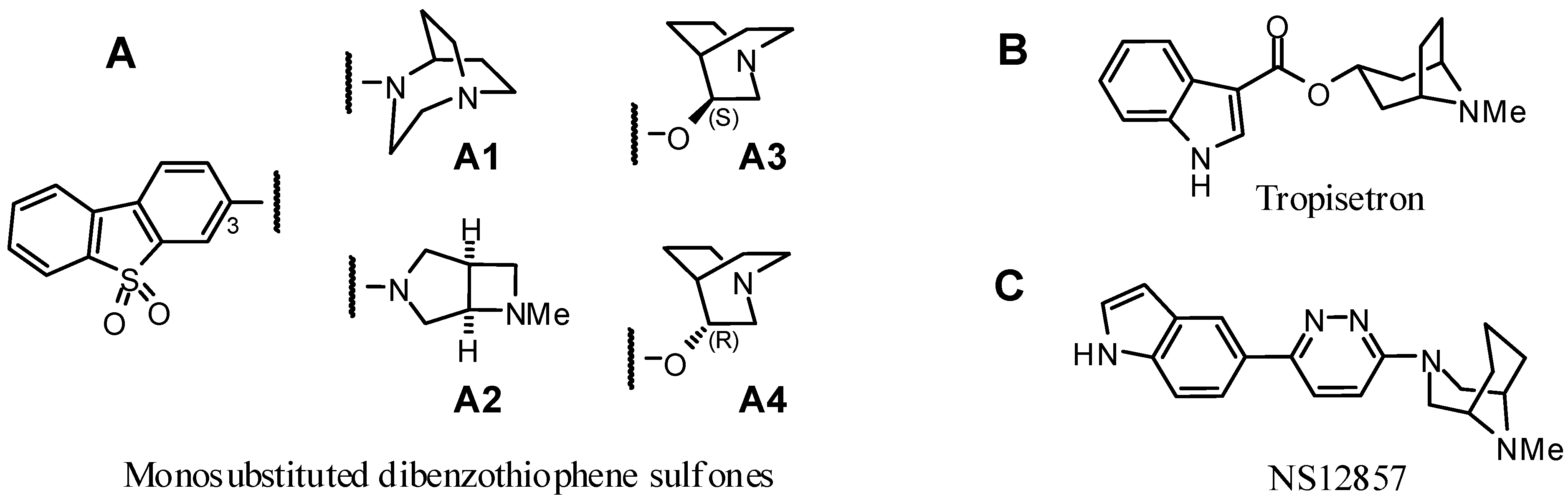
2. Results and Discussion
2.1. Chemistry
2.1.1. Rationale


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | X1 | X2 | X3 |
|---|---|---|---|---|
| 10 |  | H | H | H |
| 10a | | H | F | H |
| 10b | | F | H | H |
| 10c | | H | H | F |
| 12a |  | H | F | H |
| 12b | | F | H | H |
| 13b | 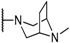 | F | H | H |
| 14b | 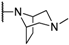 | F | H | H |
2.1.2. Synthesis of α7 nAChR Ligands
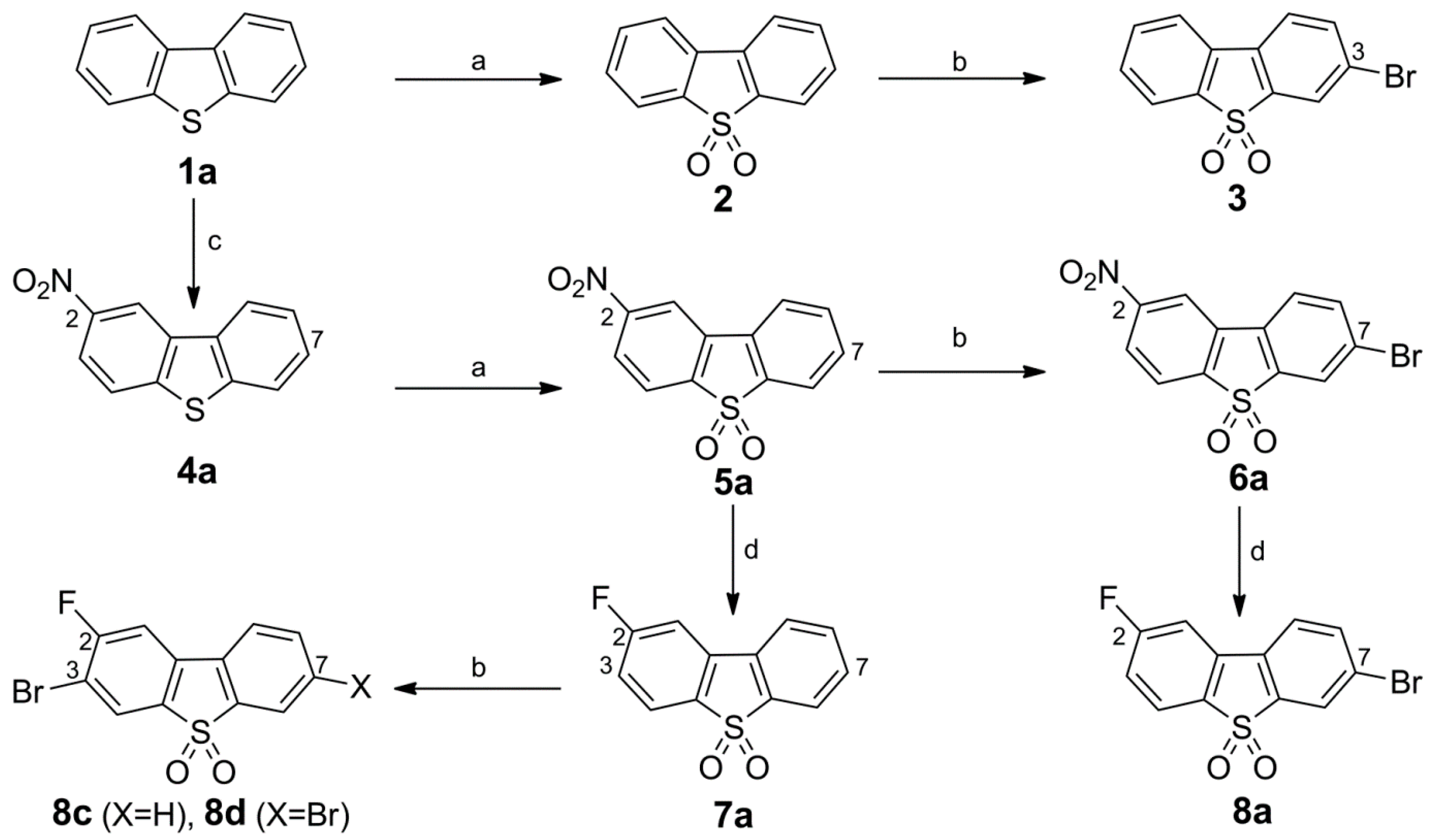
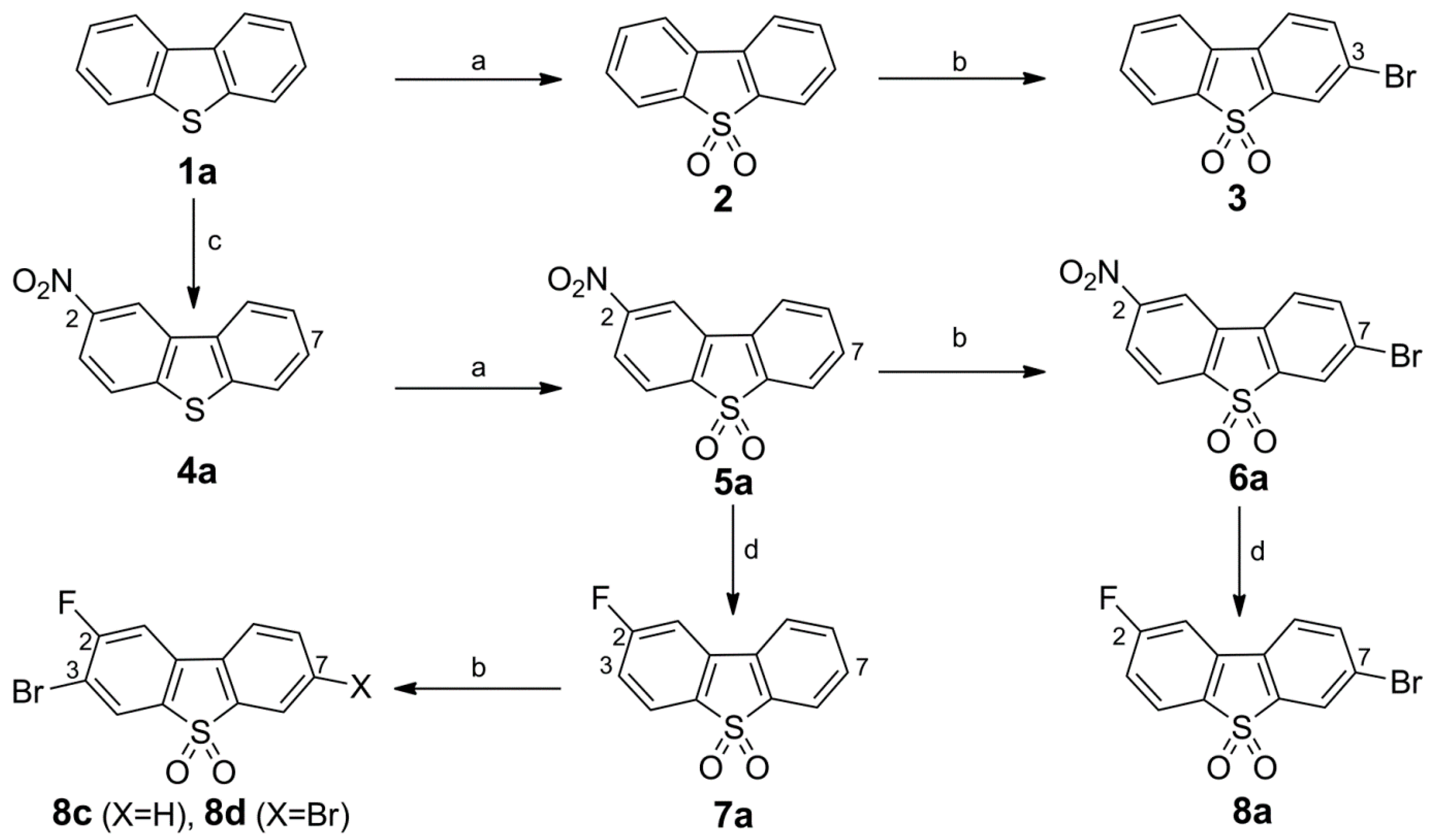
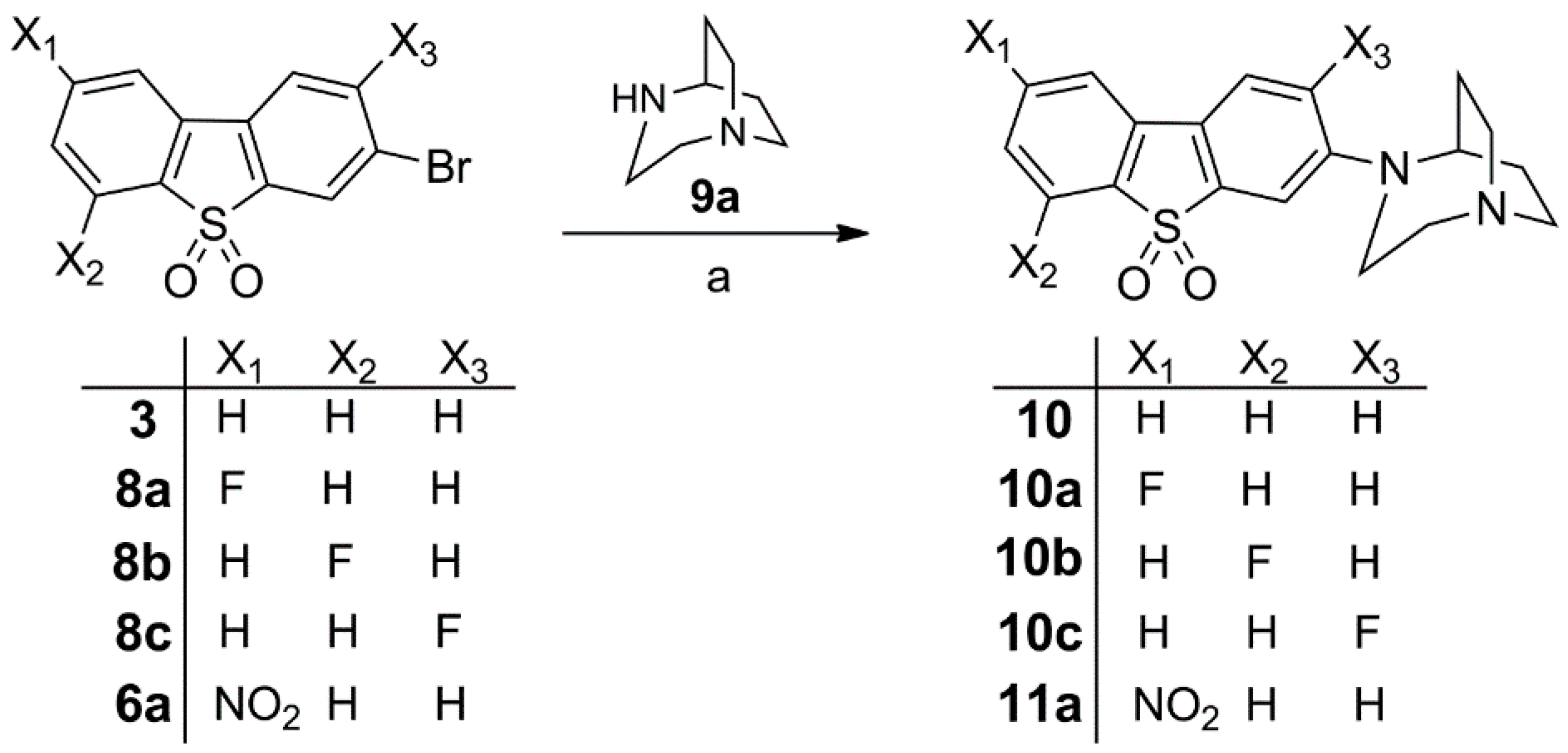
Synthesis of Bromo Dibenzothiophene Intermediates 3, 6a and 8a–8c
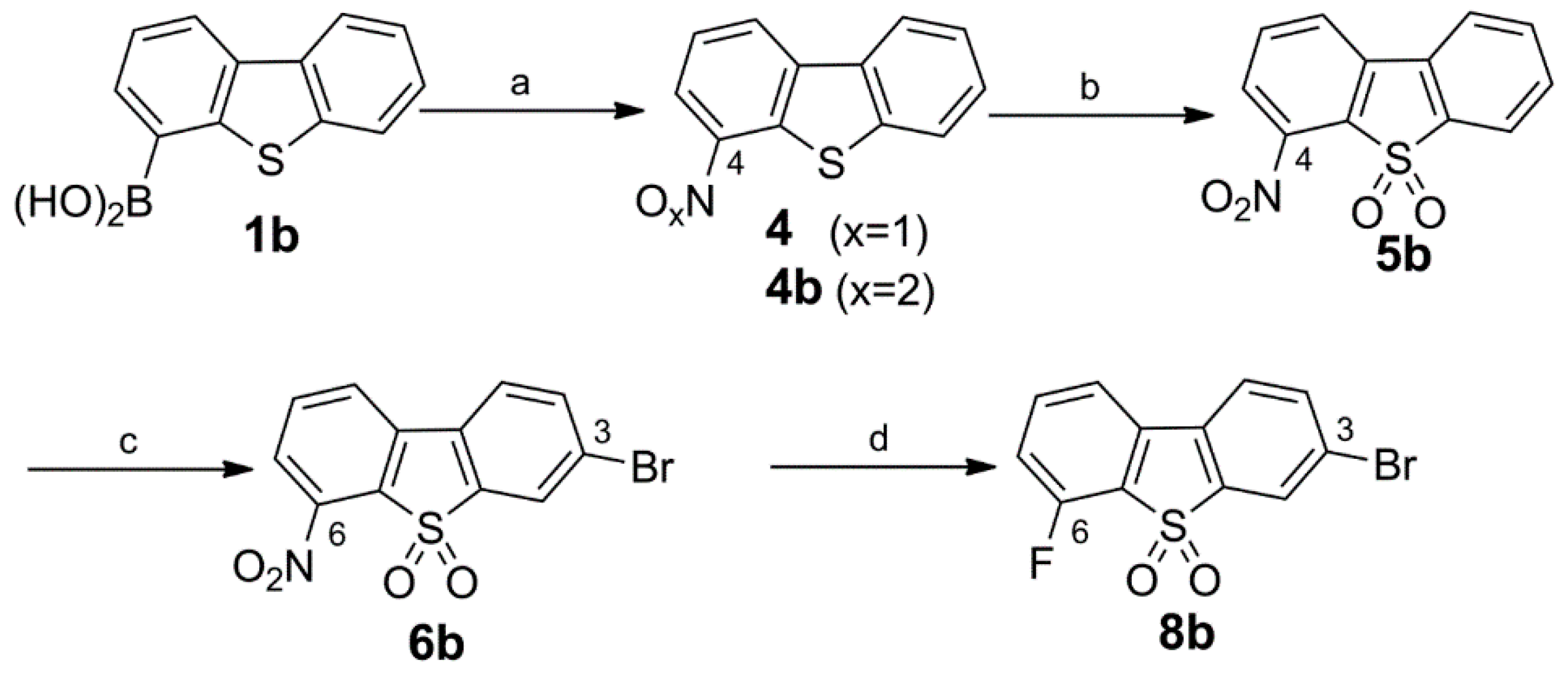
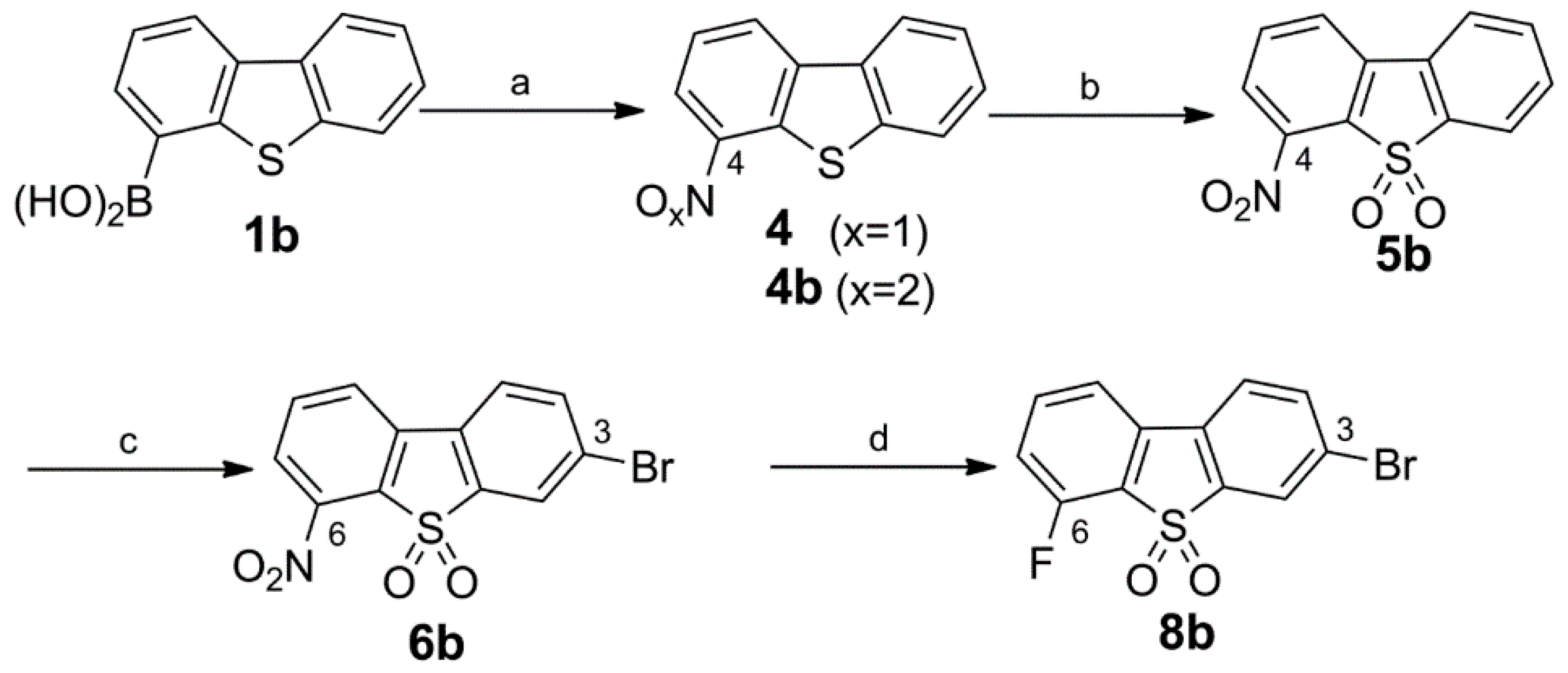
Synthesis of the 3-Bromo Dibenzothiophene Intermediates 6b and 8b
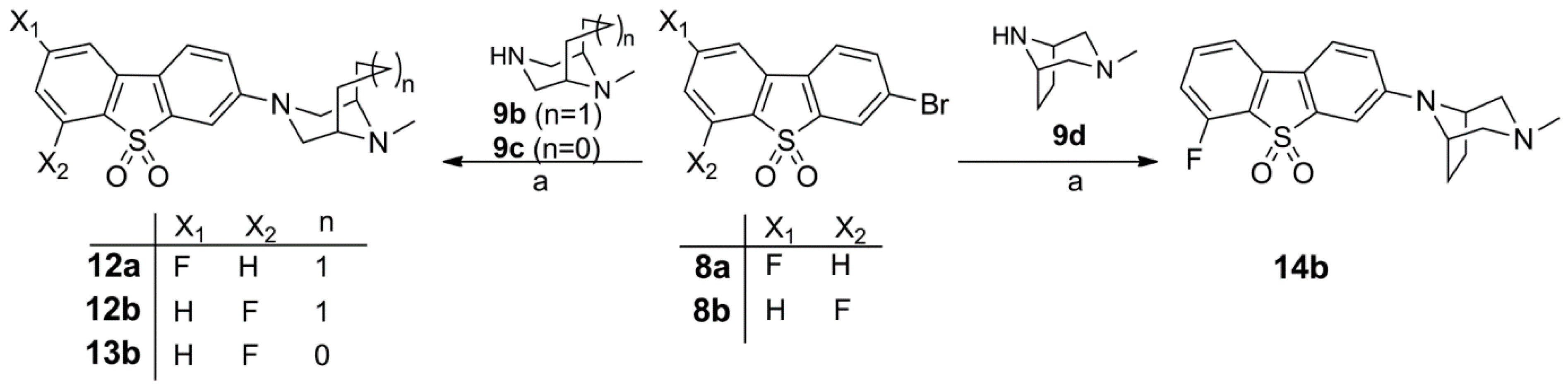
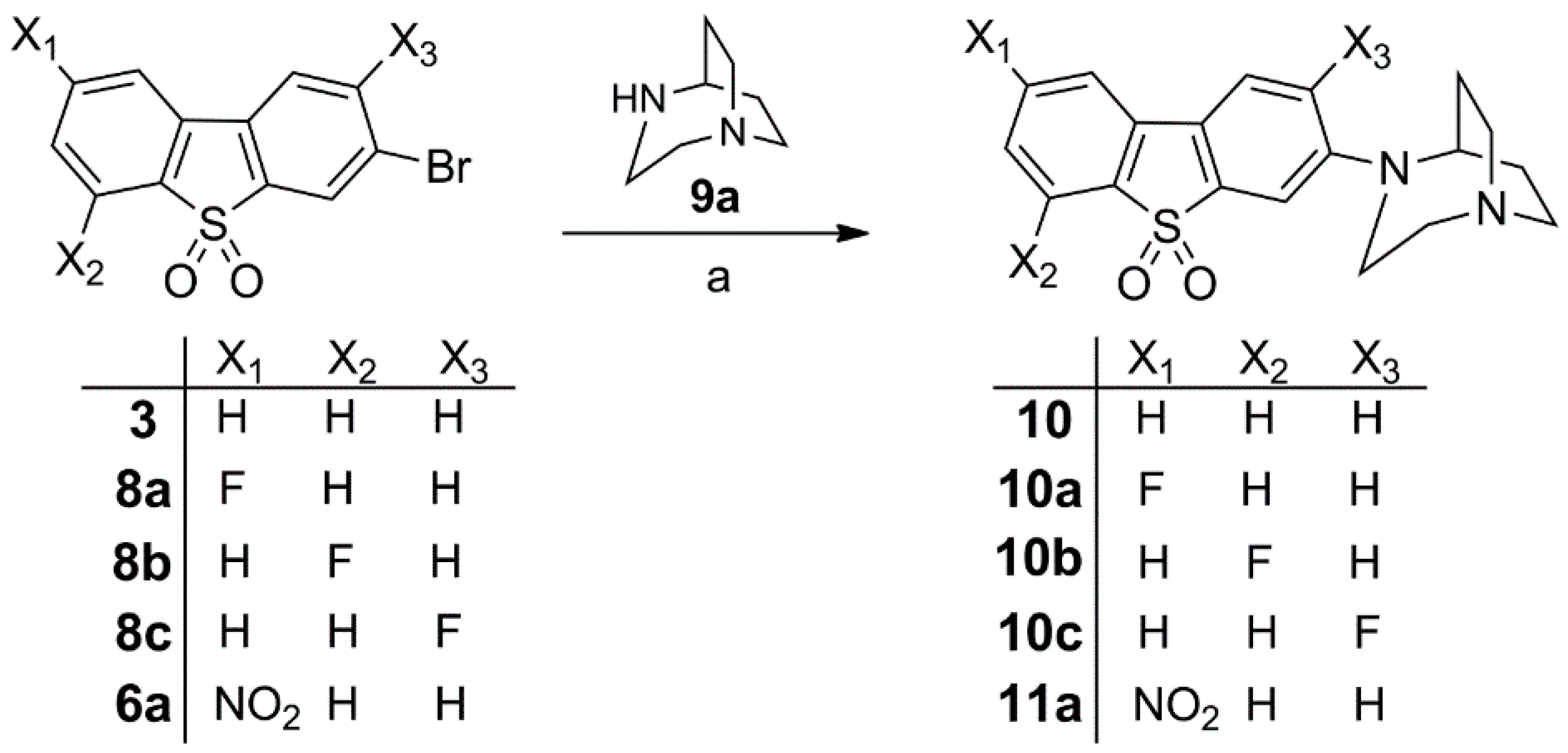
Synthesis of 10, Fluorinated Reference Compounds 10a–c, and Nitro Precursor 11
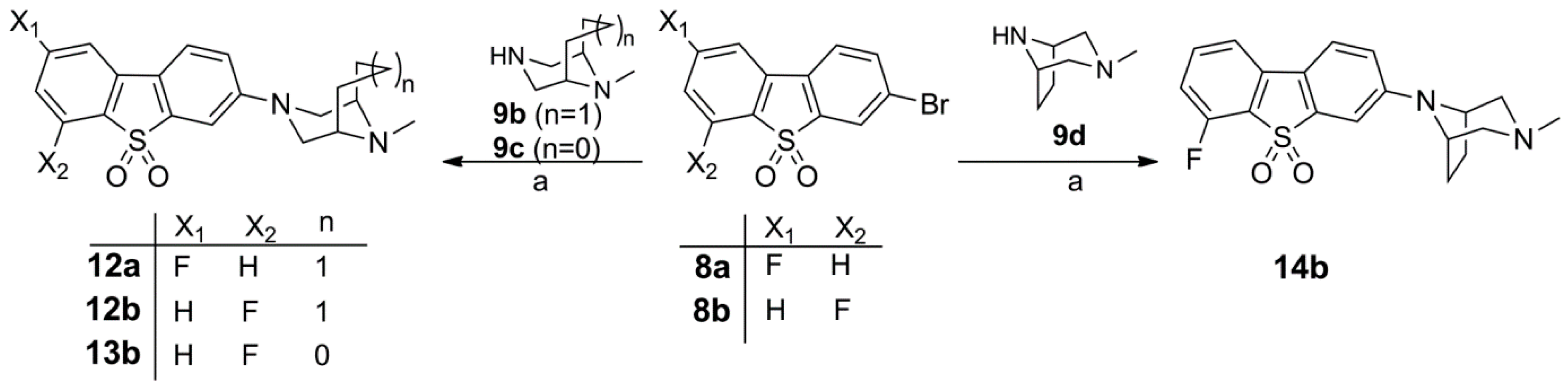
Synthesis of the Fluorinated Reference Compounds 12a,b, 13b, and 14b
2.2. In Vitro Affinity Assays
| Compound | Affinity (Ki in nM) | Selectivity (Ki ratio) | |||||
|---|---|---|---|---|---|---|---|
| nAChR Subtype | 5-HT3 d,e | ||||||
| hα7 a | hα4β2 b | hα3β4 b | rα6β2* c | α7/α4β2 | α7/α3β4 | ||
| 10 | 0.51 ± 0.32 | 318 ± 43.3 | 49.6 ± 14.7 | 517 ± 186 | (35%) | 623 | 97 |
| 10a | 0.60 ± 0.44 | 517 ± 375 | 119 ± 29.0 | 589 ± 217 | 440 (2%) | 862 | 198 |
| 10b | 0.84 ± 0.16 | 211 ± 108 | 42.3 ± 4.73 | 435 ± 152 | (42%) | 251 | 50 |
| 10c | 8.53 ± 1.74 | 507 ± 212 | 279 ± 24.4 | 1390 ± 340 | (26%) | 59 | 33 |
| 12a | 30.9 ± 8.72 | 141 ± 11.7 | 96.0 ± 1.48 | 1180 ± 360 | (10%) | 5 | 3 |
| 12b | 105 ± 23.9 | 301 ± 148 | 94.8 ± 6.48 | 1190 ± 230 | (11%) | 3 | 1 |
| 13b | 40.9 ± 7.77 | 426 ± 197 | 224 ± 51.7 | 2260 ± 540 | (10%) | 10 | 5 |
| 14b | 9.26 ± 2.23 | >4000 | >5000 | 1450 ± 370 | (4%) | >400 | >500 |
2.2.1. Affinity for α7 nAChR
2.2.2. Affinities for α4β2, α3β4, and α6β2 nAChR
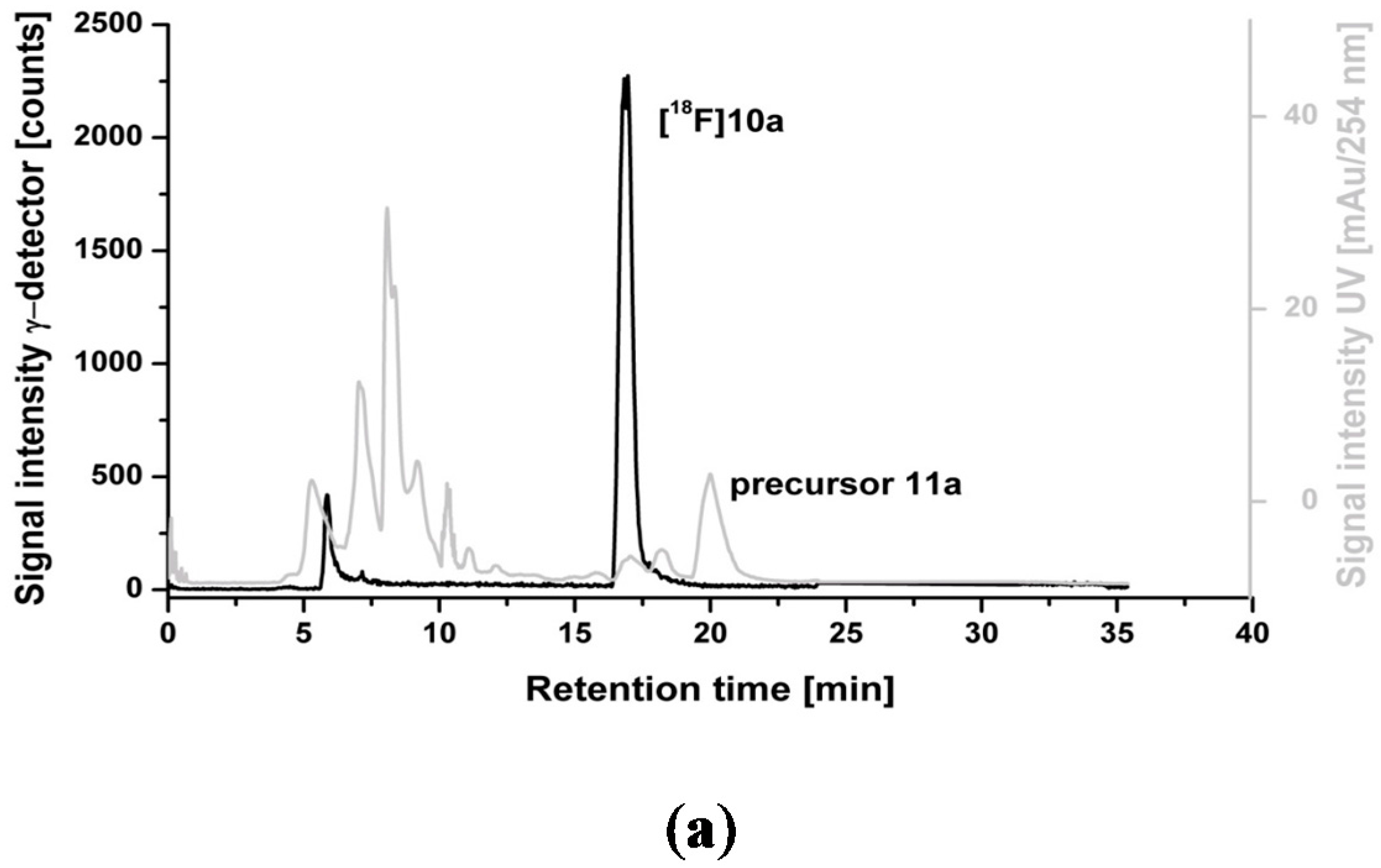
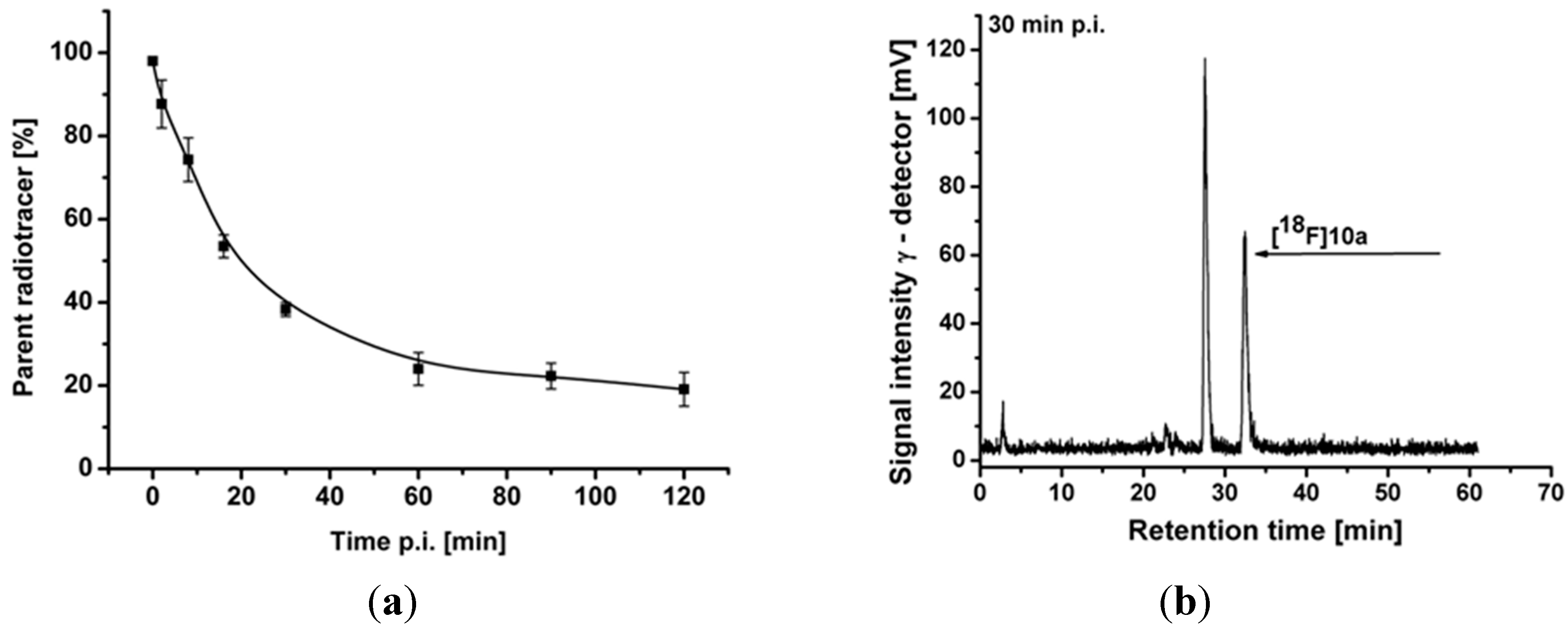
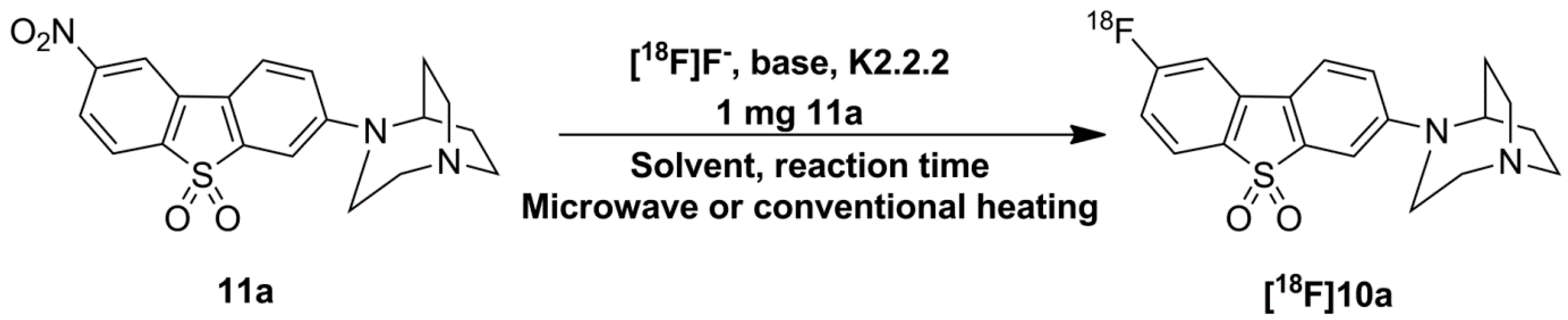
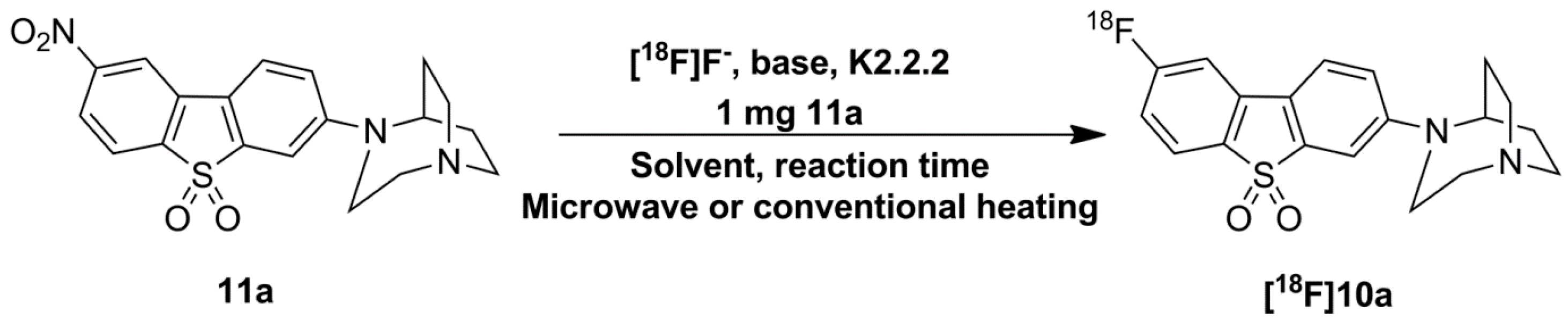
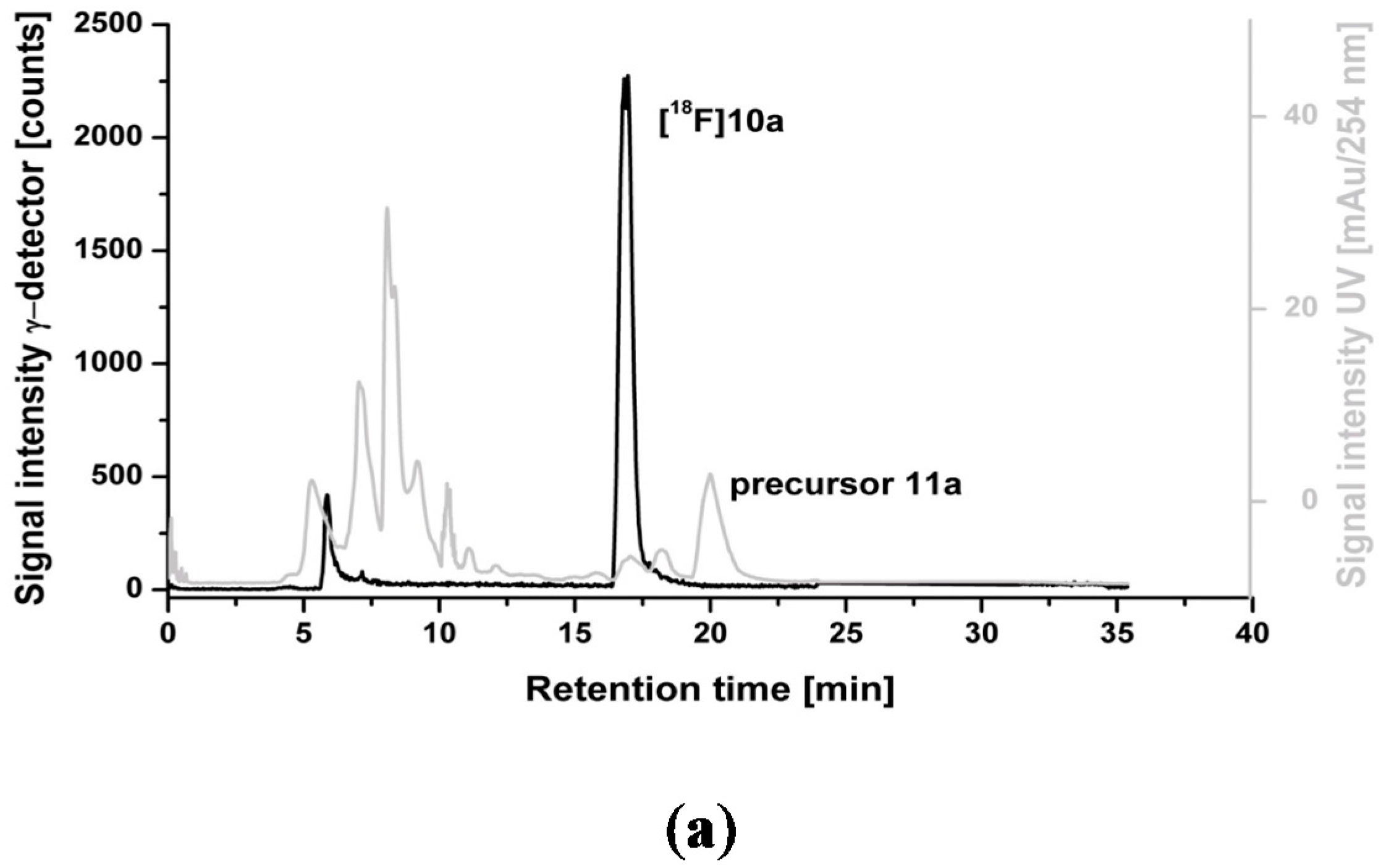
2.3. Optimization of Manual Radiosynthesis of [18F]10a for Translation to an Automated Module

2.4. Dynamic PET Studies of [18F]10a in Piglets
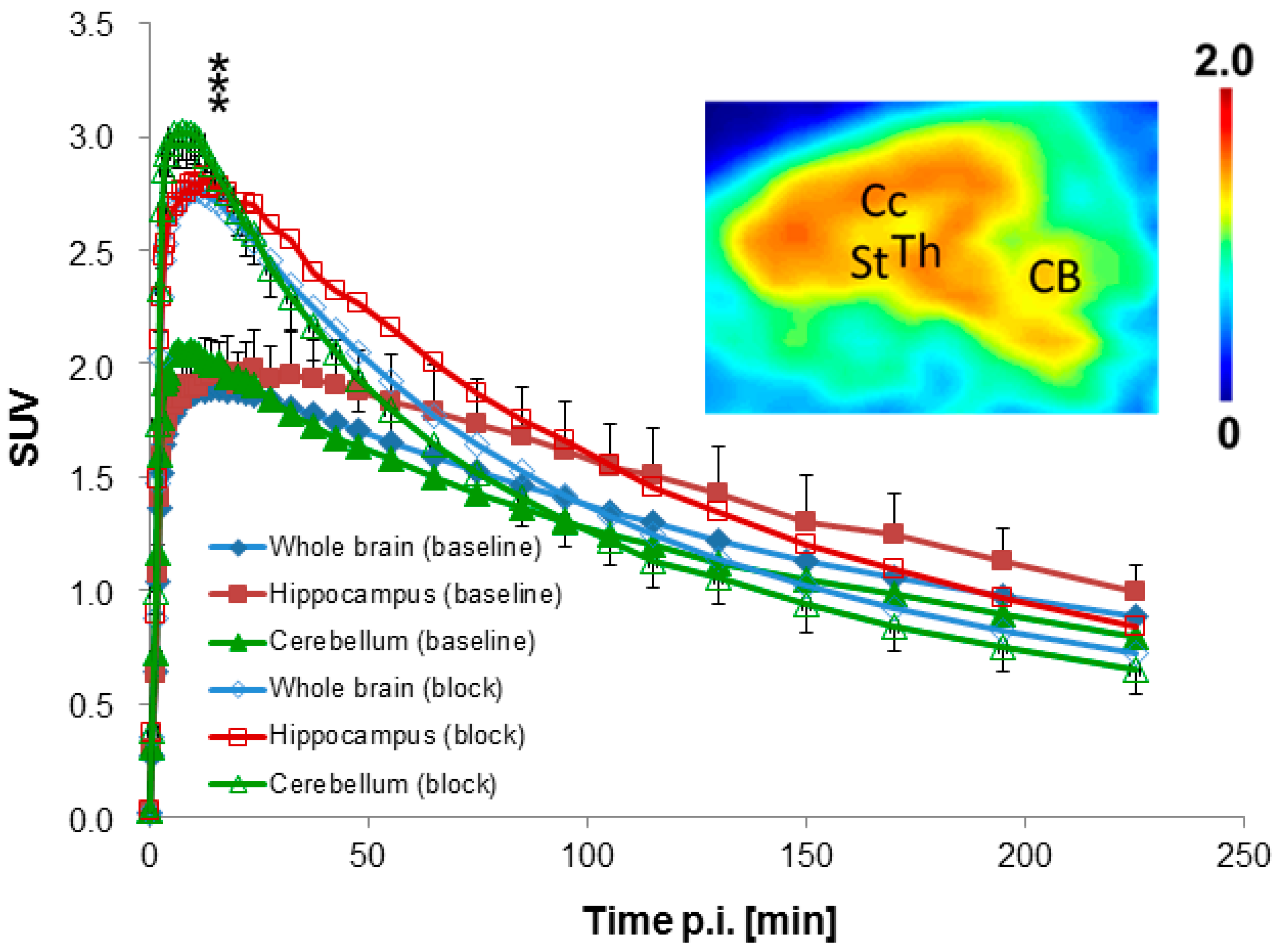
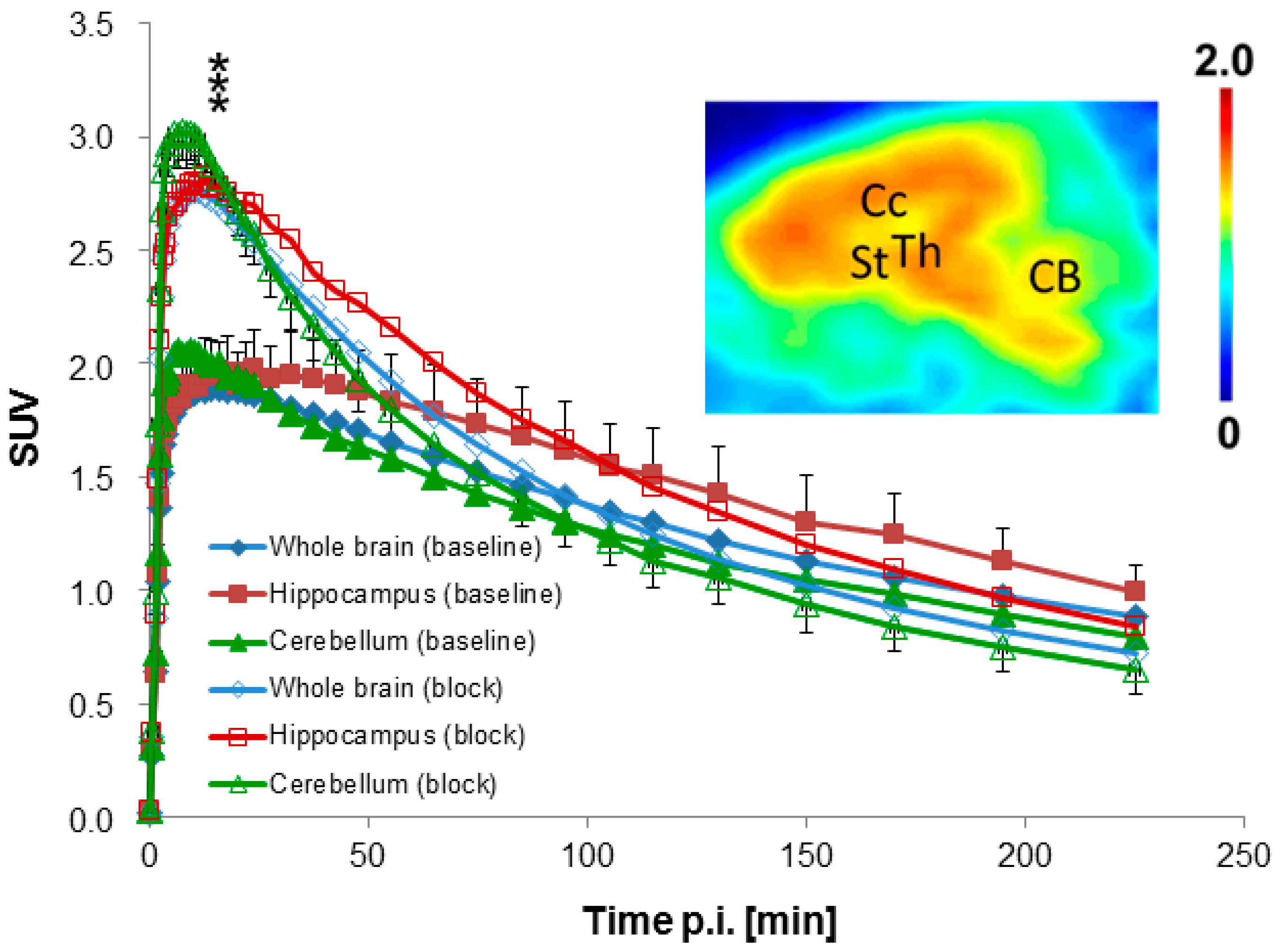
2.4.1. Baseline Studies
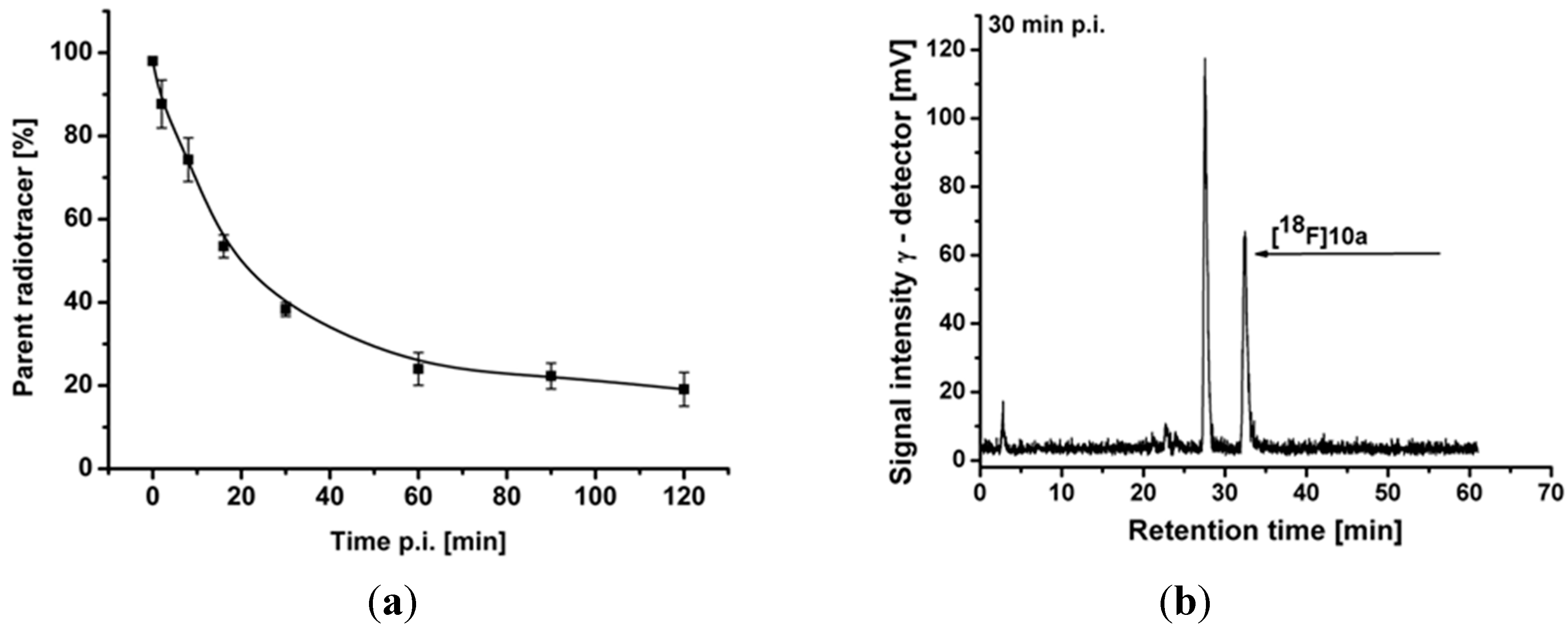
2.4.2. Metabolite Analysis
2.4.3. Blocking Studies
2.4.4. Modeling
| Brain Region | Rate Constant | VND | BPND | |||
|---|---|---|---|---|---|---|
| K1 (mL·cm−3·min−1) | k2 (min−1) | k3 (min−1) | k4 (min−1) | |||
| Whole | 0.362 ± 0.038 | 0.131 ± 0.062 | 0.093 ± 0.074 | 0.027 ± 0.018 | 3.32 ± 1.74 | 3.31 ± 1.02 |
| Frontal Cortex | 0.387 ± 0.059 | 0.182 ± 0.153 | 0.142 ± 0.165 | 0.027 ± 0.015 | 3.32 ± 2.06 | 4.22 ± 2.93 |
| Parietal Cortex | 0.399 ± 0.038 | 0.140 ± 0.089 | 0.126 ± 0.135 | 0.031 ± 0.021 | 3.88 ± 2.49 | 3.47 ± 1.65 |
| Occipital Cortex | 0.395 ± 0.053 | 0.123 ± 0.063 | 0.099 ± 0.087 | 0.030 ± 0.023 | 4.07 ± 2.23 | 3.16 ± 1.11 |
| Hippocampus | 0.400 ± 0.023 | 0.188 ± 0.083 | 0.145 ± 0.101 | 0.025 ± 0.013 | 2.43 ± 0.96 | 5.50 ± 1.01 |
| Striatum | 0.382 ± 0.028 | 0.144 ± 0.079 | 0.114 ± 0.093 | 0.027 ± 0.014 | 3.35 ± 1.81 | 3.98 ± 1.40 |
| Thalamus | 0.448 ± 0.027 | 0.150 ± 0.071 | 0.111 ± 0.085 | 0.030 ± 0.018 | 3.67 ± 2.10 | 3.51 ± 1.13 |
| Colliculi | 0.450 ± 0.072 | 0.134 ± 0.074 | 0.070 ± 0.042 | 0.027 ± 0.018 | 4.19 ± 2.37 | 2.68 ± 1.42 |
| Midbrain | 0.436 ± 0.045 | 0.162 ± 0.117 | 0.121 ± 0.132 | 0.029 ± 0.022 | 4.04 ± 3.06 | 3.40 ± 1.73 |
| Pons | 0.440 ± 0.045 | 0.149 ± 0.059 | 0.068 ± 0.042 | 0.026 ± 0.019 | 3.32 ± 1.40 | 2.76 ± 0.81 |
| Cerebellum | 0.421 ± 0.048 | 0.131 ± 0.052 | 0.067 ± 0.051 | 0.026 ± 0.020 | 3.62 ± 1.47 | 2.53 ± 0.57 |
| Brain Region | Rate Constant | VND | BPND | |||
|---|---|---|---|---|---|---|
| K1 (mL·cm−3·min−1) | k2 (min−1) | k3 (min−1) | k4 (min−1) | |||
| Whole | 0.598 ± 0.108 * | 0.062 ± 0.015 | 0.018 ± 0.022 | 0.011 ± 0.018 | 10.27 ± 3.94 * | 0.84 ± 1.07 * |
| Frontal Cortex | 0.647 ± 0.117 * | 0.055 ± 0.011 | 0.014 ± 0.019 | 0.010 ± 0.020 | 12.34 ± 4.27 * | 0.86 ± 0.92 |
| Parietal Cortex | 0.659 ± 0.120 * | 0.055 ± 0.013 | 0.015 ± 0.020 | 0.011 ± 0.021 | 12.58 ± 4.52 * | 0.82 ± 0.88 * |
| Occipital Cortex | 0.668 ± 0.103 * | 0.064 ± 0.019 | 0.022 ± 0.032 | 0.013 ± 0.023 | 11.36 ± 4.41 * | 0.95 ± 1.03 * |
| Hippocampus | 0.603 ± 0.118 * | 0.063 ± 0.023 * | 0.024 ± 0.030 | 0.011 ± 0.015 | 10.81 ± 5.02 * | 0.84 ± 1.70 * |
| Striatum | 0.624 ± 0.143 * | 0.057 ± 0.015 | 0.020 ± 0.025 | 0.011 ± 0.017 | 11.78 ± 4.91 * | 0.75 ± 1.29 * |
| Thalamus | 0.762 ± 0.129 * | 0.070 ± 0.025 | 0.023 ± 0.031 | 0.012 ± 0.021 | 12.09 ± 5.31 * | 0.88 ± 1.00 * |
| Colliculi | 0.735 ± 0.182 * | 0.066 ± 0.007 | 0.011 ± 0.013 | 0.007 ± 0.015 | 11.41 ± 3.90 * | 0.73 ± 0.74 * |
| Midbrain | 0.685 ± 0.131 * | 0.063 ± 0.010 | 0.013 ± 0.014 | 0.012 ± 0.018 | 11.17 ± 3.64 * | 0.61 ± 0.77 * |
| Pons | 0.670 ± 0.092 * | 0.084 ± 0.030 | 0.019 ± 0.023 | 0.012 ± 0.016 | 8.93 ± 3.96 * | 0.76 ± 1.16 * |
| Cerebellum | 0.711 ± 0.148 * | 0.076 ± 0.014 | 0.016 ± 0.019 | 0.011 ± 0.016 | 9.75 ± 3.57 * | 0.71 ± 1.10 * |
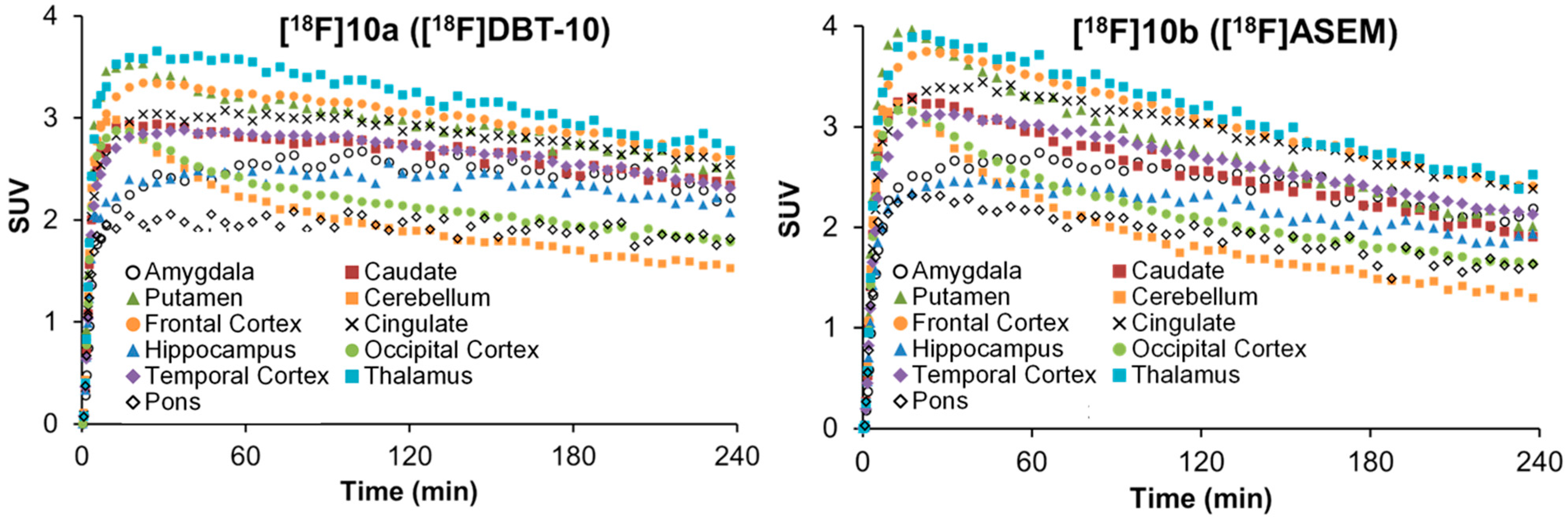
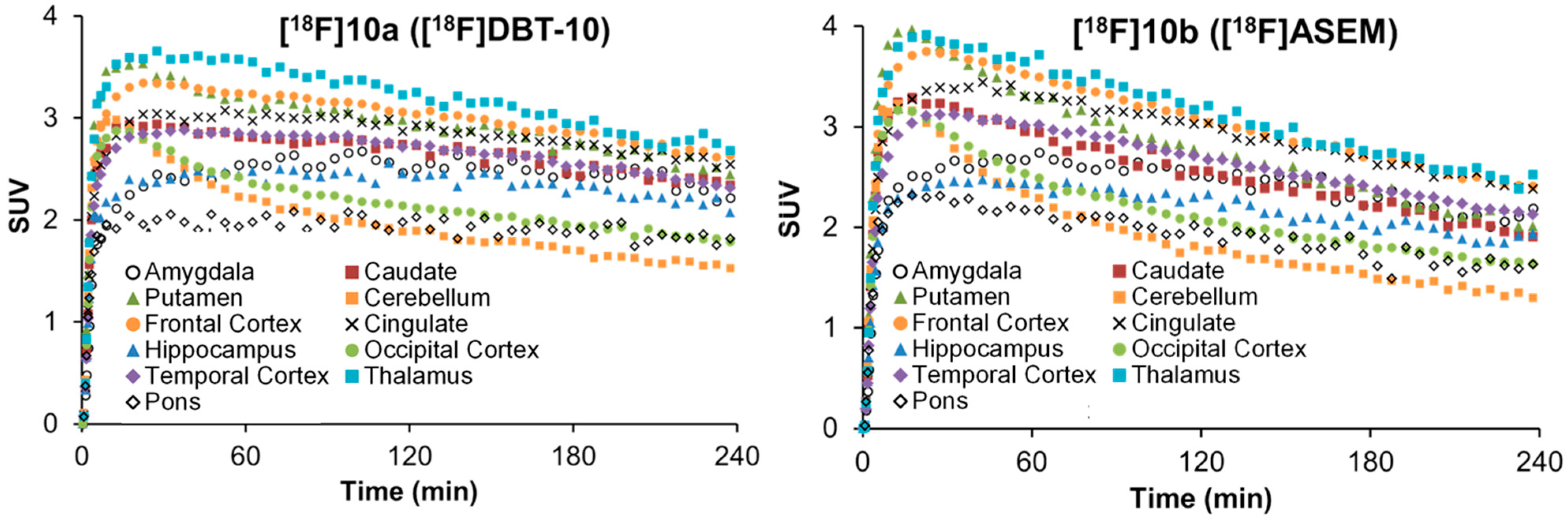
2.5. Comparative PET Study of [18F]10a ([18F]DBT-10) and [18F]10b ([18F]ASEM) in a Rhesus Monkey
2.6. Toxicity Studies in Rats
3. Experimental Section
3.1. General Information
3.2. Chemistry
3.3. Manual Radiosynthesis of [18F]10a
3.4. Automated Radiosynthesis of [18F]10a and [18F]10b ([18F]ASEM)
3.5. Quality Control
3.6. Determination of in Vitro Stability and Lipophilicity (Log D7.2)
3.7. Biological Evaluation
3.7.1. Animals
3.7.2. Binding Assays
Affinity for Human α7 nAChR
Affinity for Human α4β2 and α3β4 nAChR
Affinity for Subunit-Specific Antibodies Immobilized Native Rat α6β2* nAChR
Affinity for Human 5-HT3 Receptor
Affinity for Other Target Proteins
3.7.3. PET Studies in Piglets
PET Scanning Protocol
Arterial Input Function
Quantification of PET Data
3.7.4. PET Study in Rhesus Monkey
3.7.5. Toxicity Studies in Rats
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dineley, K.T.; Pandya, A.A.; Yakel, J.L. Nicotinic ACh receptors as therapeutic targets in CNS disorders. Trends Pharmacol. Sci. 2015, 36, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Weiland, S.; Bertrand, D.; Leonard, S. Neuronal nicotinic acetylcholine receptors: From the gene to the disease. Behav. Brain Res. 2000, 113, 43–56. [Google Scholar] [CrossRef]
- Briggs, C.A.; Gronlien, J.H.; Curzon, P.; Timmermann, D.B.; Ween, H.; Thorin-Hagene, K.; Kerr, P.; Anderson, D.J.; Malysz, J.; Dyhring, T.; et al. Role of channel activation in cognitive enhancement mediated by α7 nicotinic acetylcholine receptors. Br. J. Pharmacol. 2009, 158, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.L. Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol. 2014, 89, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Conti, L.; Roseti, C.; Limatola, C. Novel approaches to study the involvement of α7-nAChR in human diseases. Curr. Drug Targets 2012, 13, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, N.A.; Leonik, F.M.; Papke, R.L. Multiple pharmacophores for the selective activation of nicotinic α7-type acetylcholine receptors. Mol. Pharmacol. 2008, 74, 1496–1511. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, A.; Hauser, T.; Miller, C.H. Selective α7 nicotinic acetylcholine receptor ligands. Curr. Med. Chem. 2006, 13, 1567–1584. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Ding, Y.S.; Alexoff, D.; Patel, V.; Logan, J.; Lin, K.S.; Shea, C.; Muench, L.; Xu, Y.; Carter, P.; et al. Synthesis and positron emission tomography studies of C-11-labeled isotopomers and metabolites of GTS-21, a partial α7 nicotinic cholinergic agonist drug. Nucl. Med. Biol. 2007, 34, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, J.; Ishiwata, K.; Sakata, M.; Wu, J.; Nishiyama, S.; Tsukada, H.; Hashimoto, K. In vivo evaluation of α7 nicotinic acetylcholine receptor agonists [11C]A-582941 and [11C]A-844606 in mice and conscious monkeys. PLoS ONE 2010, 5, e8961. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Nishiyama, S.; Ohba, H.; Matsuo, M.; Kobashi, T.; Takahagi, M.; Iyo, M.; Kitashoji, T.; Tsukada, H. [11C]CHIBA-1001 as a novel PET ligand for α7 nicotinic receptors in the brain: A PET study in conscious monkeys. PLoS ONE 2008, 3, e3231. [Google Scholar] [CrossRef] [PubMed]
- Ettrup, A.; Mikkelsen, J.D.; Lehel, S.; Madsen, J.; Nielsen, E.Ø.; Palner, M.; Timmermann, D.B.; Peters, D.; Knudsen, G.M. 11C-NS14492 as a novel PET radioligand for imaging cerebral α7 nicotinic acetylcholine receptors: In vivo evaluation and drug occupancy measurements. J. Nucl. Med. 2011, 52, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Deuther-Conrad, W.; Fischer, S.; Hiller, A.; Stergaard Nielsen, E.; Brunicardi Timmermann, D.; Steinbach, J.; Sabri, O.; Peters, D.; Brust, P. Molecular imaging of α7 nicotinic acetylcholine receptors: Design and evaluation of the potent radioligand [18F]NS10743. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Rötering, S.; Scheunemann, M.; Fischer, S.; Hiller, A.; Peters, D.; Deuther-Conrad, W.; Brust, P. Radiosynthesis and first evaluation in mice of [18F]NS14490 for molecular imaging of α7 nicotinic acetylcholine receptors. Bioorg. Med. Chem. 2013, 21, 2635–2642. [Google Scholar] [CrossRef] [PubMed]
- Briggs, C.A.; Schrimpf, M.R.; Anderson, D.J.; Gubbins, E.J.; Grønlien, J.H.; Håkerud, M.; Ween, H.; Thorin-Hagene, K.; Malysz, J.; Li, J.; et al. α7 nicotinic acetylcholine receptor agonist properties of tilorone and related tricyclic analogues. Br. J. Pharmacol. 2008, 153, 1054–1061. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, M.R.; Sippy, K.B.; Briggs, C.A.; Anderson, D.J.; Li, T.; Ji, J.; Frost, J.M.; Surowy, C.S.; Bunnelle, W.H.; Gopalakrishnan, M.; et al. SAR of α7 nicotinic receptor agonists derived from tilorone: Exploration of a novel nicotinic pharmacophore. Bioorg. Med. Chem. Lett. 2012, 22, 1633–1638. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, R.; Deuther-Conrad, W.; Rötering, S.; Scheunemann, M.; Patt, M.; Donat, C.K.; Wenzel, B.; Peters, D.; Sabri, O.; Brust, P. Comparative evaluation of two novel fluorine-18 PET radiotracers for the α7 nicotinic acetylcholine receptor. J. Nucl. Med. 2014, 55, 1099. [Google Scholar]
- Gao, Y.; Kellar, K.J.; Yasuda, R.P.; Tran, T.; Xiao, Y.; Dannals, R.F.; Horti, A.G. Derivatives of dibenzothiophene for positron emission tomography imaging of α7-nicotinic acetylcholine receptors. J. Med. Chem. 2013, 56, 7574–7589. [Google Scholar] [CrossRef] [PubMed]
- Horti, A.G.; Gao, Y.; Kuwabara, H.; Wang, Y.; Abazyan, S.; Yasuda, R.P.; Tran, T.; Xiao, Y.; Sahibzada, N.; Holt, D.P.; et al. 18F-ASEM, a radiolabeled antagonist for imaging the α7-nicotinic acetylcholine receptor with PET. J. Nucl. Med. 2014, 55, 672–677. [Google Scholar] [CrossRef] [PubMed]
- Horti, A.G. Development of [18F]ASEM, a specific radiotracer for quantification of the α7-nAChR with positron-emission tomography. Biochem. Pharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wong, D.F.; Kuwabara, H.; Pomper, M.; Holt, D.P.; Brasic, J.R.; George, N.; Frolov, B.; Willis, W.; Gao, Y.; Valentine, H.; et al. Human brain imaging of α7 nAChR with [18F]ASEM: A new PET radiotracer for neuropsychiatry and determination of drug occupancy. Mol. Imaging Biol. 2014, 16, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Deuther-Conrad, W.; Fischer, S.; Hiller, A.; Becker, G.; Cumming, P.; Xiong, G.; Funke, U.; Sabri, O.; Peters, D.; Brust, P. Assessment of α7 nicotinic acetylcholine receptor availability in juvenile pig brain with [18F]NS10743. European J. Nucl. Med. Mol. Imaging 2011, 38, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Toma, L.; Quadrelli, P.; Bunnelle, W.H.; Anderson, D.J.; Meyer, M.D.; Cignarella, G.; Gelain, A.; Barlocco, D. 6-Chloropyridazin-3-yl derivatives active as nicotinic agents: Synthesis, binding, and modeling studies. J. Med. Chem. 2002, 45, 4011–4017. [Google Scholar] [CrossRef] [PubMed]
- Eibl, C.; Tomassoli, I.; Munoz, L.; Stokes, C.; Papke, R.L.; Gündisch, D. The 3,7-diazabicyclo[3.3.1]nonane scaffold for subtype selective nicotinic acetylcholine receptor (nAChR) ligands. Part 1: The influence of different hydrogen bond acceptor systems on alkyl and (hetero)aryl substituents. Bioorg. Med. Chem. 2013, 21, 7283–7308. [Google Scholar] [CrossRef] [PubMed]
- Papke, R.L.; Schiff, H.C.; Jack, B.A.; Horenstein, N.A. Molecular dissection of tropisetron, an α7 nicotinic acetylcholine receptor-selective partial agonist. Neurosci. Lett. 2005, 378, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Lehel, S.; Madsen, J.; Ettrup, A.; Mikkelsen, J.D.; Timmermann, D.B.; Peters, D.; Knudsen, G.M. [11C]NS-12857: A novel PET ligand for α7-nicotinergic receptors. J. Labelled Compd. Rad. 2009, 52, S379. [Google Scholar]
- O’Donnell, C.J.; Peng, L.; O’Neill, B.T.; Arnold, E.P.; Mather, R.J.; Sands, S.B.; Shrikhande, A.; Lebel, L.A.; Spracklin, D.K.; Nedza, F.M. Synthesis and SAR studies of 1,4-diazabicyclo[3.2.2]nonane phenyl carbamates—Subtype selective, high affinity α7 nicotinic acetylcholine receptor agonists. Bioorg. Med. Chem. Lett. 2009, 19, 4747–4751. [Google Scholar] [CrossRef] [PubMed]
- Slowinski, F.; Ben Ayad, O.; Vache, J.; Saady, M.; Leclerc, O.; Lochead, A. Synthesis of bridgehead-substituted azabicyclo[2.2.1]heptane and -[3.3.1]nonane derivatives for the elaboration of α7 nicotinic ligands. J. Org. Chem. 2011, 76, 8336–8346. [Google Scholar] [CrossRef] [PubMed]
- Wawzonek, S.; Thelen, P.J. Preparation of N-methylgranatanine. J. Am. Chem. Soc. 1950, 72, 2118–2120. [Google Scholar] [CrossRef]
- Lambert, F.L.; Ellis, W.D.; Parry, R.J. Halogenation of aromatic compounds by N-bromo- and N-chlorosuccinimide under ionic conditions. J. Org. Chem. 1965, 30, 304–306. [Google Scholar] [CrossRef]
- Butts, C.P.; Eberson, L.; Hartshorn, M.P.; Radner, F.; Robinson, W.T.; Wood, B.R. Regiochemistry of the reaction between dibenzothiophene radical cation and nucleophiles or nitrogen dioxide. Acta Chem. Scand. 1997, 51, 839–848. [Google Scholar] [CrossRef]
- Gilman, H.; Nobis, J.F. Some aminodibenzothiophenes. J. Am. Chem. Soc. 1949, 71, 274–276. [Google Scholar] [CrossRef]
- Cullinane, N.M.; Davies, C.G.; Davies, G.I. Substitution derivatives of diphenylene sulphide and diphenylenesulphone. J. Chem. Soc. 1936, 1435–1437. [Google Scholar] [CrossRef]
- Adams, D.J.; Clark, J.H. Nucleophilic routes to selectively fluorinated aromatics. Chem. Soc. Rev. 1999, 28, 225–231. [Google Scholar] [CrossRef]
- Boechat, N.; Clark, J.H. Fluorodenitrations using tetramethylammonium fluoride. J. Chem. Soc. Chem. Commun. 1993, 11, 921–922. [Google Scholar] [CrossRef]
- Manna, S.; Maity, S.; Rana, S.; Agasti, S.; Maiti, D. Ipso-nitration of arylboronic acids with bismuth nitrate and perdisulfate. Org. Lett. 2012, 14, 1736–1739. [Google Scholar] [CrossRef] [PubMed]
- Molander, G.A.; Cavalcanti, L.N. Nitrosation of aryl and heteroaryltrifluoroborates with nitrosonium tetrafluoroborate. J. Org. Chem. 2012, 77, 4402–4413. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.F.; Schranck, J.; Neumann, H.; Beller, M. Convenient and mild synthesis of nitroarenes by metal-free nitration of arylboronic acids. Chem. Commun. 2011, 47, 12462–12463. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Hammond, P.S.; Mazurov, A.A.; Yohannes, D. Multiple interaction regions in the orthosteric ligand binding domain of the α7 neuronal nicotinic acetylcholine receptor. J. Chem. Inf. Model. 2012, 52, 3064–3073. [Google Scholar] [CrossRef] [PubMed]
- Brunzell, D.H.; McIntosh, J.M.; Papke, R.L. Diverse strategies targeting α7 homomeric and α6β2* heteromeric nicotinic acetylcholine receptors for smoking cessation. Ann. N. Y. Acad. Sci. 2006, 1327, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Baddick, C.G.; Marks, M.J. An autoradiographic survey of mouse brain nicotinic acetylcholine receptors defined by null mutants. Biochem. Pharmacol. 2011, 82, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Guiducci, S.; Tedesco, V.; Corbioli, S.; Zanetti, L.; Moretti, M.; Zanardi, A.; Rimondini, R.; Mugnaini, M.; Clementi, F.; et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: Primary role of ventral tegmental area α6β2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J. Neurosci. 2010, 30, 5311–5325. [Google Scholar] [CrossRef] [PubMed]
- Quik, M.; Polonskaya, Y.; Gillespie, A.; Jakowec, M.; Lloyd, G.K.; Langston, J.W. Localization of nicotinic receptor subunit mRNAs in monkey brain by in situ hybridization. J. Comp. Neurol. 2000, 425, 58–69. [Google Scholar] [CrossRef]
- Han, Z.Y.; Le Novere, N.; Zoli, M.; Hill, J.A., Jr.; Champtiaux, N.; Changeux, J.P. Localization of nAChR subunit mRNAs in the brain of Macaca mulatta. European J. Neurosci. 2000, 12, 3664–3674. [Google Scholar] [CrossRef]
- Exley, R.; Maubourguet, N.; David, V.; Eddine, R.; Evrard, A.; Pons, S.; Marti, F.; Threlfell, S.; Cazala, P.; McIntosh, J.M.; et al. Distinct contributions of nicotinic acetylcholine receptor subunit α4 and subunit α6 to the reinforcing effects of nicotine. Proc. Natl. Acad. Sci. USA 2011, 108, 7577–7582. [Google Scholar] [CrossRef] [PubMed]
- Faure, P.; Tolu, S.; Valverde, S.; Naudé, J. Role of nicotinic acetylcholine receptors in regulating dopamine neuron activity. Neuroscience 2014, 282, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Marks, M.J.; Millar, N.S.; Wonnacott, S. Nicotinic acetylcholine receptors: α6. Available online: http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId = 467&familyId=76&familyType=IC (accessed on 2 October 2015).
- Le Novere, N.; Grutter, T.; Changeux, J.P. Models of the extracellular domain of the nicotinic receptors and of agonist- and Ca2+-binding sites. Proc. Natl. Acad. Sci. USA 2002, 99, 3210–3215. [Google Scholar] [CrossRef] [PubMed]
- Ding, M.; Ghanekar, S.; Elmore, C.S.; Zysk, J.R.; Werkheiser, J.L.; Lee, C.M.; Liu, J.; Chhajlani, V.; Maier, D.L. [3H]Chiba-1001(methyl-SSR180711) has low in vitro binding affinity and poor in vivo selectivity to nicotinic alpha-7 receptor in rodent brain. Synapse 2012, 66, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, A.A.; Kombo, D.C.; Akireddy, S.; Murthy, S.; Hauser, T.A.; Jordan, K.G.; Gatto, G.J.; Yohannes, D. Novel nicotinic acetylcholine receptor agonists containing carbonyl moiety as a hydrogen bond acceptor. Bioorg. Med. Chem. Lett. 2013, 23, 3927–3934. [Google Scholar] [CrossRef] [PubMed]
- Girard, B.M.; Merriam, L.A.; Tompkins, J.D.; Vizzard, M.A.; Parsons, R.L. Decrease in neuronal nicotinic acetylcholine receptor subunit and PSD-93 transcript levels in the male mouse MPG after cavernous nerve injury or explant culture. Am. J. Physiol. 2013, 305, F1504–F1512. [Google Scholar] [CrossRef] [PubMed]
- Glushakov, A.V.; Voytenko, L.P.; Skok, M.V.; Skok, V. Distribution of neuronal nicotinic acetylcholine receptors containing different alpha-subunits in the submucosal plexus of the guinea-pig. Auton. Neurosci. Basic Clin. 2004, 110, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Teodoro, R.; Wenzel, B.; Oh-Nishi, A.; Fischer, S.; Peters, D.; Suhara, T.; Deuther-Conrad, W.; Brust, P. A high-yield automated radiosynthesis of the alpha-7 nicotinic receptor radioligand [18F]NS10743. Appl. Radiat. Isot. 2015, 95, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, O.; Chen, X. PET designated flouride-18 production and chemistry. Curr. Top. Med. Chem. 2010, 10, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Ravert, H.T.; Holt, D.P.; Gao, Y.; Horti, A.G.; Dannals, R.F. Microwave-assisted radiosynthesis of [18F]ASEM, a radiolabeled α7-nicotinic acetylcholine receptor antagonist. J. Labelled Compd. Radiopharm. 2015, 58, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Angiogenic activity of nicotinic acetylcholine receptors: Implications in tobacco-related vascular diseases. Pharmacol. Ther. 2009, 121, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Cooke, J.P.; Bitterman, H. Nicotine and angiogenesis: A new paradigm for tobacco-related diseases. Ann. Med. 2004, 36, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Pena, V.B.; Bonini, I.C.; Antollini, S.S.; Kobayashi, T.; Barrantes, F.J. α7-Type acetylcholine receptor localization and its modulation by nicotine and cholesterol in vascular endothelial cells. J. Cell. Biochem. 2011, 112, 3276–3288. [Google Scholar] [CrossRef] [PubMed]
- Innis, R.B.; Cunningham, V.J.; Delforge, J.; Fujita, M.; Gjedde, A.; Gunn, R.N.; Holden, J.; Houle, S.; Huang, S.C.; Ichise, M.; et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J. Cereb. Blood Flow Metab. 2007, 27, 1533–1539. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, V.J.; Rabiner, E.A.; Slifstein, M.; Laruelle, M.; Gunn, R.N. Measuring drug occupancy in the absence of a reference region: The Lassen plot re-visited. J. Cereb. Blood Flow Metab. 2010, 30, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Brust, P.; Peters, D.; Deuther-Conrad, W. Development of radioligands for the imaging of α7 nicotinic acetylcholine receptors with positron emission tomography. Curr. Drug Targets 2012, 13, 594–601. [Google Scholar] [CrossRef] [PubMed]
- Harwood, L.M. “Dry-column” flash chromatography. Aldrichimica Acta 1985, 18, 25. [Google Scholar]
- Schrimpf, M.; Sippy, K.; Ji, J.; Li, T.; Frost, J.; Briggs, C.; Bunnelle, W. Amino-Substituted Tricyclic Derivatives and Methods of Use. U.S. Patent 20,050,234,031 A1, 20 October 2005. [Google Scholar]
- Rötering, S.; Franke, K.; Zessin, J.; Brust, P.; Füchtner, F.; Fischer, S.; Steinbach, J. Convenient recycling and reuse of bombarded [18O]H2O for the production and the application of [18F]F. Appl. Radiat. Isot. 2015, 101, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Charpantier, E.; Wiesner, A.; Huh, K.H.; Ogier, R.; Hoda, J.C.; Allaman, G.; Raggenbass, M.; Feuerbach, D.; Bertrand, D.; Fuhrer, C. α7 neuronal nicotinic acetylcholine receptors are negatively regulated by tyrosine phosphorylation and Src-family kinases. J. Neurosci. 2005, 25, 9836–9849. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Michelmore, S.; Croskery, K.; Nozulak, J.; Hoyer, D.; Longato, R.; Weber, A.; Bouhelal, R.; Feuerbach, D. Study of the calcium dynamics of the human α4β2, α3β4 and α1β1γδ nicotinic acetylcholine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2002, 366, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Grady, S.R.; Moretti, M.; Zoli, M.; Marks, M.J.; Zanardi, A.; Pucci, L.; Clementi, F.; Gotti, C. Rodent habenulo-interpeduncular pathway expresses a large variety of uncommon nAChR subtypes, but only the αβ4* and αβ3β4* subtypes mediate acetylcholine release. J. Neurosci. 2009, 29, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- Pucci, L.; Grazioso, G.; Dallanoce, C.; Rizzi, L.; De Micheli, C.; Clementi, F.; Bertrand, S.; Bertrand, D.; Longhi, R.; De Amici, M.; et al. Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. FASEB J. 2011, 25, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Munson, P.J.; Rodbard, D. Ligand: A versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 1980, 107, 220–239. [Google Scholar] [CrossRef]
- Brust, P.; Patt, J.T.; Deuther-Conrad, W.; Becker, G.; Patt, M.; Schildan, A.; Sorger, D.; Kendziorra, K.; Meyer, P.; Steinbach, J.; et al. In vivo measurement of nicotinic acetylcholine receptors with [18F]norchloro-fluoro-homoepibatidine. Synapse 2008, 62, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Gillings, N.M.; Bender, D.; Falborg, L.; Marthi, K.; Munk, O.L.; Cumming, P. Kinetics of the metabolism of four PET radioligands in living minipigs. Nucl. Med. Biol. 2001, 28, 97–104. [Google Scholar] [CrossRef]
- Brust, P.; Zessin, J.; Kuwabara, H.; Pawelke, B.; Kretzschmar, M.; Hinz, R.; Bergman, J.; Eskola, O.; Solin, O.; Steinbach, J.; et al. Positron emission tomography imaging of the serotonin transporter in the pig brain using [11C](+)-McN5652 and S-[18F]fluoromethyl-(+)-McN5652. Synapse 2003, 47, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Brust, P.; Hinz, R.; Kuwabara, H.; Hesse, S.; Zessin, J.; Pawelke, B.; Stephan, H.; Bergmann, R.; Steinbach, J.; Sabri, O. In vivo measurement of the serotonin transporter with (S)-([18F]fluoromethyl)-(+)-McN5652. Neuropsychopharmacology 2003, 28, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 10a,b and 14b are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teodoro, R.; Scheunemann, M.; Deuther-Conrad, W.; Wenzel, B.; Fasoli, F.M.; Gotti, C.; Kranz, M.; Donat, C.K.; Patt, M.; Hillmer, A.; et al. A Promising PET Tracer for Imaging of α7 Nicotinic Acetylcholine Receptors in the Brain: Design, Synthesis, and in Vivo Evaluation of a Dibenzothiophene-Based Radioligand. Molecules 2015, 20, 18387-18421. https://doi.org/10.3390/molecules201018387
Teodoro R, Scheunemann M, Deuther-Conrad W, Wenzel B, Fasoli FM, Gotti C, Kranz M, Donat CK, Patt M, Hillmer A, et al. A Promising PET Tracer for Imaging of α7 Nicotinic Acetylcholine Receptors in the Brain: Design, Synthesis, and in Vivo Evaluation of a Dibenzothiophene-Based Radioligand. Molecules. 2015; 20(10):18387-18421. https://doi.org/10.3390/molecules201018387
Chicago/Turabian StyleTeodoro, Rodrigo, Matthias Scheunemann, Winnie Deuther-Conrad, Barbara Wenzel, Francesca Maria Fasoli, Cecilia Gotti, Mathias Kranz, Cornelius K. Donat, Marianne Patt, Ansel Hillmer, and et al. 2015. "A Promising PET Tracer for Imaging of α7 Nicotinic Acetylcholine Receptors in the Brain: Design, Synthesis, and in Vivo Evaluation of a Dibenzothiophene-Based Radioligand" Molecules 20, no. 10: 18387-18421. https://doi.org/10.3390/molecules201018387
APA StyleTeodoro, R., Scheunemann, M., Deuther-Conrad, W., Wenzel, B., Fasoli, F. M., Gotti, C., Kranz, M., Donat, C. K., Patt, M., Hillmer, A., Zheng, M.-Q., Peters, D., Steinbach, J., Sabri, O., Huang, Y., & Brust, P. (2015). A Promising PET Tracer for Imaging of α7 Nicotinic Acetylcholine Receptors in the Brain: Design, Synthesis, and in Vivo Evaluation of a Dibenzothiophene-Based Radioligand. Molecules, 20(10), 18387-18421. https://doi.org/10.3390/molecules201018387