
Iminoiodane- and Brønsted Base-Mediated Cross Dehydrogenative Coupling of Cyclic Ethers with 1,3-Dicarbonyl Compounds


 ,
, 
Abstract
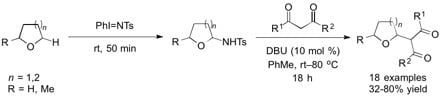
:
1. Introduction

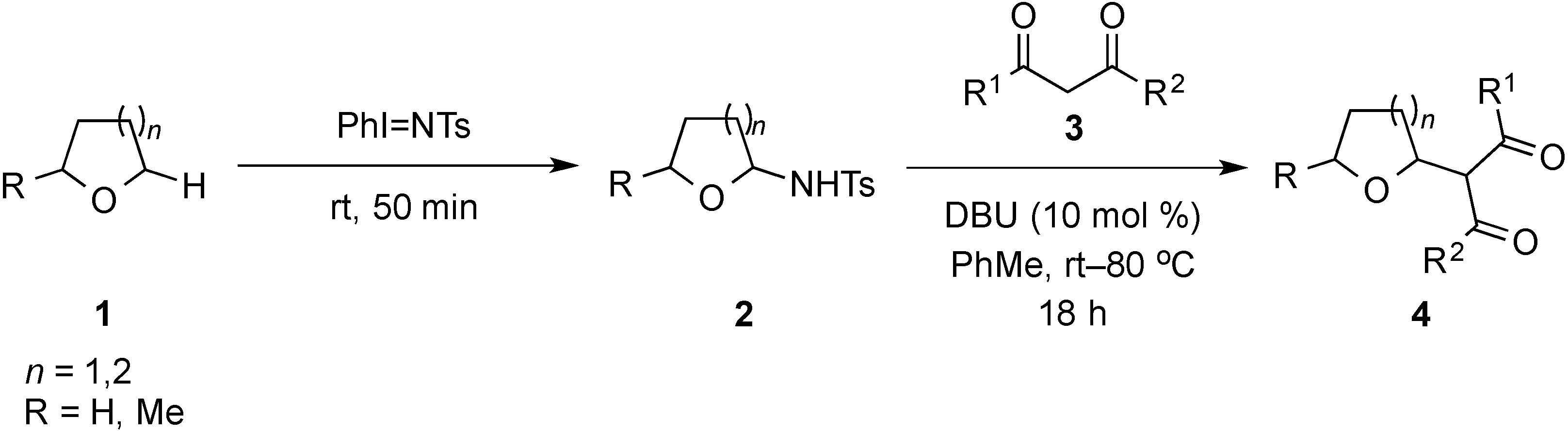
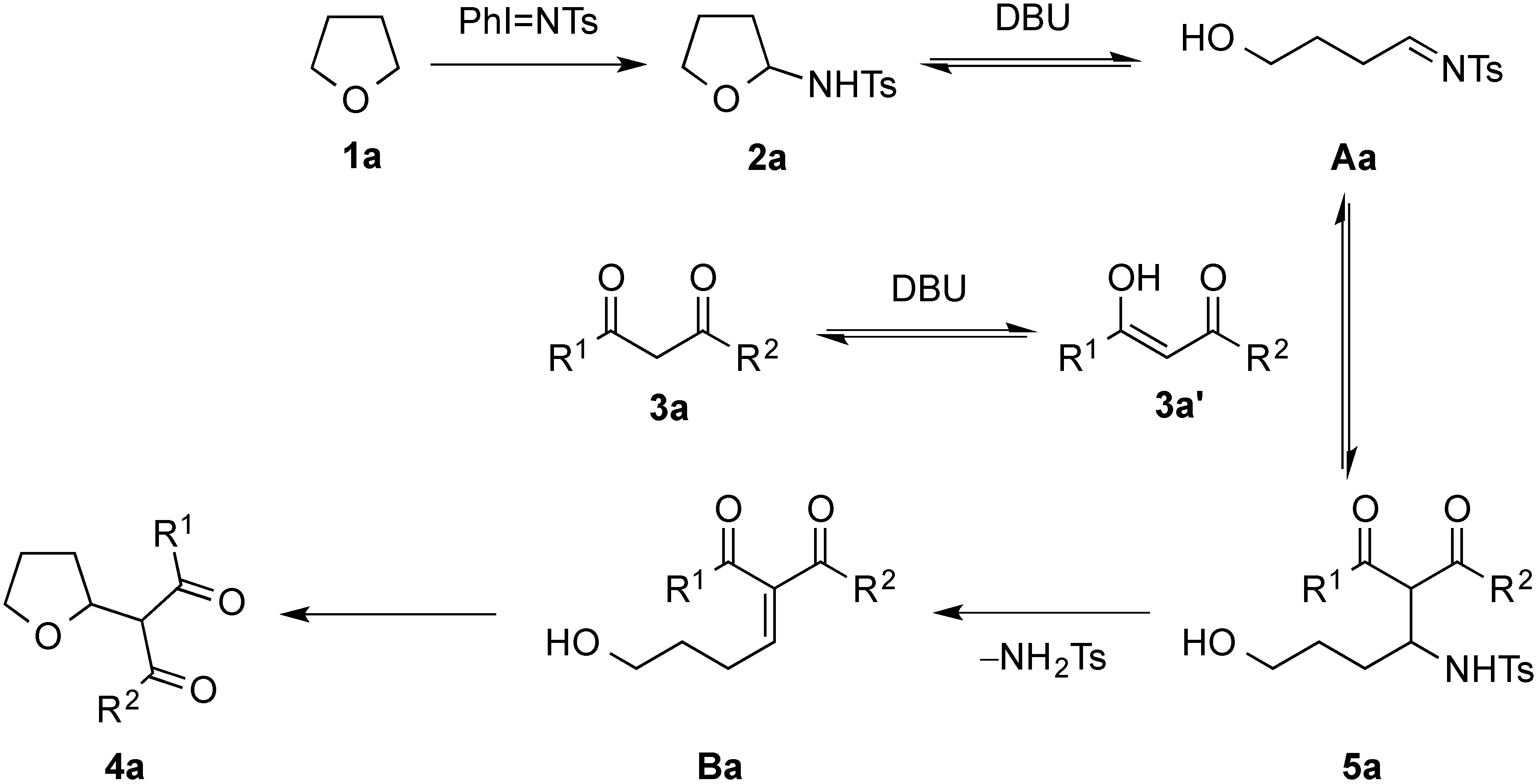
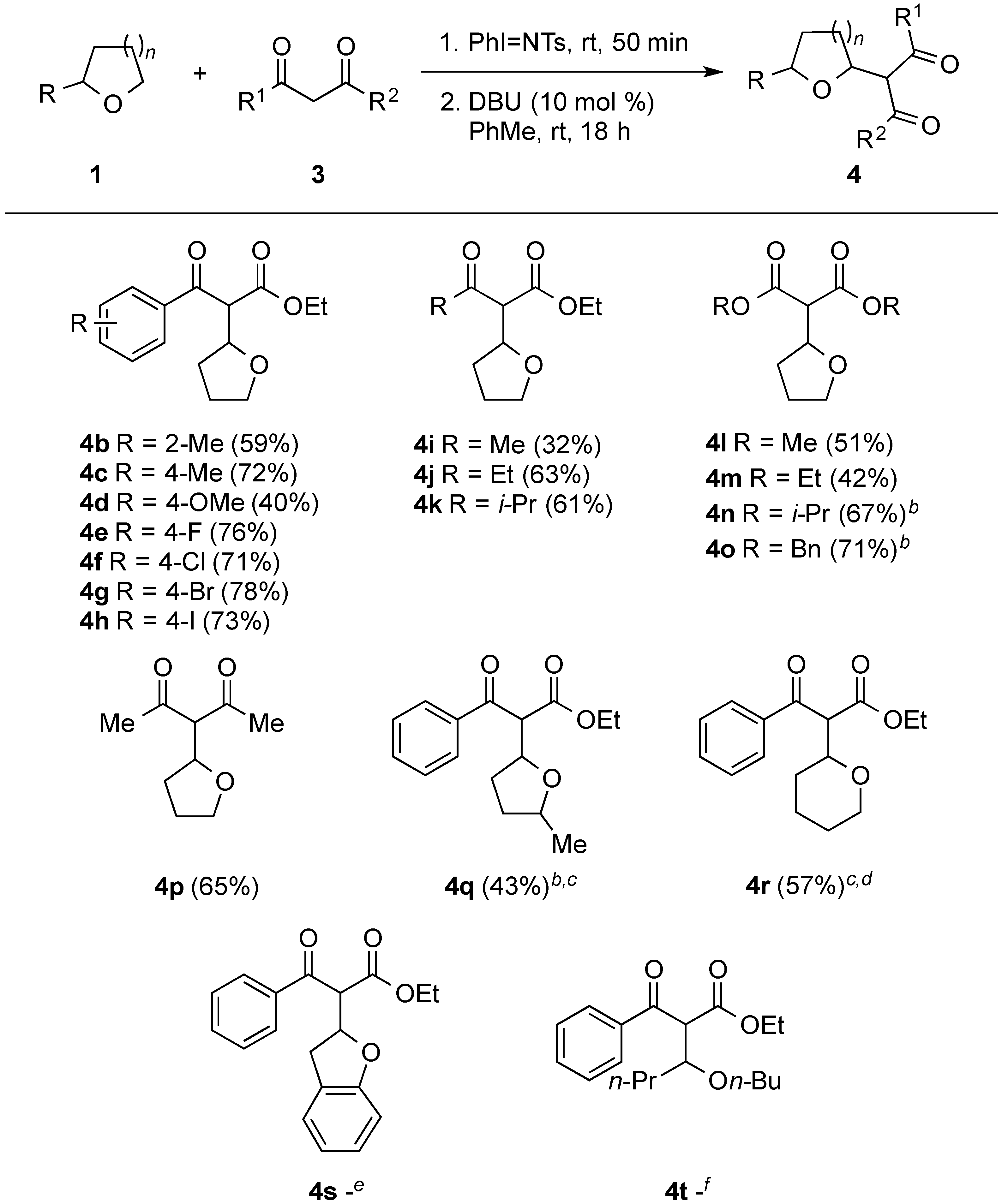
2. Results and Discussion

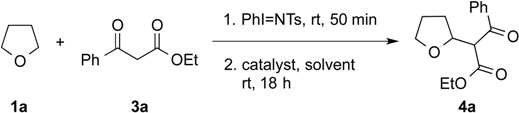
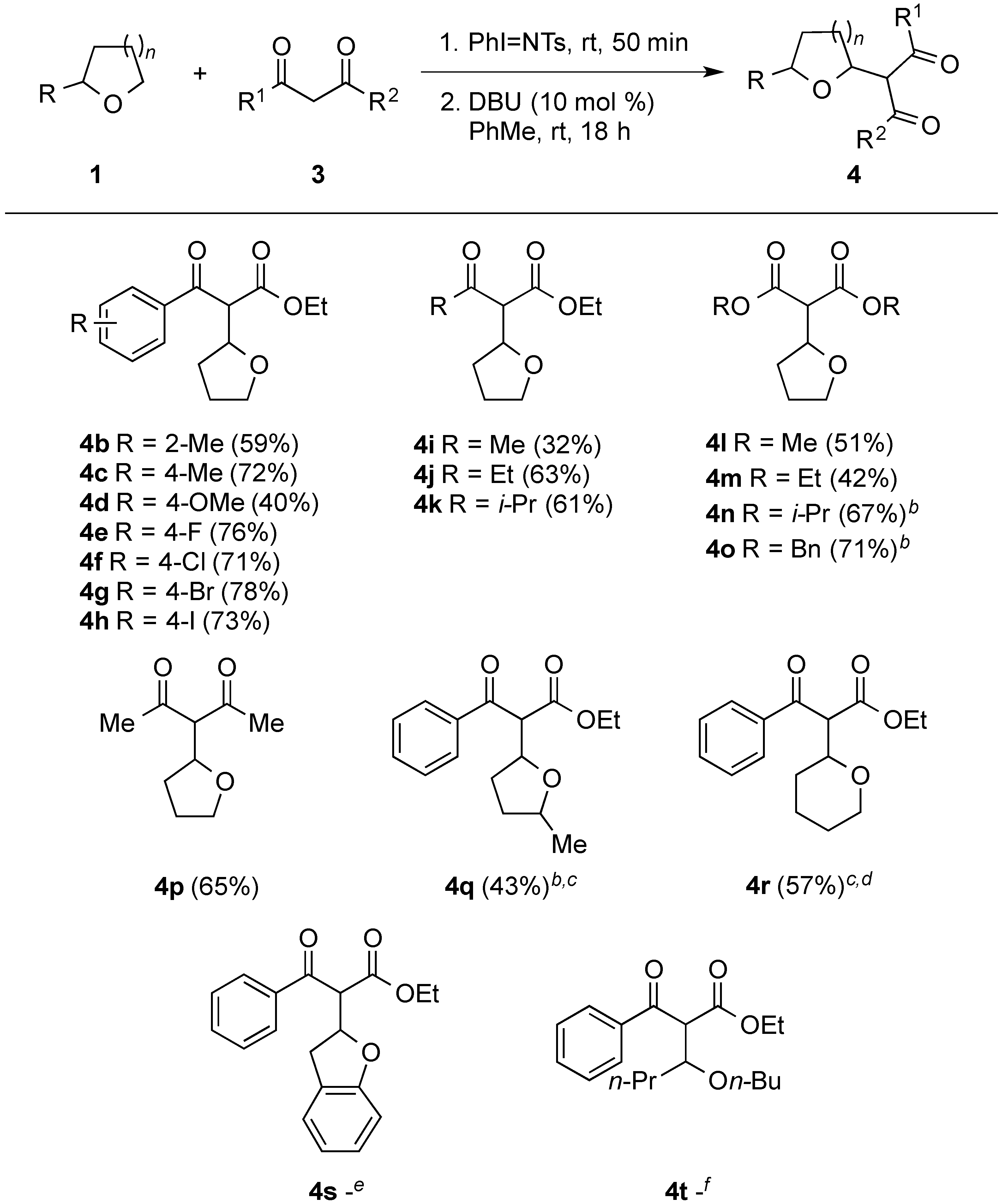
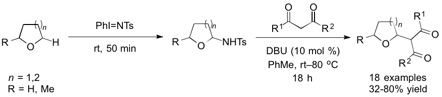{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst (mol %) | Solvent | Yield (%) b |
|---|---|---|---|
| 1 | DBU (10) | THF | 41 |
| 2 | DBU (10) | Et2O | 54 c |
| 3 | DBU (10) | CH2Cl2 | 79 |
| 4 | DBU (10) | PhMe | 78 |
| 5 | Et3N (10) | PhMe | - d |
| 6 | DABCO (10) | PhMe | - d |
| 7 | MTBD (10) | PhMe | 61 |
| 8 e | DBU (10) | PhMe | 80 |
| 9 f | DBU (10) | PhMe | 73 c |
| 10 e | DBU (5) | PhMe | 67 |
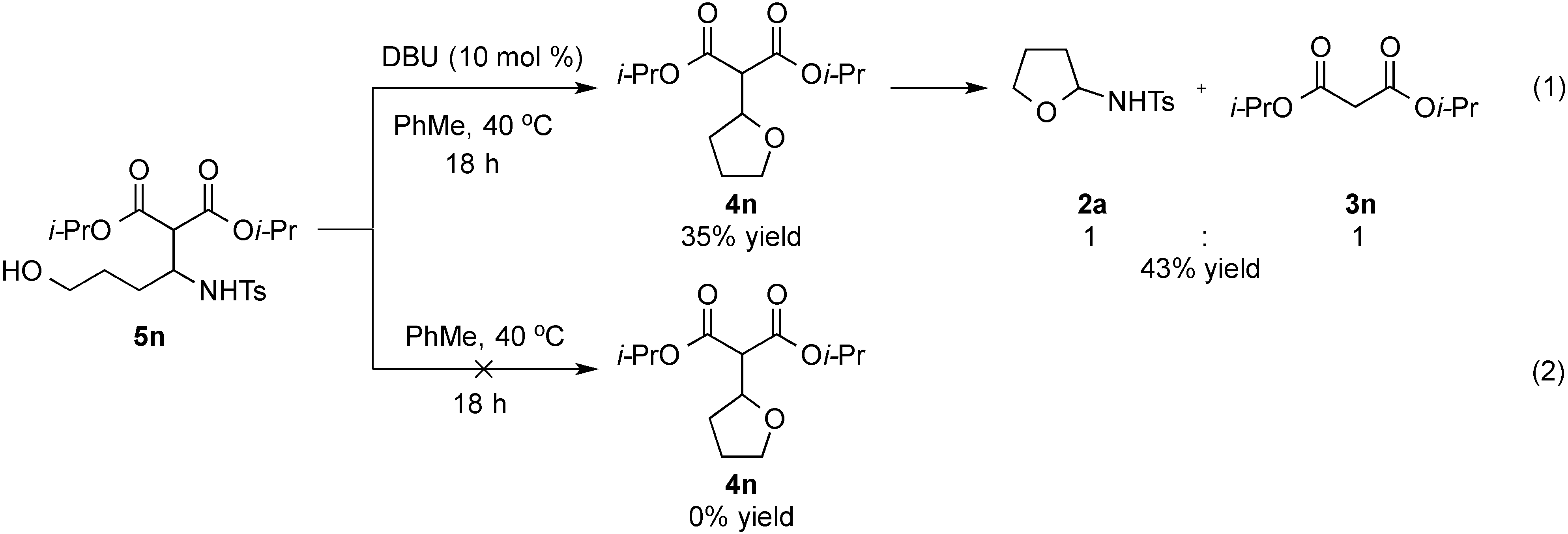
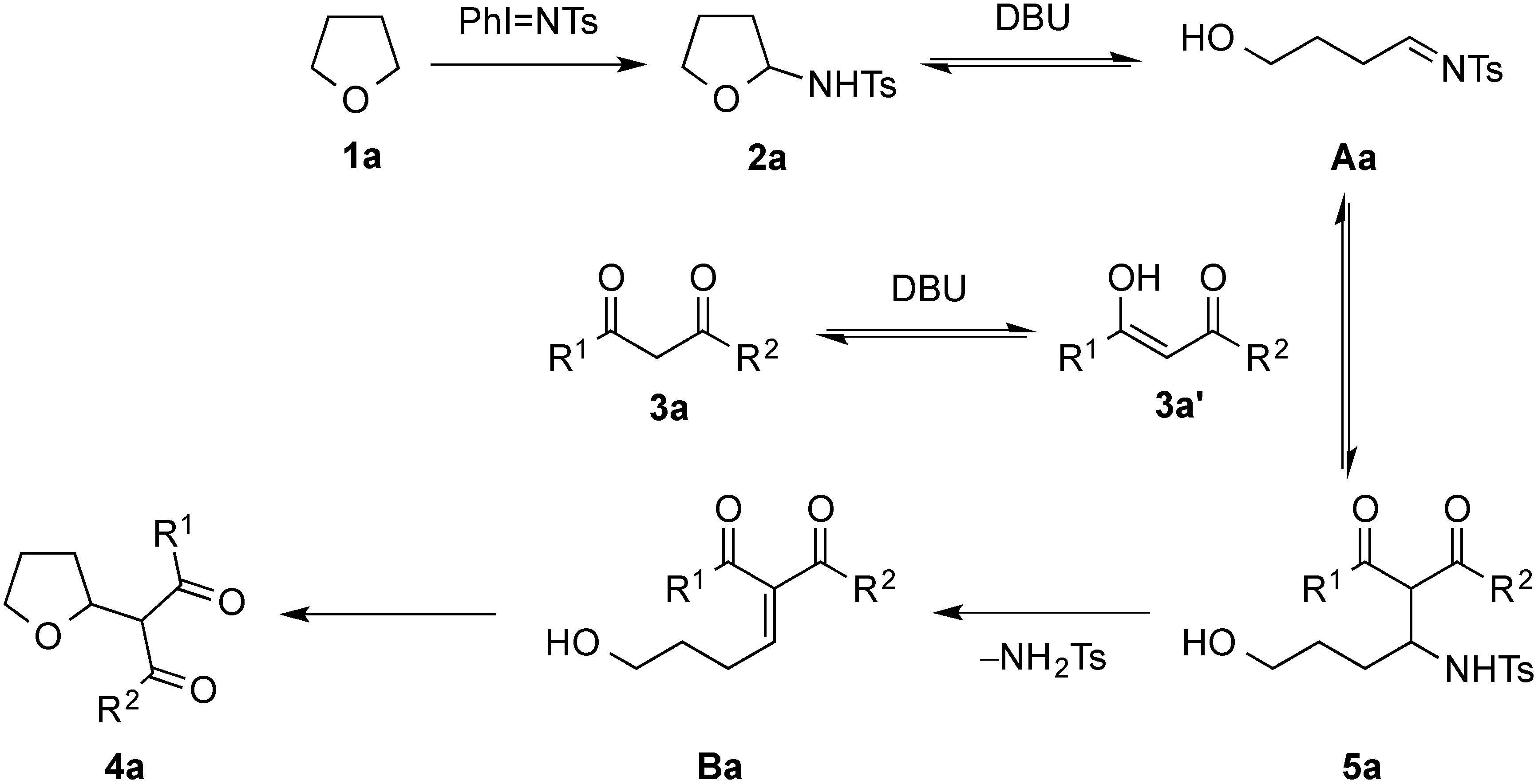
3. Experimental Section
General Information
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- For recent reviews on green and sustainable chemistry, see refs. 2–4.
- Gaich, T.; Baran, P.S. Aiming for the ideal synthesis. J. Org. Chem. 2010, 75, 4657–4673. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Trost, B.M. Green chemistry for chemical synthesis. Proc. Natl. Acad. Sci. USA 2008, 105, 13197–13202. [Google Scholar] [CrossRef] [PubMed]
- Constable, D.J.C.; Curzons, A.D.; Cunningham, V.L. Metrics to “green chemistry”—Which are the best? Green Chem. 2002, 4, 521–527. [Google Scholar] [CrossRef]
- For recent reviews on transition metal-catalyzed cross dehydrogenative coupling (CDC) for C–C bond formation, see refs. 6–9.
- Tsurugi, H.; Yamamoto, K.; Nagae, H.; Kaneko, H.; Mashima, K. Direct functionalization of unactivated C–H bonds catalyzed by group 3–5 metal alkyl complexes. Dalton Trans. 2014, 43, 2331–2343. [Google Scholar] [CrossRef] [PubMed]
- Girard, S.A.; Knauber, T.; Li, C.J. The cross-dehydrogenative coupling of Csp3–H bonds: A versatile strategy for C–C bond formations. Angew. Chem. Int. Ed. 2014, 53, 74–100. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; Kim, J.Y.; Kwak, J.; Chang, S. Recent advances in the transition metal-catalyzed twofold oxidative C–H bond activation strategy for C–C and C–N bond formation. Chem. Soc. Rev. 2011, 40, 5068–5083. [Google Scholar] [CrossRef] [PubMed]
- Yeung, C.S.; Dong, V.M. Catalytic dehydrogenative cross-coupling: Forming carbon-carbon bonds by oxidizing two carbon-hydrogen bonds. Chem. Rev. 2011, 111, 1215–1292. [Google Scholar] [CrossRef] [PubMed]
- For selected examples of Pd-catalyzed CDC for C–C bond formation, see refs. 11–23.
- Szabo, F.; Simko, D.; Novak, Z. A one-pot process for palladium catalyzed direct C–H acylation of anilines in water using a removable ortho directing group. RSC. Adv. 2014, 4, 3883–3886. [Google Scholar] [CrossRef]
- Jafarpour, F.; Hazrati, H.; Mohasselyazdi, N.; Khoobi, M.; Shafiee, A. Palladium catalyzed dehydrogenative arylation of coumarins: An unexpected switch in regioselectivity. Chem. Commun. 2013, 49, 10935–10937. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Jie, X.; Zhou, J.; Hu, P.; Huang, S.; Su, W. Pd-Catalyzed C–H olefination of (hetero)arenes by using saturated ketones as an olefin source. Angew. Chem. Int. Ed. 2013, 52, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, X.; Glorius, F. Palladium-catalyzed selective dehydrogenative cross-couplings of heteroarenes. Angew. Chem. Int. Ed. 2011, 50, 7479–7481. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, J.; Sun, C.L.; Li, B.J.; Shi, Z.J. Palladium-catalyzed cross-coupling of polyfluoroarenes with simple arenes. Org. Lett. 2011, 13, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yeung, C.S.; Dong, V.M. Palladium-catalyzed ortho-arylation of o-phenylcarbamates with simple arenes and sodium persulfate. J. Am. Chem. Soc. 2010, 132, 5837–5844. [Google Scholar] [CrossRef] [PubMed]
- Wasa, M.; Engle, K.M.; Yu, J.Q. Pd(II)-Catalyzed Olefination of sp3 C–H Bonds. J. Am. Chem. Soc. 2010, 132, 3680–3681. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.H.; Engle, K.M.; Shi, B.F.; Yu, J.Q. Ligand-enabled reactivity and selectivity in a synthetically versatile aryl C–H olefination. Science 2010, 327, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Cui, X.; Chen, L.; Jiang, G.; Wu, Y. Palladium-catalyzed alkenylation of quinoline-N-oxides via C–H activation under external-oxidant-free conditions. J. Am. Chem. Soc. 2009, 131, 1388–1389. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Mei, T.S.; Yu, J.Q. Synthesis of indolines and tetrahydroisoquinolines from arylethylamines by PdII-catalyzed C–H activation reactions. Angew. Chem. Int. Ed. 2008, 47, 6452–6455. [Google Scholar] [CrossRef] [PubMed]
- Houlden, C.E.; Bailey, C.D.; Gair Ford, J.; Gagné, M.R.; Lloyd-Jones, G.C.; Booker-Milburn, K.I. Distinct reactivity of Pd(OTs)2: The intermolecular Pd(II)-catalyzed 1,2-carboamination of dienes. J. Am. Chem. Soc. 2008, 130, 10066–10067. [Google Scholar] [CrossRef] [PubMed]
- Li, B.J.; Tian, S.L.; Fang, Z.; Shi, Z.J. Multiple C–H activations to construct biologically active molecules in a process completely free of organohalogen and organometallic components. Angew. Chem. Int. Ed. 2008, 47, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- Hull, K.L.; Sanford, M.S. Catalytic and highly regioselective cross-coupling of aromatic C–H substrates. J. Am. Chem. Soc. 2007, 129, 11904–11905. [Google Scholar] [CrossRef] [PubMed]
- For selected examples on CDC C–C bond formation catalyzed by other transition metal, see refs. 25–35.
- Shang, M.; Wang, H.L.; Sun, S.Z.; Dai, H.X.; Yu, J.Q. Cu(II)-mediated ortho C–H alkynylation of (hetero)arenes with terminal alkynes. J. Am. Chem. Soc. 2014, 136, 11590–11593. [Google Scholar] [CrossRef] [PubMed]
- Vora, H.U.; Silvestri, A.P.; Engelin, C.J.; Yu, J.Q. Rhodium(II)-catalyzed nondirected oxidative alkenylation of arenes: Arene loading at one equivalent. Angew. Chem. Int. Ed. 2014, 53, 2683–2686. [Google Scholar] [CrossRef] [PubMed]
- Odani, R.; Hirano, K.; Satoh, T.; Miura, M. Copper-mediated dehydrogenative biaryl coupling of naphthylamines and 1,3-azoles. J. Org. Chem. 2013, 78, 11045–11052. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Lei, Z.Q.; Wang, H.; Li, H.; Sun, J.; Shi, Z.J. Rhodium(I)-catalyzed redox-economic cross-coupling of carboxylic acids with arenes directed by N-containing groups. Angew. Chem. Int. Ed. 2013, 52, 2063–2067. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Liu, H.; Qin, D.; Wu, Q.; You, J.; Zhao, D.; Guo, Q.; Huang, X.; Lan, J. Chelation-assisted Rh(III)-catalyzed C2-selective oxidative C–H/C–H cross-coupling of indoles/pyrroles with arenes. Chem. Sci. 2013, 4, 1964–1969. [Google Scholar] [CrossRef]
- Hirano, K.; Miura, M. Copper-mediated oxidative direct C–C (hetero)aromatic cross-coupling. Chem. Commun. 2012, 48, 10704–10714. [Google Scholar] [CrossRef] [PubMed]
- Wencel-Delord, J.; Nimphius, C.; Patureau, F.W.; Glorius, F. [RhIIICp*]-Catalyzed dehydrogenative aryl-aryl bond formation. Angew. Chem. Int. Ed. 2012, 51, 2247–2251. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Song, G.Y.; Li, X.W. Rh(III)-Catalyzed tandem oxidative olefination-michael reactions between aryl carboxamides and alkenes. Org. Lett. 2010, 12, 5430–5433. [Google Scholar] [CrossRef] [PubMed]
- Patureau, F.W.; Glorius, F. Rh Catalyzed olefination and vinylation of unactivated acetanilides. J. Am. Chem. Soc. 2010, 132, 9982–9983. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Zhao, L.; Li, C.J. Ruthenium-catalyzed oxidative cross-coupling of chelating arenes and cycloalkanes. Angew. Chem. Int. Ed. 2008, 47, 6278–6282. [Google Scholar] [CrossRef] [PubMed]
- Ueura, K.; Satoh, T.; Miura, M. An efficient waste-free oxidative coupling via regioselective C–H bond cleavage: Rh/Cu-catalyzed reaction of benzoic acids with alkynes and acrylates under air. Org. Lett. 2007, 9, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- For selected examples on transition metal-catalyzed CDC C–C bond formation of amines, see refs. 37–46.
- Jin, X.; Yamaguchi, K.; Mizuno, N. Aerobic cross-dehydrogenative coupling of terminal alkynes and tertiary amines by a combined catalyst of Zn2+ and OMS-2. RSC. Adv. 2014, 4, 34712–34715. [Google Scholar] [CrossRef]
- Zhong, J.J.; Meng, Q.Y.; Liu, B.; Li, X.B.; Gao, X.W.; Lei, T.; Wu, C.J.; Li, Z.J.; Tung, C.H.; Wu, L.Z. Cross-coupling hydrogen evolution reaction in homogeneous solution without noble metals. Org. Lett. 2014, 16, 1988–1991. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Qian, S.; Wang, C.; You, J. Palladium(II)-catalyzed dehydrogenative cross-coupling between two Csp3–H bonds: Unexpected C=C bond formation. Angew. Chem. Int. Ed. 2013, 52, 7837. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.Z.; Sun, X.; Wei, W.T.; Zhang, X.J.; Yan, M.; Xiao, J.L. Unprecedented construction of C=C double bonds via Ir-catalyzed dehydrogenative and dehydrative cross-couplings. Org. Lett. 2013, 15, 2394–2397. [Google Scholar] [CrossRef] [PubMed]
- Alagiri, K.; Prabhu, K.R. C–H functionalization of tertiary amines by cross dehydrogenative coupling reactions: Solvent-free synthesis of α-iminonitriles and β-nitroamines under aerobic condition. Org. Biomol. Chem. 2012, 10, 835–842. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, H.; Zhou, J.; Cheng, Y.; Zhu, C. A Highly efficient gold-catalyzed oxidative C–C coupling from C–H bonds using air as oxidant. Angew. Chem. Int. Ed. 2012, 51, 1252–1255. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Niu, T.; Wu, J.; Zhang, Y. Copper-catalyzed oxidative cross-coupling of N,N-dimethylanilines with heteroarenes with molecular oxygen. J. Org. Chem. 2011, 76, 1759–1766. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, E.; Uchiyama, N.; Hayashi, T. Iron-catalyzed oxidative coupling of alkylamides with arenes through oxidation of alkylamides followed by Friedel-crafts alkylation. J. Org. Chem. 2011, 76, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, J.; Xie, J.; Huang, Z.Z. Copper-catalyzed dehydrogenative coupling reactions of tertiary amines with ketones or indoles. Org. Lett. 2010, 12, 5214–5217. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhou, C.Y.; Xiang, S.; Che, C.M. Highly efficient oxidative carbon–carbon coupling with SBA-15-support iron terpyridine catalyst. Chem. Commun. 2010, 46, 2739–2741. [Google Scholar] [CrossRef] [PubMed]
- For selected examples on transition metal-catalyzed CDC of ethers for C–C bond formation, see refs. 48–61.
- Pandit, R.P.; Lee, Y.R. Direct oxidative arylation of C(sp3)–H bonds adjacent to oxygen of ethers and alcohols. Adv. Synth. Catal. 2014, 356, 3171–3179. [Google Scholar] [CrossRef]
- Zhou, L.; Tang, S.; Qi, X.; Lin, C.; Lin, K.; Liu, C.; Lan, Y.; Lei, A. Transition-metal-assisted radical/radical cross-coupling: A new strategy to the oxidative C(sp3)−H/N−H cross-coupling. Org. Lett. 2014, 16, 3404–3407. [Google Scholar] [CrossRef] [PubMed]
- Rout, S.K.; Guin, S.; Ali, W.; Gogoi, A.; Patel, B.K. Copper-catalyzed esterification of alkylbenzenes with cyclic ethers and cycloalkanes via C(sp3)–H activation following cross-dehydrogenative coupling. Org. Lett. 2014, 16, 3086–3089. [Google Scholar] [CrossRef] [PubMed]
- Siddaraju, Y.; Lamani, M.; Prabhu, K.R. A Transition metal-free minisci reaction: Acylation of isoquinolines, quinolines, and quinoxaline. J. Org. Chem. 2014, 79, 3856–3865. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, C.; Li, H.; Lei, A. Copper-catalyzed oxidative C–H/C–H coupling between olefins and simple ethers. Chem. Commun. 2014, 50, 3623–3626. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Pi, C.; Cui, X.; Bai, J.; Wu, Y. Direct C-2 alkylation of quinoline N-oxides with ethers via palladium-catalyzed dehydrogenative cross-coupling reaction. Adv. Synth. Catal. 2013, 355, 1971–1976. [Google Scholar] [CrossRef]
- Xie, Z.; Cai, Y.; Hu, H.; Lin, C.; Jiang, J.; Chen, Z.; Wang, L.; Pan, Y. Cu-catalyzed cross-dehydrogenative coupling reaction of (benzo)thiazoles with cyclic ethers. Org. Lett. 2013, 15, 4600–4603. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, C.; Li, H.; Lei, A. Direct functionalization of tetrahydrofuran and 1,4-dioxane: Nickel-catalyzed oxidative C(sp3)−H arylation. Angew. Chem. Int. Ed. 2013, 52, 4453–4456. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Price, J.R.; Todd, M.H. Oxidative arylation of isochroman. J. Org. Chem. 2012, 77, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Pieber, B.; Reddy, R.; Oliver Kappe, C. Copper-catalyzed formation of C–O bonds by direct α-C–H bond activation of ethers using stoichiometric amounts of peroxide in batch and continuous-flow formats. Chem. Eur. J. 2012, 18, 6124–6128. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Shang, X.; Shao, X.F.; Liu, Z.Q. Copper-catalyzed decarboxylative alkenylation of sp3 C–H bonds with cinnamic acids via a radical process. Chem. Sci. 2012, 3, 2853–2858. [Google Scholar] [CrossRef]
- Guo, X.; Pan, S.; Liu, J.; Li, Z. One-pot synthesis of symmetric and unsymmetric 1,1-bis-indolylmethanes via tandem iron-catalyzed C–H bond oxidation and C–O bond cleavage. J. Org. Chem. 2009, 74, 8848–8851. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Yu, R.; Li, H. Iron-catalyzed C–C bond formation by direct functionalization of C–H bonds adjacent to heteroatoms. Angew. Chem. Int. Ed. 2008, 47, 7497–7500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, C.J. Highly efficient cross-dehydrogenative-coupling between ethers and active methylene compounds. Angew. Chem. Int. Ed. 2006, 45, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- For reviews on transition metal-free CDC, see refs. 63 and 64.
- Sun, C.L.; Shi, Z.J. Transition-metal-free coupling reactions. Chem. Rev. 2014, 114, 9219–9280. [Google Scholar] [CrossRef] [PubMed]
- Klussmann, M.; Sureshkumar, D. Catalytic oxidative coupling reactions for the formation of carbon–carbon bonds without carbon–metal intermediates. Synthesis 2011, 353–369. [Google Scholar] [CrossRef]
- For selected examples concerning transition metal-free CDC, see refs. 66–71.
- Tanoue, A.; Yoo, W.J.; Kobayashi, S. Sulfuryl chloride as an efficient initiator for the metal-free aerobic cross-dehydrogenative coupling reaction of tertiary amines. Org. Lett. 2014, 16, 2346–2349. [Google Scholar] [CrossRef] [PubMed]
- Dhineshkumar, J.; Lamani, M.; Alagiri, K.; Prabhu, K.R. A versatile C–H functionalization of tetrahydroisoquinolines catalyzed by iodine at aerobic conditions. Org. Lett. 2013, 15, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer-Chaput, B.; Klussmann, M. Brønsted acid catalyzed C–H functionalization of N-protected tetrahydroisoquinolines via intermediate peroxides. Eur. J. Org. Chem. 2013, 666–671. [Google Scholar] [CrossRef]
- Kumar, R.A.; Saidulu, G.; Prasad, K.R.; Kumar, G.S.; Sridhar, B.; Reddy, K.R. Transition metal-free α-C(sp3)–H bond functionalization of amines by oxidative cross dehydrogenative coupling reaction. Simple and direct access to C-4-alkylated 3,4-dihydroquinazoline derivatives. Adv. Synth. Catal. 2012, 354, 2985–2991. [Google Scholar] [CrossRef]
- Richter, H.; Froehlich, R.; Daniliuc, C.G.; Garcia Mancheño, O. Mild metal-free tandem α-alkylation/cyclization of N-benzyl carbamates with simple olefins. Angew. Chem. Int. Ed. 2012, 51, 8656–8660. [Google Scholar] [CrossRef] [PubMed]
- Alagiri, K.; Devadig, P.; Prabhu, K.R. CDC reactions of N-aryl tetrahydroisoquinolines using catalytic amounts of DDQ: C–H activation under aerobic conditions. Chem. Eur. J. 2012, 18, 5160–5164. [Google Scholar] [CrossRef] [PubMed]
- For reviews on hypervalent iodine or diaryliodonium salts in cross coupling reactions, see ref. 64 and; Merritt, E.A.; Olofsson, B. Diaryliodonium Salts: A Journey from Obscurity to Fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070. [Google Scholar]
- For selected examples on transition metal-free CDC using hypervalent iodine, see refs. 74–81.
- Narayan, R.; Antonchick, A.P. Hypervalent iodine-mediated selective oxidative functionalization of (thio)chromones with alkanes. Chem. Eur. J. 2014, 20, 4568–4572. [Google Scholar] [CrossRef] [PubMed]
- Antonchick, A.P.; Burgmann, L. Direct selective oxidative cross-coupling of simple alkanes with heteroarenes. Angew. Chem. Int. Ed. 2013, 52, 3267–3271. [Google Scholar] [CrossRef] [PubMed]
- Matcha, K.; Antonchick, A.P. Metal-free cross-dehydrogenative coupling of heterocycles with aldehydes. Angew. Chem. Int. Ed. 2013, 52, 2082–2086. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.; Fernandez, J.J.; Vicente, R.; Fananas, F.J.; Rodriguez, F. Base- and metal-free C–H direct arylations of naphthalene and other unbiased arenes with diaryliodonium salts. Chem. Commun. 2012, 48, 9089–9091. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Zhang, R.Y.; Chen, S.Y.; Zhang, J.; Yu, X.Q. Direct arylation of arene and N-heteroarenes with diaryliodonium salts without the use of transition metal catalyst. J. Org. Chem. 2012, 77, 766–771. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, L.; Dell’Acqua, M.; Fenner, S.; Vicente, R.; Sandmann, R. Metal-free direct arylations of indoles and pyrroles with diaryliodonium Salts. Org. Lett. 2011, 13, 2358–2360. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Morimoto, K.; Ito, M.; Ogawa, C.; Goto, A.; Dohi, T. Metal-free oxidative cross-coupling of unfunctionalized aromatic compounds. J. Am. Chem. Soc. 2009, 131, 1668–1669. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Ito, M.; Morimoto, K.; Iwata, M.; Kita, Y. Oxidative cross-coupling of arenes induced by single-electron transfer leading to biaryls using organoiodine(III) oxidants. Angew. Chem. Int. Ed. 2008, 47, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Selected general reviews on transition-metal-mediated imido/nitrene reactions, see refs. 83–91
- Dequirez, G.; Pons, V.; Dauban, P. Nitrene chemistry in organic synthesis: Still in its infancy? Angew. Chem. Int. Ed. 2012, 51, 7384–7395. [Google Scholar] [CrossRef] [PubMed]
- Roizen, J.L.; Harvey, M.E.; Du Bois. Metal-catalyzed nitrogen-atom transfer methods for the oxidation of aliphatic C–H bonds. Acc. Chem. Res. 2012, 45, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Collet, F.; Lescot, C.; Dauban, P. Catalytic C-H amination: The stereoselectivity issue. Chem. Soc. Rev. 2011, 40, 1926–1936. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.W.; Ton, T.M.U.; Chan, P.W.H. Transition-metal-catalyzed aminations and aziridinations of C–H and C=C bonds with iminoiodinane. Chem. Rec. 2011, 11, 331–357. [Google Scholar] [CrossRef] [PubMed]
- Collet, F.; Dodd, R.H.; Dauban, P. Catalytic C–H amination: Recent progress and future directions. Chem. Commun. 2009, 5061–5074. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Requejo, M.M.; Pérez, P.J. Coinage Metal catalyzed C–H bond functionalization of hydrocarbons. Chem. Rev. 2008, 108, 3379–3394. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M. L.; Manning, J.R. Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.M.L. Recent advances in catalytic enantioselective intermolecular C–H functionalization. Angew. Chem. Int. Ed. 2006, 45, 6422–6425. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.; Fruit, C. Enantioselective catalytic aziridinations and asymmetric nitrene insertions into C–H bonds. Chem. Rev. 2003, 103, 2905–2920. [Google Scholar] [CrossRef] [PubMed]
- For selected recent examples on transition-metal-free reactions with nitrenoid precursors, see refs. 93–103.
- Kiyokawa, K.; Kosaka, T.; Minakata, S. Metal-free aziridination of styrene derivatives with iminoiodinane catalyzed by a combination of iodine and ammonium iodide. Org. Lett. 2013, 15, 4858–4861. [Google Scholar] [CrossRef] [PubMed]
- Souto, J.A.; Martínez, C.; Velilla, I.; Muñiz, K. Defined hypervalent iodine(III) regents incorporating transferable nitrogen groups: Nucleophilic amination through electrophilic activation. Angew. Chem. Int. Ed. 2013, 52, 1324–1328. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, M.; Yamane, S.; Hoque, M.M.; Saito, M.; Miyamoto, K. Metal-free α-CH amination of ethers with hypervalent sulfonylimino-λ3-bromane that acts as an active nitrenoid. Chem. Commun. 2012, 48, 5280–5282. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, M.; Miyamoto, T.; Kaneaki, K.; Hayashi, S.; Nakanishi, W. Highly regioselective amination of unactivated alkanes by hypervalent sulfonylimino-λ3-bromane. Science 2011, 332, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Hayakawa, J.; Yano, K.; Minakata, S. Transition-metal-free benzylic C–H bond intermolecular amination utilizing chloramine-T and I2. Chem. Lett. 2012, 41, 1672–1674. [Google Scholar] [CrossRef]
- Zhang, D.H.; Wei, Y.; Shi, M. Metal-free ring expansions of methylenecyclopropanes through nitrene equivalent. Eur. J. Org. Chem. 2011, 2011, 4940–4944. [Google Scholar] [CrossRef]
- Karabal, P.U.; Chouthaiwale, P.V.; Shaikh, T.M.; Suryavanshi, G.; Sudalai, A. NIO4/LiBr-mediated aziridination of olefins using chloramine-T. Tetrahedron Lett. 2010, 51, 6460–6462. [Google Scholar] [CrossRef]
- Fang, C.; Qian, W.; Bao, W. A mild and clean method for oxidative formation of amides from aldehydes and amines. Synlett 2008, 2529–2531. [Google Scholar] [CrossRef]
- Li, J.; Chan, P.W.H.; Che, C.M. Aryl iodide mediated aziridination of alkenes. Org. Lett. 2005, 7, 5801–5804. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.W.; Ahn, K.H. The reaction of [N-(p-toluenesulfonyl)lmino]-phenyliodinane with enol silanes. Synth. Commun. 1996, 26, 3407–3412. [Google Scholar] [CrossRef]
- Tejo, C.; Yeo, H.Q.; Chan, P.W.H. Brønsted acid catalyzed amination of 1,3-dicarbonyl compounds by iminoiodanes. Synlett 2014, 25, 201–204. [Google Scholar]
- For selected recent works by our group, see refs. 103 and 105–109.
- Ton, T.M.U.; Himawan, F.; Chang, J.W.W.; Chan, P.W.H. Copper(II) triflate catalyzed amination of 1,3-dicarbonyl compounds. Chem. Eur. J. 2012, 18, 12020–12027. [Google Scholar] [CrossRef] [PubMed]
- Ton, T.M.U.; Tejo, C.; Tiong, D.L.Y.; Chan, P.W.H. Copper(II) triflate catalyzed amination and aziridination of 2-alkyl substituted 1,3-dicarbonyl compounds. J. Am. Chem. Soc. 2012, 134, 7344–7350. [Google Scholar] [CrossRef] [PubMed]
- Ton, T.M.U.; Tejo, C.; Tania, S.; Chang, J.W.W.; Chan, P.W.H. Iron(III)-catalyzed amidation of aldehydes with iminoiodanes at room temperature and under microwave-assisted conditions. J. Org. Chem. 2011, 76, 4894–4904. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.W.; Ton, T.M.U.; Tania, S.; Taylor, P.C.; Chan, P.W.H. Practical copper(I)-catalyzed amidation of aldehydes. Chem. Commun. 2010, 46, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.W.W.; Chan, P.W.H. Highly Efficient Ruthenium(II) Porphyrin-Catalyzed Amidation of Aldehydes. Angew. Chem. Int. Ed. 2008, 47, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- For reports correlating 1,3-dicarbonyl compound reactivity to pKa values, see refs. 103, 111 and 112.
- Arnett, E.M.; Maroldo, S.G.; Schilling, S.L.; Harrelson, J.A. Ion pairing and reactivity of enolate anions. 5. Thermodynamics of ionization of β-di- and tricarbonyl compounds in dimethyl sulfoxide solution and ion pairing of their alkali salts. J. Am. Chem. Soc. 1984, 106, 6759–6767. [Google Scholar] [CrossRef]
- Olmstead, W.N.; Bordwell, F.G. Ion-pair association constants in dimethyl sulfoxide. J. Org. Chem. 1980, 45, 3299–3305. [Google Scholar] [CrossRef]
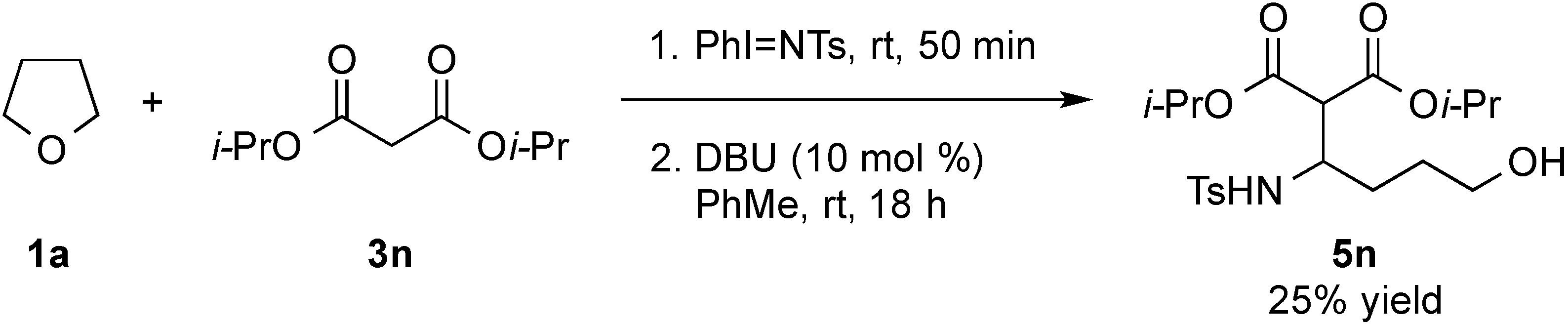
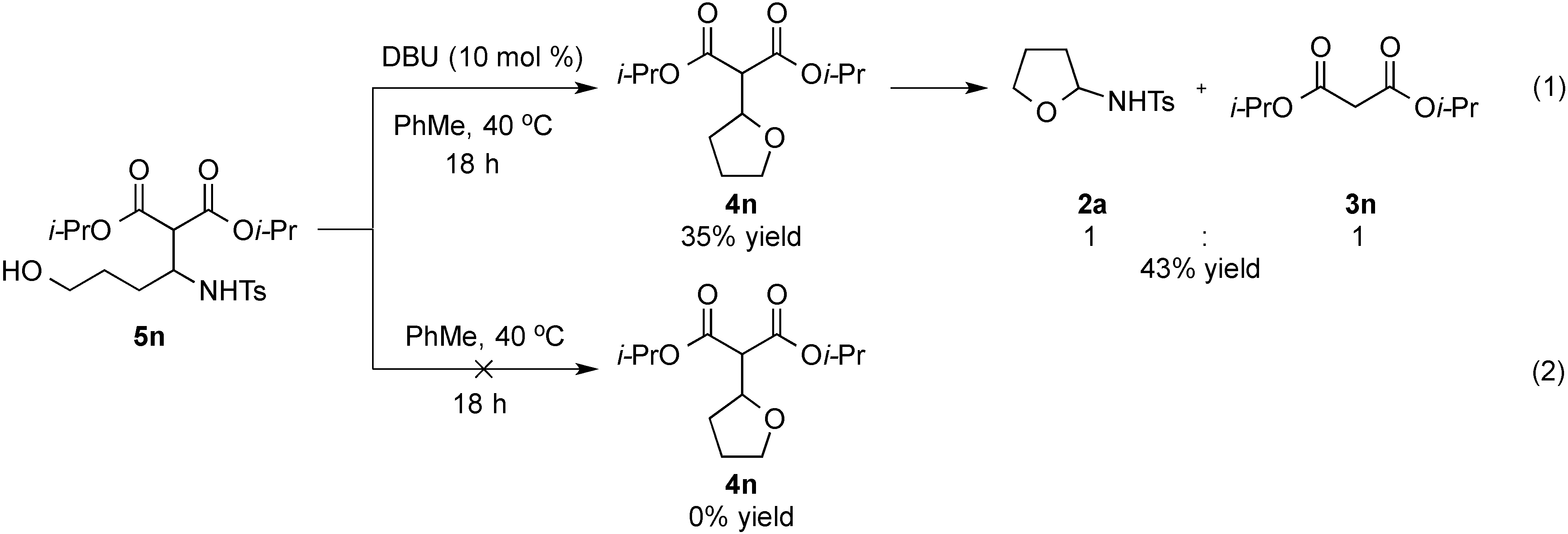
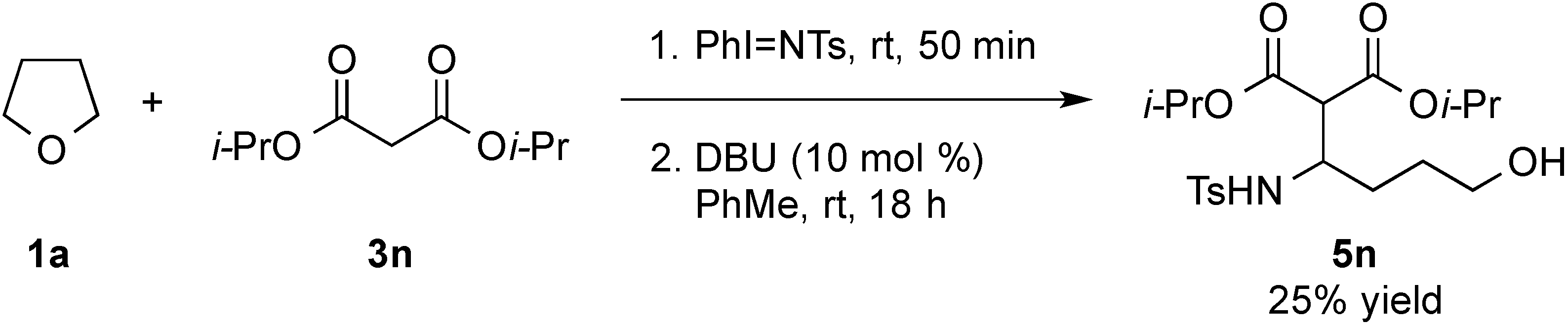
- Treatment of 5n under the optimized conditions at room temperature led to an observed 6% conversion to 2a and 3n. For selected studies on retro-Aldol or retro-Mannich reactions, see refs. 114–118.
- Gao, B.; Zhao, Y.; Hu, M.; Ni, C.; Hu, J. gem-Difluoroolefination of diaryl ketones and enolizable aldehydes with difluoromethyl 2-pyridyl sulfone. New insights into the Julia-Kocienski reaction. Chem. Eur. J. 2014, 20, 7803–7810. [Google Scholar] [CrossRef] [PubMed]
- Bartrum, H.E.; Viceriat, A.; Carret, S.; Poisson, J.F. Sulfinylimidates as chiral amide equivalents for irreversible, asymmetric aldol reactions. Org. Lett. 2014, 16, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Uematsu, R.; Maeda, S.; Taketsugu, T. Multiple reaction pathways operating in the mechanism of vinylogous Mannich-type reaction activated by a water molecule. Chem. Asian J. 2014, 9, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, D.C.; Malow, E.J.; Johnson, J.S. Three-component glycolate michael reactions of enolates, silyl glyoxylates, and α,β-enones. J. Org. Chem 2012, 77, 3246–3251. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Davydova, M.P.; Pal, R.; Gilmore, K.; Tolstikov, G.A.; Vasilevsky, S.F.; Alabugin, I.V. Dissecting alkynes: Full cleavage of polarized C≡C moiety via sequential bis-Michael Addition/Retro-Mannich cascade. J. Org. Chem. 2011, 76, 7482–7490. [Google Scholar] [CrossRef] [PubMed]
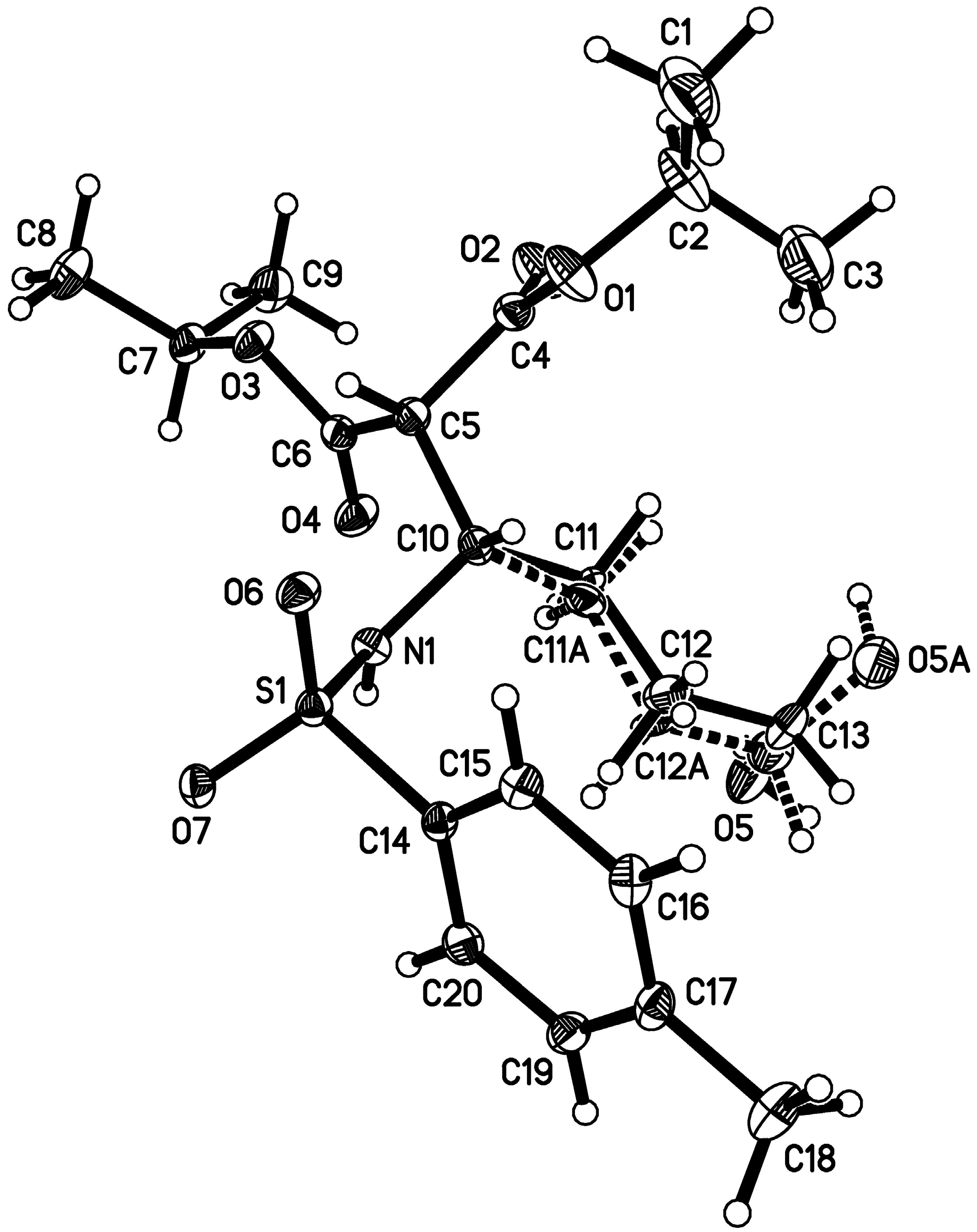
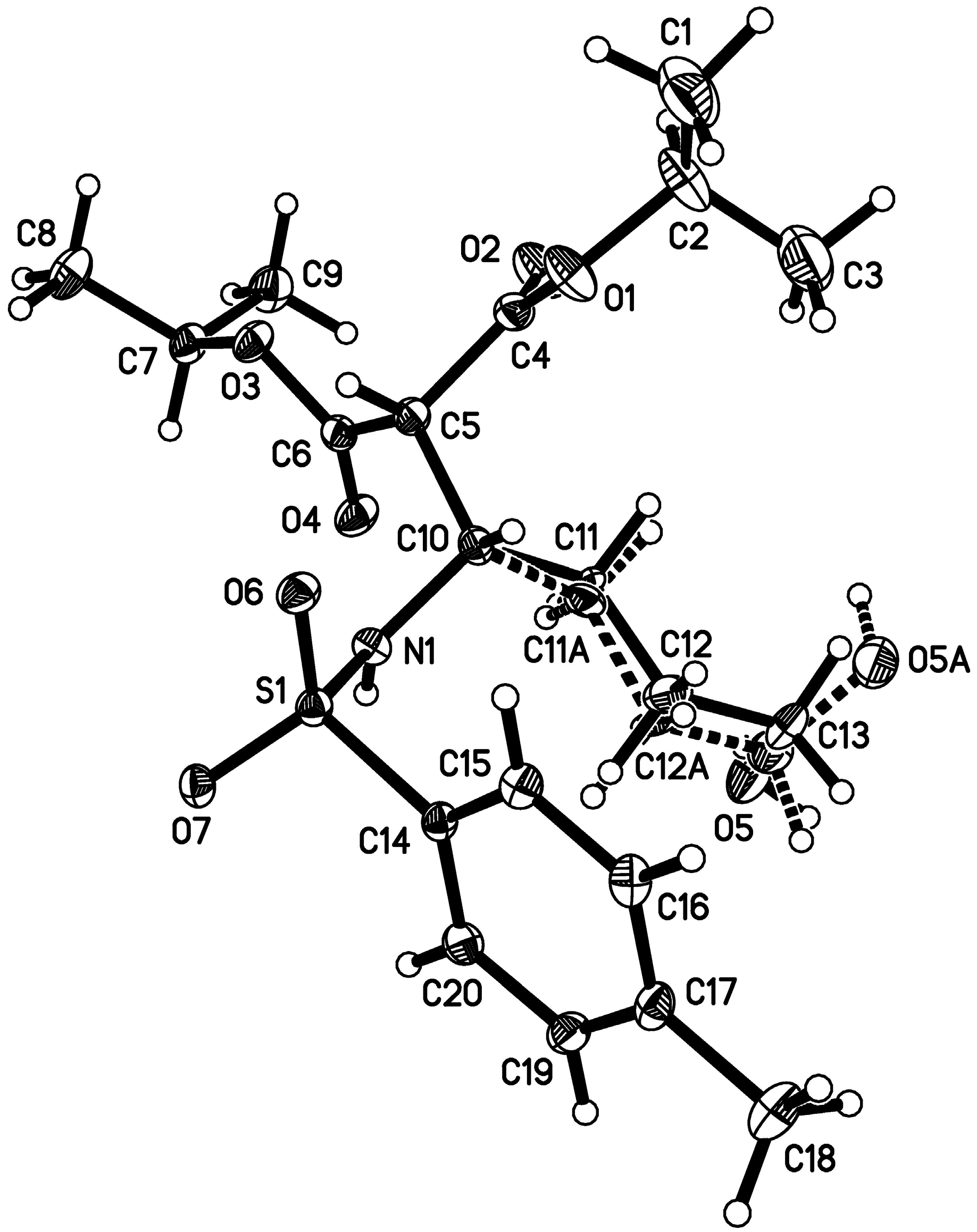
- CCDC 1059668 contains the supplementary crystallographic data for this paper. This data can be obtained free of charge from The Cambridge Crystallographic Data Centre. Available online: http://www.ccdc.cam.ac.uk/data_request/cif (accessed on 1 June 2015).
- Yamada, Y.; Yamamoto, T.; Okawara, M. Synthesis and reaction of new type iodine-nitrogen ylide, N-tosyliminoiodane. Chem. Lett. 1975, 4, 361–362. [Google Scholar] [CrossRef]
- De Godoy, L.A.F.; Camilo, N.S.; Pilli, R.A. Addition of carbon nucleophiles to cyclic N-acyliminium and oxocarbenium ions under solvent-free conditions. Tetrahedron Lett. 2006, 47, 7853–7856. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejo, C.; Sim, X.R.; Lee, B.R.; Ayers, B.J.; Leung, C.-H.; Ma, D.-L.; Chan, P.W.H. Iminoiodane- and Brønsted Base-Mediated Cross Dehydrogenative Coupling of Cyclic Ethers with 1,3-Dicarbonyl Compounds. Molecules 2015, 20, 13336-13353. https://doi.org/10.3390/molecules200713336
Tejo C, Sim XR, Lee BR, Ayers BJ, Leung C-H, Ma D-L, Chan PWH. Iminoiodane- and Brønsted Base-Mediated Cross Dehydrogenative Coupling of Cyclic Ethers with 1,3-Dicarbonyl Compounds. Molecules. 2015; 20(7):13336-13353. https://doi.org/10.3390/molecules200713336
Chicago/Turabian StyleTejo, Ciputra, Xiao Rong Sim, Bo Ra Lee, Benjamin James Ayers, Chung-Hang Leung, Dik-Lung Ma, and Philip Wai Hong Chan. 2015. "Iminoiodane- and Brønsted Base-Mediated Cross Dehydrogenative Coupling of Cyclic Ethers with 1,3-Dicarbonyl Compounds" Molecules 20, no. 7: 13336-13353. https://doi.org/10.3390/molecules200713336
APA StyleTejo, C., Sim, X. R., Lee, B. R., Ayers, B. J., Leung, C.-H., Ma, D.-L., & Chan, P. W. H. (2015). Iminoiodane- and Brønsted Base-Mediated Cross Dehydrogenative Coupling of Cyclic Ethers with 1,3-Dicarbonyl Compounds. Molecules, 20(7), 13336-13353. https://doi.org/10.3390/molecules200713336