
Pretreatment with Relaxin Does Not Restore NO-Mediated Modulation of Calcium Signal in Coronary Endothelial Cells Isolated from Spontaneously Hypertensive Rats


 ,
, 

Abstract
:
1. Introduction
2. Results
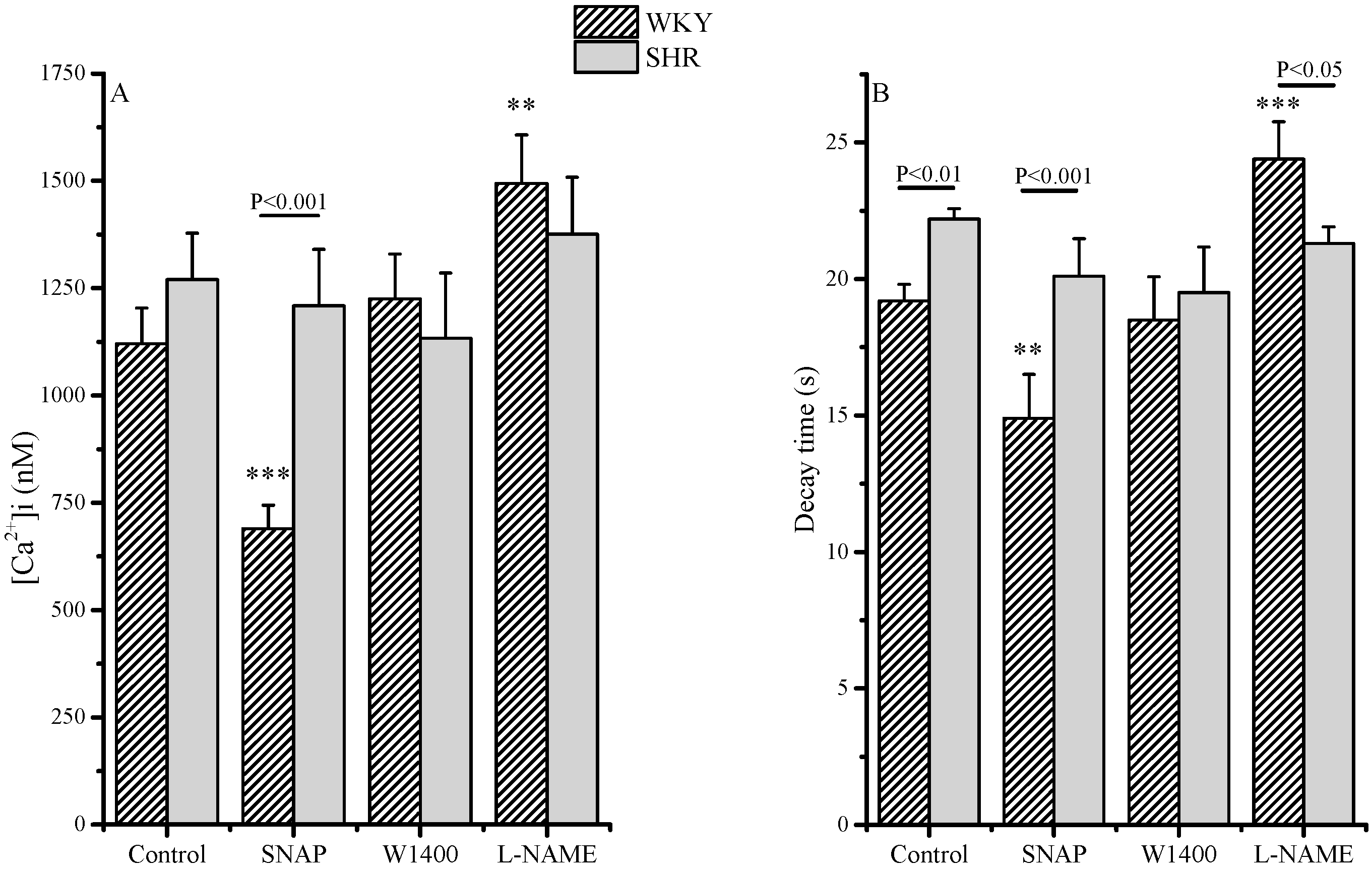
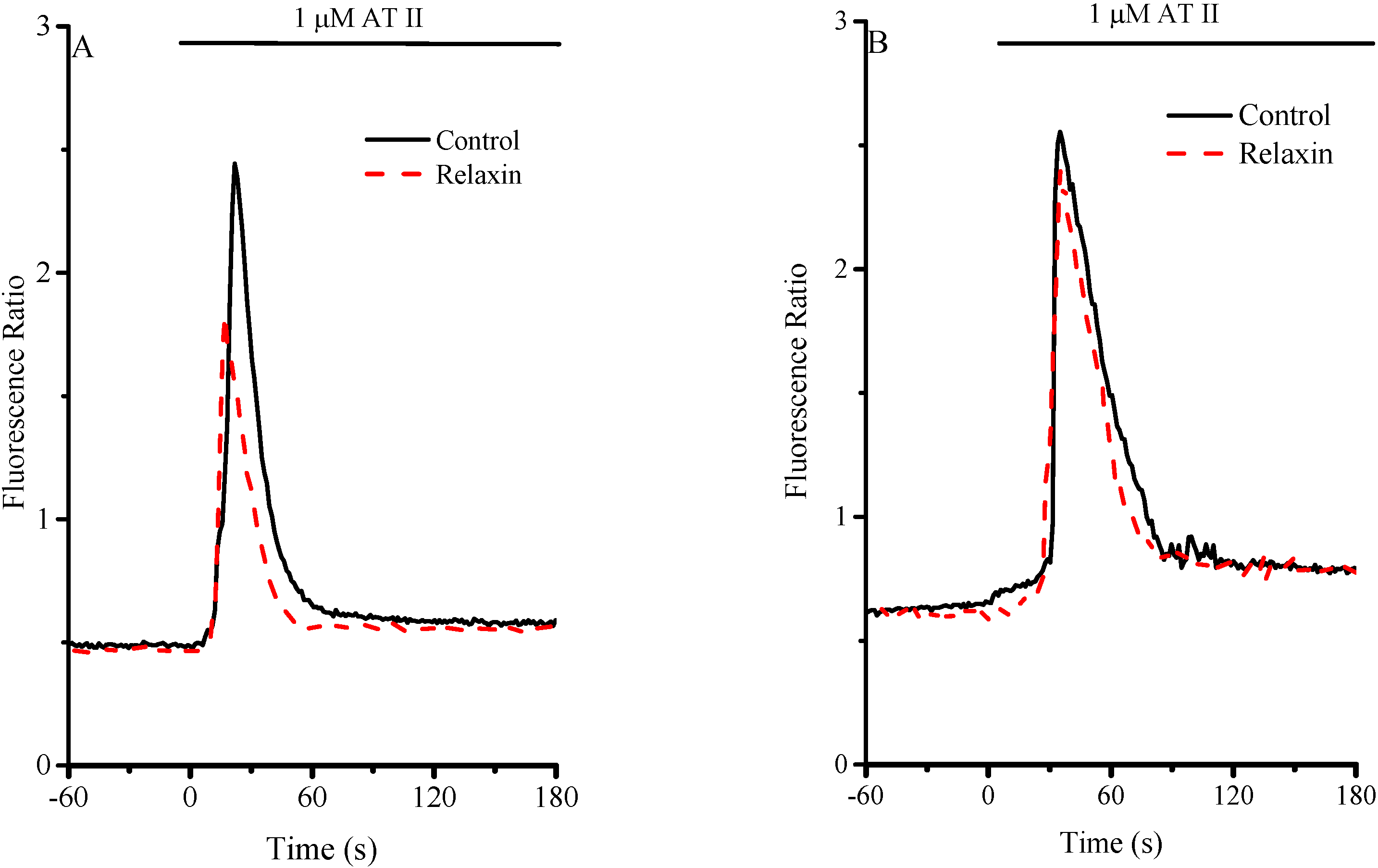
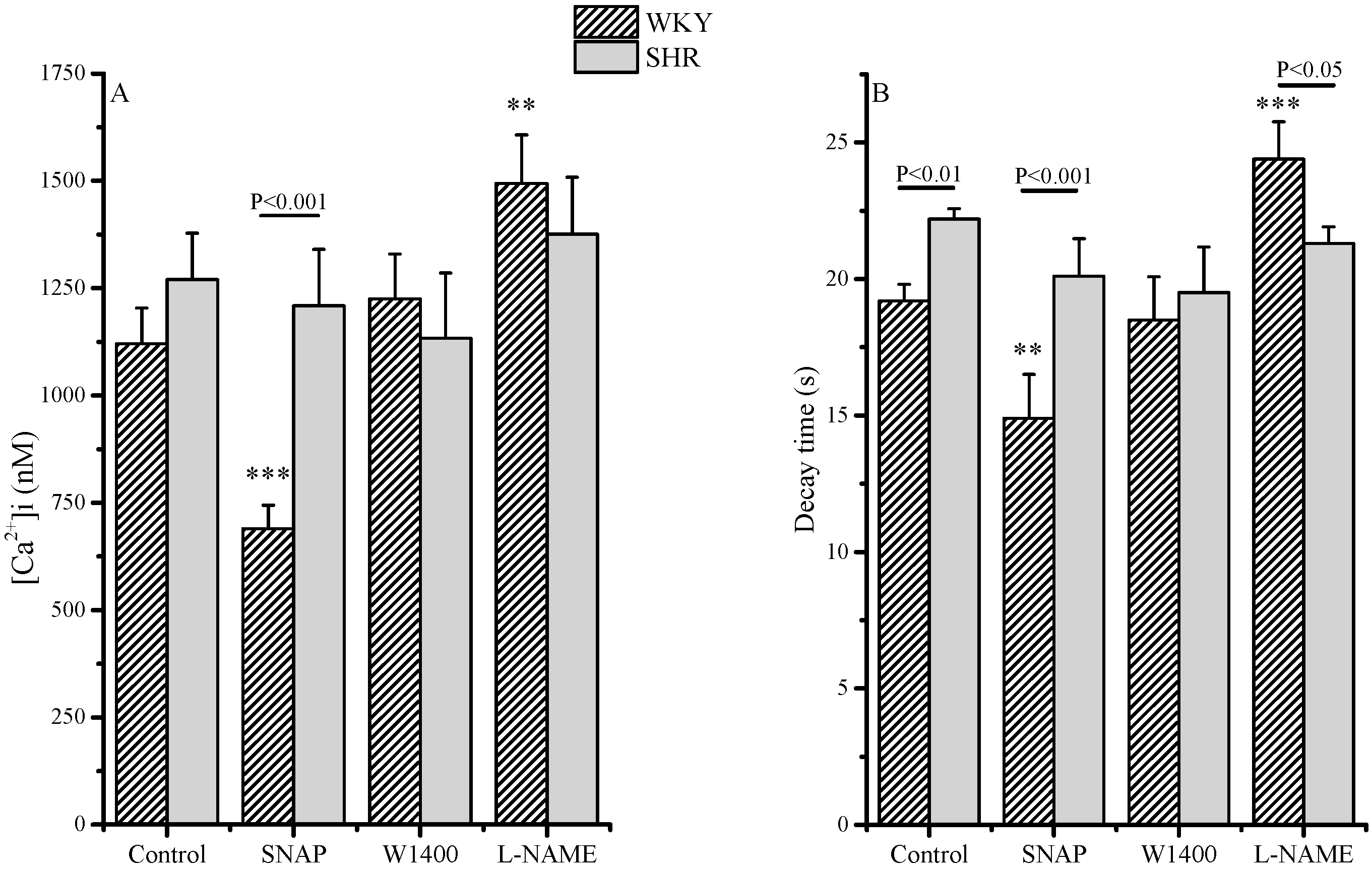
2.1. Intracellular Ca2+ Control Conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delta [Ca2+]i (nM) | ||
| WKY | SHR | |
| Control (basal) | 0 | 0 |
| SNAP | −430 | −61 |
| W1400 | +150 | −137 |
| L-NAME | +374 | +106 |
| RLX-pretreated | −223 | +3 |
| Delta Decay Time (s) | ||
| WKY | SHR | |
| Control (basal) | 0 | 0 |
| SNAP | −4.3 | −2.1 |
| W1400 | −0.7 | −2.7 |
| L-NAME | +5.2 | −0.9 |
| RLX-pretreated | −5 | −1.9 |
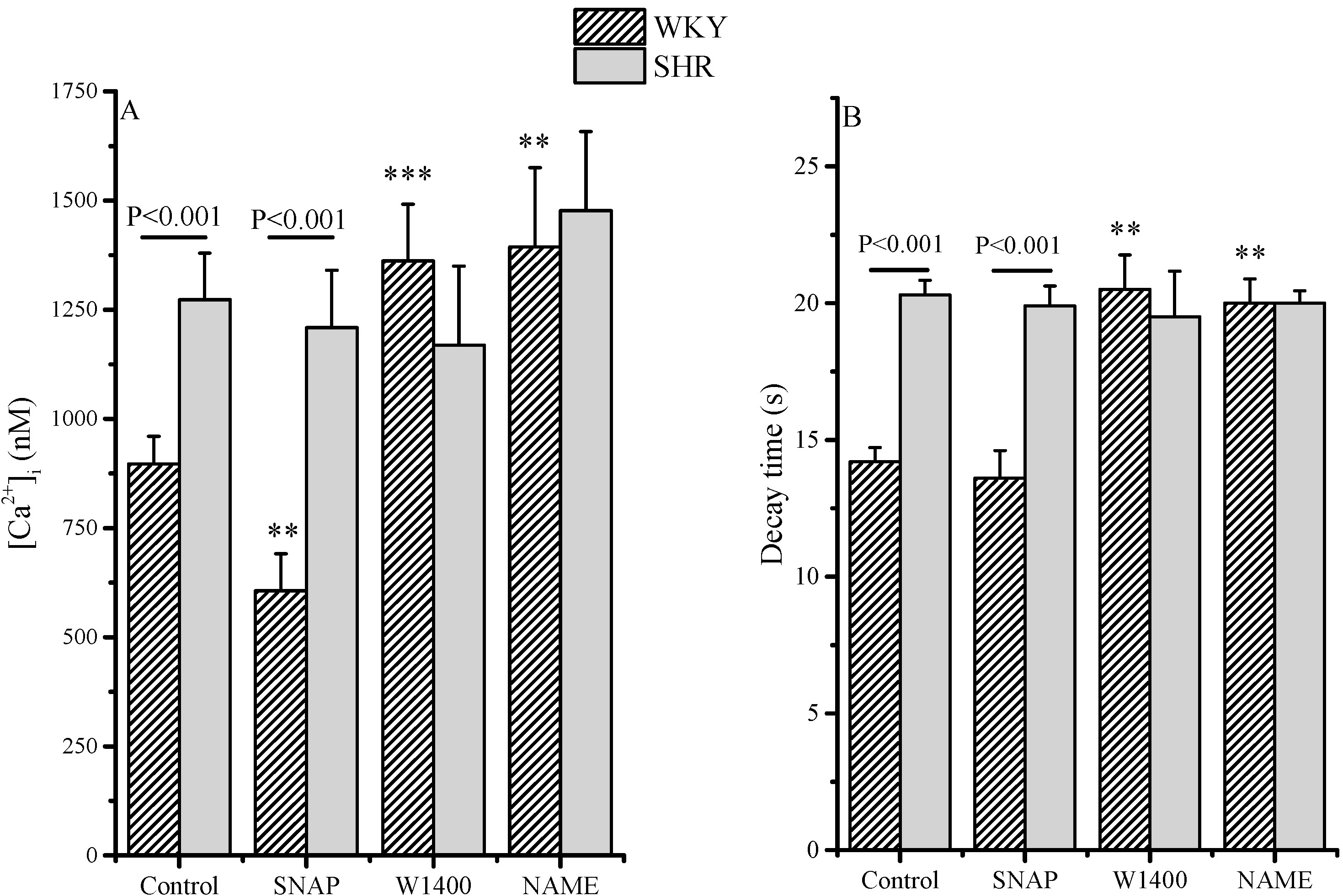
2.2. Intracellular [Ca2+]: RLX Effects

| Delta [Ca2+]i (nM) | ||
| WKY | SHR | |
| Control (RLX-pretreated) | 0 | 0 |
| SNAP | −281 | −64 |
| W1400 | +465 | −104 |
| L-NAME | +495 | +204 |
| Control (basal) | +223 | −3 |
| Delta Decay Time (s) | ||
| WKY | SHR | |
| Control (RLX-pretreated) | 0 | 0 |
| SNAP | −0.6 | −0.4 |
| W1400 | +6.3 | −0.8 |
| L-NAME | +5.8 | −0.3 |
| Control (basal) | +5 | +1.9 |
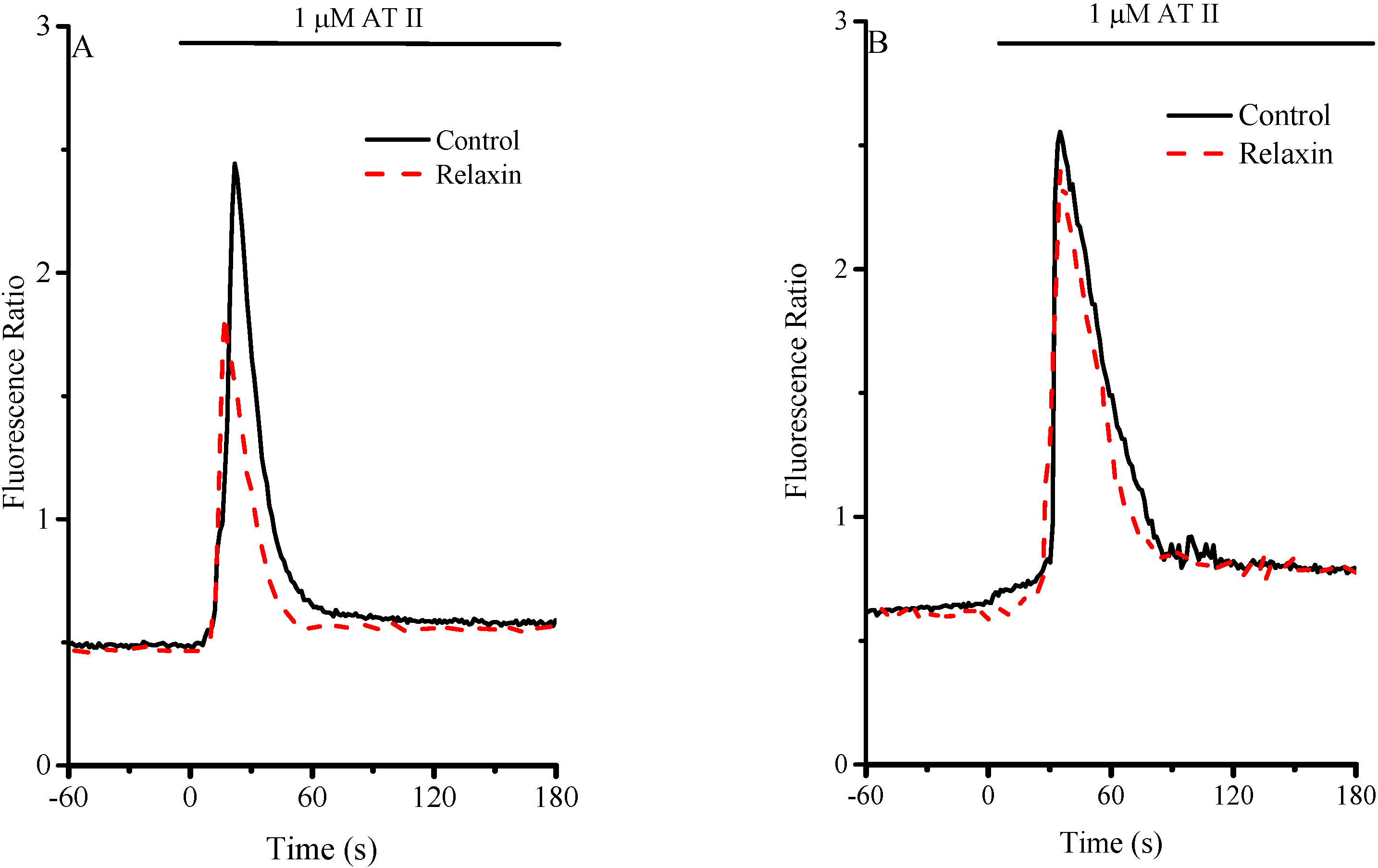
2.3. Effect of RLX on α-Thrombin-Induced Calcium Transient
| Control (basal) | WKY | SHR |
|---|---|---|
| [Ca2+]i (Maximal value) | 1820 ± 185.3 nM | 1901 ± 178.6 nM |
| Decay time | 18.1 ± 0.83 s | 19.0 ± 0.64 s |
| 24h RLX-Pretreated | ||
| [Ca2+]i (Maximal value) | 1168 ± 146.4 nM **, §§ | 1841 ± 206.1 nM |
| Decay time | 13.4 ± 1.19 s ***, §§ | 17.5 ± 2.03 s |
2.4. NO Production in WKY and SHR Endothelial Cells Pretreated with Relaxin
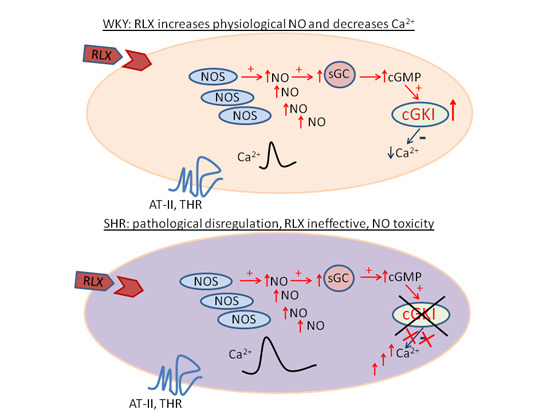
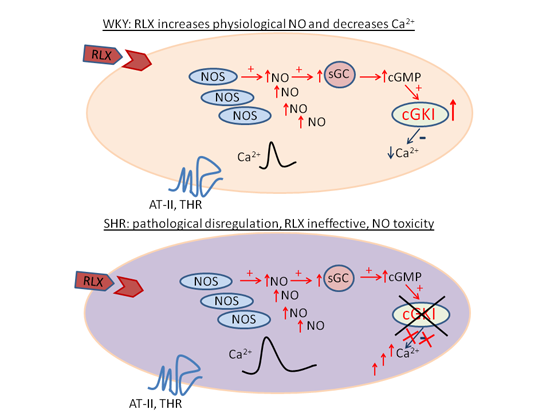
3. Discussion
4. Experimental Section
4.1. Chemicals
4.2. Isolation and Culture of Rat Coronary Endothelial (RCE) Cells
4.3. Determination of Intracellular Ca2+
4.4. Calculations and Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moncada, S.; Palmer, R.M.J.; Higgs, E.A. Nitric oxide physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991, 43, 109–142. [Google Scholar] [PubMed]
- Pacher, P.; Beckman, S.J.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Baccari, M.C.; Bani, D. Relaxin and nitric oxide signalling. Curr. Protein Pept. Sci. 2008, 9, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Nistri, S.; Bigazzi, M.; Bani, D. Relaxin as a cardiovascular hormone. Physiology, pathophysiology and therapeutic promises. Cardiovasc. Hematol. Agents Med. Chem. (CHA-MC) 2007, 5, 101–108. [Google Scholar]
- Bani, D. Relaxin as a natural agent for vascular health. Vasc. Health Risk Manag. 2008, 4, 515–524. [Google Scholar] [PubMed]
- Du, X.J.; Bathgate, R.A.D.; Samuel, C.S.; Dart, A.M.; Summers, J.R. Cardiovascular effects of relaxin: From basic science to clinical therapy. Nat. Rev. Cardiol. 2010, 7, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Chen-Izu, Y.; Chen, L.; Banyasz, T.; McCulle, S.L.; Norton, B.; Scharf, S.M.; Agarwal, A.; Patwardhan, A.; Izu, L.T.; Balke, C.W. Hypertension-induced remodeling of cardiac excitation-contraction coupling in ventricular myocytes occurs prior to hypertrophy development. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3301–H3310. [Google Scholar] [CrossRef] [PubMed]
- Ruetten, H.; Zabel, U.; Linz, W.; Schmidt, H.H. Downregulation of soluble guanylyl cyclase in young and aging spontaneously hypertensive rats. Circ. Res. 1999, 85, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.F.; Davis, C.W.; Tse, J. Depressed cardiac cyclic GMP-dependent protein kinase in spontaneously hypertensive rats and its further depression by guanethidine. Nature 1976, 261, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Ecker, T.; Göbel, C.; Hullin, R.; Rettig, R.; Seitz, G.; Hofmann, F. Decreased cardiac concentration of cGMP kinase in hypertensive animals. An index for cardiac vascularization? Circ. Res. 1989, 65, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Mazzetti, L.; Ruocco, C.; Giovannelli, L.; Ciuffi, M.; Franchi-Micheli, S.; Marra, F.; Zilletti, L.; Failli, P. Guanosine 3′,5′-cyclic monophosphate-dependent pathway alterations in ventricular cardiomyocytes of spontaneously hypertensive rats. Br. J. Pharmacol. 2001, 134, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Nistri, S.; di Cesare Mannelli, L.; Mazzetti, L.; Feil, R.; Bani, D.; Failli, P. Restoring nitric oxide cytosolic calcium regulation by cyclic guanosine monophosphate protein kinase I alpha transfection in coronary endothelial cells of spontaneously hypertensive rats. J. Vasc. Res. 2012, 49, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare Mannelli, L.; Nistri, S.; Mazzetti, L.; Bani, D.; Feil, R.; Failli, P. Altered nitric oxide calcium responsiveness of aortic smooth muscle cells in spontaneously hypertensive rats depends on low expression of cyclic guanosine monophosphate-dependent protein kinase type I. J. Hypertens. 2009, 27, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- St-Louis, J.; Massicotte, G. Chronic decrease of blood pressure by rat relaxin in spontaneously hypertensive rats. Life Sci. 1985, 37, 1351–1357. [Google Scholar] [CrossRef]
- Massicotte, G.; Parent, A.; St-Louis, J. Blunted responses to vasoconstrictors in mesenteric vasculature but not in portal vein of spontaneously hypertensive rats treated with relaxin. Proc. Soc. Exp. Biol. Med. 1989, 190, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Chakravorty, A.; Bathgate, R.A.; Dart, A.M.; Du, X.J. Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. Hypertension 2010, 55, 1260–1266. [Google Scholar] [CrossRef] [PubMed]
- Failli, P.; Nistri, S.; Quattrone, S.; Mazzetti, L.; Bigazzi, M.; Sacchi, T.B.; Bani, D. Relaxin up-regulates inducible nitric oxide synthase expression and nitric oxide generation in rat coronary endothelial cells. FASEB J. 2002, 16, 252–254. [Google Scholar] [CrossRef] [PubMed]
- Bayraktutan, U.; Ulker, S. Effects of angiotensin II on nitric oxide generation in proliferating and quiescent rat coronary microvascular endothelial cells. Hypertens. Res. 2003, 26, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Failli, P.; Fazzini, A.; Ruocco, C.; Mazzetti, L.; Cecchi, E.; Giovannelli, L.; Marra, F.; Milani, S.; Giotti, A. Lack of nitric oxide- and guanosine 3′:5′-cyclic monophosphate-dependent regulation of alpha thrombin-induced calcium transient in endothelial cells of spontaneously hypertensive rat hearts. Br. J. Pharmacol. 2000, 130, 1468–1476. [Google Scholar] [CrossRef] [PubMed]
- Failli, P.; Nistri, S.; Mazzetti, L.; Chiappini, L.; Bani, D. Effects of relaxin on vascular smooth muscle and endothelial cells in normotensive and hypertensive rats. Ann. N. Y. Acad. Sci. 2005, 1041, 311–313. [Google Scholar] [CrossRef] [PubMed]
- Lekgabe, E.D.; Kiriazis, H.; Zhao, C.; Xu, Q.; Moore, X.L.; Su, Y.; Bathgate, R.A.; Du, X.J.; Samuel, C.S. Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension 2005, 46, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Halls, M.L.; Bathgate, R.A.; Summers, R.J. Relaxin family peptide receptors RXFP1 and RXFP2 modulate cyclic AMP signaling by distinct mechanisms. Mol. Pharmacol. 2006, 70, 214–226. [Google Scholar] [PubMed]
- Bani, D.; Failli, P.; Bello, M.G.; Thiemermann, C.; Bani Sacchi, T.; Bigazzi, M.; Masini, E. Relaxin activates the l-arginine-nitric oxide pathway in vascular smooth muscle cells in culture. Hypertension 1998, 31, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Bani, D.; Baccari, M.C.; Nistri, S.; Calamai, F.; Bigazzi, M.; Bani Sacchi, T. Relaxin upregulates the nitric oxide biosynthetic pathway in the mouse uterus. Involvement in the inhibition of myometrial contractility. Endocrinology 1999, 140, 4434–4441. [Google Scholar] [PubMed]
- Bani, D.; Baccari, M.C.; Quattrone, S.; Nistri, S.; Calamai, F.; Bigazzi, M.; Bani Sacchi, T. Relaxin depresses small bowel motility through a nitric oxide-mediated mechanism. Studies in mice. Biol. Reprod. 2002, 66, 778–784. [Google Scholar] [CrossRef]
- Baccari, M.C.; Nistri, S.; Quattrone, S.; Bigazzi, M.; Bani Sacchi, T.; Calamai, F.; Bani, D. Depression by relaxin of neurally-induced contractile responses in the mouse gastric fundus. Biol. Reprod. 2004, 70, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Citterio, L.; Simonini, M.; Zagato, L.; Salvi, E.; Delli Carpini, S.; Lanzani, C.; Messaggio, E.; Casamassima, N.; Frau, F.; D’Avila, F.; Cusi, D.; Barlassina, C.; Manunta, P. Genes Involved in Vasoconstriction and Vasodilation System Affect Salt-Sensitive Hypertension. PLoS ONE 2011, 6, e19620. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, T.; Citterio, L.; Zagato, L.; Delli Carpini, S.; Thijs, L.; Casamassima, N.; D’hooge, J.; Bianchi, G.; Manunta, P.; Staessen, J.A. Left ventricular radial function associated with genetic variation in the cGMP-dependent protein kinase. Hypertension 2013, 62, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Bani, D.; Masini, E.; Bello, M.G.; Bigazzi, M.; Bani Sacchi, T. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am. J. Pathol. 1998, 152, 1367–1376. [Google Scholar] [PubMed]
- Garvey, E.P.; Oplinger, J.A.; Furfine, E.S.; Kiff, R.J.; Laszlo, F.; Whittle, B.J.; Knowles, R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric oxide synthase in vitro and in vivo. J. Biol. Chem. 1997, 272, 4959–4963. [Google Scholar] [CrossRef] [PubMed]
- Nistri, S.; Mazzetti, L.; Failli, P.; Bani, D. High-Yield Method for Isolation and Culture of Endothelial Cells from Rat Coronary Blood Vessels Suitable for Analysis of Intracellular Calcium and Nitric Oxide Biosynthetic Pathways. Biol. Proced. Online 2002, 4, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nistri, S.; Di Cesare Mannelli, L.; Ghelardini, C.; Zanardelli, M.; Bani, D.; Failli, P. Pretreatment with Relaxin Does Not Restore NO-Mediated Modulation of Calcium Signal in Coronary Endothelial Cells Isolated from Spontaneously Hypertensive Rats. Molecules 2015, 20, 9524-9535. https://doi.org/10.3390/molecules20069524
Nistri S, Di Cesare Mannelli L, Ghelardini C, Zanardelli M, Bani D, Failli P. Pretreatment with Relaxin Does Not Restore NO-Mediated Modulation of Calcium Signal in Coronary Endothelial Cells Isolated from Spontaneously Hypertensive Rats. Molecules. 2015; 20(6):9524-9535. https://doi.org/10.3390/molecules20069524
Chicago/Turabian StyleNistri, Silvia, Lorenzo Di Cesare Mannelli, Carla Ghelardini, Matteo Zanardelli, Daniele Bani, and Paola Failli. 2015. "Pretreatment with Relaxin Does Not Restore NO-Mediated Modulation of Calcium Signal in Coronary Endothelial Cells Isolated from Spontaneously Hypertensive Rats" Molecules 20, no. 6: 9524-9535. https://doi.org/10.3390/molecules20069524
APA StyleNistri, S., Di Cesare Mannelli, L., Ghelardini, C., Zanardelli, M., Bani, D., & Failli, P. (2015). Pretreatment with Relaxin Does Not Restore NO-Mediated Modulation of Calcium Signal in Coronary Endothelial Cells Isolated from Spontaneously Hypertensive Rats. Molecules, 20(6), 9524-9535. https://doi.org/10.3390/molecules20069524