
Chemical Characterization of Different Sumac and Pomegranate Extracts Effective against Botrytis cinerea Rots

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Total Anthocyanins and Total Phenolics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Plant Materials | 50% Ethanol/Water Extracts (1:1) | 80% Ethanol/Water Extracts (4:1) | Concentrated Aqueous Extract from 80% Ethanol/Water | Water at 40 °C Extracts |
|---|---|---|---|---|---|
| Pomegranate | Peels | PGE-50 | PGE-80 | PGE-C | PGE-H2O |
| Sumac | Fruits | SUF-50 | SUF-80 | SUF-C | SUF-H2O |
| Sumac | Leaves | SUL-50 | SUL-80 | SUL-C | SUL-H2O |
| Extracts ¥ | Total Anthocyanins § (mg CGE/kg) | Total Phenolics § (g GAE/kg) |
|---|---|---|
| PGE-C | 21.64 ± 0.03 e | 66.97 ± 0.67 a |
| PGE-50 | n.q. Ω | 9.86 ± 0.20 gf |
| PGE-80 | n.q. | 10.36 ± 0.31 f |
| PGE-H2O | n.q. | 7.35 ± 0.08 h |
| SUF-C | 171.96 ± 1.51 a | 9.47 ± 0.01 g |
| SUF-50 | 39.73 ± 0.04 d | 5.34 ± 0.43 i |
| SUF-80 | 41.97 ± 0.02 c | 5.42 ± 0.34 i |
| SUF-H2O | 94.92 ± 0.16 b | 2.80 ± 0.01 j |
| SUL-C | - | 29.38 ± 0.24 b |
| SUL-50 | - | 27.16 ± 0.31 d |
| SUL-80 | - | 27.84 ± 0.23 c |
| SUL-H2O | - | 15.22 ± 0.13 e |
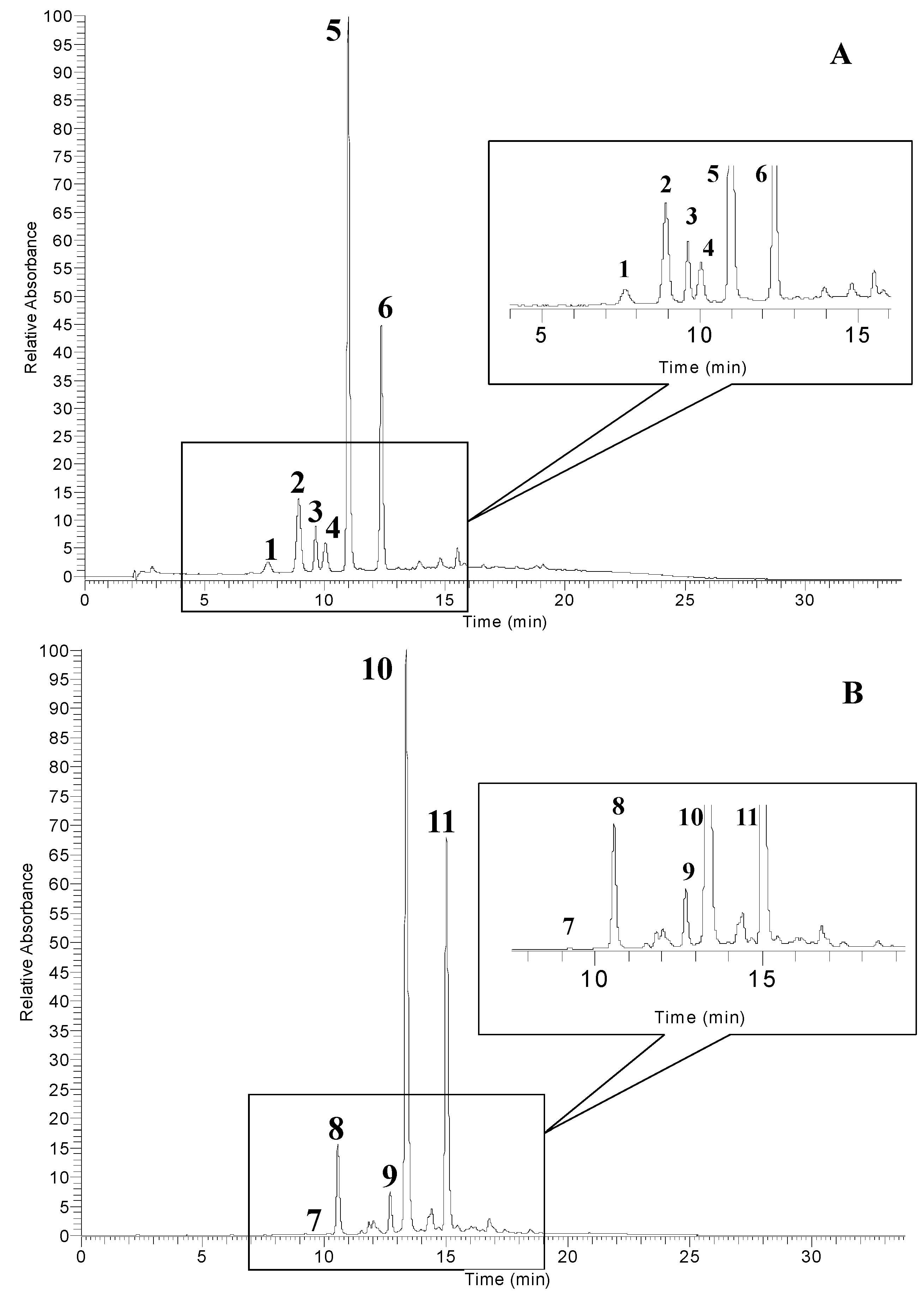
2.2. Identification of Anthocyanins by UPLC-PDA-ESI/MSn
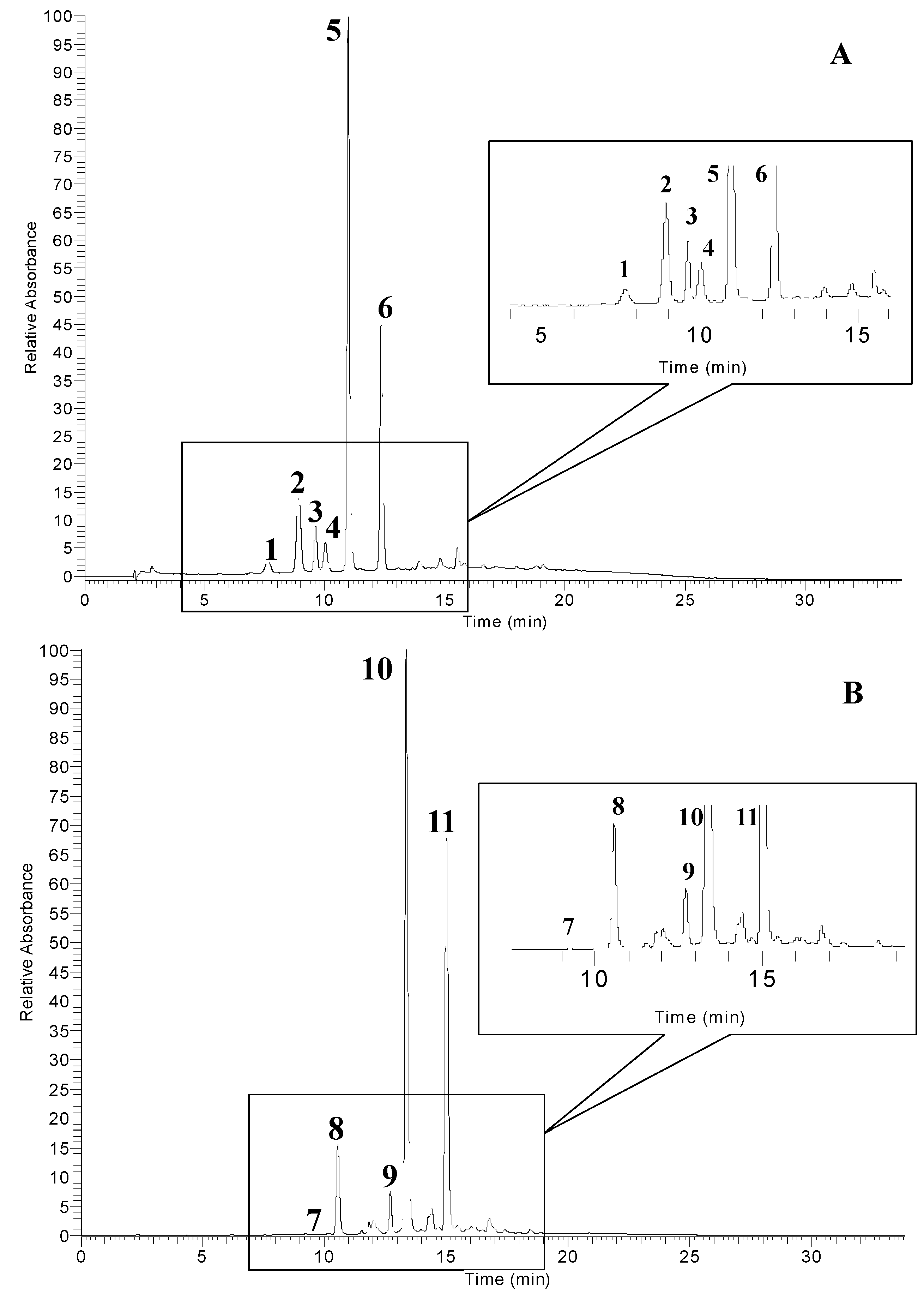
| Peak No. a | RT (min) | [M]+ (m/z) | MSn (m/z) | Anthocyanins | Relative Compositions b (%) |
|---|---|---|---|---|---|
| Pomegranate peel | |||||
| 1 | 7.6 | 627 | 465/303 | delphinidin 3,5-diglucoside | 3.37 |
| 2 | 9.1 | 611 | 449/287 | cyanidin 3,5-diglucoside | 12.41 |
| 3 | 10.0 | 595 | 433/271 | pelargonidin 3,5-diglucoside | 5.62 |
| 4 | 10.3 | 465 | 303 | delphinidin 3-glucoside | 4.62 |
| 5 | 11.0 | 449 | 287 | cyanidin 3-glucoside | 49.36 |
| 6 | 12.6 | 433 | 271 | pelargonidin 3-glucoside | 24.62 |
| Sumac fruit | |||||
| 7 | 10.2 | 465 | 303 | delphinidin 3-glucoside | 0.28 |
| 8 | 10.9 | 449 | 287 | cyanidin 3-glucoside | 7.84 |
| 9 | 12.7 | 601 | 287 | cyanidin 3-(2′′-galloyl) galactoside | 3.83 |
| 10 | 13.4 | 463 | 301 | 7-methyl-cyanidin 3-galactoside | 52.92 |
| 11 | 15.3 | 615 | 301 | 7-methyl-cyanidin 3-(2′′-galloyl)galactoside | 35.14 |
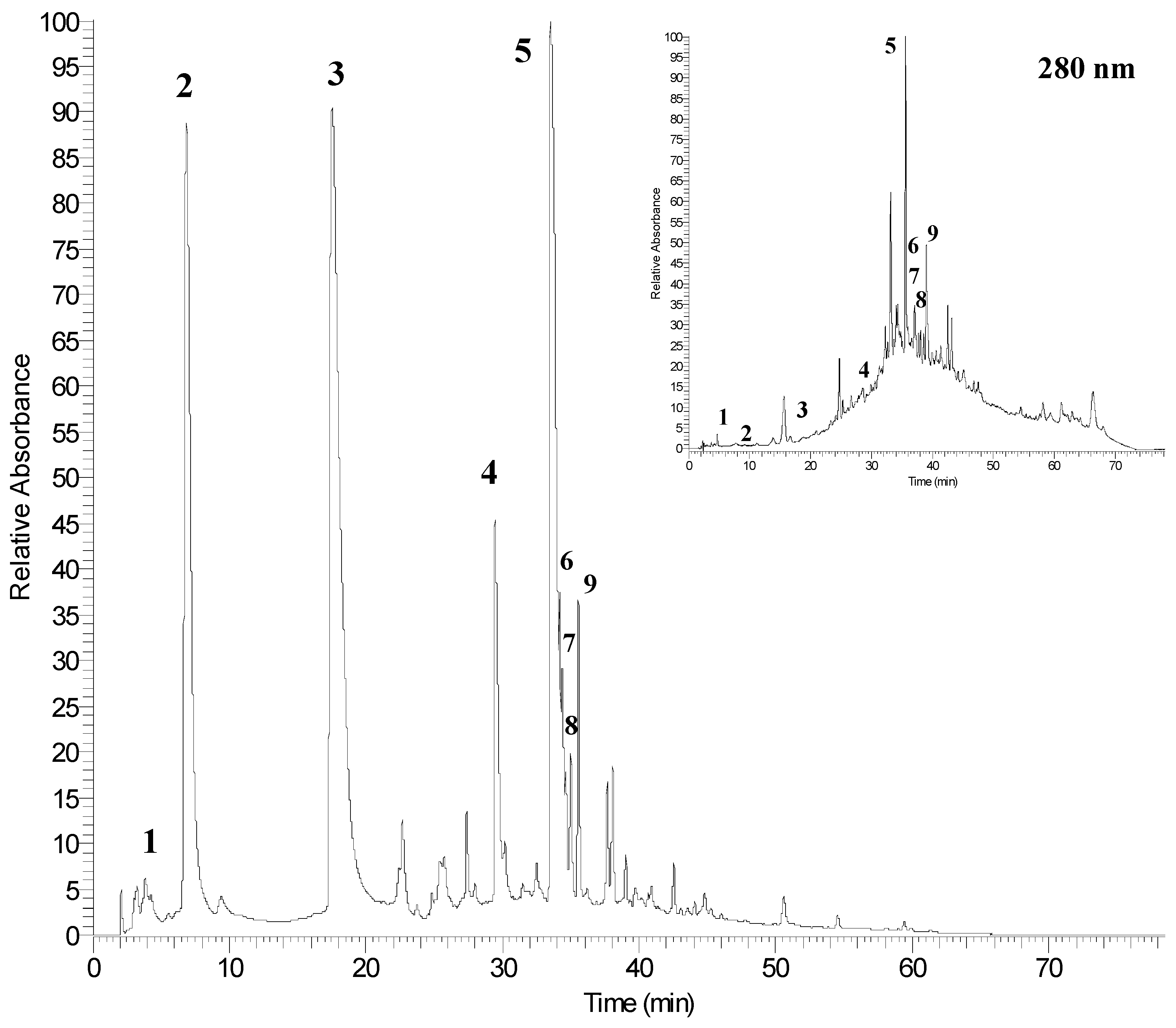
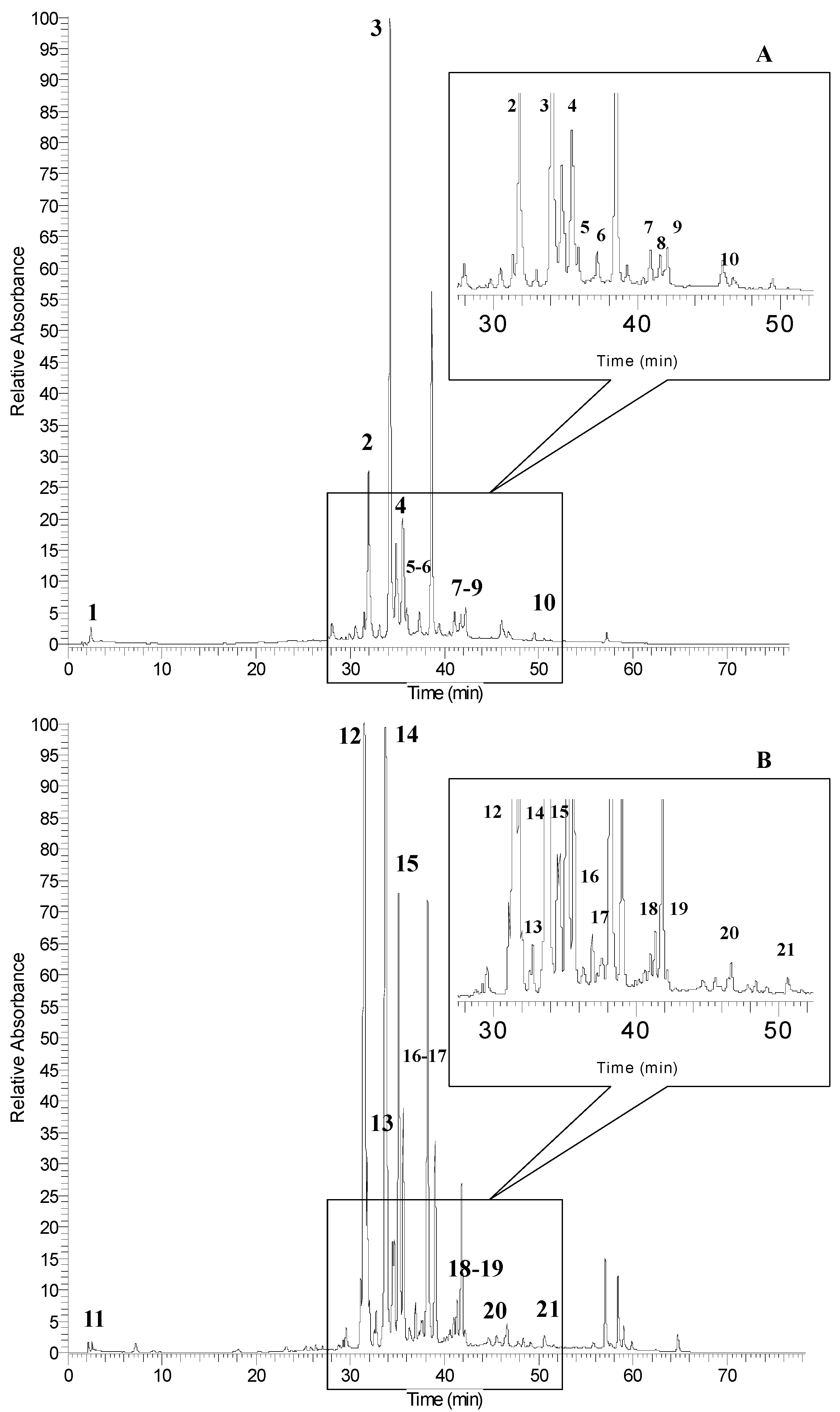
2.3. Identification of Phenolic Compounds by UPLC-PDA-ESI/MSn
| Peak No. a | RT (min) | [M − H]− (m/z) | MSn (m/z) | λmax | Phenolic Compounds |
|---|---|---|---|---|---|
| 1 | 2.5 | 169 | 125 | 269, 310 | gallic acid |
| 2 | 6.9 | 1083 | 781/601 | 258, 378 | punicalagin A |
| 3 | 17.5 | 1083 | 781/601 | 258, 378 | punicalagin B |
| 4 | 29.4 | 951 | 933/613 | 260, 365 | granatin B |
| 5 | 33.5 | 301 | 229/185 | 254, 367 | ellagic acid |
| 6 | 34.2 | - | 301 | 254, 361 | ellagic acid derivative |
| 7 | 34.6 | - | 301 | 254, 360 | ellagic acid derivative |
| 8 | 34.9 | - | 301 | 255, 362 | ellagic acid derivative |
| 9 | 35.5 | - | 301 | 254, 363 | ellagic acid derivative |

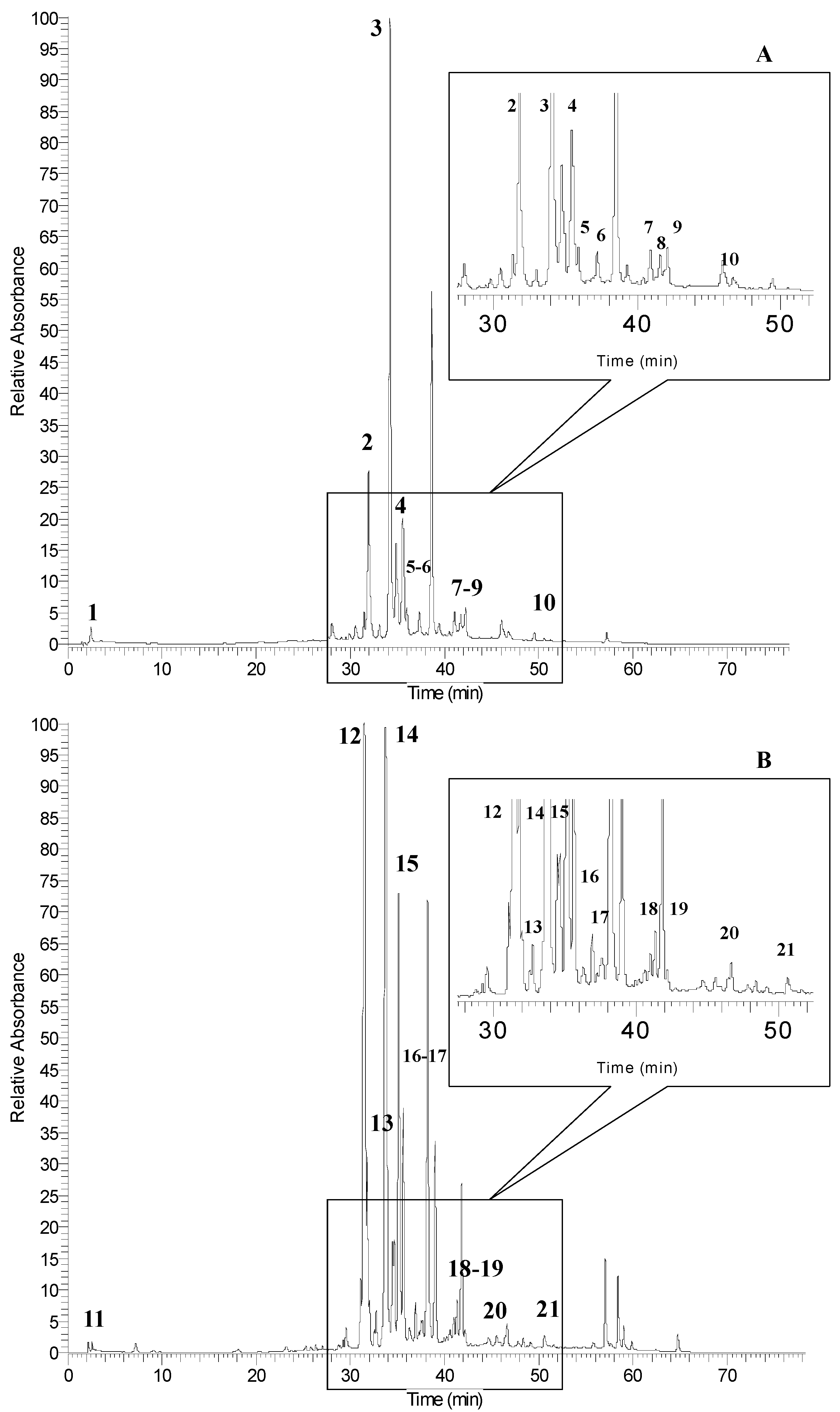
| Peak No. a | RT (min) | [M − H]− (m/z) | MSn (m/z) | λmax | Phenolic Compounds |
|---|---|---|---|---|---|
| Sumac Fruit | |||||
| 1 | 2.6 | 169 | 125 | 269, 310 | gallic acid |
| 2 | 31.9 | - | 301 | 255, 354 | quercetin derivative |
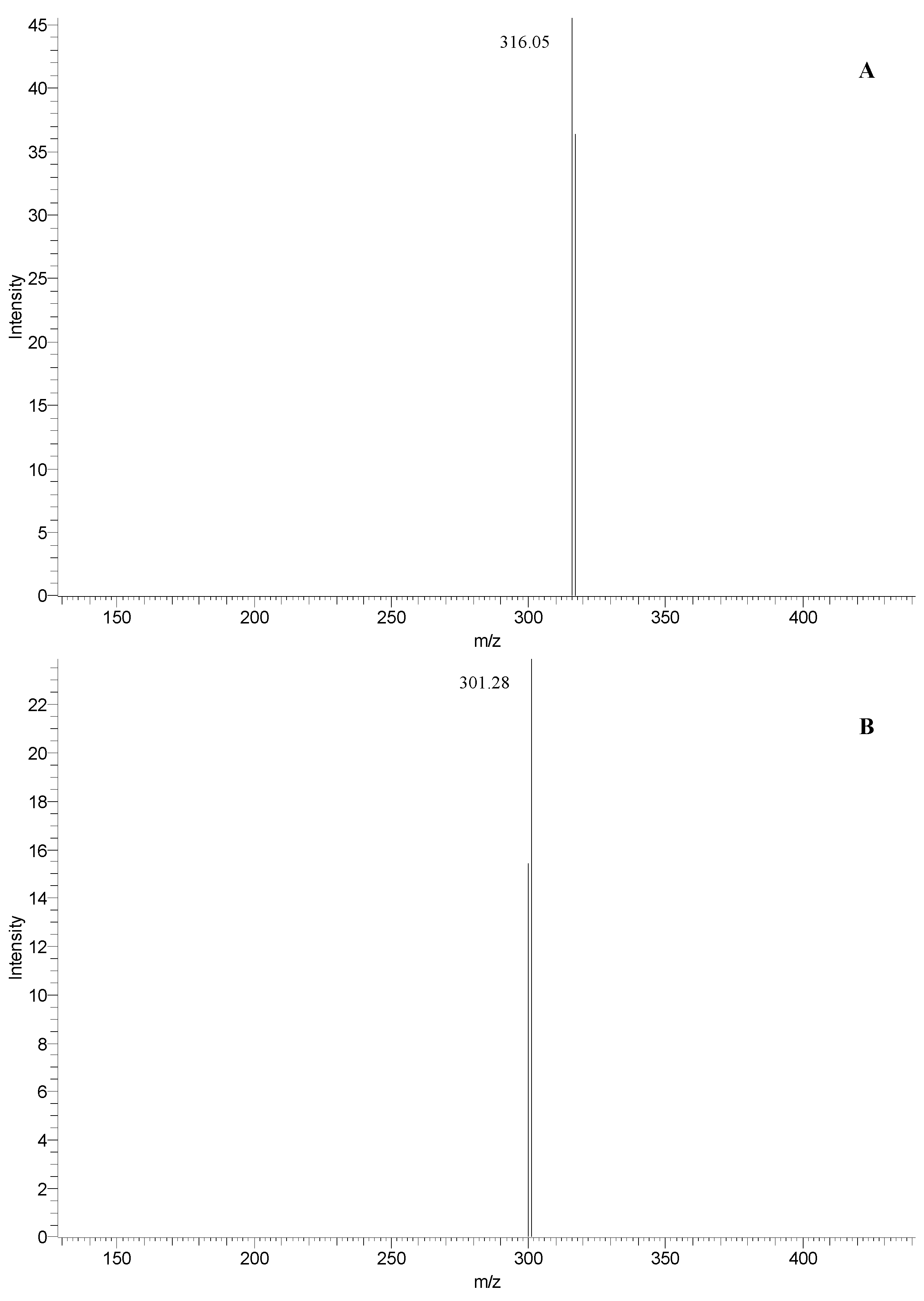
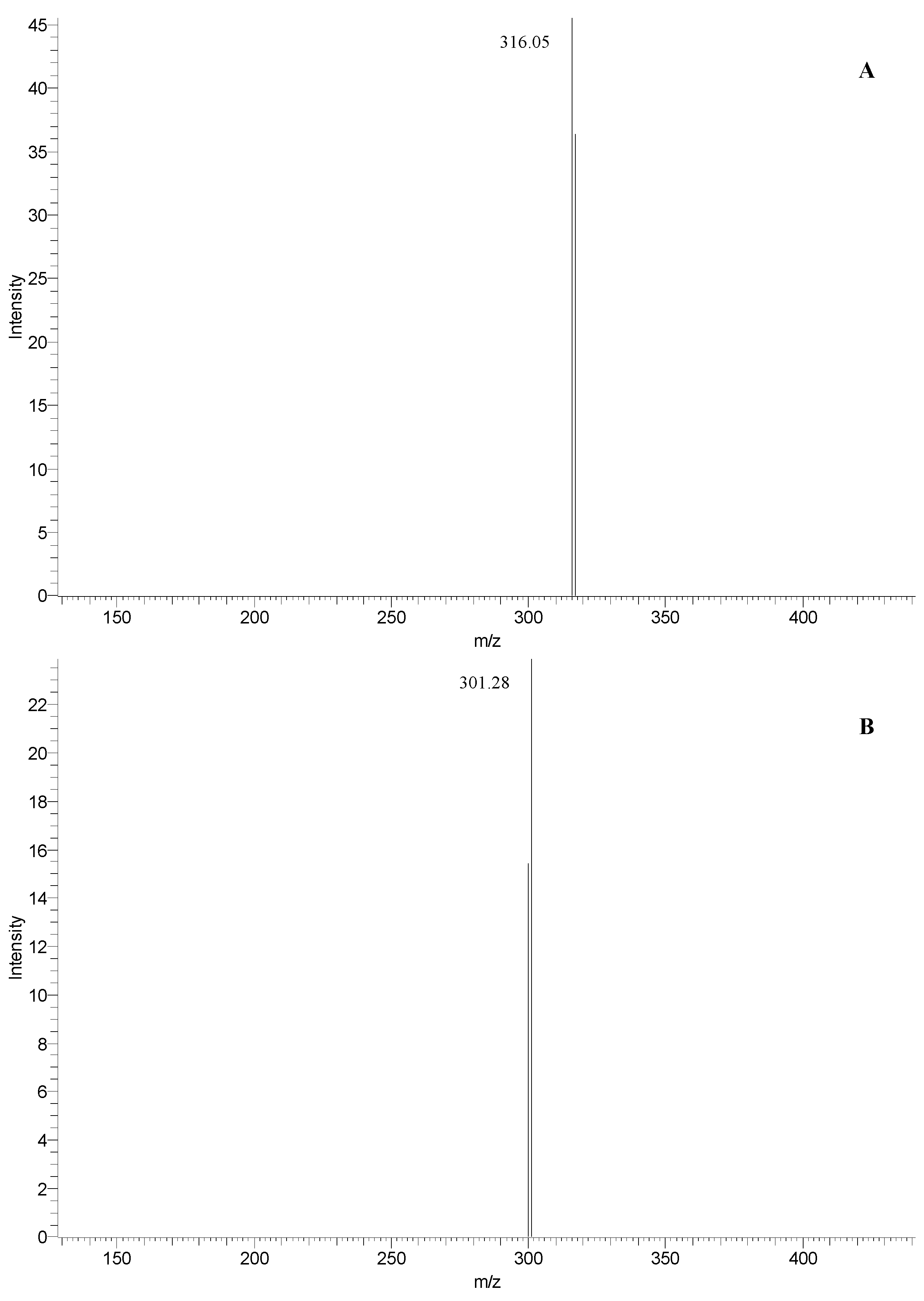
| 3 | 33.9 | 463 | 316 | 257, 366 | myricetin 3-rhamnoside (see Figure 4A) |
| 4 | 35.3 | 463 | 301 | 255, 351 | quercetin 3-glucoside (see Figure 4B) |
| 5 | 37.3 | 939 | 921/787/169 | 280 | pentagalloyl-glucoside |
| 6 | 39.9 | 1091 | 939/169 | 282 | hexagalloyl-glucoside |
| 7 | 41.9 | 1243 | 1091/169 | 281 | heptagalloyl-glucoside |
| 8 | 43.5 | 1395 | 1243/169 | 285 | octagalloyl-glucoside |
| 9 | 45.5 | 1547 | 1395/169 | 283 | nonagalloyl-glucoside |
| 10 | 50.5 | 1699 | 1547/169 | 278 | decagalloyl-glucoside |
| Sumac leaves | |||||
| 11 | 2.5 | 169 | 125 | 269, 310 | gallic acid |
| 12 | 31.4 | - | 316 | 255, 366 | myricetin derivative |
| 13 | 31.8 | - | 301 | 255, 346 | quercetin derivative |
| 14 | 33.9 | 463 | 316 | 255, 366 | myricetin 3-rhamnoside (see Figure 4A) |
| 15 | 35.3 | 463 | 301 | 255, 352 | quercetin 3-glucoside (see Figure 4B) |
| 16 | 37.1 | 939 | 921/787/169 | 280 | pentagalloyl-glucoside |
| 17 | 39.7 | 1091 | 939/169 | 280 | hexagalloyl-glucoside |
| 18 | 41.7 | 1243 | 1091/169 | 281 | heptagalloyl-glucoside |
| 19 | 43.2 | 1395 | 1243/169 | 285 | octagalloyl-glucoside |
| 20 | 45.4 | 1547 | 1395/169 | 284 | nonagalloyl-glucoside |
| 21 | 50.6 | 1699 | 1547/169 | 277 | decagalloyl-glucoside |
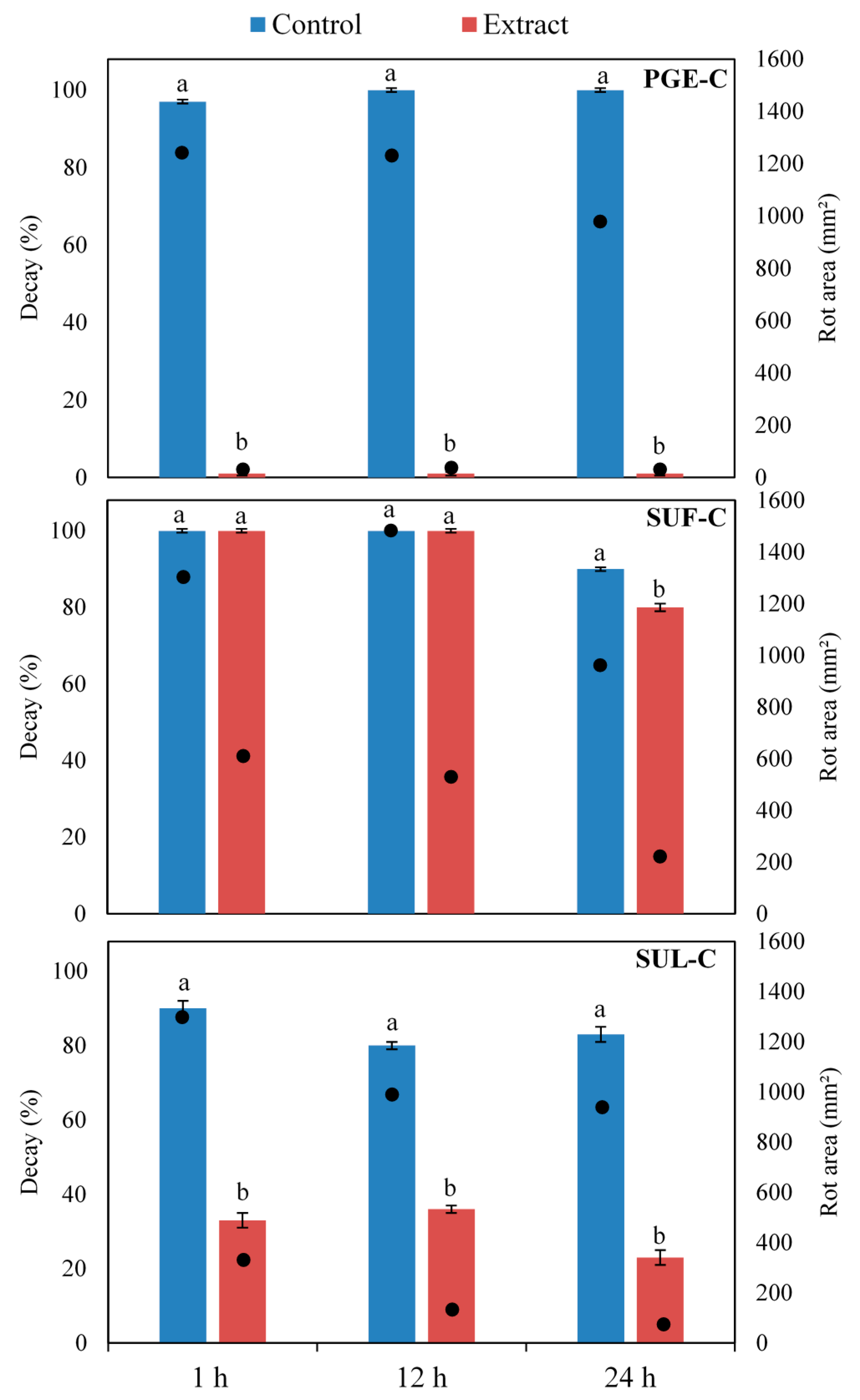
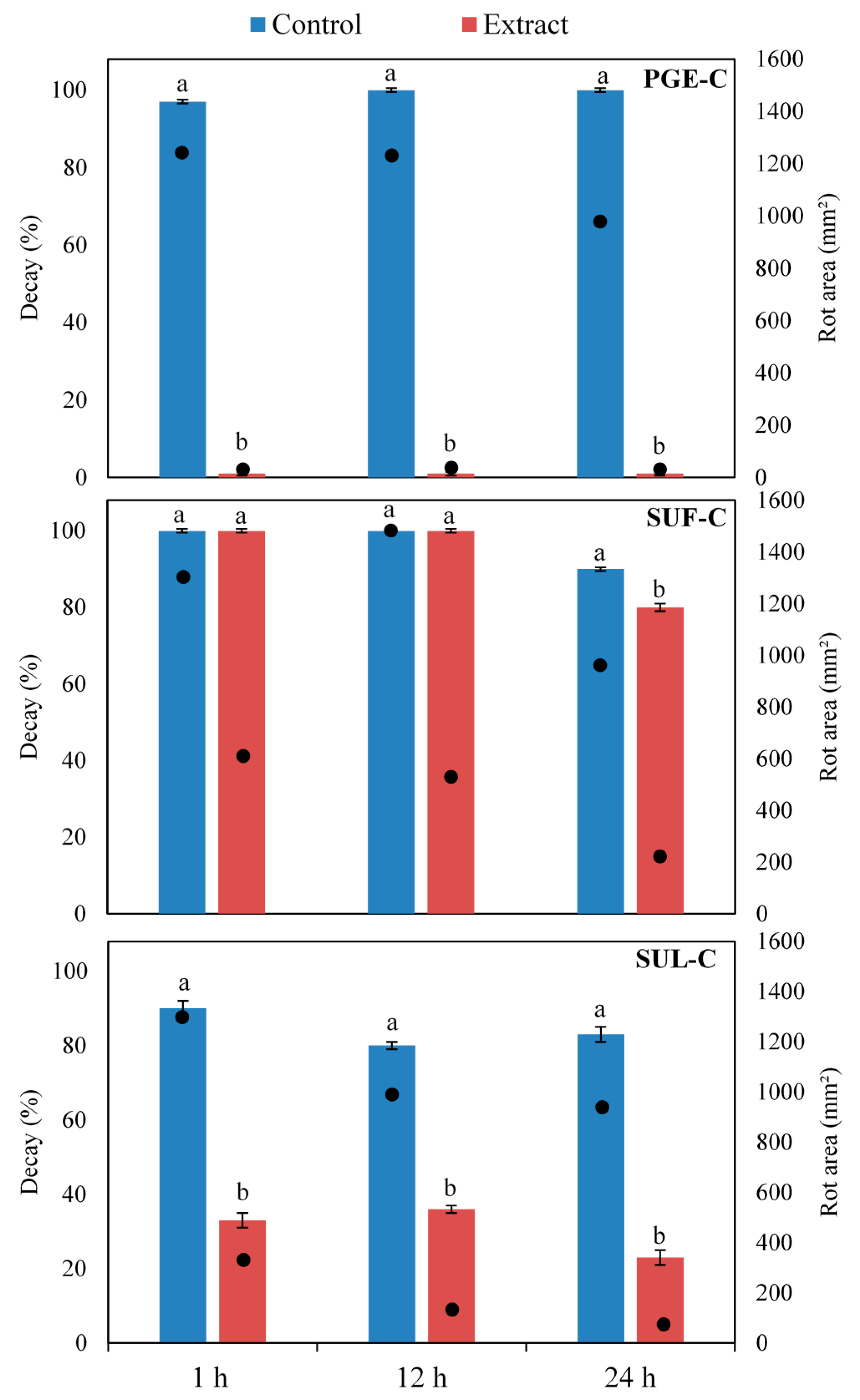
2.4. Efficacy of Sumac and Pomegranate Extracts against Botrytis cinerea on Artificially Inoculated Grape Berries
3. Experimental Section
3.1. Pomegranate and Sumac Extracts
3.2. Determination of Total Anthocyanins and Total Phenolic Content of the Plant Extracts
3.3. UPLC-PDA-ESI/MSn Analyses
3.4. Preparation of the Pathogen Inoculum
3.5. In-Vivo Assays of Extracts on Artificially Inoculated Grape Berries
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Glazer, I.; Masaphy, S.; Marciano, P.; Bar-Ilan, I.; Holland, D.; Kerem, Z.; Amir, R. Partial Identification of Antifungal Compounds from Punica granatum Peel Extracts. J. Agric. Food Chem. 2012, 60, 4841–4848. [Google Scholar] [CrossRef] [PubMed]
- Janisiewicz, W.J.; Korsten, L. Biological control of postharvest diseases of fruits. Ann. Rev. Phytopathol. 2002, 40, 411–441. [Google Scholar] [CrossRef] [PubMed]
- Dayan, F.E.; Cantrell, C.L.; Duke, S.O. Natural products in crop protection. Bioorg. Med. Chem. 2009, 17, 4022–4034. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Banos, S. Postharvest Decay: Control Strategies, 1st ed.; Academic Press, Elsevier: San Diego, CA, USA, 2014. [Google Scholar]
- Feliziani, E.; Romanazzi, G. Preharvest application of synthetic fungicides and alternative treatments to control postharvest decay of fruit. Stewart Postharvest Rev. 2013, 9, 1–6. [Google Scholar] [CrossRef]
- Smilanick, J.L.; Harstell, P.I.; Henson, D.; Fouse, D.C.; Assemi, M.; Harris, C.M. Inhibitory activity of sulphur dioxide on the germination of spores of Botrytis cinerea. Phytopathology 1990, 80, 217–220. [Google Scholar] [CrossRef]
- Gao, H.; Hu, X.; Liu, L.; Zhang, H.; Wang, S. Study on sensitivity of table grapes to SO2. Acta Hortic. 2003, 628, 541–545. [Google Scholar]
- Zoffoli, J.P.; Latorre, B.A.; Rodriguez, J.; Aguilera, J.M. Biological indicators to estimate the prevalence of gray mold and hairline cracks on table grapes cv. Thompson Seedless after cold storage. Postharvest Biol. Technol. 2009, 52, 126–133. [Google Scholar] [CrossRef]
- Timbo, B.; Koehler, K.M.; Wolyniak, C.; Klontz, K.C. Sulfites—A food and drug administration review of recalls and reported adverse events. J. Food Protect. 2004, 67, 1806–1811. [Google Scholar]
- Sanzani, S.M.; Schena, L.; de Cicco, V.; Ippolito, A. Early detection of Botrytis cinerea latent infections as a tool to improve postharvest quality of table grapes. Postharvest Biol. Technol. 2012, 68, 64–71. [Google Scholar] [CrossRef]
- Strano, M.C.; Calandra, M.; Aloisi, V.; Rapisarda, P.; Strano, T.; Ruberto, G. Hot water dipping treatments on Tarocco orange fruit and their effects on peel essential oil. Postharvest Biol. Technol. 2014, 94, 26–34. [Google Scholar] [CrossRef]
- Al-Zoreky, N.S. Antimicrobial activity of pomegranate (Punica granatum L.) fruit peels. Int. J. Food Microbiol. 2009, 134, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Machado, T.; Pinto, A.; Pinto, M.; Leal, I.; Silva, M.; Amaral, A.; Kuster, R.; Netto dosSantos, K. In vitro activity of Brazilian medicinal plants, naturally occurring naphthoquinones and their analogues, against methicillin-resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 2003, 21, 279–284. [Google Scholar] [CrossRef]
- Akhtar, S.; Ismail, T.; Fraternale, D.; Sestili, P. Pomegranate peel and peel extracts: Chemistry and food features. Food Chem. 2015, 174, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Fischer, U.A.; Carle, R.; Kammerer, D.R. Identification and quantification of phenolic compounds from pomegranate (Punica granatum L.) peel, mesocarp, aril and differently produced juices by HPLC-DAD-ESI/MSn. Food Chem. 2011, 127, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Dora, J.; Kumar, A.; Kumar, A. Pomegranate (Punica granatum)—Overview. Int. J. Pharm. Chem. Sci. 2012, 1, 1218–1222. [Google Scholar]
- Nonaka, G.; Nishioka, I.; Nishizawa, M.; Yamagishi, T.; Kashiwada, Y.; Dutschman, G.E.; Bodner, A.J.; Kilkuskie, R.E.; Cheng, Y.C.; Lee, K.H. Anti-AIDS agents, 2: Inhibitory effects of tannins on HIV reverse transcriptase and HIV replication in H9 lymphocyte cells. J. Nat. Prod. 1990, 53, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Chen, L.G.; Liang, W.L.; Wang, C.C. Anti-inflammatory effects of Punica granatum Linne in vitro and in vivo. Food Chem. 2010, 118, 315–322. [Google Scholar] [CrossRef]
- Tehranifar, A.; Selahvarzi, Y.; Kharrazi, M.; Bakhsh, V.J. High potential of agro-industrial by-products of pomegranate (Punica granatum L.) as the powerful antifungal and antioxidant substances. Ind. Crops Prod. 2011, 34, 1523–1527. [Google Scholar] [CrossRef]
- Wetherilt, H.; Pala, M. Herbs and spices indigenous to Turkey. In Spices, Herbs and Edible Fungi. Developments in Food Science; Charalambous, G., Ed.; Elsevier: Amsterdam, The Netherlands, 1994; Volume 34, pp. 285–307. [Google Scholar]
- Rayne, S.; Mazza, G. Biological Activities of Extracts from Sumac (Rhus spp.): A Review. Plant Foods Hum. Nutr. 2007, 62, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, A. Rhus coriaria Linn, a plant of medicinal, nutritional and industrial importance: A review. J. Anim. Plant Sci. 2012, 22, 505–512. [Google Scholar]
- Schena, L.; Nigro, F.; Ippolito, A. Natural antimicrobials to improve storage and shelf-life of fresh fruit, vegetables and cut flowers. In Microbial Biotechnology in Horticulture; Ray, R.C., Ward, O.P., Eds.; Oxford & IBH Publishing Co.: New Delhi, India, 2007; Volume 2, pp. 259–303. [Google Scholar]
- Romeo, F.V.; de Luca, S.; Piscopo, A.; de Salvo, E.; Poiana, M. Effect of some essential oils as natural food preservatives on commercial grated carrots. J. Essent. Oil Res. 2010, 22, 283–287. [Google Scholar] [CrossRef]
- Campolo, O.; Romeo, F.V.; Malacrinò, A.; Laudani, F.; Carpinteri, G.; Fabroni, S.; Rapisarda, P.; Palmeri, V. Effects of inert dusts applied alone and in combination with sweet orange essential oil against Rhyzopertha dominica (Coleoptera: Bostrichidae) and wheat microbial population. Ind. Crops Prod. 2014, 61, 361–369. [Google Scholar] [CrossRef]
- Sanzani, S.M.; Schena, L.; Ippolito, A. Effectiveness of Phenolic Compounds against Citrus Green Mould. Molecules 2014, 19, 12500–12508. [Google Scholar] [CrossRef] [PubMed]
- Bakkiyaraj, D.; Nandhini, J.R.; Malathy, B.; Pandian, S.K. The anti-biofilm potential of pomegranate (Punica granatum L.) extract against human bacterial and fungal pathogens. Biofouling 2013, 29, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Tayel, A.A.; El-Baz, A.F.; Salem, M.F.; El-Hadary, M.H. Potential applications of pomegranate peel extract for the control of citrus green mould. J. Plant Dis. Protect. 2009, 116, 252–256. [Google Scholar]
- Panico, A.; Cardile, V.; Santagati, N.A.; Messina, R. Antioxidant and protective effects of sumac leaves on chondrocytes. J. Med. Plants Res. 2009, 3, 855–861. [Google Scholar]
- Kosar, M.; Bozan, B.; Temelli, F.; Baser, K.H.C. Antioxidant activity and phenolic composition of sumac (Rhus coriaria L.) extracts. Food Chem. 2007, 103, 952–959. [Google Scholar] [CrossRef]
- Pala, C.U.; Toklucu, A.K. Effect of UV-C light on anthocyanin content and other quality parameters of pomegranate juice. J. Food Compos. Anal. 2011, 24, 790–795. [Google Scholar] [CrossRef]
- Qu, W.; Breksa, A.P., III; Pan, Z.; Ma, H. Quantitative determination of major polyphenol constituents in pomegranate products. Food Chem. 2012, 132, 1585–1591. [Google Scholar] [CrossRef]
- Borges, G.; Crozier, A. HPLC-PDA-MS fingerprinting to assess the authenticity of pomegranate beverages. Food Chem. 2012, 135, 1863–1867. [Google Scholar] [CrossRef] [PubMed]
- Mena, P.; Calani, L.; Dall’Asta, C.; Galaverna, G.; Garcia-Viguera, C.; Bruni, R.; Crozier, A.; del Rio, D. Rapid and comprehensive evaluation of (poly)phenolic compounds in pomegranate (Punica granatum L.) juice by UHPLC-MSn. Molecules 2012, 17, 14821–14840. [Google Scholar] [CrossRef] [PubMed]
- Abu-Reidah, I.M.; Ali-Shtayeh, M.S.; Jamous, R.M.; Arraez-Roman, D.; Segura-Carretero, A. HPLC-DAD-ESI-MS/MS screening of bioactive components from Rhus coriaria L. (sumac) fruits. Food Chem. 2015, 166, 179–191. [Google Scholar] [CrossRef] [PubMed]
- Kirby, C.W.; Wu, T.; Tsao, R.; McCallum, J.L. Isolation and structural characterization of unusual pyranoanthocyanins and related anthocyanins from Staghorn sumac (Rhus typhina L.) via UPLC-ESI-MS, 1H, 13C, and 2D NMR spectroscopy. Phytochemistry 2013, 94, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Gil, M.I.; Tomas-Barberan, F.A.; Hess-Pierce, B.; Holcroft, D.M.; Kader, A.A. Antioxidant activity of pomegranate juice and its relationship with phenolic composition and processing. J. Agric. Food Chem. 2000, 48, 4581–4589. [Google Scholar] [CrossRef] [PubMed]
- Regazzoni, L.; Arlandini, E.; Garzon, D.; Santagati, N.A.; Beretta, G.; Maffei Facino, R. A rapid profiling of gallotannins and flavonoids of the aqueous extract of Rhus coriaria L. by flow injection analysis with high-resolution mass spectrometry assisted with database searching. J. Pharm. Biomed. 2013, 72, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Berardini, N.; Carle, R.; Schieber, A. Characterization of gallotannins and benzophenone derivatives from mango (Mangifera indica L. cv. Tommy Atkins) peels, pulp and kernels by high-performance liquid chromatography/electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Sanzani, S.M.; Schena, L.; Nigro, F.; de Girolamo, A.; Ippolito, A. Effect of quercetin and umbelliferone on the transcript level of Penicillium expansum genes involved in patulin biosynthesis. Eur. J. Plant Pathol. 2009, 125, 223–233. [Google Scholar] [CrossRef]
- Sanzani, S.M.; Schena, L.; de Girolamo, A.; Ippolito, A.; González-Candelas, L. Characterization of genes associated with induced resistance against Penicillium expansum in apple fruit treated with quercetin. Postharvest Biol. Technol. 2010, 56, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rapisarda, P.; Fanella, F.; Maccarone, E. Reliability of analytical methods for determining anthocyanins in blood orange juices. J. Agric. Food Chem. 2000, 48, 2249–2252. [Google Scholar] [CrossRef] [PubMed]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventos, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of Folin-Ciocalteau reagent. Methods Enzymol. 1999, 299, 152–178. [Google Scholar]
- Sample Availability: Samples of the pomegranate peel and sumac fruit and leaf extracts are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romeo, F.V.; Ballistreri, G.; Fabroni, S.; Pangallo, S.; Nicosia, M.G.L.D.; Schena, L.; Rapisarda, P. Chemical Characterization of Different Sumac and Pomegranate Extracts Effective against Botrytis cinerea Rots. Molecules 2015, 20, 11941-11958. https://doi.org/10.3390/molecules200711941
Romeo FV, Ballistreri G, Fabroni S, Pangallo S, Nicosia MGLD, Schena L, Rapisarda P. Chemical Characterization of Different Sumac and Pomegranate Extracts Effective against Botrytis cinerea Rots. Molecules. 2015; 20(7):11941-11958. https://doi.org/10.3390/molecules200711941
Chicago/Turabian StyleRomeo, Flora V., Gabriele Ballistreri, Simona Fabroni, Sonia Pangallo, Maria Giulia Li Destri Nicosia, Leonardo Schena, and Paolo Rapisarda. 2015. "Chemical Characterization of Different Sumac and Pomegranate Extracts Effective against Botrytis cinerea Rots" Molecules 20, no. 7: 11941-11958. https://doi.org/10.3390/molecules200711941
APA StyleRomeo, F. V., Ballistreri, G., Fabroni, S., Pangallo, S., Nicosia, M. G. L. D., Schena, L., & Rapisarda, P. (2015). Chemical Characterization of Different Sumac and Pomegranate Extracts Effective against Botrytis cinerea Rots. Molecules, 20(7), 11941-11958. https://doi.org/10.3390/molecules200711941