
Probing the Relationship between Anti-Pneumocystis carinii Activity and DNA Binding of Bisamidines by Molecular Dynamics Simulations


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Relative DNA Binding Affinity: Changes of DNA Melting Temperature Tm

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chemical Structure | (Tm ± SDTm) [°C] | ΔTm [°C] | (IC50 ± SDIC50) [µM] | ΔGbind [kcal/mol] |
|---|---|---|---|---|---|
| 1 | 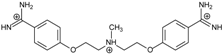 | 56.41 ± 0.23 | 3.12 | 1.73 ± 0.93 | −35.5 |
| 2 | 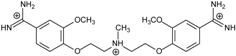 | 55.41 ± 0.62 | 2.12 | 3.41 ± 2.00 | −25.6 |
| 3 | 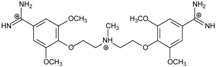 | 54.76 ± 0.42 | 1.47 | 6.26 ± 2.57 | −20.2 |
| 4 | 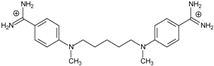 | 54.46 ± 0.22 | 1.17 | 1.99 ± 1.00 | −19.9 |
| 5 | 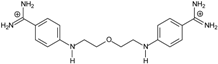 | 59.13 ± 0.06 | 5.84 | 1.11 ± 0.21 | −47.3 |
| 6 | 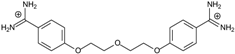 | 56.21 ± 0.28 | 2.92 | 2.13 ± 0.71 | −26.9 |
| 7 |  | 55.11 ± 0.83 | 1.82 | 4.21 ± 3.47 | −22.0 |
| 8 | 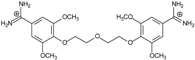 | 53.39 ± 0.85 | 0.10 | 12.99 ± 2.83 | −10.7 |
| 9 | 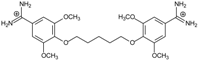 | 55.64 ± 0.72 | 2.35 | 3.00 ± 0.66 | −27.2 |
| 10 | 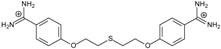 | 58.11 ± 0.22 | 4.83 | 1.18 ± 0.09 | −44.3 |
| 11 | 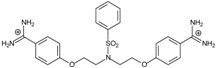 | 56.54 ± 0.62 | 3.25 | 1.33 ± 0.20 | −39.7 |
| 12 | 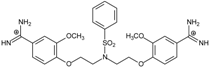 | 55.74 ± 0.72 | 2.45 | 2.66 ± 1.21 | −27.3 |
| 13 |  | 55.16 ± 0.81 | 1.87 | 4.16 ± 1.13 | −22.4 |
| PN | 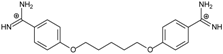 | 57.94 ± 0.21 | 4.65 | 0.51 [31] | −42.7 |
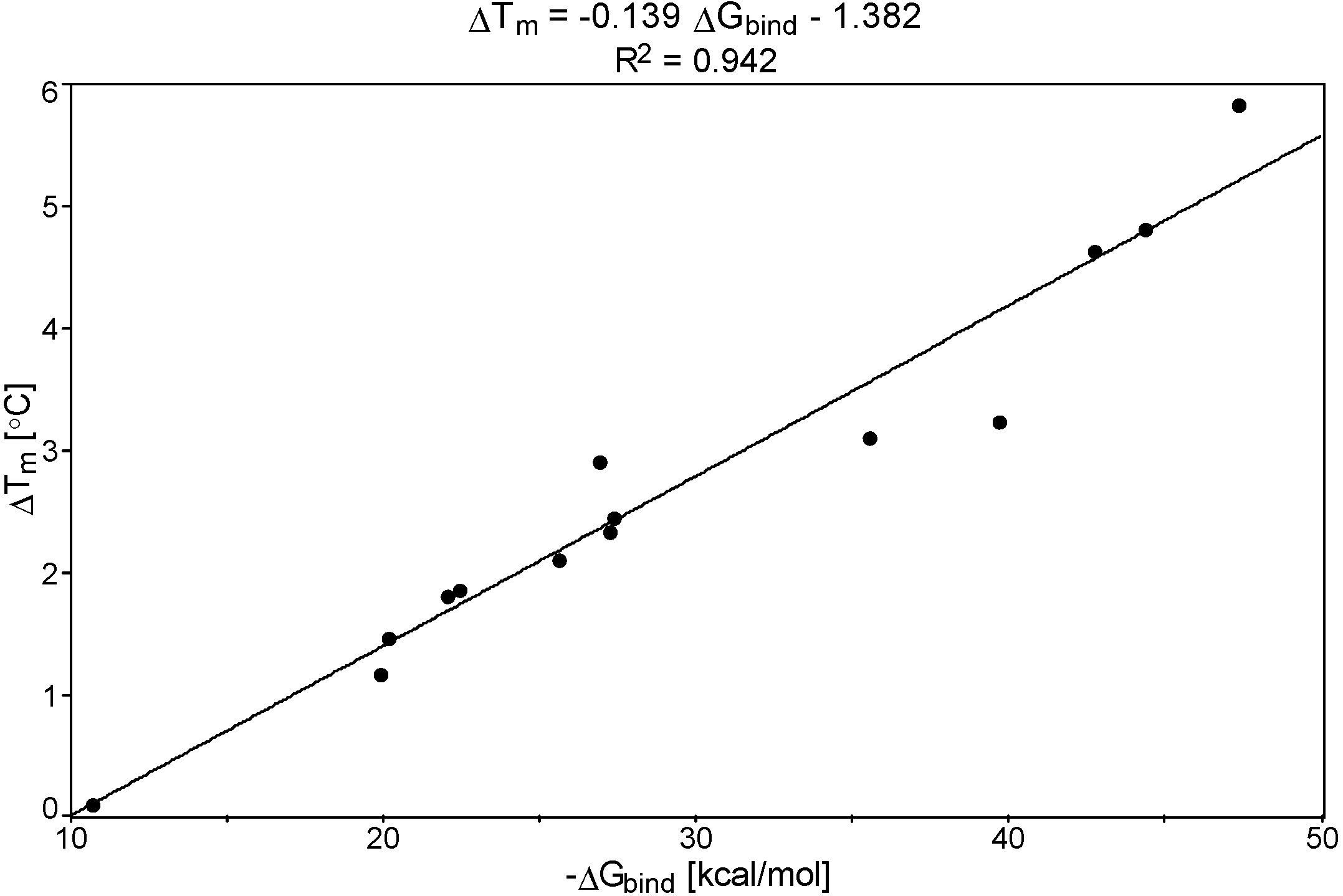
2.2. Relationship between the DNA Free Energy of Binding ΔGbind and ΔTm
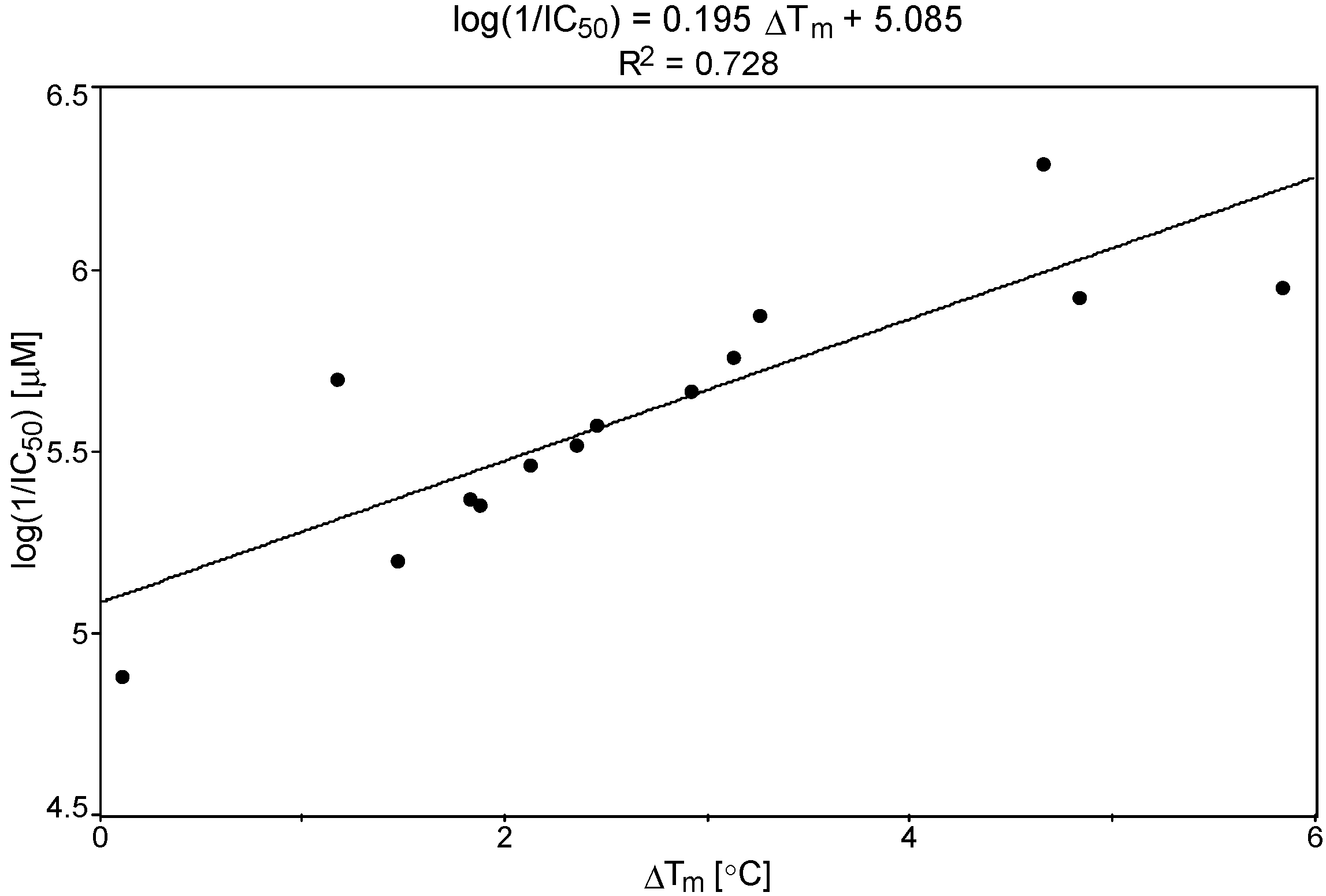
2.3. Relationship between IC50 vs. ΔGbind and ΔTm

2.4. Evaluation of Theoretical Models
| Name | Chemical Structure | (Tm ± SDTm) [°C] | ΔTm [°C] | (IC50 ± SDIC50) [µM] | ΔGbind [kcal/mol] |
|---|---|---|---|---|---|
| TC1 | 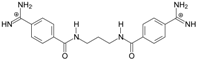 | 55.66 ± 0.22 | 2.37 Predicted: 2.31 | 2.99 ± 1.76 Predicted: 3.97 | −26.6 |
| TC2 | 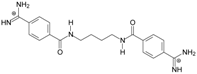 | - | - | 3.58 ± 0.02 Predicted: 4.65 | −21.1 |
| TC3 | 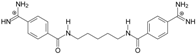 | 50.21 ± 0.71 | −3.08 Predicted: −2.46 | 14.33 ± 0.42 Predicted: 10.75 | 7.7 |
| TC4 | 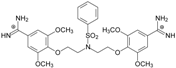 | - | - | 4.40 ± 1.39 Predicted: 4.93 | −19.2 |
| TC5 | 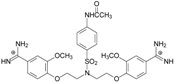 | - | - | 3.71 ± 1.19 Predicted: 4.37 | −23.3 |
2.5. Intermolecular Interactions of Ligands in DNA Minor Groove

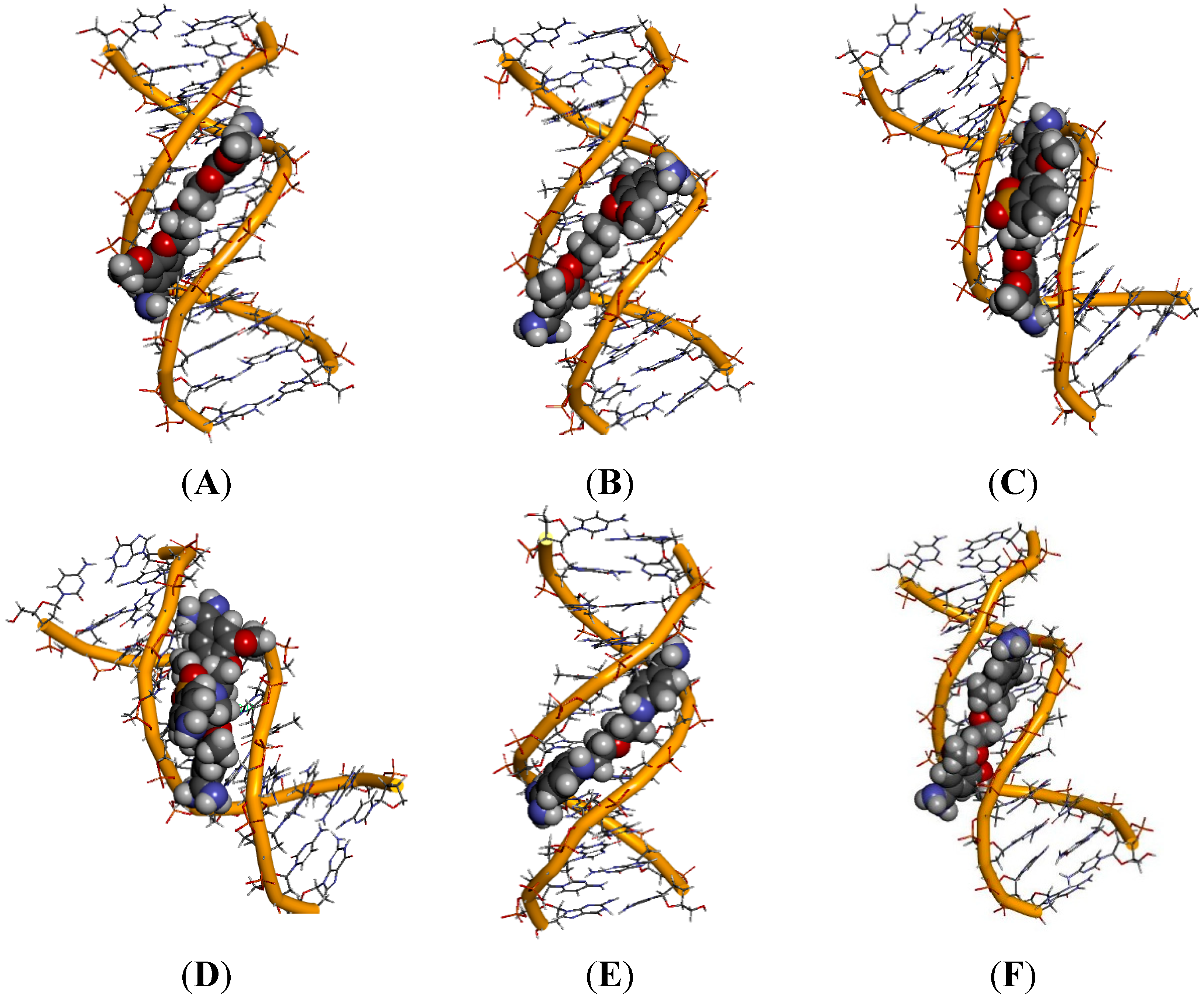
2.6. Hydration at the DNA-Ligand Complexes
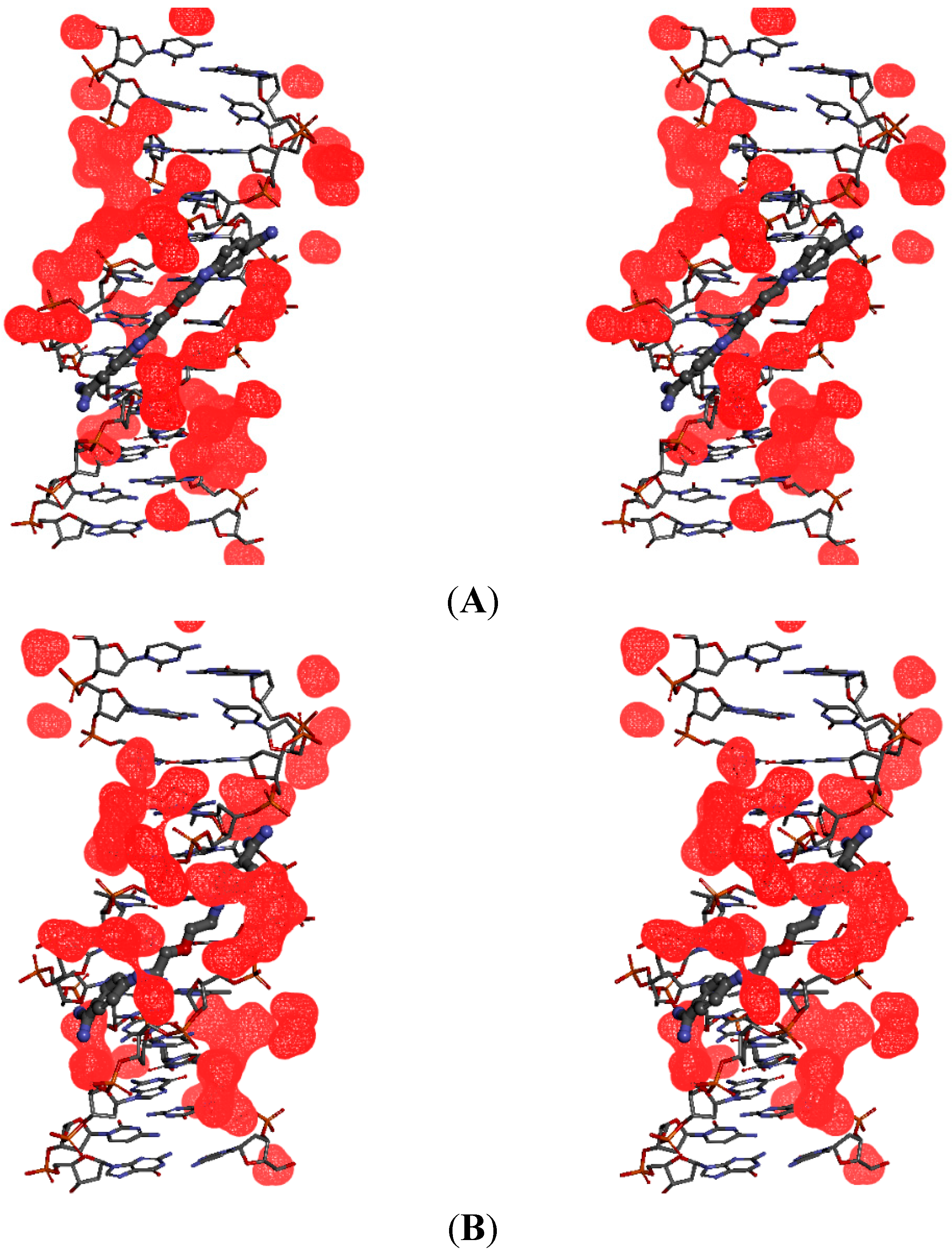
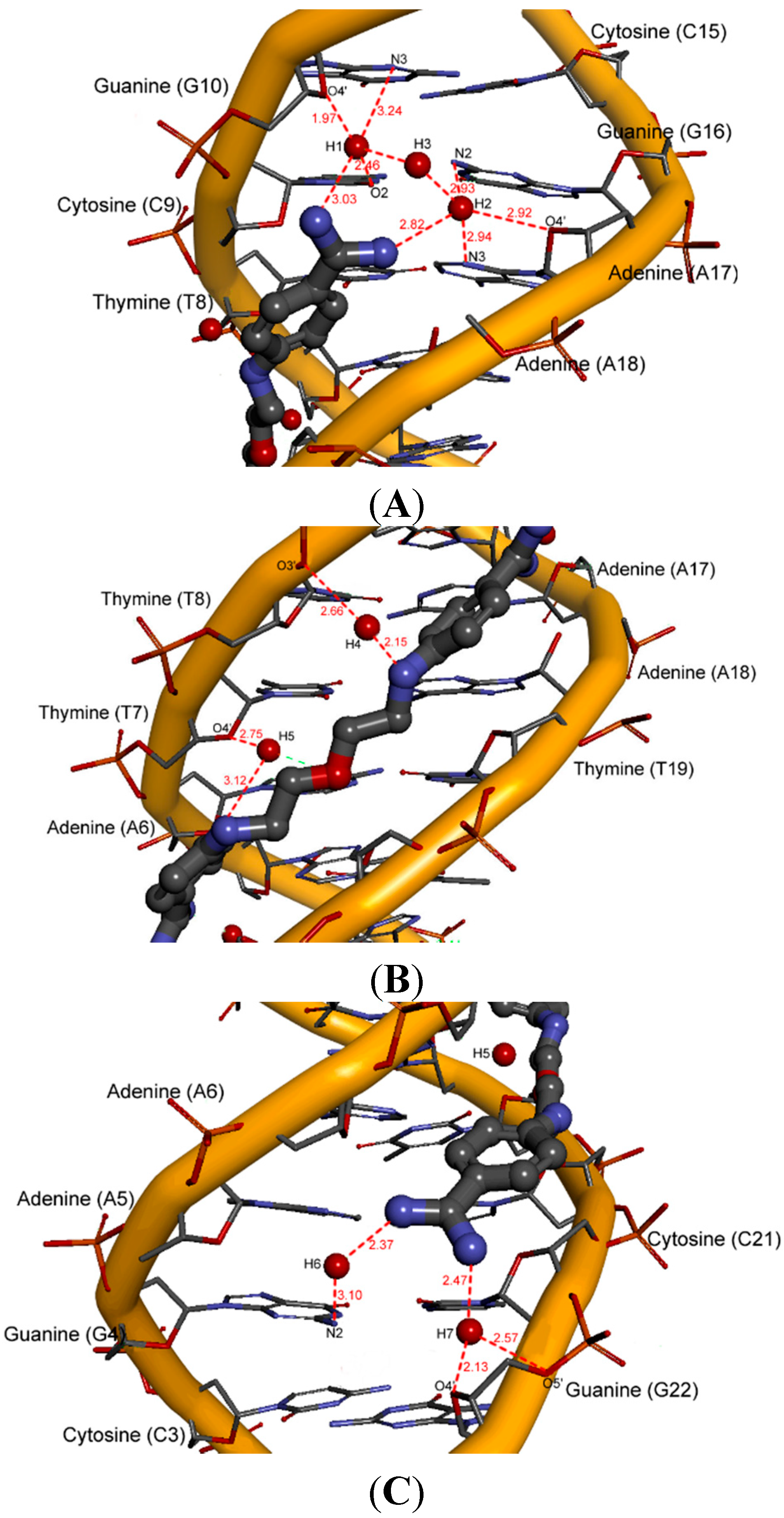
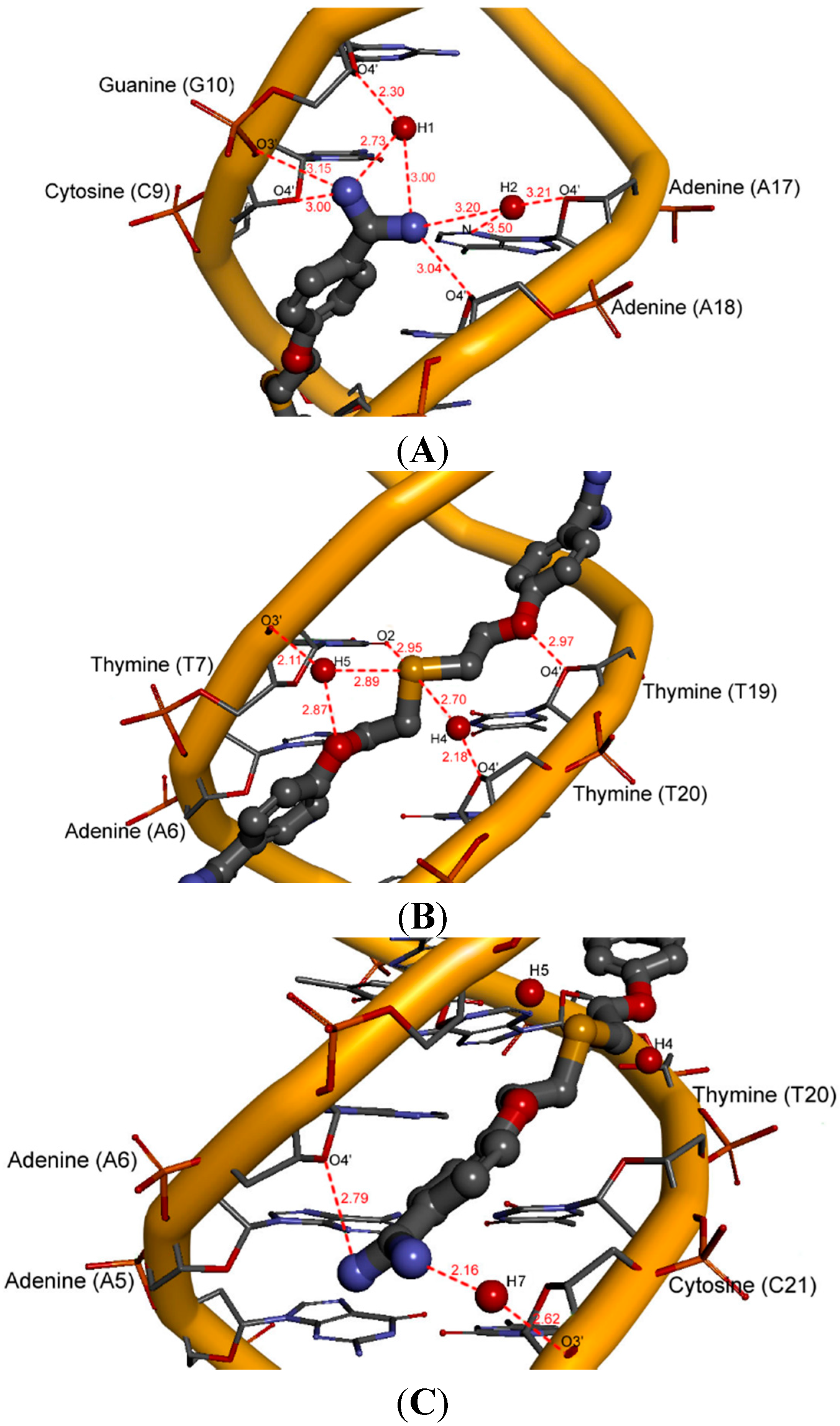
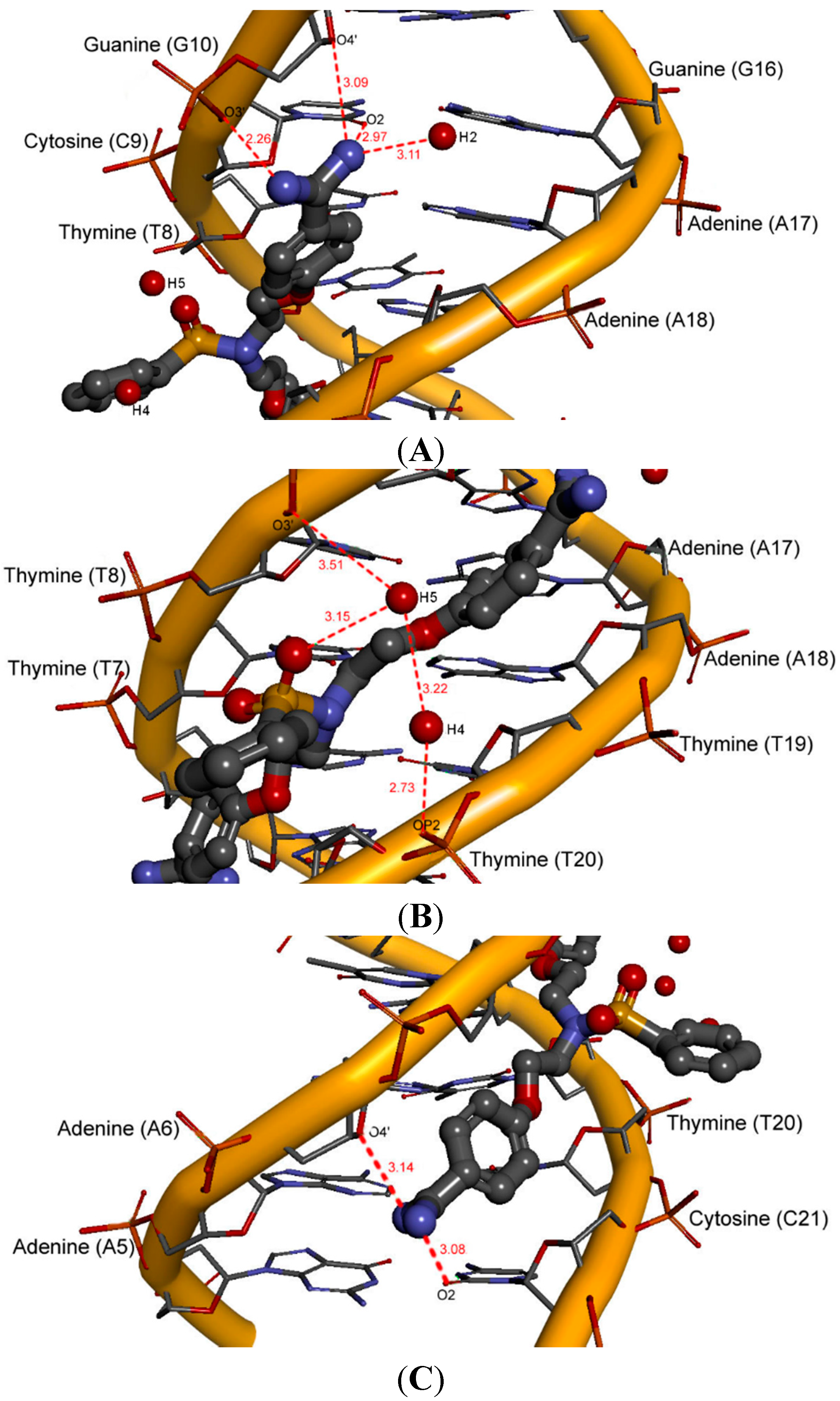
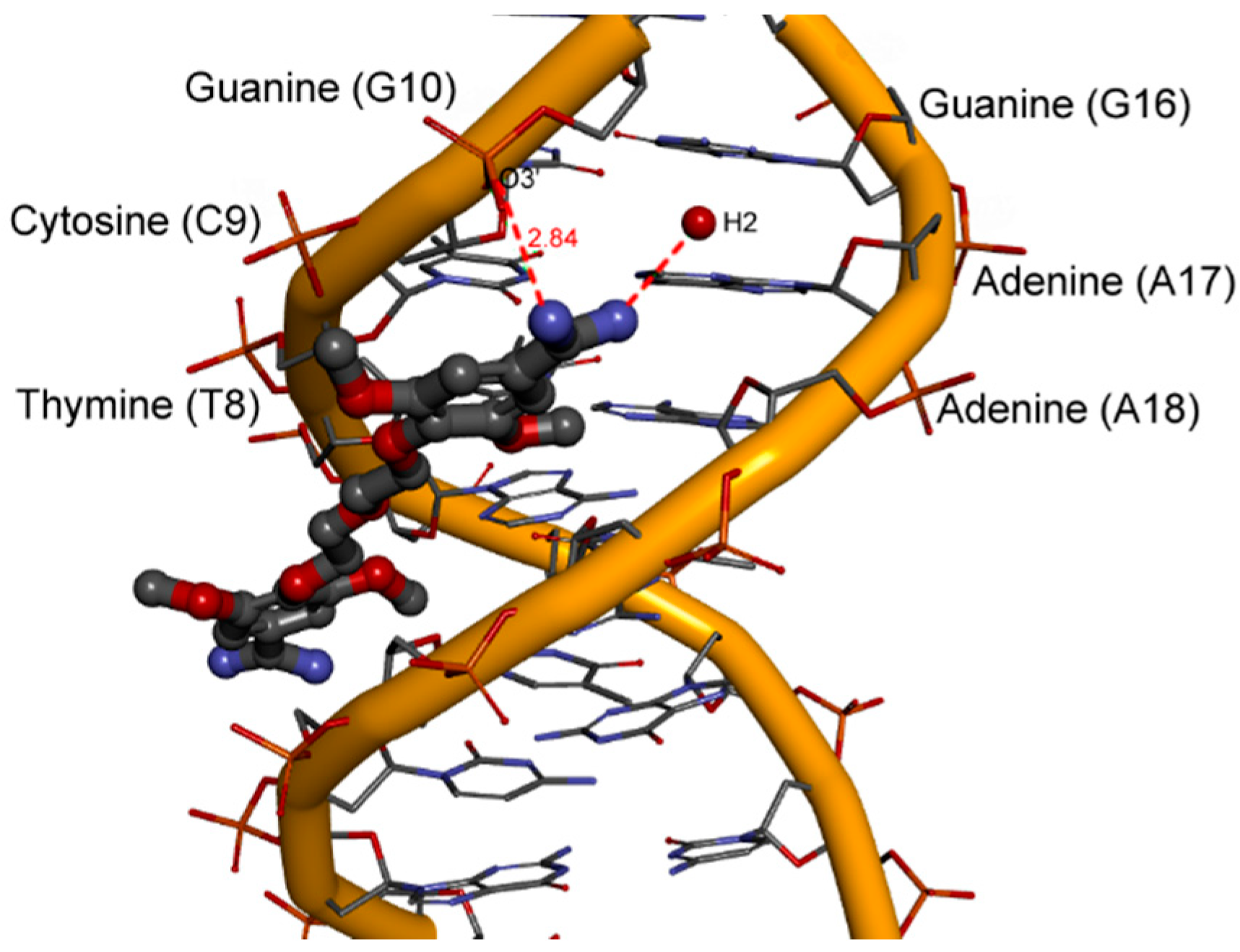
3. Experimental Section
3.1. DNA Melting Point Measurements (Tm)
3.2. ATP Bioluminescent Assay for Bisamidines
3.3. Computational Methods
3.4. Molecular Dynamics Simulations
3.5. Binding Free Energy Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tidwell, R.R.; Jones, S.K.; Geratz, J.D.; Ohemeng, K.A.; Cory, M.; Hall, J.E. Analogues of 1,5-bis(4-amidinophenoxy)pentane (pentamidine) in the treatment of experimental Pneumocystis carinii pneumonia. J. Med. Chem. 1990, 33, 1252–1257. [Google Scholar] [CrossRef] [PubMed]
- Fishman, J.A. Treatment of infection due to Pneumocystis carinii. Antimicrob. Agents Chemother. 1998, 42, 1309–1314. [Google Scholar] [PubMed]
- Ismail, M.A.; Arafa, R.K.; Brun, R.; Wenzler, T.; Miao, Y.; Wilson, W.D.; Bridges, A.; Hall, J.E.; Boykin, D.W. Synthesis, DNA affinity, and antiprotozoal activity of linear dications: Terphenyl diamidines and analogues. J. Med. Chem. 2006, 49, 5324–5332. [Google Scholar] [CrossRef] [PubMed]
- Mathis, A.M.; Holman, J.L.; Sturk, L.M.; Ismail, M.A.; Boykin, D.W.; Tidwell, R.R.; Hall, J.E. Acumulation and intracelular distribution of Antitrypasomal diamidine compounds DB75 and DB820 in African Trypanosomes. Antimicrob. Agents Chemother. 2006, 50, 2185–2191. [Google Scholar] [CrossRef] [PubMed]
- Brendle, J.J.; Outlaw, A.; Kumar, A.; Boykin, D.W.; Patrick, D.A.; Tidwell, R.R.; Werbovetz, K.A. Antileishmanial activities of several classes of aromatic dications. Antimicrob. Agents Chemother. 2002, 46, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Eynde, J.J.V.; Mayence, A.; Collins, M.S.; Cushion, M.T.; Rattendi, D.; Londono, I.; Mazumder, L.; Bacchi, C.J.; Yarlett, N. Synthesis and SAR of alkanediamidine-linked bisbenzamidines with anti-trypanosomal and antipneumocystis activity. Bioorg. Med. Chem. Lett. 2009, 19, 5884–5886. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Barrett, M.P.; Urbina, J.A. Chemotherapy of trypanosomiases and leishmaniasis. Trends Parasit. 2005, 21, 508–512. [Google Scholar] [CrossRef]
- Soeiro, M.N.C.; Souza, E.M.D.; Stephens, C.E.; Boykin, D.W. Aromatic diamidines as antiparasitic agents. Expert Opin. Investig. Drugs 2005, 14, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Queener, S.F. New drug developments for opportunistic infections in immunosuppressed patients: Pneumocystis carinii. J. Med. Chem. 1995, 38, 4739–4759. [Google Scholar] [CrossRef] [PubMed]
- Rolain, J.M.; Fenollar, F.; Raoult, D. In vitro activity of pentamidine against Tropheryma whipplei. Int. J. Antimicrob. Agents 2011, 38, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Minnick, M.F.; Hicks, L.D.; Battisti, J.M.; Raghavan, R. Pentamidine inhibits Coxiella burnetti growth and 23S rRNA intron splicing in vitro. Int. J. Antimicrob. Agents 2010, 36, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Walzer, P.D.; Perl, D.P.; Krogstad, D.J.; Rawson, P.G.; Schultz, M.G. Pneumocystis carinii pneumonia in the united states. Epidemiologic, diagnostic, and clinical feature. Ann. Intern. Med. 1974, 80, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska, D.; Żabiński, J.; Kaźmierczak, P.; Rezler, M.; Krassowska-Świebocka, B.; Collins, M.C.; Cushion, M.T. Analogs of pentamidine as potential anti-Pneumocystis chemotherapeutics. Eur. J. Med. Chem. 2012, 48, 164–173. [Google Scholar] [CrossRef]
- Daliry, A.; Pires, M.Q.; Silva, C.F.; Pacheco, R.S.; Munde, M.; Stephens, C.E.; Kumar, A.; Ismail, M.A.; Liu, Z.; Farahat, A.A.; et al. The Trypanocidal Activity of Amidine Compounds Does Not Correlate with Their Binding Affinity to Trypanosoma cruzi Kinetoplast DNA. Antimicrob. Agents Chemother. 2011, 55, 4765–4773. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.L.; Mayence, A.; Eynde, J.J.V. Some non-conventional biomolecular targets for diamidines. Bioorg. Med. Chem. 2014, 22, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.D.; Nguyen, B.; Tanious, F.A.; Mathis, A.M.; Hall, J.E.; Stephens, C.E.; Boykin, D.W. Dications that target the DNA minor groove: Compound design and preparation, DNA interactions, cellular distribution and biological activity. Curr. Med. Chem. Anti-Canc. Agents 2005, 5, 389–408. [Google Scholar] [CrossRef]
- Edwards, K.J.; Jenkins, T.C.; Neidle, S. Crystal structure of a pentamidine-oligonucleotide complex: Implications for DNA—Binding properties. Biochemistry 1992, 31, 7104–7109. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.C.; Lane, A.N.; Neidle, S.; Brown, D.G. NMR and modeling studies of the interaction of berenil and pentamidine with d(CGCAAATTTGCG)2. Eur. J. Biochem. 1993, 213, 1175–1184. [Google Scholar] [CrossRef] [PubMed]
- Nunn, C.M.; Jenkins, T.C.; Neidle, S. Crystal Structure of γ-Oxapentamidine Complexed with d(CGCGAATTCGCG)2: The Effects of Drug Structural Change on DNA Minor-Groove Recognition. Eur. J. Biochem. 1994, 226, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Tidwell, R.R.; Boykin, D.W. DNA and RNA Binders: From Small Molecules to Drugs; Wiley-VCH: New York, NY, USA, 2003; pp. 414–460. [Google Scholar]
- Laughton, C.A.; Tanious, F.A.; Nunn, C.M.; Boykin, D.W.; Wilson, W.D.; Neidle, S. A crystalographic and spectroscopic study of the complex between d(CGCGAATTCGCG)2 and 2,5-bis(4-guanylphenyl)furan, an analogue of berenil. Structural orgins of enhanced DNA-binding affinity. Biochemistry 1996, 35, 5655–5661. [Google Scholar] [CrossRef] [PubMed]
- Greenidge, P.A.; Jenkins, T.C.; Neidle, S. DNA minor groove recognition properties of pentamidine and its analogs: A molecular modeling study. Mol. Pharmacol. 1993, 43, 982–988. [Google Scholar] [PubMed]
- Montanari, C.A.; Tute, M.S.; Beezer, A.E.; Mithcell, J.C. Determination of receptor-bound drug conformations by QSAR using flexible fitting to derive a molecular similarity index. J. Comput. Aided Mol. Des. 1996, 10, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Simpson, I.J.; Michael, I.; Kumar, A.; Boykin, D.W.; Neidle, S. DNA minor groove interactions and biological activity of 2,5-bis-[4-(N-alkylamidino)phenyl]furans. Bioorg. Med. Chem. Lett. 2000, 10, 2593–2597. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.M.D.; Custodio, F.B.; Donnici, C.L.; Montanari, C.A. QSAR and molecular modeling studies of B-DNA recognition of minor groove binders. Eur. J. Med. Chem. 2003, 38, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Breslauer, K.J. Methods for obtaining thermodynamic data on oligonucleotide transitions. In Thermodynamic Data for Biochemistry and Biotechnology; Hinz, H.J., Ed.; Springer: New York, NY, USA, 1986; pp. 402–427. [Google Scholar]
- Żołek, T.; Maciejewska, D. Theoretical models of pentamidine analogs based on their DNA minor groove complexes. Eur. J. Med. Chem. 2010, 45, 1991–1999. [Google Scholar] [CrossRef]
- Nguyen, B.; Lee, M.P.H.; Hamelberg, D.; Joubert, A.; Bailly, C.; Brun, R.; Neidle, S.; Wilson, W.D. Strong binding in DNA minor groove by an aromatic diamidine with a shape that does not math the curvature of the groove. J. Am. Chem. Soc. 2002, 124, 13680–13681. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for the simulation of liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Guerri, A.; Simpson, I.J.; Neidle, S. Visualisation of extensive water ribbons and networks in a DNA minor-groove drug complex. Nucleic Acids Res. 1998, 26, 2873–2878. [Google Scholar] [CrossRef] [PubMed]
- Cushion, M.T.; Walzer, P.D.; Collins, M.C.; Rebholz, S.; Vanden Eynde, J.J.; Mayence, A.; Huang, T.L. Highly active anti-Pneumocystis carinii compounds in a library of novel piperazine-linked bisbenzamidines and related compounds. Antimicrob. Agents Chemother. 2004, 48, 4209–4209. [Google Scholar] [CrossRef] [PubMed]
- Spackova, N.; Cheatham, T.E., III; Ryjacek, F.; Lankas, F.; Meervelt, L.; Hobza, P.; Sponer, J. Molecular dynamics simulations and thermodynamics analysis of DNA-drug complexes. Minor groove binding between 4',6-diamidino-2-phenylidole and DNA duplexes in solution. J. Am. Chem. Soc. 2003, 125, 1759–1769. [Google Scholar] [CrossRef]
- Madhumalar, A.; Bansal, M. Structural insights into the effect of hydration and Ions on A-tract DNA: A molecular dynamics study. Biophys. J. 2003, 85, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Żabiński, J.; Wolska, I.; Maciejewska, D. Comparsion of structure in solid state of new 1,5-bis(4-cyano-2,6-dimethoxyphenoxy)alkanes by means of 13C CP/MAS NMR and X-ray diffraction. J. Mol. Struct. 2007, 883, 74–81. [Google Scholar] [CrossRef]
- Maciejewska, D.; Kaźmierczak, P.; Żabiński, J.; Wolska, I.; Popis, S. Pentamidine analogs: Syntheses, structures. In solid state by 13C CP/MAS NMR spectroscopy, and X-ray crystallography and their preliminary biological screening against human cancer. Monatshefte fuer Chemie 2006, 137, 1225–1240. [Google Scholar] [CrossRef]
- Cushion, M.T.; Walzer, P.D.; Ashbaugh, A.; Rebholz, S.; Brubaker, R.; Vandes Eynde, J.J.; Mayence, A.; Huang, T.L. In vitro selection and in vivo efficacy of piperazine- and alkanediamide-linked bisbenzamidines against Pneumocystis Pneumonia in mice. Antimicrob. Agents Chemother. 2006, 50, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Accelrys Software Inc. Discovery Studio Modeling Environment; Release 3.1; Accelrys Software Inc.: San Diego, CA, USA, 2011. [Google Scholar]
- Frish, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Zakrzewski, V.G.; Montgomery, J.A.J.; Stratmann, R.E.; Burant, J.C.; et al. Gaussian 03; Revision C.02; Gaussian, Inc.: Pittsburgh, PA, USA, 2009. [Google Scholar]
- Breneman, C.M.; Wiberg, K.B. Determing atom-centered monopoles from molecular electrostatic potentials. The need for high sampling in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Brooks, B.R.; Bruccoleri, R.E.; Olafson, B.D.; States, D.J.; Swaminathan, S.; Karplus, M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem. 1983, 4, 187–217. [Google Scholar] [CrossRef]
- Foloppe, N.; MacKerell, A.D., Jr. All-atom empirical force field for nucleic acids: I. Parameter optimization based on small molecule and condensed phase macromolecular target data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- MacKerell, A.D.J.; Banavali, N.K. All-atom empirical force field for nucleic acids: II. Application to molecular dynamics simulations of DNA and RNA in solution. J. Comput. Chem. 2000, 21, 105–120. [Google Scholar] [CrossRef]
- Aqvist, J. Ion-water interaction potentials derived from free energy perturbation simulations. J. Phys. Chem. 1990, 94, 8021–8024. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Brerkowitz, M.L.; Darden, T.; Lee, H.; Pedesen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Sagui, C.; Darden, T.A. Molecular dynamics simulations of biomolecules: Long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155–179. [Google Scholar] [CrossRef] [PubMed]
- Steinbach, P.J.; Brooks, B.R. New spherical-cutoff methods for long-range forces in macromolecular simulation. J. Comput. Chem. 1994, 15, 667–683. [Google Scholar] [CrossRef]
- Norberg, J.; Nilsson, L. On the truncation of long-range electrostatic interactions in DNA. Biophys. J. 2000, 79, 1537–1553. [Google Scholar] [CrossRef] [PubMed]
- Trieb, M.; Rauch, C.; Wellenzohn, B.; Wibowo, F.; Loerting, T.; Liedl, K.R. Dynamics of DNA: BI and BII Phosphate Backbone Transitions. J. Phys. Chem. B 2004, 108, 12258–12258. [Google Scholar] [CrossRef]
- Lei, H.; Wang, X.; Wu, C. Early stage intercalation of doxorubicin to DNA fragments observed in molecular dynamics binding simulations. J. Mol. Graph. Model. 2012, 38, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Dolenc, J.; Oostenbrink, C.; Koller, J.; Gunsteren, W.F. Molecular dynamics simulations and free energy calculations of netropsin and distamycin binding to an AAAAA DNA binding sit. Nucl. Acids Res. 2005, 33, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Johnson, T.L.; Clugston, S.; Huang, H.; Butenhof, K.J.; Stanton, R.V. molecular dynamics simulation and binding energy calculation for estimation of oligonucleotide duplex thermostability in RNA-based therapeutics. J. Chem. Inf. Model 2011, 51, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Verlet, L. Computer “Experiments” on classical fluids. I. Thermodynamical properties of Lennard-Jones molecules. Phys. Rev. 1967, 159, 98–100. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Orozco, M.; Laughton, C.A.; Herzyk, P.; Neidle, S. Molecular- mechanics modelling of drug-DNA structure; the effects of differing dielectric treatment on helix parameters and comparison with a fully solvated structural model. J. Biomol. Struct. Dyn. 1990, 8, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Fogolari, F.; Tosatto, S.C.E. Application of MM/PBSA colony free energy to loop decoy discrimination: Towards corelation between energy and root mean square deviation. Protein Sci. 2005, 14, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Treesuwan, W.; Wittayanarakul, K.; Anthony, N.G.; Huchet, G.; Alniss, H.; Hannongbua, S.; Khalaf, A.I.; Suckling, C.J.; Parkinson, J.A.; Mackay, S.P. A detailed binding free energy study of 2:1 ligand-DNA complex formation by experiment and simulation. Phys. Chem. Chem. Phys. 2009, 11, 10682–10693. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.; Hamelberg, D.; Bailly, C.; Colson, P.; Stanek, J.; Brun, R.; Neidle, S.; Wilson, W.D. Characterization of novel DNA minor-groove complex. Biophys. J. 2004, 86, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żołek, T.; Maciejewska, D.; Żabiński, J.; Kaźmierczak, P.; Rezler, M. Probing the Relationship between Anti-Pneumocystis carinii Activity and DNA Binding of Bisamidines by Molecular Dynamics Simulations. Molecules 2015, 20, 5942-5964. https://doi.org/10.3390/molecules20045942
Żołek T, Maciejewska D, Żabiński J, Kaźmierczak P, Rezler M. Probing the Relationship between Anti-Pneumocystis carinii Activity and DNA Binding of Bisamidines by Molecular Dynamics Simulations. Molecules. 2015; 20(4):5942-5964. https://doi.org/10.3390/molecules20045942
Chicago/Turabian StyleŻołek, Teresa, Dorota Maciejewska, Jerzy Żabiński, Paweł Kaźmierczak, and Mateusz Rezler. 2015. "Probing the Relationship between Anti-Pneumocystis carinii Activity and DNA Binding of Bisamidines by Molecular Dynamics Simulations" Molecules 20, no. 4: 5942-5964. https://doi.org/10.3390/molecules20045942
APA StyleŻołek, T., Maciejewska, D., Żabiński, J., Kaźmierczak, P., & Rezler, M. (2015). Probing the Relationship between Anti-Pneumocystis carinii Activity and DNA Binding of Bisamidines by Molecular Dynamics Simulations. Molecules, 20(4), 5942-5964. https://doi.org/10.3390/molecules20045942