
A Three-Enzyme-System to Degrade Curcumin to Natural Vanillin


Abstract
:1. Introduction
2. Results and Discussion
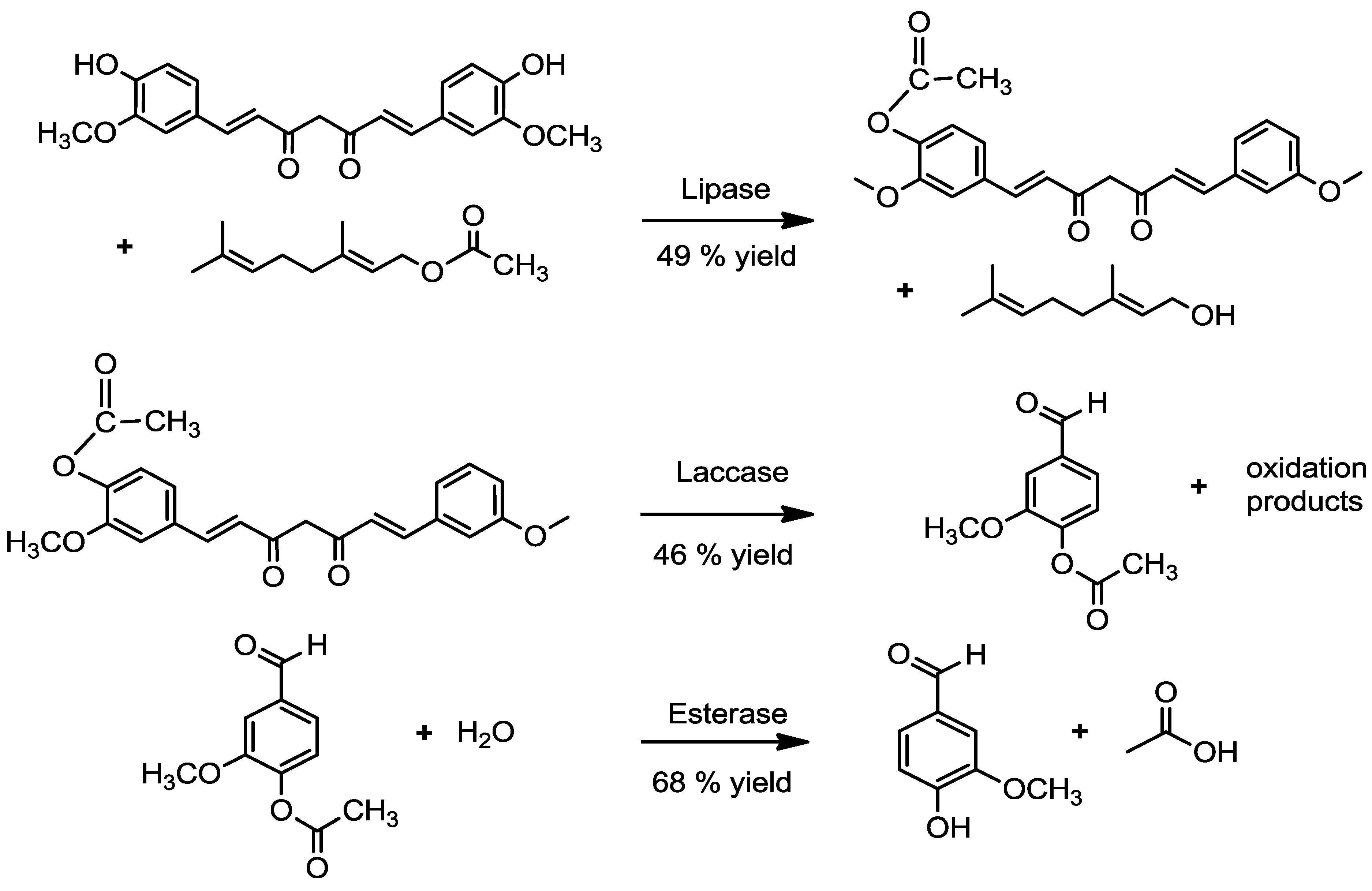
2.1. Acetylation of Curcumin
2.1.1. By a Chemical Route
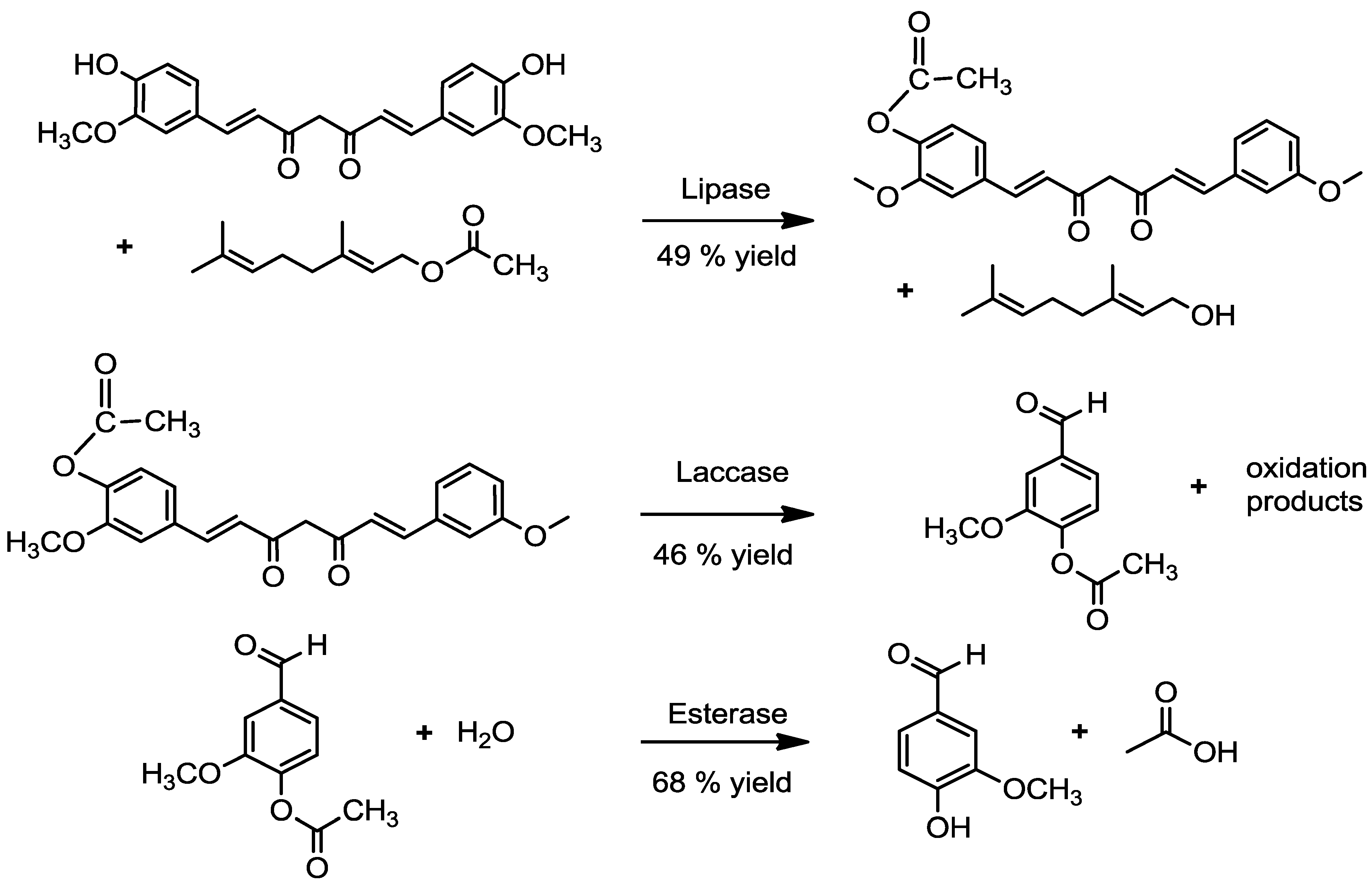
2.1.2. By an Enzymatic Route
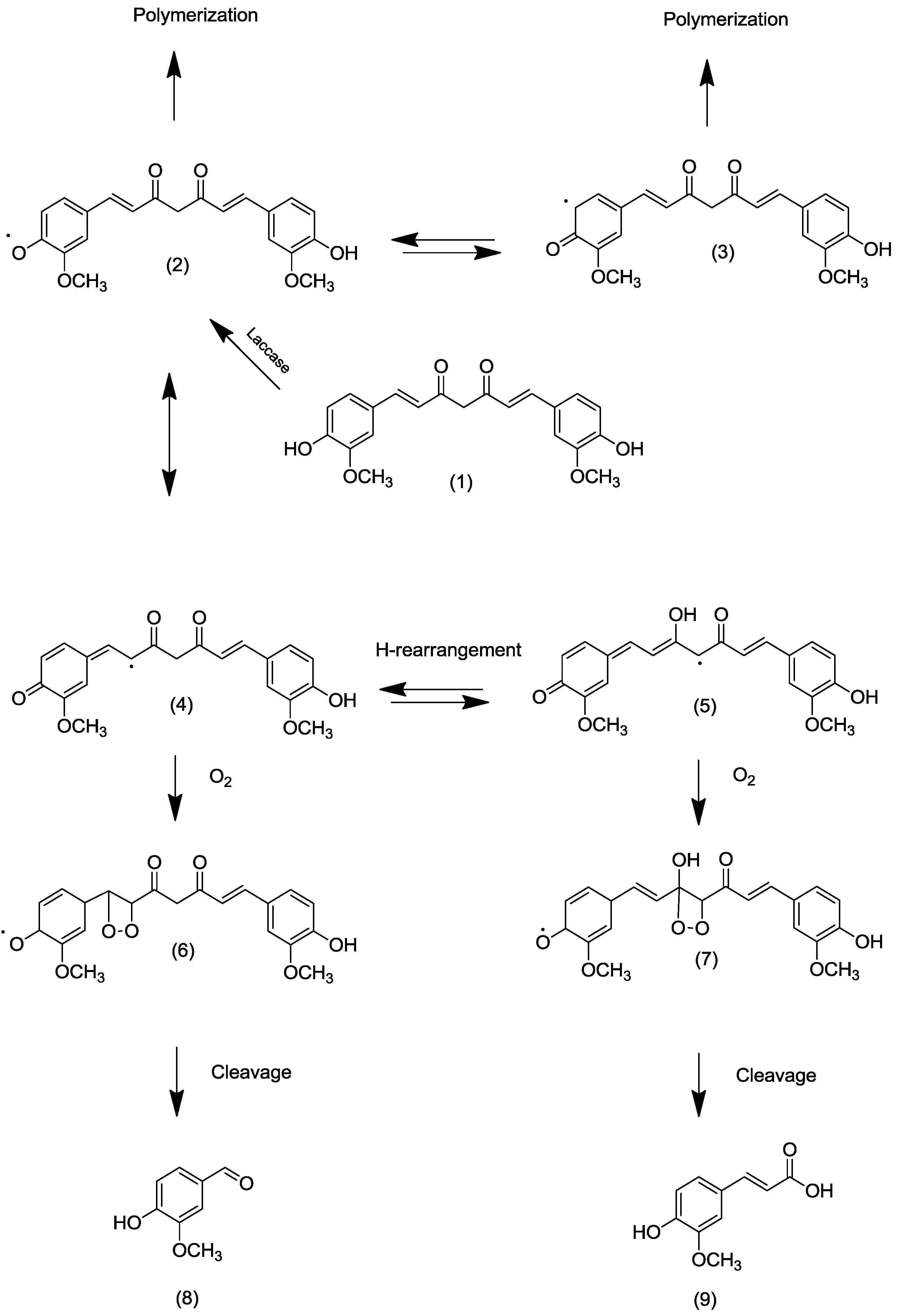
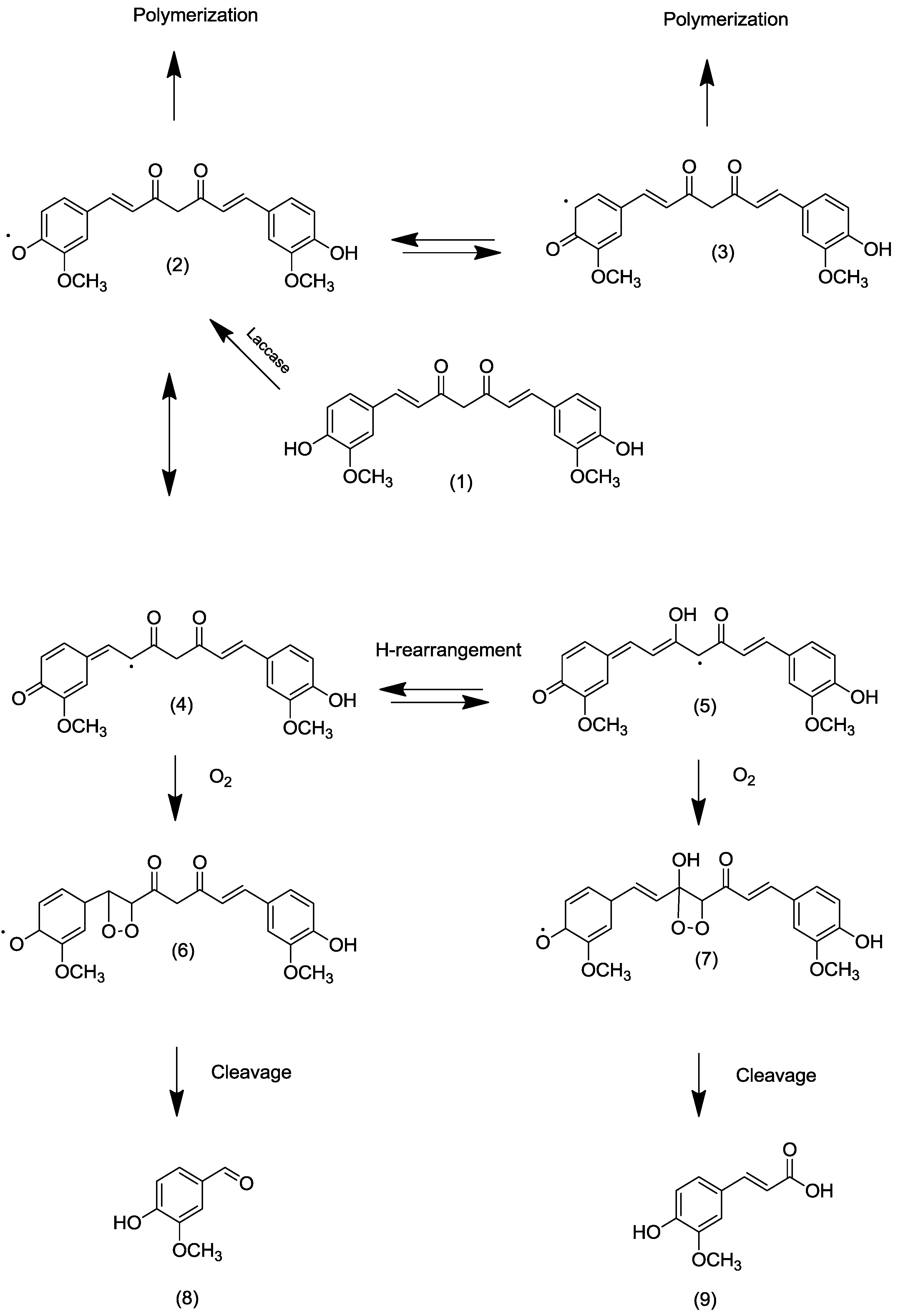
2.2. Transformation of Acetyl Curcumin by Laccases

{kind=link}
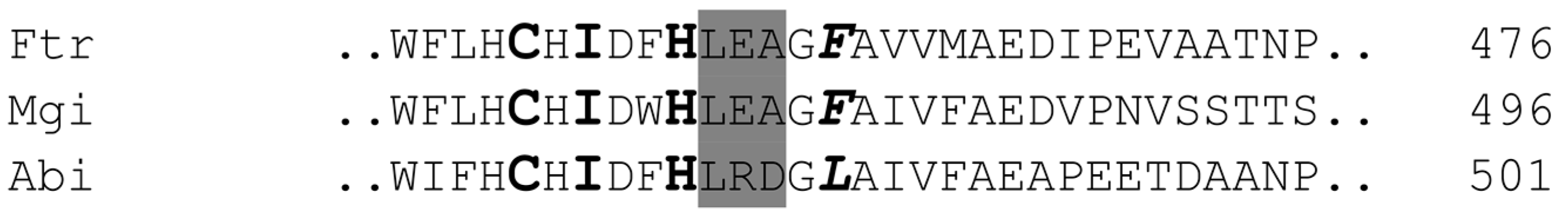{kind=link}
{kind=link}
| Laccase | Origin | Redox Potential a | pI | pH Optimum b | Temperature Optimum b (°C) |
|---|---|---|---|---|---|
| LccAbi | A. bisporus | Middle (0.47–0.71 V) | 3.5 | 4.5–5 | 30–40 |
| LccMgi | M. giganteus | High (0.73–0.78 V) | 3.1 | 5–5.5 | 30–40 |
| LccFtr | F. trogii | High (0.73–0.78 V) | 3.8 | 4.5–5 | 30–40 |
| Laccase | Vanillin Acetate (mg·L−1) | Vanillin Acetate (mM) | Molar Product Yield (%) * |
|---|---|---|---|
| LccAbi | 6.4 | 0.032 | 6.4 |
| LccMgi | 15.1 | 0.078 | 15.6 |
| LccFtr | 45.02 | 0.23 | 46 |
2.3. Alignment of the Laccases
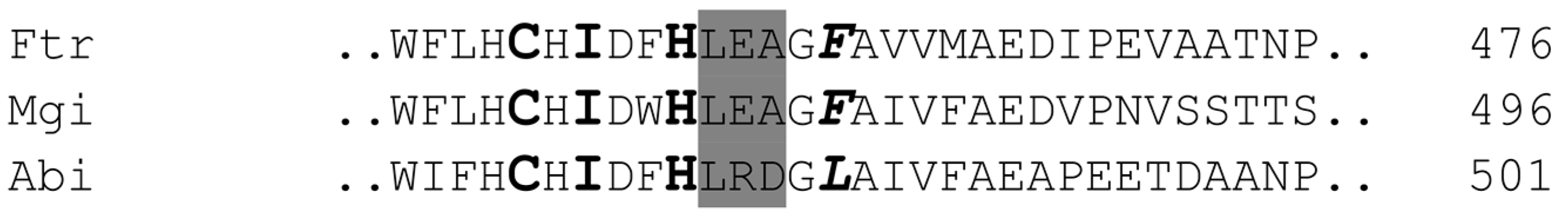
2.4. Enzymatic Deacetylation of Acetyl Vanillin
| Esterase | Vanillin (mg·L−1) * | Vanillin (mM) | Molar Product Yield (%) |
|---|---|---|---|
| UmChlE | 0 | 0 | 0 |
| Porcine liver | 75.5 | 0.50 | 50 |
| PeFaeA | 103 | 0.68 | 68 |
3. Experimental Section
3.1. Materials
3.2. Enzymes
3.3. Cultivation of Fungi
3.4. Laccase Isolation and Purification
3.5. Analysis of Amino Acid Sequence
3.6. Enzyme Assays
3.6.1. Laccase Activity
3.6.2. Esterase Activity
3.6.3. Lipase Activity
3.7. Curcumin Transformation
3.7.1. Chemical Acetylation of Curcumin
3.7.2. Lipase-Catalyzed Acetylation of Curcumin
3.7.3. Monoacetyl Curcumin Degradation
3.7.4. Esterase-Catalyzed Deacetylation of Vanillin Acetate
3.8. Gas Chromatography
3.8.1. Gas Chromatography/Flame Ionization Detection
3.8.2. Gas Chromatography/Mass Spectrometry
3.9. Liquid Chromatography/Mass Spectrometry
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Berger, R.G. Biotechnology as a source of natural volatile flavours. Curr. Opin. Food Sci. 2015, 1, 38–43. [Google Scholar] [CrossRef]
- Mayer, A.M.; Staples, R.C. Laccase new functions for an old enzyme. Phytochemistry 2002, 60, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis; Wiley-VCH: New York, NY, USA, 2000; pp. 1–11. [Google Scholar]
- Negishi, O.; Sugiura, K.; Negishi, Y. Biosynthesis of vanillin via ferulic acid in Vanilla planifolia. J. Agric. Food Chem. 2009, 57, 9956–9961. [Google Scholar] [CrossRef] [PubMed]
- Sindhwani, G.; Uk, I.; Aeri, V. Microbial transformation of eugenol to vanillin. J. Microbiol. Biotechnol. Res. 2012, 2, 313–318. [Google Scholar]
- Walton, N.J.; Narbad, A.; Faulds, C.B.; Williamson, G. Novel approach to the biosynthesis of vanillin. Curr. Opin. Biotechnol. 2000, 11, 490–496. [Google Scholar] [CrossRef] [PubMed]
- Korthou, H.; Verpoorte, R. Vanilla. In Flavours and Fragrances; Berger, R.G., Ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 203–217. [Google Scholar]
- Kumar Singh, P.; Wani, K.; Kaul-Ghanekar, R.; Prabhune, A.; Ogale, S. From micron to nano-curcumin by sophorolipid co-processing highly enhanced bioavailability, fluorescence and anti-cancer efficacy. RSC Adv. 2014, 4, 60334–60341. [Google Scholar]
- Bharti, A.; Nagpour, A.L.; Gupta, R.K. Biotransformation of curcumin to vanillin. Indian J. Chem. 2011, 50B, 1119–1122. [Google Scholar]
- Priefert, H.; Rabenhorst, J.; Steinbuechel, A. Biotechnological production of vanillin. Appl. Microbiol. Biotechnol. 2001, 56, 296–314. [Google Scholar] [CrossRef] [PubMed]
- Gordon, O.N.; Schneider, C. Vanillin and ferulic acid: Not the major degradation products of curcumin. Trends Mol. Med. 2012, 18, 361–363. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Godinez, G.; Tellez-Tellez, M.; Diaz, R.; Sanchez, C. Applications of Microbial Genes in Enzyme Technology, 1st ed.; Gupta, V.K., Touhy, M.G., Eds.; Nova Science Publishers Inc: New York, NY, USA, 2013; pp. 19–47. [Google Scholar]
- Kruegener, S.; Schaper, C.; Krings, U.; Berger, R.G. Pleurotus species convert monoterpenes to furanoterpenoids through 1,4-endoperoxides. Bioresour. Technol. 2009, 100, 2855–2860. [Google Scholar] [CrossRef] [PubMed]
- Strong, P.P.; Claus, H. Laccase a review of its past and its future in bioremediation. Crit. Rev. Env. Sci. Technol. 2011, 41, 373–434. [Google Scholar] [CrossRef]
- Gunne, M.; Hoeppner, A.; Hagedoorn, P.L.; Urlacher, V.B. Structural and redox properties of the small laccase Ssl1 from Streptomyces sviceus. FEBS J. 2014, 281, 4307–4318. [Google Scholar] [CrossRef] [PubMed]
- Uzan, E.; Nousiainen, P.; Balland, V.; Sipila, J.; Piumi, F.; Navarro, D.; Asther, M.; Record, E.; Lomascolo, A. High redox potential laccases from the ligninolytic fungi Pycnoporus coccineus and Pycnoporus sanguineus suitable for white biotechnology from gene coning to enzyme characterization and application. J. Appl. Microbiol. 2010, 108, 2199–2213. [Google Scholar] [PubMed]
- Rodgers, C.J.; Blanford, C.F.; Giddens, S.R.; Skamnioti, P.; Armstrong, F.A.; Gurr, S.J. Designer laccases: A vogue for high-potentail fungal enzyme. Trends Biotechnol. 2009, 28, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.; Krings, U.; Nimtz, M.A.; Berger, R.G. A surfactant tolerant laccase of Meripilus giganteus. World J. Microbiol. Biotechnol. 2012, 28, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Plagemann, I.; Krings, U.; Berger, R.G. Isolation and characterization of wild-type lipoxygenase LOXPsa1 from Pleurotus sapidus. Z. Naturforschung 2014, 69c, 149–154. [Google Scholar]
- Linke, D.; Matthes, R.; Nimtz, M.; Zorn, H.; Bunzel, M.; Berger, R.G. An esterase from the basidiomycetes Pleurotus sapidus hydrolyzes feruloylated saccharides. Appl. Microbiol. Biotechnol. 2013, 97, 7241–7251. [Google Scholar] [CrossRef] [PubMed]
- Haase-Aschoff, P.; Linke, D.; Berger, R.G. Detection of feruloyl- and cinnamoyl esterases from basidiomycetes in the presence of interfering laccase. Bioresour. Technol. 2013, 130, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Escorcia, A.M.; Molina, D.; Daza, M.C.; Doerr, M. Acetylation of (R,S)-propranolol catalyzed by Candida antarctica lipase B, An experimental and computational study. J. Mol. Catal. B Enzym. 2013, 98, 21–29. [Google Scholar] [CrossRef]
- Wells, A.; Meyer, H.P. Biocatalysis as a Strategic Green Technology for the Chemical Industry. ChemCatChem 2014, 6, 918–920. [Google Scholar] [CrossRef]
- Kunamneni, A.; Polu, F.J.; Ballesteros, A.; Alcalde, M. Uses of laccases in food industry. Pat. Biotechnol. 2008, 2, 10–24. [Google Scholar] [CrossRef]
- Sample Availability: Acetylated curcumin, vanillin acetate and vanillin are available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esparan, V.; Krings, U.; Struch, M.; Berger, R.G. A Three-Enzyme-System to Degrade Curcumin to Natural Vanillin. Molecules 2015, 20, 6640-6653. https://doi.org/10.3390/molecules20046640
Esparan V, Krings U, Struch M, Berger RG. A Three-Enzyme-System to Degrade Curcumin to Natural Vanillin. Molecules. 2015; 20(4):6640-6653. https://doi.org/10.3390/molecules20046640
Chicago/Turabian StyleEsparan, Vida, Ulrich Krings, Marlene Struch, and Ralf G. Berger. 2015. "A Three-Enzyme-System to Degrade Curcumin to Natural Vanillin" Molecules 20, no. 4: 6640-6653. https://doi.org/10.3390/molecules20046640
APA StyleEsparan, V., Krings, U., Struch, M., & Berger, R. G. (2015). A Three-Enzyme-System to Degrade Curcumin to Natural Vanillin. Molecules, 20(4), 6640-6653. https://doi.org/10.3390/molecules20046640