
A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2012

 ,
, 
Abstract
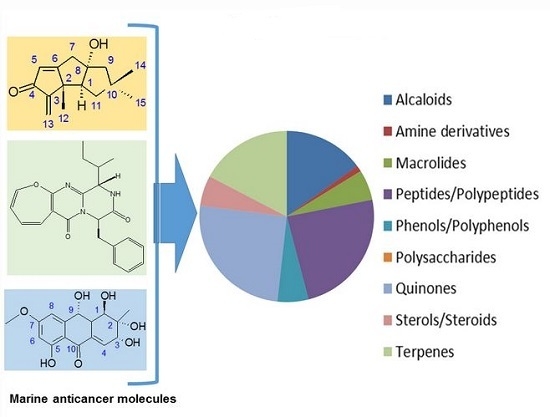
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Marine Anticancer Molecules Reported in 2012
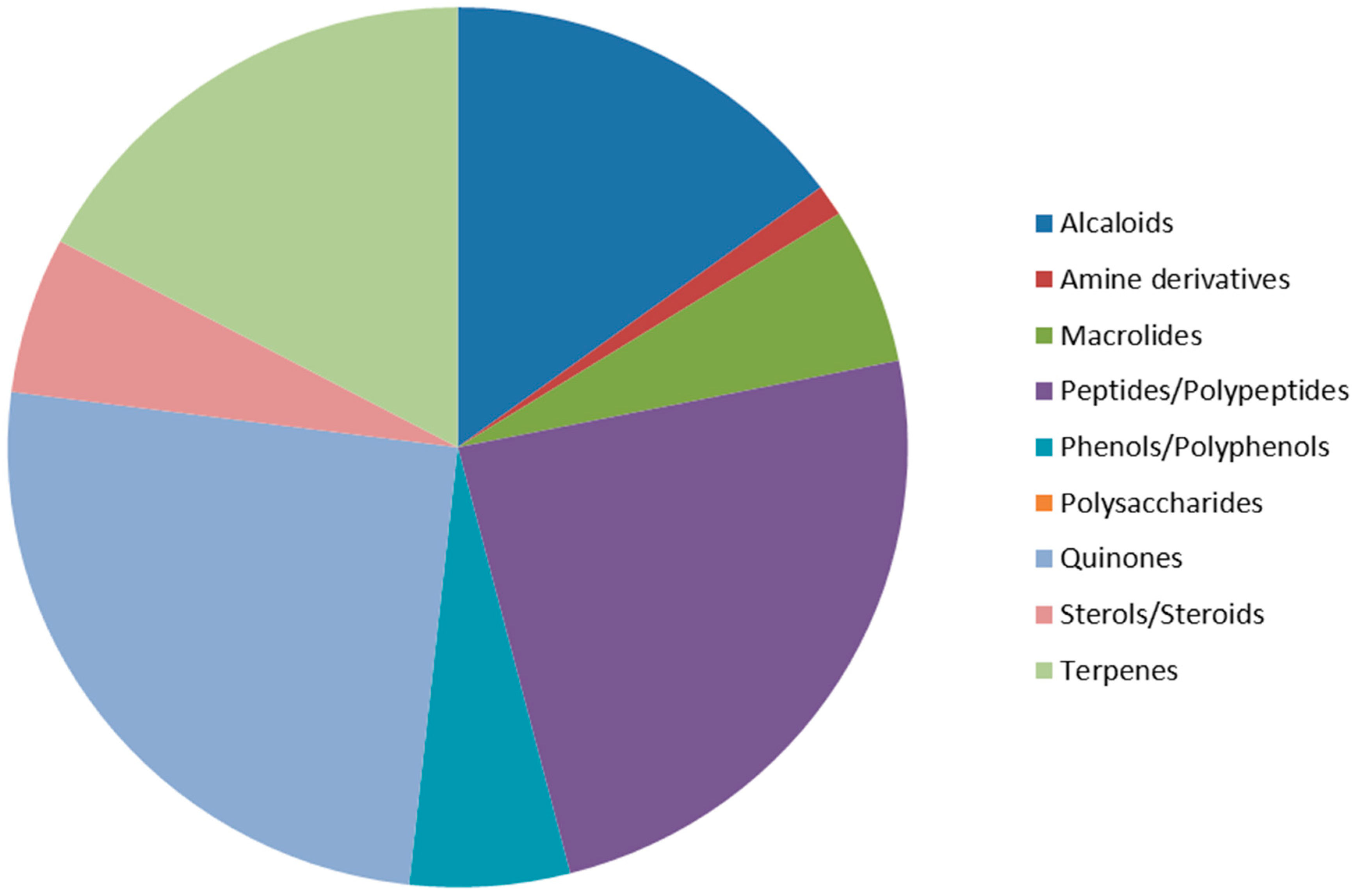
2.1. Alkaloids
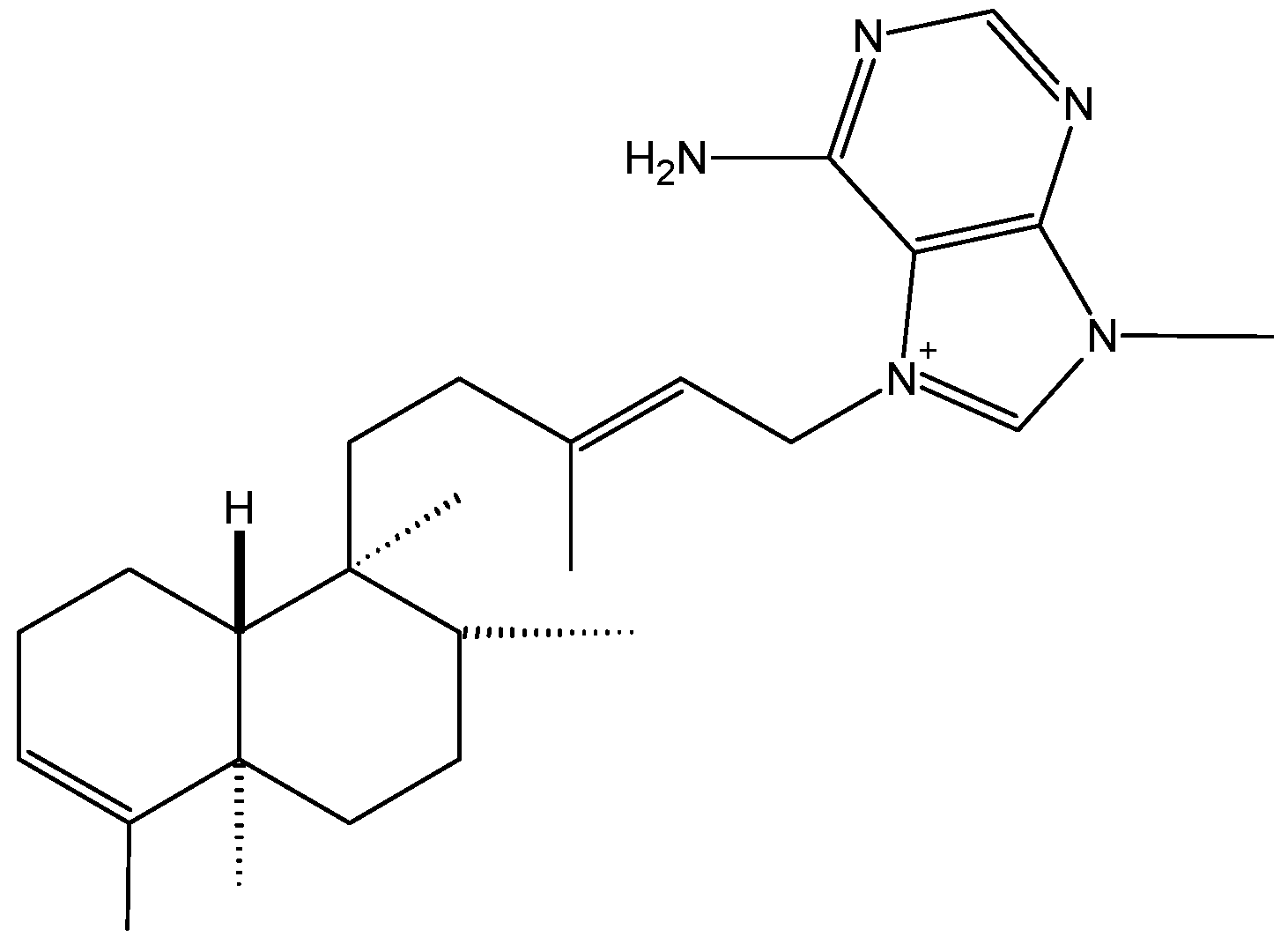
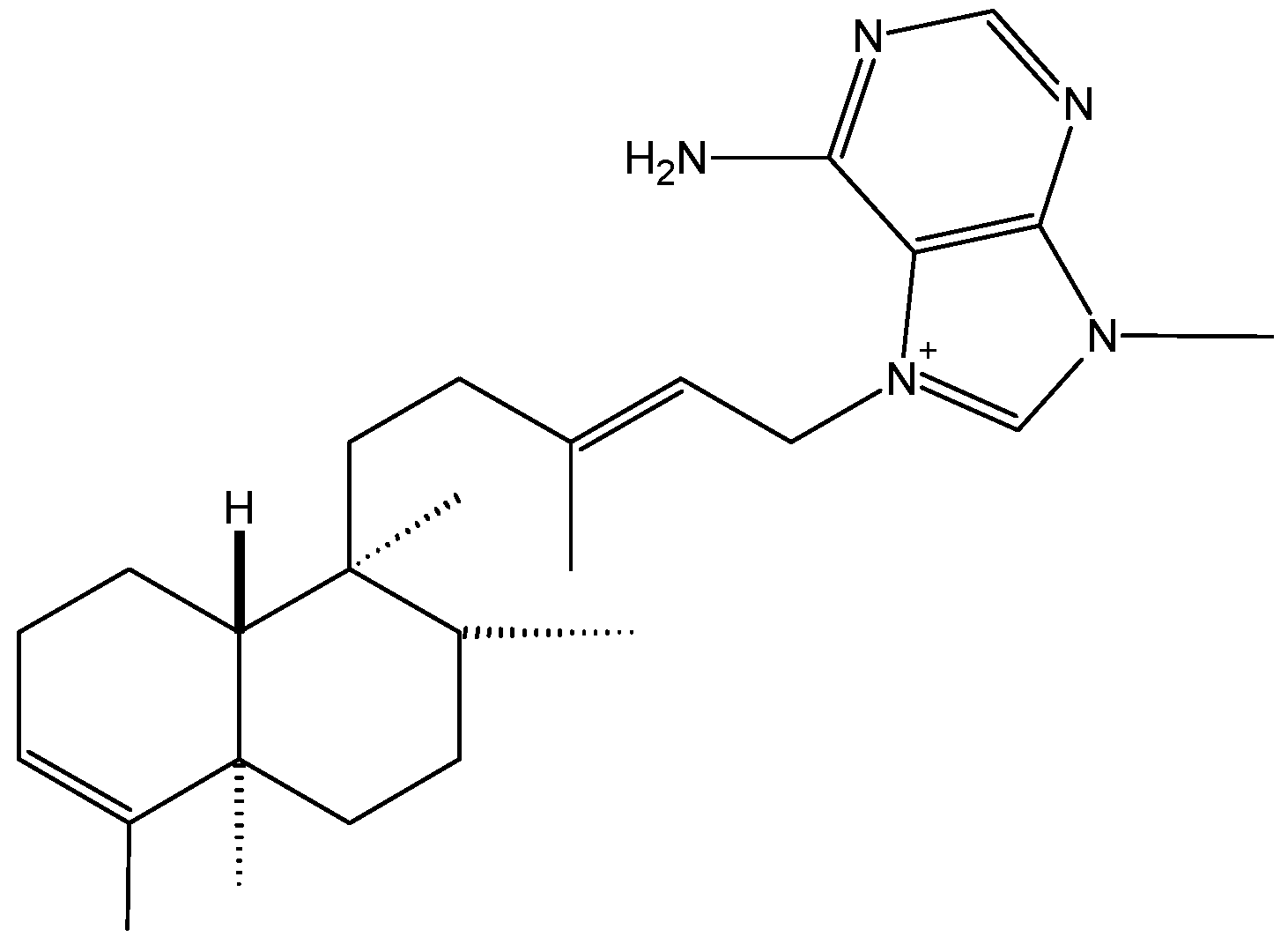
2.1.1. Agelasine B
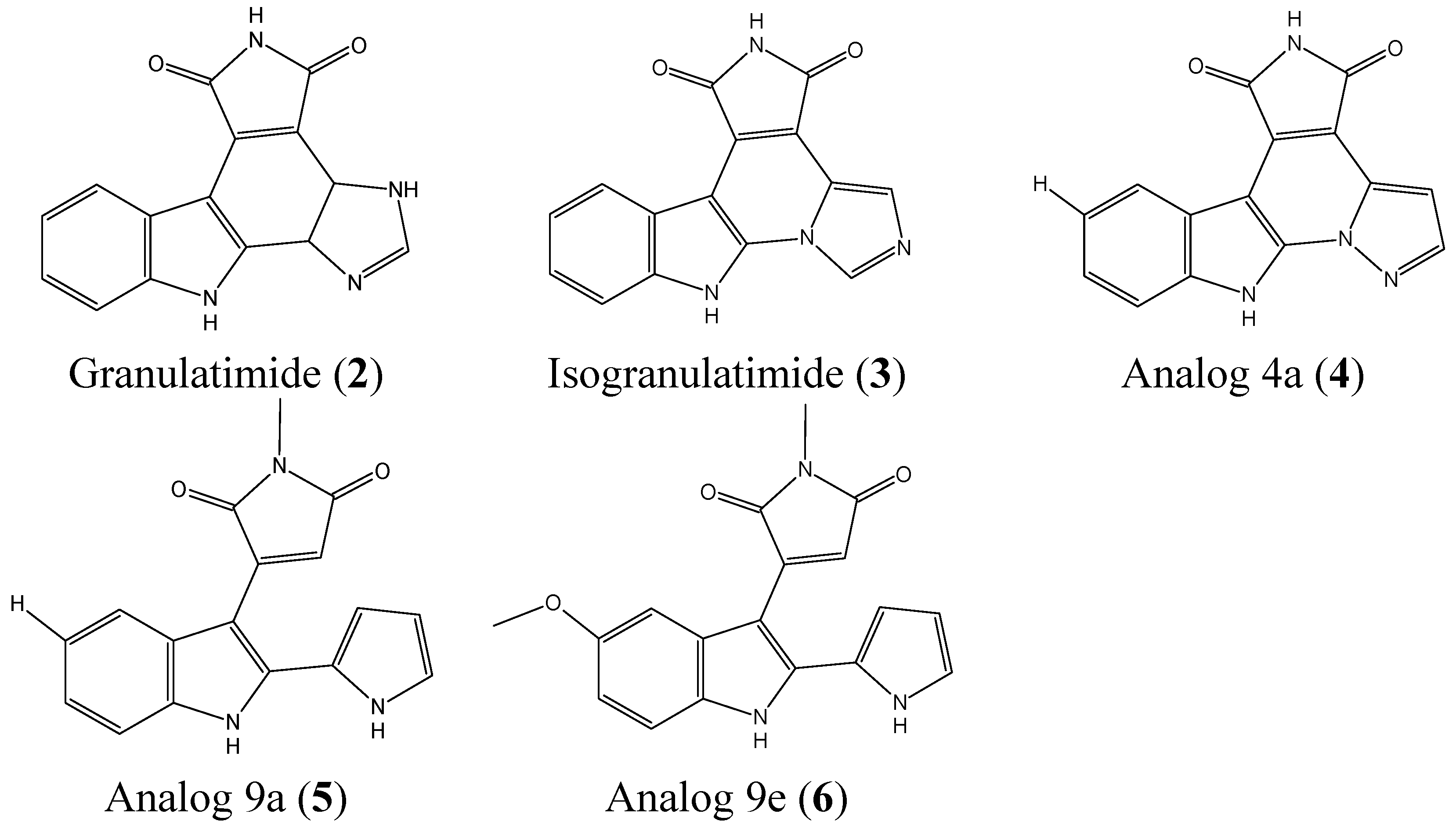
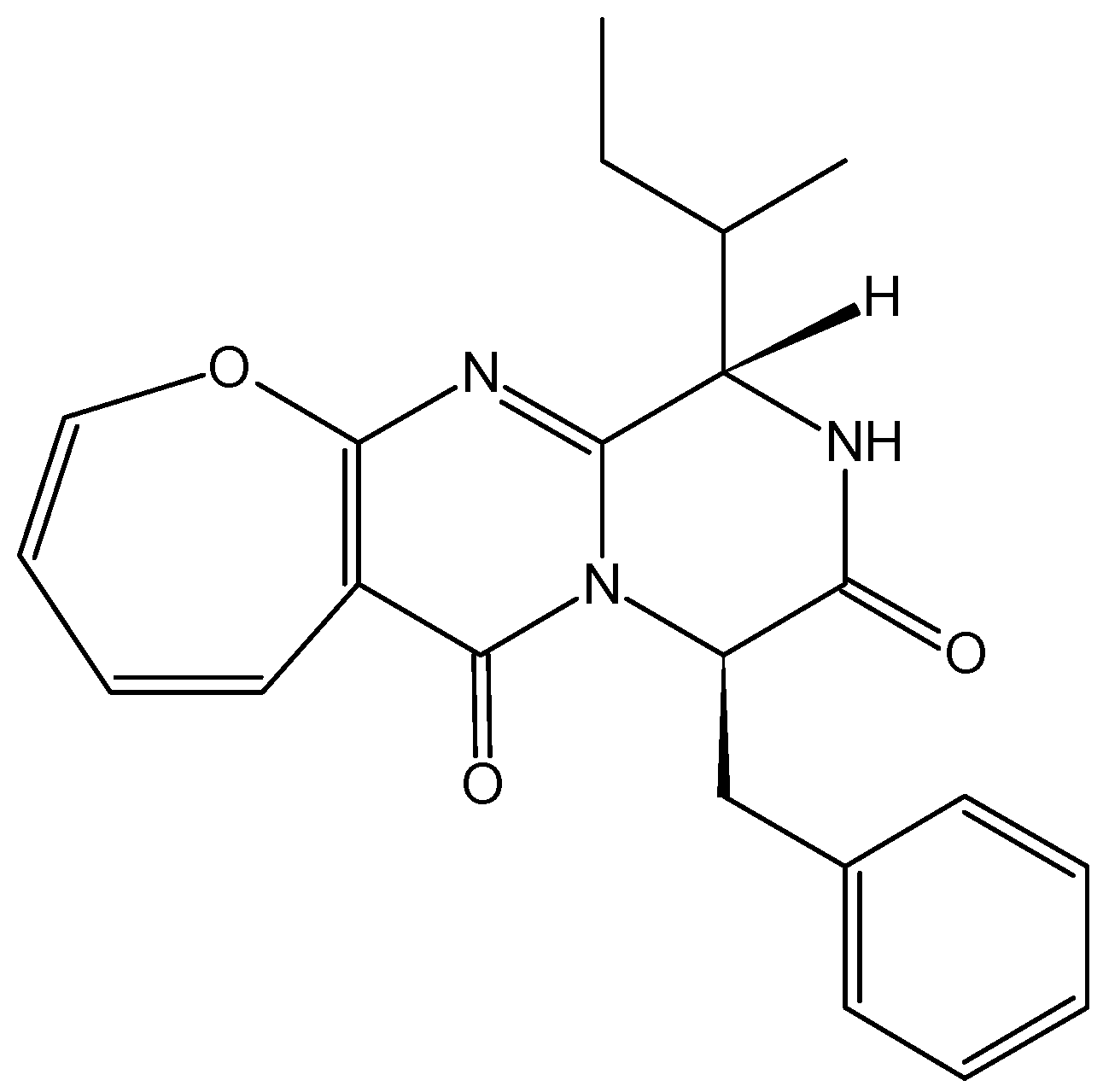
2.1.2. Granulatimide and Isogranulatimide Analogs
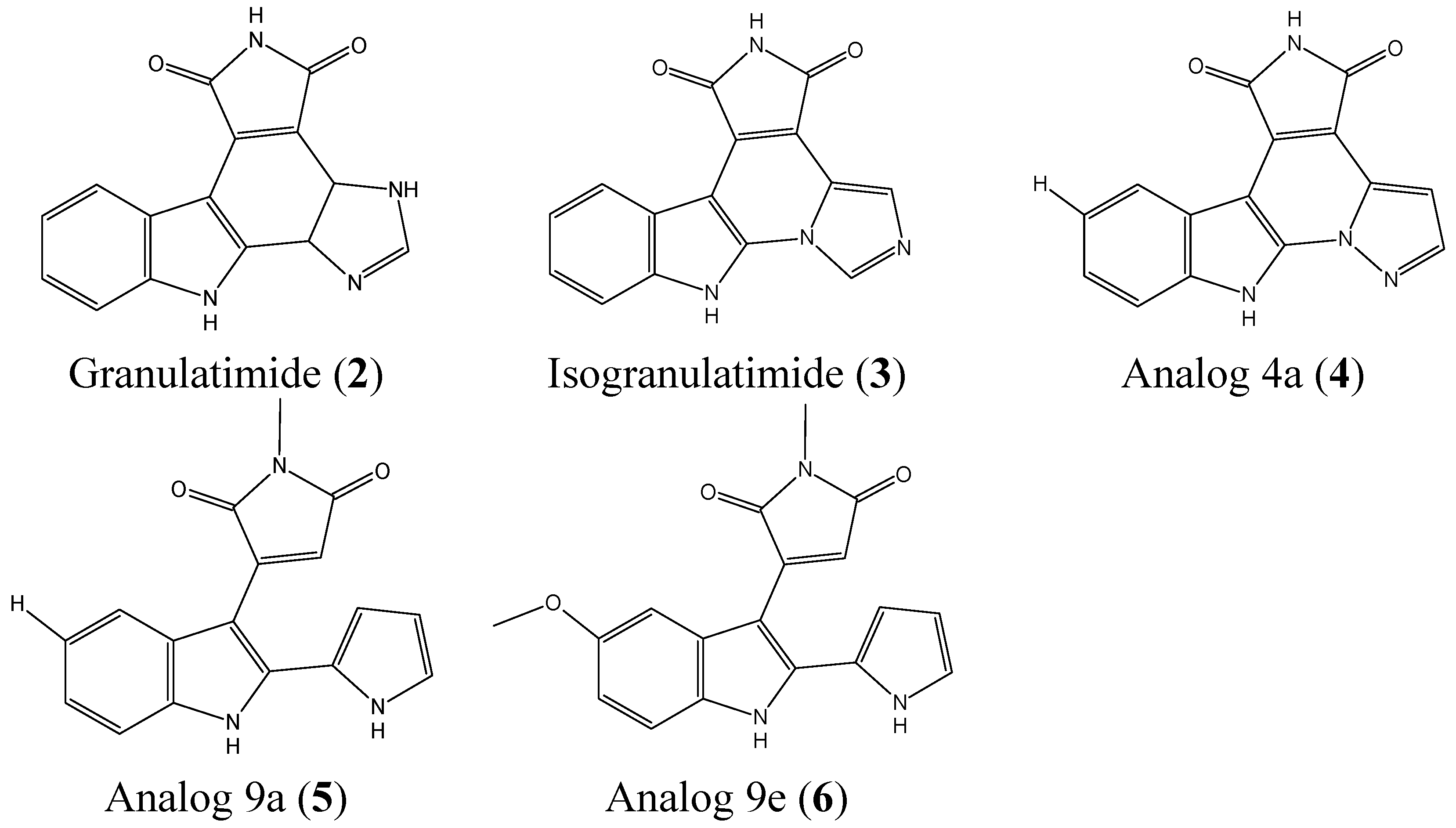
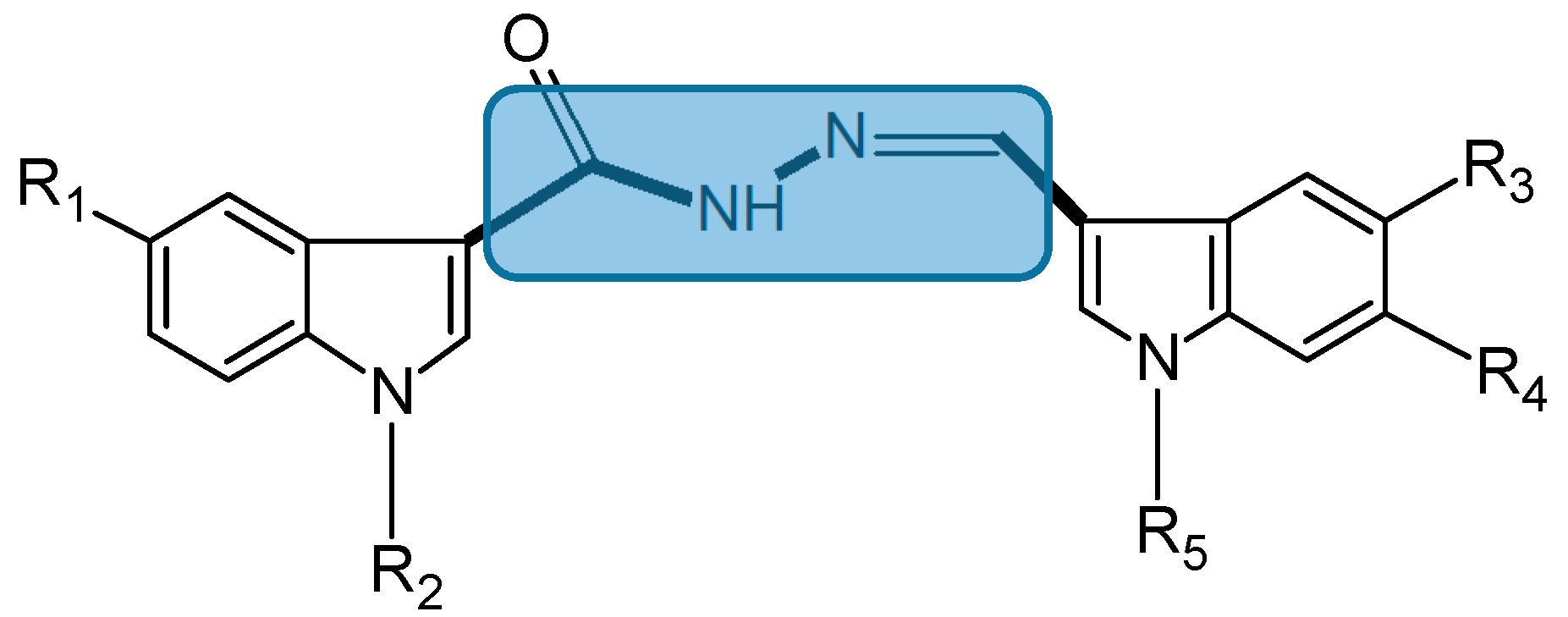
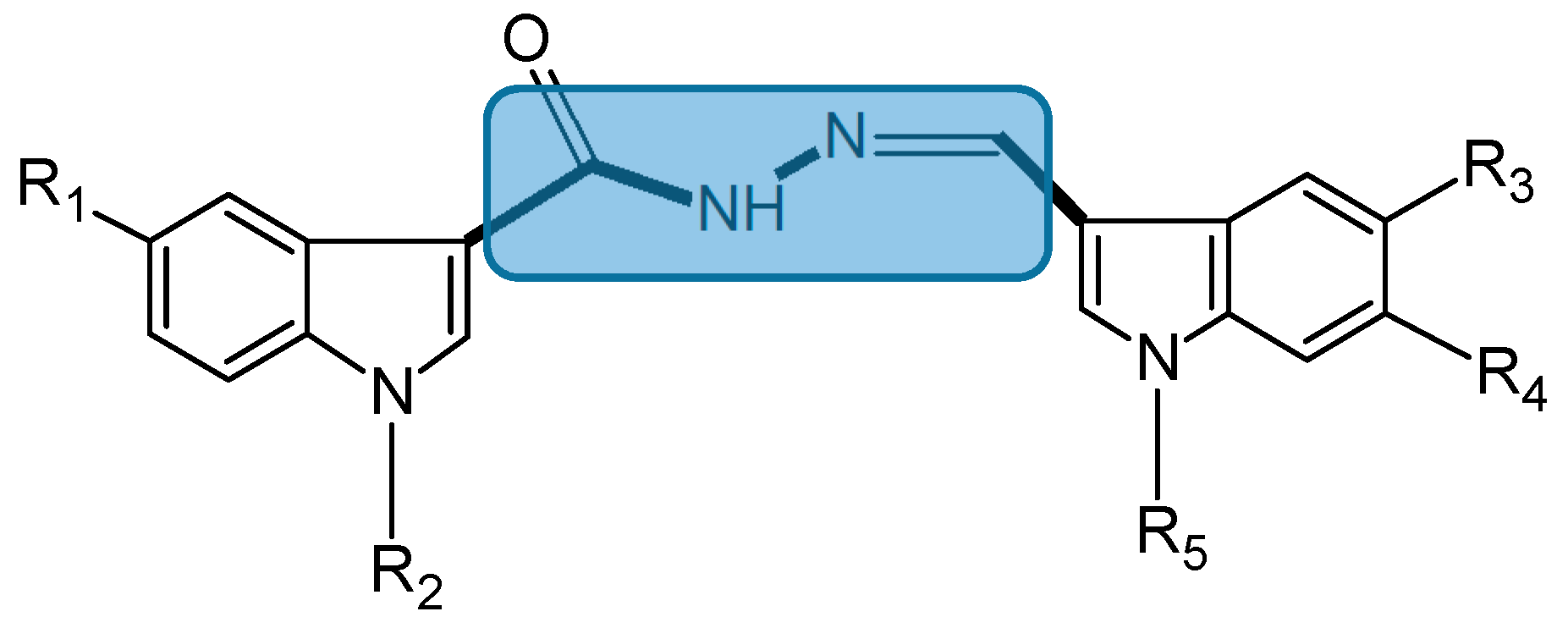
2.1.3. Bis(indolyl)hydrazide-hydrazone Analogs
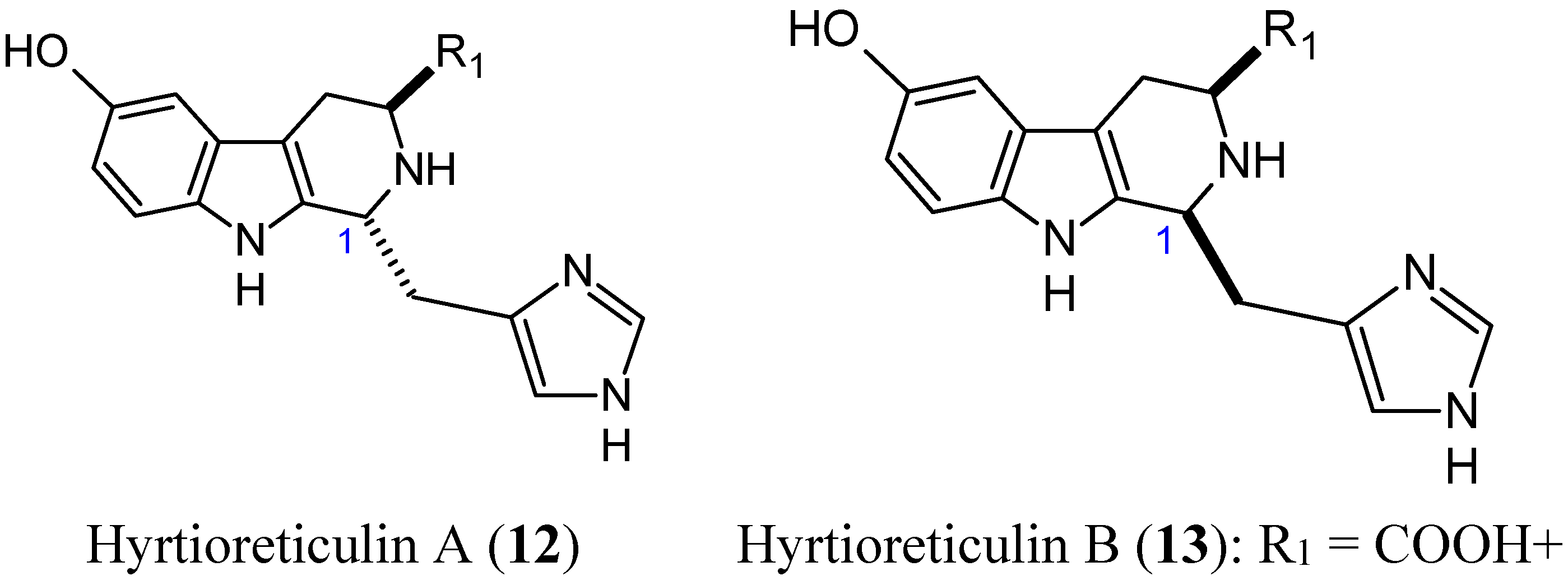
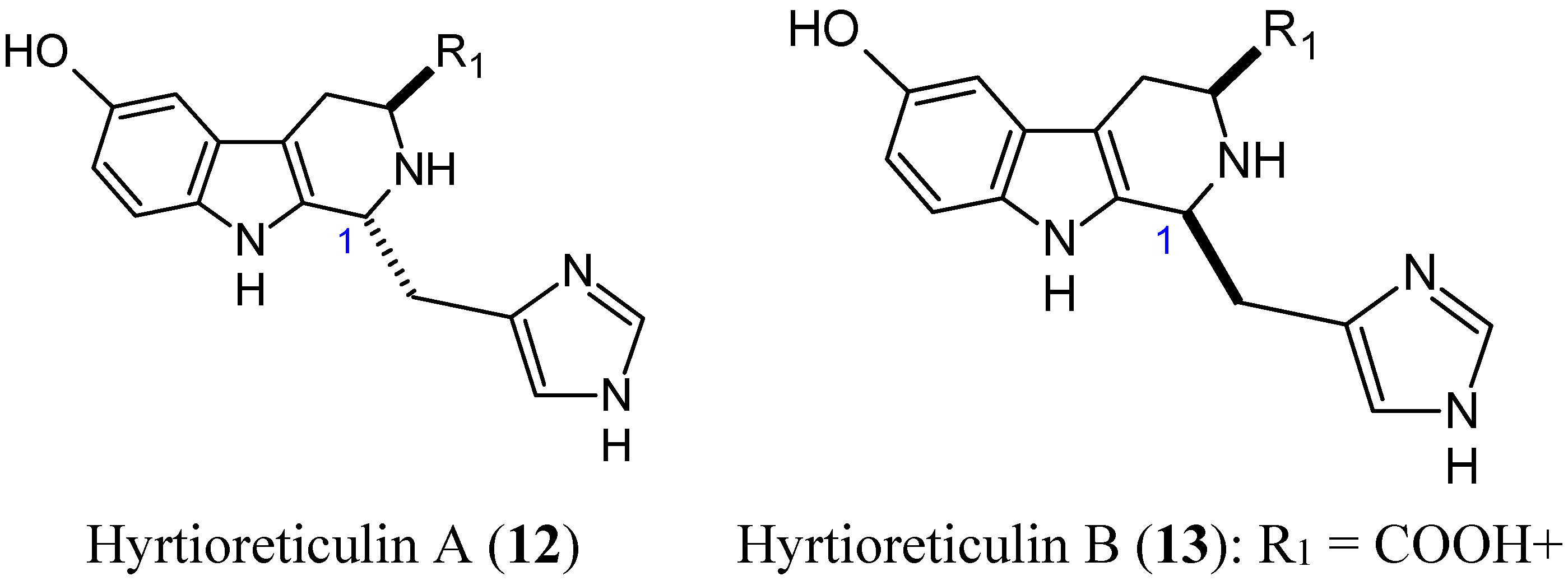
2.1.4. Hyrtioreticulins A and B
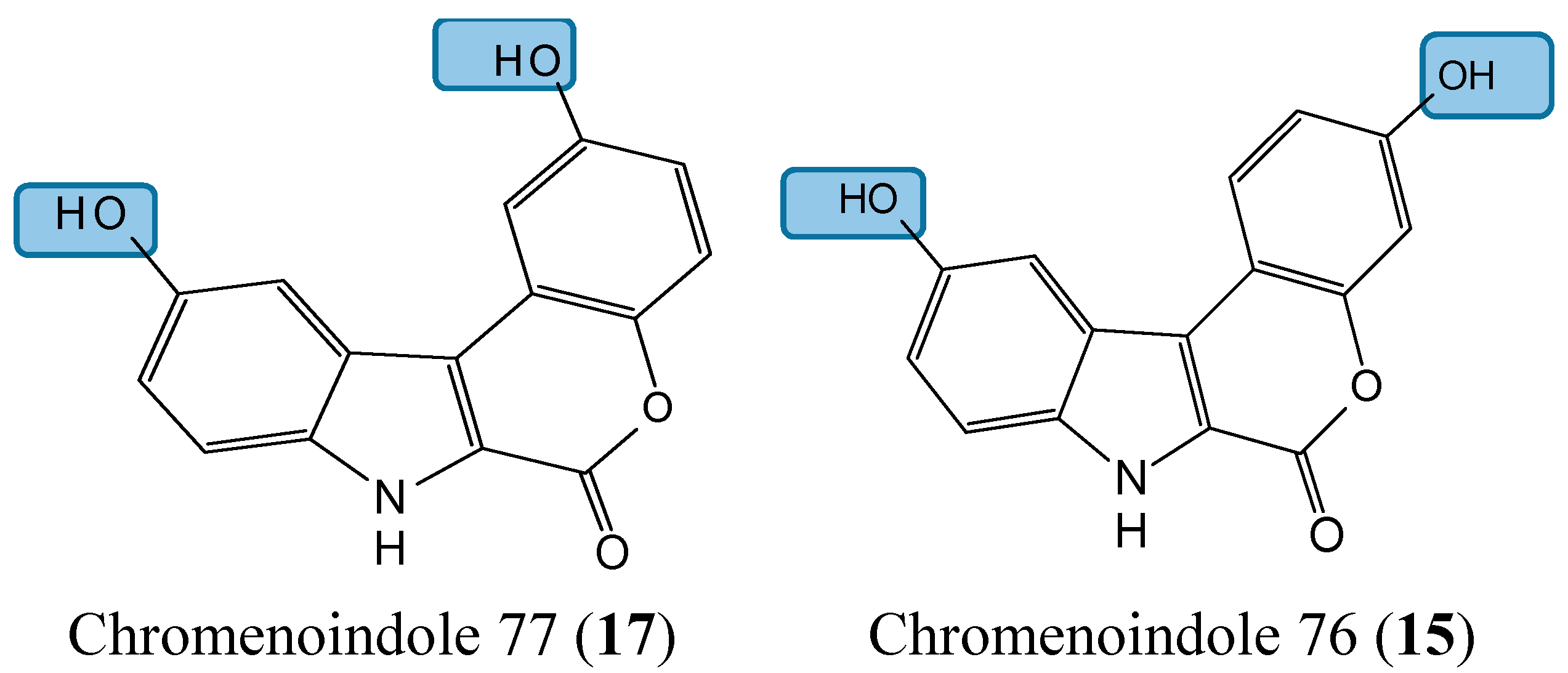
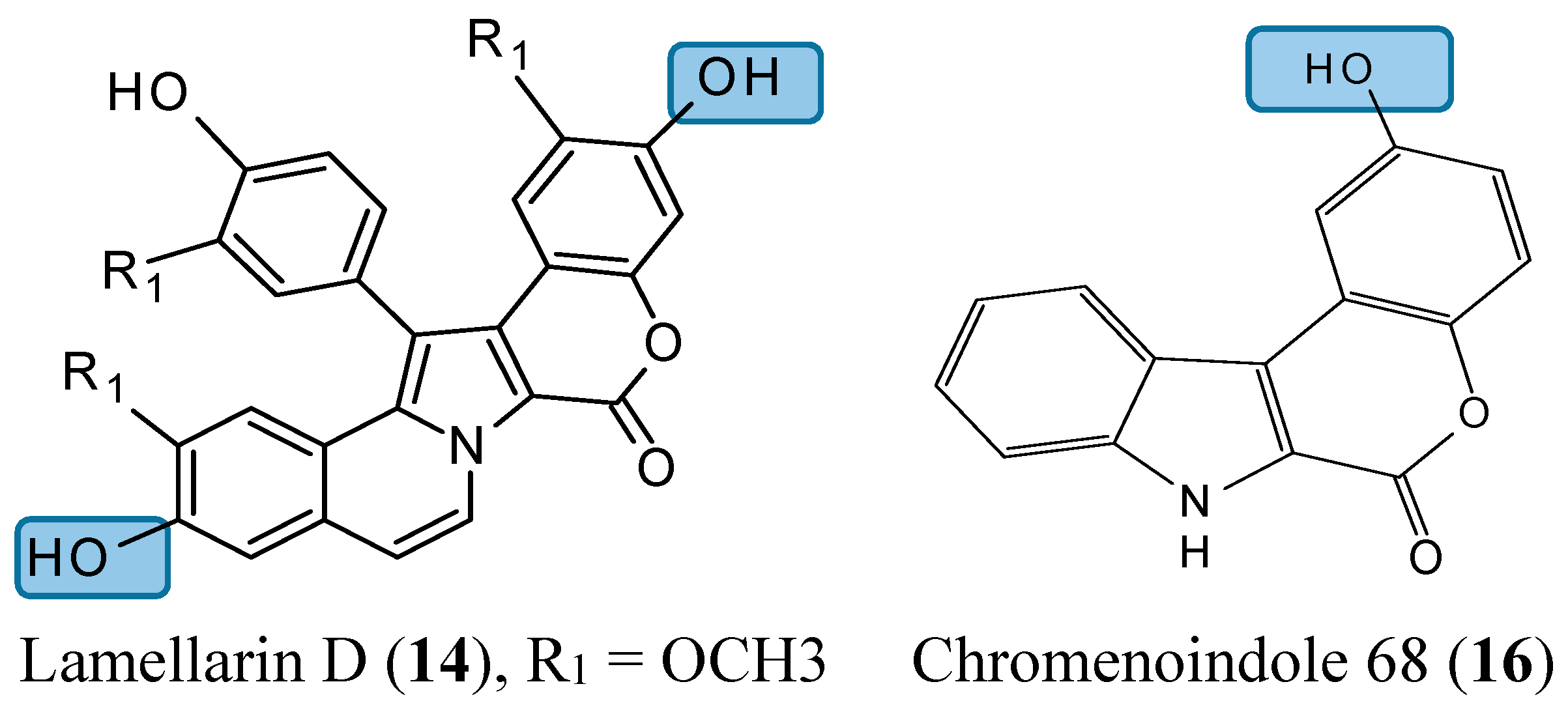
2.1.5. Lamellarin D Analogs
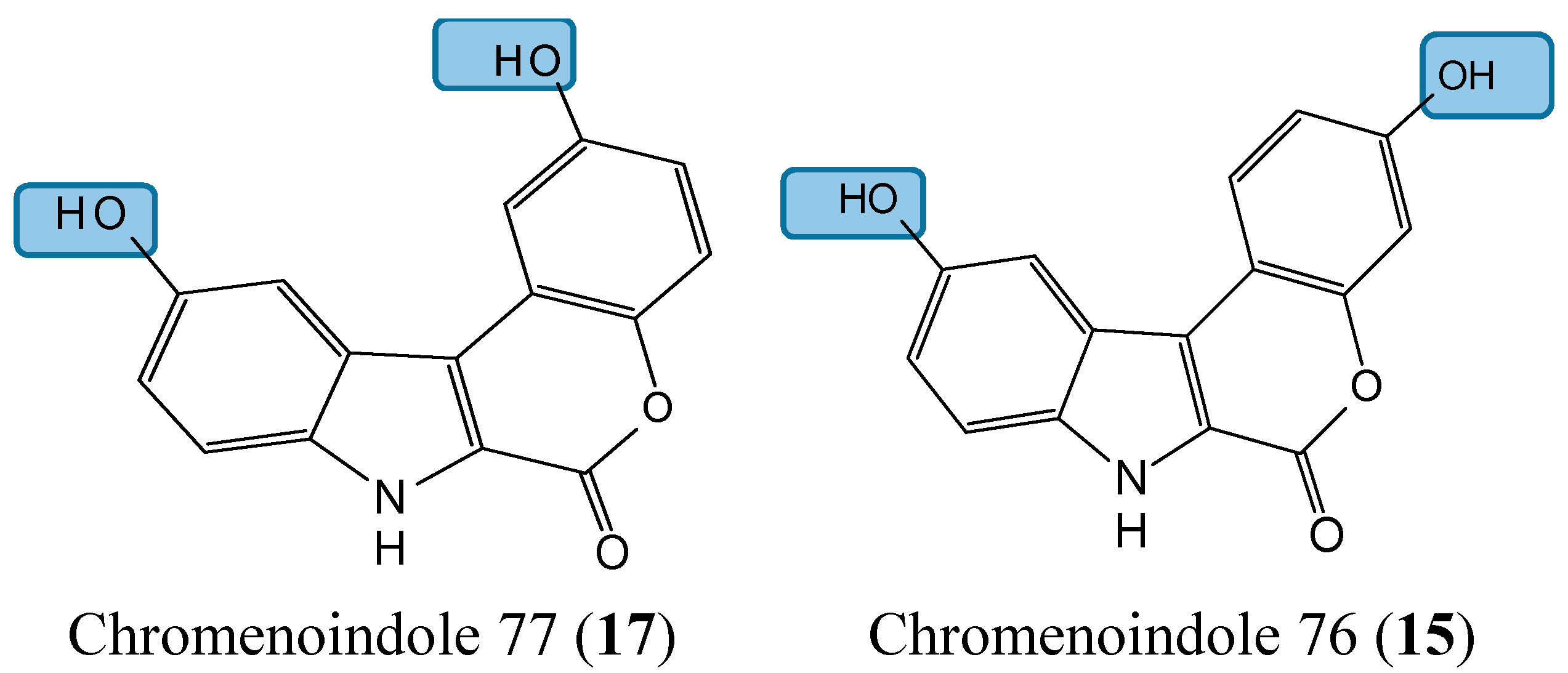
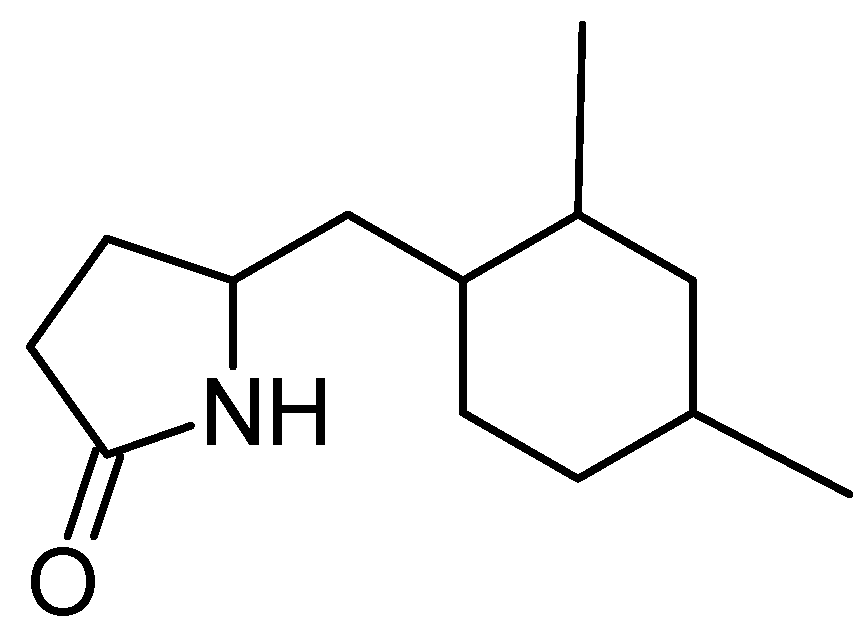
2.2. Amine Derivatives
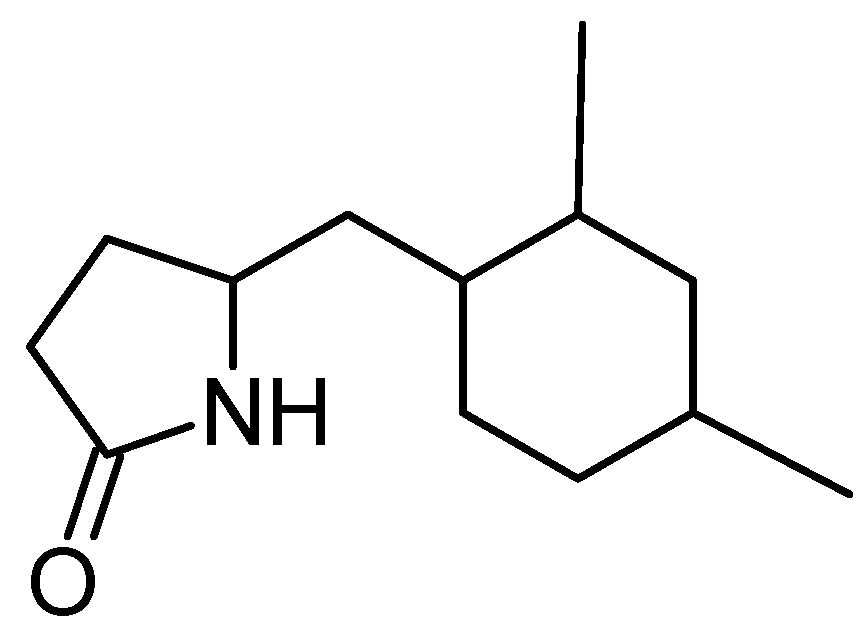
5-(2,4-Dimethylbenzyl) pyrrolidin-2-one (DMBPO)
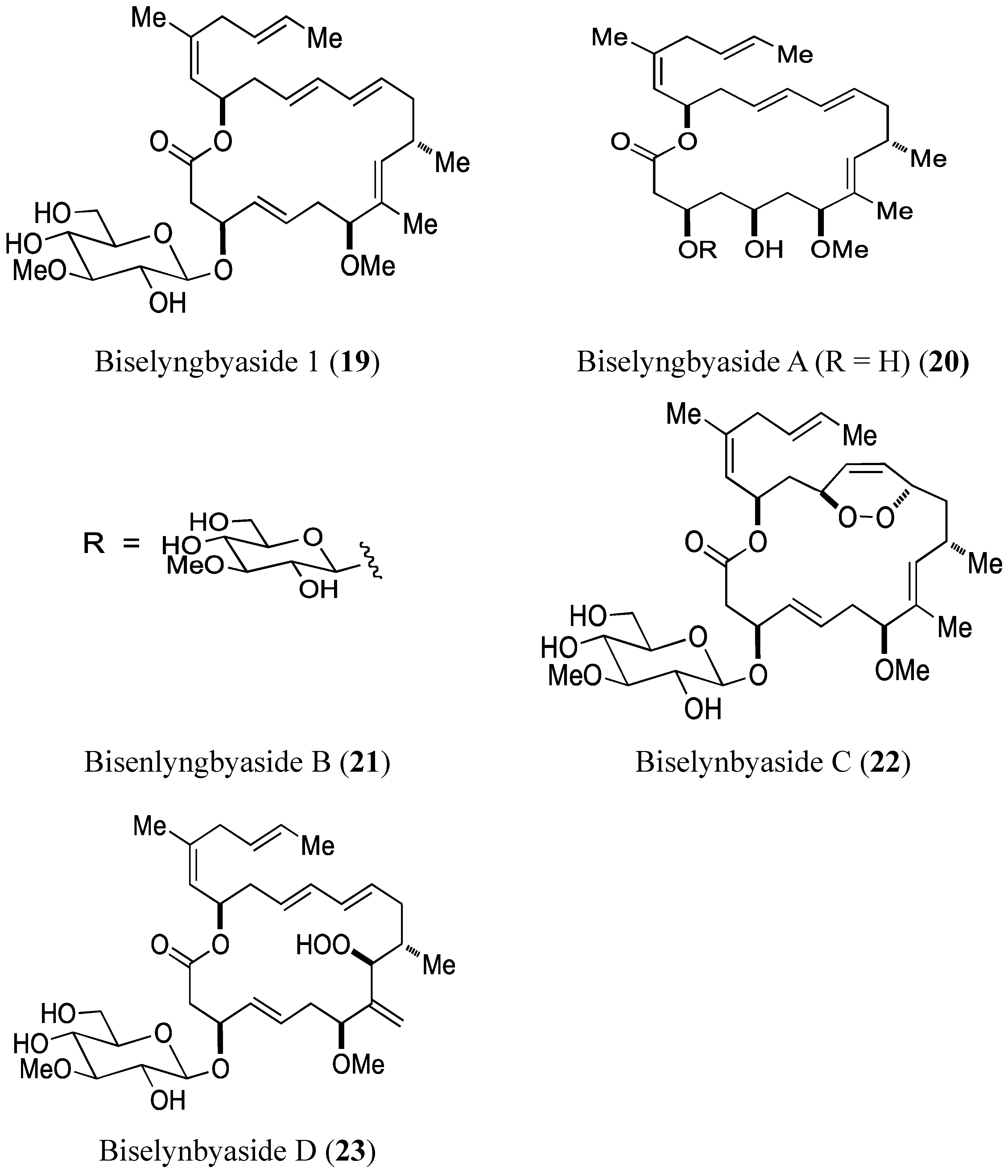
2.3. Macrolides
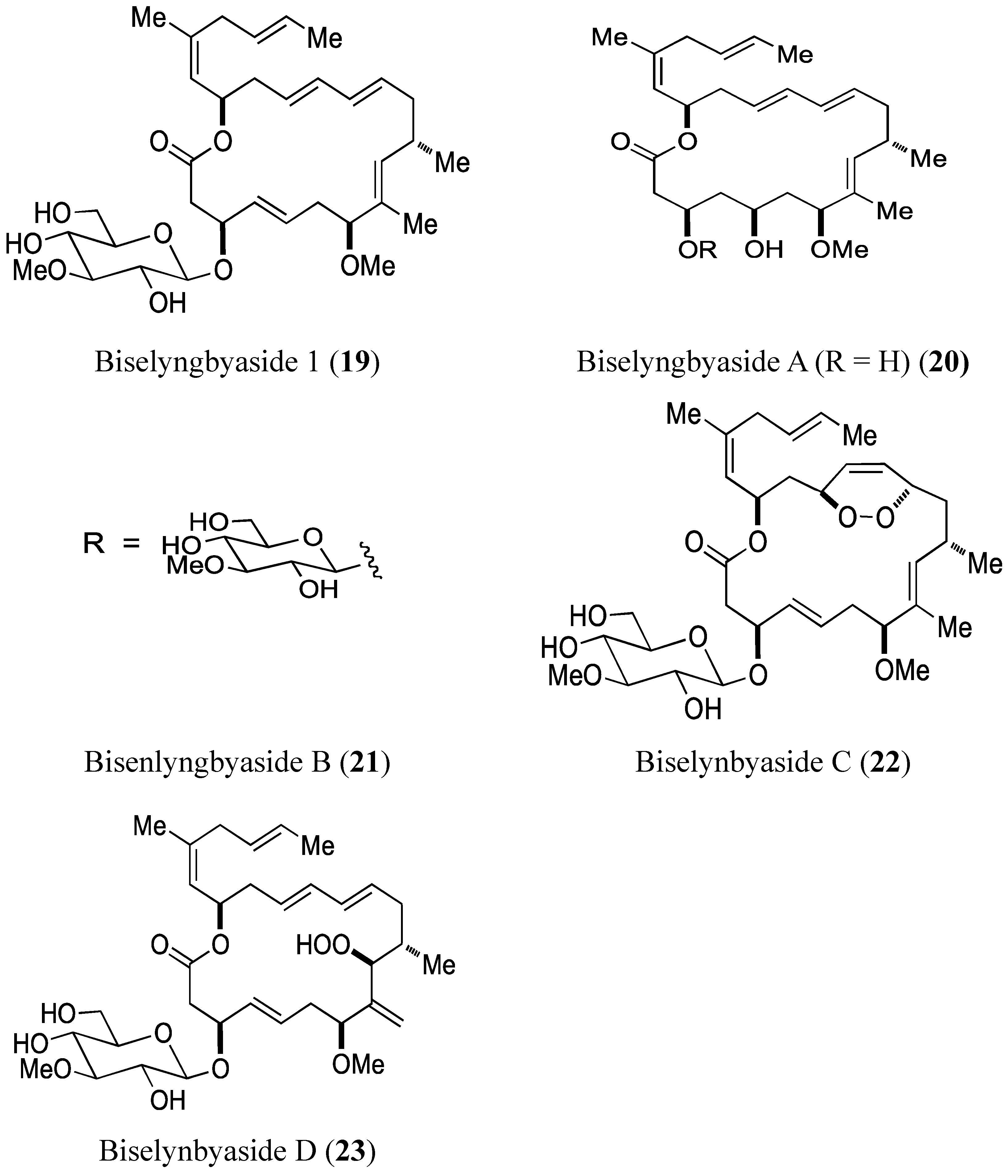
2.3.1. Biselyngbyasides
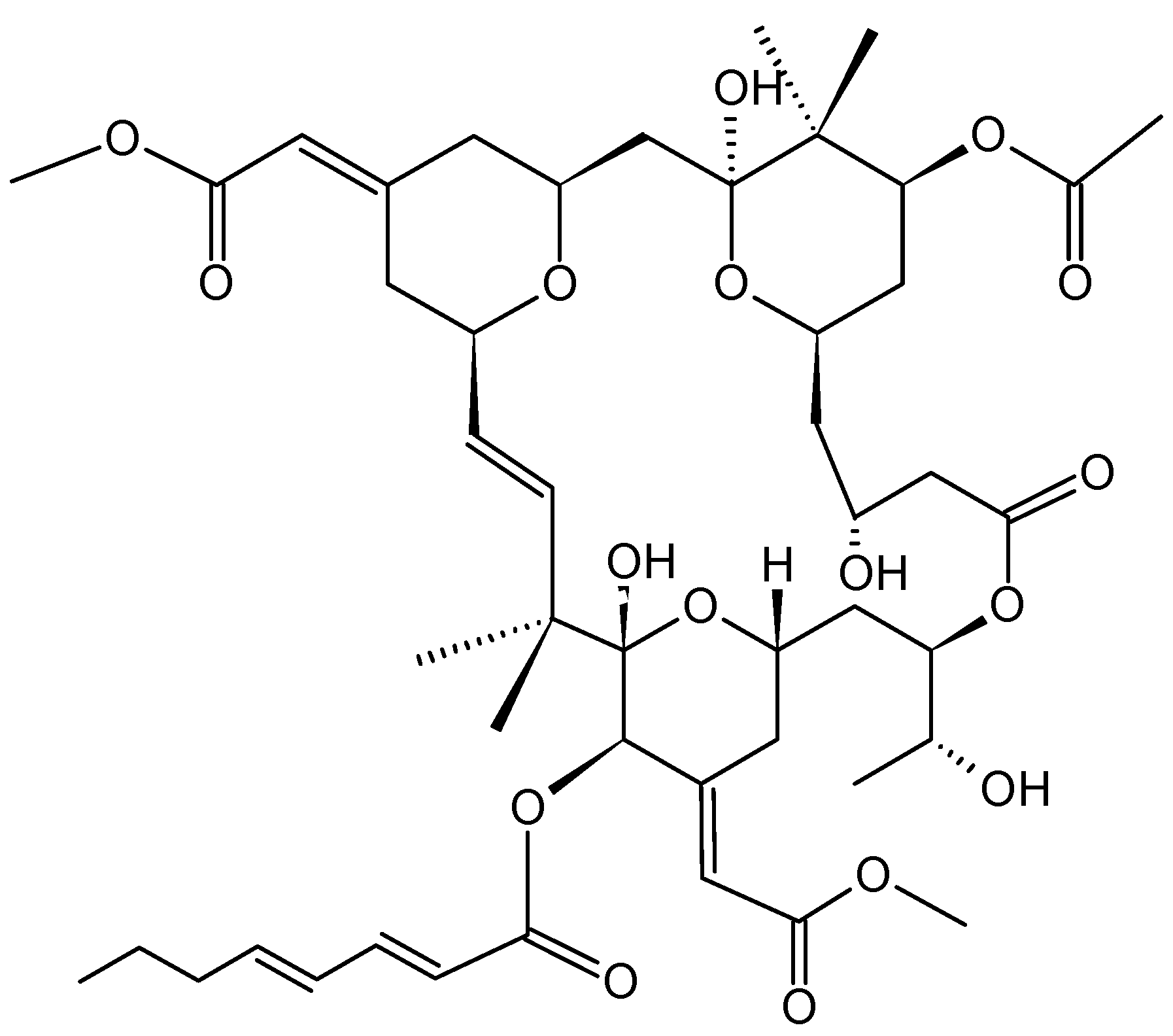
2.3.2. Bryostatin-1
2.3.3. Eribulin
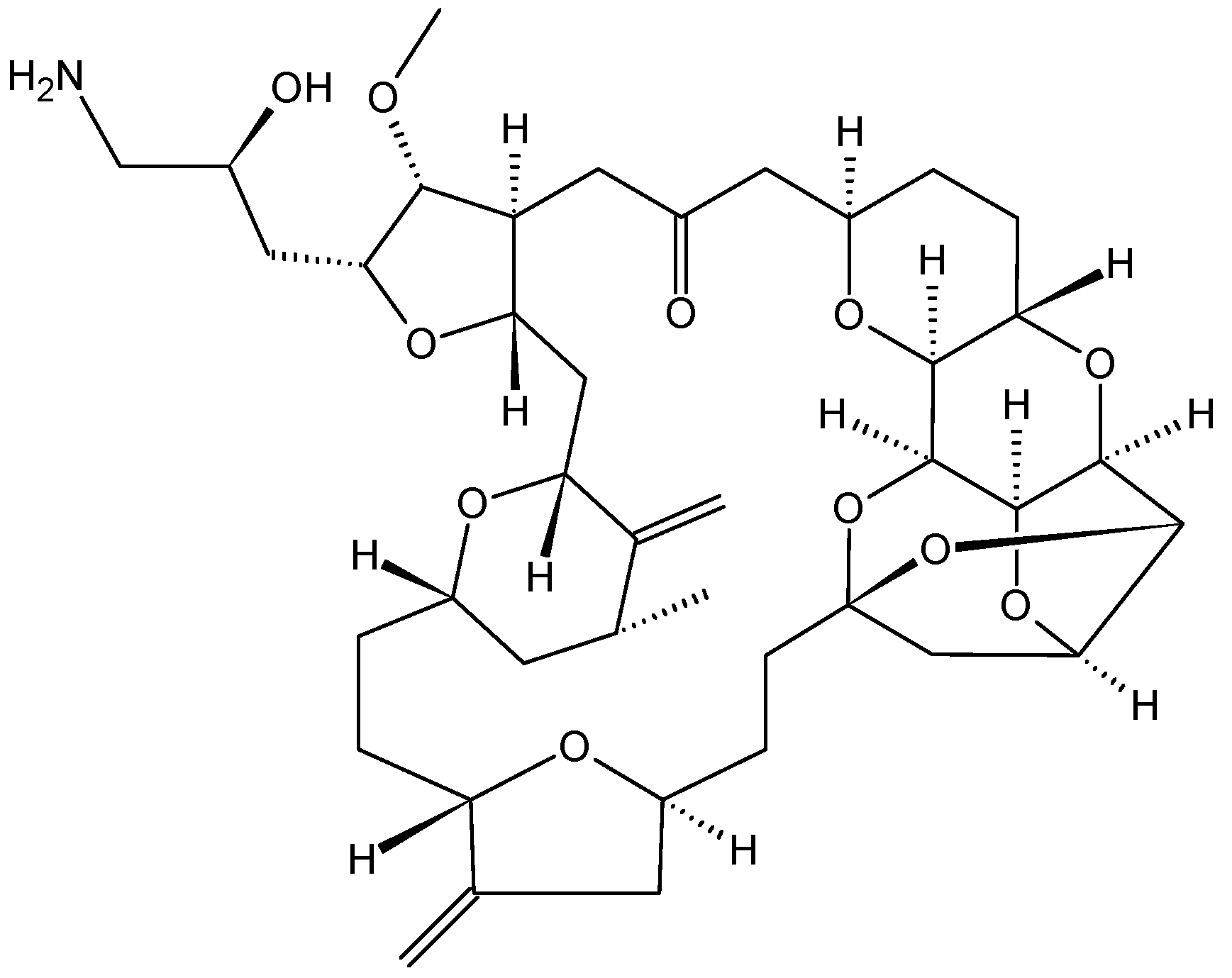
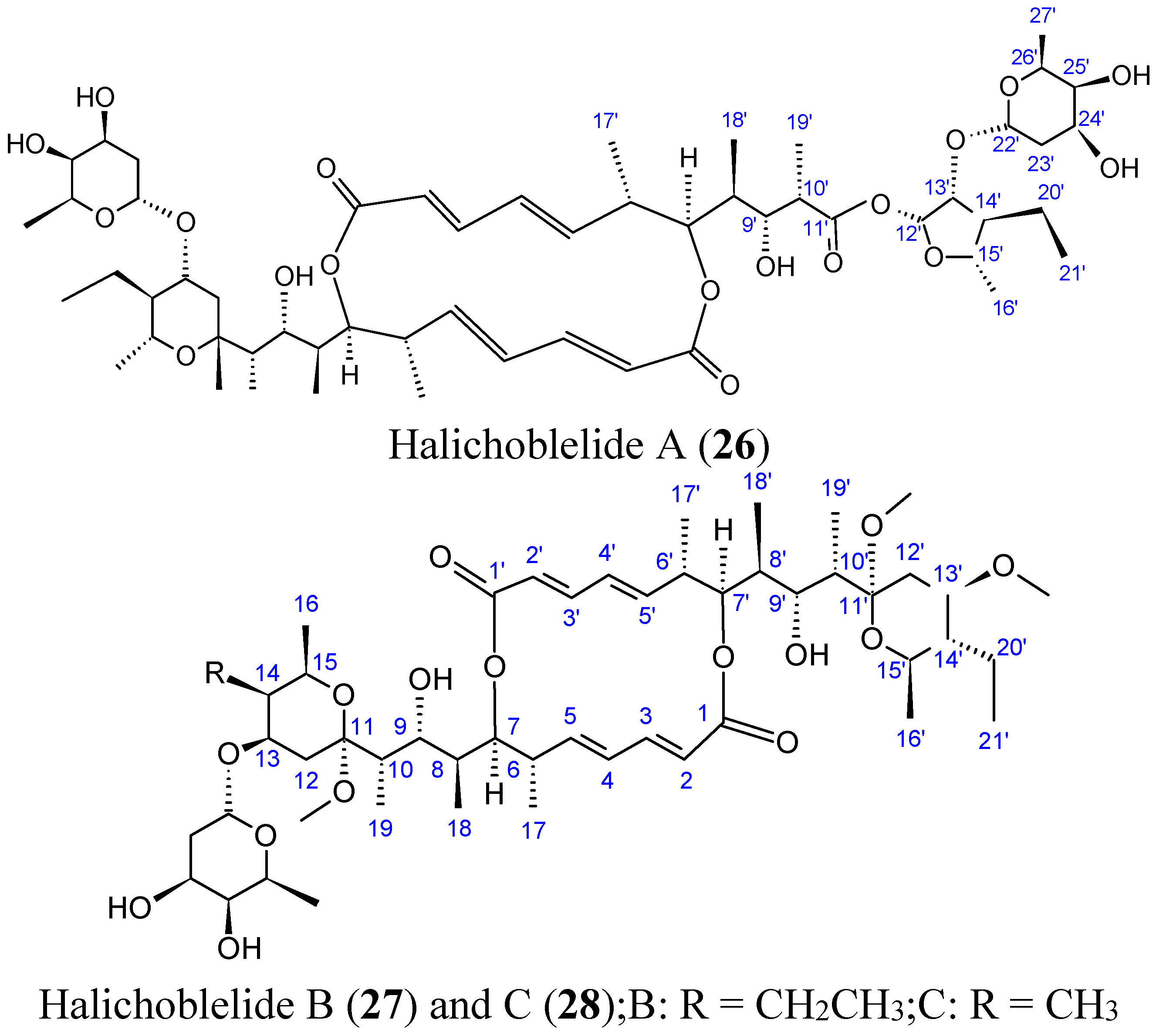
2.3.4. Halichoblelides
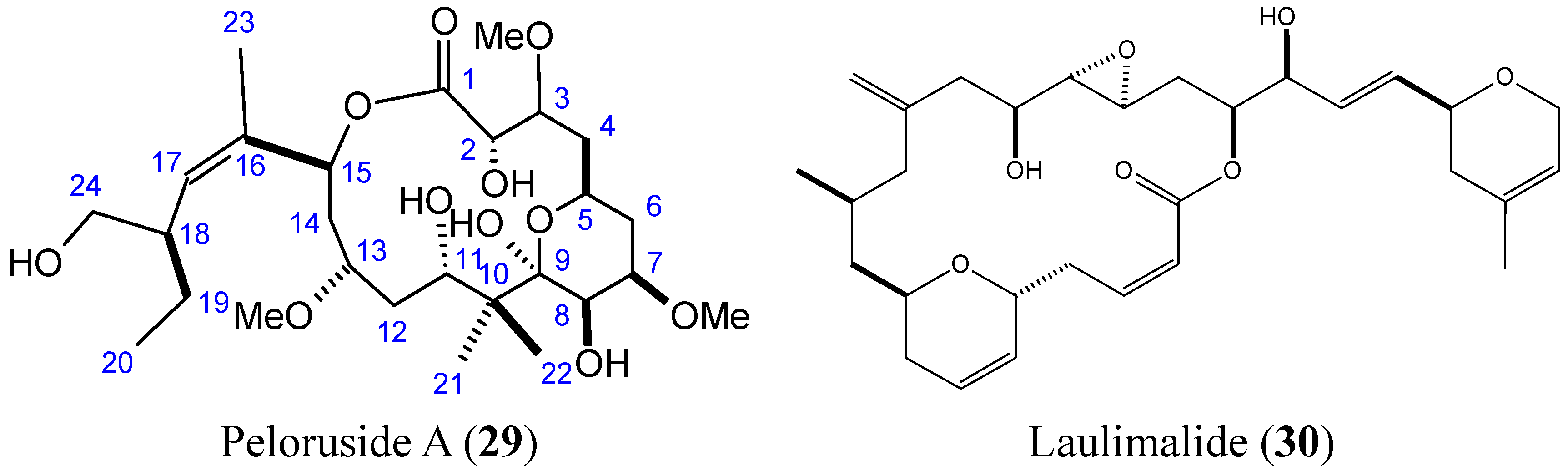
2.3.5. Peloruside A and Laulimalide
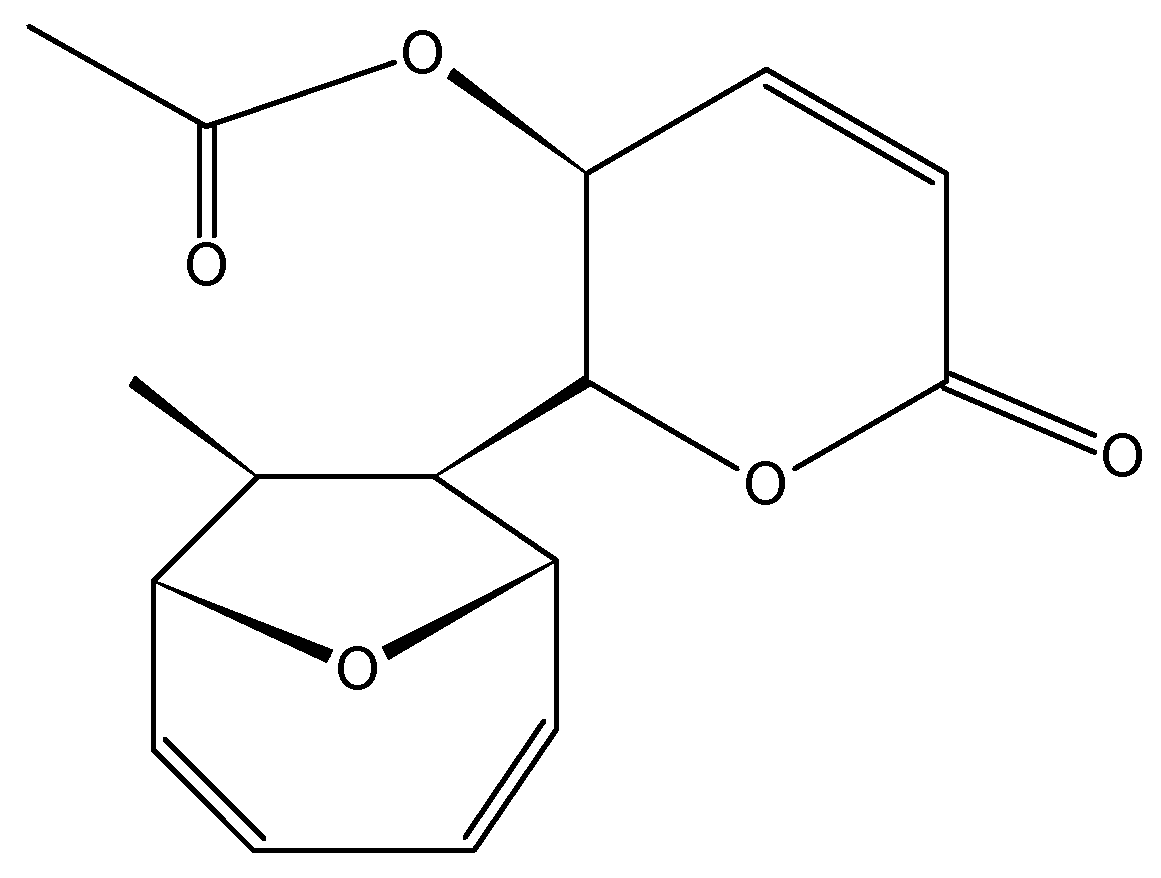
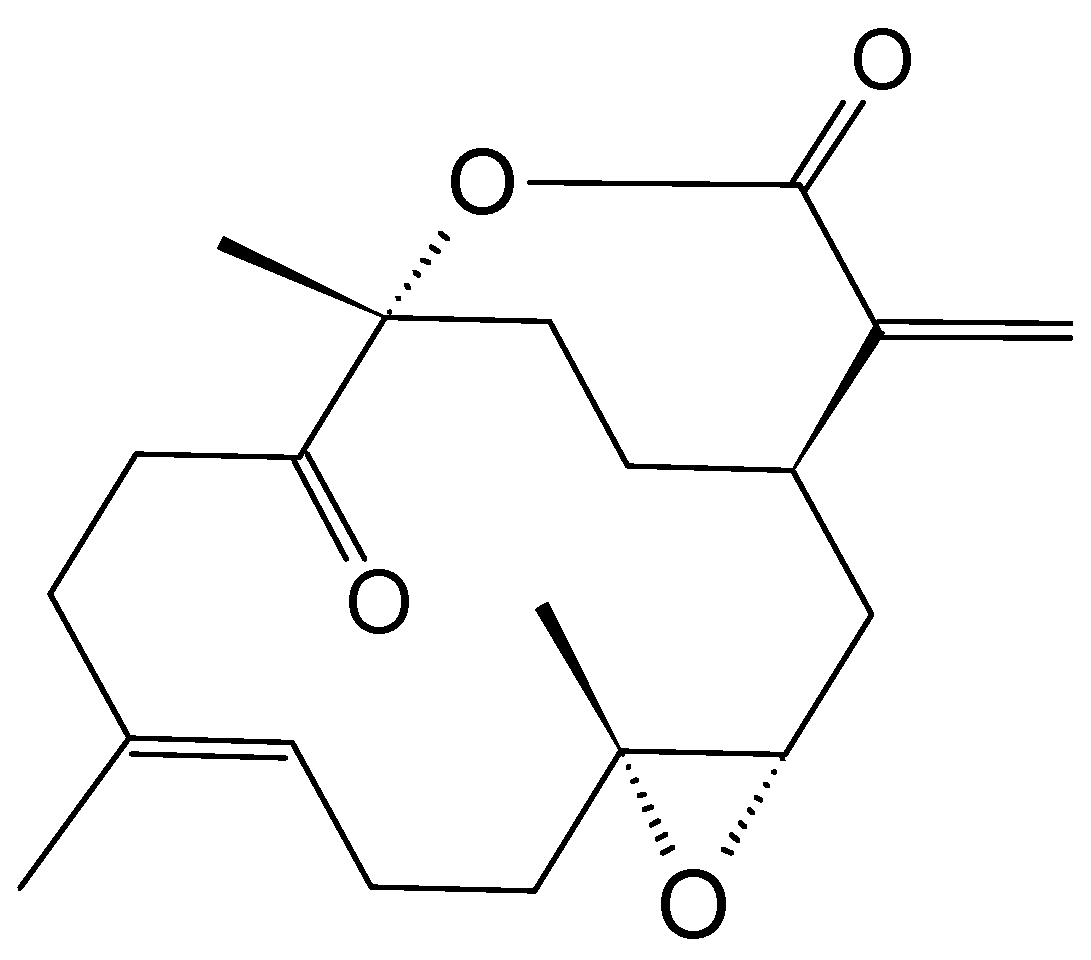
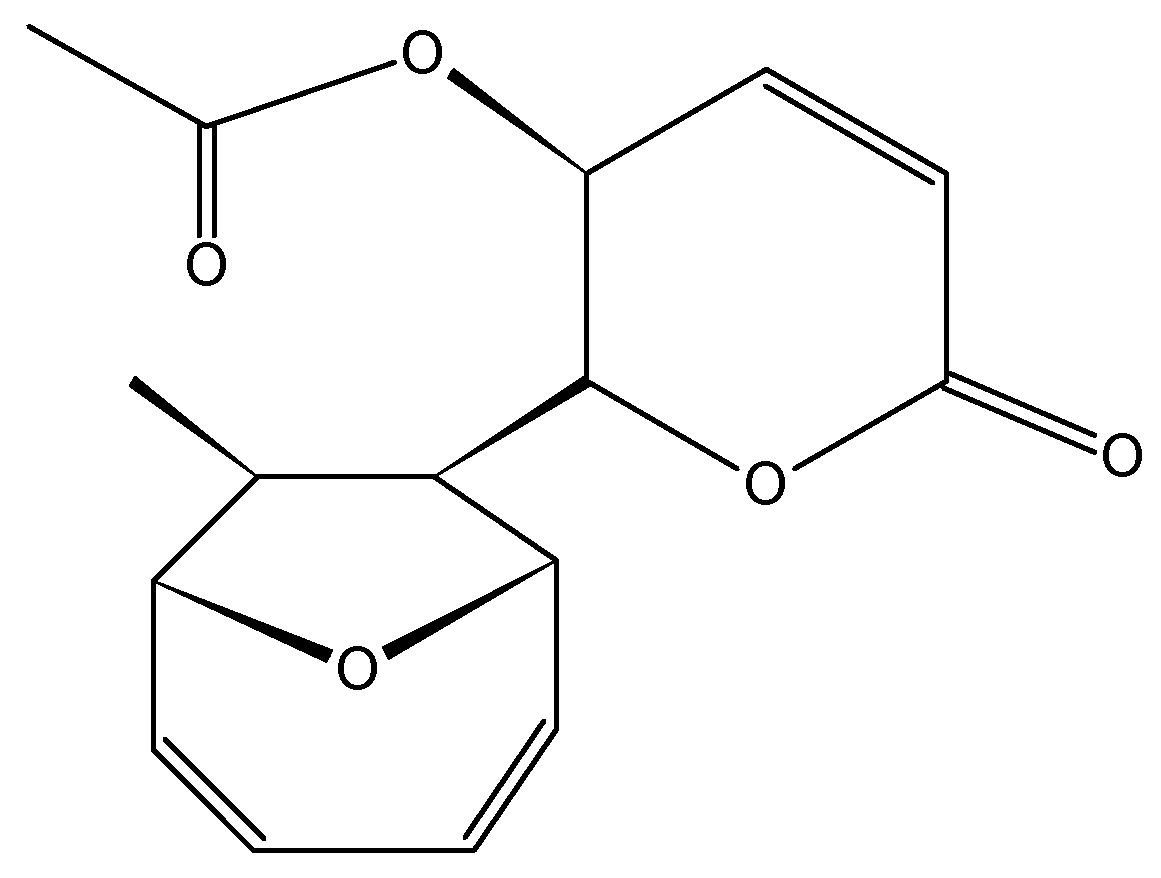
2.3.6. Mycoepoxydiene
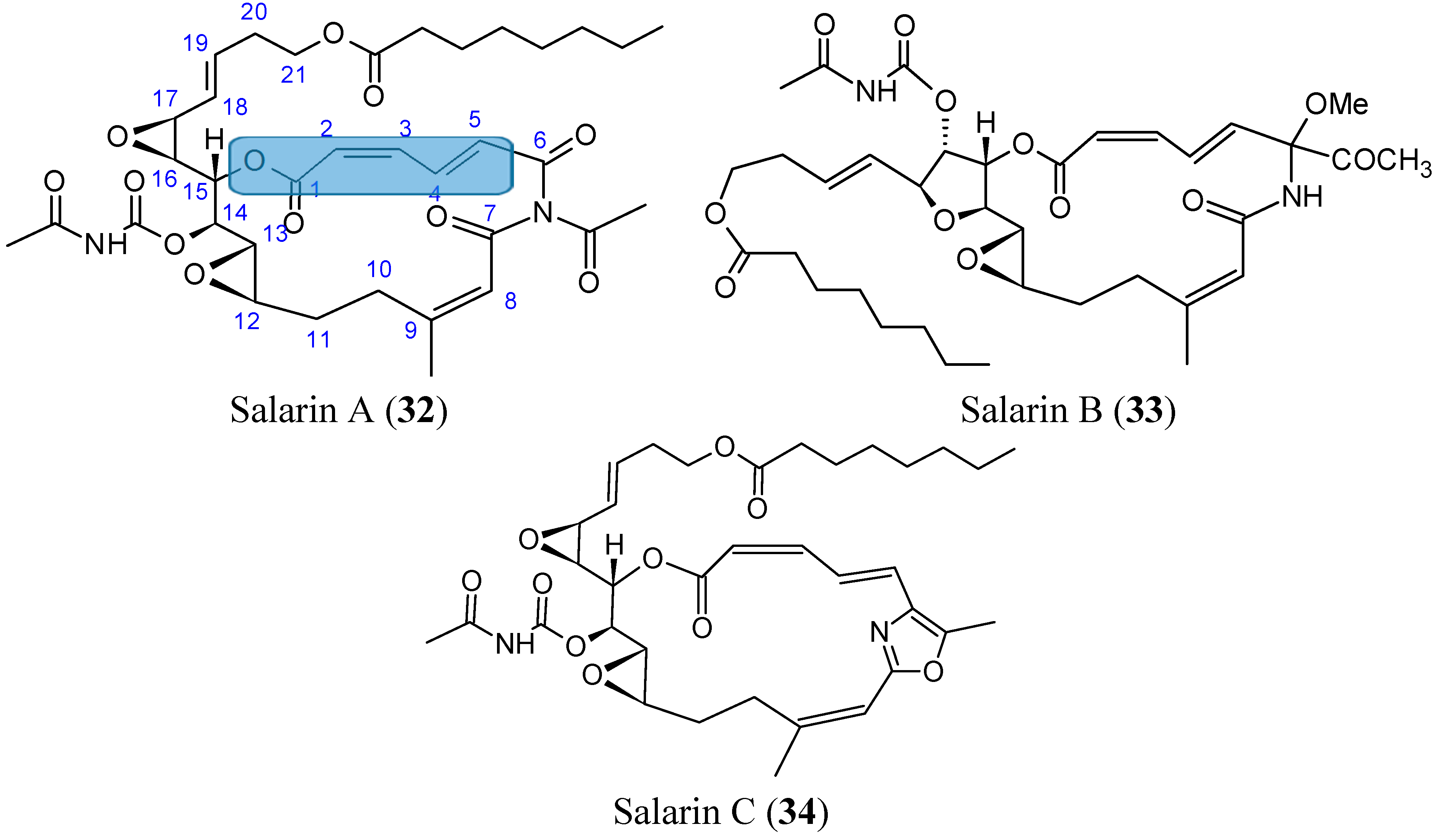
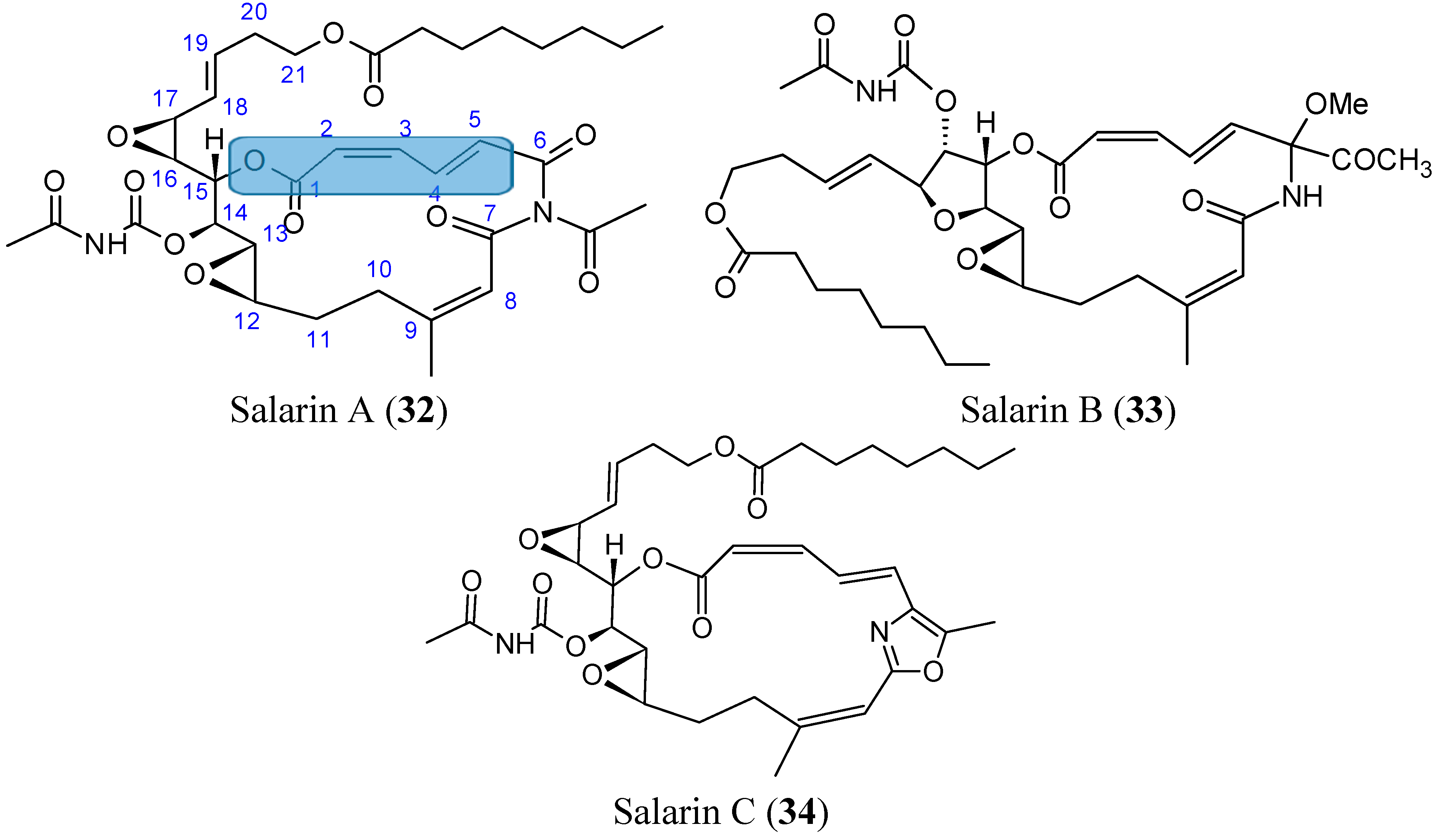
2.3.7. Salarins
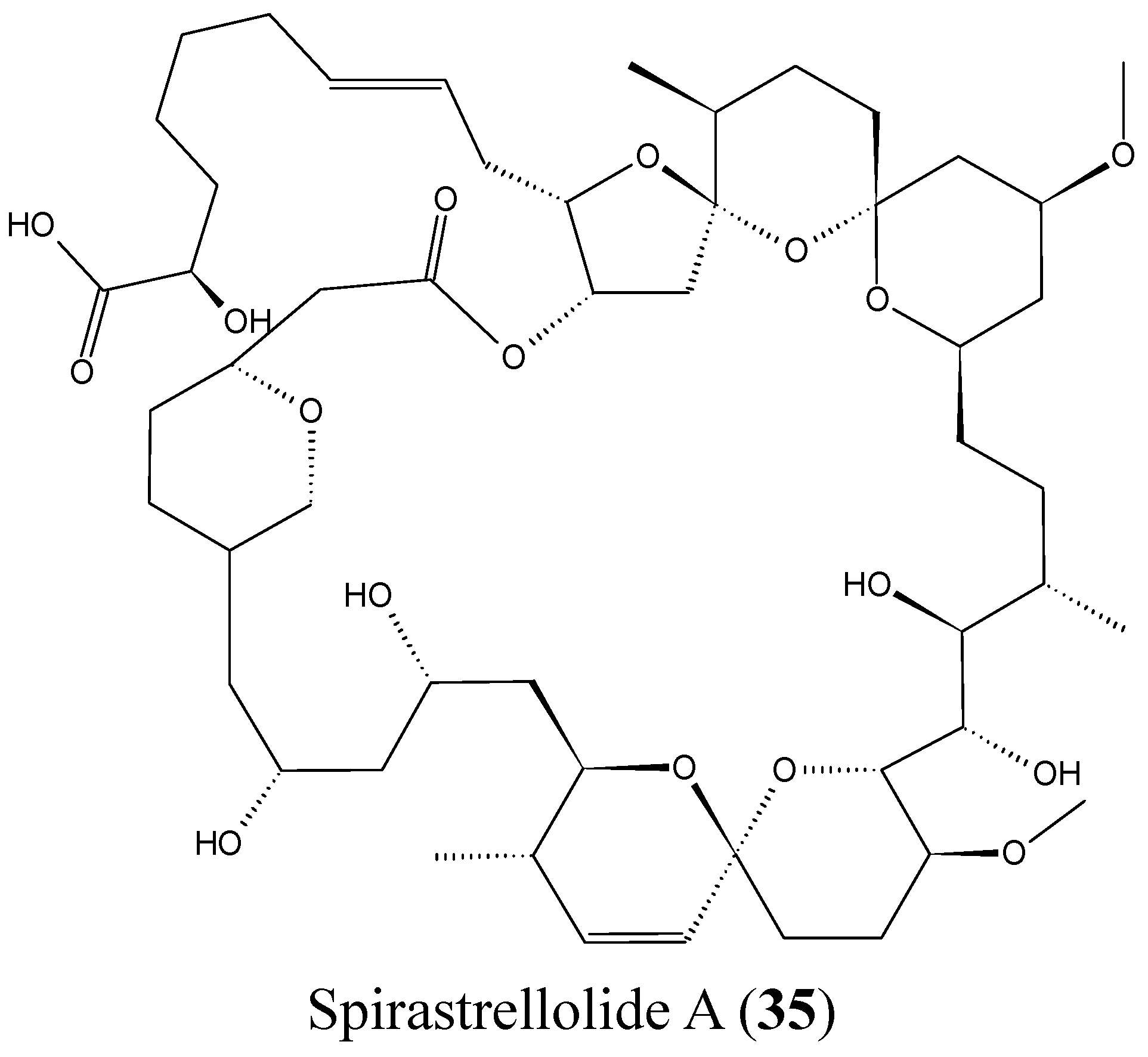
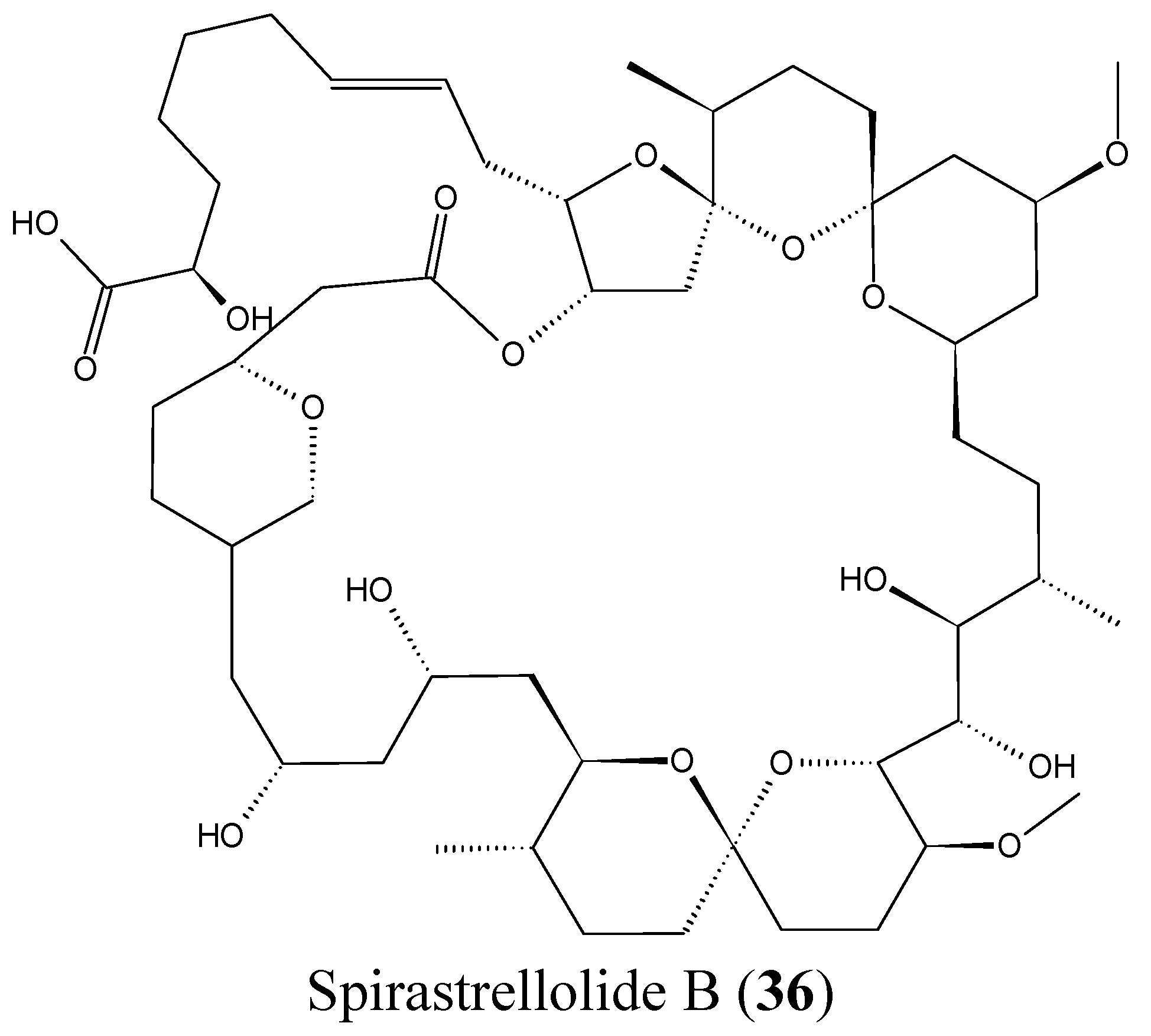
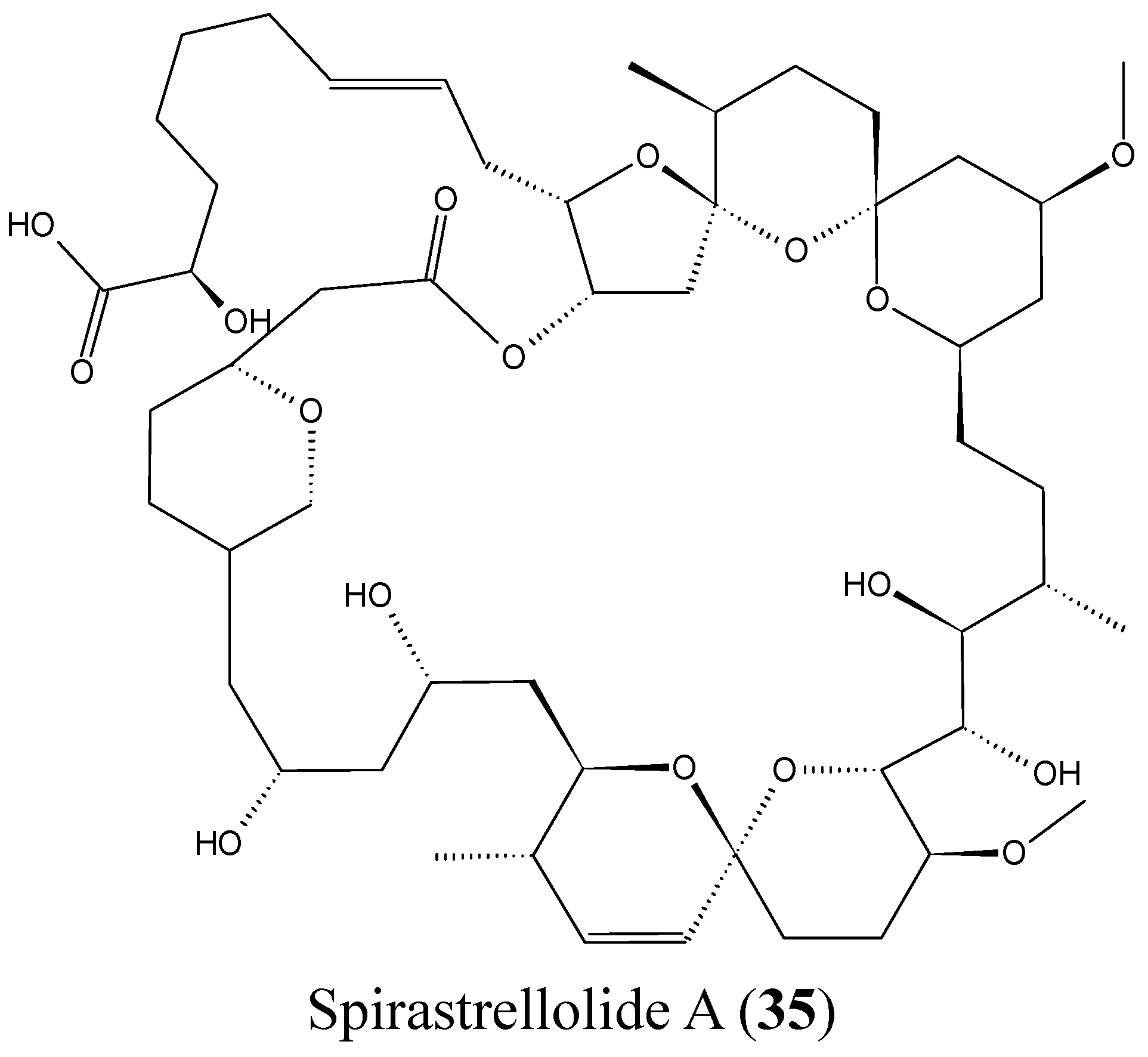
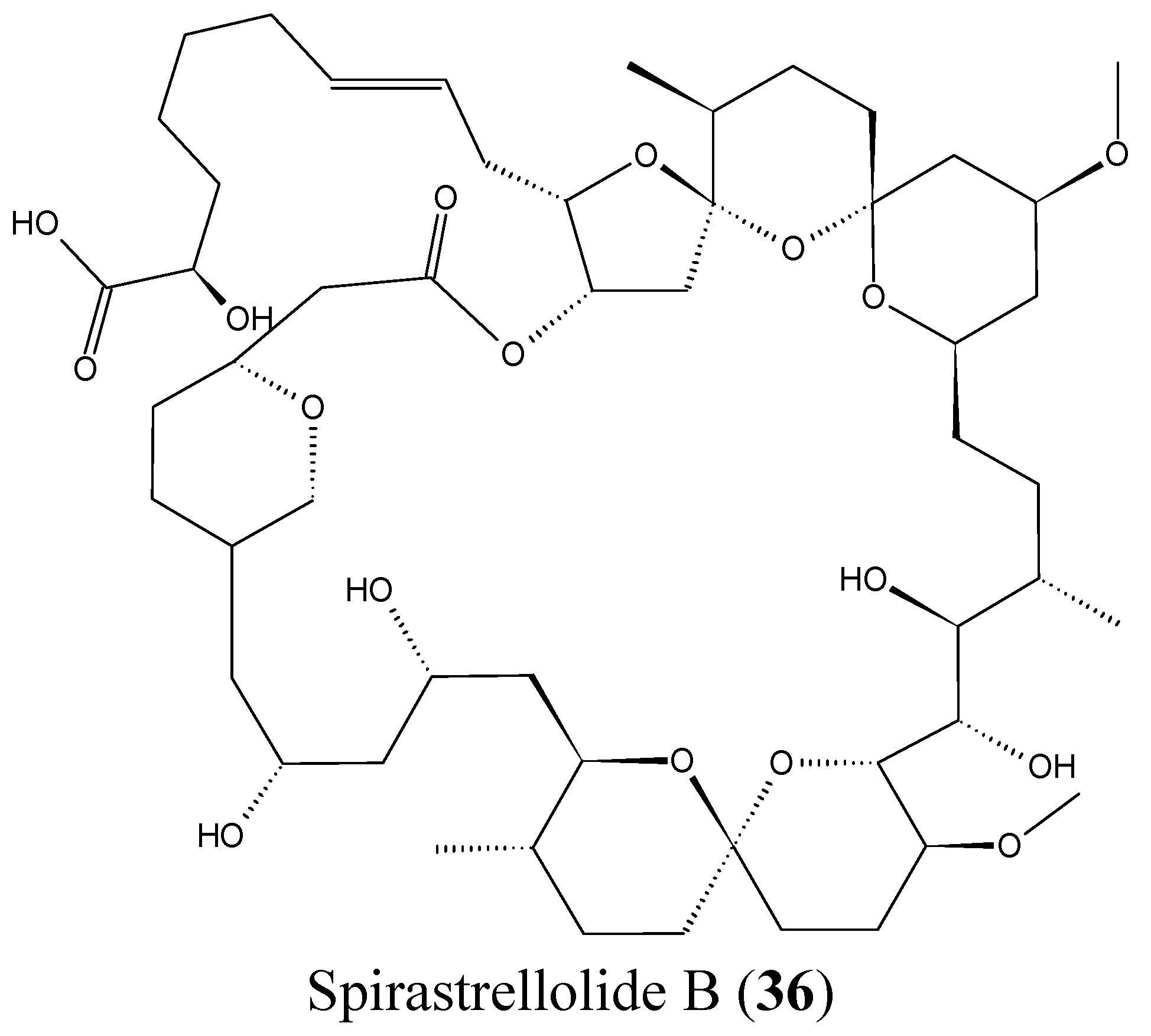
2.3.8. Spirastrellolides
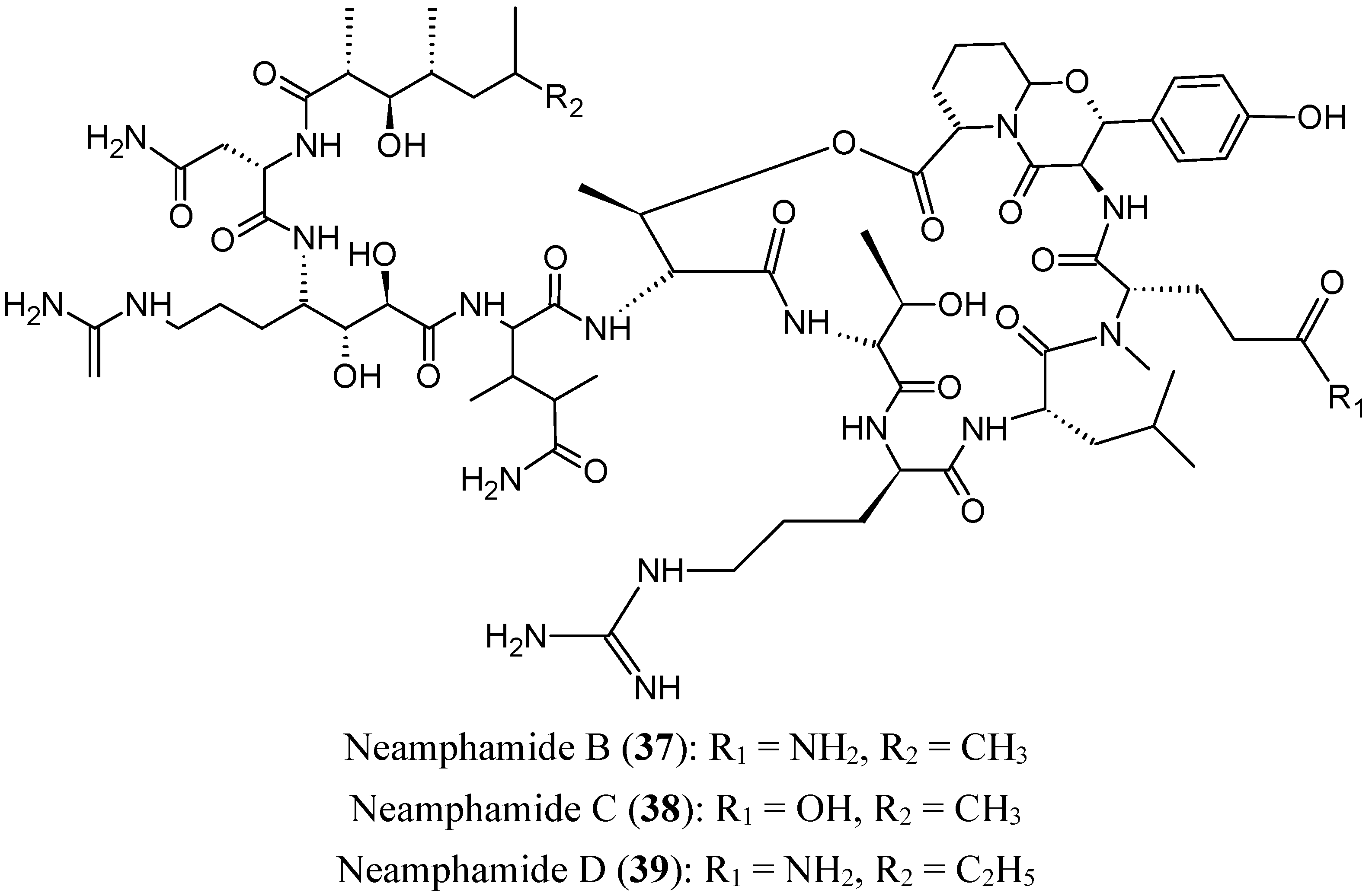
2.4. Peptides/Polypeptides
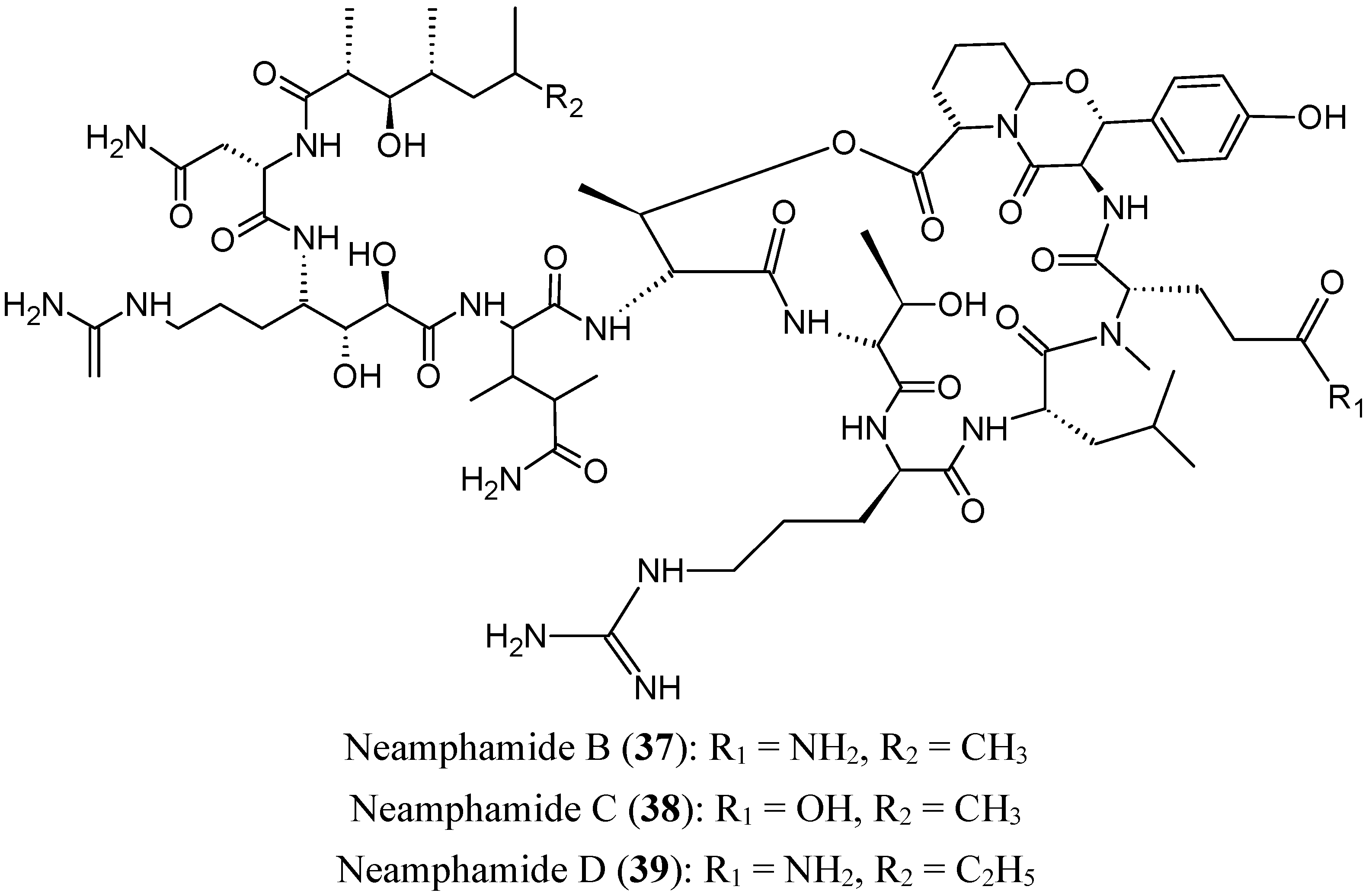
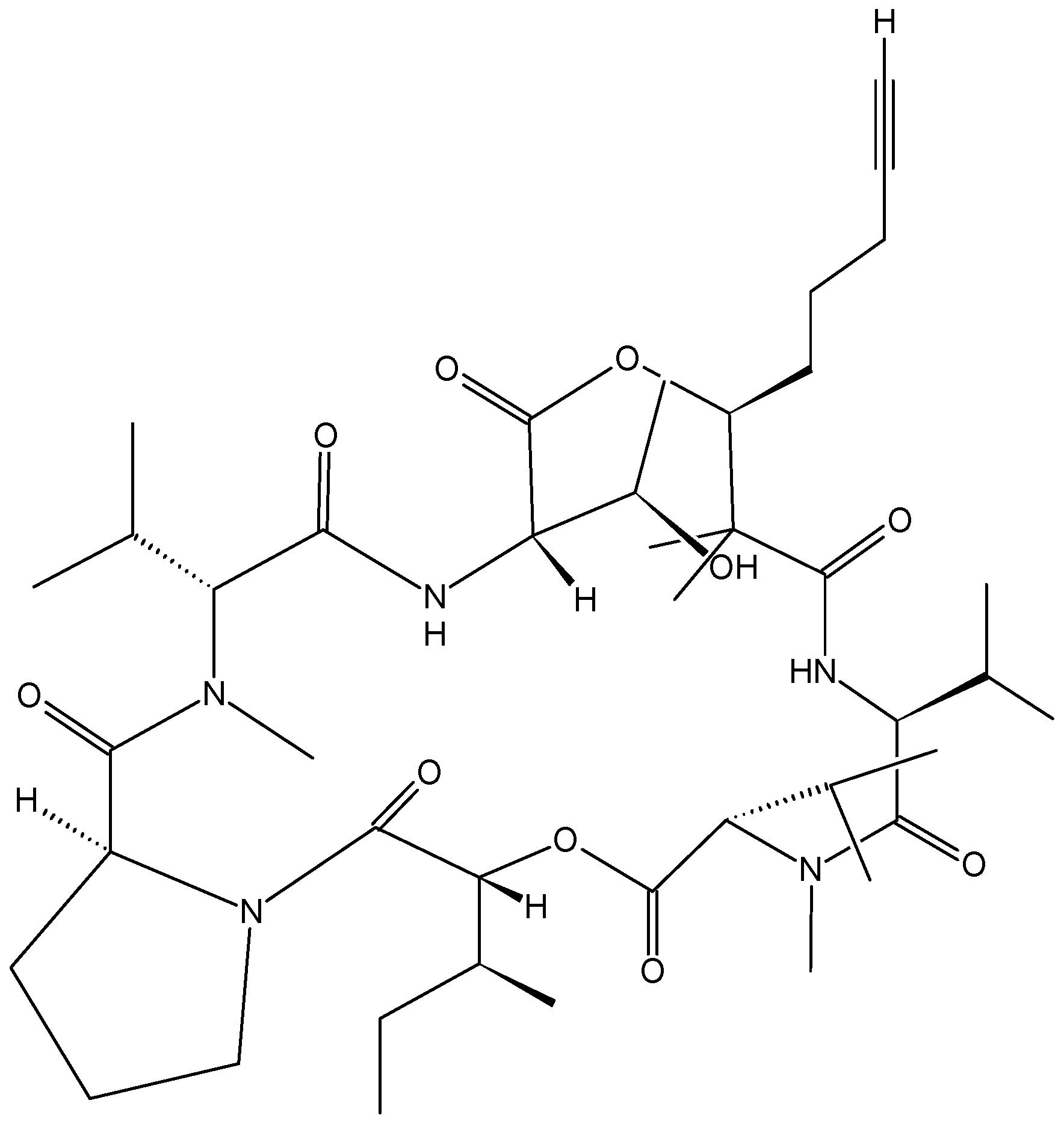
2.4.1. Cyclic Depsipeptides
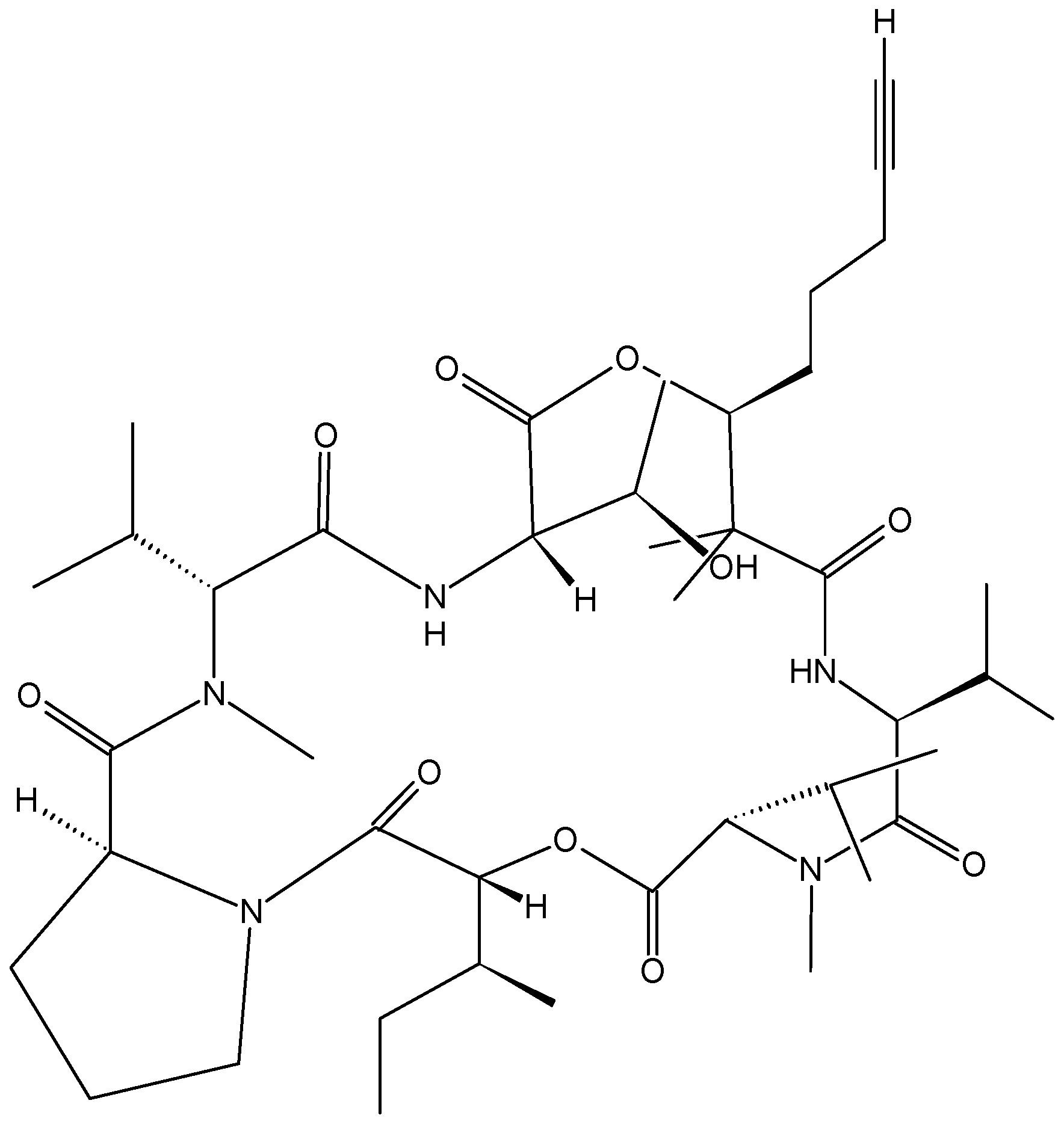
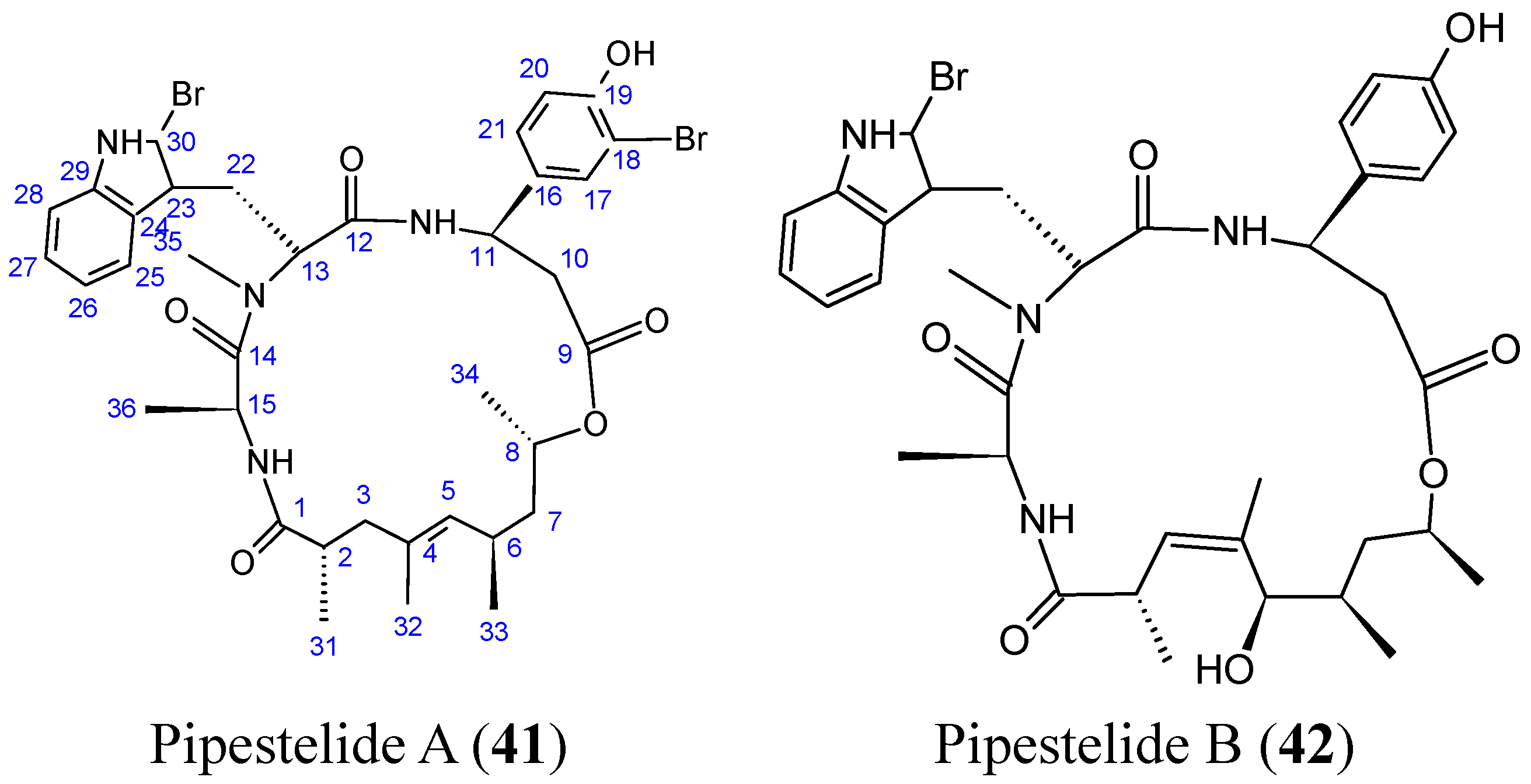
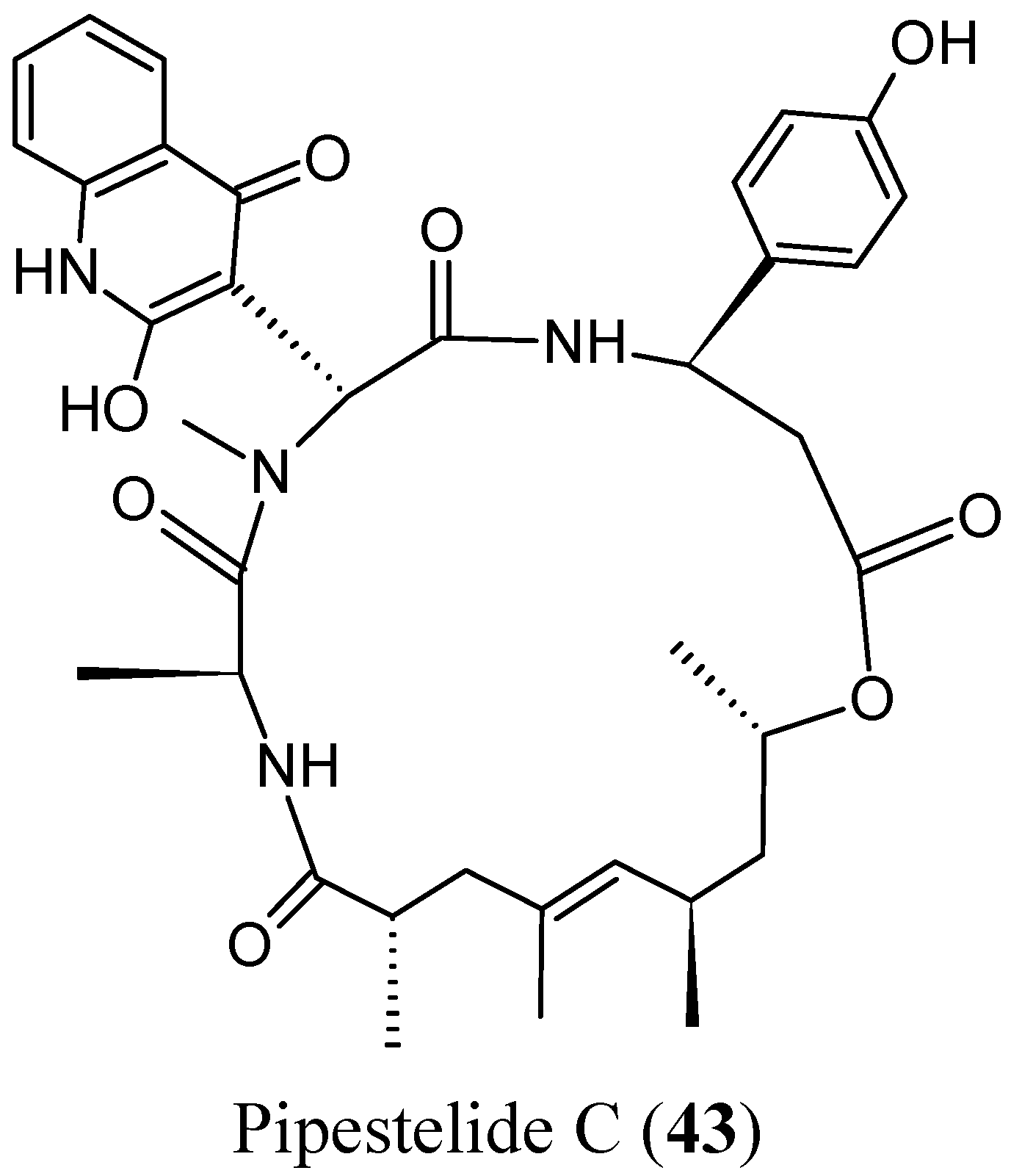
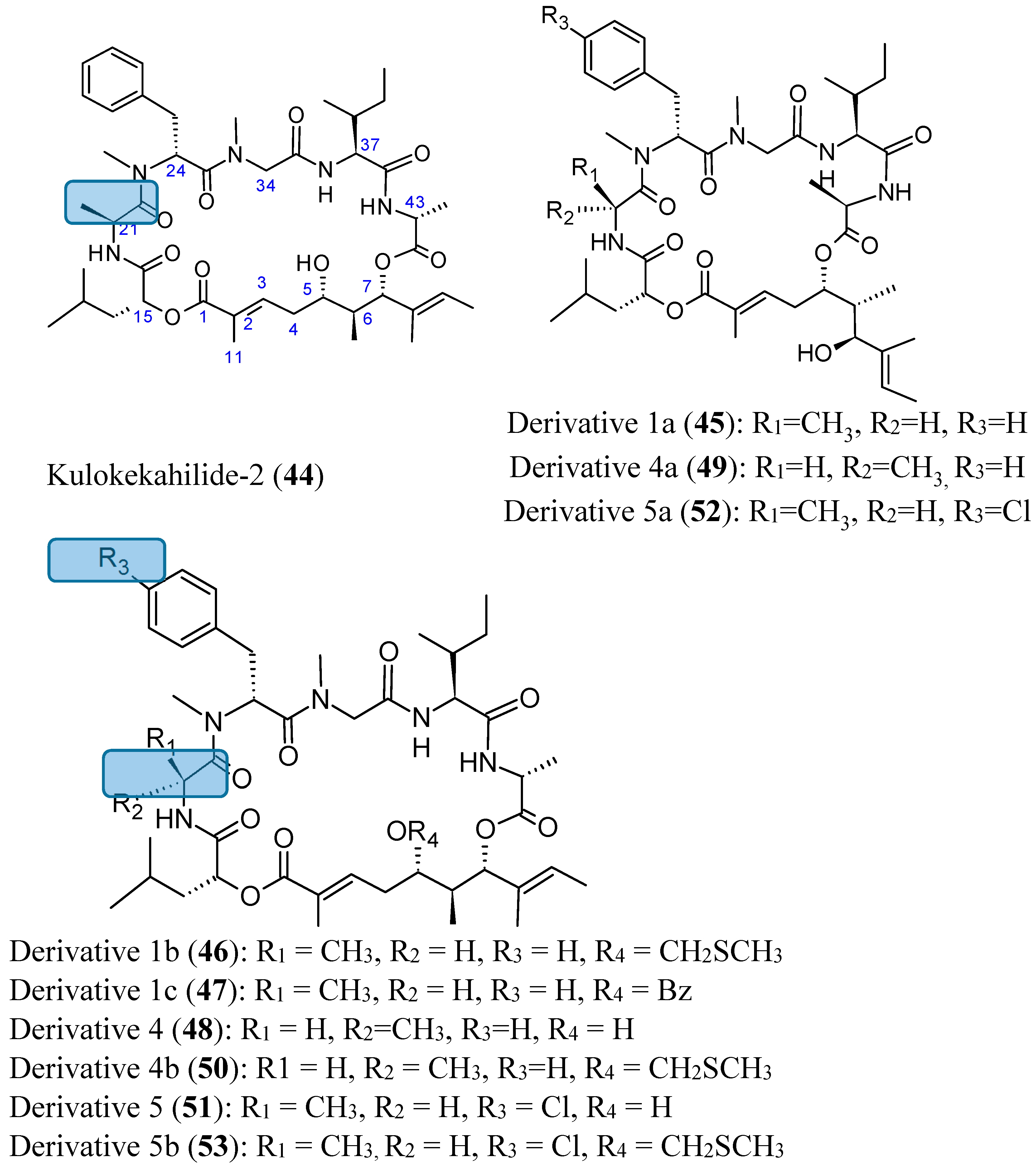
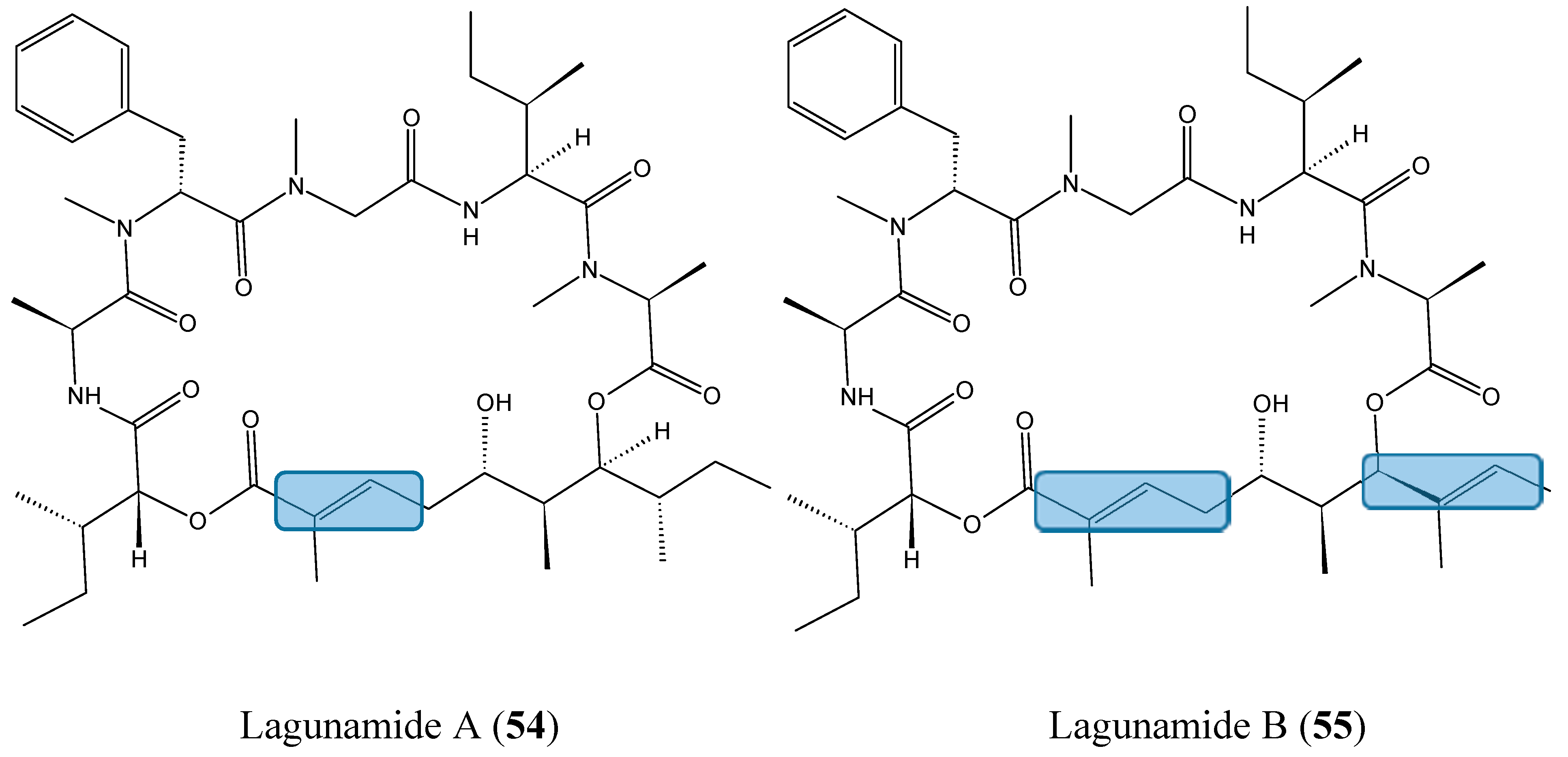
2.4.2. Cyclic heptapeptides
2.4.3. Sepia Ink Oligopeptide (SIO)
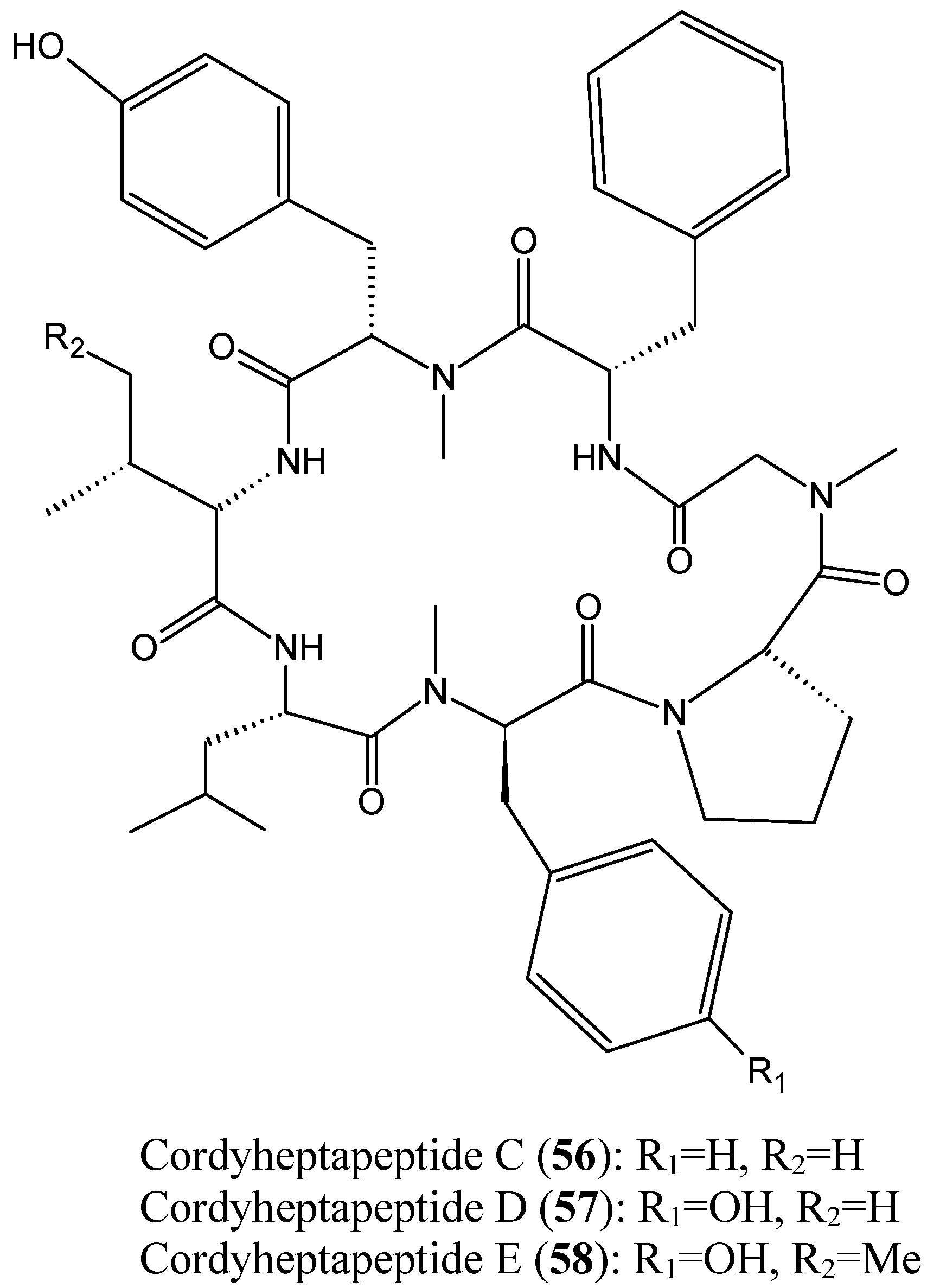
2.4.4. Hoiamide D
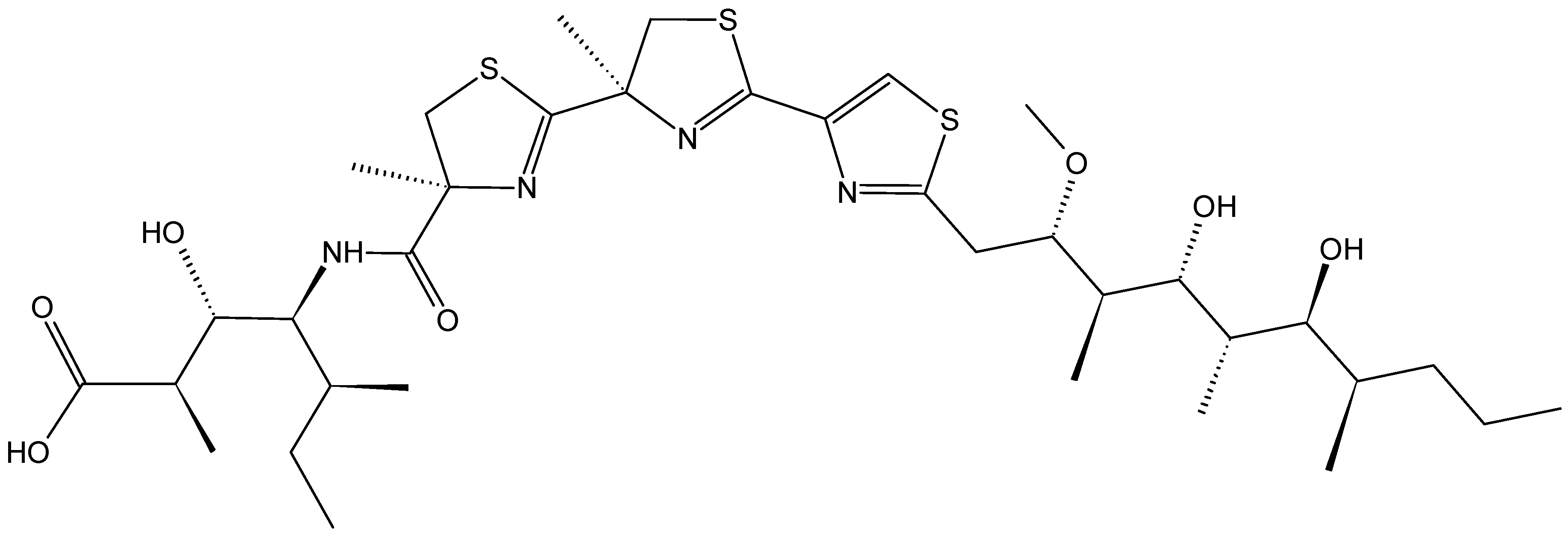
2.4.5. Protuboxepin A
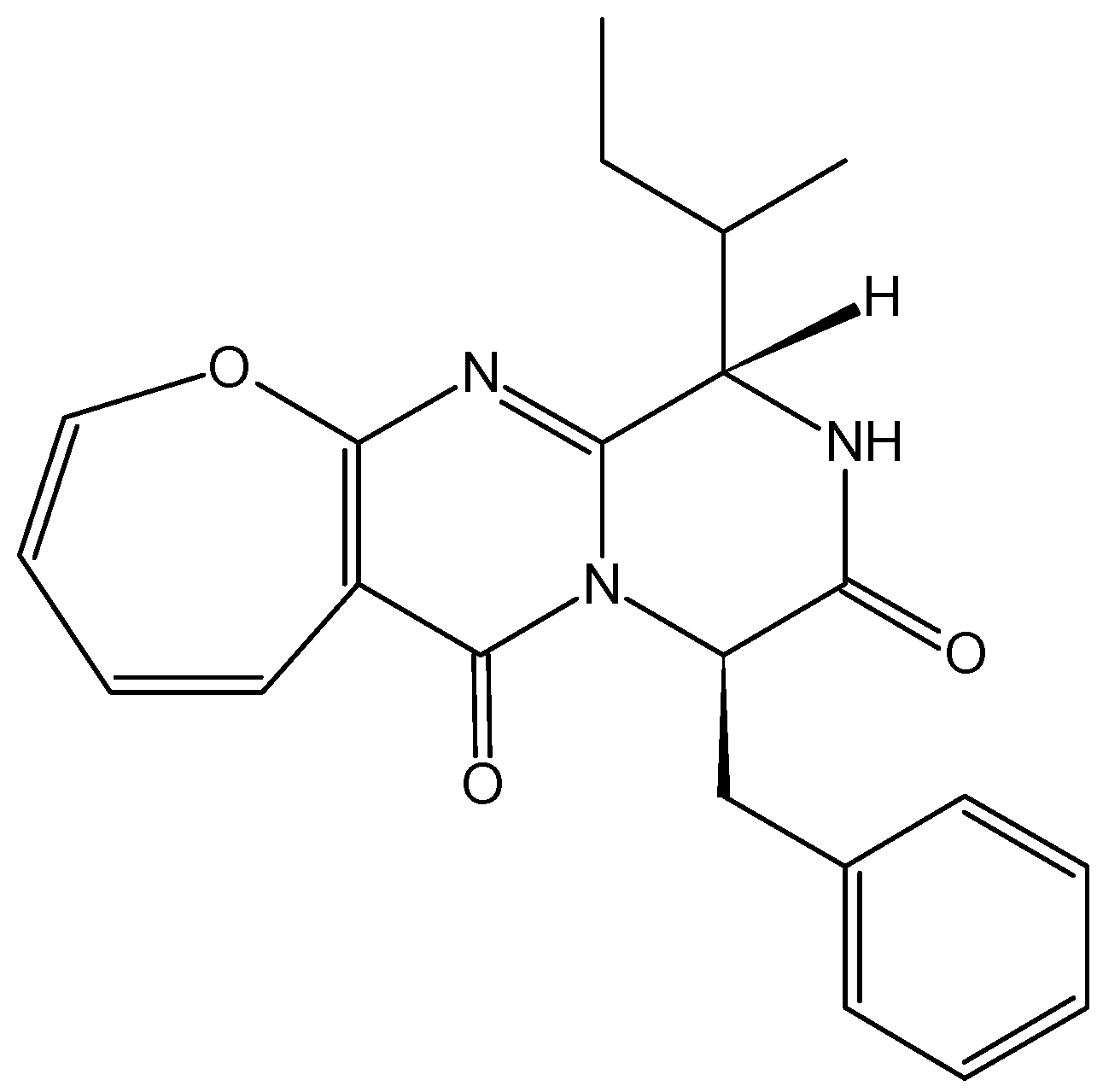
2.4.6. Polypeptide CS5931
2.5. Phenols/Polyphenols
2.5.1. Aeroplysinin-1
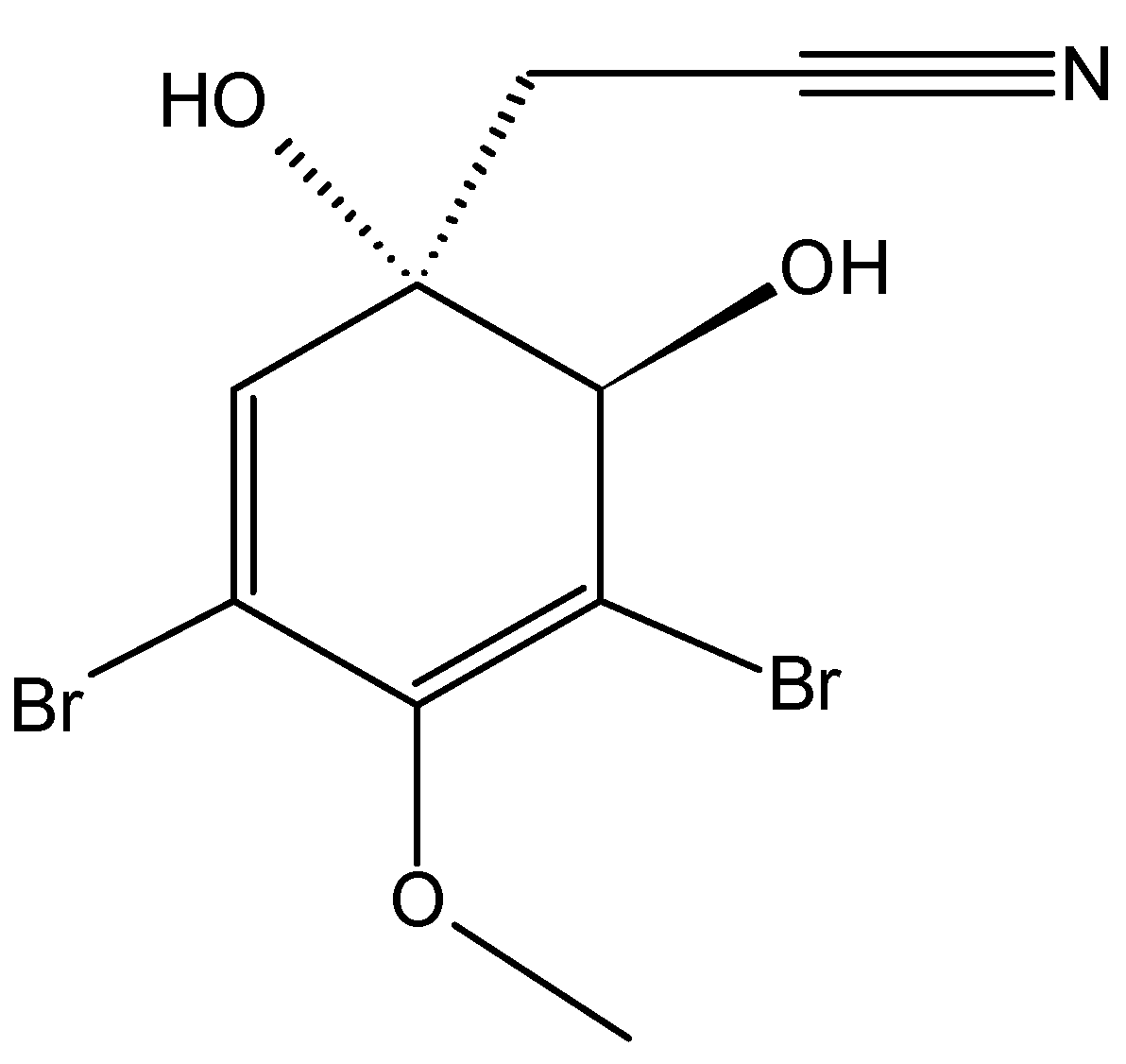
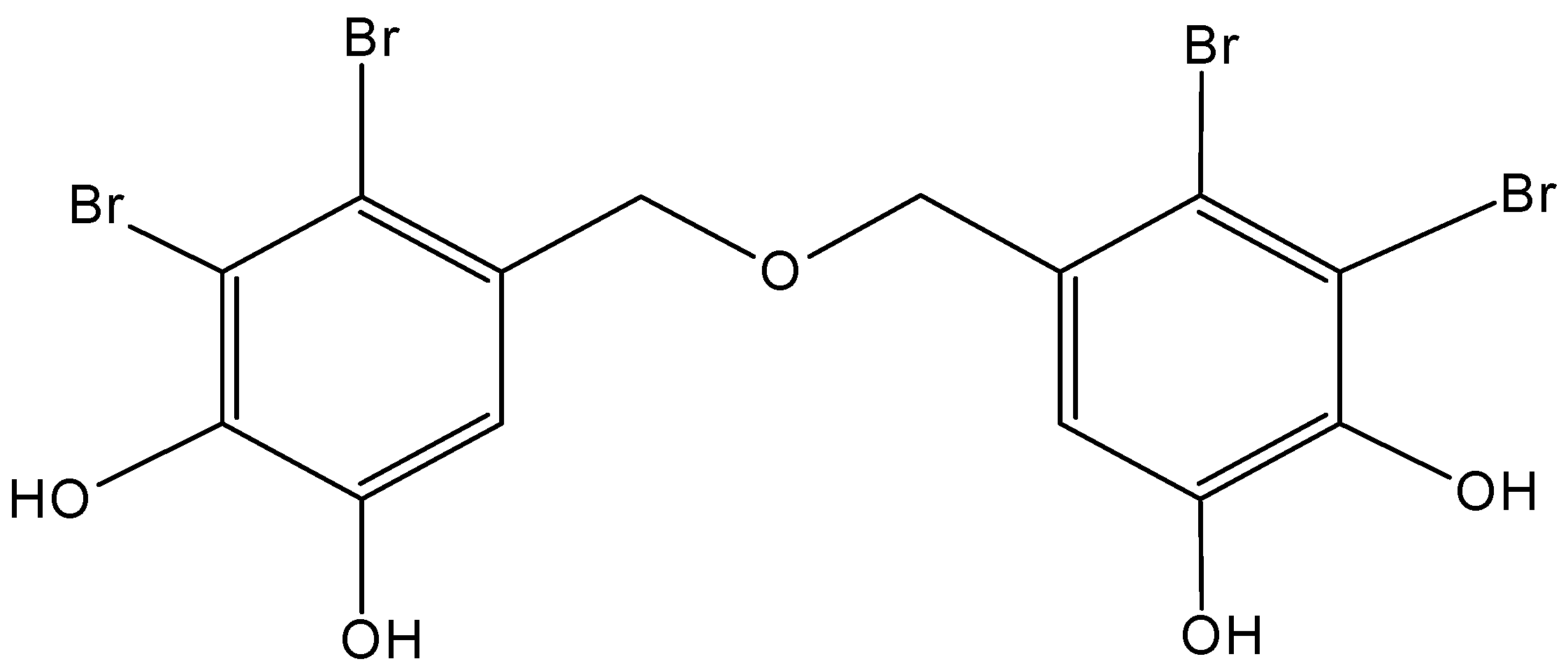
2.5.2. Bromophenol Bis (2,3-dibromo-4,5-dihydroxybenzyl) ether (BDDE)
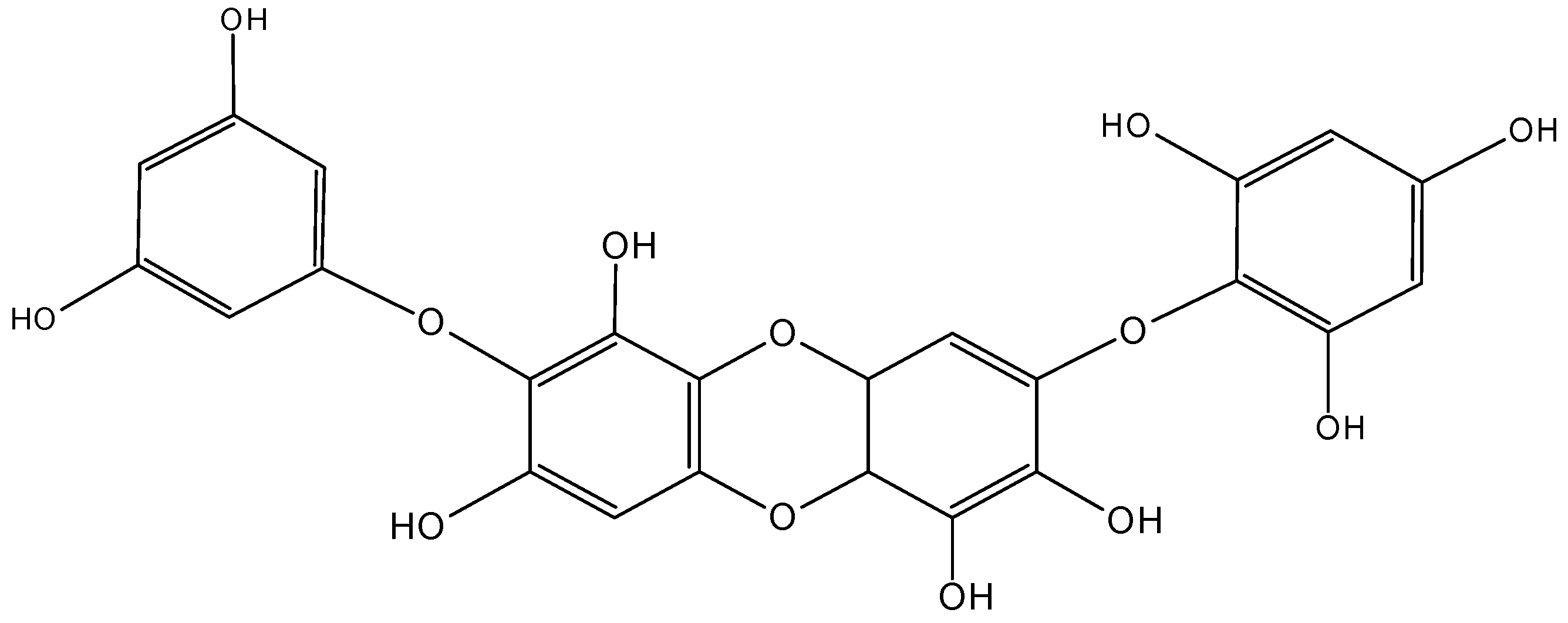
2.5.3. Diphlorethohydroxycarmalol
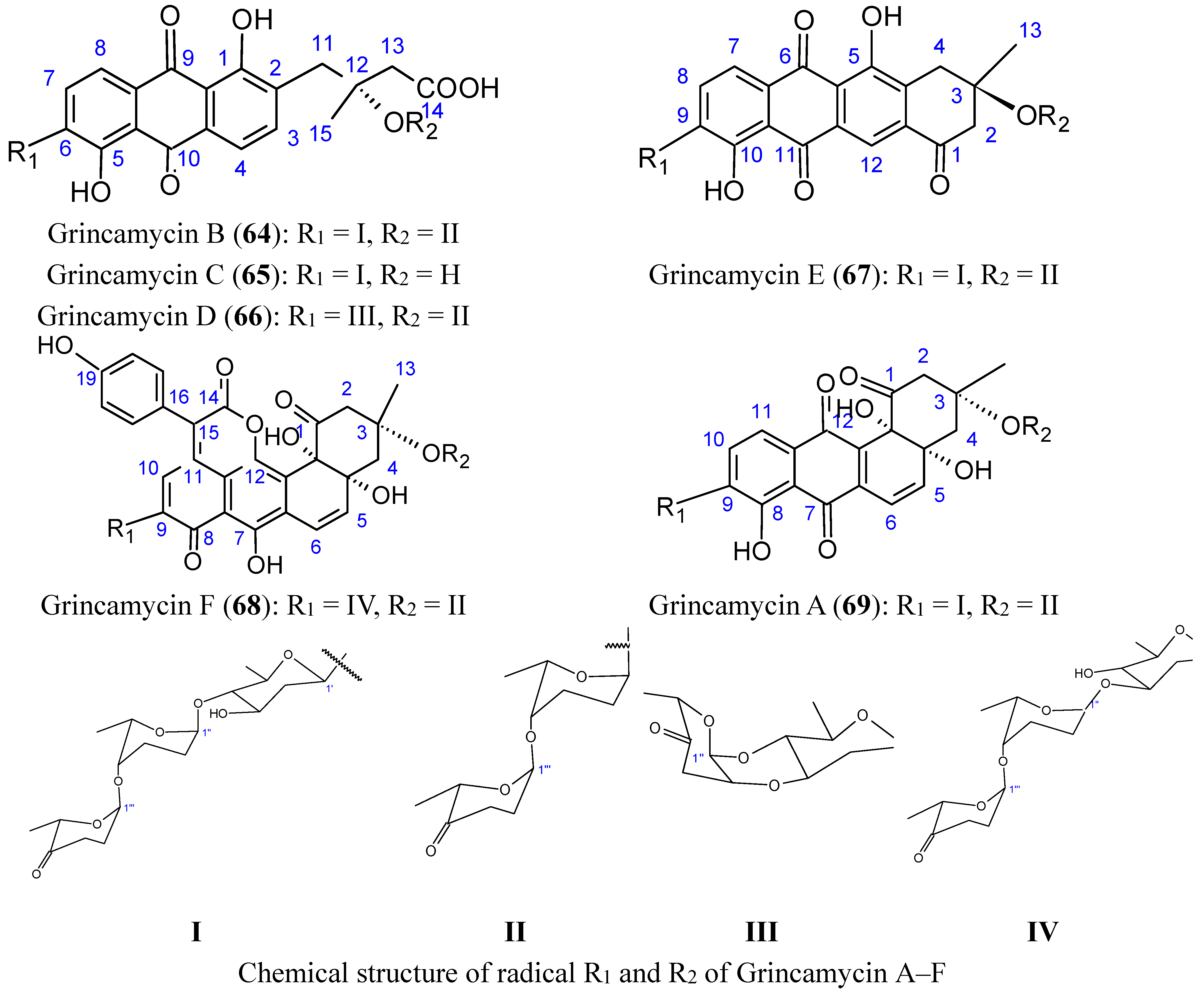
2.5.4. C-glycoside Angucyclines
2.6. Polysaccharides
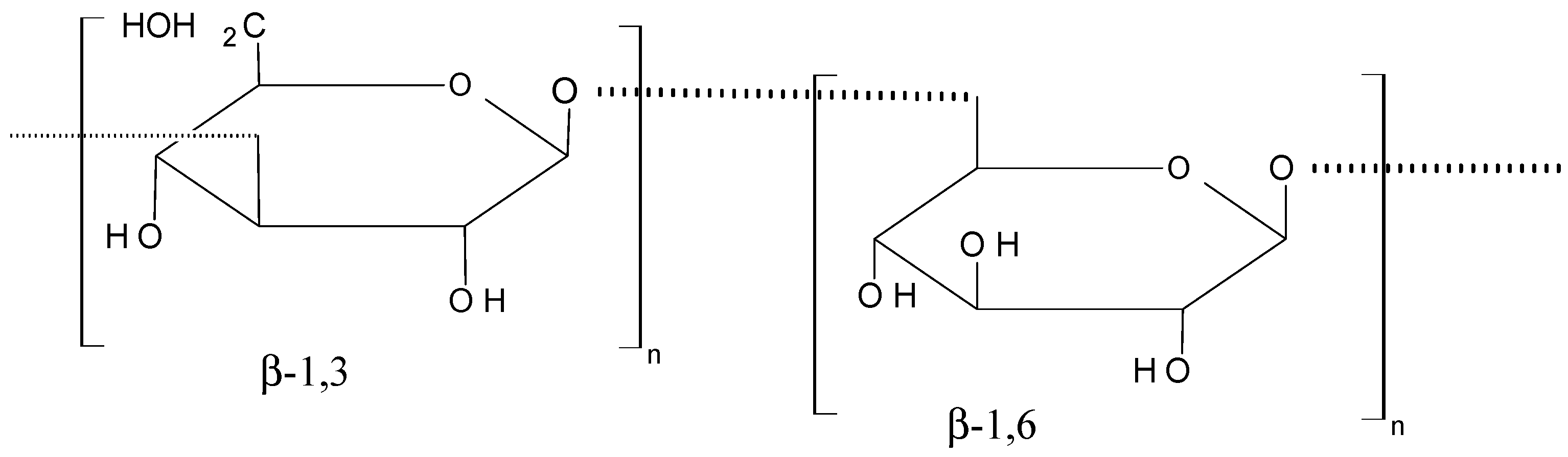
2.7. Quinones
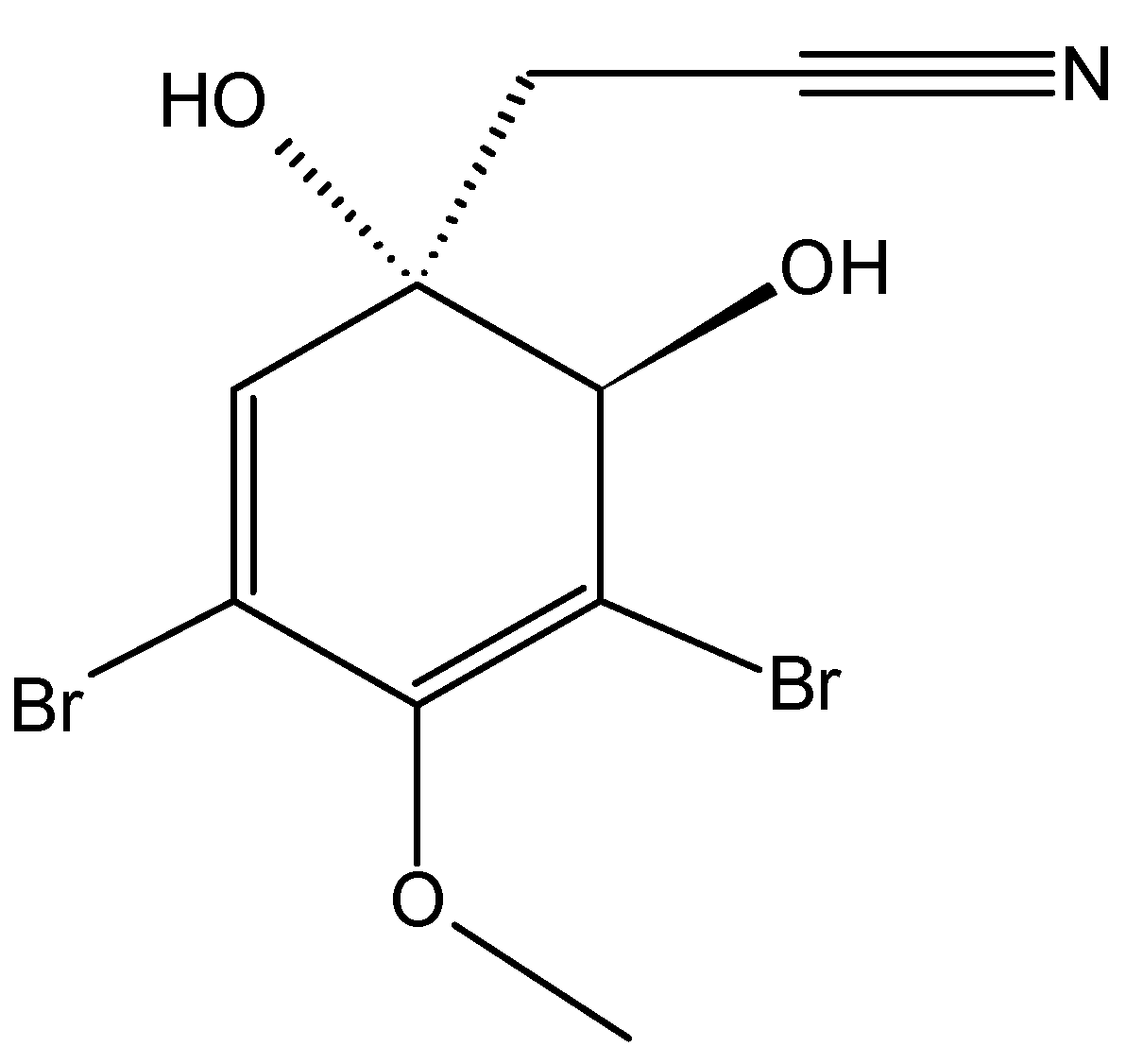
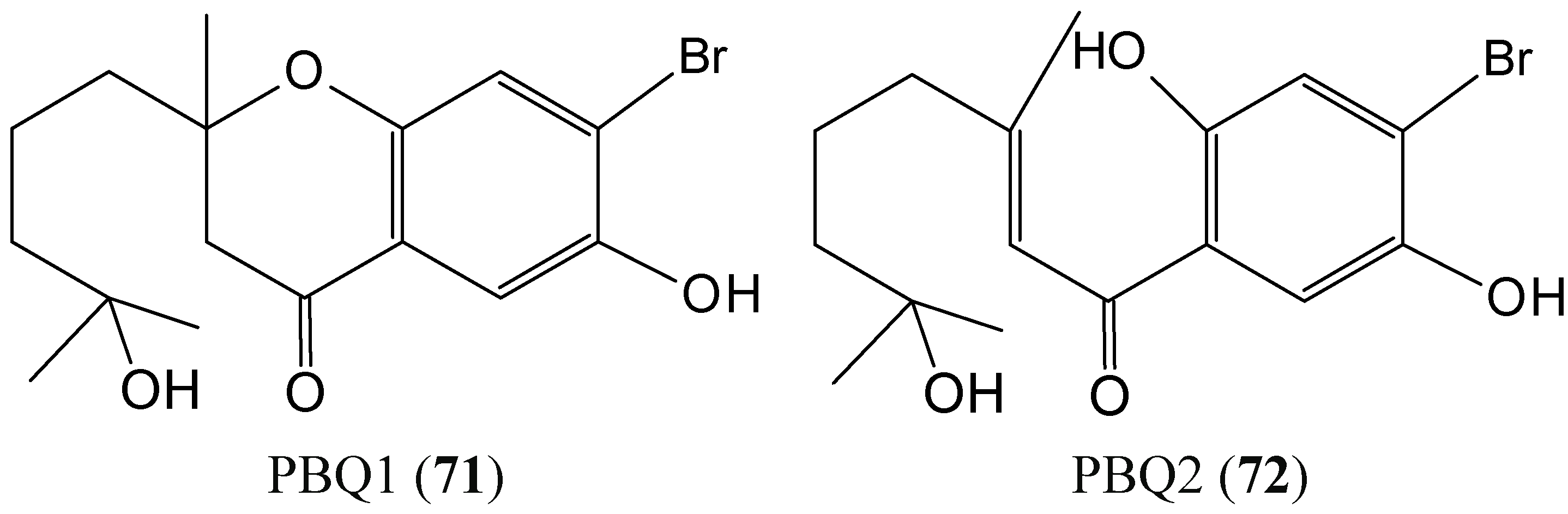
2.7.1. Prenylated Bromohydroquinones
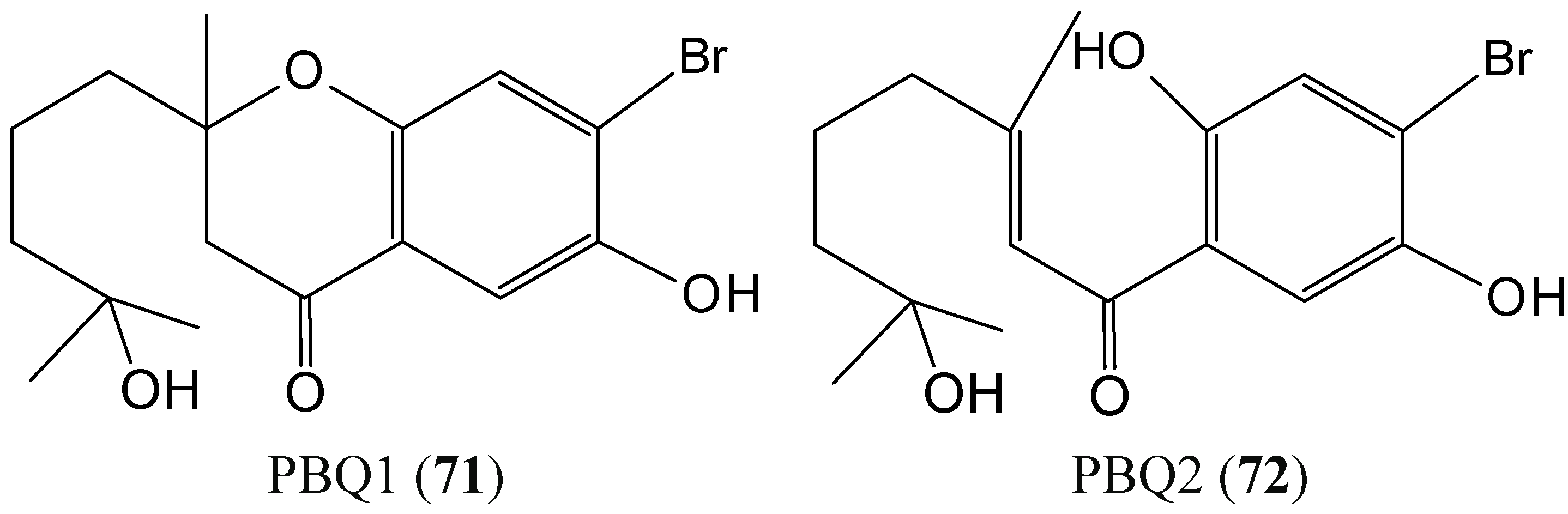
2.7.2. SZ-685C
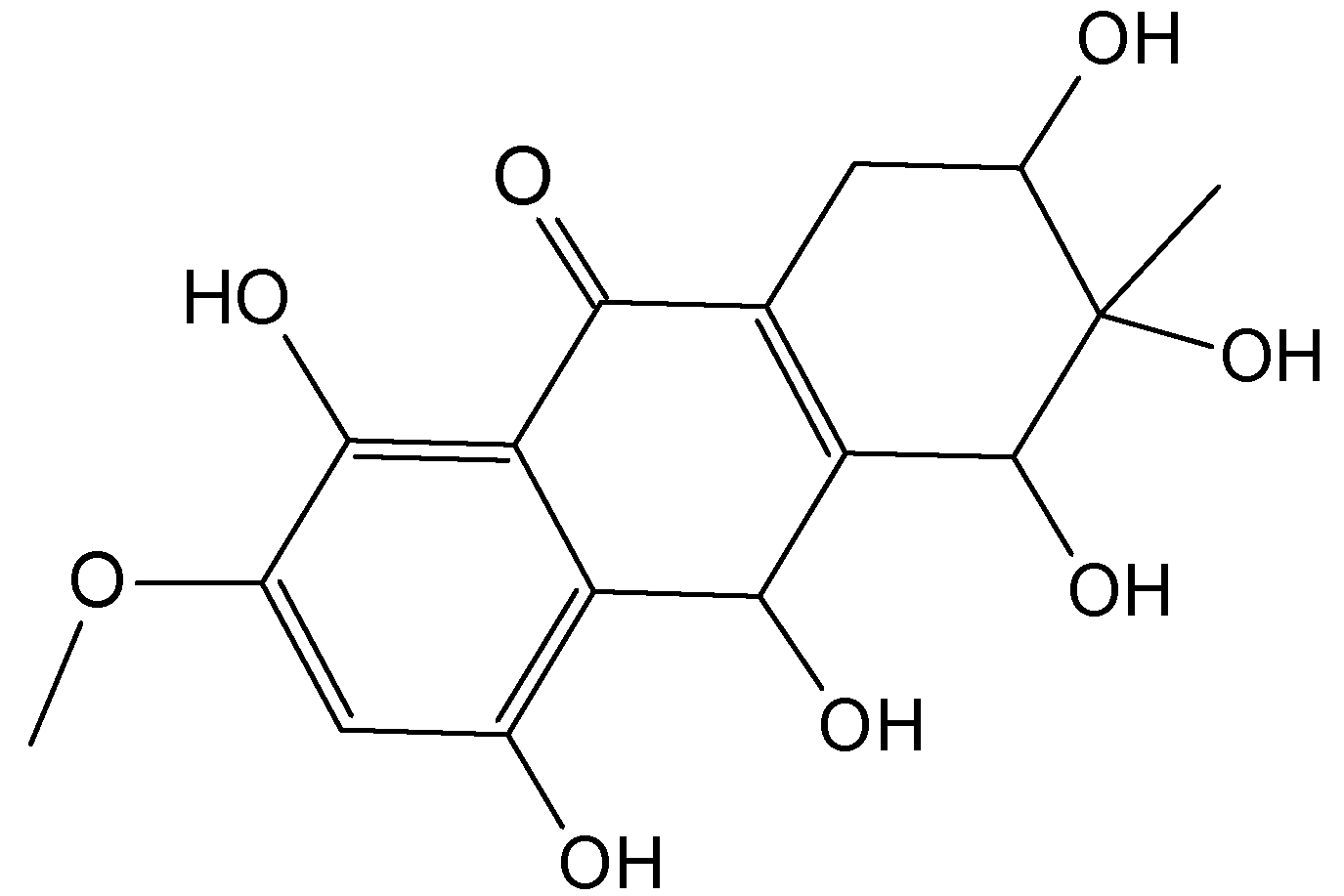
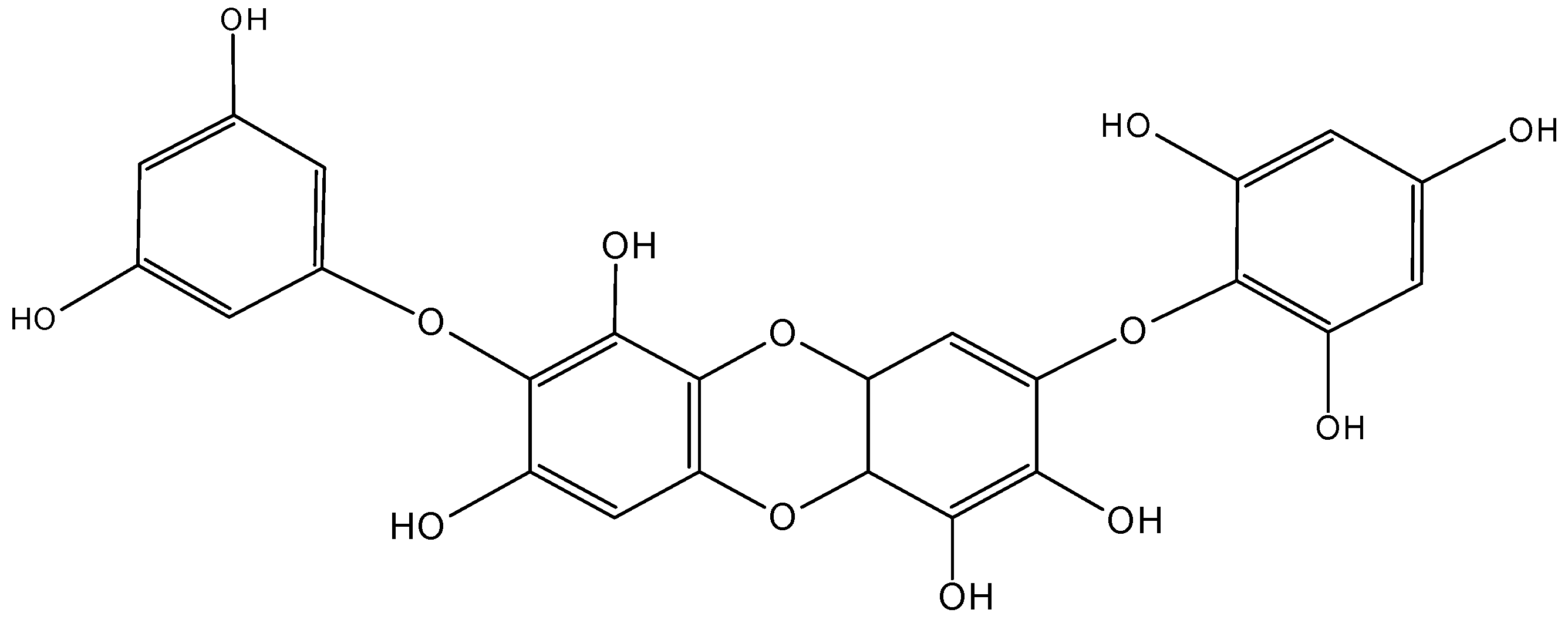
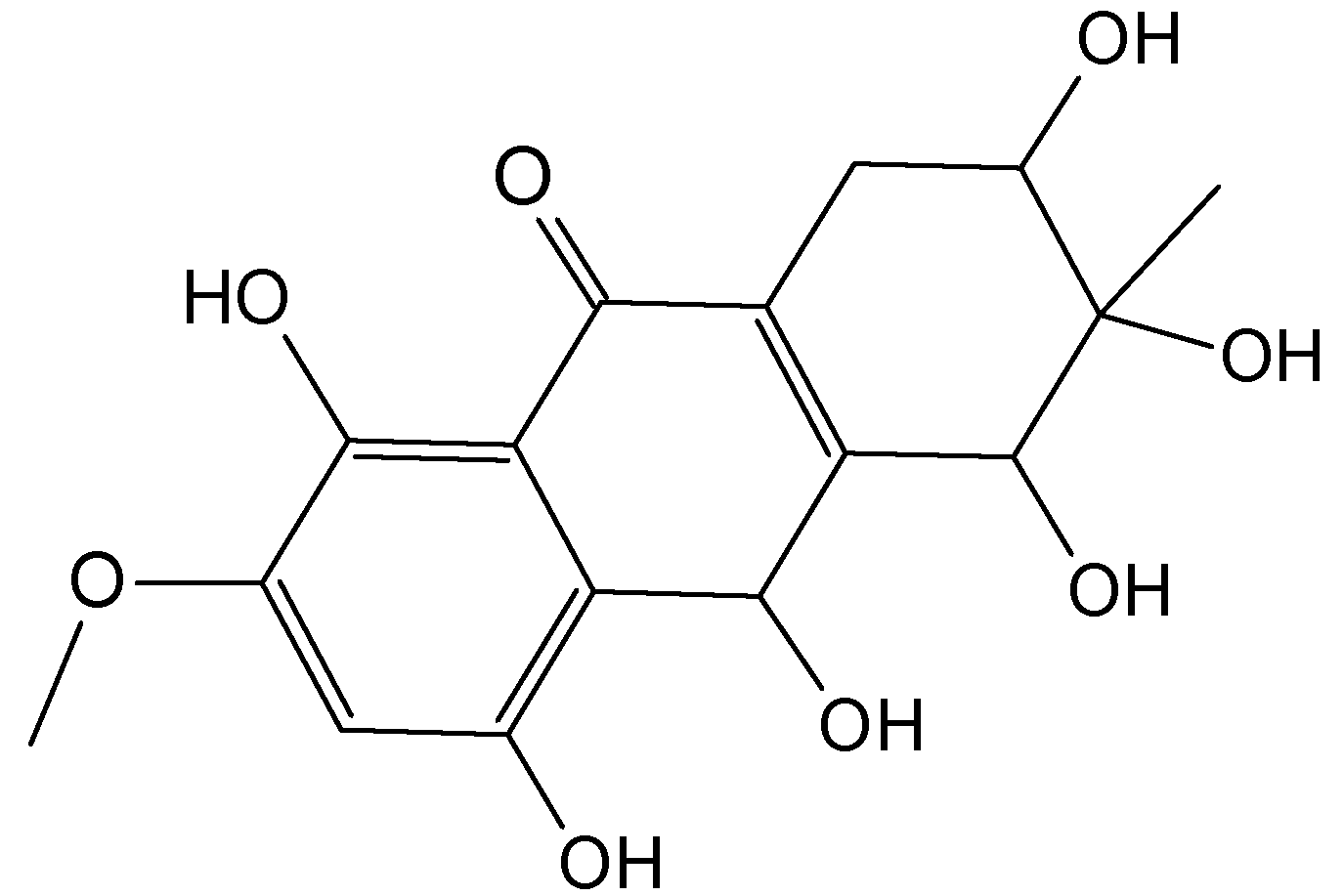
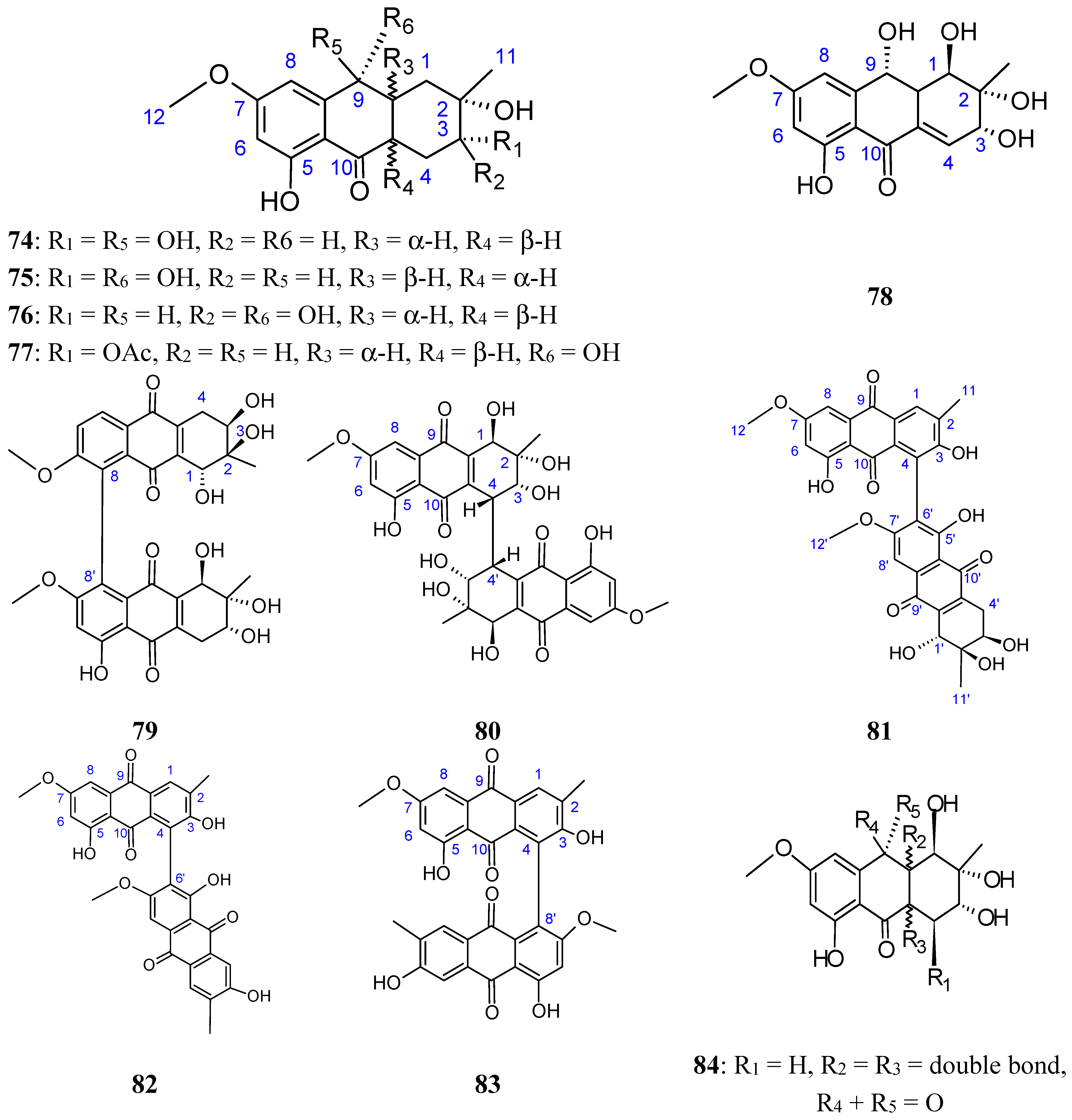
2.7.3. Hydroanthraquinones and Anthraquinones Dimers
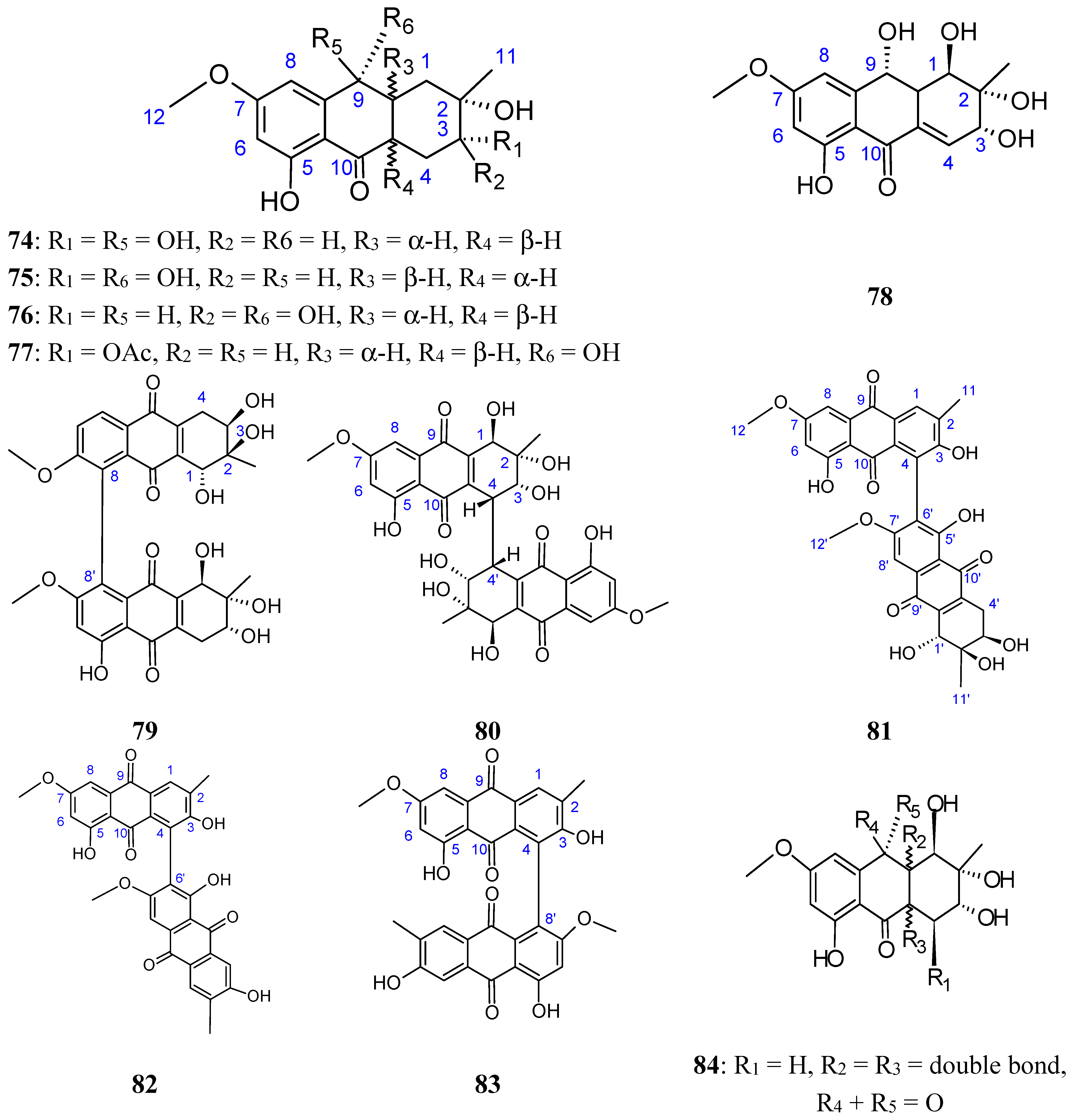
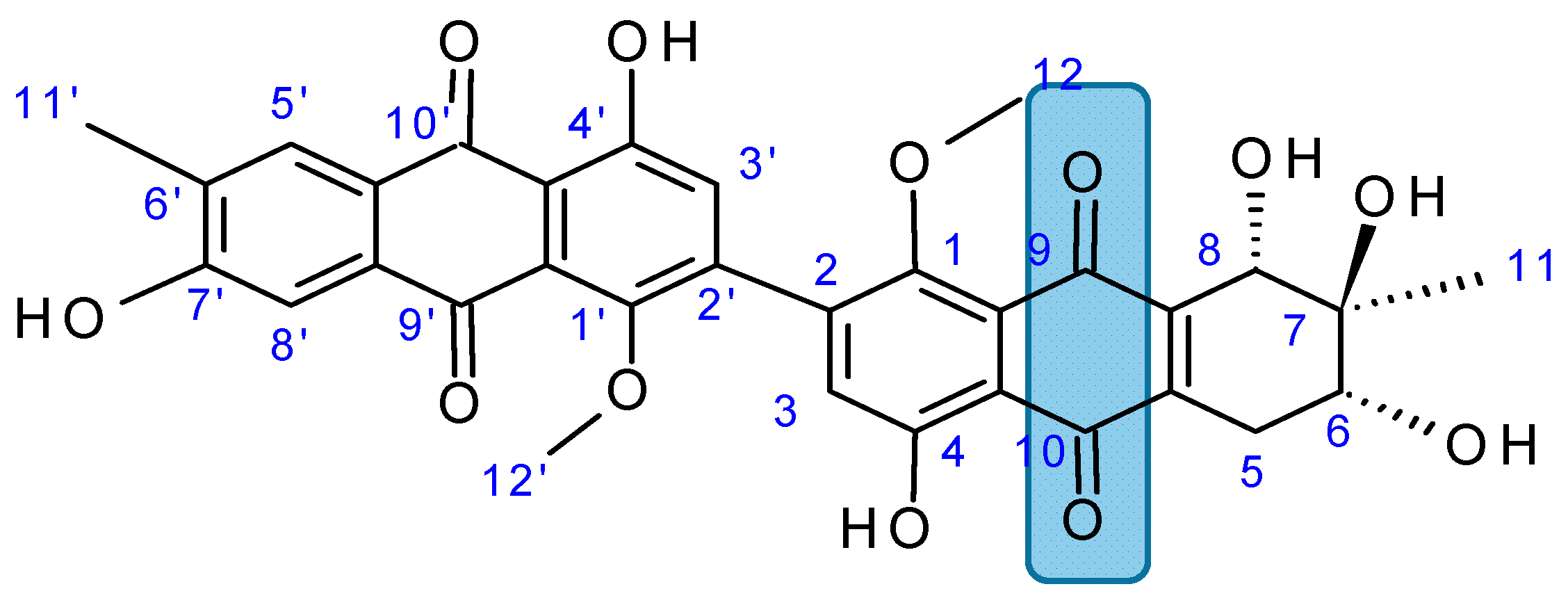
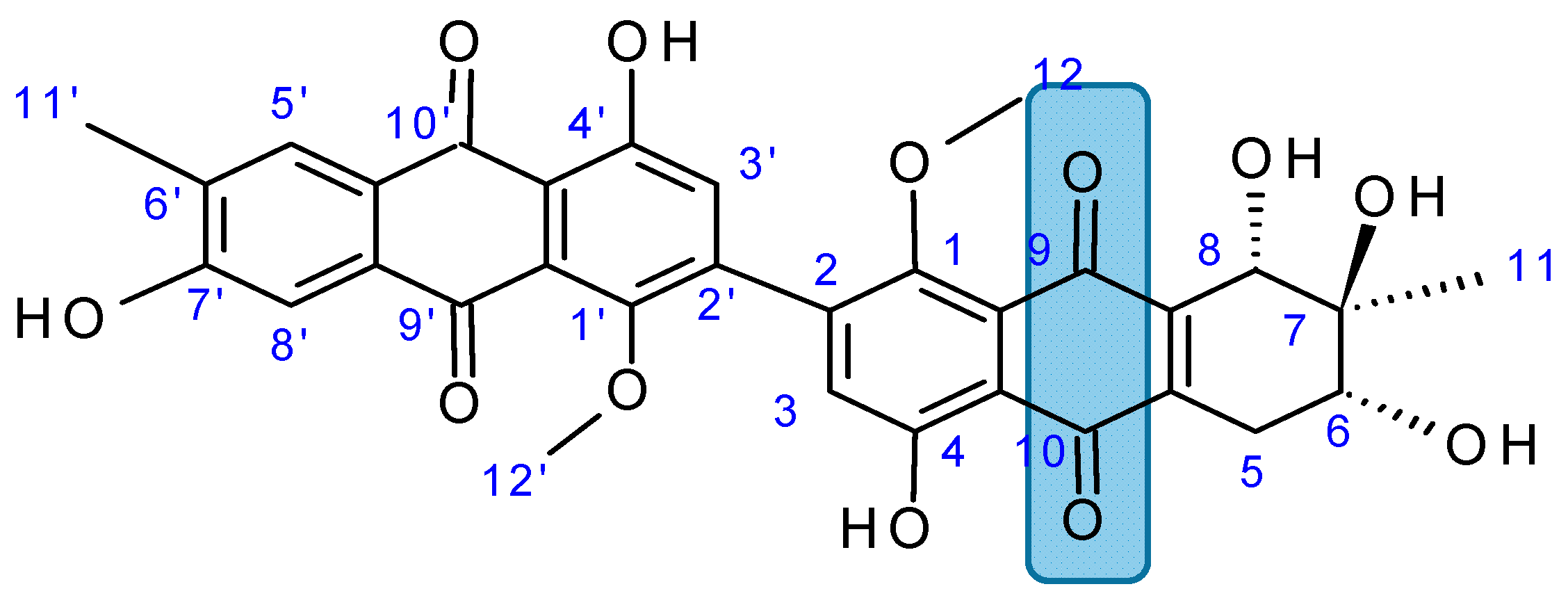
2.7.4. Alterporriol L
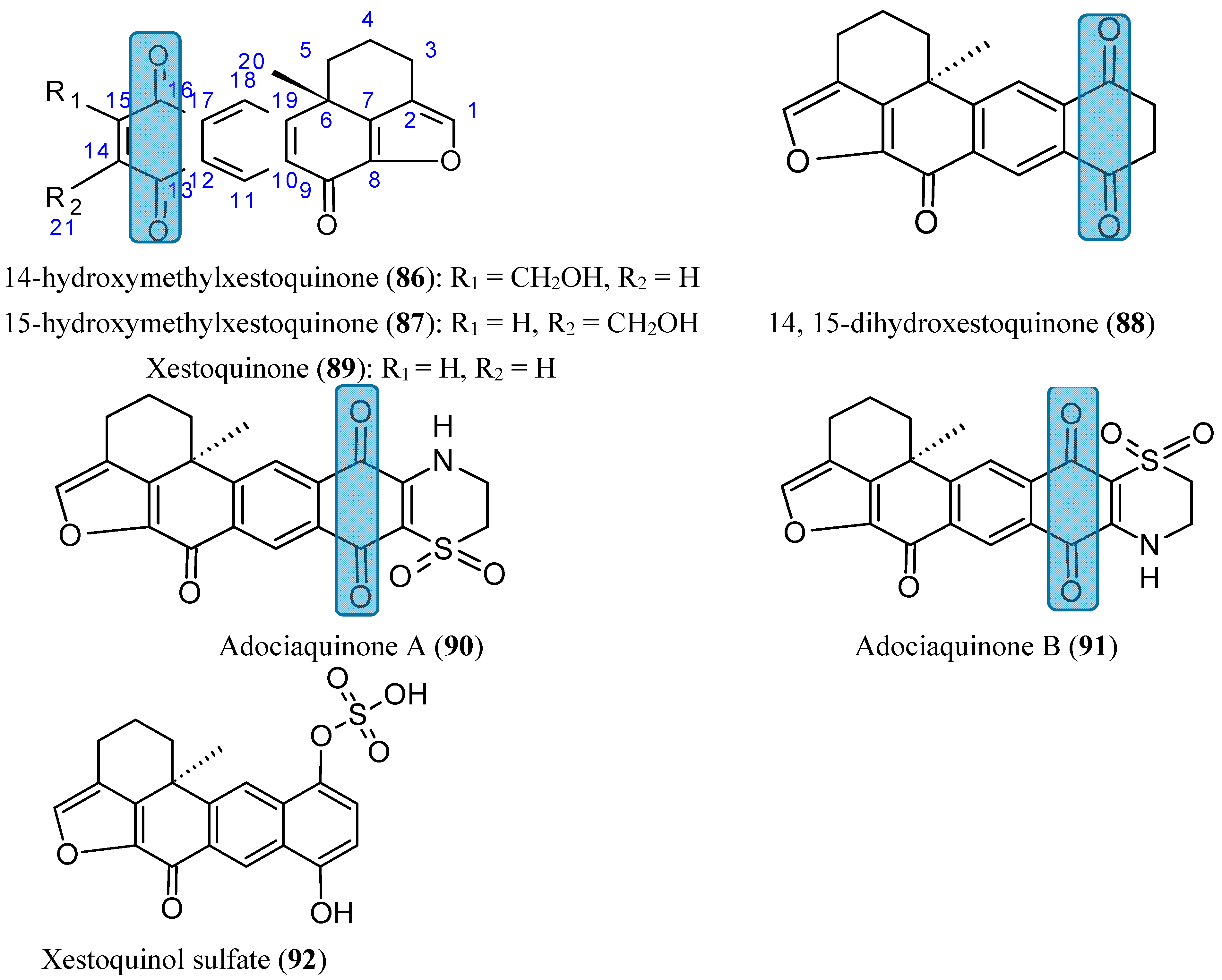
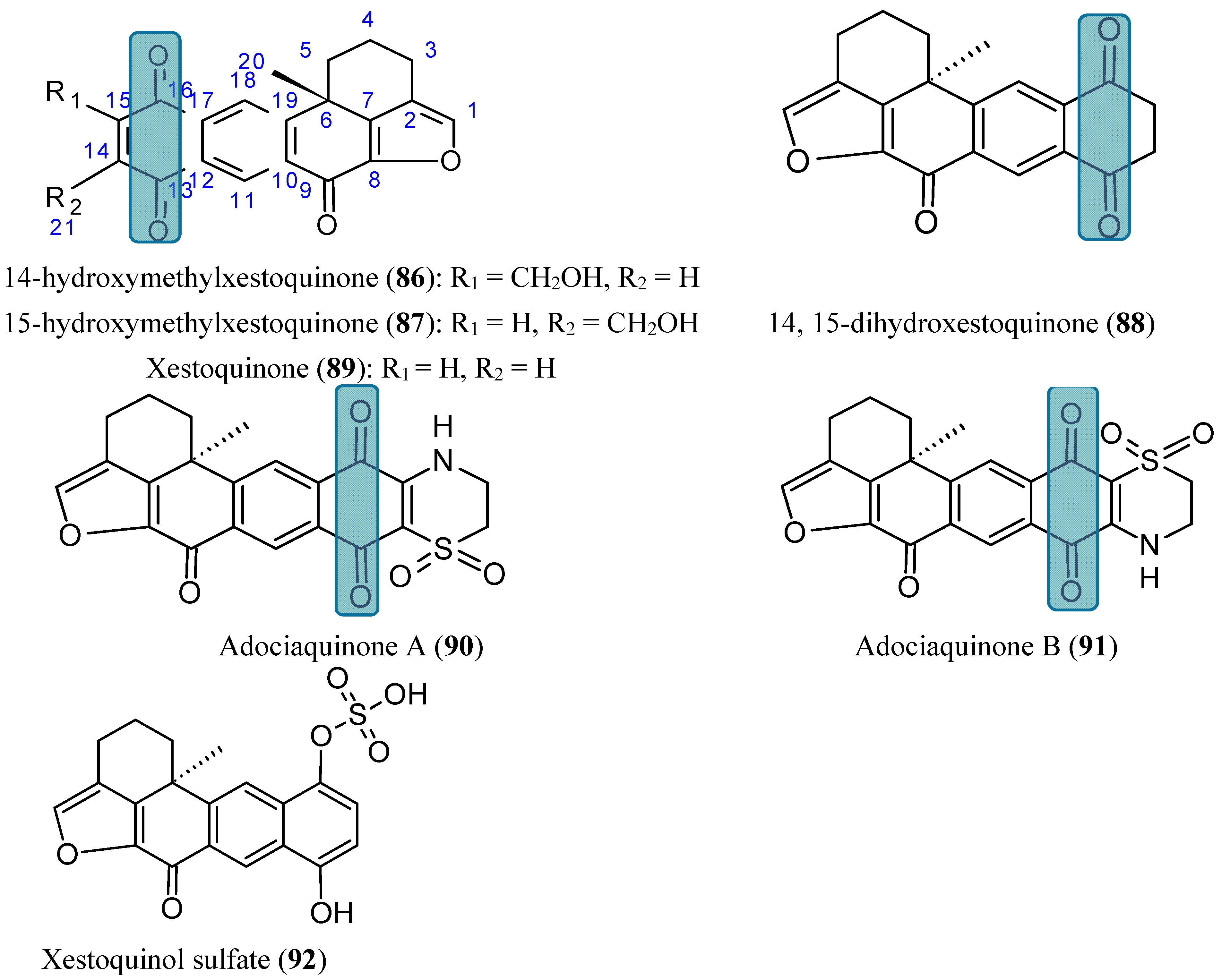
2.7.5. Xestoquinones
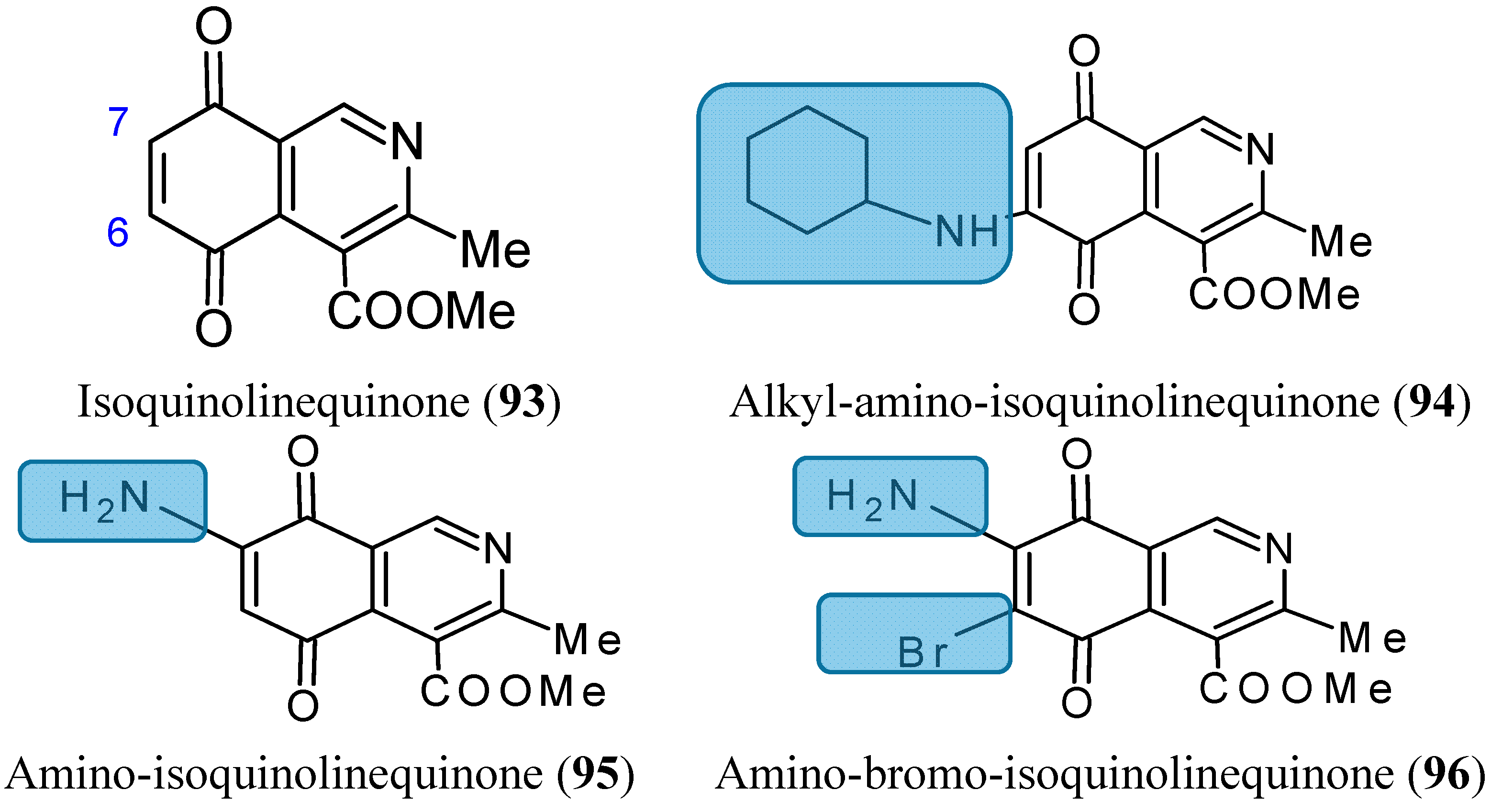
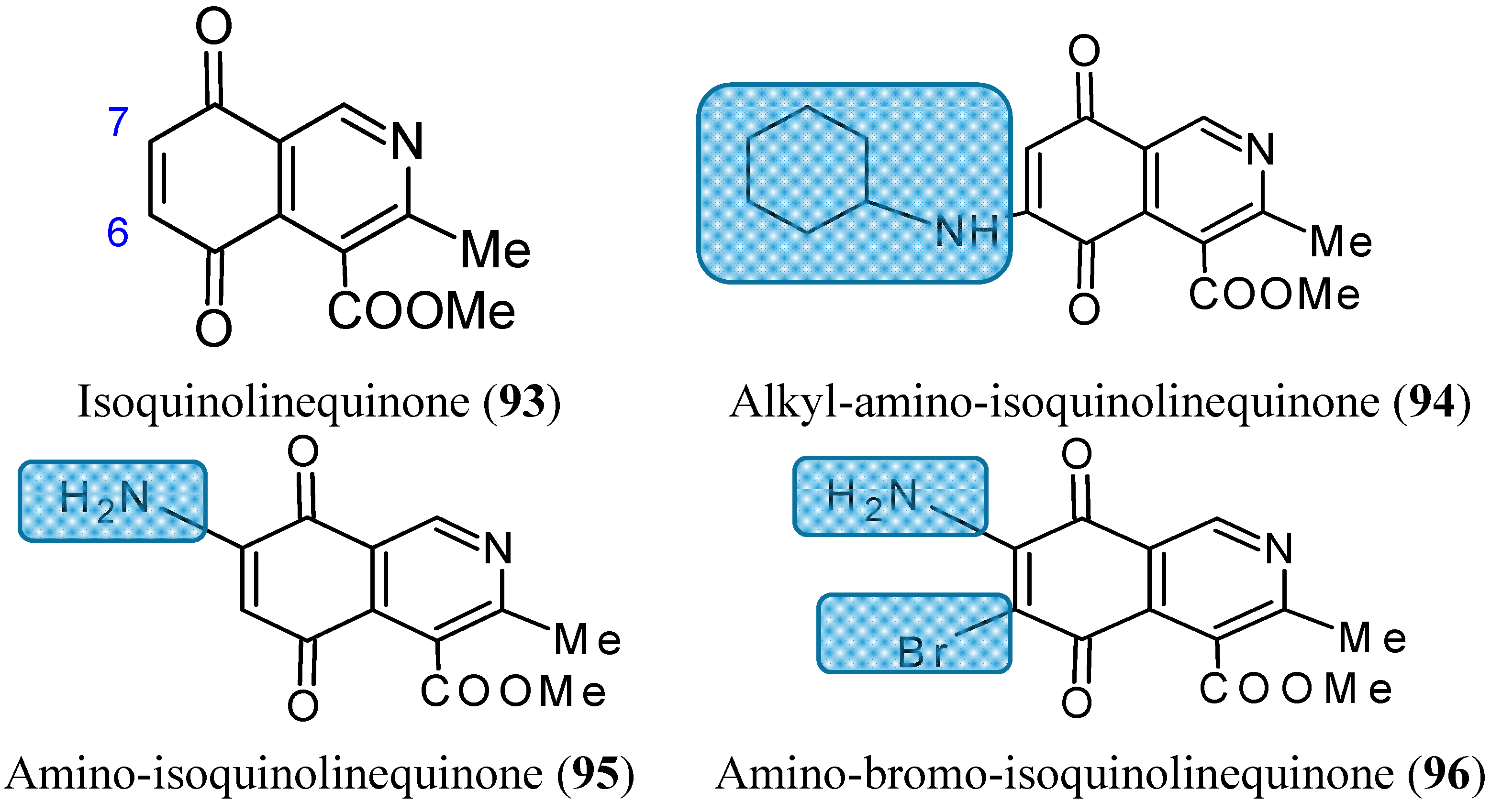
2.7.6. Aminoquinones
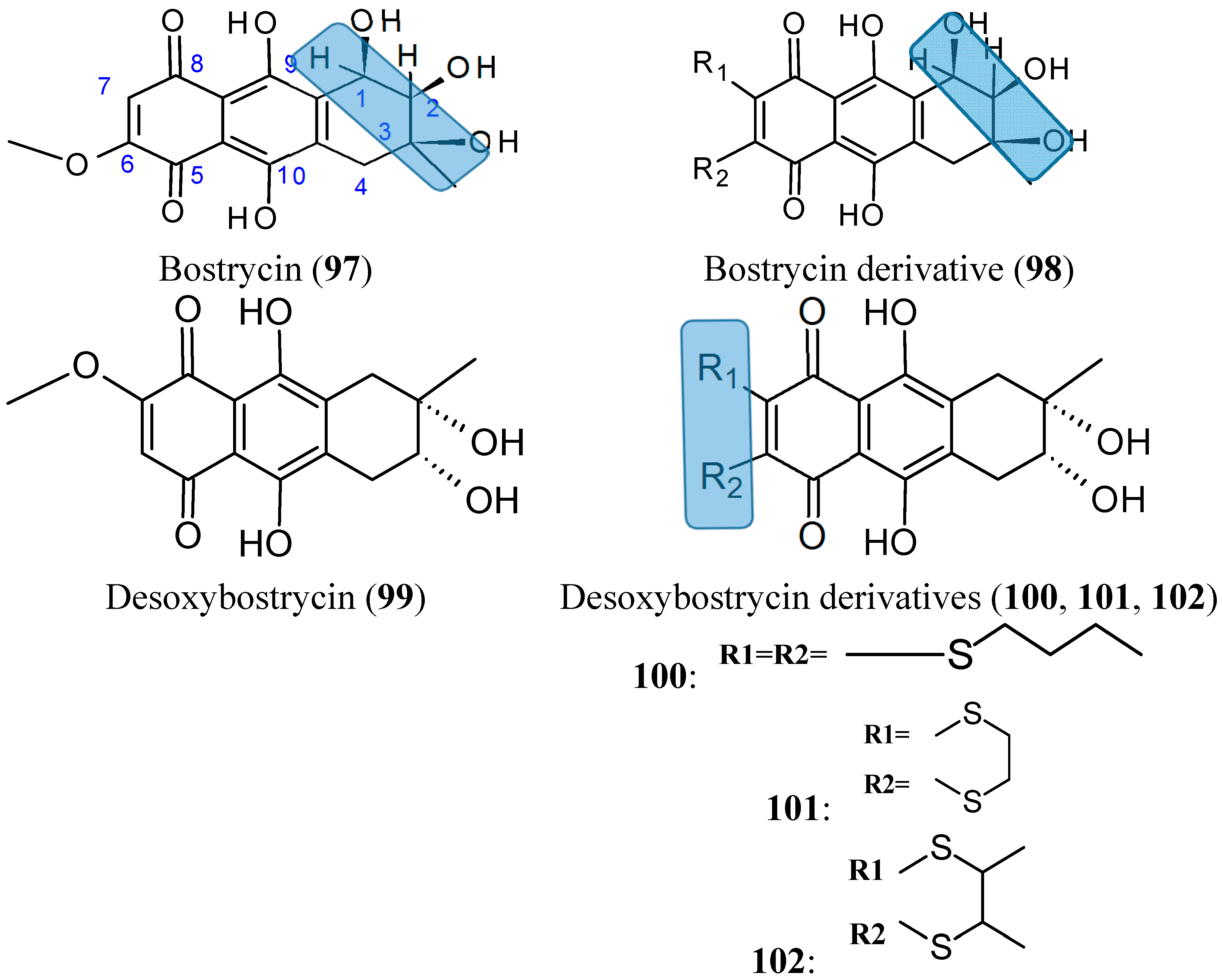
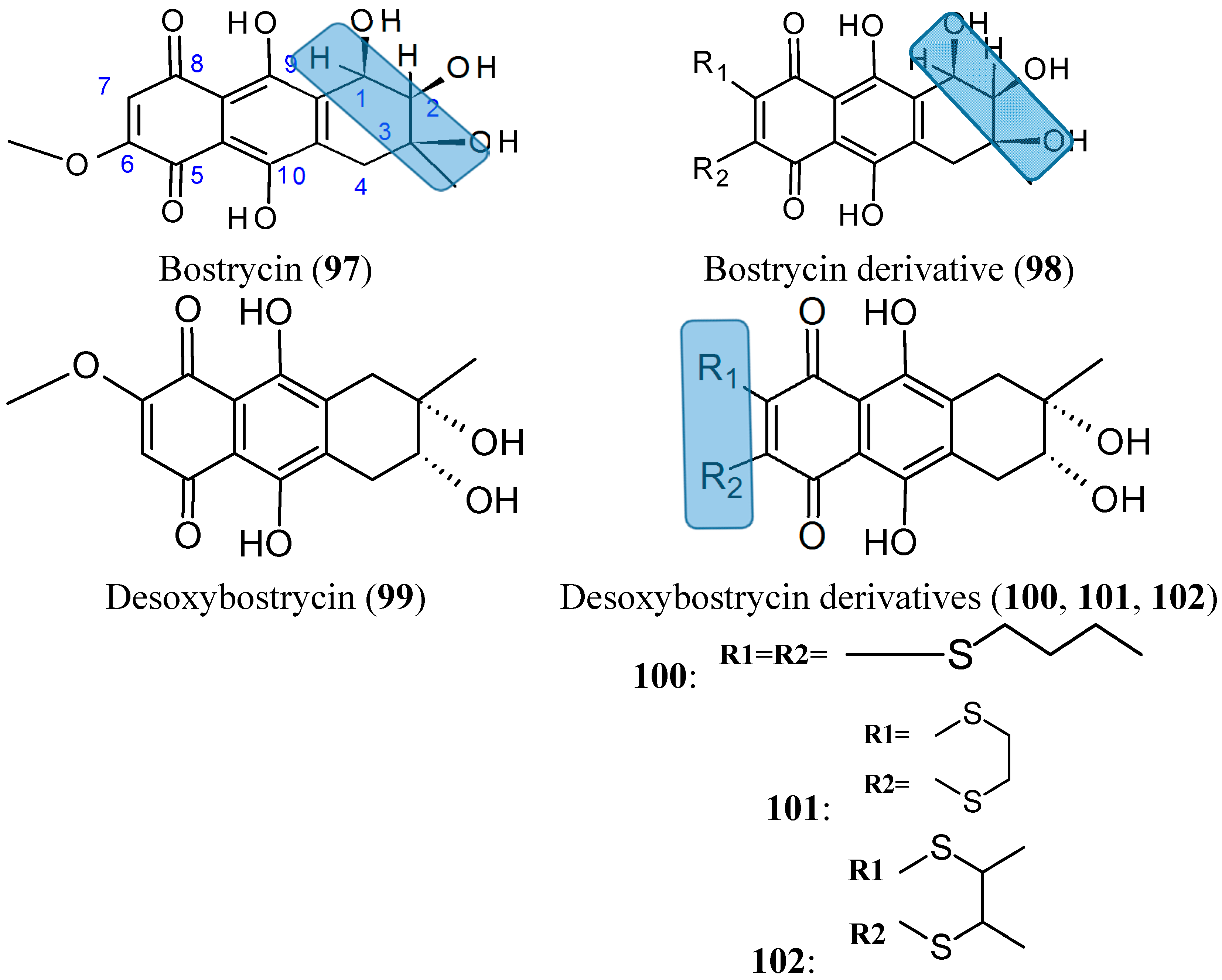
2.7.7. Bostrycin and Deoxybostrycin analogs
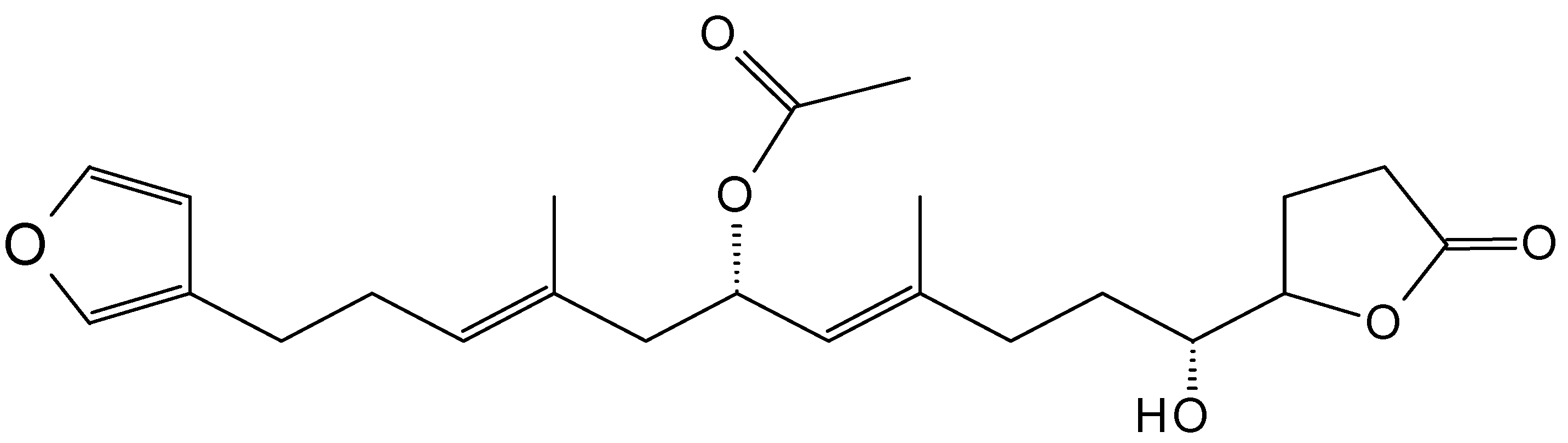
2.8. Sterols and Steroids
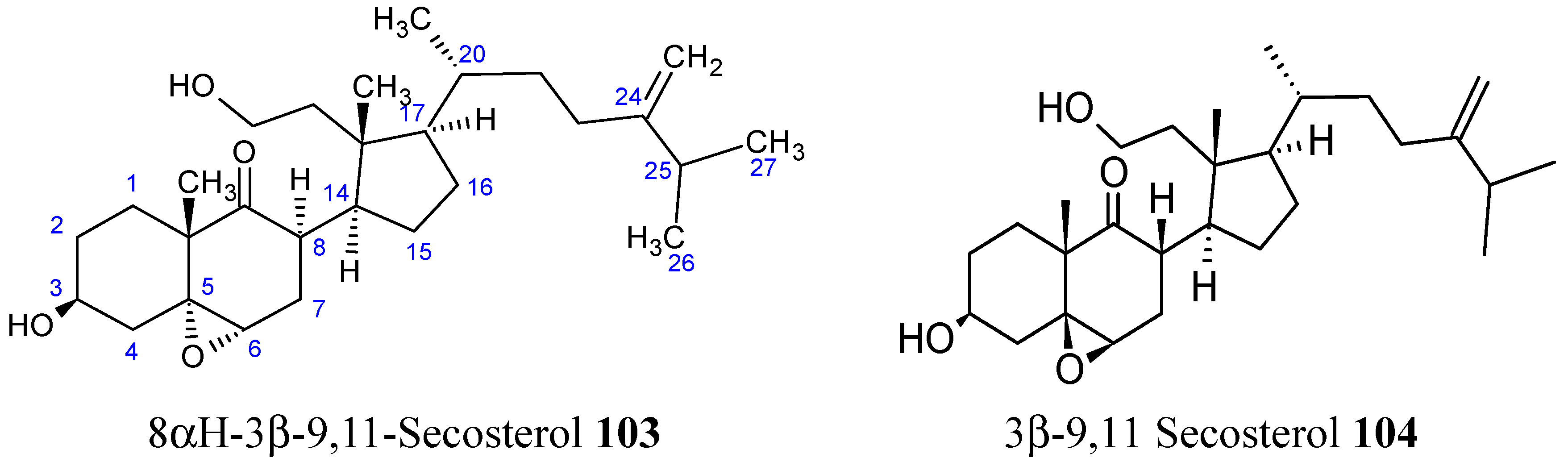
2.8.1. 9,11-Secosterol
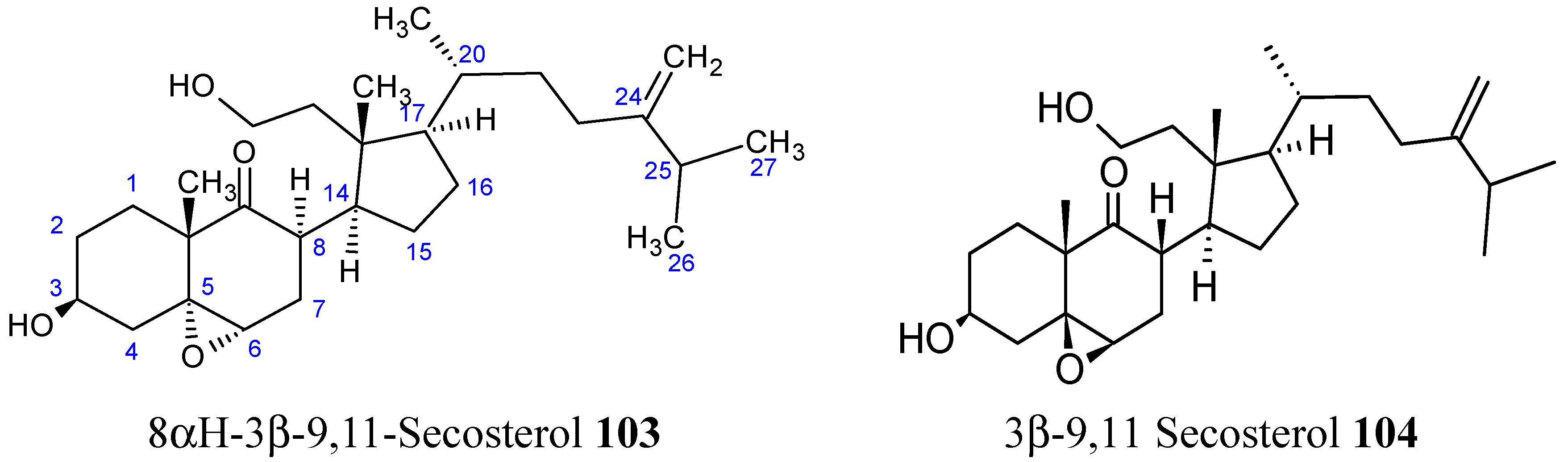
2.8.2. Polyoxygenated Steroids
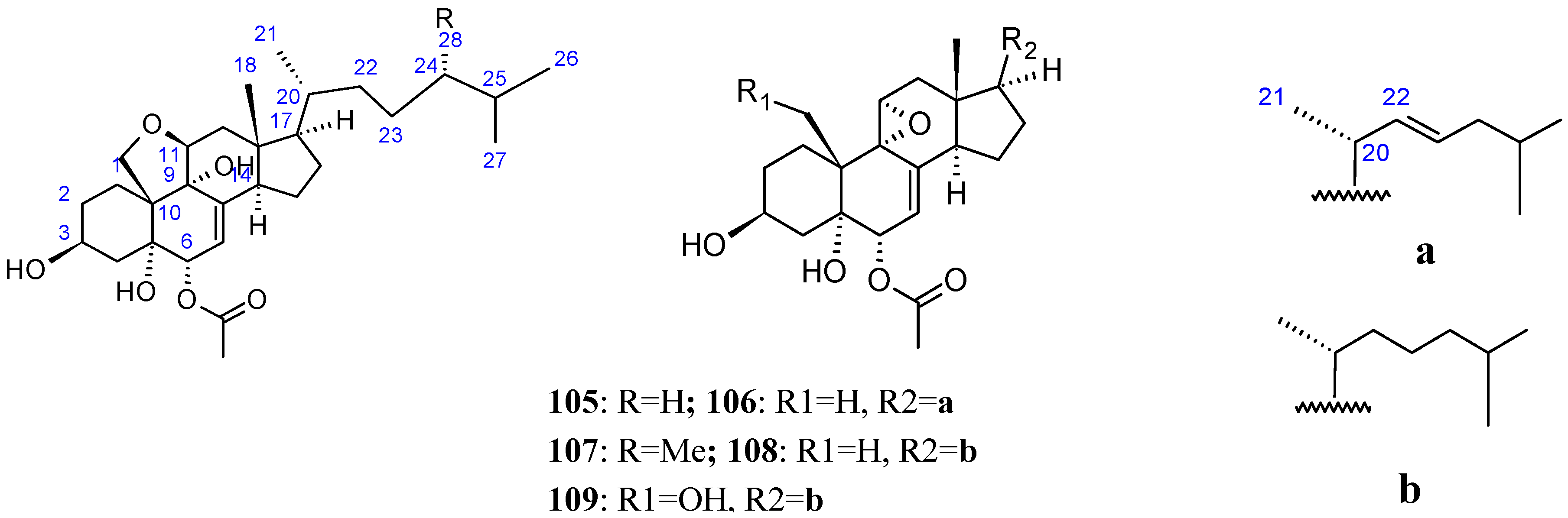
2.8.3. 11-Dehydrosinulariolide
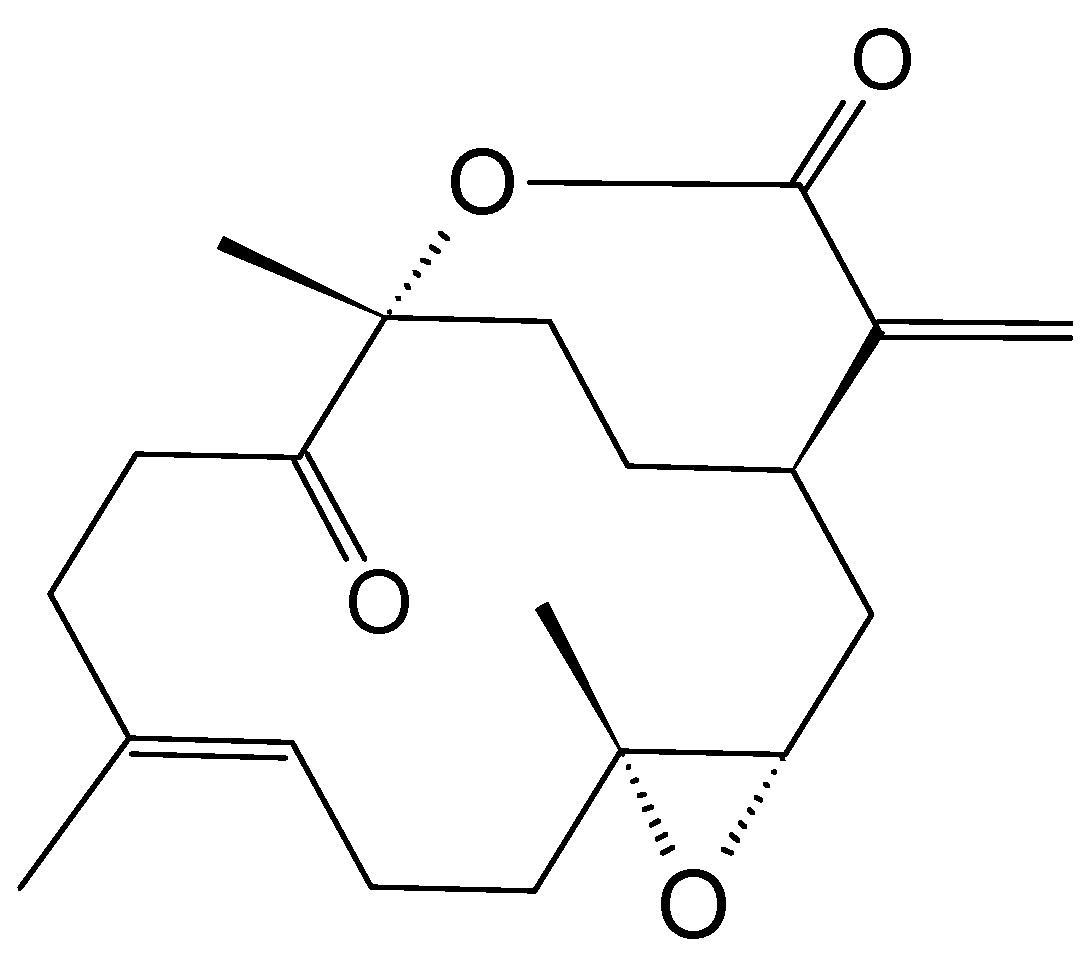
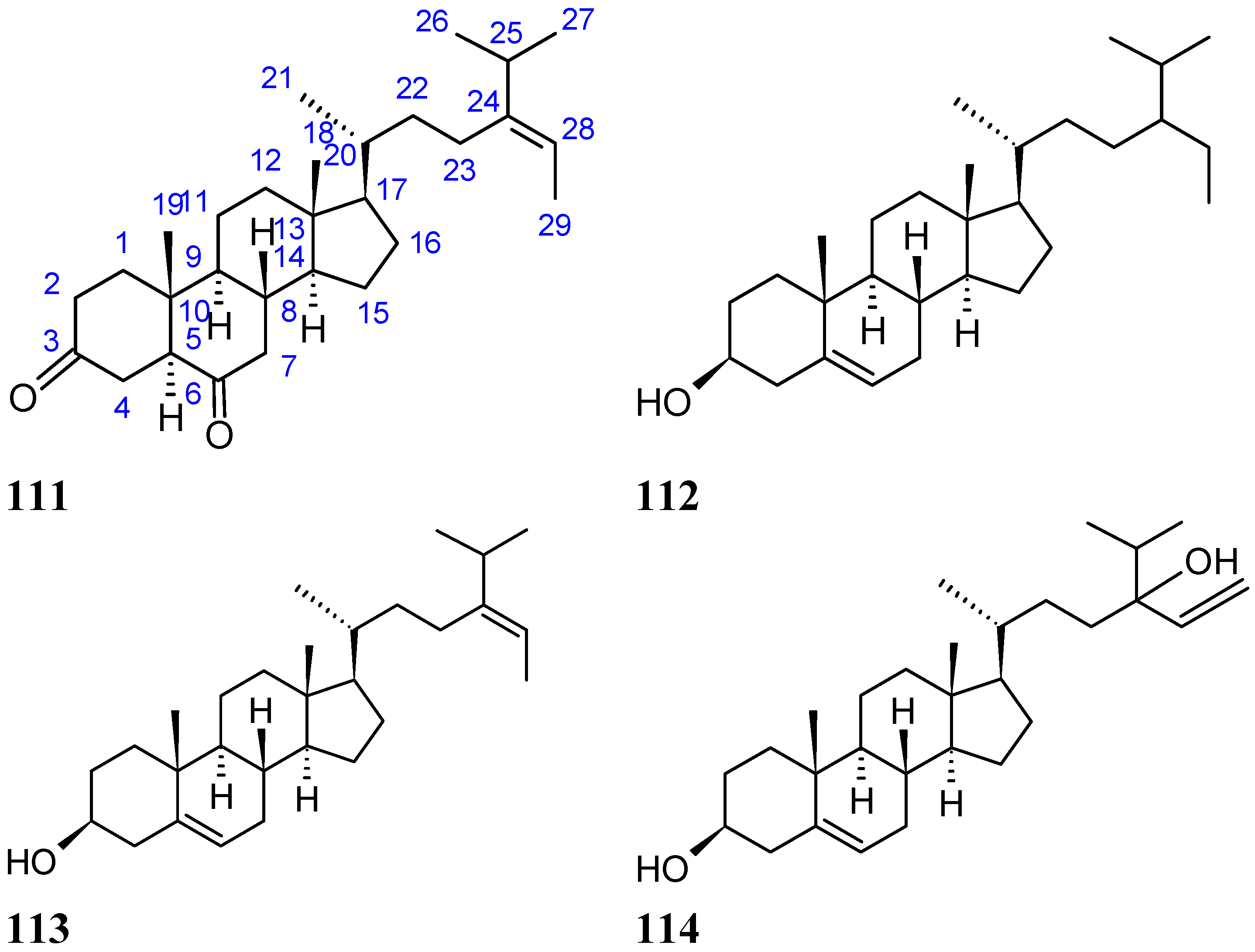
2.8.4. Diketosteroid (E)-Stigmasta-24(28)-en-3,6-dione
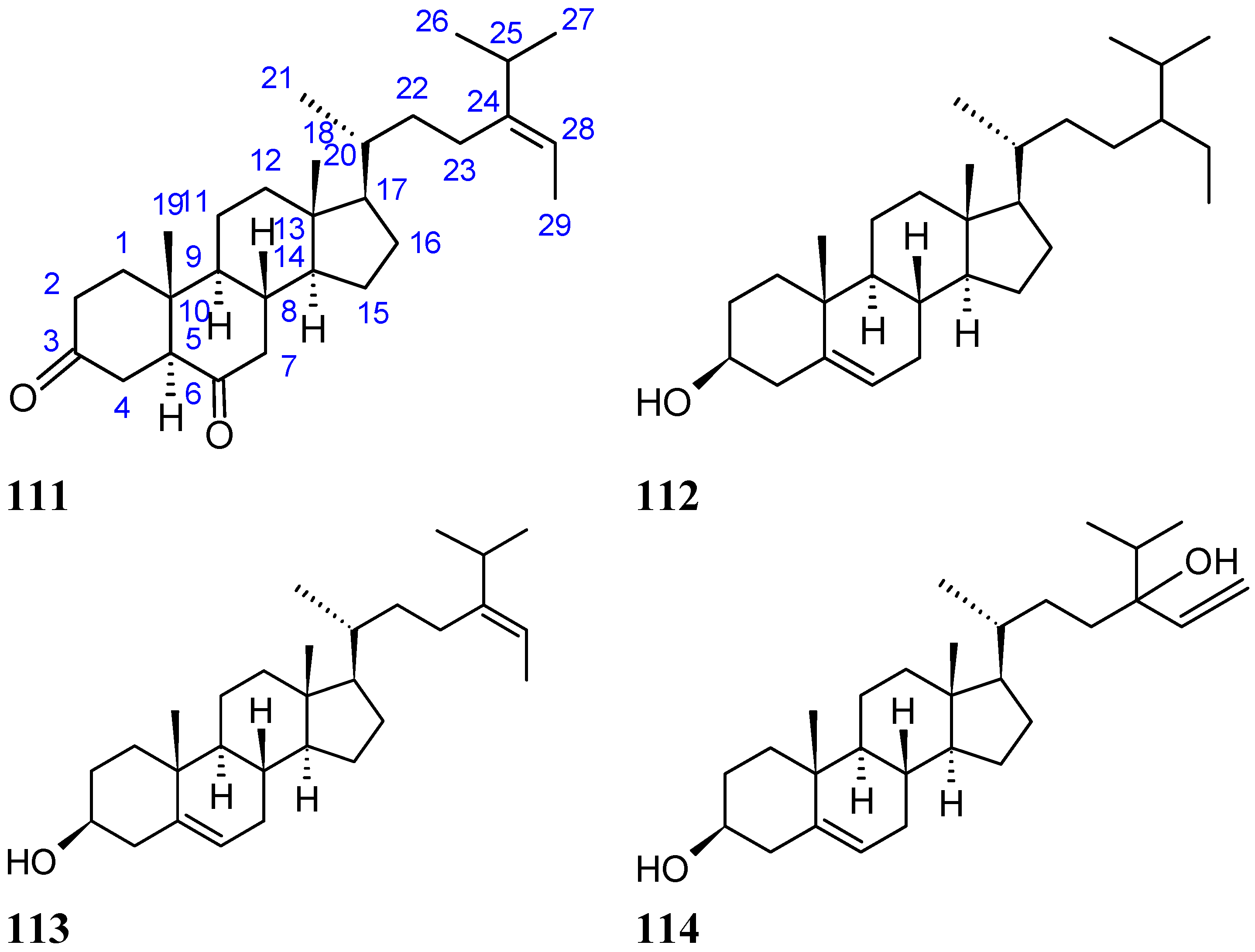
2.9. Terpenes and Terpenoids
2.9.1. 10-Acetylirciformonin B
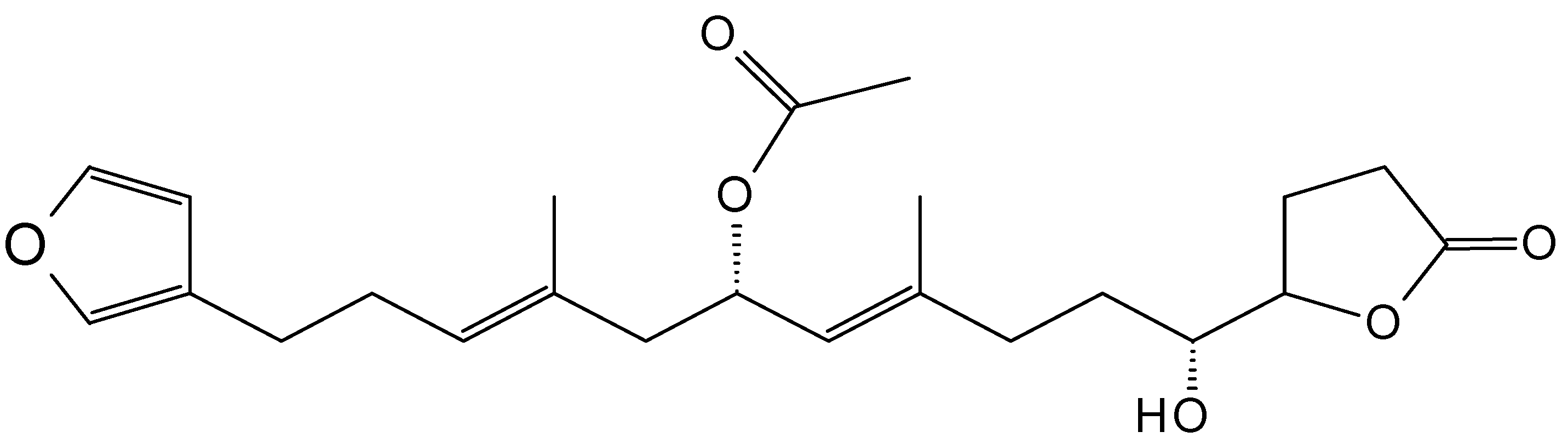
2.9.2. Phenazine Analogs
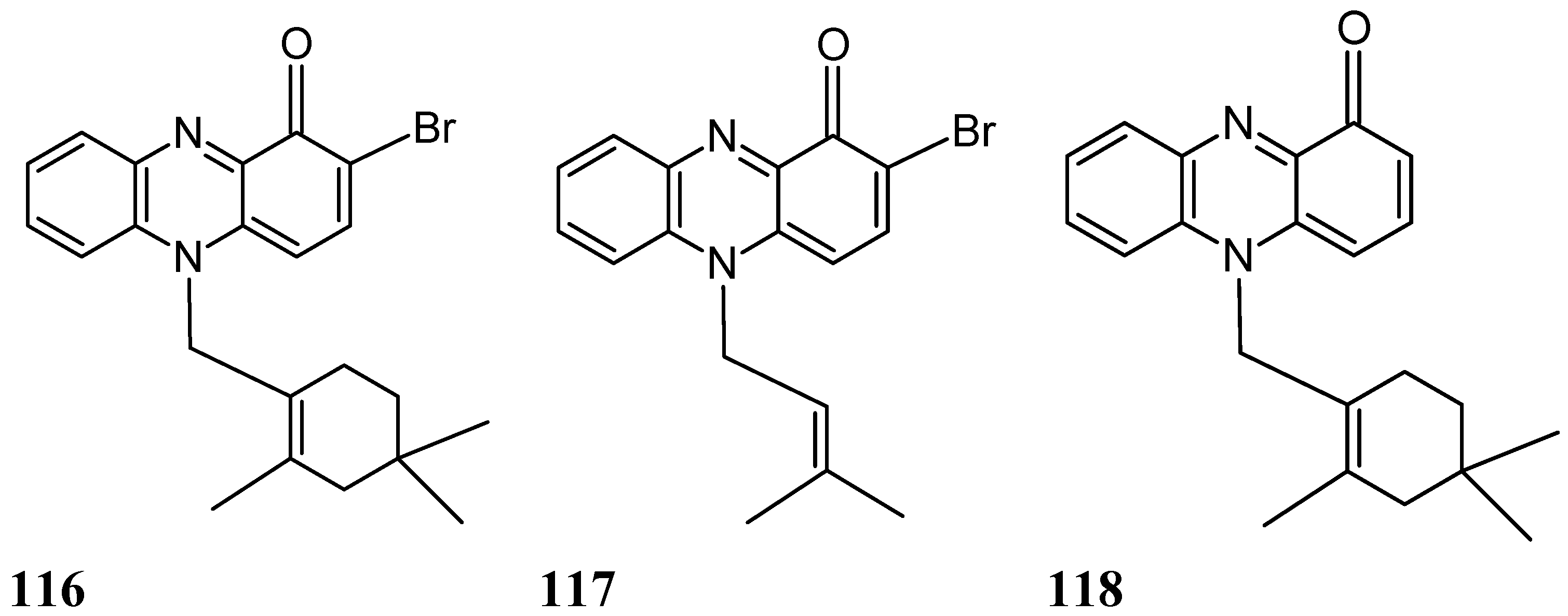
2.9.3. Diterpenoid Compounds
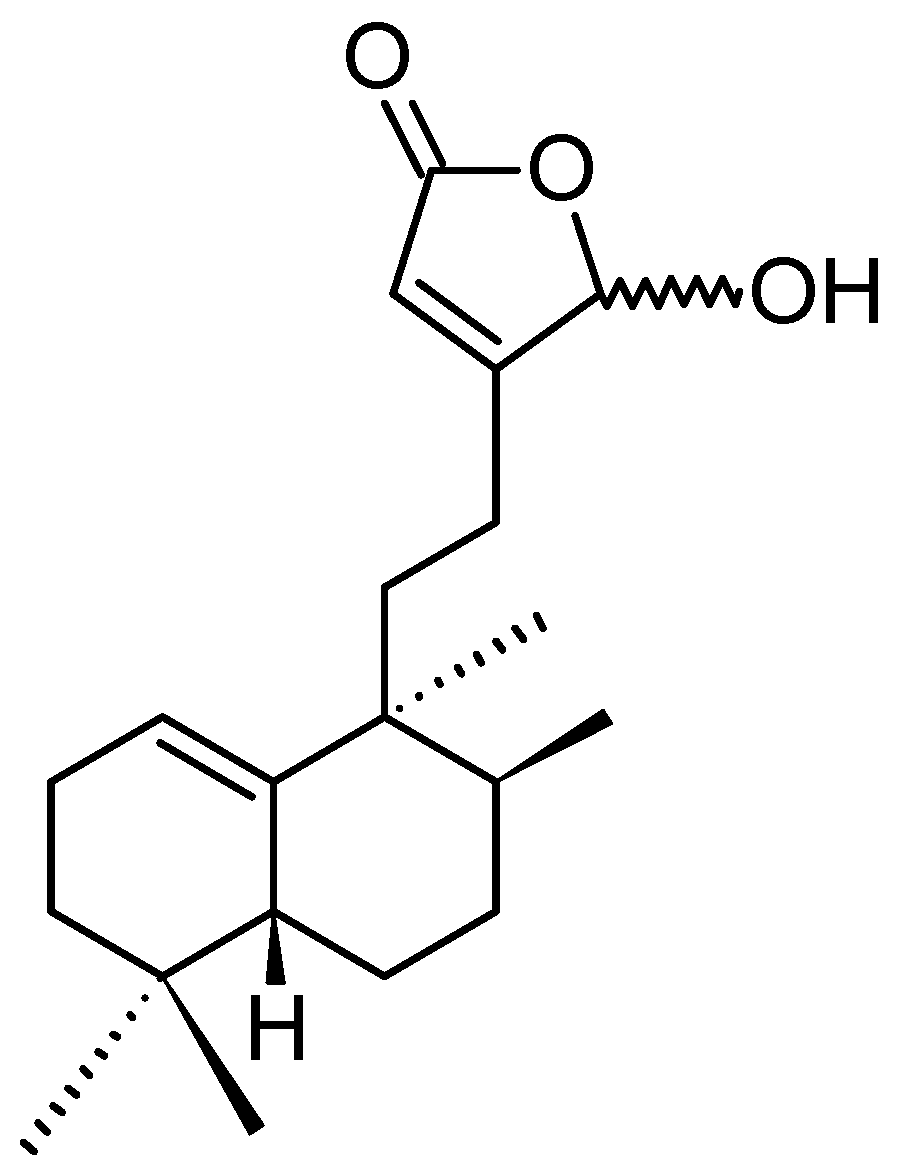
Echinohalimane A
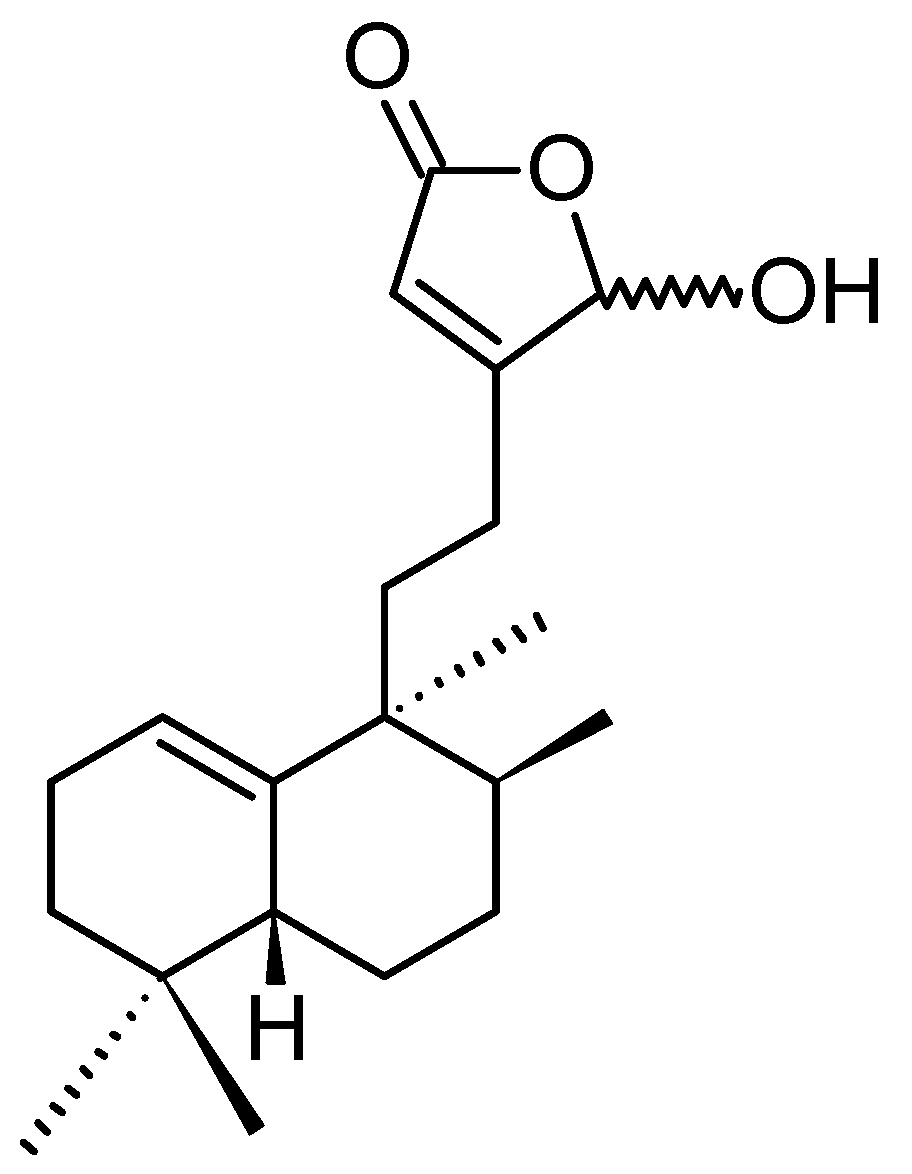
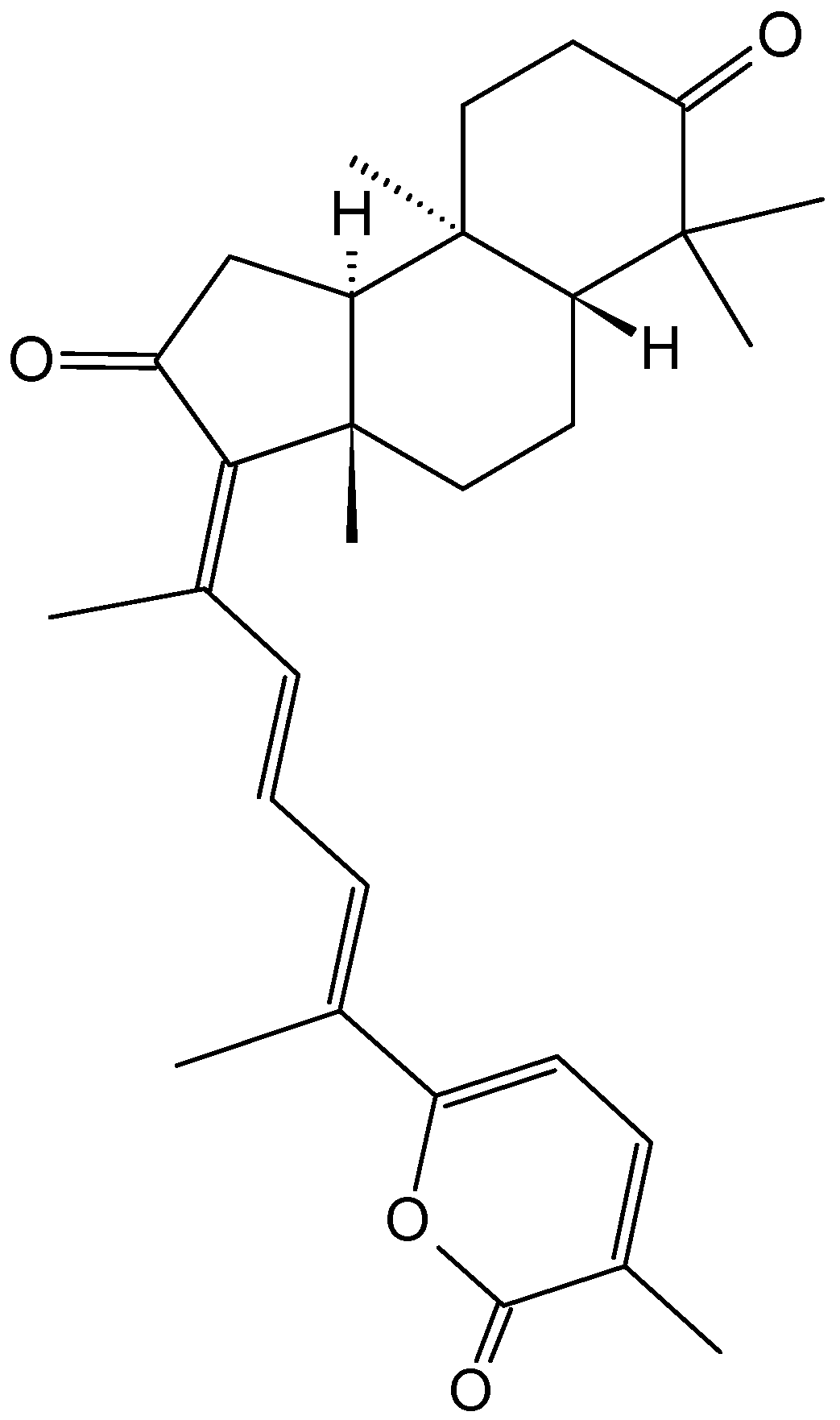
(1S,2S,3E,7E,11E)-3,7,11,15-Cembratetraen-17,2-olide
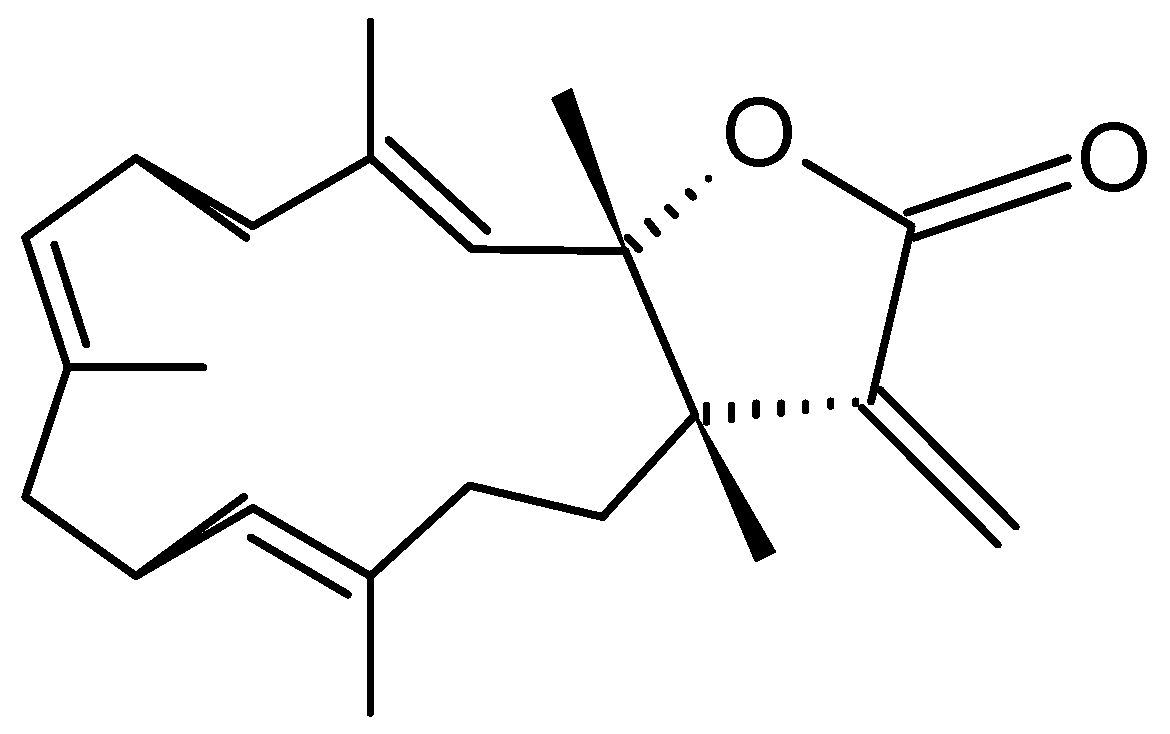
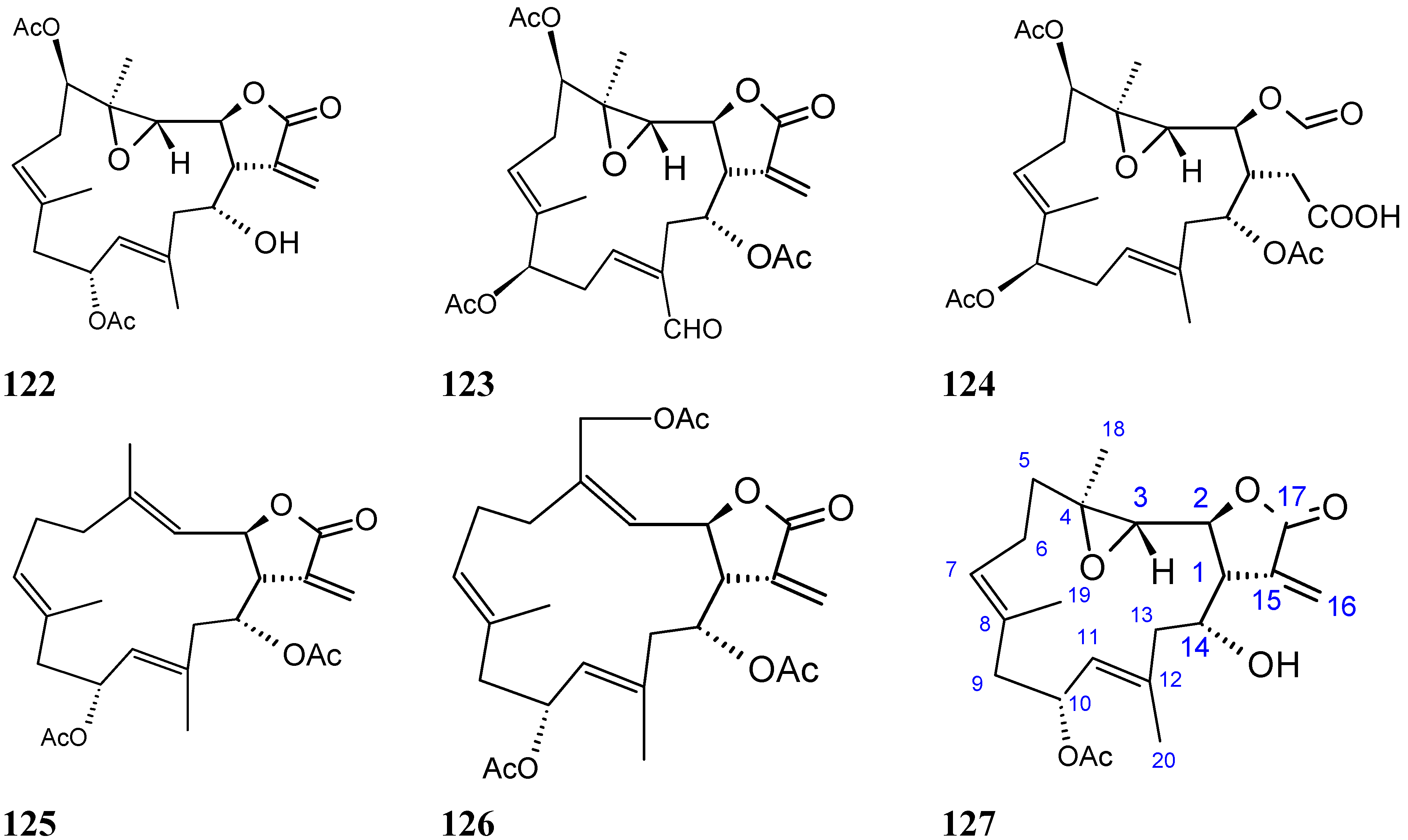
Michaolides
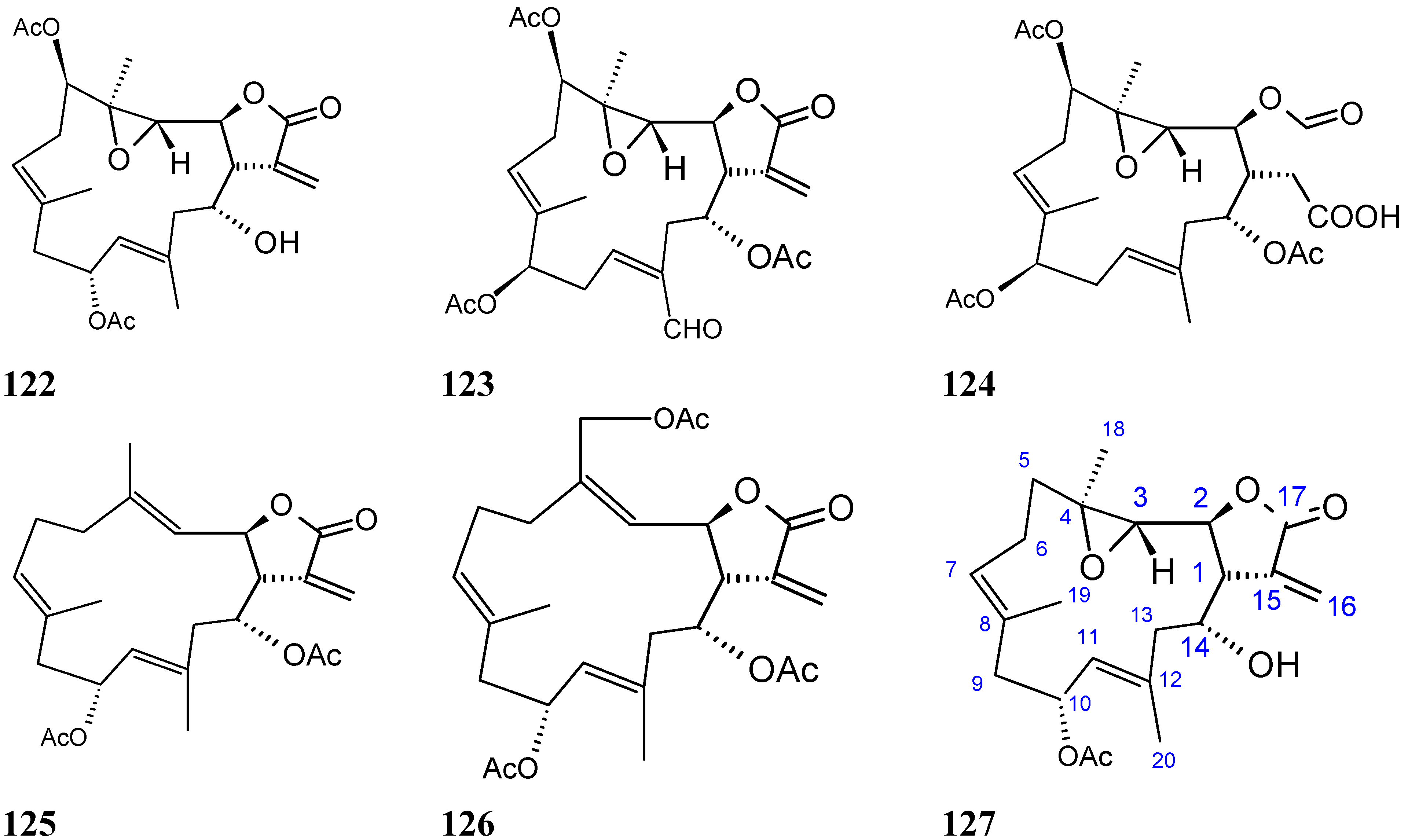
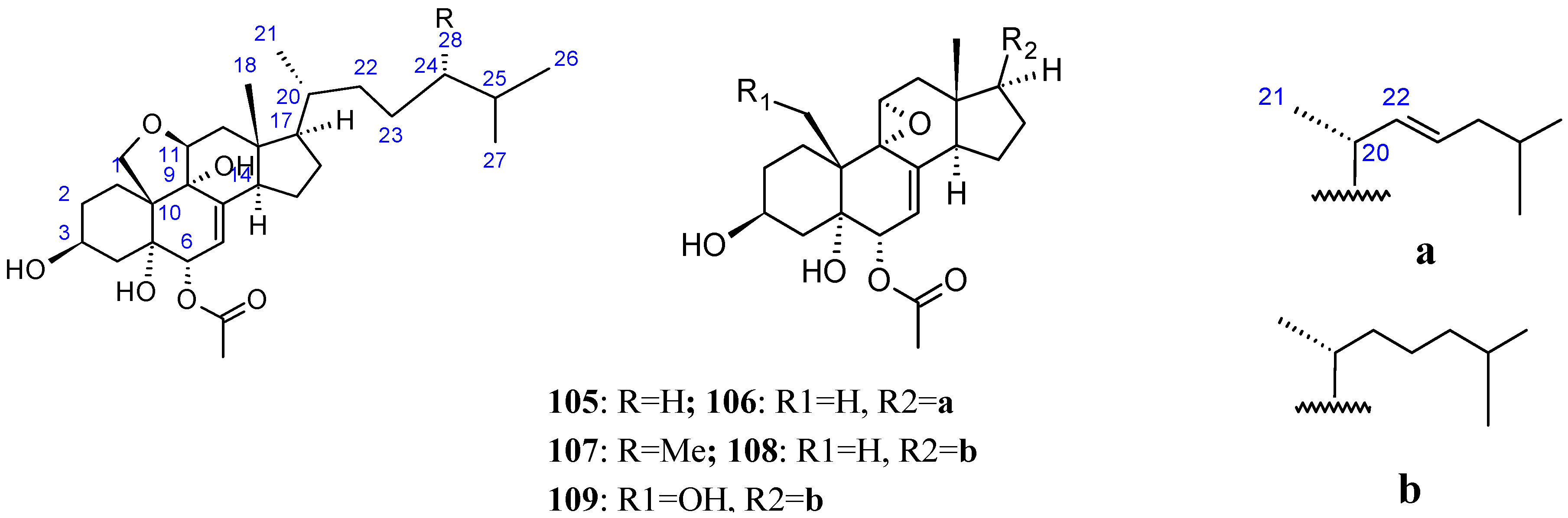
2.9.4. Triterpenoid Compounds
Stellettin A
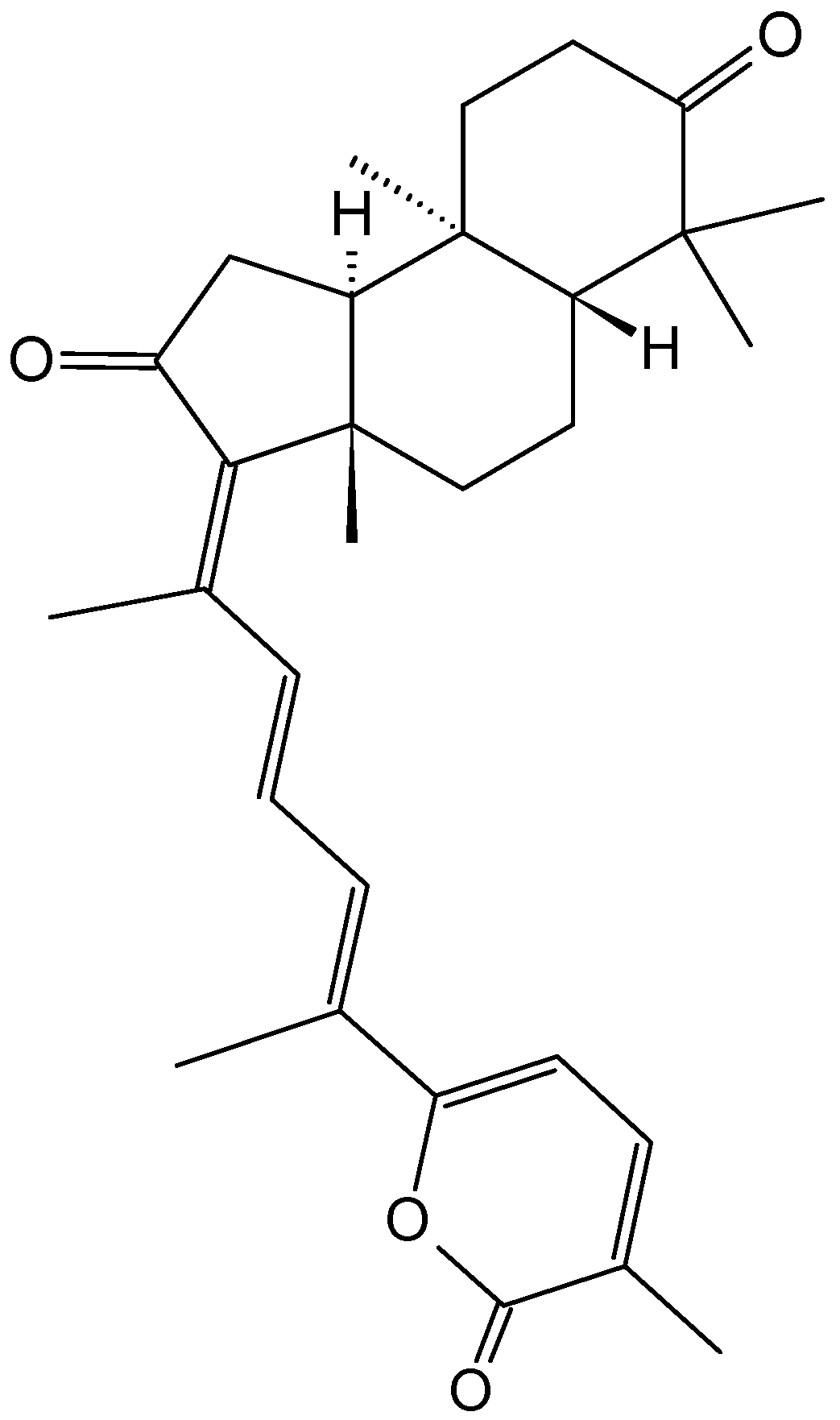
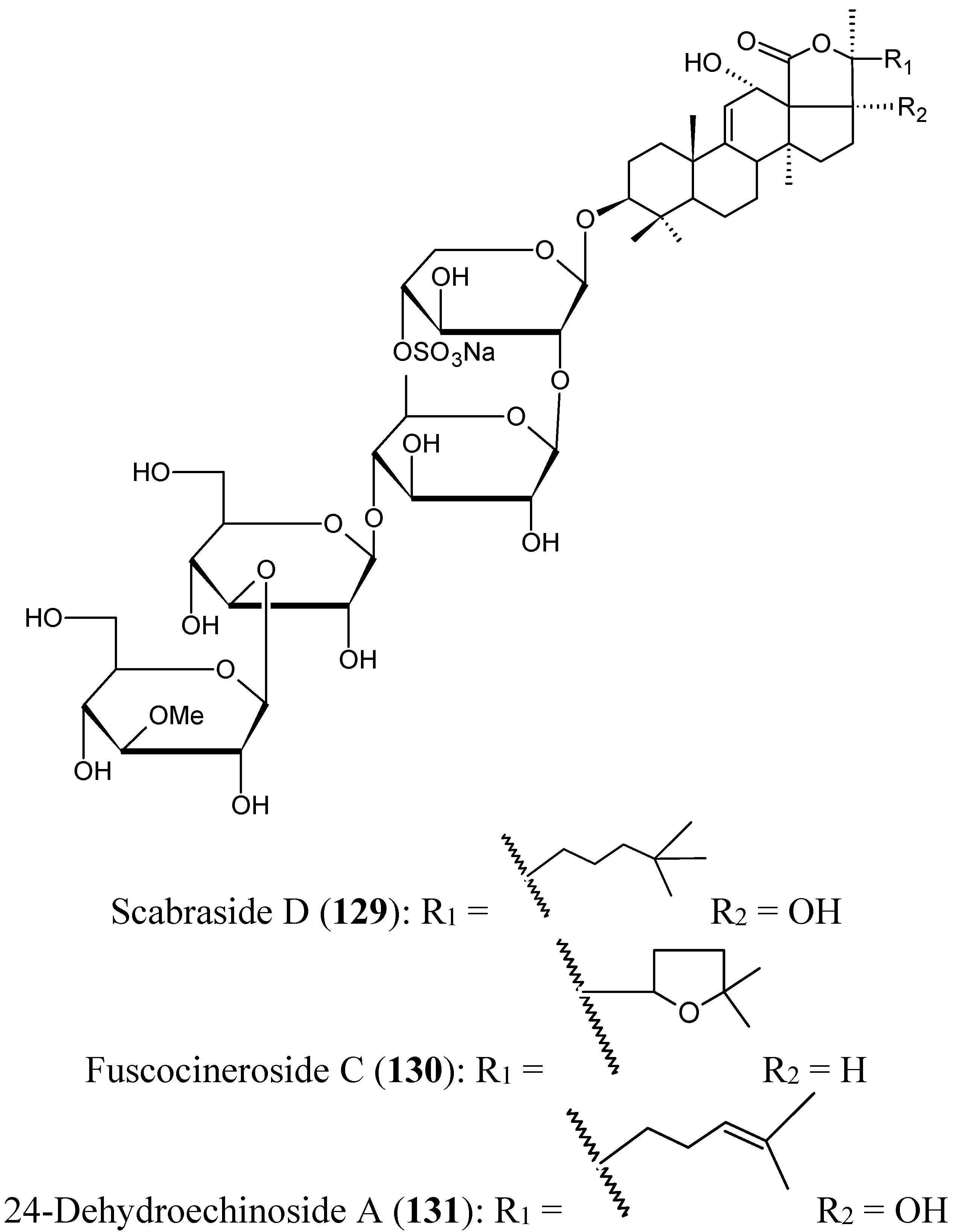
Triterpene glycosides
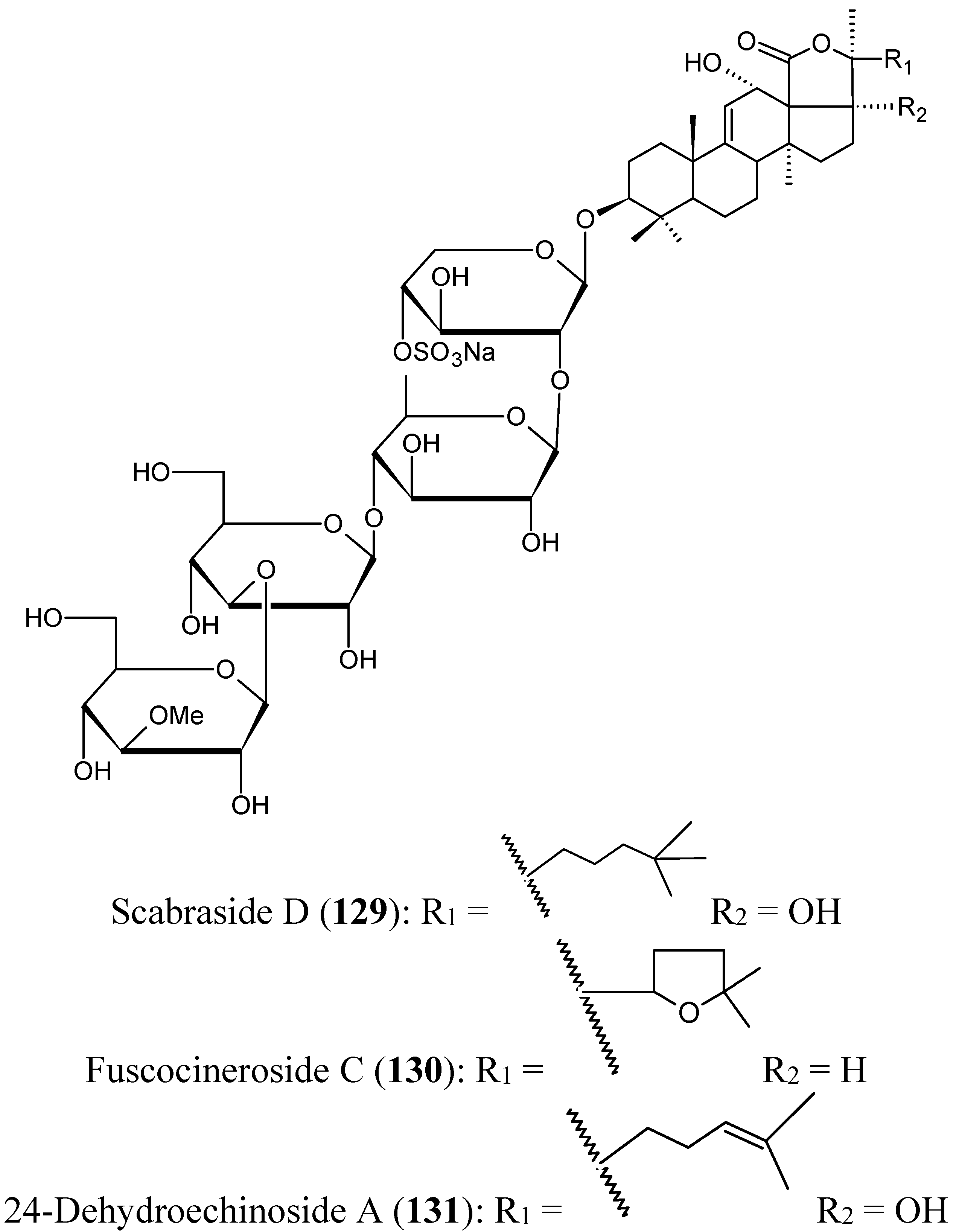
2.9.5. Sesquiterpene Compounds
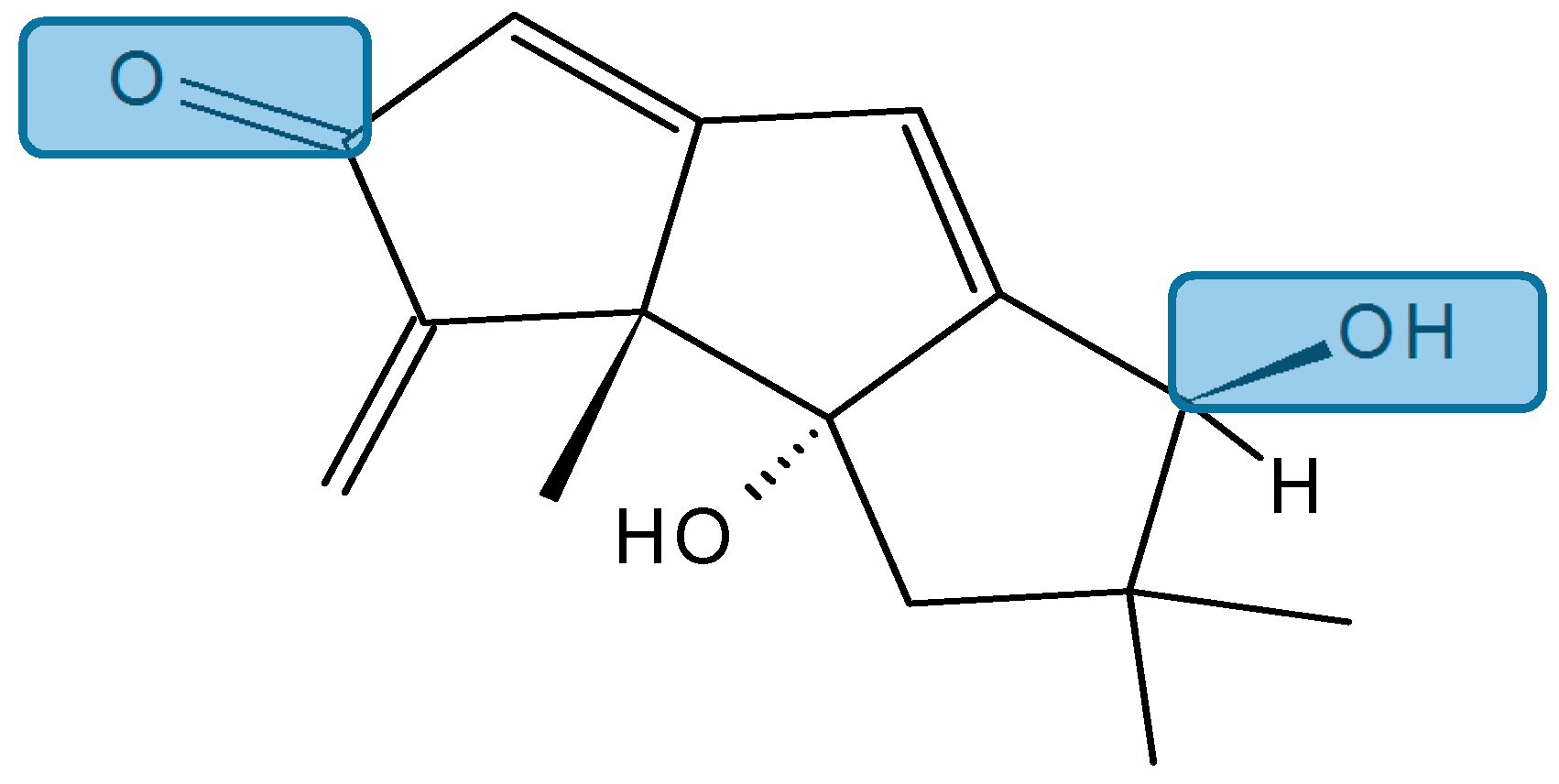
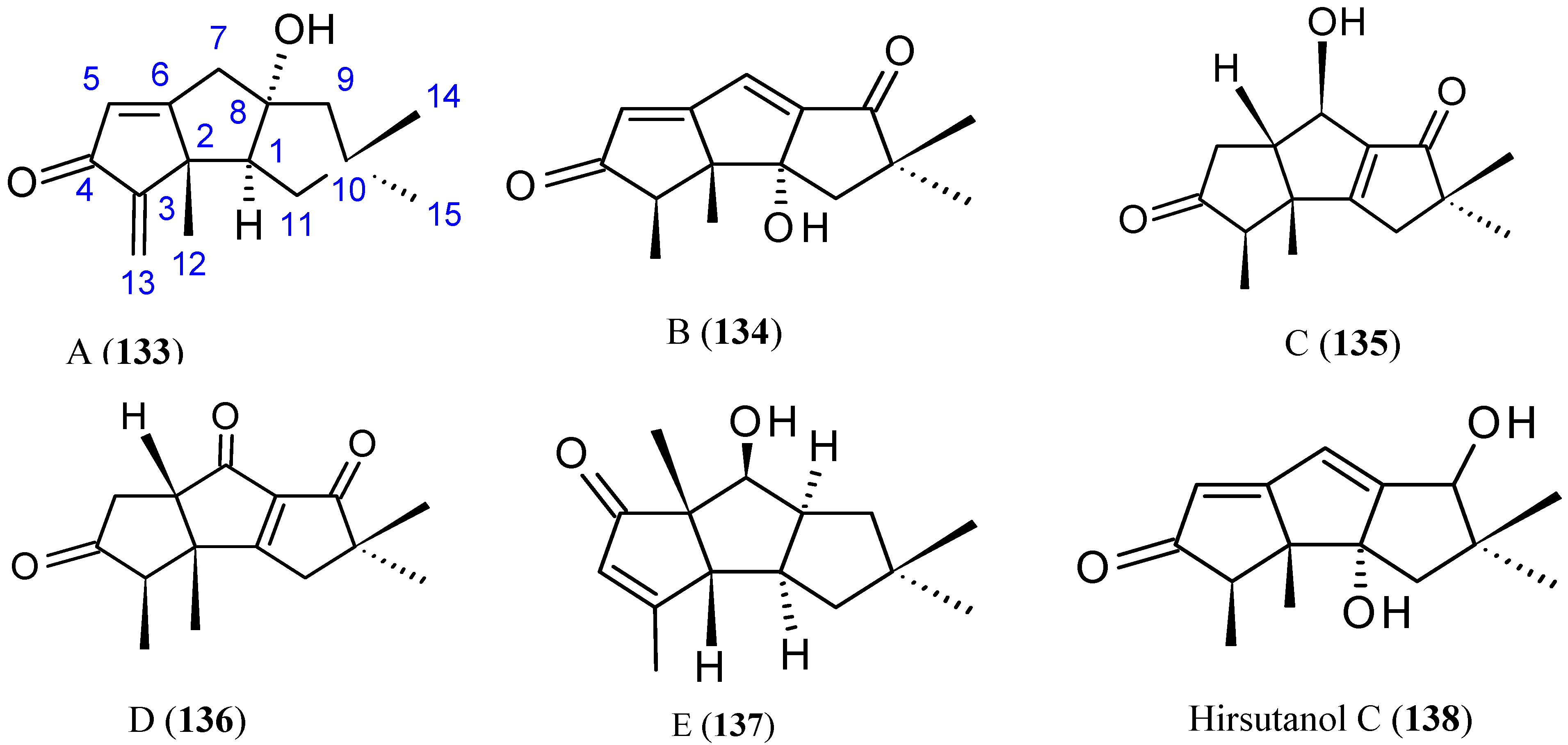
Hirsutanol A

Chondrosterins
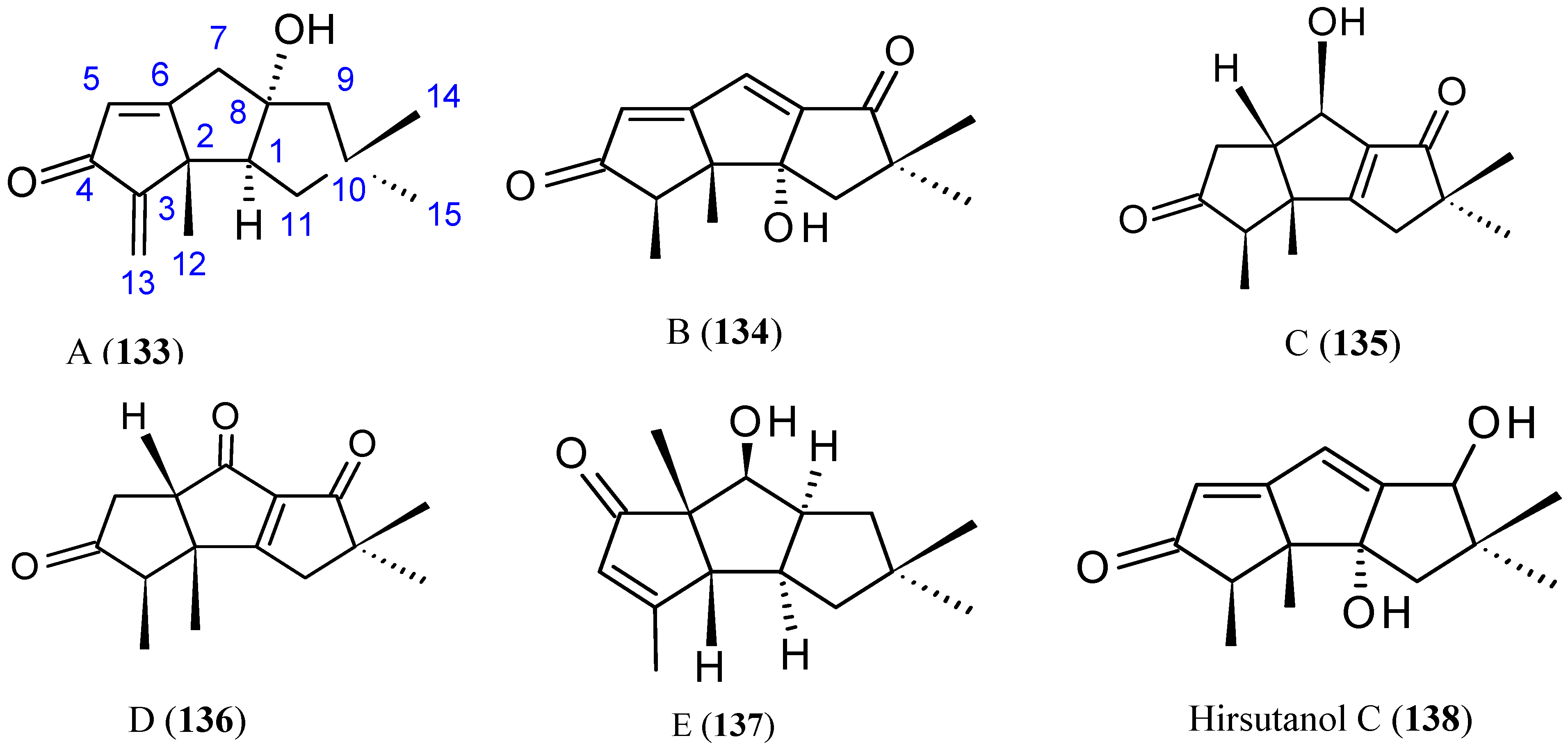
3. Discussion
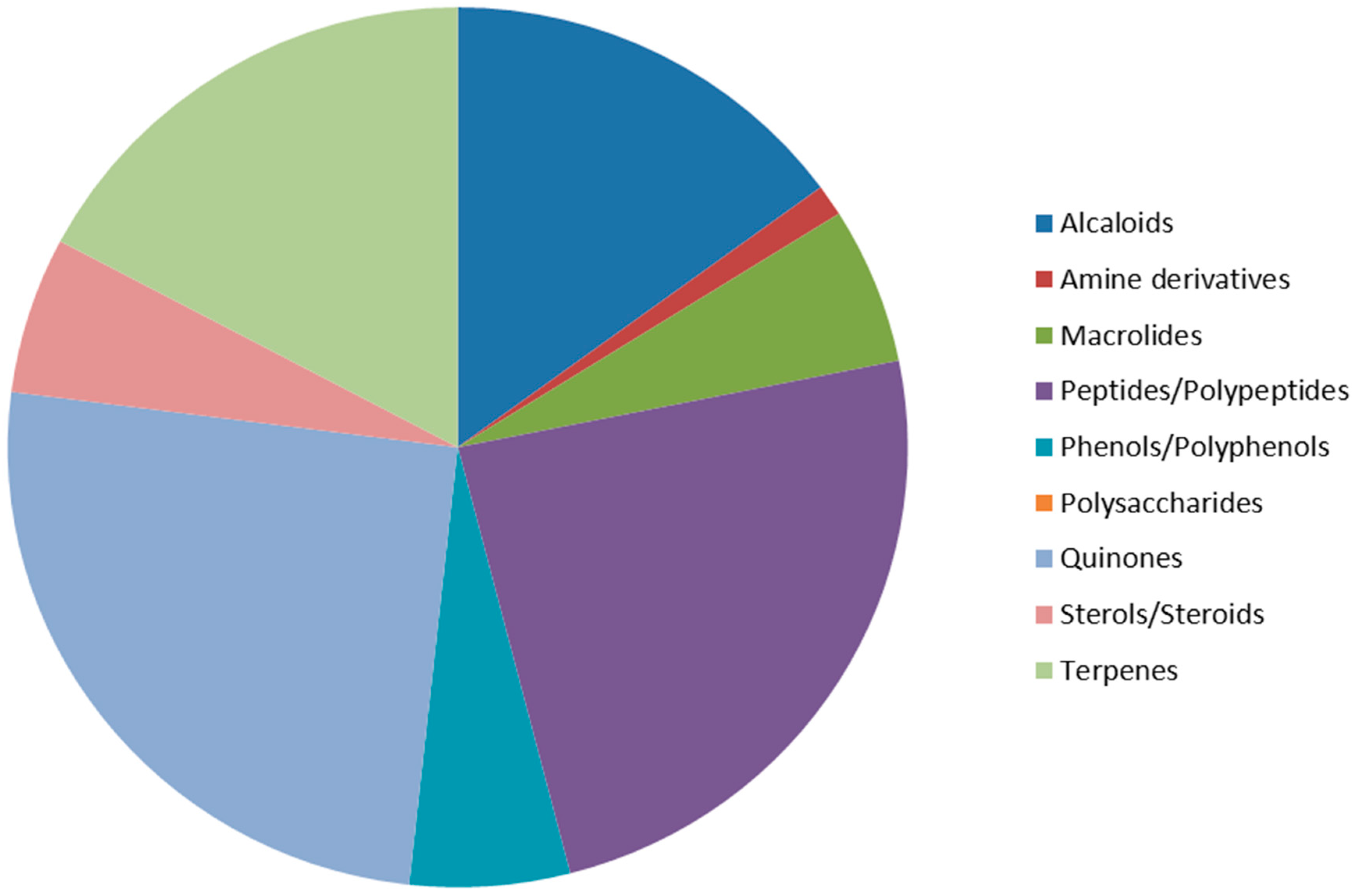
4. Conclusions
Acknowledgments
Author Contributions
Abbreviations
Cell lines
| A549 | Lung carcinoma cell line |
| AGS | Human stomach adenocarcinoma cell line |
| B16 | Mouse melanoma cell line |
| BEL-7402 | Human hepatoma cancer cell line |
| CEM | leukemia cell |
| CNE2 | Human nasopharyngeal carcinoma cell line |
| Daoy | Human medulloblastoma cells |
| DLCL | Diffuse large cell line |
| DU-145 | Prostate cancer cell line |
| H1299 | Lung cancer cells |
| HCC | Human hepatocellular carcinoma cell line |
| HCT116 | Human colon carcinoma cell line |
| HCT116 | Human colon carcinoma cell line |
| HCT-8 | Human colon cancer cell line |
| HEL | Human embryonic lung |
| HEL | Human embryonic lung cell line |
| HeLa | Human cervical epithelial carcinoma |
| HEp 2 | Human laryngeal carcinoma cells |
| HepG2 | Human hepatocellular liver carcinoma cell line |
| HL-60 | Human promyelocytic leukemia cell line |
| HT-29 | Human colon adenocarcinoma cell line |
| HT29 | Human colon carcinoma cells |
| IGROV1 | Human ovarian carcinoma cells |
| IGROV1-DXR | Doxorubicin-resistant cell line |
| J82 | Bladder carcinoma cells |
| K562 | Human chronic myeloid leukemia cell line |
| KB | Human nasopharynx carcinoma cell line |
| LNCaP | Prostate cancer cell line |
| LoVo | Colon cancer cell line |
| MCF-10A | Human breast epithelial cell line |
| MCF-7 | Human breast adenocarcinoma cell line |
| MDA-MB231 | Mammary carcinoma cell line |
| MDA-MB-231 | Mammary carcinoma cell line |
| MiaPaCa2 | Pancreatic cancer cells |
| MKN-28 | Gastric cancer cells |
| MRC-5 | Healthy lung fibroblasts |
| NCI-H460 | Human lung cancer cell line |
| P-388 | Murine lymphocytic leukemia cell line |
| PBMC | Peripheral blood mononuclear cells |
| PC-3 | Prostate cancer cells |
| S180 | Murine sarcoma cell line |
| SF-268 | Human glioblastoma cells |
| SK-MES-1 | Lung cancer cell line |
| SNU-423 | Hepatocellular carcinoma cell line |
| SW-1990 | Human pancreatic cell line |
| UCI-101 | Ovarian cancer cells |
Other
| ABC transporters | ATP-binding cassette transporters |
| ADR | Adriamycin |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | Bcl-2-associated X protein |
| BDDE | Bis (2, 3-dibromo-4, 5-dihydroxybenzyl) ether; BF65 and BF78: Hemiasterlin derivatives |
| cDDP | Cisplatin |
| C-L | Caspase-like |
| CLL | Chronic lymphocytic leukemia |
| COX-2 | Cyclooxygenase 2 |
| CT-L | Chymotrypsin-like |
| DC | Diphlorethohydroxycarmalol |
| DMBPO | 5-(2, 4-dimethylbenzyl) pyrrolidin-2-one |
| EGFR | Epidermal Growth Factor Receptor |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| HDAC | Histone deacetylase |
| IGF-IR | Insulin-like growth factor-I receptor |
| JNK | c-Jun N-terminal kinase |
| MDR | Multi-Drug Resistance |
| MED | Mycoepoxydiene |
| MMP | Mitochondrial membrane potential |
| MTD | Maximum tolerated dose |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| NF-κB | Nuclear factor-kappa B |
| NO | Nitric oxide |
| PARP | Poly (ADP-ribose) polymerase |
| PGE2 | Prostaglandin E2 |
| P-gp | P-glycoprotein |
| PKC | Protein kinase C |
| PLA and Lau | Peloruside A and Laulimalide |
| PP2A | Protein phosphatase 2A |
| ROS | Reactive oxygen species |
| SAR | Structure-activity relationships |
| SERCA | Sarcoplasmic-ER Ca2+-ATPase |
| T-L | Trypsin-like |
| TNF-α | Tumor necrosis factor |
| TRP-1 | Tyrosinase-related protein 1 |
| TYR | Tyrosinase |
| VEGFR 2 | Vascular Endothelial Growth Factor Receptor 2 |
Conflicts of Interest
References
- IARC. Cancer Incidence and Mortality Worlwide; International Agency for Research on Cancer: Lyon, France, 2011. [Google Scholar]
- WHO. Global Status Report on Noncommunicable Diseases 2010; WHO: Geneva, Switzerland, 2011; pp. 11–15. [Google Scholar]
- WHO. Cancer: Fact Sheet N°297. Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 8 October 2014).
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Sarfaraj, H.M.; Sheeba, F.; Saba, A.; Mohd, S.K. Marine natural products: A lead for anticancer. Indian J. Geo-Mar. Sci. 2012, 41, 27–39. [Google Scholar]
- Sawadogo, W.R.; Schumacher, M.; Teiten, M.H.; Cerella, C.; Dicato, M.; Diederich, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2011. Molecules 2013, 18, 3641–3673. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Kelkel, M.; Dicato, M.; Diederich, M. A survey of marine natural compounds and their derivatives with anti-cancer activity reported in 2010. Molecules 2011, 16, 5629–5646. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Wu, H.; Ohizumi, Y.; Hirata, Y. Agelasine-A, -B, -C and -D, novel bicyclic diterpenoids with a 9-methyladeninium unit possessing inhibitory effects on na,K-atpase from the okinawa sea sponge Agelas sp.1). Tetrahedron Lett. 1984, 25, 2989–2992. [Google Scholar] [CrossRef]
- Roggen, H.; Charnock, C.; Burman, R.; Felth, J.; Larsson, R.; Bohlin, L.; Gundersen, L.L. Antimicrobial and antineoplastic activities of agelasine analogs modified in the purine 2-position. Arch. Pharm. 2011, 344, 50–55. [Google Scholar] [CrossRef]
- Pimentel, A.A.; Felibertt, P.; Sojo, F.; Colman, L.; Mayora, A.; Silva, M.L.; Rojas, H.; Dipolo, R.; Suarez, A.I.; Compagnone, R.S.; et al. The marine sponge toxin agelasine B increases the intracellular Ca2+ concentration and induces apoptosis in human breast cancer cells (MCF-7). Cancer Chemother. Pharmacol. 2012, 69, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim. Biophys. Acta 2009, 1787, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Nutt, L.K.; Pataer, A.; Pahler, J.; Fang, B.; Roth, J.; McConkey, D.J.; Swisher, S.G. Bax and Bak promote apoptosis by modulating endoplasmic reticular and mitochondrial Ca2+ stores. J. Biol. Chem. 2002, 277, 9219–9225. [Google Scholar] [CrossRef] [PubMed]
- Rasola, A.; Bernardi, P. Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell. Calcium 2011, 50, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK regulation of endoplasmic reticulum Ca2+: A control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Roberge, M.; Berlinck, R.G.; Xu, L.; Anderson, H.J.; Lim, L.Y.; Curman, D.; Stringer, C.M.; Friend, S.H.; Davies, P.; Vincent, I.; et al. High-throughput assay for G2 checkpoint inhibitors and identification of the structurally novel compound isogranulatimide. Cancer Res. 1998, 58, 5701–5706. [Google Scholar] [PubMed]
- Jiang, X.; Zhao, B.; Britton, R.; Lim, L.Y.; Leong, D.; Sanghera, J.S.; Zhou, B.B.; Piers, E.; Andersen, R.J.; Roberge, M. Inhibition of Chk1 by the G2 DNA damage checkpoint inhibitor isogranulatimide. Mol. Cancer Ther. 2004, 3, 1221–1227. [Google Scholar] [PubMed]
- Hugon, B.; Anizon, F.; Bailly, C.; Golsteyn, R.M.; Pierre, A.; Leonce, S.; Hickman, J.; Pfeiffer, B.; Prudhomme, M. Synthesis and biological activities of isogranulatimide analogues. Bioorg. Med. Chem. 2007, 15, 5965–5980. [Google Scholar] [CrossRef] [PubMed]
- Deslandes, S.; Lamoral-Theys, D.; Frongia, C.; Chassaing, S.; Bruyere, C.; Lozach, O.; Meijer, L.; Ducommun, B.; Kiss, R.; Delfourne, E. Synthesis and biological evaluation of analogs of the marine alkaloids granulatimide and isogranulatimide. Eur. J. Med. Chem. 2012, 54, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3'-indolyl)-furans and 3,5-bis(3'-indolyl)-isoxazoles, nortopsentin analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; Barraja, P.; Spano, V.; Cirrincione, G.; Diana, P.; Maier, A.; Kelter, G.; Fiebig, H.H. Synthesis and Antiproliferative Activity of 2,5-bis(3'-Indolyl)pyrroles, Analogues of the Marine Alkaloid Nortopsentin. Mar. Drugs 2013, 11, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Maruthi Kumar, N.; Ghosh, S.; Shah, K. Novel bis(indolyl)hydrazide-hydrazones as potent cytotoxic agents. Bioorg. Med. Chem. Lett. 2012, 22, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Yamanokuchi, R.; Imada, K.; Miyazaki, M.; Kato, H.; Watanabe, T.; Fujimuro, M.; Saeki, Y.; Yoshinaga, S.; Terasawa, H.; Iwasaki, N.; et al. Hyrtioreticulins A-E, indole alkaloids inhibiting the ubiquitin-activating enzyme, from the marine sponge Hyrtios reticulatus. Bioorg. Med. Chem. 2012, 20, 4437–4442. [Google Scholar] [CrossRef] [PubMed]
- Bagola, K.; von Delbruck, M.; Dittmar, G.; Scheffner, M.; Ziv, I.; Glickman, M.H.; Ciechanover, A.; Sommer, T. Ubiquitin Binding by a CUE Domain Regulates Ubiquitin Chain Formation by ERAD E3 Ligases. Mol. Cell. 2013, 50, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [PubMed]
- Cananzi, S.; Merlini, L.; Artali, R.; Beretta, G.L.; Zaffaroni, N.; Dallavalle, S. Synthesis and topoisomerase I inhibitory activity of a novel diazaindeno[2,1-b]phenanthrene analogue of Lamellarin D. Bioorg. Med. Chem. 2011, 19, 4971–4984. [Google Scholar] [CrossRef] [PubMed]
- Neagoie, C.; Vedrenne, E.; Buron, F.; Merour, J.Y.; Rosca, S.; Bourg, S.; Lozach, O.; Meijer, L.; Baldeyrou, B.; Lansiaux, A.; et al. Synthesis of chromeno[3,4-b]indoles as Lamellarin D analogues: a novel DYRK1A inhibitor class. Eur. J. Med. Chem. 2012, 49, 379–396. [Google Scholar] [CrossRef] [PubMed]
- Guerrant, W.; Patil, V.; Canzoneri, J.C.; Yao, L.P.; Hood, R.; Oyelere, A.K. Dual-acting histone deacetylase-topoisomerase I inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 3283–3287. [Google Scholar] [CrossRef] [PubMed]
- Pozo, N.; Zahonero, C.; Fernandez, P.; Linares, J.M.; Ayuso, A.; Hagiwara, M.; Perez, A.; Ricoy, J.R.; Hernandez-Lain, A.; Sepulveda, J.M.; et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J. Clin. Investig. 2013, 123, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Saurav, K.; Kannabiran, K. Cytotoxicity and antioxidant activity of 5-(2,4-dimethylbenzyl)pyrrolidin-2-one extracted from marine Streptomyces VITSVK5 spp. Saudi J. Biol. Sci. 2012, 19, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Ohno, O.; Teruya, T.; Yamori, T.; Inuzuka, T.; Suenaga, K. Isolation and structures of biselyngbyasides B, C, and D from the marine cyanobacterium Lyngbya sp., and the biological activities of biselyngbyasides. Tetrahedron 2012, 68, 5984–5990. [Google Scholar] [CrossRef]
- Morgan, R.J., Jr.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Investig. New Drugs 2012, 30, 723–728. [Google Scholar] [CrossRef]
- Pean, E.; Klaar, S.; Berglund, E.G.; Salmonson, T.; Borregaard, J.; Hofland, K.F.; Ersboll, J.; Abadie, E.; Giuliani, R.; Pignatti, F. The European medicines agency review of eribulin for the treatment of patients with locally advanced or metastatic breast cancer: Summary of the scientific assessment of the committee for medicinal products for human use. Clin. Cancer Res. 2012, 18, 4491–4497. [Google Scholar] [CrossRef] [PubMed]
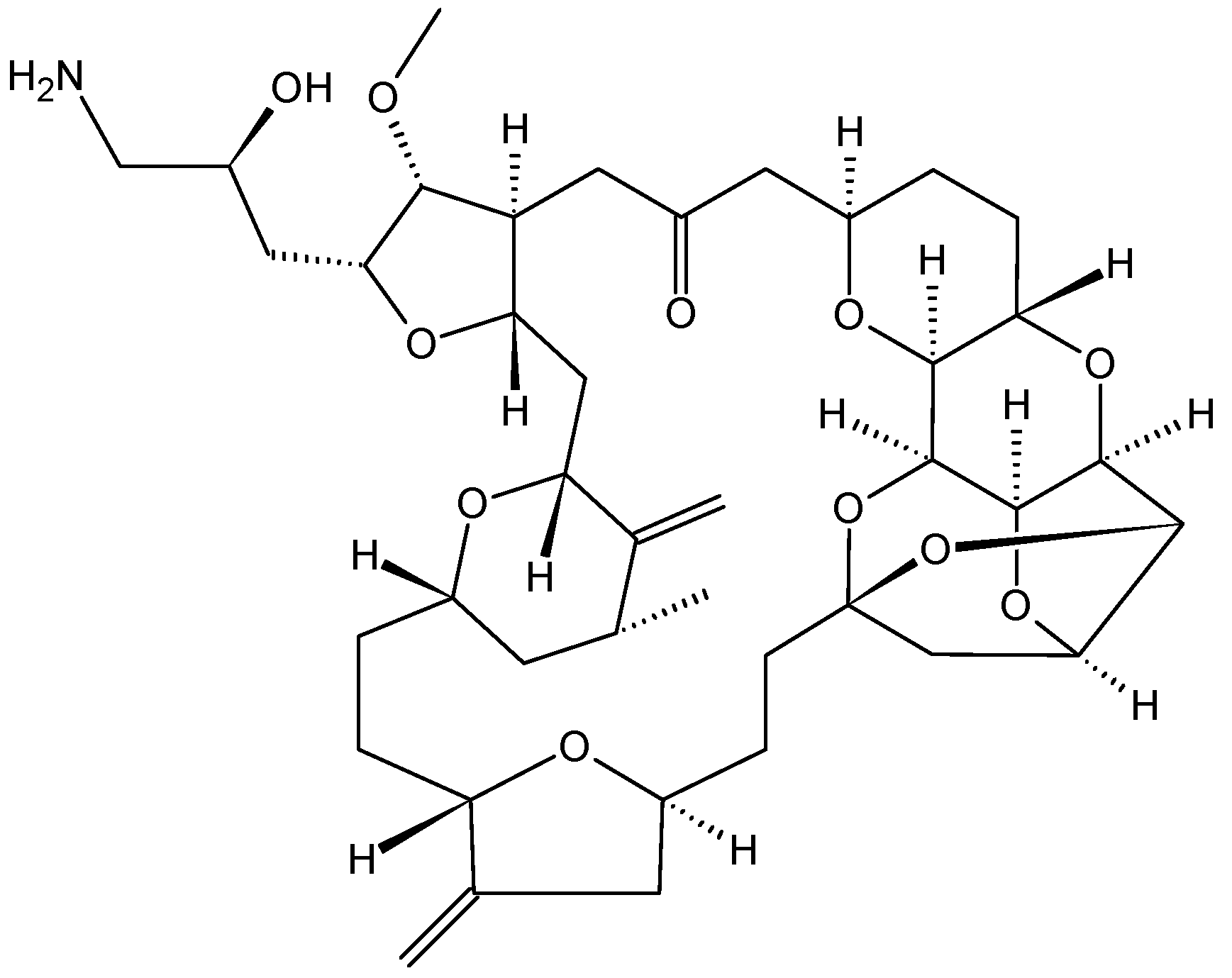
- Renouf, D.J.; Tang, P.A.; Major, P.; Krzyzanowska, M.K.; Dhesy-Thind, B.; Goffin, J.R.; Hedley, D.; Wang, L.; Doyle, L.; Moore, M.J. A phase II study of the halichondrin B analog eribulin mesylate in gemcitabine refractory advanced pancreatic cancer. Investig. New Drugs 2012, 30, 1203–1207. [Google Scholar] [CrossRef]
- Mukohara, T.; Nagai, S.; Mukai, H.; Namiki, M.; Minami, H. Eribulin mesylate in patients with refractory cancers: a Phase I study. Investig. New Drugs 2012, 30, 1926–1933. [Google Scholar] [CrossRef]
- Spira, A.I.; Iannotti, N.O.; Savin, M.A.; Neubauer, M.; Gabrail, N.Y.; Yanagihara, R.H.; Zang, E.A.; Cole, P.E.; Shuster, D.; Das, A. A phase II study of eribulin mesylate (E7389) in patients with advanced, previously treated non-small-cell lung cancer. Clin. Lung Cancer 2012, 13, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Vahdat, L.; Blum, J.L.; Twelves, C.; Campone, M.; Roche, H.; Bachelot, T.; Awada, A.; Paridaens, R.; Goncalves, A.; et al. Phase II study of the halichondrin B analog eribulin mesylate in patients with locally advanced or metastatic breast cancer previously treated with an anthracycline, a taxane, and capecitabine. J. Clin. Oncol. 2010, 28, 3922–3928. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Kikuchi, T.; Tanaka, R.; Numata, A. Halichoblelides B and C, potent cytotoxic macrolides from a Streptomyces species separated from a marine fish. Tetrahedron Lett. 2012, 53, 2842–2846. [Google Scholar] [CrossRef]
- Yamada, T.; Minoura, K.; Numata, A. Halichoblelide, a potent cytotoxic macrolide from a Streptomyces species separated from a marine fish. Tetrahedron Lett. 2002, 43, 1721–1724. [Google Scholar] [CrossRef]
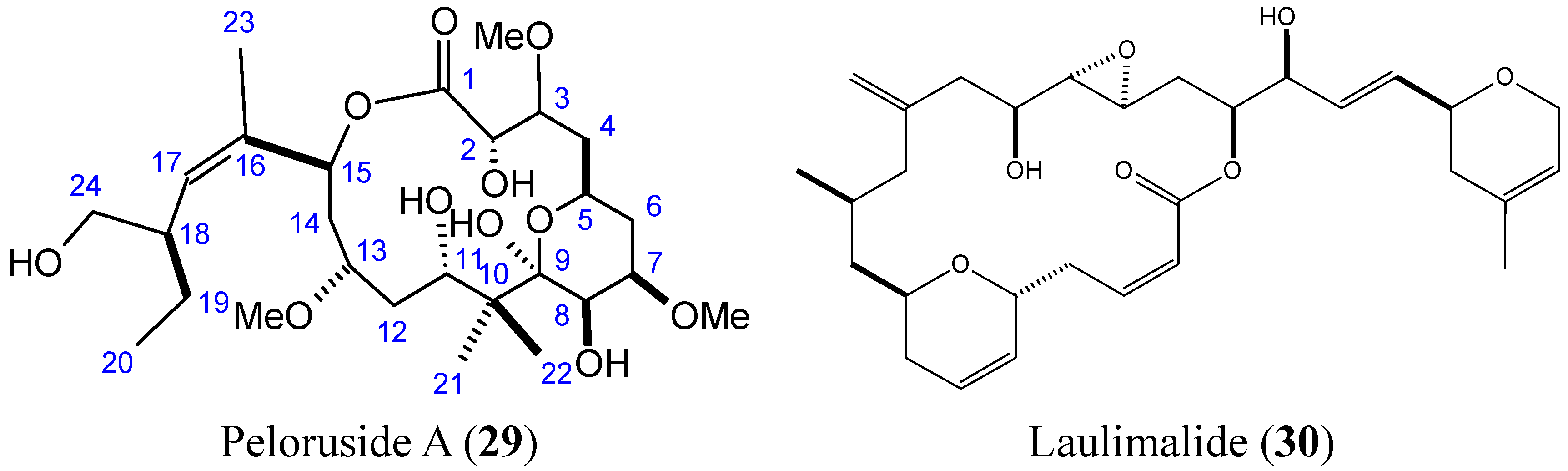
- Hood, K.A.; West, L.M.; Rouwe, B.; Northcote, P.T.; Berridge, M.V.; Wakefield, S.J.; Miller, J.H. Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule- stabilizing activity. Cancer Res. 2002, 62, 3356–3360. [Google Scholar] [PubMed]
- Liu, J.; Towle, M.J.; Cheng, H.; Saxton, P.; Reardon, C.; Wu, J.; Murphy, E.A.; Kuznetsov, G.; Johannes, C.W.; Tremblay, M.R.; et al. In vitro and in vivo anticancer activities of synthetic (−)-laulimalide, a marine natural product microtubule stabilizing agent. Anticancer Res. 2007, 27, 1509–1518. [Google Scholar] [PubMed]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar] [PubMed]
- Pera, B.; Razzak, M.; Trigili, C.; Pineda, O.; Canales, A.; Buey, R.M.; Jimenez-Barbero, J.; Northcote, P.T.; Paterson, I.; Barasoain, I.; et al. Molecular recognition of peloruside A by microtubules. The C24 primary alcohol is essential for biological activity. ChemBioChem 2010, 11, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Kanakkanthara, A.; Rawson, P.; Northcote, P.T.; Miller, J.H. Acquired resistance to peloruside A and laulimalide is associated with downregulation of vimentin in human ovarian carcinoma cells. Pharm. Res. 2012, 29, 3022–3032. [Google Scholar] [CrossRef] [PubMed]
- Hamel, E.; Day, B.W.; Miller, J.H.; Jung, M.K.; Northcote, P.T.; Ghosh, A.K.; Curran, D.P.; Cushman, M.; Nicolaou, K.C.; Paterson, I.; et al. Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol. Pharmacol 2006, 70, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Huang, Y.; Fang, M.; Wang, J.; Zheng, Z.; Su, W. Cytotoxic and antimicrobial metabolites from marine lignicolous fungi, Diaporthe sp. FEMS Microbiol. Lett. 2005, 251, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Chen, T.; Li, W.; Zhang, W.; Zhu, J.; Li, Y.; Huang, Y.; Shen, Y.; Yu, C. Mycoepoxydiene inhibits lipopolysaccharide-induced inflammatory responses through the suppression of TRAF6 polyubiquitination [corrected]. PLoS ONE 2012, 7, e44890. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, B.; Yi, Y.; Zhang, W.; Wu, X.; Zhang, L.; Shen, Y. Mycoepoxydiene, a fungal polyketide inhibits MCF-7 cells through simultaneously targeting p53 and NF-kappaB pathways. Biochem. Pharmacol. 2012, 84, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Kashman, Y.; Bishara, A.; Aknin, M. Recent N-atom containing compounds from indo-pacific invertebrates. Mar. Drugs 2010, 8, 2810–2836. [Google Scholar] [CrossRef] [PubMed]
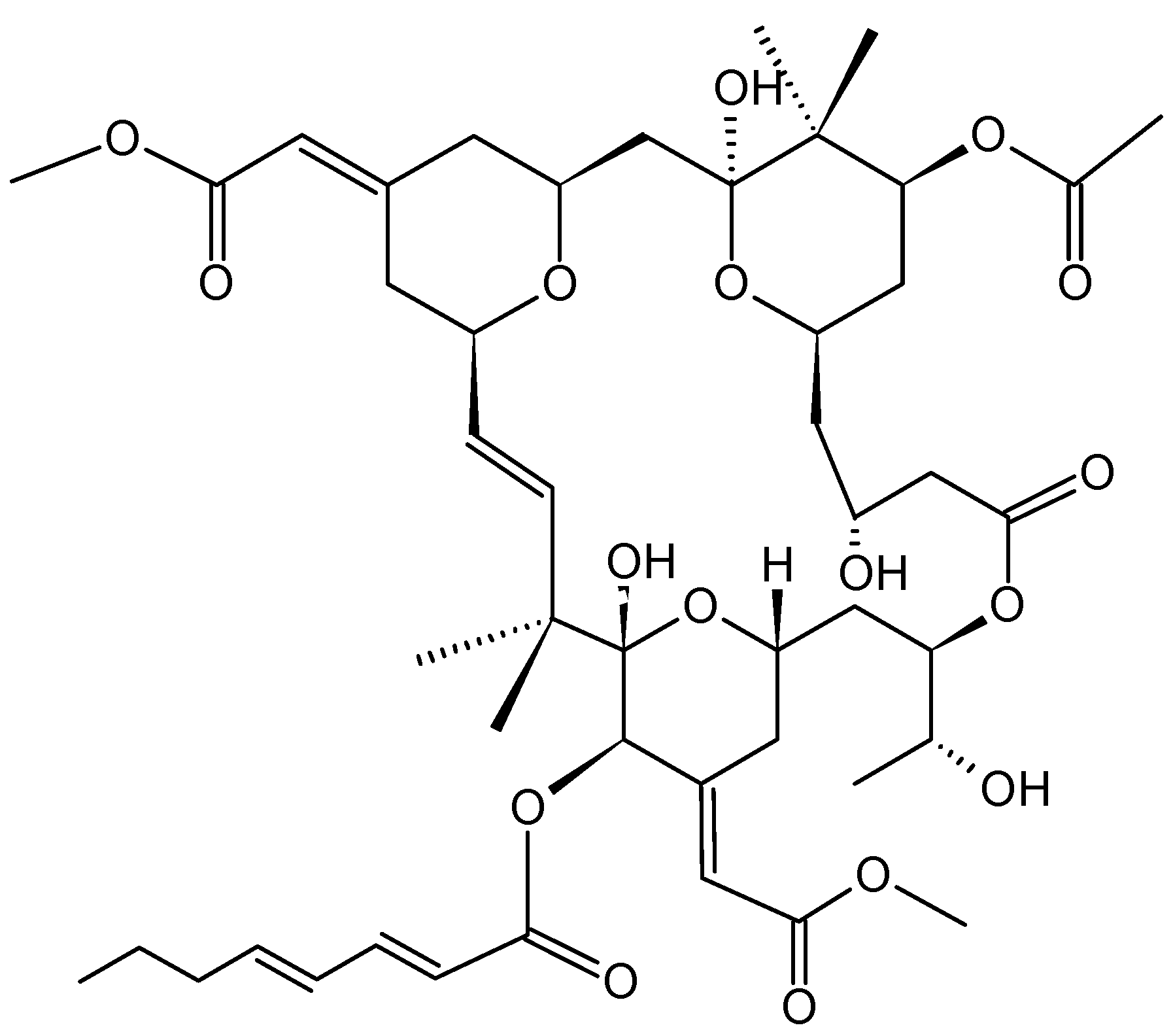
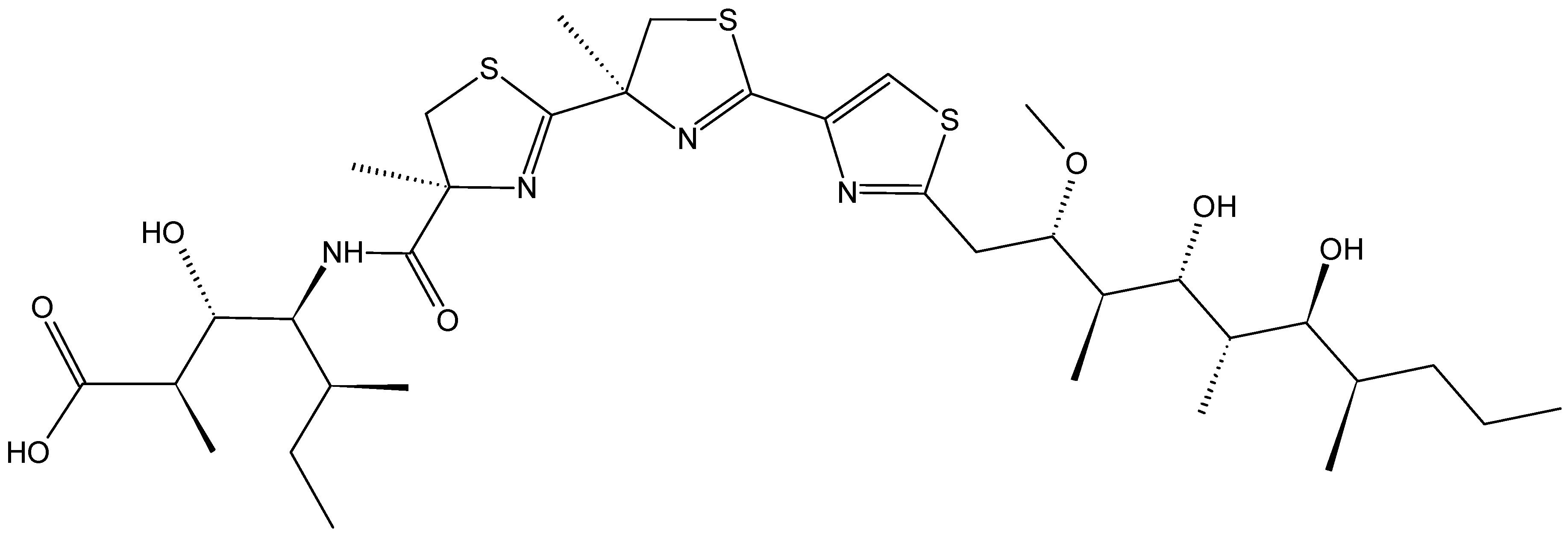
- Ben-Califa, N.; Bishara, A.; Kashman, Y.; Neumann, D. Salarin C, a member of the salarin superfamily of marine compounds, is a potent inducer of apoptosis. Investig. New Drugs 2012, 30, 98–104. [Google Scholar] [CrossRef]
- Paterson, I.; Dalby, S.M. Synthesis and stereochemical determination of the spirastrellolides. Nat. Prod. Rep. 2009, 26, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Ueoka, R.; Takada, K.; Okada, S.; Ohtsuka, S.; Ise, Y.; Matsunaga, S. Isolation of spirastrellolides A and B from a marine sponge Epipolasis sp. and their cytotoxic activities. J. Nat. Prod. 2012, 75, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Pham, N.B.; Fechner, G.; Zencak, D.; Vu, H.T.; Hooper, J.N.; Quinn, R.J. Cytotoxic cyclic depsipeptides from the Australian marine sponge Neamphius huxleyi. J. Nat. Prod. 2012, 75, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, P.D.; Byrum, T.; Liu, W.T.; Dorrestein, P.C.; Gerwick, W.H. Viequeamide A, a cytotoxic member of the kulolide superfamily of cyclic depsipeptides from a marine button cyanobacterium. J. Nat. Prod. 2012, 75, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
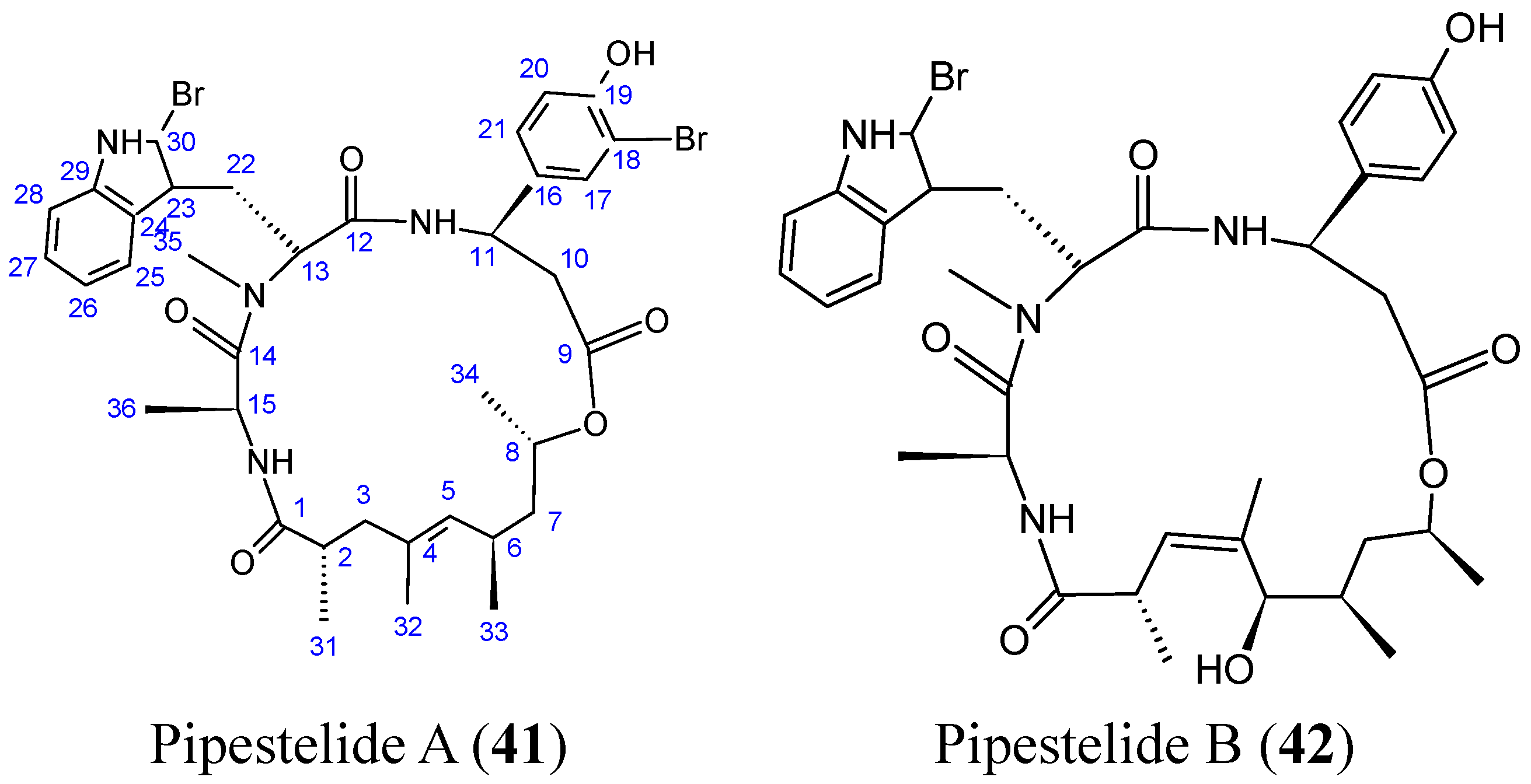
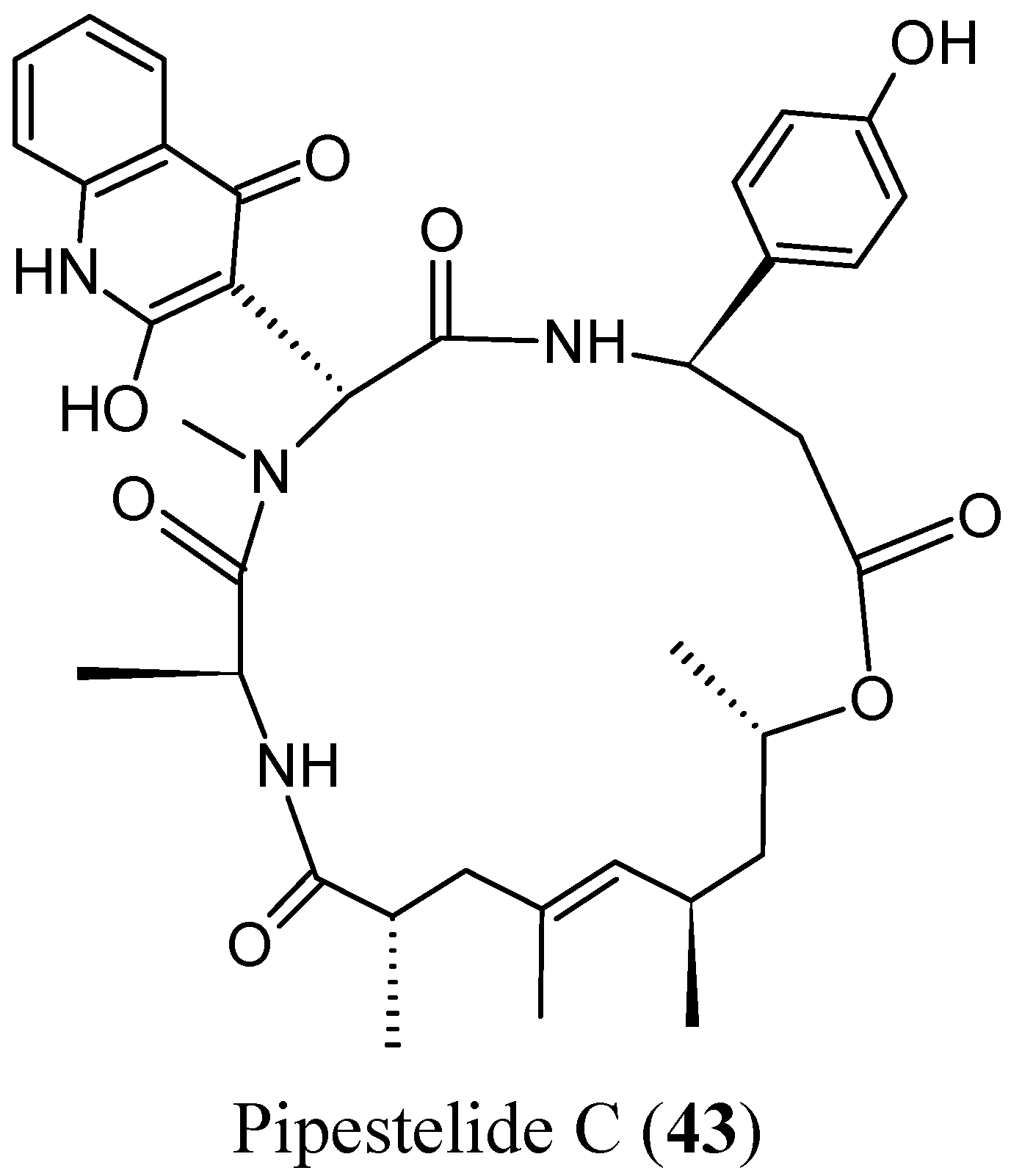
- Sorres, J.; Martin, M.T.; Petek, S.; Levaique, H.; Cresteil, T.; Ramos, S.; Thoison, O.; Debitus, C.; Al-Mourabit, A. Pipestelides A–C: cyclodepsipeptides from the Pacific marine sponge Pipestela candelabra. J. Nat. Prod. 2012, 75, 759–763. [Google Scholar] [CrossRef] [PubMed]
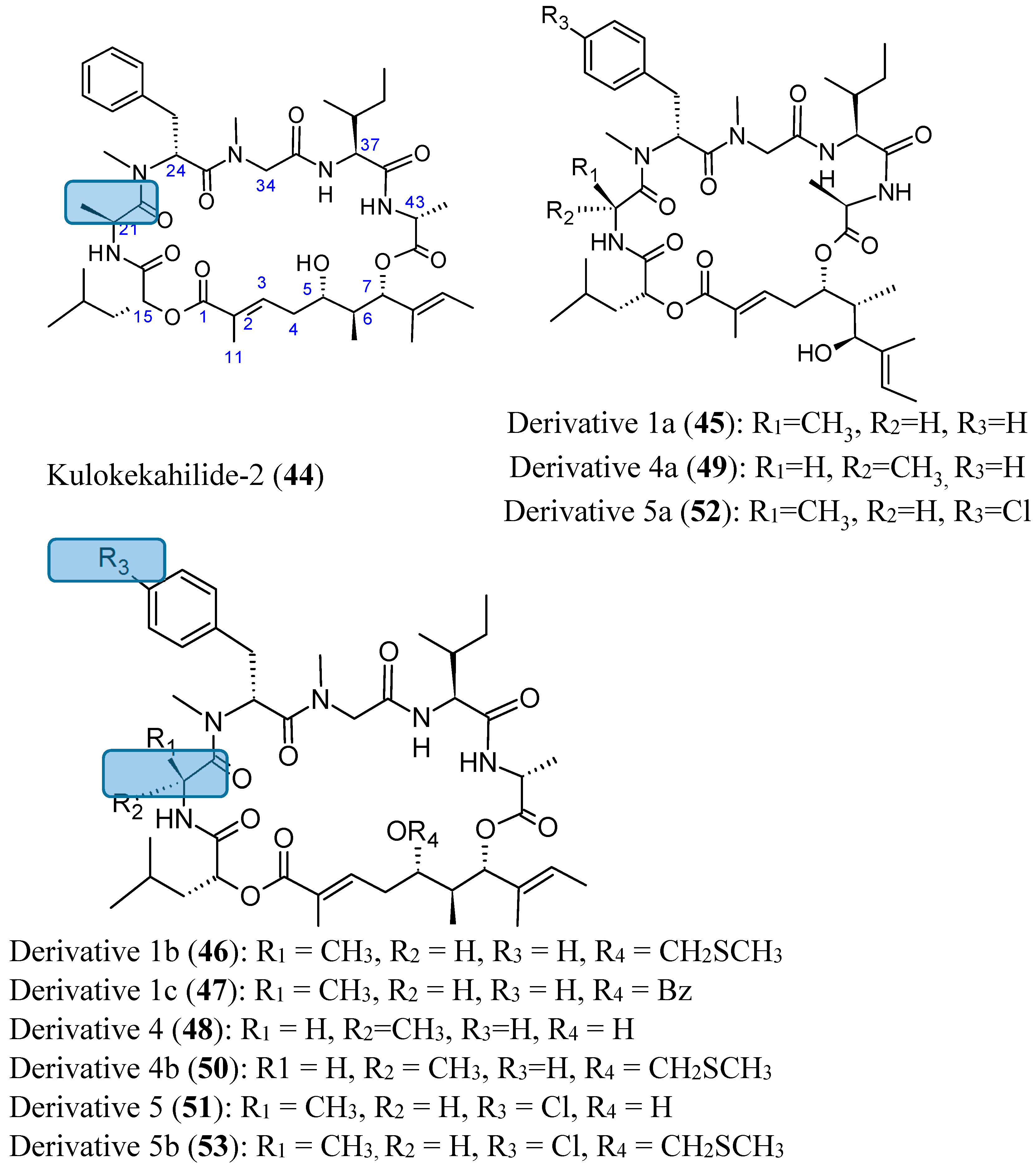
- Umehara, M.; Negishi, T.; Tashiro, T.; Nakao, Y.; Kimura, J. Structure-related cytotoxic activity of derivatives from kulokekahilide-2, a cyclodepsipeptide in Hawaiian marine mollusk. Bioorg. Med. Chem. Lett. 2012, 22, 7422–7425. [Google Scholar] [CrossRef] [PubMed]
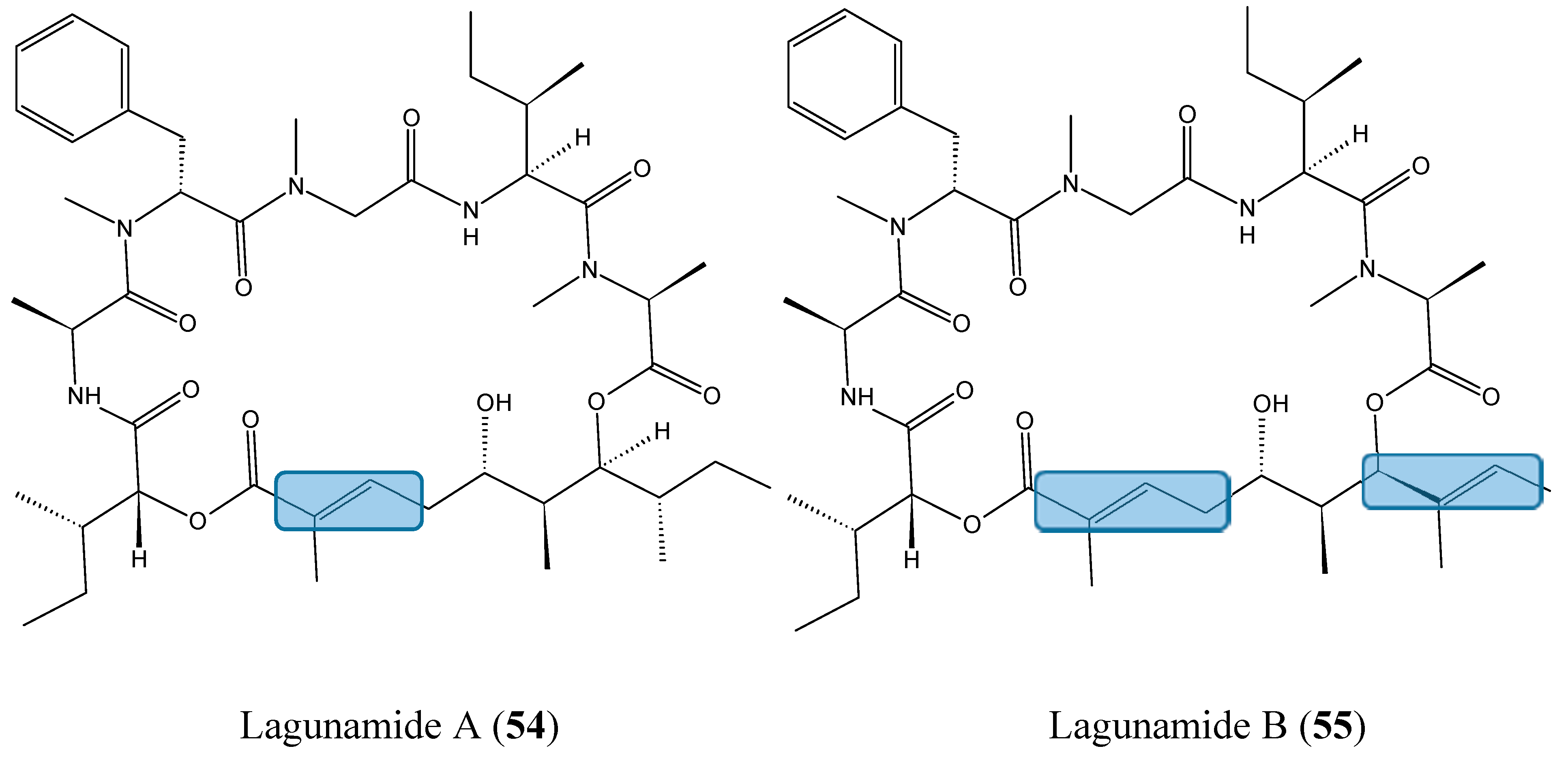
- Tripathi, A.; Fang, W.; Leong, D.T.; Tan, L.T. Biochemical studies of the lagunamides, potent cytotoxic cyclic depsipeptides from the marine cyanobacterium Lyngbya majuscula. Mar. Drugs 2012, 10, 1126–1137. [Google Scholar] [CrossRef] [PubMed]
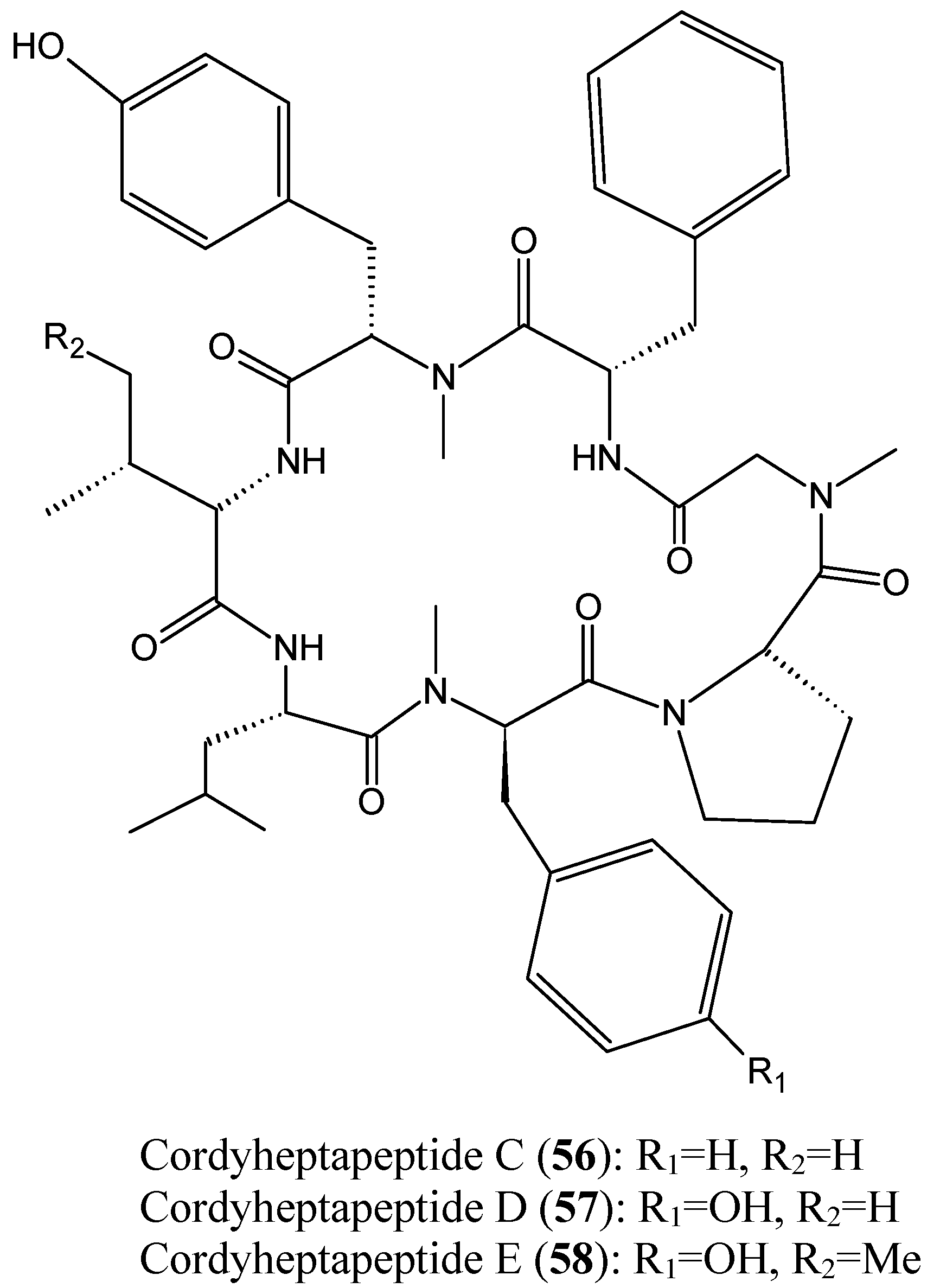
- Chen, Z.; Song, Y.; Chen, Y.; Huang, H.; Zhang, W.; Ju, J. Cyclic heptapeptides, cordyheptapeptides C–E, from the marine-derived fungus Acremonium persicinum SCSIO 115 and their cytotoxic activities. J. Nat. Prod. 2012, 75, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Yang, Z.; Yu, D.; Wang, J.; Li, R.; Ding, G. Sepia ink oligopeptide induces apoptosis in prostate cancer cell lines via caspase-3 activation and elevation of Bax/Bcl-2 ratio. Mar. Drugs 2012, 10, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Malloy, K.L.; Choi, H.; Fiorilla, C.; Valeriote, F.A.; Matainaho, T.; Gerwick, W.H. Hoiamide D, a marine cyanobacteria-derived inhibitor of p53/MDM2 interaction. Bioorg. Med. Chem. Lett. 2012, 22, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Asami, Y.; Jang, J.H.; Soung, N.K.; He, L.; Moon, D.O.; Kim, J.W.; Oh, H.; Muroi, M.; Osada, H.; Kim, B.Y.; et al. Protuboxepin A, a marine fungal metabolite, inducing metaphase arrest and chromosomal misalignment in tumor cells. Bioorg. Med. Chem. 2012, 20, 3799–3806. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Wang, C.; Liu, H.; Wang, F.; Zheng, L.; Zhao, J.; Chu, E.; Lin, X. A novel polypeptide extracted from Ciona savignyi induces apoptosis through a mitochondrial-mediated pathway in human colorectal carcinoma cells. Clin. Colorectal Cancer 2012, 11, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Poveda, B.; Rodriguez-Nieto, S.; Garcia-Caballero, M.; Medina, M.A.; Quesada, A.R. The antiangiogenic compound aeroplysinin-1 induces apoptosis in endothelial cells by activating the mitochondrial pathway. Mar. Drugs 2012, 10, 2033–2046. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, W.; Wei, J.; Qiu, L.; Lin, X. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, induces mitochondrial apoptosis in K562 cells and inhibits topoisomerase I in vitro. Toxicol. Lett. 2012, 211, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-M.; Kim, A.-D.; Heo, S.-J.; Kim, K.-N.; Lee, S.-H.; Ko, S.-C.; Jeon, Y.-J. Induction of apoptosis by diphlorethohydroxycarmalol isolated from brown alga, Ishige okamurae. J. Funct. Foods 2012, 4, 433–439. [Google Scholar] [CrossRef]
- Lee, S.-H.; Choi, J.-I.; Heo, S.-J.; Park, M.-H.; Park, P.-J.; Jeon, B.-T.; Kim, S.-K.; Han, J.-S.; Jeon, Y.-J. Diphlorethohydroxycarmalol isolated from Pae (Ishige okamurae) protects high glucose-induced damage in RINm5F pancreatic β cells via its antioxidant effects. Food Sci. Biotechnol. 2012, 21, 239–246. [Google Scholar] [CrossRef]
- Huang, H.; Yang, T.; Ren, X.; Liu, J.; Song, Y.; Sun, A.; Ma, J.; Wang, B.; Zhang, Y.; Huang, C.; et al. Cytotoxic angucycline class glycosides from the deep sea actinomycete Streptomyces lusitanus SCSIO LR32. J. Nat. Prod. 2012, 75, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Kim, I.H.; Kim, J.; Nam, T.J. Induction of apoptosis by laminarin, regulating the insulin-like growth factor-IR signaling pathways in HT-29 human colon cells. Int. J. Mol. Med. 2012, 30, 734–738. [Google Scholar] [PubMed]
- Ji, Y.B.; Ji, C.F.; Zhang, H. Laminarin induces apoptosis of human colon cancer LOVO cells through a mitochondrial pathway. Molecules 2012, 17, 9947–9960. [Google Scholar] [CrossRef] [PubMed]
- Badal, S.; Gallimore, W.; Huang, G.; Tzeng, T.R.; Delgoda, R. Cytotoxic and potent CYP1 inhibitors from the marine algae Cymopolia barbata. Org. Med. Chem. Lett. 2012, 2, 21. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhu, X.; Li, Q.; Gu, M.; He, Z.; Wu, J.; Li, J.; Lin, Y.; Li, M.; She, Z.; et al. SZ-685C, a marine anthraquinone, is a potent inducer of apoptosis with anticancer activity by suppression of the Akt/FOXO pathway. Br. J. Pharmacol. 2010, 159, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; He, Z.; Wu, J.; Yuan, J.; Wen, W.; Hu, Y.; Jiang, Y.; Lin, C.; Zhang, Q.; Lin, M.; et al. A marine anthraquinone SZ-685C overrides adriamycin-resistance in breast cancer cells through suppressing Akt signaling. Mar. Drugs 2012, 10, 694–711. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.J.; Shao, C.L.; Guo, Z.Y.; Chen, J.F.; Deng, D.S.; Yang, K.L.; Chen, Y.Y.; Fu, X.M.; She, Z.G.; Lin, Y.C.; et al. Bioactive hydroanthraquinones and anthraquinone dimers from a soft coral-derived Alternaria sp. fungus. J. Nat. Prod. 2012, 75, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Jin, H.; Song, B.; Zhu, X.; Zhao, H.; Cai, J.; Lu, Y.; Chen, B.; Lin, Y. The cytotoxicity and anticancer mechanisms of alterporriol L, a marine bianthraquinone, against MCF-7 human breast cancer cells. Appl. Microbiol. Biotechnol. 2012, 93, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Mahdi, F.; Datta, S.; Jekabsons, M.B.; Zhou, Y.D.; Nagle, D.G. Structures and mechanisms of antitumor agents: xestoquinones uncouple cellular respiration and disrupt HIF signaling in human breast tumor cells. J. Nat. Prod. 2012, 75, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Delgado, V.; Ibacache, A.; Theoduloz, C.; Valderrama, J.A. Synthesis and in vitro cytotoxic evaluation of aminoquinones structurally related to marine isoquinolinequinones. Molecules 2012, 17, 7042–7056. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhong, L.; Long, Y.; Li, J.; Wu, J.; Liu, L.; Chen, S.; Lin, Y.; Li, M.; Zhu, X.; et al. Studies on the synthesis of derivatives of marine-derived bostrycin and their structure-activity relationship against tumor cells. Mar. Drugs 2012, 10, 932–952. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhu, X.; Zhong, L.L.; Yang, B.; Li, J.; Wu, J.H.; Chen, S.P.; Lin, Y.C.; Long, Y.; She, Z.G. Synthesis and antitumor activities of derivatives of the marine mangrove fungal metabolite deoxybostrycin. Mar. Drugs 2012, 10, 2715–2728. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.Y.; Su, J.H.; Duh, C.Y.; Chen, B.W.; Wen, Z.H.; Kuo, Y.H.; Sheu, J.H. A new 9,11-secosterol from the soft coral Sinularia granosa. Bioorg. Med. Chem. Lett. 2012, 22, 4373–4376. [Google Scholar] [CrossRef] [PubMed]
- Govindam, S.V.; Choi, B.K.; Yoshioka, Y.; Kanamoto, A.; Fujiwara, T.; Okamoto, T.; Ojika, M. Novel cytotoxic polyoxygenated steroids from an Okinawan sponge Dysidea sp. Biosci Biotechnol. Biochem. 2012, 76, 999–1002. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.I.; Chen, C.C.; Chen, J.C.; Su, J.H.; Huang, H.H.; Chen, J.Y.; Wu, Y.J. Proteomic analysis of anti-tumor effects of 11-dehydrosinulariolide on CAL-27 cells. Mar. Drugs 2011, 9, 1254–1272. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.I.; Wang, R.Y.; Lin, J.J.; Su, J.H.; Chiu, C.C.; Chen, J.C.; Chen, J.Y.; Wu, Y.J. Proteomic profiling of the 11-dehydrosinulariolide-treated oral carcinoma cells Ca9–22: effects on the cell apoptosis through mitochondrial-related and ER stress pathway. J. Proteomics 2012, 75, 5578–5589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Tian, H.Y.; Li, J.; Jin, L.; Luo, C.; Ye, W.C.; Jiang, R.W. Steroids with inhibitory activity against the prostate cancer cells and chemical diversity of marine alga Tydemania expeditionis. Fitoterapia 2012, 83, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Chang, W.B.; Chen, H.M.; El-Shazly, M.; Du, Y.C.; Kung, T.H.; Chen, Y.C.; Sung, P.J.; Ho, Y.S.; Kuo, F.W.; et al. 10-acetylirciformonin B, a sponge furanoterpenoid, induces DNA damage and apoptosis in leukemia cells. Molecules 2012, 17, 11839–11848. [Google Scholar] [CrossRef] [PubMed]
- Kondratyuk, T.P.; Park, E.J.; Yu, R.; van Breemen, R.B.; Asolkar, R.N.; Murphy, B.T.; Fenical, W.; Pezzuto, J.M. Novel marine phenazines as potential cancer chemopreventive and anti-inflammatory agents. Mar. Drugs 2012, 10, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.M.; Hu, L.C.; Yen, W.H.; Su, J.H.; Lu, M.C.; Hwang, T.L.; Wang, W.H.; Sung, P.J. Echinohalimane A, a bioactive halimane-type diterpenoid from a Formosan gorgonian Echinomuricea sp. (Plexauridae). Mar. Drugs 2012, 10, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.Y.; Boo, H.J.; Kang, J.I.; Kim, M.K.; Yoo, E.S.; Hyun, J.W.; Koh, Y.S.; Kim, G.Y.; Maeng, Y.H.; Hyun, C.L.; et al. (1S,2S,3E,7E,11E)-3,7,11,15-Cembratetraen-17,2-olide, a cembrenolide diterpene from soft coral Lobophytum sp., inhibits growth and induces apoptosis in human colon cancer cells through reactive oxygen species generation. Biol. Pharm. Bull. 2012, 35, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.K.; Duh, C.Y. New cytotoxic cembranolides from the soft coral Lobophytum michaelae. Mar. Drugs 2012, 10, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.K.; Ling, Y.H.; Cheung, F.W.; Che, C.T. Stellettin A induces endoplasmic reticulum stress in murine B16 melanoma cells. J. Nat. Prod. 2012, 75, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Li, L.; Yi, Y.H.; Wang, X.H.; Pan, M.X. Triterpene Glycosides from Sea Cucumber Holothuria scabra with Cytotoxic Activity. Chin. Herbal Med. 2012, 4, 183–188. [Google Scholar]
- Yang, F.; Chen, W.D.; Deng, R.; Li, D.D.; Wu, K.W.; Feng, G.K.; Li, H.J.; Zhu, X.F. Hirsutanol A induces apoptosis and autophagy via reactive oxygen species accumulation in breast cancer MCF-7 cells. J. Pharmacol. Sci. 2012, 119, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Xie, Y.L.; Xie, Z.L.; Chen, Y.; Lam, C.K.; Lan, W.J. Chondrosterins A-E, triquinane-type sesquiterpenoids from soft coral-associated fungus Chondrostereum sp. Mar. Drugs 2012, 10, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Zips, D.; Thames, H.D.; Baumann, M. New anticancer agents: in vitro and in vivo evaluation. In Vivo 2005, 19, 1–7. [Google Scholar] [PubMed]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawadogo, W.R.; Boly, R.; Cerella, C.; Teiten, M.H.; Dicato, M.; Diederich, M. A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2012. Molecules 2015, 20, 7097-7142. https://doi.org/10.3390/molecules20047097
Sawadogo WR, Boly R, Cerella C, Teiten MH, Dicato M, Diederich M. A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2012. Molecules. 2015; 20(4):7097-7142. https://doi.org/10.3390/molecules20047097
Chicago/Turabian StyleSawadogo, Wamtinga Richard, Rainatou Boly, Claudia Cerella, Marie Hélène Teiten, Mario Dicato, and Marc Diederich. 2015. "A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2012" Molecules 20, no. 4: 7097-7142. https://doi.org/10.3390/molecules20047097
APA StyleSawadogo, W. R., Boly, R., Cerella, C., Teiten, M. H., Dicato, M., & Diederich, M. (2015). A Survey of Marine Natural Compounds and Their Derivatives with Anti-Cancer Activity Reported in 2012. Molecules, 20(4), 7097-7142. https://doi.org/10.3390/molecules20047097