
Characterization and Pharmacokinetic Study of Aprepitant Solid Dispersions with Soluplus®

Abstract
:1. Introduction
2. Results and Discussion
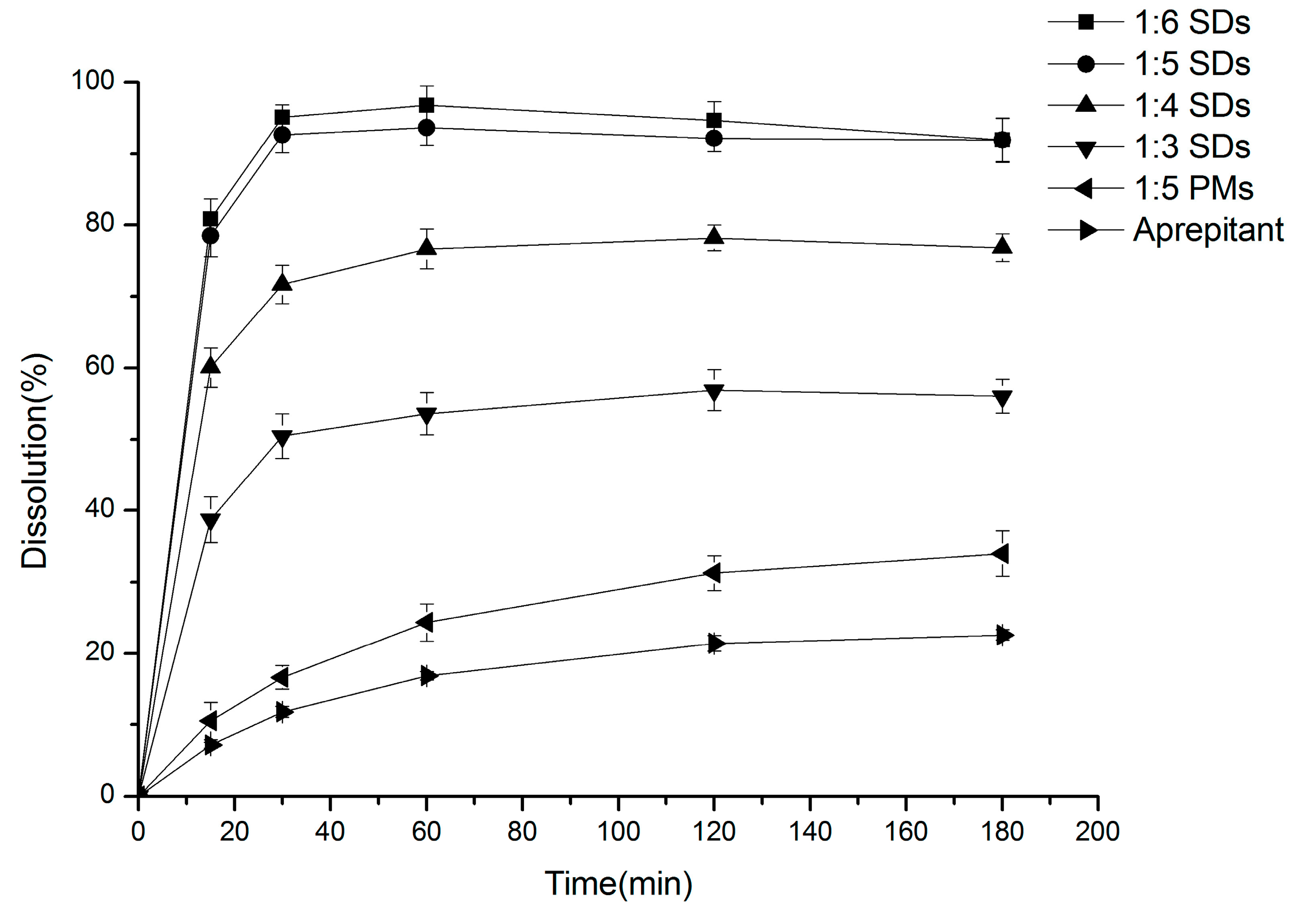
2.1. Dissolution Studies
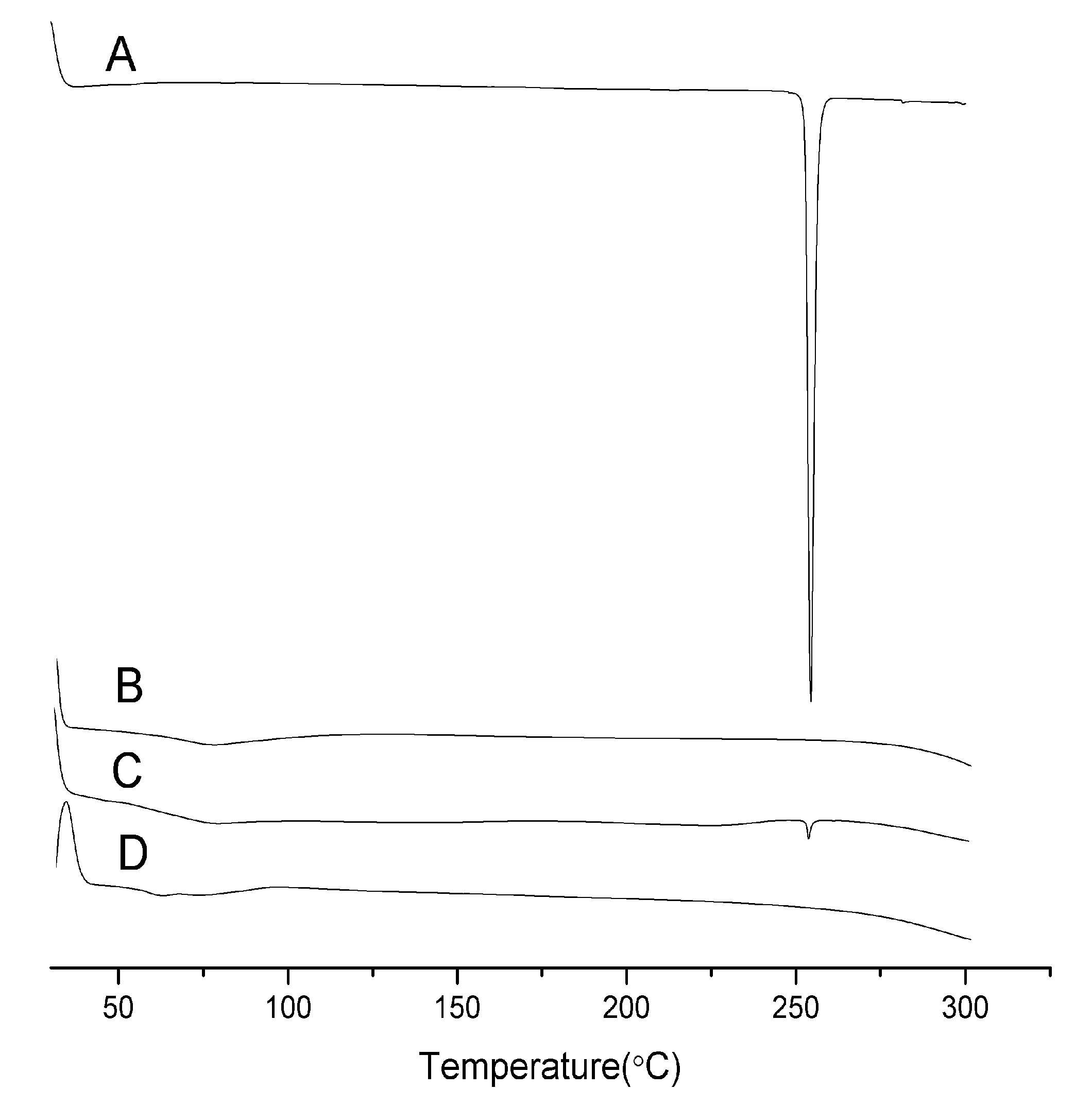
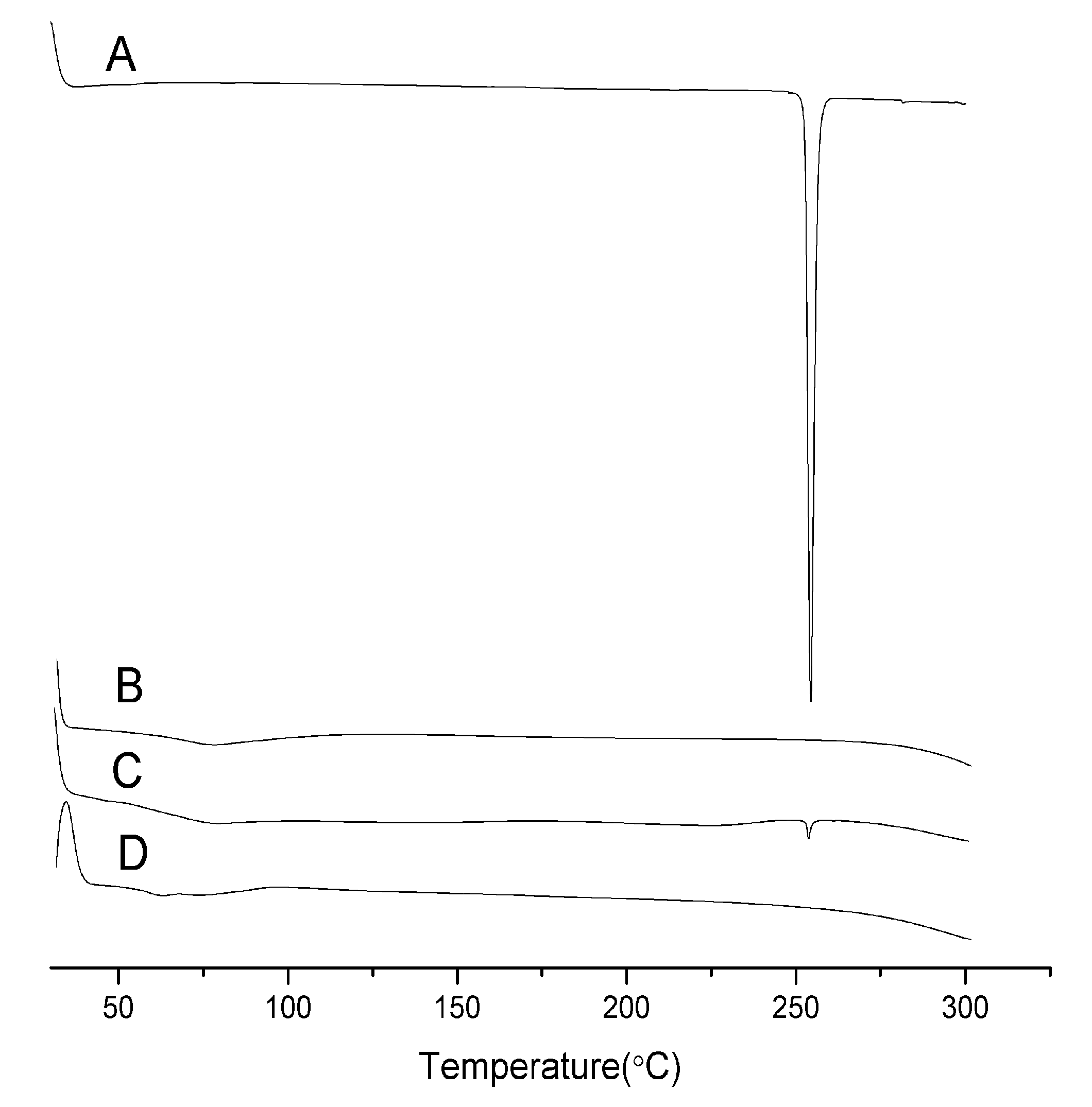
2.2. Differential Scanning Calorimetry (DSC)
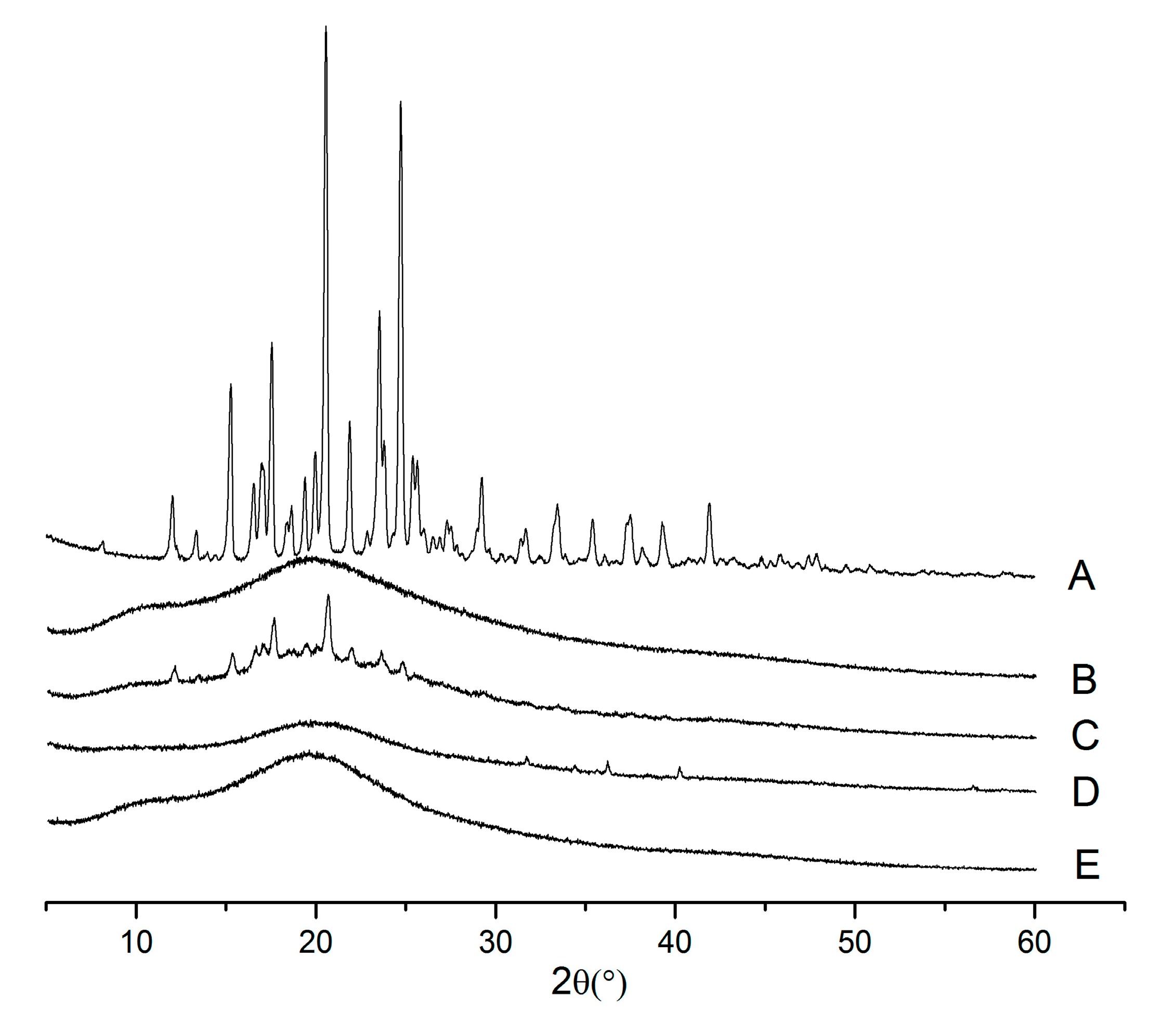
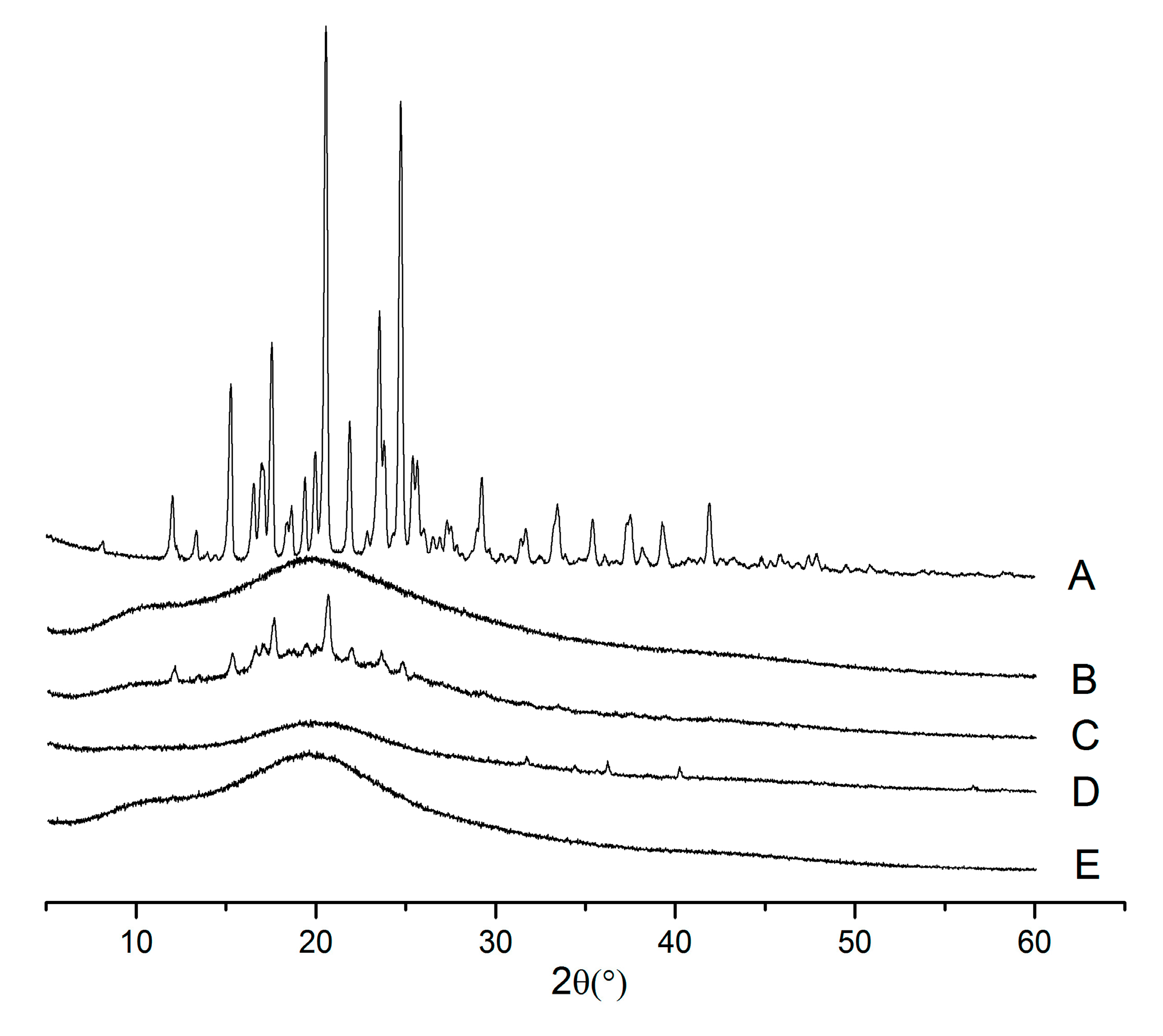
2.3. X-ray Powder Diffraction (XRPD)
2.4. Scanning Electron Microscopy (SEM)

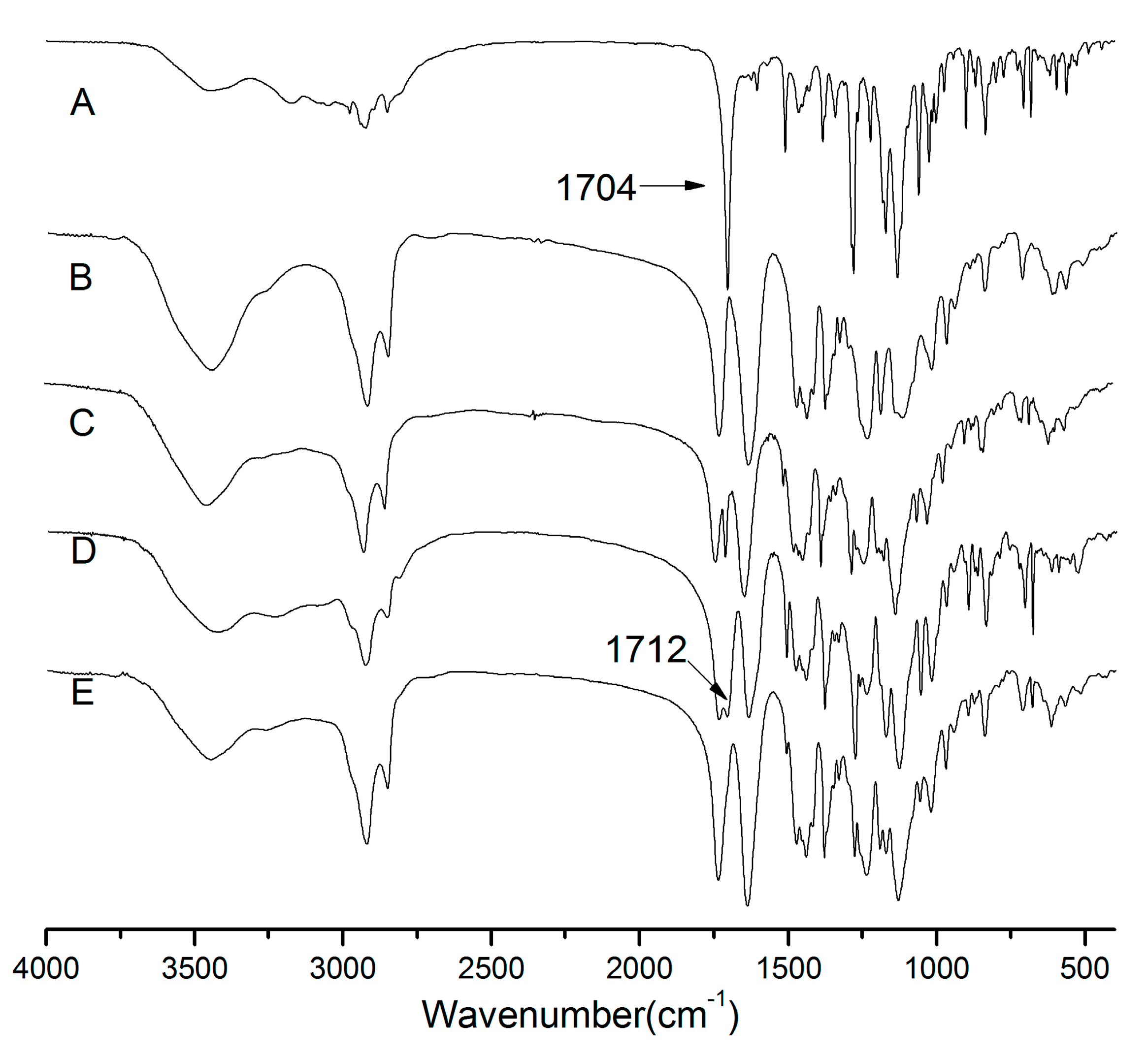
2.5. Fourier Transform Infrared Spectroscopy (FTIR)

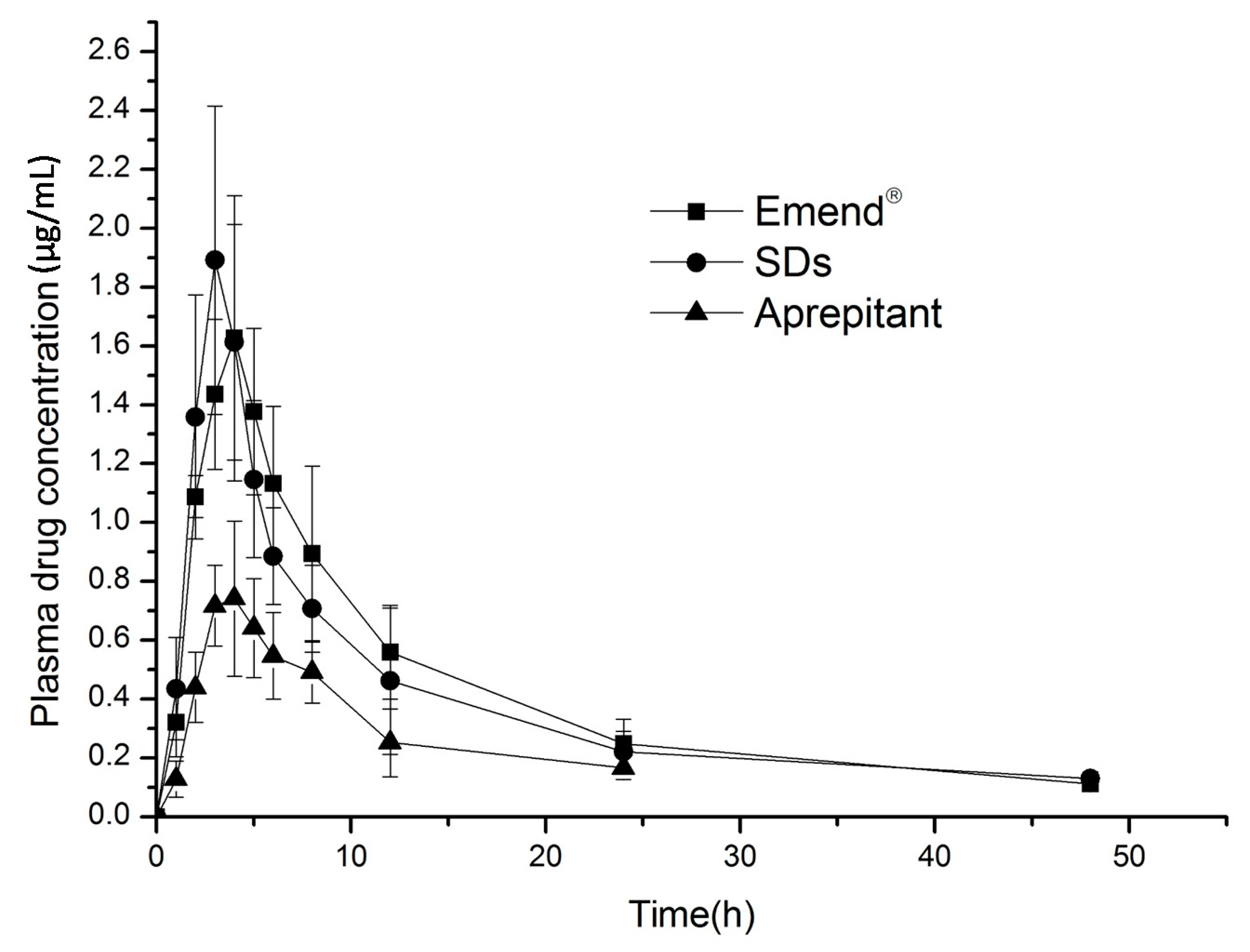
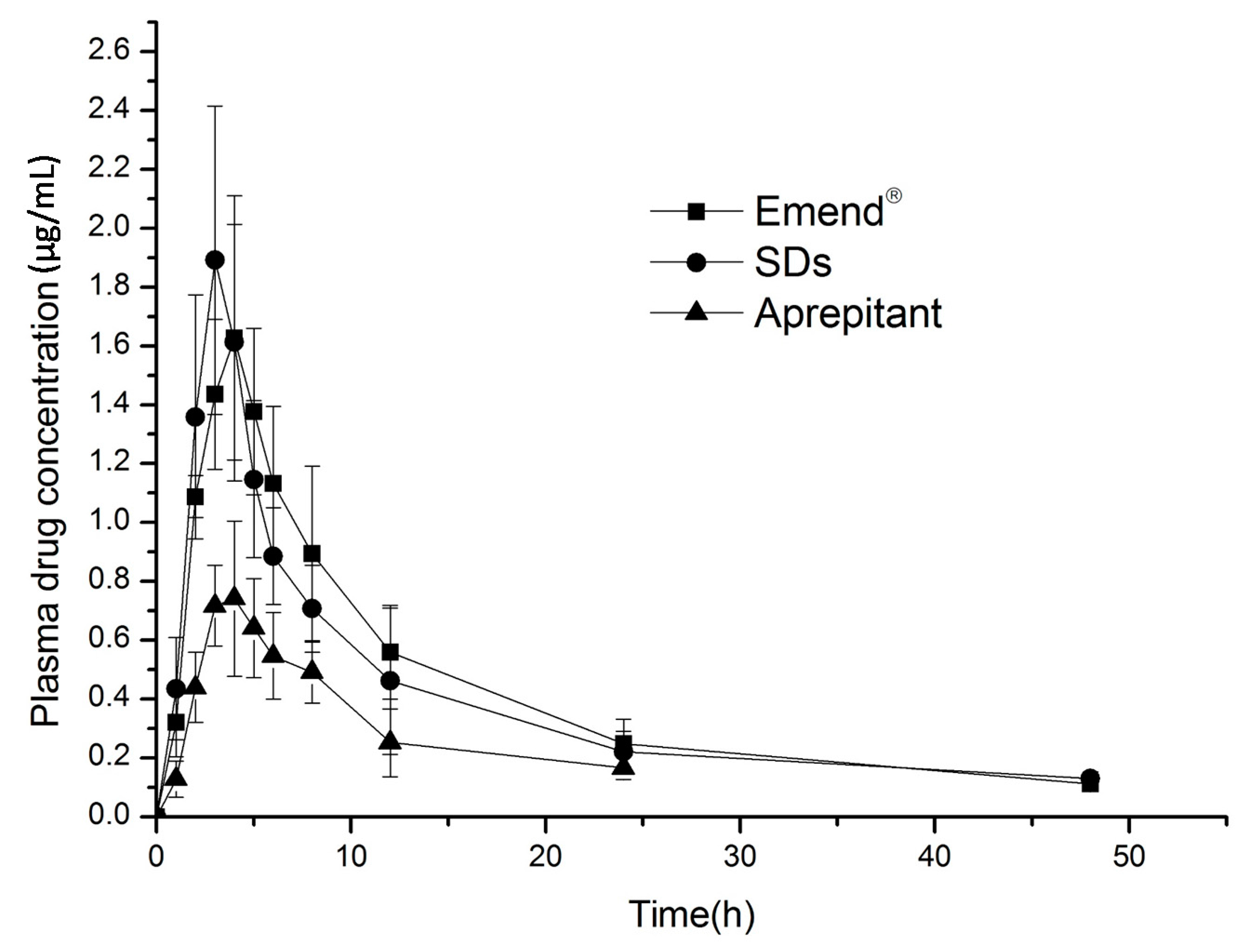
2.6. In Vivo Evaluation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Emend® | Solid Dispersions | Aprepitant |
|---|---|---|---|
| AUC0–48 h μg·h/mL | 20.51 ± 3.60 | 19.10 ± 3.10 * | 7.97 ± 1.67 |
| AUC0–∞ μg·h/mL | 22.21 ± 3.96 | 20.09 ± 3.93 | 9.42 ± 2.29 |
| Cmax μg/mL | 1.71 ± 0.40 | 2.085 ± 0.30 | 0.82 ± 0.14 |
| Tmax h | 4.33 ± 0.52 | 3 ± 0.63 | 3.67 ± 0.52 |
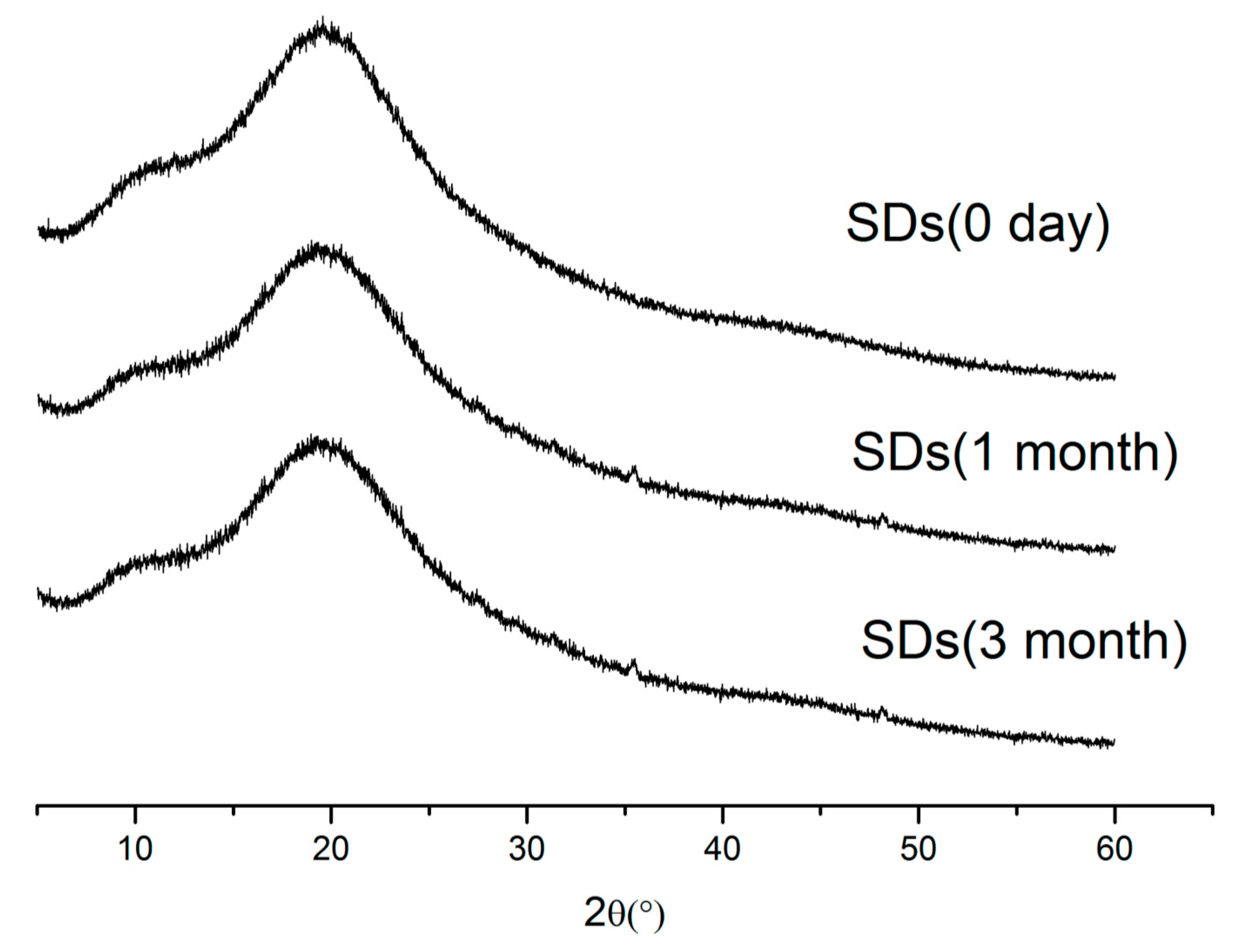
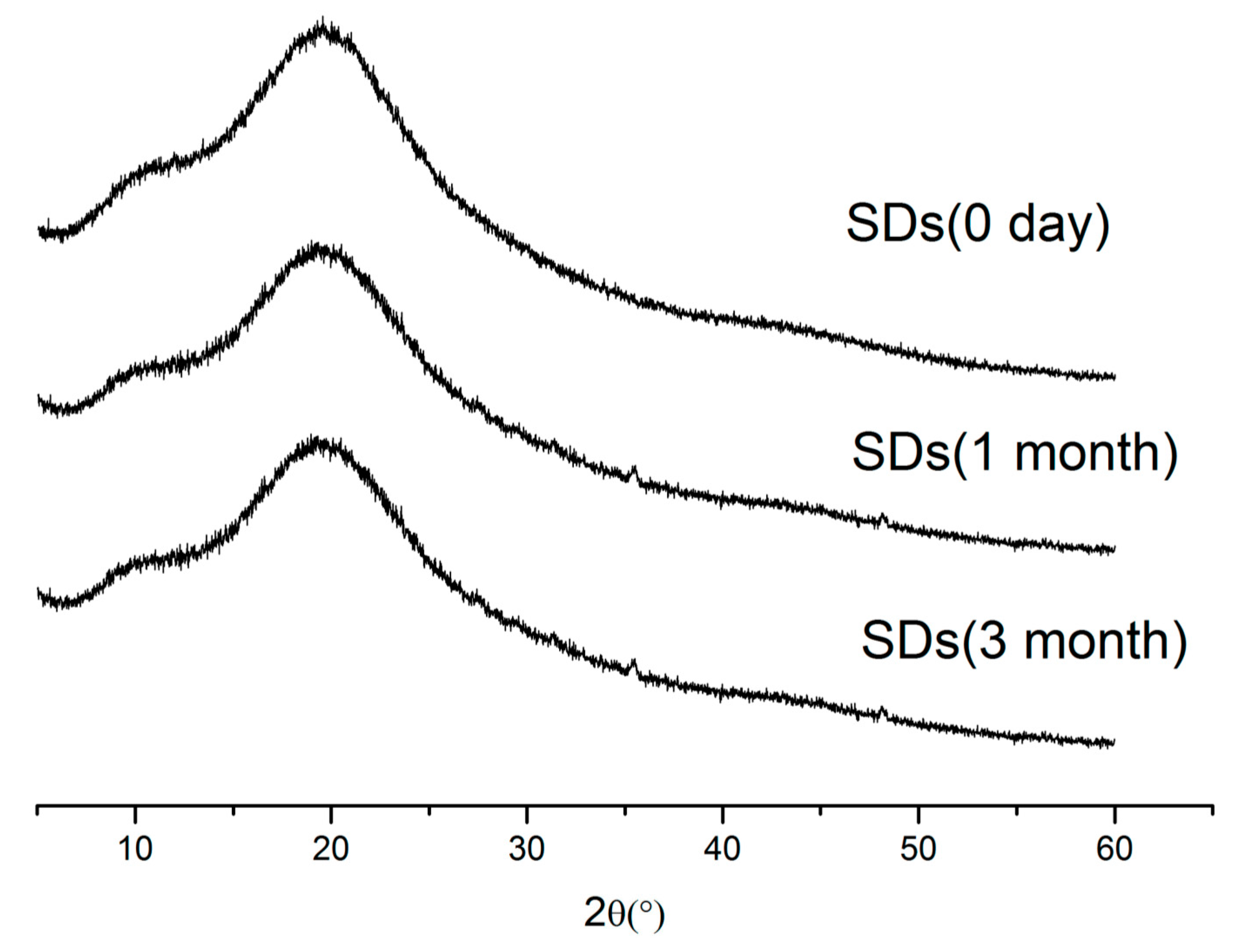
2.7. Stability Study
3. Experimental Section
3.1. Materials
3.2. Preparation of the Solid Dispersions
3.3. Dissolution Studies
3.4. Differentinal Scanning Calorimetry (DSC)
3.5. Analysis of X-ray Powder Diffraction (XRPD)
3.6. Scanning Electron Microscopy (SEM)
3.7. Fourier Transform Infrared Spectroscocopy (FTIR)
3.8. In Vivo Experiments
3.8.1. Animal Experiments
3.8.2. Plasma Processing and Analysis
3.8.3. Stability Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wu, D.; Paul, D.J.; Zhao, X.G.; Douglas, S.D.; Barrett, J.S. A sensitive and rapid charomatography-tandem mass spectrometry method for the quantification of the novel neurokinin-1 receptor antagonist aprepitant in rhesus macaque plasma, and cerebral spinal fluid, and human plasma with application in translational NeuroAIDs research. Eur. J. Pharm. Biopharm. 2009, 49, 739–745. [Google Scholar]
- Navari, R.M.; Reinhardt, R.R.; Gralla, R.J.; Kris, M.G.; Hesketh, P.J.; Khojasteh, A.; Grote, T.H.; Pendergrass, K.; Grunberg, S.M.; Carides, A.D.; et al. Reduction of cisplatin-indued emesis by a selective neurokinin-1-recptor antagonist. N. Engl. J. Med. 1999, 340, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Sorbera, L.A.; Castaner, J.; Bayes, M.; Silvestre, J. Drugs of the Future. Drugs Future 2002, 27, 211–216. [Google Scholar] [CrossRef]
- Nama, S.; Chandu, B.R.; Awen, Z.B.; Khagga, M. Development and validation of a new RP-HPLC method for the determination of aprepitant in solid dosage forms. Trop. J. Pharm. Res. 2011, 1, 491–497. [Google Scholar] [CrossRef]
- Cocquyt, V.; van, B.S.; Reinhardt, R.R.; Decramer, M.L.; O’Brien, M.; Schellens, J.H.; Verbeke, L.; van, A.F.; de, S.M.; Carides, A.D.; et al. Comparison of l-758,298, a prodrug for the selective neurokinin-1 antagonist, l-754,030, with ondansetron for the prevention of cisplatin-induced emesis. Eur. J. Cancer 2001, 37, 835–842. [Google Scholar] [CrossRef]
- Wu, Y.; Loper, A.; Landis, E.; Hettrick, L.; Novak, L.; Lynn, K.; Chen, C.; Thompson, K.; Higgins, R.; Batra, U.; et al. The role of biopharmaceutics in the development of a clinical nanoparticle formulation of MK-0869: A Beagle dog model predicts improved bioavailability and diminished food effect on absorption in human. Int. J. Pharm. 2004, 285, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Olver, L.; Shelukar, S.; Thompson, K.C. Nanomedicines in the treatment of emesis during chemotherapy: Focus on aprepitant. Int. J. Nanome 2007, 2, 13–18. [Google Scholar] [CrossRef]
- Yasushi, S.; Ekarat, J.; Filippos, K.; Christos, R.; Jennifer, B.D. Forecasting in vivo oral absorption and food effect of micronized and nanosized aprepitant formulations in humans. Eur. J. Pharm. Biopharm. 2010, 76, 95–104. [Google Scholar]
- Dhananjay, S.S.; Seshasai, M.M.; Gowthamrajanb, K.; Giriraj, K.T.; Rajesh, V.; Parchuri, S.R. Optimization of formulation and process variable of nanosuspension: An industrial perspective. Int. J. Pharm. 2010, 402, 213–220. [Google Scholar]
- Ridhurkar, D.N.; Ansari, K.A.; Kumar, D.; Kaul, N.S.; Krishnamurthy, T.; Dhawan, S.; Pillai, R. Inclusion complex of aprepitant with cyclodextrin:evaluation of physico-chemical and pharmacokininetic properties. Drug Dev. Ind. Pharm. 2013, 39, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Müller, R.H.; Jacobs, C.; Kayser, O. Nanosuspensions as particulate drug formulations in therapy. Rationale for development and what we can expect for the future. Adv. Drug Deliv. Rev. 2001, 47, 3–19. [Google Scholar] [CrossRef]
- Basa, S.; Muniyappan, T.; Karatgi, P.; Prabhu, R.; Pillai, R. Production and in vitro characterization of solid dosage form incorporating drug nanoparticles. Drug Dev. Ind. Pharm. 2008, 34, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Humberstone, A.J.; Charman, W.N. Lipid-based vehicled for the oral delivery of poorly water soluble drugs. Adv. Drug Deliv. Rev. 1997, 25, 103–128. [Google Scholar] [CrossRef]
- Pouton, C.W. Lipid formulations for oral administration of drugs: Non-emulsifying, self-emulsifying and “selfmicroemulsifying” drug delivery systems. Eur. J. Pharm. Sci. 2000, 2, 93–98. [Google Scholar] [CrossRef]
- Wang, X.P.; Li, L.; Hou, W.; Hou, L.L.; Zhao, Z.Y.; Li, W.G. Characterization and stability of tanshinone IIA solid dispersions with hydroxyapatite. Materials 2013, 6, 805–816. [Google Scholar] [CrossRef]
- Eloy, J.O.; Marchetti, J.M. Solid dispersions containing ursolic acid in Poloxamer407 and PEG6000: A comparative study of fusion and solvent methods. Powder Technol. 2014, 253, 98–106. [Google Scholar] [CrossRef]
- Maulvi, F.A.; Dalwadi, S.J.; Thakkar, V.T.; Soni, T.G.; Gohel, M.C.; Gandhi, T.R. Improvement of dissolution rate of aceclofenac by solid dispersion technique. Powder Technol. 2011, 207, 47–54. [Google Scholar] [CrossRef]
- Frizon, F.; Eloy, J.O.; Donaduzzi, C.M.; Mitsui, M.L.; Marchetti, J.M. Dissolution rate enhancement of loratadine in polyvinylpyrrolidone K-30 solid dispersions by solvent methods. Powder Technol. 2013, 235, 532–539. [Google Scholar] [CrossRef]
- Babu, G.; Prasad, C.; Murthy, K. Evaluation of modified gum karaya as carrier for the dissolute enhancement of poorly water-soluble drug nimodipine. Int. J. Pharm. 2002, 234, 1–17. [Google Scholar] [CrossRef]
- Linna, M.; Collnot, E.M.; Djuric, D.; Hempel, K.; Fabian, E.; Kolter, K.; Lehr, C.M. Soluplus as an effective absorption enhancer of poorly soluble drugs in vitro and in vivo. Eur. J. Pharm. Sci. 2012, 45, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Hardung, H.D.; Djuric, D.; Ali, S. Combining HME & Solubilization: Soluplus®—the solid solution. Drug Dev. Technol. 2010, 3, 20–27. [Google Scholar]
- Vasconcelos, T.; Sarmento, B.; Costa, P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 23–24. [Google Scholar] [CrossRef] [PubMed]
- Dangprasirt, P.; Ritthidej, G.C. Development of diclofenac sodium controlled release solid dispersions by spray drying using optimization strategy I. Drug Dev. Ind. Pharm. 1995, 21, 2323–2337. [Google Scholar] [CrossRef]
- Verheyen, S.; Blaton, N.; Kinget, R.; van den Mooter, G. Mechanism of increased dissolution of diazepam and temazepam from polyethylene glycol 6000 solid dispersions. Int. J. Pharm. 2002, 249, 45–58. [Google Scholar] [CrossRef]
- Riekes, M.K.; Kuminek, G.; Rauber, G.S.; Bortoluzzi, A.J.; Stulzer, H.K. HPMC as a potential enhancer of nimodipine biopharmaceutical properties via ball-milled solid dispersions. Carbohydr. Polym. 2014, 99, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Hintz, R.J.; Johnson, K.C. The effect of particle size distribution on dissolution rate and oral absorption. Int. J. Pharm. 1989, 51, 9–17. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds aprepitant are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Zou, M.; Piao, H.; Liu, Y.; Tang, B.; Gao, Y.; Ma, N.; Cheng, G. Characterization and Pharmacokinetic Study of Aprepitant Solid Dispersions with Soluplus®. Molecules 2015, 20, 11345-11356. https://doi.org/10.3390/molecules200611345
Liu J, Zou M, Piao H, Liu Y, Tang B, Gao Y, Ma N, Cheng G. Characterization and Pharmacokinetic Study of Aprepitant Solid Dispersions with Soluplus®. Molecules. 2015; 20(6):11345-11356. https://doi.org/10.3390/molecules200611345
Chicago/Turabian StyleLiu, Jinwen, Meijuan Zou, Hongyu Piao, Yi Liu, Bo Tang, Ying Gao, Ning Ma, and Gang Cheng. 2015. "Characterization and Pharmacokinetic Study of Aprepitant Solid Dispersions with Soluplus®" Molecules 20, no. 6: 11345-11356. https://doi.org/10.3390/molecules200611345
APA StyleLiu, J., Zou, M., Piao, H., Liu, Y., Tang, B., Gao, Y., Ma, N., & Cheng, G. (2015). Characterization and Pharmacokinetic Study of Aprepitant Solid Dispersions with Soluplus®. Molecules, 20(6), 11345-11356. https://doi.org/10.3390/molecules200611345