
Gaseous Mediators Nitric Oxide and Hydrogen Sulfide in the Mechanism of Gastrointestinal Integrity, Protection and Ulcer Healing




Abstract
:1. Introduction
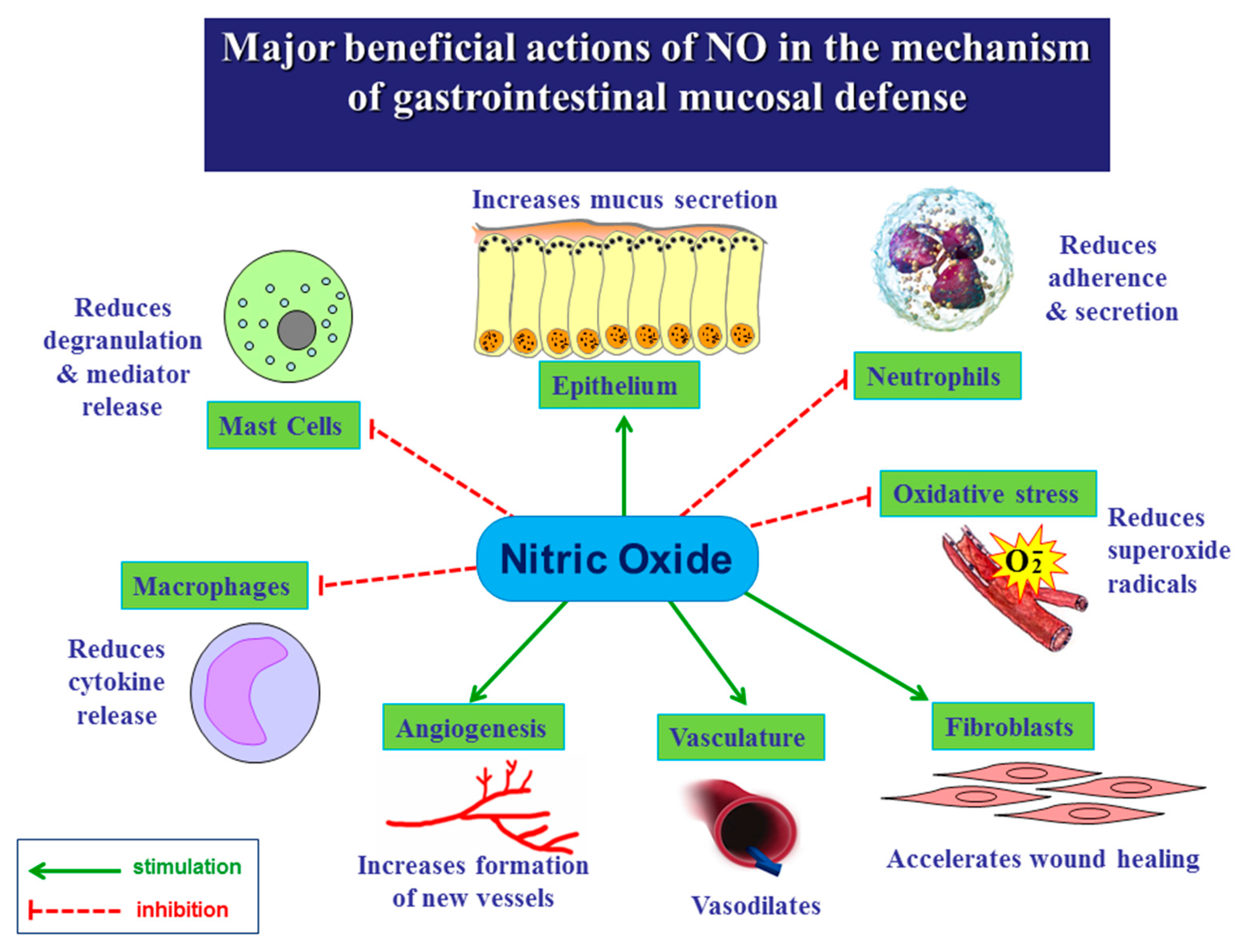
2. Biosynthesis of NO and Its Major Functions in Various Body Systems
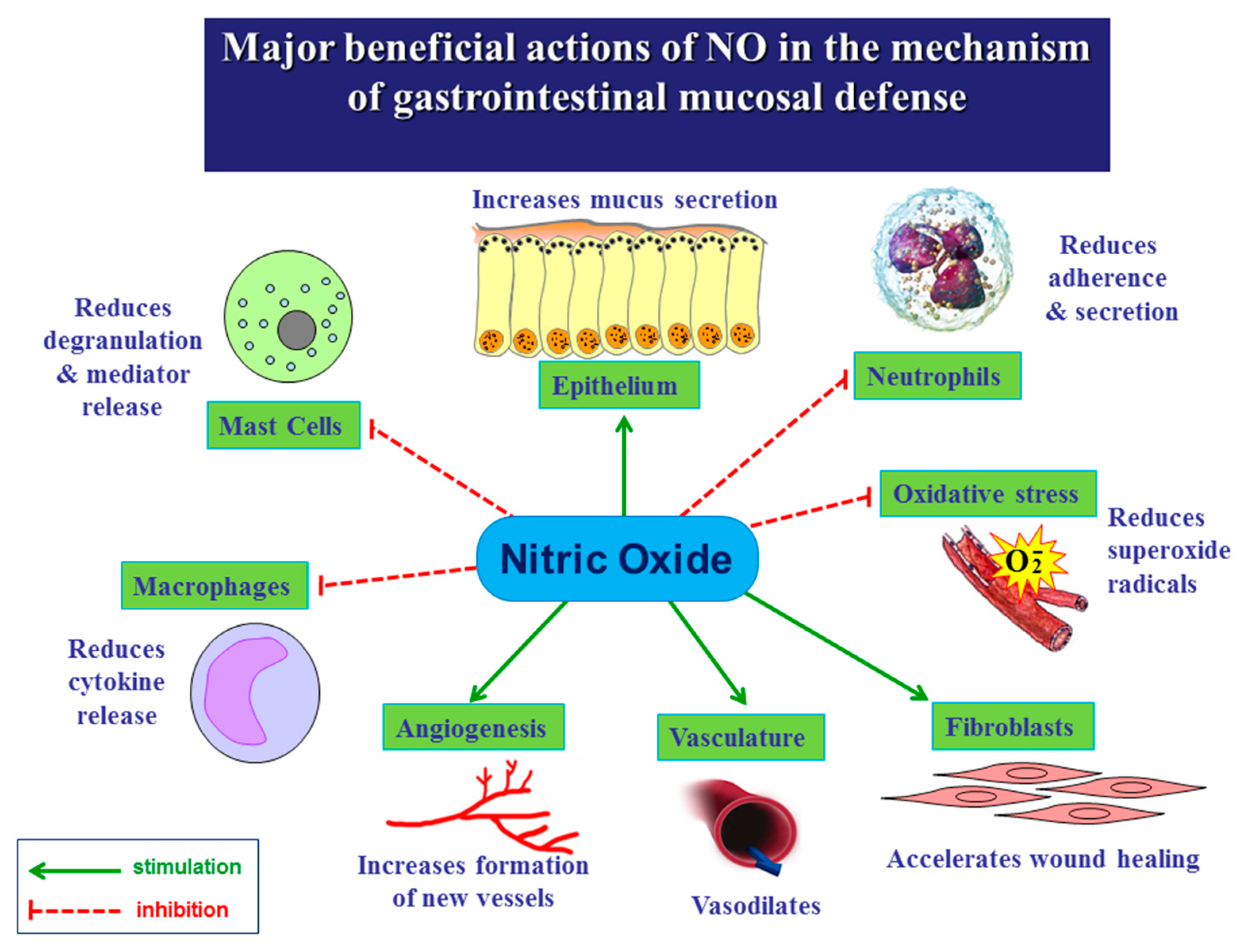
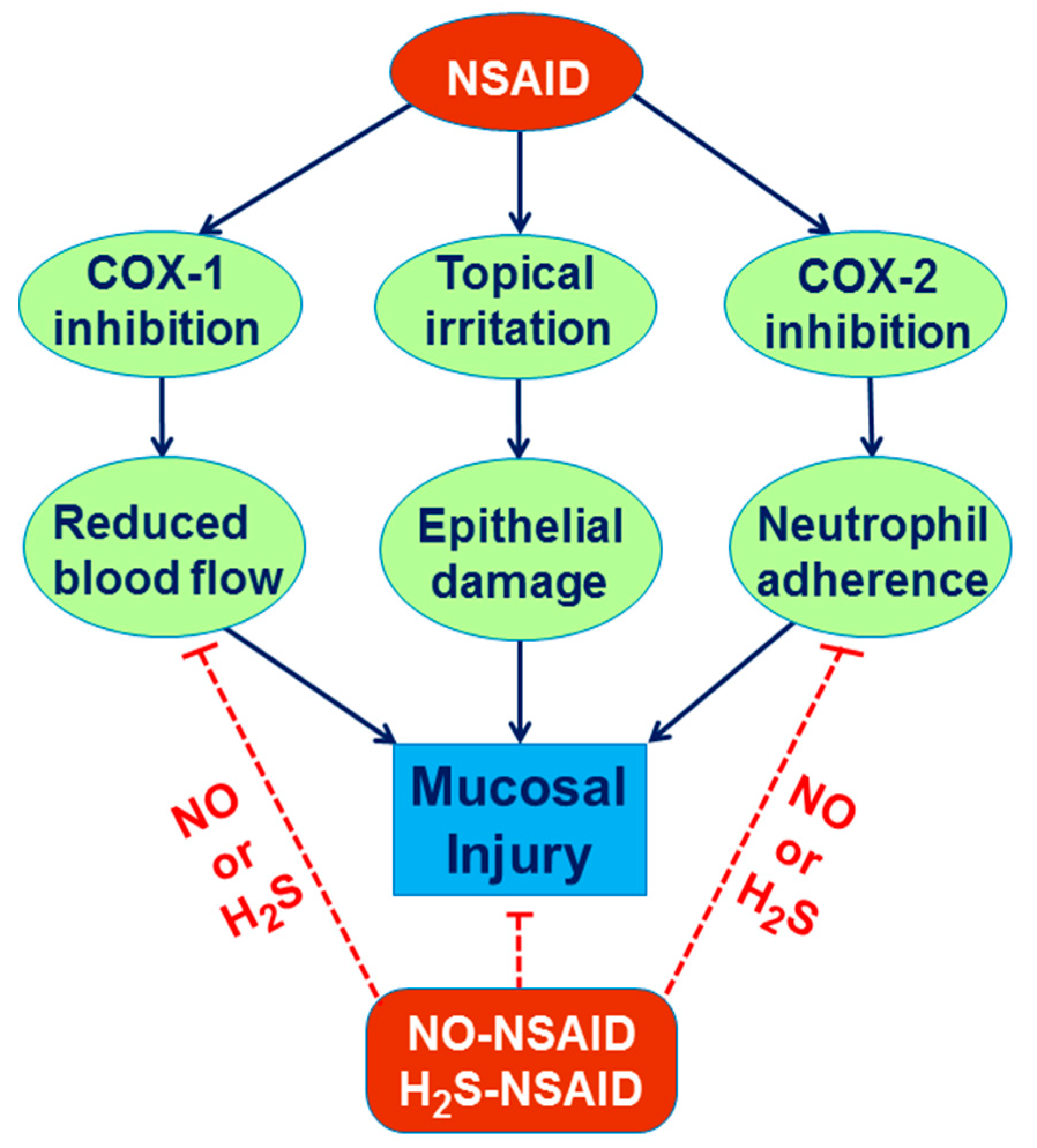
3. Role of NO in the Mechanism of Gastric Integrity, Protection and Ulcer Healing
4. Role of NO in the Esophageal and Intestinal Protection
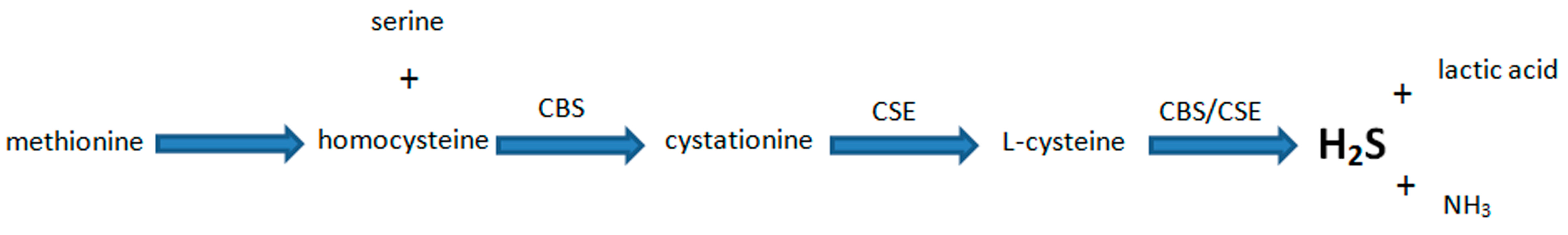
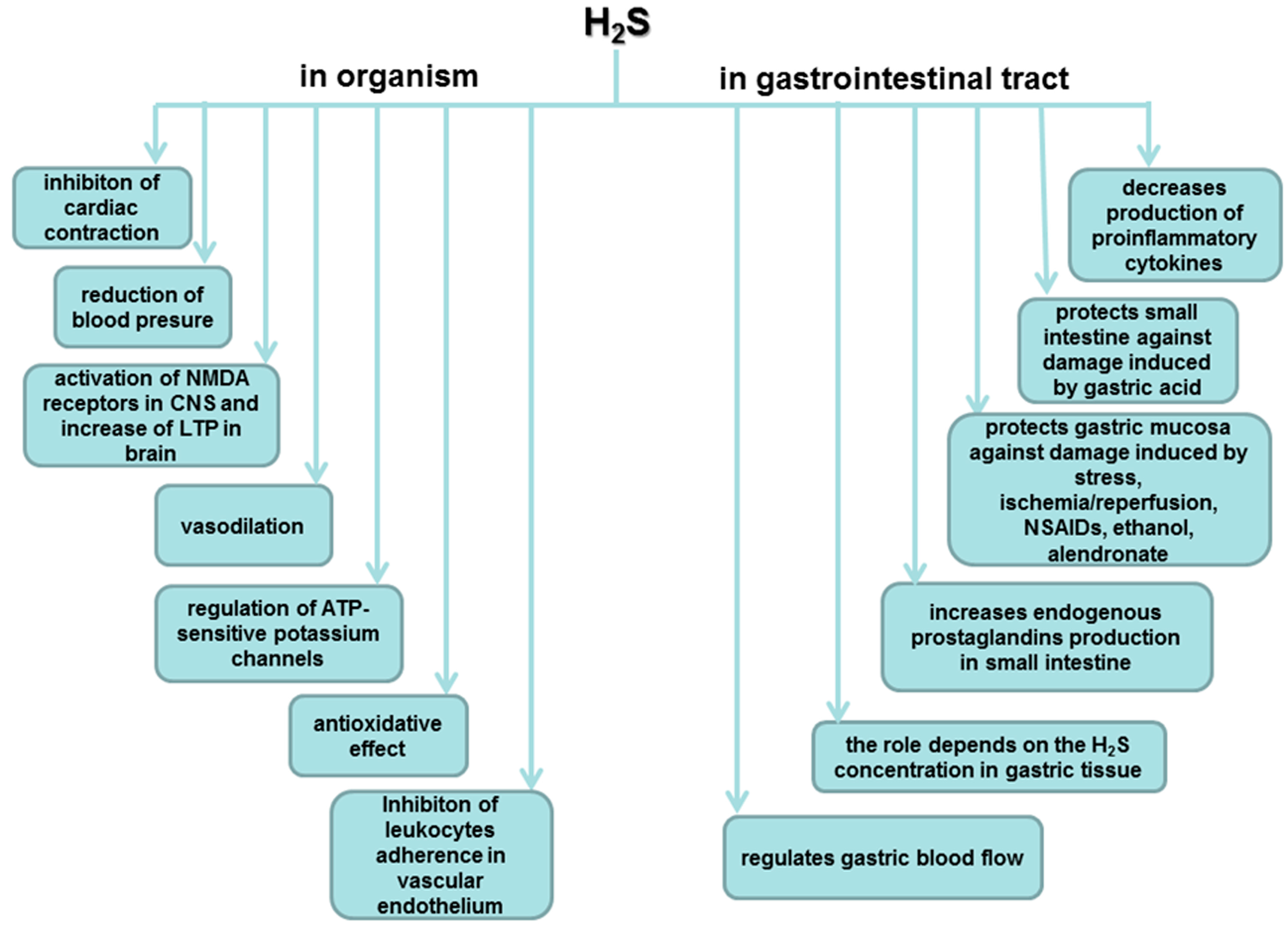
5. Biosynthesis of H2S and Its Major Functions in Various Body Systems
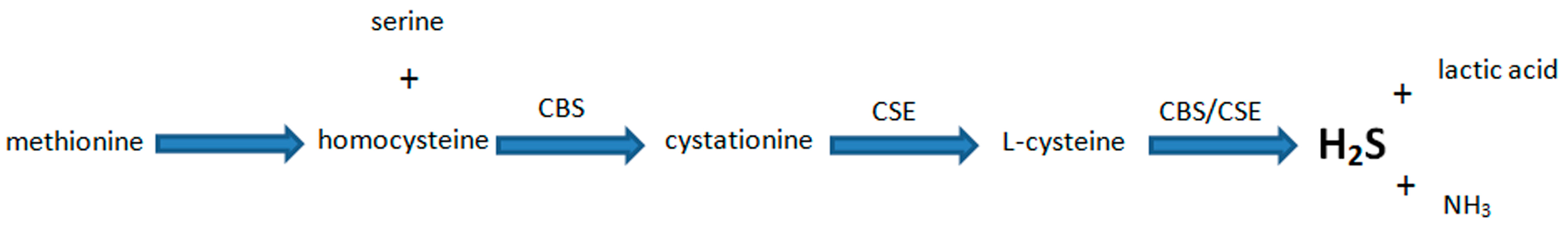
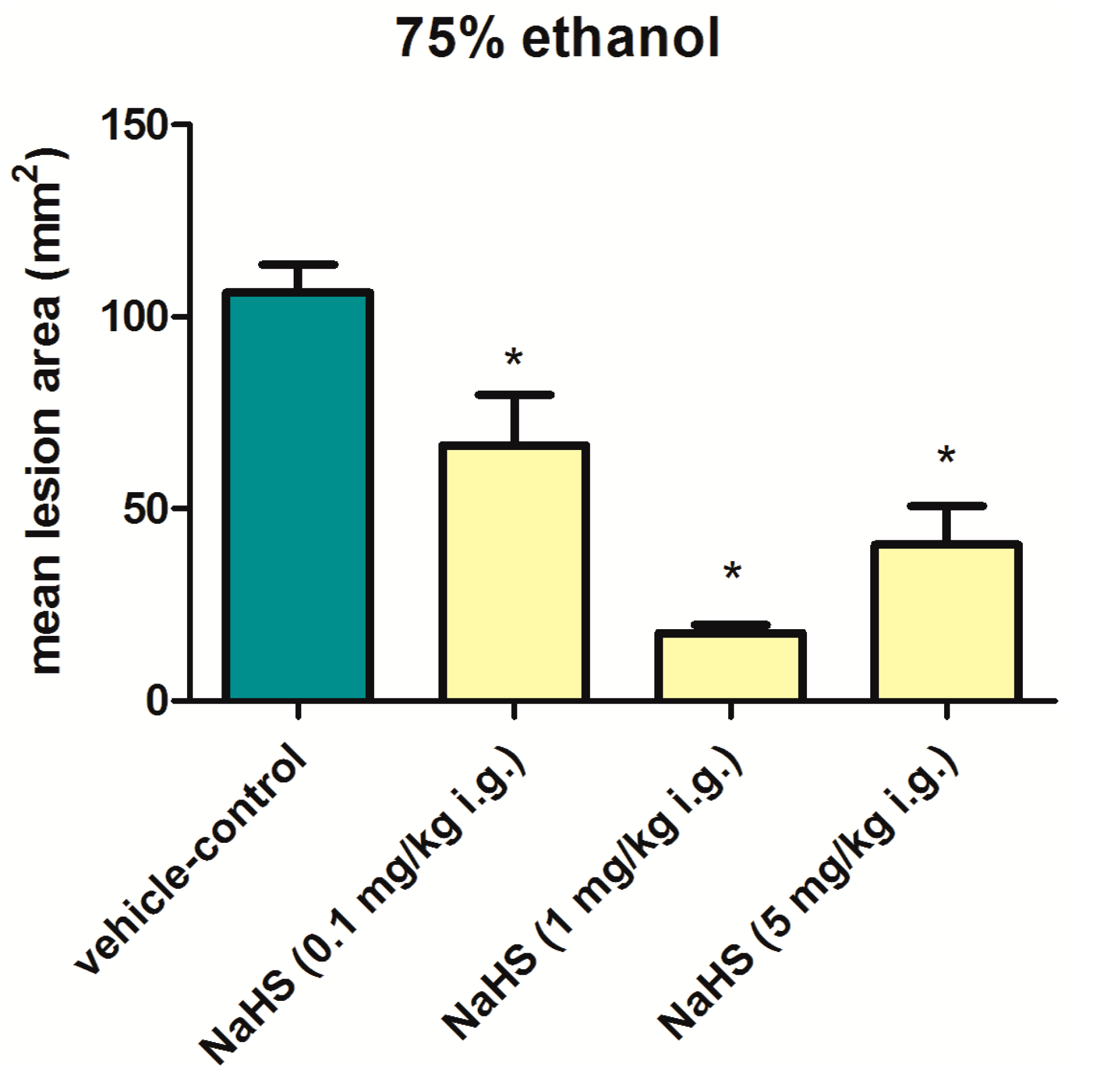
6. Involvement of H2S in the Mechanism of Gastroprotection and Ulcer Healing

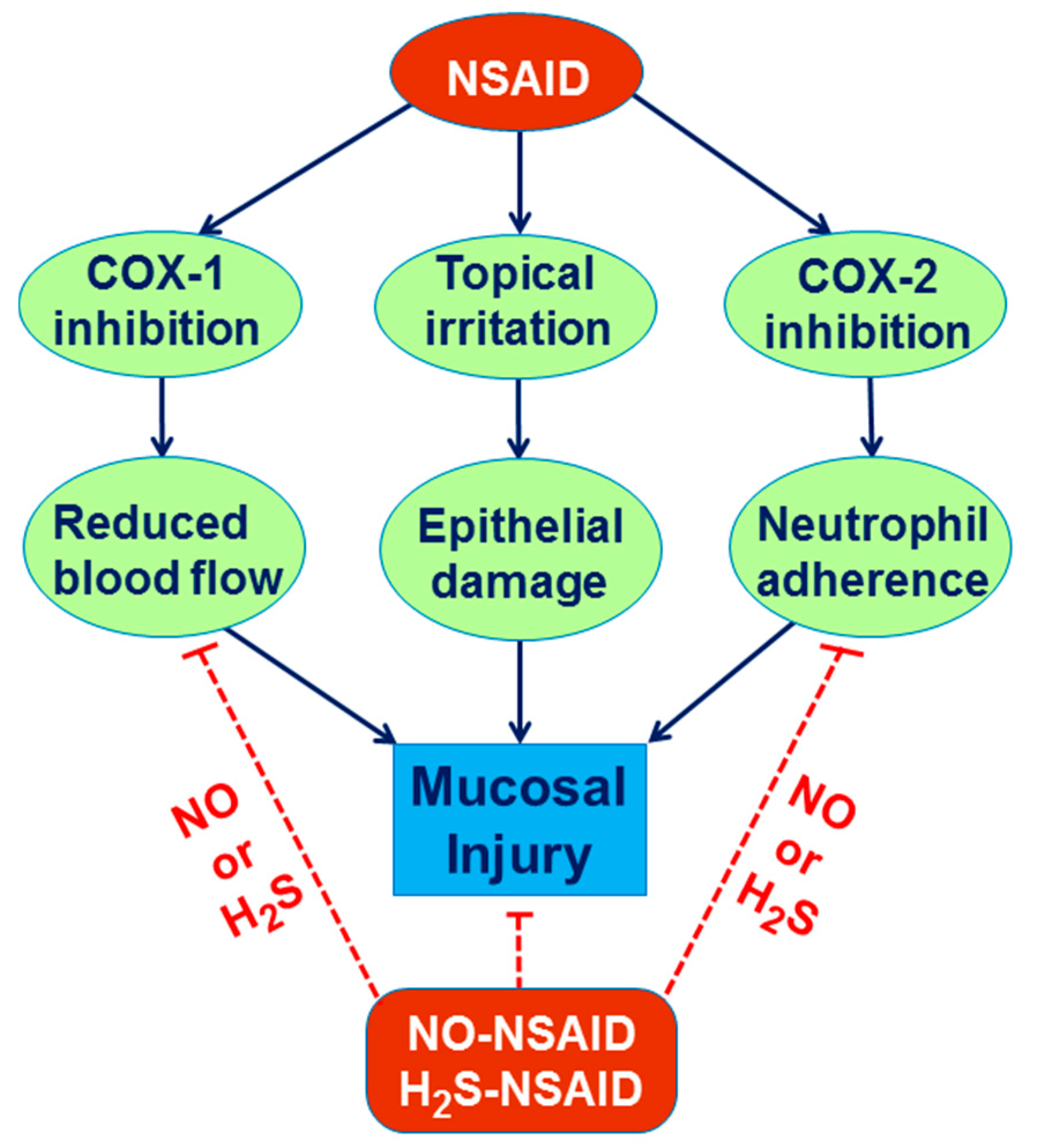
7. Role of H2S in the Esophageal and Intestinal Protection
8. Experimental Section
9. Conclusions
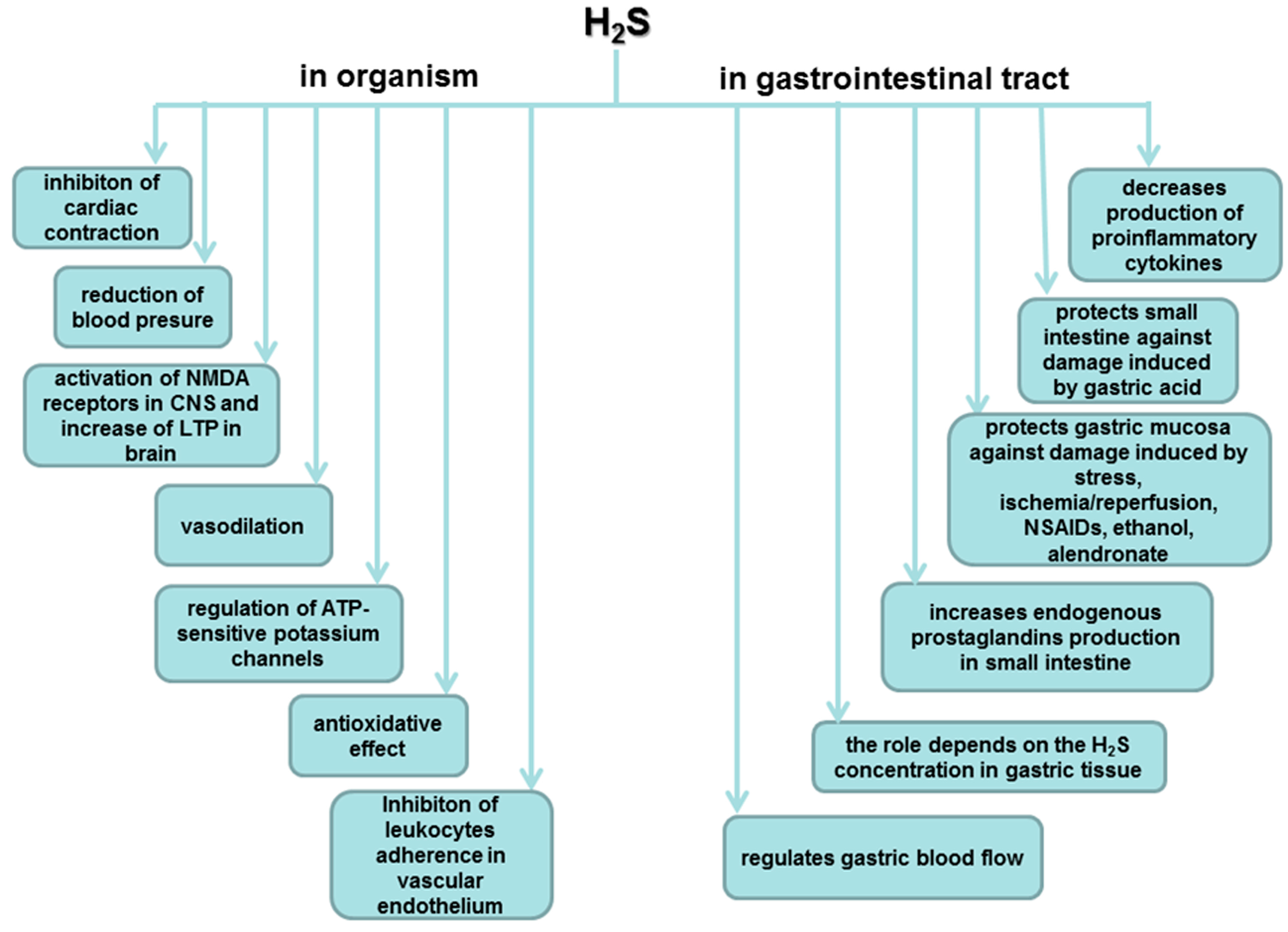

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | Reference |  | Reference | |
|---|---|---|---|---|
| Physiological concentrations | ||||
| serum | 1 nM | [132] | 30–100 μM | |
| brain/tissue | 100–250 nM | [133] | 50–160 μM | [106] |
| toxic | 0.5 µM | [134] | 250 μM | |
| Biochemical properties | ||||
| Half-life | Seconds—minutes | [135] | Seconds | [135] |
| Physiological forms | NO exists as a free radical | [136] | 20% exist as H2S, 80% as HS−, trace amounts of S2− | [137] |
| Crosstalk interaction on catalyzing enzymes | ||||
 |  | |||
| NO donor increases the expression and activity of CSE in cultured aortic smooth muscle cells (SMCs) | [97] | NaHS inhibits iNOS expression and NO production in macrophage cells (RAW264.7) | [138] | |
| NO cooperates with H2S via activation of guanylyl cyclase and increase of cGMP | [139] | NaHS treatment reduces eNOS activity and expression but not nNOS and iNOS in isolated rat aortas | [140] | |
| |  | |||
| NO does not increase the expression of H2S-generating enzymes and the H2S level in endothelial cells. | [141] | NaHS/Na2S profoundly increases the expression or/and the activity of eNOS | [141,142,143,144] | |
| H2S interacts with NO synthase to transform NO to nitroxyl (HNO) ↓ NO → ↑HNO | [145] | Na2S augmented NO production in chronically ischemic tissues, by influencing iNOS and nNOS expression and stimulating nitrite reduction to NO via xanthine oxidase (XO) under hypoxic condition | [146] | |
| Potent mechanisms of gastroprotection | ||||
| I/R injury | ↑ gastric blood flow ↓ lipid peroxidation ↓ free radicals | [147] | ↓ plasma level of IL-1β and TNF-α mRNA expression | [114] |
| WRS injury | ↓ lipid peroxidation ↑ SOD activity ↑ GSH concentration | [58] | ↓ acid output, ↑ gastric juice pH and mucin concentration, ↑GSH, CAT and SOD enzymes activities | [148] |
| ↓ lipid peroxidation products | [110] | |||
| Ethanol injury | ↓ free radicals ↑prostaglandins production | [149] | Involvement of KATP channels, capsaicin-sensitive nerve fibers and TRPV1 receptors | [2] |
| Gastric ulcers healing | ||||
| NO inhibits oxidative stress leading to acceleration of chronic gastric ulcers healing | [150] | Beneficial effect is not dependent on NO synthesis and do not occur through activation of ATP-sensitive K+ channels | [90] | |
Acknowledgments
Conflicts of Interest
References
- Brzozowski, T.; Konturek, P.C.; Sliwowski, Z.; Kwiecien, S.; Drozdowicz, D.; Pawlik, M.; Mach, K.; Konturek, S.J.; Pawlik, W.W. Interaction of nonsteroidal anti-inflammatory drugs (NSAID) with Helicobacter pylori in the stomach of humans and experimental animals. J. Physiol. Pharmacol. 2006, 57, 67–79. [Google Scholar] [PubMed]
- Medeiros, J.V.R.; Bezerra, V.H.; Gomes, A.S.; Barbosa, A.L.R.; Lima-Júnior, R.C.P.; Soares, P.M.G.; Brito, G.A.C.; Ribeiro, R.A.; Cunha, F.Q.; Souza, M.H.L.P. Hydrogen sulfide prevents ethanol-induced gastric damage in mice: Role of ATP-sensitive potassium channels and capsaicin-sensitive primary afferents neurons. J. Pharmacol. Exp. Ther. 2009, 330, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Piña, A.E.; Tapia-Alvarez, G.R.; Navarrete, A. Inhibition of endogenous hydrogen sulfide synthesis by PAG protects against ethanol-induced gastric damage in the rat. Eur. J. Pharmacol. 2010, 630, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L. Prostaglandins, NSAIDs, and gastric mucosal protection: Why doesn’t the stomach digest itself? Physiol. Rev. 2008, 88, 1547–1565. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Tandon, R.K. Stress and the gastrointestinal tract. J. Gastroenterol. Hepatol. 2005, 20, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Kaunitz, J. Gastroduodenal Mucosal Defense. Curr. Gastroenterol. Rep. 2008, 10, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Kwiecien, S.; Konturek, P.C.; Sliwowski, Z.; Mitis-Musiol, M.; Pawlik, M.W.; Brzozowski, B.; Jasnos, K.; Magierowski, M.; Konturek, S.J.; Brzozowski, T. Interaction between selective cyclooxygenase inhibitors and capsaicin-sensitive afferent sensory nerves in pathogenesis of stress-induced gastric lesions. Role of oxidative stress. J. Physiol. Pharmacol. 2012, 63, 143–151. [Google Scholar]
- Robert, A.; Kane, G.; Reele, S.B. Dose response inhibition in man of meal-stimulated gastric acid secretion by 15(R)-15-methyl prostaglandin E2, given orally. Gut 1981, 22, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Leung, F.W.; Guth, P.H. Morphological and functional gastric cytoprotection by prostaglandin in rats receiving absolute ethanol orally. Gut 1992, 33, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Laine, L.; Takeuchi, K.; Tarnawski, A. Gastric mucosal defense and cytoprotection: Bench to bedside. Gastroenterology 2008, 135, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K. Gastric cytoprotection by prostaglandin E2 and prostacyclin: Relationship to EP1 and IP receptors. J. Physiol. Pharmacol. 2014, 65, 3–14. [Google Scholar] [PubMed]
- Mita, M.; Satoh, M.; Shimada, A.; Okajima, M.; Azuma, S.; Suzuki, J.S.; Sakabe, K.; Hara, S.; Himeno, S. Metallothionein is a crucial protective factor against Helicobacter pylori induced gastric erosive lesions in a mouse model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G877–G884. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Konturek, S.J. Gut clock: Implication of circadian rhythms in the gastrointestinal tract. J. Physiol. Pharmacol. 2011, 62, 139–150. [Google Scholar] [PubMed]
- El Eter, E.; Al Tuwaijiri, A.; Hagar, H.; Arafa, M. In vivo and in vitro antioxidant activity of ghrelin: Attenuation of gastric ischemic injury in the rat. J. Gastroenterol. Hepatol. 2007, 22, 1791–1799. [Google Scholar] [CrossRef] [PubMed]
- Bülbül, M.; Tan, R.; Gemici, B.; Ongüt, G.; Izgüt-Uysal, V.N. Effect of orexin-a on ischemia-reperfusion-induced gastric damage in rats. J. Gastroenterol. 2008, 43, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Motawi, T.K.; Abd Elgawad, H.M.; Shahin, N.N. Gastroprotective effect of leptin in indomethacin-induced gastric injury. J. Biomed. Sci. 2008, 15, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. The gasotransmitter role of hydrogen sulfide. Antioxid. Redox Signal. 2003, 5, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Jasnos, K.; Magierowski, M.; Kwiecien, S.; Brzozowski, T. Carbon monoxide in human physiology-its role in the gastrointestinal tract. Postepy Hig. Med. Dosw. 2014, 68, 101–109. [Google Scholar] [CrossRef]
- Napoli, C.; Ignarro, L.J. Nitric oxide and pathogenic mechanisms involved in the development of vascular diseases. Arch. Phrm. Res. 2009, 32, 1103–1108. [Google Scholar] [CrossRef]
- Bryan, N.S.; Bian, K.; Murad, F. Discovery of the nitric oxide signaling pathway and targets for drug development. Front. Biosci. 2009, 14, 1–18. [Google Scholar] [CrossRef]
- Bian, K.; Murad, F. sGC-cGMP signaling: Target for anticancer therapy. Adv. Exp. Med. Biol. 2014, 814, 5–13. [Google Scholar] [PubMed]
- Wallace, J.L.; Del Soldato, P.; Cirino, G.; Muscará, M.N. Nitric oxide-releasing NSAIDs: GI-safe antithrombotics. Drugs 1999, 2, 321–326. [Google Scholar]
- Fiorucci, S.; Santucci, L. Distrutti E NSAIDs, coxibs, CINOD and H2S-releasing NSAIDs: What lies beyond the horizon. Dig. Liver Dis. 2007, 39, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Ruddock, M.W.; Hirst, D.G. Pentoxifylline inhibits agonist-induced vasoconstriction in vascular smooth muscle and spontaneous peristalsis in isolated ileum. Oncol. Res. 2005, 15, 81–86. [Google Scholar] [PubMed]
- Kuo, P.; Gentilcore, D.; Nair, N.; Stevens, J.E.; Wishart, J.M.; Lange, K.; Gilja, O.H.; Hausken, T.; Horowitz, M.; Jones, K.L.; et al. The nitric oxide synthase inhibitor, Ng-nitro-l-arginine-methyl-ester, attenuates the delay in gastric emptying induced by hyperglycaemia in healthy humans. Neurogastroenterol. Motil. 2009, 21. [Google Scholar] [CrossRef]
- Konturek, S.J.; Brzozowski, T.; Majka, J.; Pytko-Polonczyk, J.; Stachura, J. Inhibition of nitric oxide synthase delays healing of chronic gastric ulcers. Eur. J. Pharmacol. 1993, 239, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Breuillard, C.; Cynober, L.; Moinard, C. Citrulline and nitrogen homeostasis: An overview. Amino Acids 2015, 47, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Mu, J.J.; Fang, Y.; Yuan, Z.Y.; Liu, F.Q. Impact of High Salt Independent of Blood Pressure on PRMT/ADMA/DDAH Pathway in the Aorta of Dahl Salt-Sensitive Rats. Int. J. Mol. Sci. 2013, 14, 8062–8072. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H.; Bode-Böger, S.M.; Szuba, A.; Tsao, P.S.; Chan, J.R.; Tangphao, O.; Blaschke, T.F.; Cooke, J.P. Asymmetric dimethylarginine (ADMA): A novel risk factor for endothelial dysfunction: Its role in hypercholesterolemia. Circulation 1998, 98, 1842–1847. [Google Scholar] [CrossRef] [PubMed]
- Böger, R.H. The emerging role of ADMA as a novel cardiovascular risk factor. Cardiovasc. Res. 2003, 59, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Briet, M.; Burns, K.D. Chronic kidney disease and vascular remodeling: Molecular mechanisms and clinical implications. Clin. Sci. 2012, 123, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N.; Repossi, G.; Eynard, A.R. l-arginine, NO and asymmetrical dimethylarginine in hypertension and type 2 diabetes. Front. Biosci. 2011, 16, 13–20. [Google Scholar] [CrossRef]
- Magierowski, M.; Jasnos, K.; Sliwowski, Z.; Surmiak, M.; Krzysiek-Maczka, G.; Ptak-Belowska, A.; Kwiecien, S.; Brzozowski, T. Exogenous asymmetric dimethylarginine (ADMA) in pathogenesis of ischemia-reperfusion-induced gastric lesions: Interaction with protective nitric oxide (NO) and calcitonin gene-related peptide (CGRP). Int. J. Mol. Sci. 2014, 15, 4946–4964. [Google Scholar] [CrossRef] [PubMed]
- Kwiecień, S.; Ptak-Belowska, A.; Krzysiek-Mączka, G.; Targosz, A.; Jasnos, K.; Magierowski, M.; Szczyrk, U.; Brzozowski, B.; Konturek, S.J.; Konturek, P.C.; et al. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, interacts with gastric oxidative metabolism and enhances stress-induced gastric lesions. J. Physiol. Pharmacol. 2012, 63, 515–524. [Google Scholar]
- Mourad, F.H.; Khuri, M.; Shouaib, F.; Nassar, C.F. Protective effect of the nitric oxide donor molsidomine on indomethacin and aspirin-induced gastric injury in rats. Eur. J. Gastroenterol. Hepatol. 2000, 12, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Wybrańska, I.; Miszczuk-Jamska, B.; Baczyńska, E.; Goldsztajn, P.; Sowa, G.; Przewłocki, R.; Gryglewski, R.; Dembińska-Kieć, A. Influence of SIN-1 and sodium nitroprusside (NANP) on OX-LDL metabolism in macrophages. J. Physiol. Pharmacol. 1994, 45, 387–397. [Google Scholar] [PubMed]
- Styś, T.; Styś, A.; Paczwa, P.; Szczepańska-Sadowska, E.; Lipkowski, A.W. Decreased hypotensive respinsiveness to nitric oxide donor S-nitroso-N-acetyl d,l-penicillamine (SNAP) in spontaneously hypertensive (SHR) rats. J. Physiol. Pharmacol. 1998, 49, 37–49. [Google Scholar] [PubMed]
- Konturek, S.J.; Konturek, P.C. Role of nitric oxide in digestive system. Digestion 1995, 56, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Szlachcic, A.; Bilski, R.; Dziaduś-Sokolowska, A.; Michalski, J.; Mroczka, J. The effect of nitric oxide donors and l-arginine on the gastric electrolyte barrier. J. Physiol. Pharmacol. 2001, 52, 211–220. [Google Scholar] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Meixner, H.; Ptak, A.; Hahn, E.G.; Konturek, S.J. Central and peripheral neural aspects of gastroprotective and ulcer healing effects of lipopolysaccharides. J. Physiol. Pharmacol. 2001, 52, 611–623. [Google Scholar] [PubMed]
- Jackson, L.M.; Wu, K.C.; Mahida, Y.R. Cyclooxygenase (COX)-1 and COX-2 in normal, inflamed and ulcerated human gastric mucosa. Gut 2000, 47, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Peskar, B.M. Role of cyclooxygenase isoforms in gastric mucosal defense. J. Physiol. Pharmacol. 2000, 95, 3–9. [Google Scholar]
- Hawkey, C.J. COX-1 and COX-2 inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef] [PubMed]
- Ambrosini, M.V.; Mariucci, G.; Rambotti, M.G.; Tantucci, M.; Covarelli, C.; de Angelis, L.; del Soldato, P. Ultrastructural investigations on protective effects of NCX 4016 (nitroaspirin) on macrovascular endothelium in diabetic Wistar rats. J. Submicrosc. Cytol. Pathol. 2005, 37, 205–213. [Google Scholar] [PubMed]
- Fiorucci, S.; Mencarelli, A.; Meneguzzi, A.; Lechi, A.; Renga, B.; del Soldato, P.; Morelli, A.; Minuz, P. Co-administration of nitric oxide-aspirin (NCX-4016) and aspirin prevents platelet and monocyte activation and protects against gastric damage induced by aspirin in humans. J. Am. Coll. Cardiol. 2004, 44, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Emanueli, C.; van Linthout, S.; Salis, M.B.; Monopoli, A.; del Soldato, P.; Ongini, E.; Madeddu, P. Nitric oxide-releasing aspirin derivative, NCX 4016, promotes reparative angiogenesis and prevents apoptosis and oxidative stress in a mouse model of peripheral ischemia. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, T.; Konturek, P.C.; Konturek, S.J.; Sliwowski, Z.; Drozdowicz, D.; Kwiecień, S.; Pajdo, R.; Ptak, A.; Pawlik, M.; Hahn, E. Gastroprotective and ulcer healing effects of nitric oxide-releasing non-steroidal anti-inflammatory drugs. Dig. Liver Dis. 2000, 32, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Santucci, L.; Federici, B.; Antonelli, E.; Distrutti, E.; Morelli, O.; Renzo, G.D.; Coata, G.; Cirino, G.; Soldato, P.D.; et al. Nitric oxide-releasing NSAIDs inhibit interleukin-1β converting enzyme-like cysteine proteases and protect endothelial cells from apoptosis induced by TNFα. Aliment Pharmacol. Ther. 1999, 13, 421–435. [Google Scholar]
- Takeuchi, K.; Ukawa, H.; Konaka, A.; Kitamura, M.; Sugawa, Y. Effect of nitric oxide-releasing aspirin derivative on gastric functional and ulcerogenic responses in rats: Comparison with plain aspirin. J. Pharmacol. Exp. Ther. 1998, 286, 115–121. [Google Scholar] [PubMed]
- Konturek, P.C.; Brzozowski, T.; Ptak, A.; Kania, J.; Kwiecień, S.; Hahn, E.G.; Konturek, S.J. Nitric oxide releasing aspirin protects the gastric mucosa against stress and promotes healing of stress-induced gastric mucosal damage: Role of heat shock protein 70. Digestion 2002, 66, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; McKnight, W.; Wilson, T.L.; del Soldato, P.; Cirino, G. Reduction of shock-induced gastric damage by a nitric oxide-releasing aspirin derivative: Role of neutrophils. Am. J. Physiol. 1997, 273, G1246–G1251. [Google Scholar] [PubMed]
- Naito, Y.; Takagi, T.; Handa, O.; Yoshikawa, T. Lipid hydroperoxide-derived modification of proteins in gastrointestinal tract. Subcell Biochem. 2014, 77, 137–148. [Google Scholar] [PubMed]
- Guzik, T.J.; Olszanecki, R.; Sadowski, J.; Kapelak, B.; Rudziński, P.; Jopek, A.; Kawczyńska, A.; Ryszawa, N.; Loster, J.; Jawień, J.; et al. Superoxide dismutase activity and expression in human venous and arterial bypass graft vessels. J. Physiol. Pharmacol. 2005, 56, 313–323. [Google Scholar]
- Konaka, A.; Nishijima, M.; Tanaka, A.; Kunikata, T.; Kato, S.; Takeuchi, K. Nitric oxide, superoxide radicals and mast cells in pathogenesis of indomethacin-induced small intestinal lesions in rats. J. Physiol. Pharmacol. 1999, 50, 25–38. [Google Scholar] [PubMed]
- Kwiecień, S.; Pawlik, M.W.; Brzozowski, T.; Konturek, P.C.; Śliwowski, Z.; Pawlik, W.W.; Konturek, S.J. Nitric oxide (NO)-Releasing aspirin and other (NO) donors in protection of gastric mucosa against stress. J. Physiol. Pharmacol. 2008, 59, 103–115. [Google Scholar] [PubMed]
- Wada, K.; Kamisaki, Y.; Kitano, M.; Kishimoto, Y.; Nakamoto, K.; Itoh, T. A new gastric ulcer model induced by ischemia-reperfusion in the rat; role of leukocytes on ulceration in rat stomach. Life Sci. 1996, 59, 295–301. [Google Scholar] [CrossRef]
- Kourie, J.I. Interaction of reactive oxygen species with ion transport mechanisms. Am. J. Physiol. 1998, 275, 1–24. [Google Scholar]
- Kwiecień, S.; Jasnos, K.; Magierowski, M.; Śliwowski, Z.; Pajdo, R.; Brzozowski, B.; Mach, T.; Wójcik, D.; Brzozowski, T. Lipid peroxidation, reactive oxygen species and antioxidative factors in the pathogenesis of gastric mucosal lesions and mechanism of protection against oxidative stress-induced gastric injury. J. Physiol. Pharmacol. 2014, 65, 613–622. [Google Scholar] [PubMed]
- Orlando, R.C.; Lacy, E.R.; Tobey, N.A.; Cowart, K. Barriers to paracellular permeability in rabbit esophageal epithelium. Gastroenterology 1992, 102, 910–923. [Google Scholar] [PubMed]
- Elias, P.M.; McNutt, N.S.; Friend, D.S. Membrane alterations during cornification of mammalian squamous epithelia: A freeze-fracture, tracer, and thin-section study. Anat. Rec. 1977, 189, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Tobey, N.A.; Argote, C.M.; Awayda, M.S.; Vanegas, X.C.; Orlando, R.C. Effect of luminal acidity on the apical cation channel in rabbit esophageal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G796–G805. [Google Scholar] [CrossRef] [PubMed]
- Farré, R. Pathophysiology of gastro-esophageal reflux disease: A role for mucosa integrity? Neurogastroenterol. Motil. 2013, 25, 783–799. [Google Scholar] [PubMed]
- Tobey, N.A.; Koves, G.; Orlando, R.C. Human esophageal epithelial cells possess an Na+/H+ exchanger for H+ extrusion. Am. J. Gastroenterol. 1998, 93, 2075–2081. [Google Scholar] [CrossRef] [PubMed]
- Tobey, N.A.; Reddy, S.P.; Khalbuss, W.E.; Silvers, S.M.; Cragoe, E.J., Jr.; Orlando, R.C. Na+-dependent and -independent Cl−/HCO3− exchangers in cultured rabbit esophageal epithelial cells. Gastroenterology 1993, 104, 185–195. [Google Scholar] [PubMed]
- Layden, T.J.; Schmidt, L.; Agnone, L.; Lisitza, P.; Brewer, J.; Goldstein, J.L. Rabbit esophageal cell cytoplasmic pH regulation: Role of Na+-H+ antiport and Na+-dependent HCO3− transport systems. Am. J. Physiol. 1992, 263, G407–G413. [Google Scholar] [PubMed]
- Orlando, R.C. The integrity of the esophageal mucosa. Balance between offensive and defensive mechanisms. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 873–882. [Google Scholar] [CrossRef]
- Lanas, A.I.; Blas, J.M.; Ortego, J.; Soria, J.; Sáinz, R. Adaptation of esophageal mucosa to acid- and pepsin-induced damage: Role of nitric oxide and epidermal growth factor. Dig. Dis. Sci. 1997, 42, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Lanas, A.; Soteras, F.; Jimenez, P.; Fiteni, I.; Piazuelo, E.; Royo, Y.; Ortego, J.; Iñarrea, P.; Esteva, F. Superoxide anion and nitric oxide in high-grade esophagitis induced by acid and pepsin in rabbits. Dig. Dis. Sci. 2001, 46, 2733–2743. [Google Scholar] [CrossRef] [PubMed]
- Zayachkivska, O.; Pshyk-Titko, I.; Hrycevych, N.; Savytska, M. New insight into oseophageal injury and protection in physiologically relevant animal models. J. Physiol. Pharmacol. 2014, 65, 295–307. [Google Scholar] [PubMed]
- Konturek, P.C.; Brzozowska, I.; Targosz, A.; Pawlik, M.; Kania, J.; Hess, T.; Kwiecien, S.; Konturek, S.J.; Reiter, R.J.; Brzozowski, T. Esophagoprotection mediated by exogenous and endogenous melatonin in an experimental model of reflux esophagitis. J. Pineal Res. 2013, 55, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, M.W.; Kwiecien, S.; Pajdo, R.; Ptak-Belowska, A.; Brzozowski, B.; Krzysiek-Maczka, G.; Strzalka, M.; Konturek, S.J.; Brzozowski, T. Esophagoprotective activity of angiotensin-(1–7) in experimental model of acute reflux esophagitis. Evidence for the role of nitric oxide, sensory nerves, hypoxia-inducible factor-1alpha and proinflammatory cytokines. J. Physiol. Pharmacol. 2014, 65, 809–822. [Google Scholar]
- Ishiyama, F.; Iijima, K.; Asanuma, K.; Ara, N.; Yoshitake, J.; Abe, Y.; Koike, T.; Imatani, A.; Ohara, S.; Shimosegawa, T. Exogenous luminal nitric oxide exacerbates esophagus tissue damage in a reflux esophagitis model of rats. Scand. J. Gastroenterol. 2009, 44, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Seenan, J.P.; Wirz, A.A.; Robertson, E.V.; Clarke, A.T.; Manning, J.J.; Kelman, A.W.; Gillen, G.; Ballantyne, S.; Derakhshan, M.H.; McColl, K.E. Effect of nitrite delivered in saliva on postprandial gastro-esophageal function. Scand. J. Gastroenterol. 2012, 47, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, M.; Pajdo, R.; Kwiecien, S.; Ptak-Belowska, A.; Sliwowski, Z.; Mazurkiewicz-Janik, M.; Konturek, S.J.; Pawlik, W.W.; Brzozowski, T. Nitric oxide (NO)-releasing aspirin exhibits a potent esophagoprotection in experimental model of acute reflux esophagitis. Role of nitric oxide and proinflammatory cytokines. J. Physiol. Pharmacol. 2011, 62, 75–86. [Google Scholar]
- Gyires, K.; Toth, E.V.; Zadori, S.Z. Gut inflammation: Current update on pathophysiology, molecular mechanism and pharmacological treatment modalities. Curr. Pharm. Des. 2014, 20, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- Szkudlapski, D.; Labuzek, K.; Pokora, Z.; Smyla, N.; Gonciarz, M.; Mularczyk, A.; Maluch, P.; Okopien, B. The emering role of helminths in treatment of the inflammatory bowel disorders. J. Physiol. Pharmacol. 2014, 65, 741–751. [Google Scholar] [PubMed]
- Lamine, F.; Fioramonti, J.; Bueno, L.; Nepveu, F.; Cauquil, E.; Lobysheva, I.; Eutamène, H.; Théodorou, V. Nitric oxide released by Lactobacillus farciminis improves TNBS-induced colitis in rats. Scand. J. Gastroenterol. 2004, 39, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Kankuri, E.; Hämäläinen, M.; Hukkanen, M.; Salmenperä, P.; Kivilaakso, E.; Vapaatalo, H.; Moilanen, E. Suppression of pro-inflammatory cytokine release by selective inhibition of inducible nitric oxide synthase in mucosal explants from patients with ulcerative colitis. Scand. J. Gastroenterol. 2003, 38, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Jädert, C.; Phillipson, M.; Holm, L.; Lundberg, J.O.; Borniquel, S. Preventive and therapeutic effects of nitrite supplementation in experimental inflammatory bowel disease. Redox. Biol. 2013, 2, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, Y.; Kubo, M.; Zhang, R.; Matsumoto, K.; Yan, F.; Setiawan, H.; Takahashi, H.; Fujikura, Y.; Ogino, K. Inhibition of arginase ameliorates experimental ulcerative colitis in mice. Free Radic. Res. 2013, 47, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E. Review article: Nitric oxide from dysbiotic bacterial respiration of nitrate in the pathogenesis and as a target for therapy of ulcerative colitis. Aliment. Pharmacol. Ther. 2008, 27, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymatic capacity for cysteine desulphydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [PubMed]
- Swaroop, M.; Bradley, K.; Ohura, T.; Tahara, T.; Roper, M.D.; Rosenberg, L.E.; Kraus, J.P. Rat cystathionine beta-synthase. Gene organization and alternative splicing. J. Biol. Chem. 1992, 267, 11455–11461. [Google Scholar]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. Biochem. J. 2009, 146, 623–626. [Google Scholar] [CrossRef]
- Shibuya, N.; Tanaka, M.; Yoshida, M.; Ogasawara, Y.; Togawa, T.; Ishii, K.; Kimura, H. 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal. 2009, 11, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Dello Russo, C.; Tringali, G.; Ragazzoni, E.; Maggiano, N.; Menini, E.; Vairano, M.; Preziosi, P.; Navarra, P. Evidence that hydrogen sulfide can modulate hypothalamo-pituitary-adrenal axis function: in vitro and in vivo studies in the rat. J. Neuroendocrinol. 2000, 12, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Hydrogen sulfide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L. Physiological and pathophysiological roles of hydrogen sulfide in the gastrointestinal tract. Antioxid. Redox Signal. 2010, 12, 1125–1133. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Dicay, M.; McKnight, W.; Martin, G.R. Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 2007, 21, 4070–4076. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [PubMed]
- Qu, K.; Chen, C.P.; Halliwell, B.; Moore, P.K.; Wong, P.T. Hydrogen sulfide is a mediator of cerebral ischemic damage. Stroke 2006, 37, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, J.; Ao, G.; Hu, L.; Liu, H.; Xiao, Y.; Du, H.; Alkayed, N.J.; Liu, C.F.; Cheng, J. Hydrogen sulfide protects blood-brain barrier integrity following cerebral ischemia. J. Neurochem. 2014, 129, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.; Oh, M.Y.; Kim, Y.J.; Choi, I.Y.; Yang, H.S.; Ryu, W.S.; Lee, S.H.; Yoon, B.W. Hydrogen sulfide treatment induces angiogenesis after cerebral ischemia. J. Neurosci. Res. 2014, 92, 1520–1528. [Google Scholar] [CrossRef] [PubMed]
- Mok, Y.Y.P.; Moore, P.K. Hydrogen sulfide is pro-inflammatory in haemorrhagic shock. Inflamm. Res. 2008, 57, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Antonelli, E.; Distrutti, E.; Rizzo, G.; Mencarelli, A.; Orlandi, S.; Zanardo, R.; Renga, B.; Di Sante, M.; Morelli, A.; et al. Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 2005, 129, 1210–1224. [Google Scholar]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M. Role of hydrogen sulfide in the pathology of inflammation. Scientifica (Cairo) 2012, 2012, 159680. [Google Scholar] [CrossRef]
- Pálinkás, Z.; Furtmüller, P.G.; Nagy, A.; Jakopitsch, C.; Pirker, K.F.; Magierowski, M.; Jasnos, K.; Wallace, J.L.; Obinger, C.; Nagy, P. Interactions of hydrogen sulfide with myeloperoxidase. Br. J. Pharmacol. 2015, 172, 1516–1532. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [PubMed]
- Collin, M.; Anuar, F.B.; Murch, O.; Bhatia, M.; Moore, P.K.; Thiemermann, C. Inhibition of endogenous hydrogen sulfide formation reduces the organ injury caused by endotoxemia. Br. J. Pharmacol. 2005, 146, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Wong, F.L.; Fu, D.; Lau, H.Y.; Moochhala, S.M.; Moore, P.K. Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 2005, 19, 623–625. [Google Scholar] [PubMed]
- Zeng, J.; Lin, X.; Fan, H.; Li, C. Hydrogen sulfide attenuates the inflammatory response in a mouse burn injury model. Mol. Med. Rep. 2013, 8, 1204–1208. [Google Scholar] [PubMed]
- Wallace, J.L.; Vong, L.; McKnight, W.; Dicay, M.; Martin, G.R. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009, 137, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol. Sci. 2007, 28, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Blackler, R.; Syer, S.; Bolla, M.; Ongini, E.; Wallace, J.L. Gastrointestinal-sparing effects of novel NSAIDs in rats with compromised mucosal defence. PLoS ONE 2012, 7, e35196. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Cui, J.; Song, C.J.; Bian, J.S.; Sparatore, A.; Soldato, P.D.; Wang, X.Y.; Yan, C.D. H2S-Releasing Aspirin Protects against Aspirin-Induced Gastric Injury via Reducing Oxidative Stress. PLoS ONE 2012, 7, e46301. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, S.; Mencarelli, A.; Bruno, A.; Renga, B.; Distrutti, E.; Santucci, L.; Baldelli, F.; Fiorucci, S. Activation of the bile acid receptor GPBAR1 protects against gastrointestinal injury caused by non-steroidal anti-inflammatory drugs and aspirin in mice. Br. J. Pharmacol. 2013, 168, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Lou, L.X.; Geng, B.; Du, J.B.; Tang, C.S. Hydrogen sulfide-induced hypothermia attenuates stress-related ulceration in rats. Clin. Exp. Pharmacol. Physiol. 2008, 35, 223–228. [Google Scholar] [PubMed]
- Magierowski, M.; Jasnos, K.; Kwiecien, S.; Drozdowicz, D.; Surmiak, M.; Strzalka, M.; Ptak-Belowska, A.; Wallace, J.L.; Brzozowski, T. Endogenous prostaglandins and afferent sensory nerves in gastroprotective effect of hydrogen sulfide against stress-induced gastric lesions. PLoS ONE. 2015, 10, e0118972. [Google Scholar] [CrossRef] [PubMed]
- Yonezawa, D.; Sekiguchi, F.; Miyamoto, M.; Taniquchi, E.; Honjo, M.; Masuko, T.; Nishikawa, H.; Kawabata, A. A protective role of hydrogen sulfide against oxidative stress in rat gastric mucosal epithelium. Toxicology 2007, 241, 11–18. [Google Scholar]
- Kubo, S.; Kajiwara, M.; Kawabata, A. Dual modulation of the tension of isolated gastric artery and gastric mucosal circulation by hydrogen sulfide in rats. Inflammopharmacology 2007, 15, 288–292. [Google Scholar] [CrossRef] [PubMed]
- Mard, S.A.; Neisi, N.; Solgi, G.; Hassanpour, M.; Darbor, M.; Maleki, M. Gastroprotective effect of NaHS against mucosal lesions induced by ischemia—Reperfusion injury in rat. Dig. Dis. Sci. 2012, 57, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liang, F.; Shah Masood, W.; Yan, X. Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injurybyKeap1s-sulfhydration, MAPK dependent anti-apoptosis and NF-κB dependent anti-inflammation pathway. Eur. J. Pharmacol. 2014, 725, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Liu, L.; Zou, J.; Qiao, W.; Liu, H.; Qi, Y.; Yan, C. Protective effect of endogenous hydrogen sulfide against oxidative stress in gastric ischemia-reperfusion injury. Exp. Ther. Med. 2013, 5, 689–694. [Google Scholar] [PubMed]
- Mard, S.; Askari, H.; Neisi, N.; Veisi, A. Antisecretory effect of hydrogen sulfide on gastric acid secretion and the involvement of nitric oxide. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Ise, F.; Takahashi, K.; Aihara, E.; Hayashi, S. H2S-induced HCO3− secretion in the rat stomach—involvement of nitric oxide, prostaglandins, and capsaicin-sensitive sensory neurons. Nitric Oxide 2014, 46, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Nicolau, L.A.; Silva, R.O.; Damasceno, S.R.; Carvalho, N.S.; Costa, N.R.; Aragão, K.S.; Barbosa, A.L.; Soares, P.M.; Souza, M.H.; Medeiros, J.V. The hydrogen sulfide donor, Lawesson’s reagent prevents alendronate-induced gastric damage in rats. Braz. J. Med. Biol. Res. 2013, 46, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br. J. Pharmacol. 2010, 159, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Ferraz, J.G.; Muscara, M.N. Hydrogen Sulfide: An Endogenous Mediator of Resolution of Inflammation and Injury. Antioxid. Redox Signal. 2012, 17, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Shafigullin, M.Y.; Zefirov, R.A.; Sabirullina, G.I.; Zefirov, A.L.; Sitdikova, G.F. Effects of a hydrogen sulfide donor on spontaneous contractile activity of rat stomach and jejunum. Bull. Exp. Biol. Med. 2014, 157, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Pouokam, E.; Diener, M. Mechanisms of actions of hydrogen sulphide on rat distal colonic epithelium. Br. J. Pharmacol. 2011, 162, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Ise, F.; Takasuka, H.; Hayashi, S.; Takahashi, K.; Koyama, M.; Aihara, E.; Takeuchi, K. Stimulation of duodenal HCO3− secretion by hydrogen sulfide in rats: Relation to prostaglandins, nitric oxide and sensory neurons. Acta Physiol. 2011, 201, 117–126. [Google Scholar] [CrossRef]
- Hirata, I.; Naito, Y.; Takagi, T.; Mizushima, K.; Suzuki, T.; Omatsu, T.; Handa, O.; Ichikawa, H.; Ueda, H.; Yoshikawa, T. Endogenous hydrogen sulfide is an anti-inflammatory molecule in dextran sodium sulfate-induced colitis in mice. Dig. Dis. Sci. 2011, 56, 1379–1386. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, M.; Tarui, T.; Mitani, K.; Nagasawa, K.; Fukushima, O.; Okubo, K.; Yoshida, S.; Takemura, M.; Kawabata, A. Luminal hydrogen sulfide plays a pro-nociceptive role in mouse colon. Gut 2009, 58, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Agbor, T.A.; Motta, J.P.; Ferraz, J.G.; Wang, R.; Buret, A.G.; Wallace, J.L. Proresolution effects of hydrogen sulfide during colitis are mediated through hypoxia-inducible factor-1α. FASEB J. 2015, 29. [Google Scholar] [CrossRef] [PubMed]
- Fasolino, I.; Izzo, A.A.; Clavel, T.; Romano, B.; Haller, D.; Borrelli, F. Orally administered allyl sulfides from garlic ameliorate murine colitis. Mol. Nutr. Food Res. 2015, 59, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Motta, J.P.; Flannigan, K.L.; Agbor, T.A.; Beatty, J.K.; Blackler, R.W.; Workentine, M.L.; da Silva, G.J.; Wang, R.; Buret, A.G.; Wallace, J.L. Hydrogen sulfide protects from colitis and restores intestinal microbiota biofilm and mucus production. Inflamm. Bowel Dis. 2015, 21, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Agbor, T.A.; Blackler, R.W.; Kim, J.J.; Khan, W.I.; Verdu, E.F.; Ferraz, J.G.P.; Wallace, J.L. Impaired hydrogen sulfide synthesis and IL-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proc. Natl. Acad. Sci. USA 2014, 111, 13559–13564. [Google Scholar] [CrossRef] [PubMed]
- Zayachkivska, O.; Havryluk, O.; Hrycevych, N.; Bula, N.; Grushka, O.; Wallace, J.L. Cytoprotective effects of hydrogen sulfide in novel rat models of non-erosive esophagitis. PLoS ONE 2014, 9, e110688. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Jaraki, O.; Osborne, J.; Simon, D.I.; Keaney, J.; Vita, J.; Singel, D.; Valeri, C.R.; Loscalzo, J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. USA 1992, 89, 7674–7677. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, M.; Fukuda, R.; Bateman, R.M.; Yamamoto, T.; Suematsu, M. Interactions of multiple gas-transducing systems: Hallmarks and uncertainties of CO, NO, and H2S gas biology. Antioxid. Redox Signal. 2010, 13, 157–192. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Q.; Pang, B.; Kiziltepe, T.; Trudel, L.J.; Engelward, B.P.; Dedon, P.C.; Wogan, G.N. Threshold Effects of Nitric Oxide-Induced Toxicity and Cellular Responses in Wild-type and p53-Null Human Lymphoblastoid Cells. Chem. Res. Toxicol. 2006, 19, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Lefer, D.J. Emergence of Hydrogen Sulfide as an Endogenous Gaseous Signaling Molecule in Cardiovascular Disease. Circ. Res. 2014, 114, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Korbut, R.; Adamek-Guzik, T. Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol. 2003, 54, 469–487. [Google Scholar] [PubMed]
- Kimura, H. Hydrogen sulfide and polysulfides as biological mediators. Molecules 2014, 19, 16146–16157. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.S.; Pae, H.O.; Lee, B.S.; Kim, B.N.; Kim, J.M.; Kim, H.R.; Jeon, S.B.; Jeon, W.K.; Chae, H.J.; Chung, H.T. Hydrogen sulfide inhibits nitric oxide production and nuclear factor-κB via heme oxygenase-1 expression in RAW264.7 macrophages stimulated with lipopolysaccharide. Free Radic. Biol. Med. 2006, 41, 106–119. [Google Scholar] [CrossRef]
- Bibli, S.I.; Yang, G.; Zhou, Z.; Wang, R.; Topouzis, S.; Papapetropoulos, A. Role of cGMP in hydrogen sulfide signaling. Nitric Oxide 2014, 29, S1089–S1103. [Google Scholar]
- Geng, B.; Cui, Y.; Zhao, J.; Yu, F.; Zhu, Y.; Xu, G.; Zhang, Z.; Tang, C.; Du, J. Hydrogen sulfide downregulates the aortic l-arginine/nitric oxide pathway in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R1608–R1618. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Fu, Y.S.; Wang, Y.M.; Yang, K.H.; Wang, D.L.; Huang, B. Hydrogen sulfide increases nitric oxide production and subsequent S-nitrosylation in endothelial cells. Sci. World J. 2014, 2014, 480387. [Google Scholar] [CrossRef]
- Meng, J.; Ganesan Adaikan, P.; Srilatha, B. Hydrogen sulfide promotes nitric oxide production in corpus cavernosum by enhancing expression of endothelial nitric oxide synthase. Int. J. Impot. Res. 2013, 25, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Kida, M.; Sugiyama, T.; Yoshimoto, T.; Ogawa, Y. Hydrogen sulfide increases nitric oxide production with calcium-dependent activation of endothelial nitric oxide synthase in endothelial cells. Eur. J. Pharm. Sci. 2013, 48, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Altaany, Z.; Yang, G.; Wang, R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J. Cell Mol. Med. 2013, 17, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Bełtowski, J.; Jamroz-Wiśniewska, A. Hydrogen sulfide and endothelium-dependent vasorelaxation. Molecules 2014, 19, 21183–21199. [Google Scholar] [CrossRef] [PubMed]
- Bir, S.C.; Kolluru, G.K.; McCarthy, P.; Shen, X.; Pardue, S.; Pattillo, C.B.; Kevil, C.G. Hydrogen sulfide stimulates ischemic vascular remodeling through nitric oxide synthase and nitrite reduction activity regulating hypoxia-inducible factor-1α and vascular endothelial growth factor-dependent angiogenesis. J. Am. Heart. Assoc. 2012, 1, e004093. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Kamisaki, Y.; Ohkura, T.; Kanda, G.; Nakamoto, K.; Kishimoto, Y.; Ashida, K.; Itoh, T. Direct measurement of nitric oxide release in gastric mucosa during ischemia-reperfusion in rats. Am. J. Physiol. 1998, 274, G465–G471. [Google Scholar] [PubMed]
- Aboubakr, E.M.; Taye, A.; El-Moselhy, M.A.; Hassan, M.K. Protective effect of hydrogen sulfide against cold restraint stress-induced gastric mucosal injury in rats. Arch. Pharm. Res. 2013, 36, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, T.; Konturek, P.C.; Drozdowicz, D.; Konturek, S.J.; Zayachivska, O.; Pajdo, R.; Kwiecień, S.; Pawlik, W.W.; Hahn, E.G. Grapefruit-seed extract attenuates ethanol- and stress-induced gastric lesions via activation of prostaglandin, nitric oxide and sensory nerve pathways. World J. Gastroenterol. 2005, 11, 6450–6458. [Google Scholar] [PubMed]
- Brzozowski, T.; Kwiecień, S.; Konturek, P.C.; Konturek, S.J.; Ptak, A.; Mitis-Musioł, M.; Duda, A.; Bielański, W.; Hahn, E.G. Comparison of nitric oxide-releasing NSAID and vitamin C with classic NSAID in healing of chronic gastric ulcers; involvement of reactive oxygen species. Med. Sci. Monit. 2001, 7, 592–599. [Google Scholar] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magierowski, M.; Magierowska, K.; Kwiecien, S.; Brzozowski, T. Gaseous Mediators Nitric Oxide and Hydrogen Sulfide in the Mechanism of Gastrointestinal Integrity, Protection and Ulcer Healing. Molecules 2015, 20, 9099-9123. https://doi.org/10.3390/molecules20059099
Magierowski M, Magierowska K, Kwiecien S, Brzozowski T. Gaseous Mediators Nitric Oxide and Hydrogen Sulfide in the Mechanism of Gastrointestinal Integrity, Protection and Ulcer Healing. Molecules. 2015; 20(5):9099-9123. https://doi.org/10.3390/molecules20059099
Chicago/Turabian StyleMagierowski, Marcin, Katarzyna Magierowska, Slawomir Kwiecien, and Tomasz Brzozowski. 2015. "Gaseous Mediators Nitric Oxide and Hydrogen Sulfide in the Mechanism of Gastrointestinal Integrity, Protection and Ulcer Healing" Molecules 20, no. 5: 9099-9123. https://doi.org/10.3390/molecules20059099
APA StyleMagierowski, M., Magierowska, K., Kwiecien, S., & Brzozowski, T. (2015). Gaseous Mediators Nitric Oxide and Hydrogen Sulfide in the Mechanism of Gastrointestinal Integrity, Protection and Ulcer Healing. Molecules, 20(5), 9099-9123. https://doi.org/10.3390/molecules20059099