
Role and Regulation of Glutathione Metabolism in Plasmodium falciparum

Abstract
:1. Introduction
2. Glutathione
3. The Functions of GSH in Plasmodium
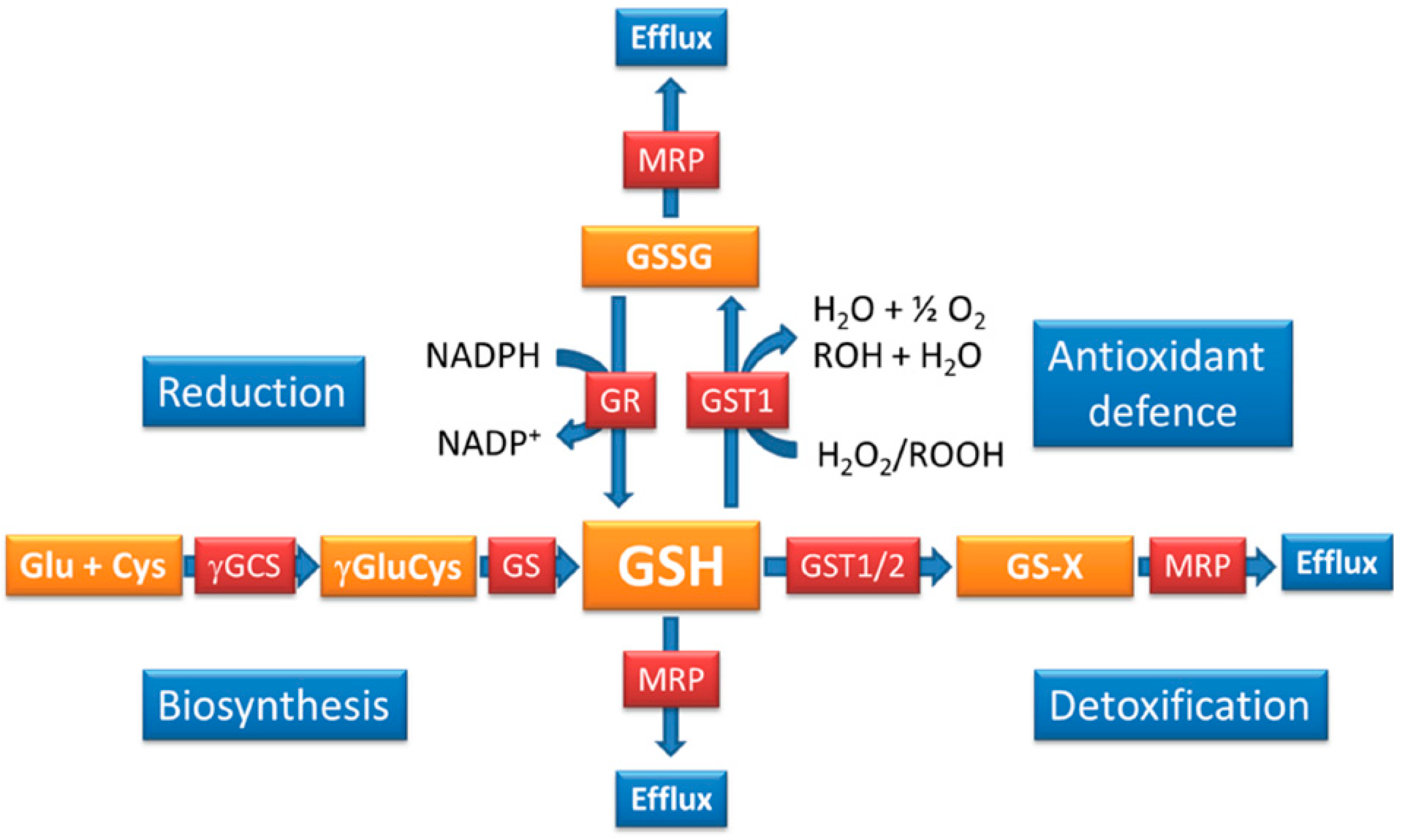
4. How is the GSH Redox Balance Regulated in Plasmodium?

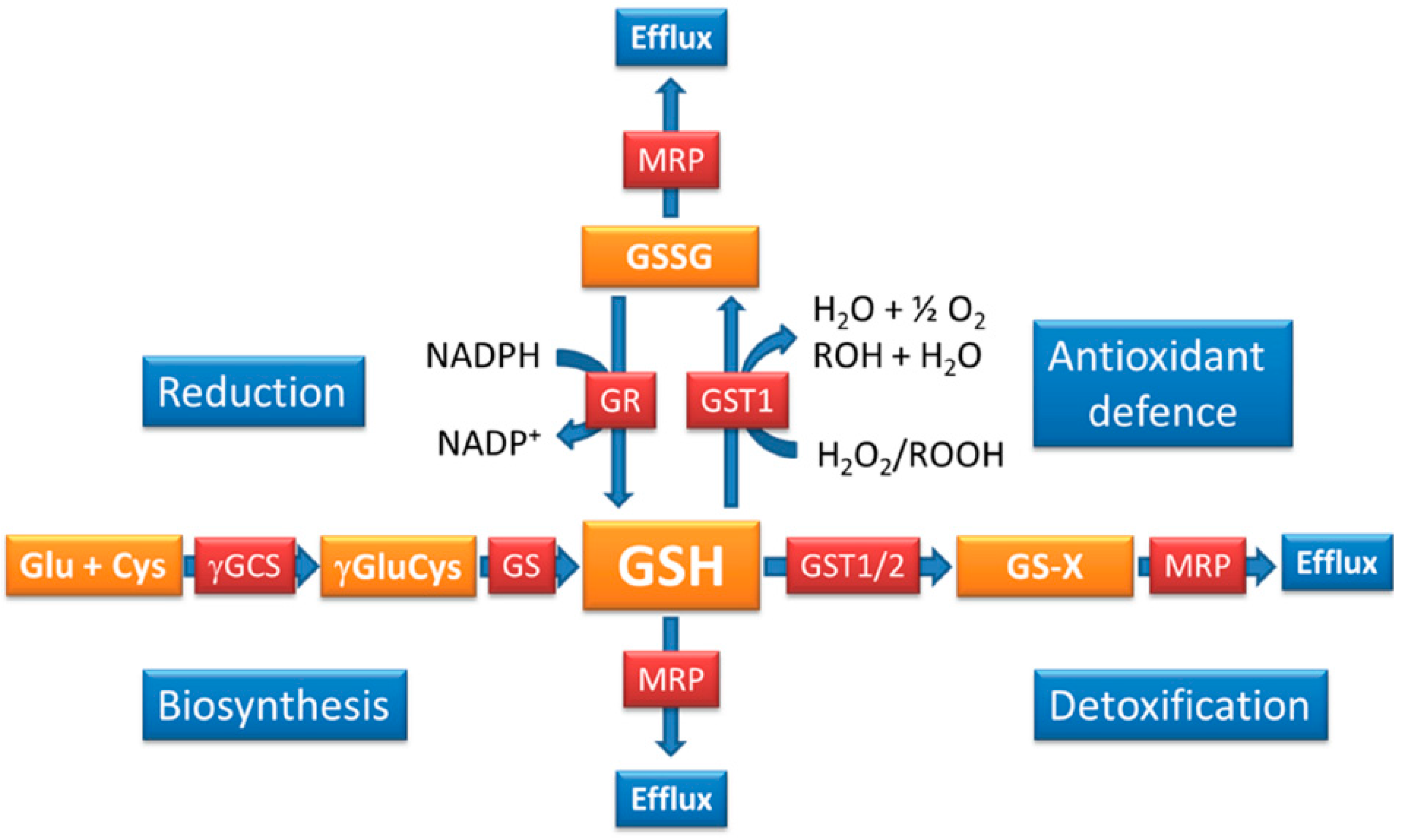{kind=link}
| P. falciparum | P. berghei | |
|---|---|---|
| Protein/Gene ID | Characteristics | Characteristics |
| Glutathione reductase (GR) Pf3D7_1419800.1; PBANKA_102340 | Genetic validation: not done Chemical validation: essential in RBC stages [73] Location: cytoplasm and apicoplast [39] | Genetic validation: essential for sexual development [72] Chemical validation: not conclusive Location: not analysed |
| Thioredoxin reductase (TrxR) | Genetic validation: only possible in presence of | Genetic validation: not essential in any life cycle stage |
| Pf3D7_0923800.1; PBANKA_082470 | expression plasmid [74] | |
| Chemical validation: essential in RBC stages [75] | Chemical validation: | |
| Location: cytoplasm and mitochondrion [39] | Location: not investigated | |
| Glutathione S-transferase 1 (GST1) | Genetic validation: not done | Genetic validation: not done |
| Pf3D7_1419300; PBANKA_102390 | Chemical validation: essential in RBC stages [76] | Chemical validation: not conclusive [76,77] |
| Location: predicted to be cytoplasmic | Location: not analysed | |
| No increase of activity with drug resistance [56] | Increased activity correlates with drug resistance [78,79,80] | |
| Has antioxidant activity [56] | Not assessed | |
| Glutathione S-transferase 2 (EXP1) | Location: parasitophorous vacuolar membrane [54] | Protein was not studied with respect to GSH metabolism |
| Pf3D7_1121600; PBANKA_092670 | Involved in heme and drug detoxification | |
| Antioxidant activity not determined | ||
| Essentiality not determined | ||
| γ-Glutamylcysteine synthetase (γGCS) | Genetic validation: impossible to KO gene [44] | Genetic validation: essential for sexual development [67] |
| Pf3D7_0918900; PBANKA_081980 | Chemical validation: specific inhibition with BSO is | Chemical validation: specific inhibition does not affect |
| lethal in RBC stages | parasite viability—not essential in RBC stages | |
| Location: cytoplasm | Location: not studied | |
| GSH and drug resistance | GSH levels elevated in some drug resistant parasites [41,81] | GSH levels elevated in drug resistant parasites [78,79] |
| GSH levels reduced in isogenic drug resistant | ||
| parasites [45] |
5. Glutathione and Its Role in Drug Resistance
6. Glutathione Metabolism of Plasmodium—A Drug Target?
Acknowledgments
List of Abbreviations
| BSO | buthionine sulfoximine |
| PfCRT | Plasmodium falciparum chloroquine resistance transporter |
| DV | digestive vacuole |
| ER | endoplasmatic reticulum |
| EXP1 | exported protein 1 |
| γ-GCS | γ-glutamylcysteine synthetase |
| GSH | reduced glutathione |
| GSSG | glutathione disulphide |
| GPx | glutathione peroxidase |
| GST | glutathione S-transferase |
| GR | glutathione reductase |
| GS | glutathione synthetase |
| PPP | pentose phosphate shunt |
| RBC | red blood cells |
Conflicts of Interest
References
- WHO. World Malaria Report 2014; WHO Global Malaria Programme: Geneva, Switzerland, 2014. [Google Scholar]
- Haldane, J.B. Suggestions as to quantitative measurement of rates of evolution. Evolution 1949, 3, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Giribaldi, G.; Ulliers, D.; Mannu, F.; Arese, P.; Turrini, F. Growth of Plasmodium falciparum induces stage-dependent haemichrome formation, oxidative aggregation of band 3, membrane deposition of complement and antibodies, and phagocytosis of parasitized erythrocytes. Br. J. Haematol. 2001, 113, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Akide-Ndunge, O.B.; Ayi, K.; Arese, P. The Haldane malaria hypothesis: facts, artifacts, and a prophecy. Redox Rep. 2003, 8, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Arese, P.; Turrini, F.; Schwarzer, E. Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell. Physiol. Biochem. 2005, 16, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.M.; Cerami, C.; Fairhurst, R.M. Hemoglobinopathies: Slicing the Gordian knot of Plasmodium falciparum malaria pathogenesis. PLoS Pathog. 2013, 9, e1003327. [Google Scholar] [CrossRef] [PubMed]
- Turrini, F.; Giribaldi, G.; Carta, F.; Mannu, F.; Arese, P. Mechanisms of band 3 oxidation and clustering in the phagocytosis of Plasmodium falciparum-infected erythrocytes. Redox Rep. 2003, 8, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, A.; Giribaldi, G.; Mannu, F.; Arese, P.; Turrini, F. Naturally occurring anti-band 3 antibodies and red blood cell removal under physiological and pathological conditions. Autoimmun. Rev. 2008, 7, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Uyoga, S.; Skorokhod, O.A.; Opiyo, M.; Orori, E.N.; Williams, T.N.; Arese, P.; Schwarzer, E. Transfer of 4-hydroxynonenal from parasitized to non-parasitized erythrocytes in rosettes. Proposed role in severe malaria anemia. Br. J. Haematol. 2012, 157, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.; Stocker, R. Oxidative stress and the redox status of malaria infected erythrocytes. Blood Cells 1990, 16, 499–526. [Google Scholar] [PubMed]
- Ginsburg, H.; Atamna, H. The redox status of malaria-infected erythrocytes: An overview with an emphasis on unresolved problems. Parasite 1994, 1, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, E.; Kuehn, H.; Arese, P. The hidden faces of hemozoin and its dangerous midwives. Trends Parasitol. 2003, 19, 197–198. [Google Scholar] [CrossRef]
- Schwarzer, E.; Kuhn, H.; Valente, E.; Arese, P. Malaria-parasitized erythrocytes and hemozoin nonenzymatically generate large amounts of hydroxy fatty acids that inhibit monocyte functions. Blood 2003, 101, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Winograd, E.; Prudhomme, J.G.; Sherman, I.W. Band 3 clustering promotes the exposure of neoantigens in Plasmodium falciparum-infected erythrocytes. Mol. Biochem. Parasitol. 2005, 142, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Skorokhod, O.A.; Caione, L.; Marrocco, T.; Migliardi, G.; Barrera, V.; Arese, P.; Piacibello, W.; Schwarzer, E. Inhibition of erythropoiesis in malaria anemia: Role of hemozoin and hemozoin-generated 4-hydroxynonenal. Blood 2010, 116, 4328–4337. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, R.; Marrocco, T.; Skorokhod, O.A.; Barbosa, A.; Nhabomba, A.; Manaca, M.N.; Guinovart, C.; Quinto, L.; Arese, P.; Alonso, P.L.; et al. Blood oxidative stress markers and Plasmodium falciparum malaria in non-immune African children. Br. J. Haematol. 2014, 164, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Tilley, L.; Vennerstrom, J.L.; Roberts, D.; Rogerson, S.; Ginsburg, H. Oxidative stress in malaria parasite-infected erythrocytes: Host-parasite interactions. Int. J. Parasitol. 2004, 34, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Müller, S. Redox and antioxidant systems of the malaria parasite Plasmodium falciparum. Mol. Microbiol. 2004, 53, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Rahlfs, S.; Nickel, C.; Schirmer, R.H. Glutathione—Functions and metabolism in the malarial parasite Plasmodium falciparum. Biol. Chem. 2003, 384, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Jortzik, E.; Becker, K. Thioredoxin and glutathione systems in Plasmodium falciparum. Int. J. Med. Microbiol. 2012, 302, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Liebau, E.; Walter, R.D.; Krauth-Siegel, R.L. Thiol-based redox metabolism of protozoan parasites. Trends Parasitol. 2003, 19, 320–328. [Google Scholar] [CrossRef]
- Bozdech, Z.; Ginsburg, H. Data mining of the transcriptome of Plasmodium falciparum: The pentose phosphate pathway and ancillary processes. Malar. J. 2005, 4. [Google Scholar] [CrossRef] [PubMed]
- Bozdech, Z.; Ginsburg, H. Antioxidant defense in Plasmodium falciparum—Data mining of the transcriptome. Malar. J. 2004, 3. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Preuss, J.; Jortzik, E.; Becker, K. Glucose-6-phosphate metabolism in Plasmodium falciparum. IUBMB Life 2012, 64, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Preuss, J.; Maloney, P.; Peddibhotla, S.; Hedrick, M.P.; Hershberger, P.; Gosalia, P.; Milewski, M.; Li, Y.L.; Sugarman, E.; Hood, B.; et al. Discovery of a Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase inhibitor (R,Z)-N-((1-ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxamide (ML276) that reduces parasite growth in vitro. J. Med. Chem. 2012, 55, 7262–7272. [Google Scholar] [PubMed]
- Jortzik, E.; Mailu, B.M.; Preuss, J.; Fischer, M.; Bode, L.; Rahlfs, S.; Becker, K. Glucose-6-phosphate dehydrogenase-6-phosphogluconolactonase: A unique bifunctional enzyme from Plasmodium falciparum. Biochem. J. 2011, 436, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Storm, J.; Perner, J.; Aparicio, I.; Patzewitz, E.M.; Olszewski, K.; Llinas, M.; Engel, P.C.; Muller, S. Plasmodium falciparum glutamate dehydrogenase a is dispensable and not a drug target during erythrocytic development. Malar. J. 2011, 10. [Google Scholar] [CrossRef] [PubMed]
- Zocher, K.; Fritz-Wolf, K.; Kehr, S.; Fischer, M.; Rahlfs, S.; Becker, K. Biochemical and structural characterization of Plasmodium falciparum glutamate dehydrogenase 2. Mol. Biochem. Parasitol. 2012, 183, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Wrenger, C.; Müller, S. Isocitrate dehydrogenase of Plasmodium falciparum. Eur. J. Biochem. 2003, 270, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Krauth-Siegel, R.L.; Muller, J.G.; Lottspeich, F.; Schirmer, R.H. Glutathione reductase and glutamate dehydrogenase of Plasmodium falciparum, the causative agent of tropical malaria. Eur. J. Biochem. 1996, 235, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Hudson, D.A.; Gannon, S.A.; Thorpe, C. Oxidative protein folding: From thiol-disulfide exchange reactions to the redox poise of the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 80, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Mullineaux, P.M.; Rausch, T. Glutathione, photosynthesis and the redox regulation of stress-responsive gene expression. Photosynth. Res. 2005, 86, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [CrossRef] [PubMed]
- Farber, P.M.; Becker, K.; Muller, S.; Schirmer, R.H.; Franklin, R.M. Molecular cloning and characterization of a putative glutathione reductase gene, the PfGR2 gene, from Plasmodium falciparum. Eur. J. Biochem. 1996, 239, 655–661. [Google Scholar] [CrossRef] [PubMed]
- Krauth- Siegel, R.L.; Lohrer, H.; Buecheler, U.S.; Schirmer, R.H. The Antioxidant Enzymes Glutathione Reductase and Trypanotione Reductase as Drug Targets; Taylor & Francis: London, UK; New York, NY, USA; Philadelphia, PA, USA, 1991; pp. 493–505. [Google Scholar]
- Farber, P.M.; Arscott, L.D.; Williams, C.H., Jr.; Becker, K.; Schirmer, R.H. Recombinant Plasmodium falciparum glutathione reductase is inhibited by the antimalarial dye methylene blue. FEBS Lett. 1998, 422, 311–314. [Google Scholar] [CrossRef]
- Kasozi, D.; Mohring, F.; Rahlfs, S.; Meyer, A.J.; Becker, K. Real-time imaging of the intracellular glutathione redox potential in the malaria parasite Plasmodium falciparum. PLoS Pathog. 2013, 9, e1003782. [Google Scholar] [CrossRef] [PubMed]
- Kehr, S.; Sturm, N.; Rahlfs, S.; Przyborski, J.M.; Becker, K. Compartmentation of redox metabolism in malaria parasites. PLoS Pathog. 2011, 6, e1001242. [Google Scholar] [CrossRef] [PubMed]
- Lüersen, K.; Walter, R.D.; Müller, S. Plasmodium falciparum-infected red blood cells depend on a functional glutathione de novo synthesis attributable to an enhanced loss of glutathione. Biochem. J. 2000, 346, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Meierjohann, S.; Walter, R.D.; Müller, S. Regulation of intracellular glutathione levels in erythrocytes infected with chloroquine-sensitive and chloroquine-resistant Plasmodium falciparum. Biochem. J. 2002, 368, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Lüersen, K.; Walter, R.D.; Müller, S. The putative gamma-glutamylcysteine synthetase from Plasmodium falciparum contains large insertions and a variable tandem repeat. Mol. Biochem. Parasitol. 1999, 98, 131–142. [Google Scholar] [CrossRef]
- Meierjohann, S.; Walter, R.D.; Müller, S. Glutathione synthetase from Plasmodium falciparum. Biochem. J. 2002, 363, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Patzewitz, E.M.; Wong, E.H.; Müller, S. Dissecting the role of glutathione biosynthesis in Plasmodium falciparum. Mol. Microbiol. 2012, 83, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Patzewitz, E.M.; Salcedo-Sora, J.E.; Wong, E.H.; Sethia, S.; Stocks, P.A.; Maughan, S.C.; Murray, J.A.; Krishna, S.; Bray, P.G.; Ward, S.A.; et al. Glutathione transport: A new role for PfCRT in chloroquine resistance. Antioxid. Redox Signal. 2013, 19, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Barrand, M.A.; Winterberg, M.; Ng, F.; Nguyen, M.; Kirk, K.; Hladky, S.B. Glutathione export from human erythrocytes and Plasmodium falciparum malaria parasites. Biochem. J. 2012, 448, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Lehane, A.M.; McDevitt, C.A.; Kirk, K.; Fidock, D.A. Degrees of chloroquine resistance in Plasmodium—Is the redox system involved? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.K.; Mu, J.; Jiang, H.; Kabat, J.; Singh, S.; Sullivan, M.; Fay, M.P.; McCutchan, T.F.; Su, X.Z. Disruption of a Plasmodium falciparum multidrug resistance-associated protein (PfMRP) alters its fitness and transport of antimalarial drugs and glutathione. J. Biol. Chem. 2009, 284, 7687–7696. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Ginsburg, H. The malaria parasite supplies glutathione to its host cell—Investigation of glutathione transport and metabolism in human erythrocytes infected with Plasmodium falciparum. Eur. J. Biochem. 1997, 250, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E. Hemoglobin degradation. Curr. Top. Microbiol. Immunol. 2005, 295, 275–291. [Google Scholar] [PubMed]
- Sigala, P.A.; Goldberg, D.E. The peculiarities and paradoxes of Plasmodium heme metabolism. Annu. Rev. Microbiol. 2014, 68, 259–278. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Ginsburg, H. Heme degradation in the presence of glutathione. A proposed mechanism to account for the high levels of non-heme iron found in the membranes of hemoglobinopathic red blood cells. J. Biol. Chem. 1995, 270, 24876–24883. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Miller, S.; Foley, M.; Tilley, L. Inhibition of the peroxidative degradation of haem as the basis of action of chloroquine and other quinoline antimalarials. Biochem. J. 1999, 339, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Lisewski, A.M.; Quiros, J.P.; Ng, C.L.; Adikesavan, A.K.; Miura, K.; Putluri, N.; Eastman, R.T.; Scanfeld, D.; Regenbogen, S.J.; Altenhofen, L.; et al. Supergenomic network compression and the discovery of EXP1 as a glutathione transferase inhibited by artesunate. Cell 2014, 158, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Klokouzas, A.; Tiffert, T.; van Schalkwyk, D.; Wu, C.P.; van Veen, H.W.; Barrand, M.A.; Hladky, S.B. Plasmodium falciparum expresses a multidrug resistance-associated protein. Biochem. Biophys. Res. Commun. 2004, 321, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Harwaldt, P.; Rahlfs, S.; Becker, K. Glutathione S-transferase of the malarial parasite Plasmodium falciparum: Characterization of a potential drug target. Biol. Chem. 2002, 383, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Wolf, K.; Becker, A.; Rahlfs, S.; Harwaldt, P.; Schirmer, R.H.; Kabsch, W.; Becker, K. X-ray structure of glutathione S-transferase from the malarial parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2003, 100, 13821–13826. [Google Scholar] [CrossRef] [PubMed]
- Liebau, E.; Bergmann, B.; Campbell, A.M.; Teesdale-Spittle, P.; Brophy, P.M.; Luersen, K.; Walter, R.D. The glutathione S-transferase from Plasmodium falciparum. Mol. Biochem. Parasitol. 2002, 124, 85–90. [Google Scholar] [CrossRef]
- Perbandt, M.; Burmeister, C.; Walter, R.D.; Betzel, C.; Liebau, E. Native and inhibited structure of a Mu class-related glutathione S-transferase from Plasmodium falciparum. J. Biol. Chem. 2004, 279, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M.; Becker, K. Glutathione S-transferase from malarial parasites: Structural and functional aspects. Methods Enzymol. 2005, 401, 241–253. [Google Scholar] [PubMed]
- Sztajer, H.; Gamain, B.; Aumann, K.D.; Slomianny, C.; Becker, K.; Brigelius-Flohe, R.; Flohe, L. The putative glutathione peroxidase gene of Plasmodium falciparum codes for a thioredoxin peroxidase. J. Biol. Chem. 2001, 276, 7397–7403. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Kawazu, S.I.; Komaki-Yasuda, K.; Oku, H.; Kano, S. Peroxiredoxins in malaria parasites: Parasitologic aspects. Parasitol. Int. 2008, 57, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M.; Rahlfs, S.; Becker, K. Peroxiredoxin systems of protozoal parasites. SubCell. Biochem. 2007, 44, 219–229. [Google Scholar] [PubMed]
- Ayi, K.; Cappadoro, M.; Branca, M.; Turrini, F.; Arese, P. Plasmodium falciparum glutathione metabolism and growth are independent of glutathione system of host erythrocyte. FEBS Lett. 1998, 424, 257–261. [Google Scholar] [CrossRef]
- Pastrana-Mena, R.; Dinglasan, R.R.; Franke-Fayard, B.; Vega-Rodriguez, J.; Fuentes-Caraballo, M.; Baerga-Ortiz, A.; Coppens, I.; Jacobs-Lorena, M.; Janse, C.J.; Serrano, A.E. Glutathione reductase-null malaria parasites have normal blood stage growth but arrest during development in the mosquito. J. Biol. Chem. 2010, 285, 27045–27056. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rodriguez, J.; Franke-Fayard, B.; Dinglasan, R.R.; Janse, C.J.; Pastrana-Mena, R.; Waters, A.P.; Coppens, I.; Rodriguez-Orengo, J.F.; Srinivasan, P.; Jacobs-Lorena, M.; et al. The glutathione biosynthetic pathway of Plasmodium is essential for mosquito transmission. PLoS Pathog. 2009, 5, e1000302. [Google Scholar] [CrossRef] [PubMed]
- Tietze, F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: Applications to mammalian blood and other tissues. Anal. Biochem. 1969, 27, 502–522. [Google Scholar] [CrossRef]
- Urscher, M.; Alisch, R.; Deponte, M. The glyoxalase system of malaria parasites—Implications for cell biology and general glyoxalase research. Semin. Cell Dev. Biol. 2011, 22, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Deponte, M.; Becker, K.; Rahlfs, S. Plasmodium falciparum glutaredoxin-like proteins. Biol. Chem. 2005, 386, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Kehr, S.; Jortzik, E.; Delahunty, C.; Yates, J.R., III; Rahlfs, S.; Becker, K. Protein S-glutathionylation in malaria parasites. Antioxid. Redox Signal. 2011, 15, 2855–2865. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, K.; Putrianti, E.D.; Rahlfs, S.; Schirmer, R.H.; Becker, K.; Matuschewski, K. Molecular genetics evidence for the in vivo roles of the two major NADPH-dependent disulfide reductases in the malaria parasite. J. Biol. Chem. 2010, 285, 37388–37395. [Google Scholar] [CrossRef] [PubMed]
- Belorgey, D.; Lanfranchi, D.A.; Davioud-Charvet, E. 1,4-naphthoquinones and other NADPH-dependent glutathione reductase-catalyzed redox cyclers as antimalarial agents. Curr. Pharm. Des. 2013, 19, 2512–2528. [Google Scholar] [CrossRef] [PubMed]
- Krnajski, Z.; Gilberger, T.W.; Walter, R.D.; Cowman, A.F.; Müller, S. Thioredoxin reductase is essential for the survival of Plasmodium falciparum erythrocytic stages. J. Biol. Chem. 2002, 277, 25970–25975. [Google Scholar] [CrossRef] [PubMed]
- Andricopulo, A.D.; Akoachere, M.B.; Krogh, R.; Nickel, C.; McLeish, M.J.; Kenyon, G.L.; Arscott, L.D.; Williams, C.H., Jr.; Davioud-Charvet, E.; Becker, K. Specific inhibitors of Plasmodium falciparum thioredoxin reductase as potential antimalarial agents. Bioorg. Med. Chem. Lett. 2006, 16, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Sturm, N.; Hu, Y.; Zimmermann, H.; Fritz-Wolf, K.; Wittlin, S.; Rahlfs, S.; Becker, K. Compounds structurally related to ellagic acid show improved antiplasmodial activity. Antimicrob. Agents Chemother. 2009, 53, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Jortzik, E.; Fritz-Wolf, K.; Sturm, N.; Hipp, M.; Rahlfs, S.; Becker, K. Redox regulation of Plasmodium falciparum ornithine delta-aminotransferase. J. Mol. Biol. 2010, 402, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.L.; Platel, D.F.; Pauly, G.; Tribouley-Duret, J. Plasmodium berghei: Implication of intracellular glutathione and its related enzyme in chloroquine resistance in vivo. Exp. Parasitol. 1995, 81, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Platel, D.F.; Mangou, F.; Tribouley-Duret, J. Role of glutathione in the detoxification of ferriprotoporphyrin IX in chloroquine resistant Plasmodium berghei. Mol. Biochem. Parasitol. 1999, 98, 215–223. [Google Scholar] [CrossRef]
- Srivastava, P.; Puri, S.K.; Kamboj, K.K.; Pandey, V.C. Glutathione-S-transferase activity in malarial parasites. Trop. Med. Int. Health 1999, 4, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, H.; Famin, O.; Zhang, J.; Krugliak, M. Inhibition of glutathione-dependent degradation of heme by chloroquine and amodiaquine as a possible basis for their antimalarial mode of action. Biochem. Pharmacol. 1998, 56, 1305–1313. [Google Scholar] [CrossRef]
- Krauth-Siegel, R.L.; Bauer, H.; Schirmer, R.H. Dithiol proteins as guardians of the intracellular redox milieu in parasites: Old and new drug targets in trypanosomes and malaria-causing plasmodia. Angew. Chem. Int. Ed. Engl. 2005, 44, 690–715. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Gilberger, T.W.; Farber, P.M.; Becker, K.; Schirmer, R.H.; Walter, R.D. Recombinant putative glutathione reductase of Plasmodium falciparum exhibits thioredoxin reductase activity. Mol. Biochem. Parasitol. 1996, 80, 215–219. [Google Scholar] [CrossRef]
- Gilberger, T.W.; Schirmer, R.H.; Walter, R.D.; Muller, S. Deletion of the parasite-specific insertions and mutation of the catalytic triad in glutathione reductase from chloroquine-sensitive Plasmodium falciparum 3D7. Mol. Biochem. Parasitol. 2000, 107, 169–179. [Google Scholar] [CrossRef]
- Sarma, G.N.; Savvides, S.N.; Becker, K.; Schirmer, M.; Schirmer, R.H.; Karplus, P.A. Glutathione reductase of the malarial parasite Plasmodium falciparum: Crystal structure and inhibitor development. J. Mol. Biol. 2003, 328, 893–907. [Google Scholar] [CrossRef]
- Savvides, S.N.; Scheiwein, M.; Bohme, C.C.; Arteel, G.E.; Karplus, P.A.; Becker, K.; Schirmer, R.H. Crystal structure of the antioxidant enzyme glutathione reductase inactivated by peroxynitrite. J. Biol. Chem. 2002, 277, 2779–2784. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.X.; Greetham, D.; Raeth, S.; Grant, C.M.; Dawes, I.W.; Perrone, G.G. The thioredoxin-thioredoxin reductase system can function in vivo as an alternative system to reduce oxidized glutathione in Saccharomyces cerevisiae. J. Biol. Chem. 2010, 285, 6118–6126. [Google Scholar] [CrossRef] [PubMed]
- Kanzok, S.M.; Fechner, A.; Bauer, H.; Ulschmid, J.K.; Muller, H.M.; Botella-Munoz, J.; Schneuwly, S.; Schirmer, R.; Becker, K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science 2001, 291, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.R.; Das, S.; Wong, E.H.; Andenmatten, N.; Stallmach, R.; Hackett, F.; Herman, J.P.; Muller, S.; Meissner, M.; Blackman, M.J. Robust inducible Cre recombinase activity in the human malaria parasite Plasmodium falciparum enables efficient gene deletion within a single asexual erythrocytic growth cycle. Mol. Microbiol. 2013, 88, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, G.; Giustarini, D.; Milzani, A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem. Sci. 2009, 34, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [PubMed]
- Grant, C.M.; MacIver, F.H.; Dawes, I.W. Glutathione synthetase is dispensable for growth under both normal and oxidative stress conditions in the yeast Saccharomyces cerevisiae due to an accumulation of the dipeptide gamma-glutamylcysteine. Mol. Biol. Cell 1997, 8, 1699–1707. [Google Scholar] [CrossRef] [PubMed]
- Grant, C.M.; MacIver, F.H.; Dawes, I.W. Glutathione is an essential metabolite required for resistance to oxidative stress in the yeast Saccharomyces cerevisiae. Curr. Genet. 1996, 29, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, B.; Ingavale, S.; Bachhawat, A.K. apd1+, a gene required for red pigment formation in ade6 mutants of Schizosaccharomyces pombe, encodes an enzyme required for glutathione biosynthesis: A role for glutathione and a glutathione-conjugate pump. Genetics 1997, 145, 75–83. [Google Scholar] [PubMed]
- Huynh, T.T.; Huynh, V.T.; Harmon, M.A.; Phillips, M.A. Gene knockdown of gamma-glutamylcysteine synthetase by RNAi in the parasitic protozoa Trypanosoma brucei demonstrates that it is an essential enzyme. J. Biol. Chem. 2003, 278, 39794–39800. [Google Scholar] [CrossRef] [PubMed]
- Baek, Y.U.; Kim, Y.R.; Yim, H.S.; Kang, S.O. Disruption of gamma-glutamylcysteine synthetase results in absolute glutathione auxotrophy and apoptosis in Candida albicans. FEBS Lett. 2004, 556, 47–52. [Google Scholar] [CrossRef]
- Shi, Z.Z.; Osei-Frimpong, J.; Kala, G.; Kala, S.V.; Barrios, R.J.; Habib, G.M.; Lukin, D.J.; Danney, C.M.; Matzuk, M.M.; Lieberman, M.W. Glutathione synthesis is essential for mouse development but not for cell growth in culture. Proc. Natl. Acad. Sci. USA 2000, 97, 5101–5106. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Anderson, M.E.; Meister, A. Amino acid sequence and function of the light subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 20578–20583. [Google Scholar] [PubMed]
- Huang, C.S.; Chang, L.S.; Anderson, M.E.; Meister, A. Catalytic and regulatory properties of the heavy subunit of rat kidney gamma-glutamylcysteine synthetase. J. Biol. Chem. 1993, 268, 19675–19680. [Google Scholar] [PubMed]
- Coblenz, A.; Wolf, K. Gcs1, a gene encoding gamma-glutamylcysteine synthetase in the fission yeast Schizosaccharomyces pombe. Yeast 1995, 11, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Lueder, D.V.; Phillips, M.A. Characterization of Trypanosoma brucei gamma-glutamylcysteine synthetase, an essential enzyme in the biosynthesis of trypanothione (diglutathionylspermidine). J. Biol. Chem. 1996, 271, 17485–17490. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W.; Mulcahy, R.T. The enzymes of glutathione synthesis: Gamma-glutamylcysteine synthetase. Adv. Enzymol. Relat. Areas Mol. Biol. 1999, 73, 209–267. [Google Scholar] [PubMed]
- Kelly, B.S.; Antholine, W.E.; Griffith, O.W. Escherichia coli gamma-glutamylcysteine synthetase. Two active site metal ions affect substrate and inhibitor binding. J. Biol. Chem. 2002, 277, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Backos, D.S.; Franklin, C.C.; Reigan, P. The role of glutathione in brain tumor drug resistance. Biochem. Pharmacol. 2012, 83, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shertzer, H.G.; Schneider, S.N.; Nebert, D.W.; Dalton, T.P. Glutamate cysteine ligase catalysis: Dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J. Biol. Chem. 2005, 280, 33766–33774. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging regulatory paradigms in glutathione metabolism. Adv. Cancer Res. 2014, 122, 69–101. [Google Scholar] [PubMed]
- Liu, J.; Istvan, E.S.; Gluzman, I.Y.; Gross, J.; Goldberg, D.E. Plasmodium falciparum ensures its amino acid supply with multiple acquisition pathways and redundant proteolytic enzyme systems. Proc. Natl. Acad. Sci. USA 2006, 103, 8840–8845. [Google Scholar] [CrossRef] [PubMed]
- Babbitt, S.E.; Altenhofen, L.; Cobbold, S.A.; Istvan, E.S.; Fennell, C.; Doerig, C.; Llinas, M.; Goldberg, D.E. Plasmodium falciparum responds to amino acid starvation by entering into a hibernatory state. Proc. Natl. Acad. Sci. USA 2012, 109, E3278–E3287. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [PubMed]
- MacRae, J.I.; Dixon, M.W.; Dearnley, M.K.; Chua, H.H.; Chambers, J.M.; Kenny, S.; Bottova, I.; Tilley, L.; McConville, M.J. Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol. 2013, 11, 67. [Google Scholar] [CrossRef] [PubMed]
- Storm, J.; Sethia, S.; Blackburn, G.J.; Chokkathukalam, A.; Watson, D.G.; Breitling, R.; Coombs, G.H.; Muller, S. Phosphoenolpyruvate carboxylase identified as a key enzyme in erythrocytic Plasmodium falciparum carbon metabolism. PLoS Pathog. 2014, 10, e1003876. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Adhikary, H.; Jha, S.; Jha, A.; Kumar, G.N. Remodulation of central carbon metabolic pathway in response to arsenite exposure in Rhodococcus sp. strain NAU-1. Microb. Biotechnol. 2012, 5, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Creek, D.J.; Evans, K.J.; de Souza, D.; Schofield, L.; Muller, S.; Barrett, M.P.; McConville, M.J.; Waters, A.P. Host Reticulocytes Provide Metabolic Reservoirs That Can Be Exploited by Malaria Parasites. PLoS Pathog 2015, 11, e1004882. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.R.; Bushell, E.; Schwach, F.; Girling, G.; Anar, B.; Quail, M.A.; Herd, C.; Pfander, C.; Modrzynska, K.; Rayner, J.C.; et al. A genome-scale vector resource enables high-throughput reverse genetic screening in a malaria parasite. Cell Host Microbe 2015, 17, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Bourbouloux, A.; Shahi, P.; Chakladar, A.; Delrot, S.; Bachhawat, A.K. Hgt1p, a high affinity glutathione transporter from the yeast Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 13259–13265. [Google Scholar] [CrossRef] [PubMed]
- Bachhawat, A.K.; Thakur, A.; Kaur, J.; Zulkifli, M. Glutathione transporters. Biochim. Biophys. Acta 2013, 1830, 3154–3164. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.E.; Marchetti, R.V.; Cowan, A.I.; Howitt, S.M.; Broer, S.; Kirk, K. Chloroquine transport via the malaria parasite’s chloroquine resistance transporter. Science 2009, 325, 1680–1682. [Google Scholar] [CrossRef] [PubMed]
- Lewis, I.A.; Wacker, M.; Olszewski, K.L.; Cobbold, S.A.; Baska, K.S.; Tan, A.; Ferdig, M.T.; Llinas, M. Metabolic QTL analysis links chloroquine resistance in Plasmodium falciparum to impaired hemoglobin catabolism. PLoS Genet. 2014, 10, e1004085. [Google Scholar] [CrossRef] [PubMed]
- Maughan, S.C.; Pasternak, M.; Cairns, N.; Kiddle, G.; Brach, T.; Jarvis, R.; Haas, F.; Nieuwland, J.; Lim, B.; Muller, C.; et al. Plant homologs of the Plasmodium falciparum chloroquine-resistance transporter, PfCRT, are required for glutathione homeostasis and stress responses. Proc. Natl. Acad. Sci. USA 2010, 107, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Fidock, D.A.; Nomura, T.; Talley, A.K.; Cooper, R.A.; Dzekunov, S.M.; Ferdig, M.T.; Ursos, L.M.; Sidhu, A.B.; Naude, B.; Deitsch, K.W.; et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol. Cell 2000, 6, 861–871. [Google Scholar] [CrossRef]
- Bray, P.G.; Martin, R.E.; Tilley, L.; Ward, S.A.; Kirk, K.; Fidock, D.A. Defining the role of PfCRT in Plasmodium falciparum chloroquine resistance. Mol. Microbiol. 2005, 56, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, V.; Bray, P.G.; Verdier-Pinard, D.; Johnson, D.J.; Horrocks, P.; Muhle, R.A.; Alakpa, G.E.; Hughes, R.H.; Ward, S.A.; Krogstad, D.J.; et al. A critical role for PfCRT K76T in Plasmodium falciparum verapamil-reversible chloroquine resistance. EMBO J. 2005, 24, 2294–2305. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 2005, 157, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Famin, O.; Krugliak, M.; Ginsburg, H. Kinetics of inhibition of glutathione-mediated degradation of ferriprotoporphyrin IX by antimalarial drugs. Biochem. Pharmacol. 1999, 58, 59–68. [Google Scholar] [CrossRef]
- Reed, M.B.; Saliba, K.J.; Caruana, S.R.; Kirk, K.; Cowman, A.F. Pgh1 modulates sensitivity and resistance to multiple antimalarials in Plasmodium falciparum. Nature 2000, 403, 906–909. [Google Scholar] [PubMed]
- Duraisingh, M.T.; Cowman, A.F. Contribution of the pfmdr1 gene to antimalarial drug-resistance. Acta Trop. 2005, 94, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.P.; Dave, A.; Stein, W.D.; Lanzer, M. Transporters as mediators of drug resistance in Plasmodium falciparum. Int. J. Parasitol. 2010, 40, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Ferdig, M.T.; Feng, X.; Joy, D.A.; Duan, J.; Furuya, T.; Subramanian, G.; Aravind, L.; Cooper, R.A.; Wootton, J.C.; et al. Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol. Microbiol. 2003, 49, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.J.; Nair, S.; Qin, H.; Singlam, S.; Brockman, A.; Paiphun, L.; Nosten, F. Are transporter genes other than the chloroquine resistance locus (pfcrt) and multidrug resistance gene (pfmdr) associated with antimalarial drug resistance? Antimicrob. Agents Chemother. 2005, 49, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Deharo, E.; Barkan, D.; Krugliak, M.; Golenser, J.; Ginsburg, H. Potentiation of the antimalarial action of chloroquine in rodent malaria by drugs known to reduce cellular glutathione levels. Biochem. Pharmacol. 2003, 66, 809–817. [Google Scholar] [CrossRef]
- Gligorijevic, B.; Bennett, T.; McAllister, R.; Urbach, J.S.; Roepe, P.D. Spinning disk confocal microscopy of live, intraerythrocytic malarial parasites. 2. Altered vacuolar volume regulation in drug resistant malaria. Biochemistry 2006, 45, 12411–12423. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, H.; Golenser, J. Glutathione is involved in the antimalarial action of chloroquine and its modulation affects drug sensitivity of human and murine species of Plasmodium. Redox Rep. 2003, 8, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Hang, H.; Paguio, M.; Roepe, P.D. The antimalarial drug resistance protein Plasmodium falciparum chloroquine resistance transporter binds chloroquine. Biochemistry 2004, 43, 8290–8296. [Google Scholar]
- Zhang, J.; Krugliak, M.; Ginsburg, H. The fate of ferriprotorphyrin IX in malaria infected erythrocytes in conjunction with the mode of action of antimalarial drugs. Mol. Biochem. Parasitol. 1999, 99, 129–141. [Google Scholar] [CrossRef]
- Famin, O.; Ginsburg, H. The treatment of Plasmodium falciparum-infected erythrocytes with chloroquine leads to accumulation of ferriprotoporphyrin IX bound to particular parasite proteins and to the inhibition of the parasite’s 6-phosphogluconate dehydrogenase. Parasite 2003, 10, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Famin, O.; Ginsburg, H. Differential effects of 4-aminoquinoline-containing antimalarial drugs on hemoglobin digestion in Plasmodium falciparum-infected erythrocytes. Biochem. Pharmacol. 2002, 63, 393–398. [Google Scholar] [CrossRef]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Miotto, O.; Amato, R.; Ashley, E.A.; MacInnis, B.; Almagro-Garcia, J.; Amaratunga, C.; Lim, P.; Mead, D.; Oyola, S.O.; Dhorda, M.; et al. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat. Genet. 2015, 47, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Takala-Harrison, S.; Jacob, C.G.; Arze, C.; Cummings, M.P.; Silva, J.C.; Dondorp, A.M.; Fukuda, M.M.; Hien, T.T.; Mayxay, M.; Noedl, H.; et al. Independent emergence of artemisinin resistance mutations among Plasmodium falciparum in Southeast Asia. J. Infect. Dis. 2015, 211, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Uruno, A.; Yagishita, Y.; Yamamoto, M. The Keap1-Nrf2 system and diabetes mellitus. Arch. Biochem. Biophys. 2015, 566, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, C.; Bathke, J.; Goyal, S.; Fischer, M.; Dahse, H.M.; Chacko, S.; Becker, K.; Grover, A. Targeting the intersubunit cavity of Plasmodium falciparum glutathione reductase by a novel natural inhibitor: Computational and experimental evidence. Int. J. Biochem. Cell Biol. 2015, 61, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Leroy, D.; Campo, B.; Ding, X.C.; Burrows, J.N.; Cherbuin, S. Defining the biology component of the drug discovery strategy for malaria eradication. Trends Parasitol. 2014, 30, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, T.; Flohe, L. The thiol-based redox networks of pathogens: Unexploited targets in the search for new drugs. Biofactors 2006, 27, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in cancer biology and therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, S. Role and Regulation of Glutathione Metabolism in Plasmodium falciparum. Molecules 2015, 20, 10511-10534. https://doi.org/10.3390/molecules200610511
Müller S. Role and Regulation of Glutathione Metabolism in Plasmodium falciparum. Molecules. 2015; 20(6):10511-10534. https://doi.org/10.3390/molecules200610511
Chicago/Turabian StyleMüller, Sylke. 2015. "Role and Regulation of Glutathione Metabolism in Plasmodium falciparum" Molecules 20, no. 6: 10511-10534. https://doi.org/10.3390/molecules200610511
APA StyleMüller, S. (2015). Role and Regulation of Glutathione Metabolism in Plasmodium falciparum. Molecules, 20(6), 10511-10534. https://doi.org/10.3390/molecules200610511