
Multifaceted Strategy for the Synthesis of Diverse 2,2'-Bithiophene Derivatives

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
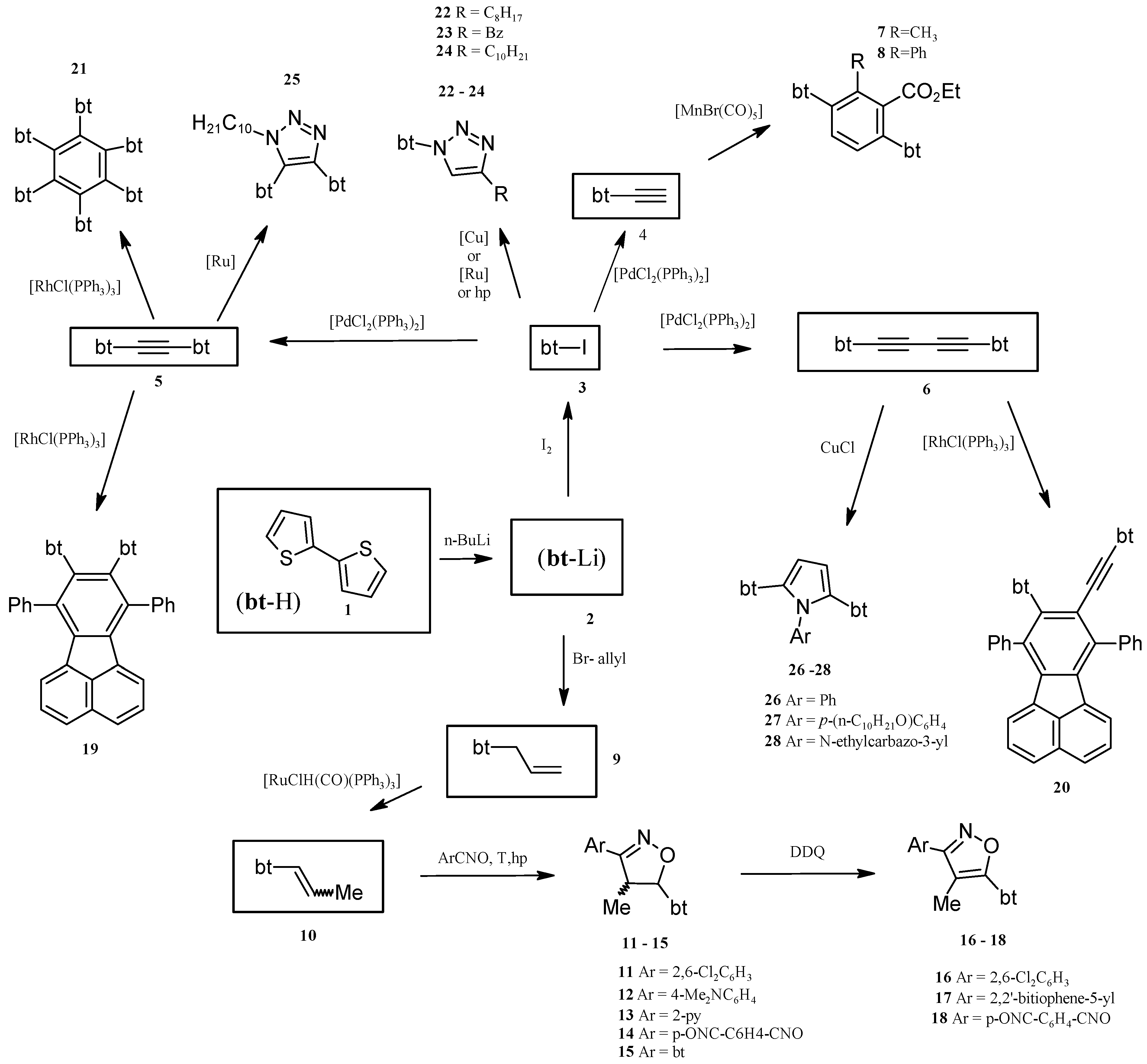
2. Results and Discussion
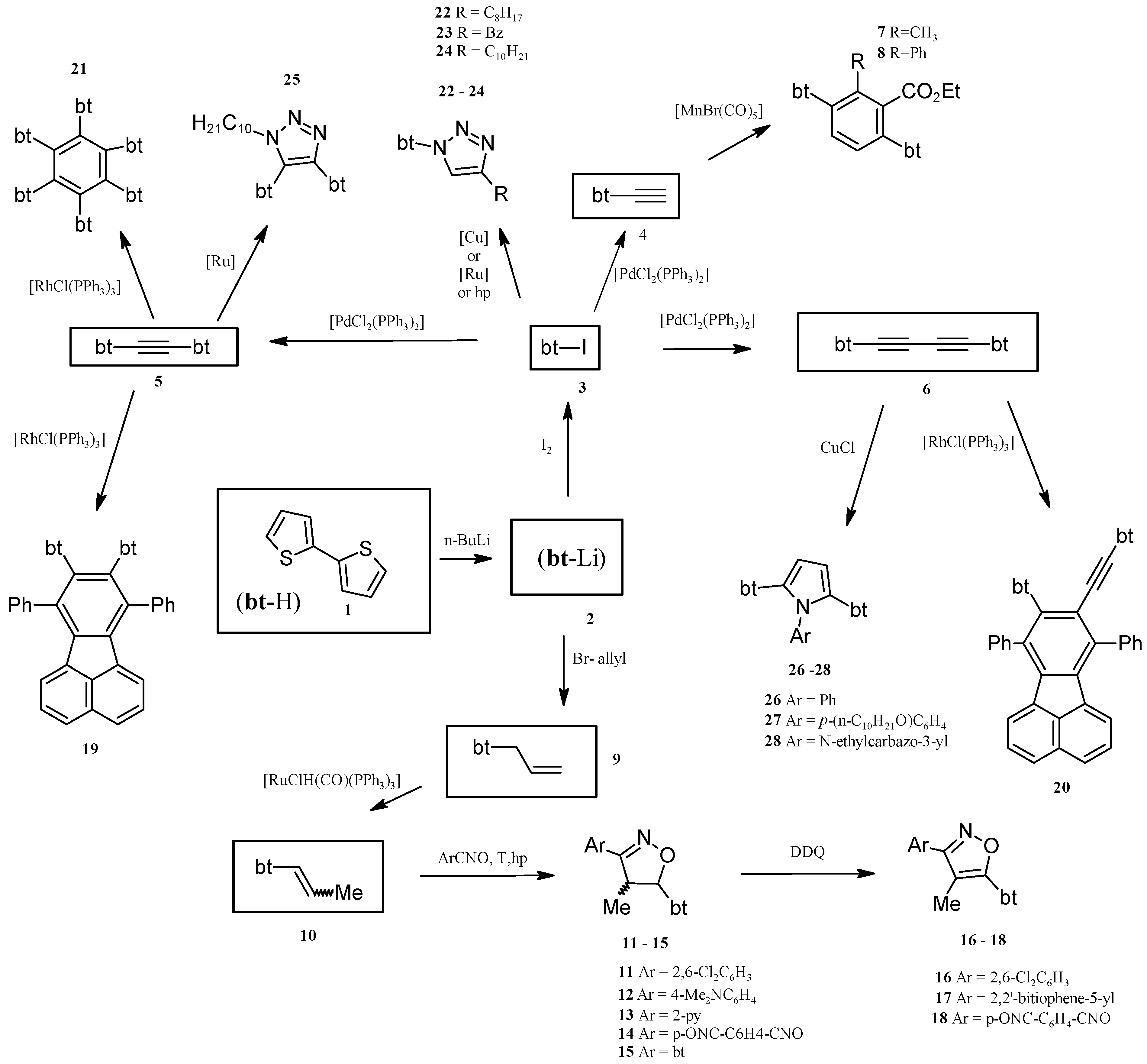
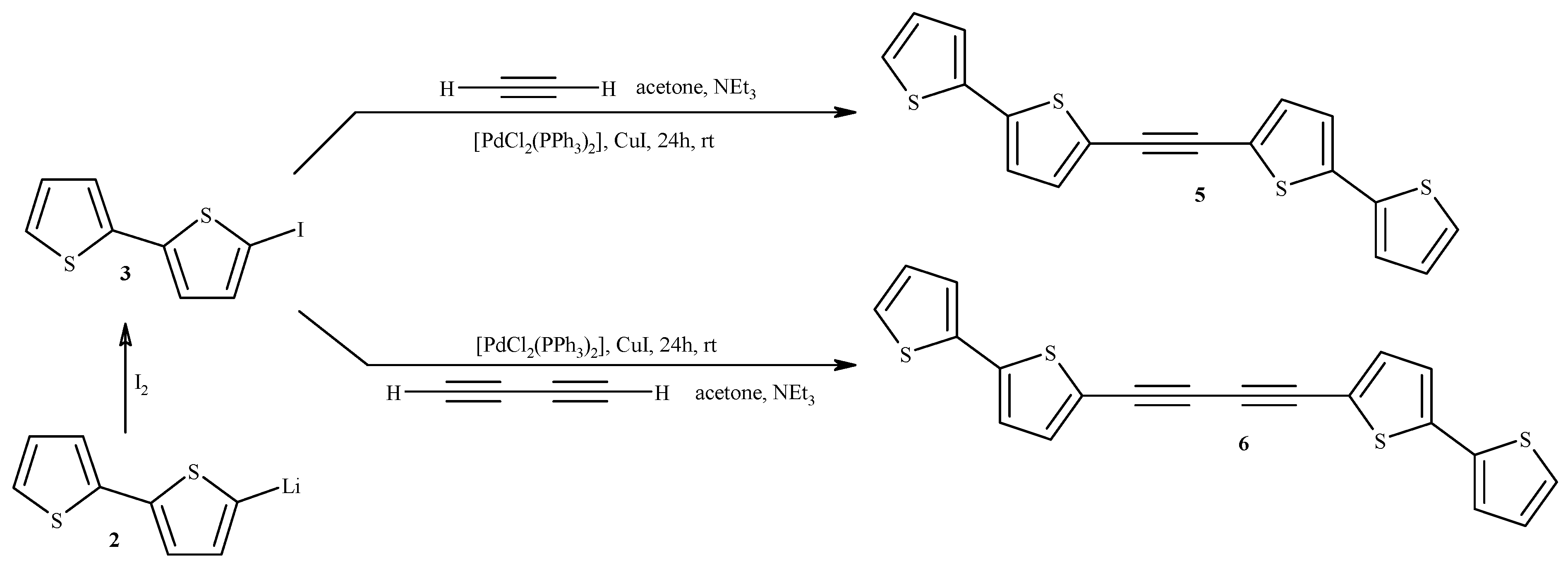
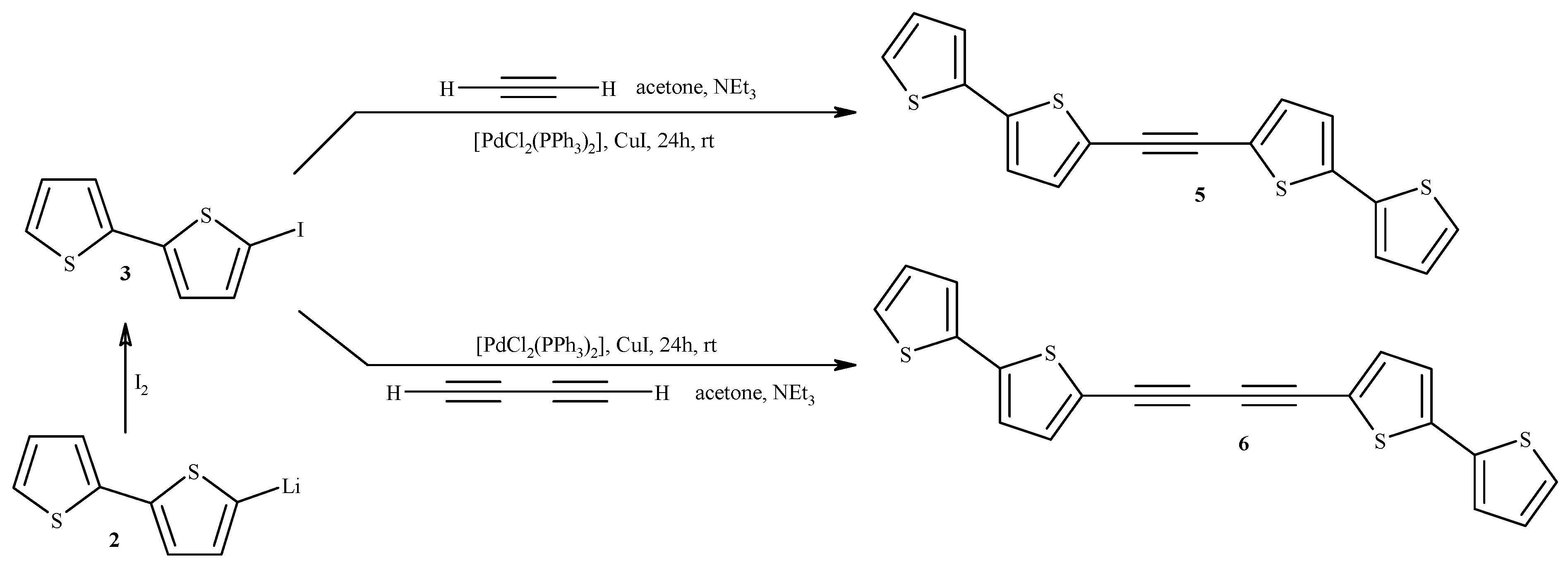
2.1. Synthesis of Substrates (for Catalytic Reactions) Containing 2,2'-Bithiophene Moiety from 2,2'-Bithiophene

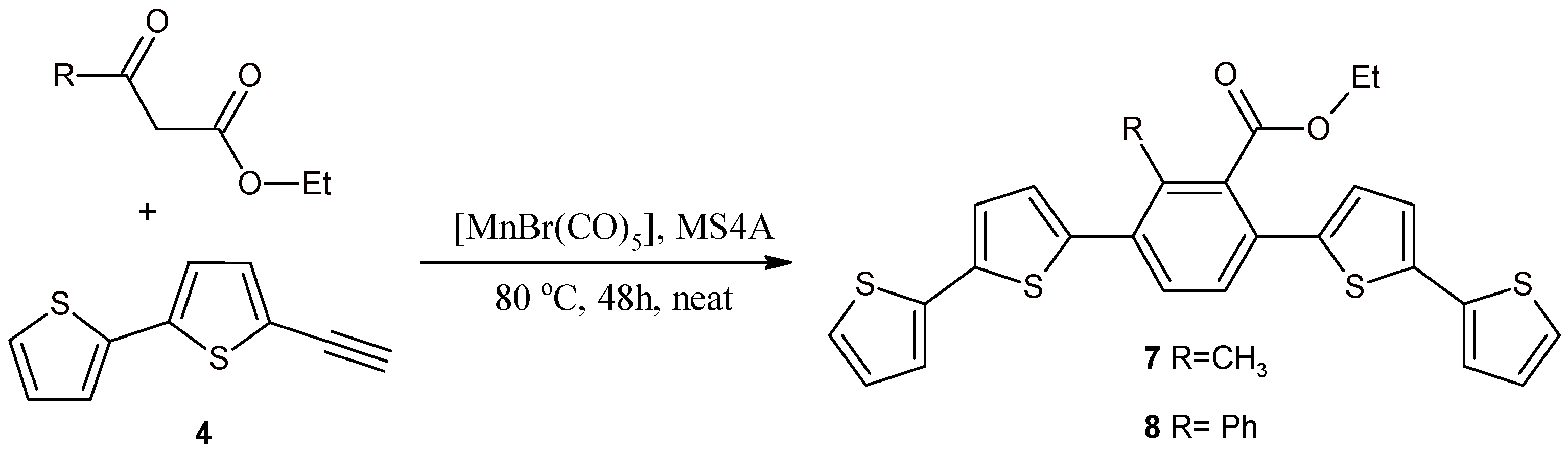
2.2. Benzene Derivatives with Two bt Moieties Prepared via Manganese-Catalyzed Reaction
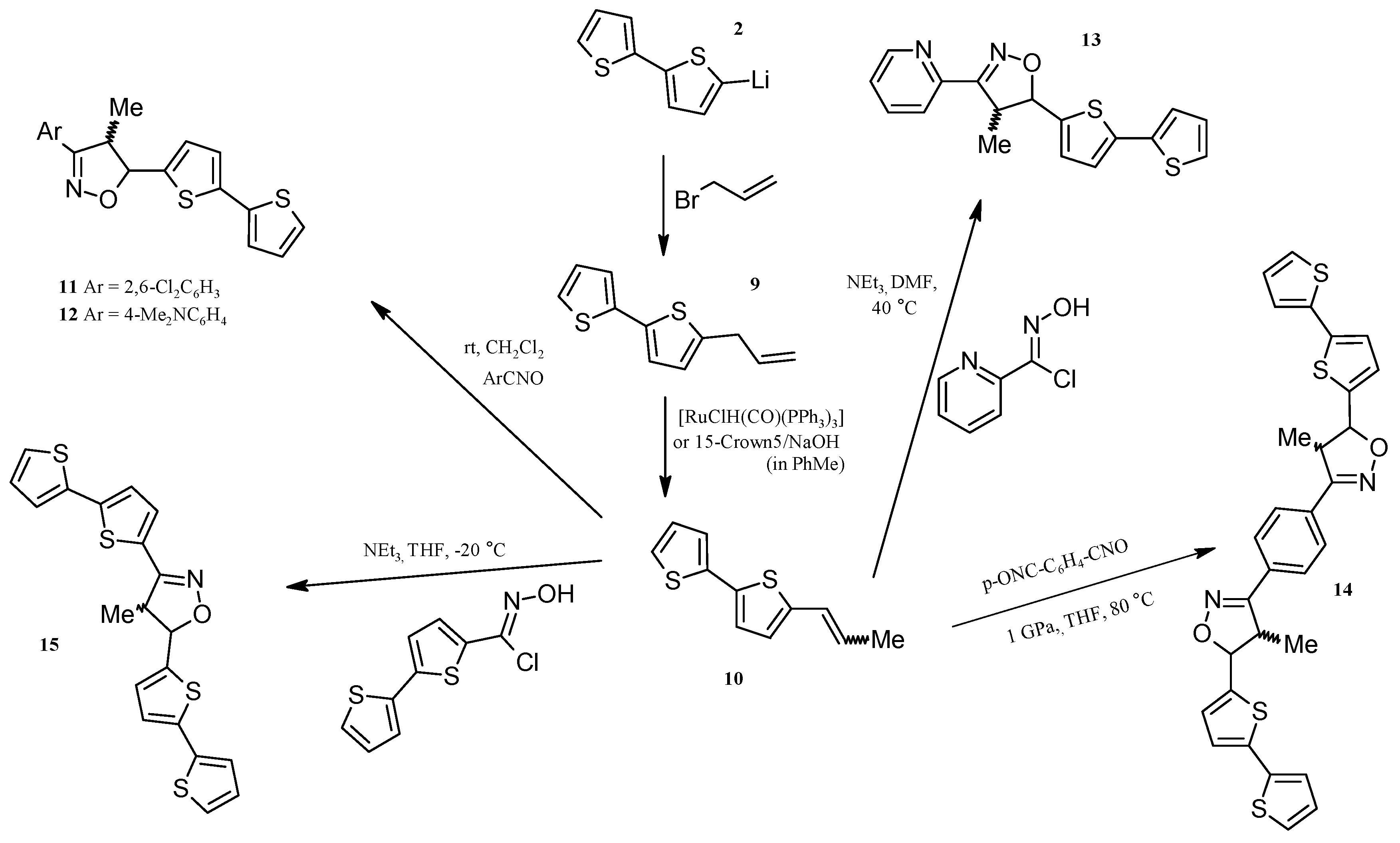
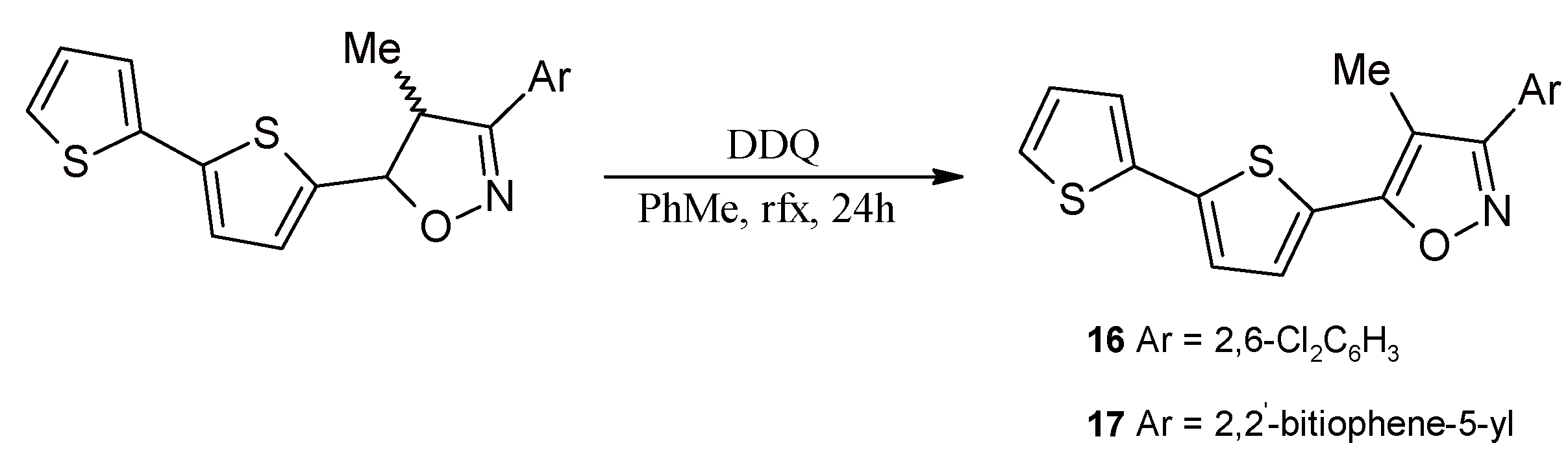
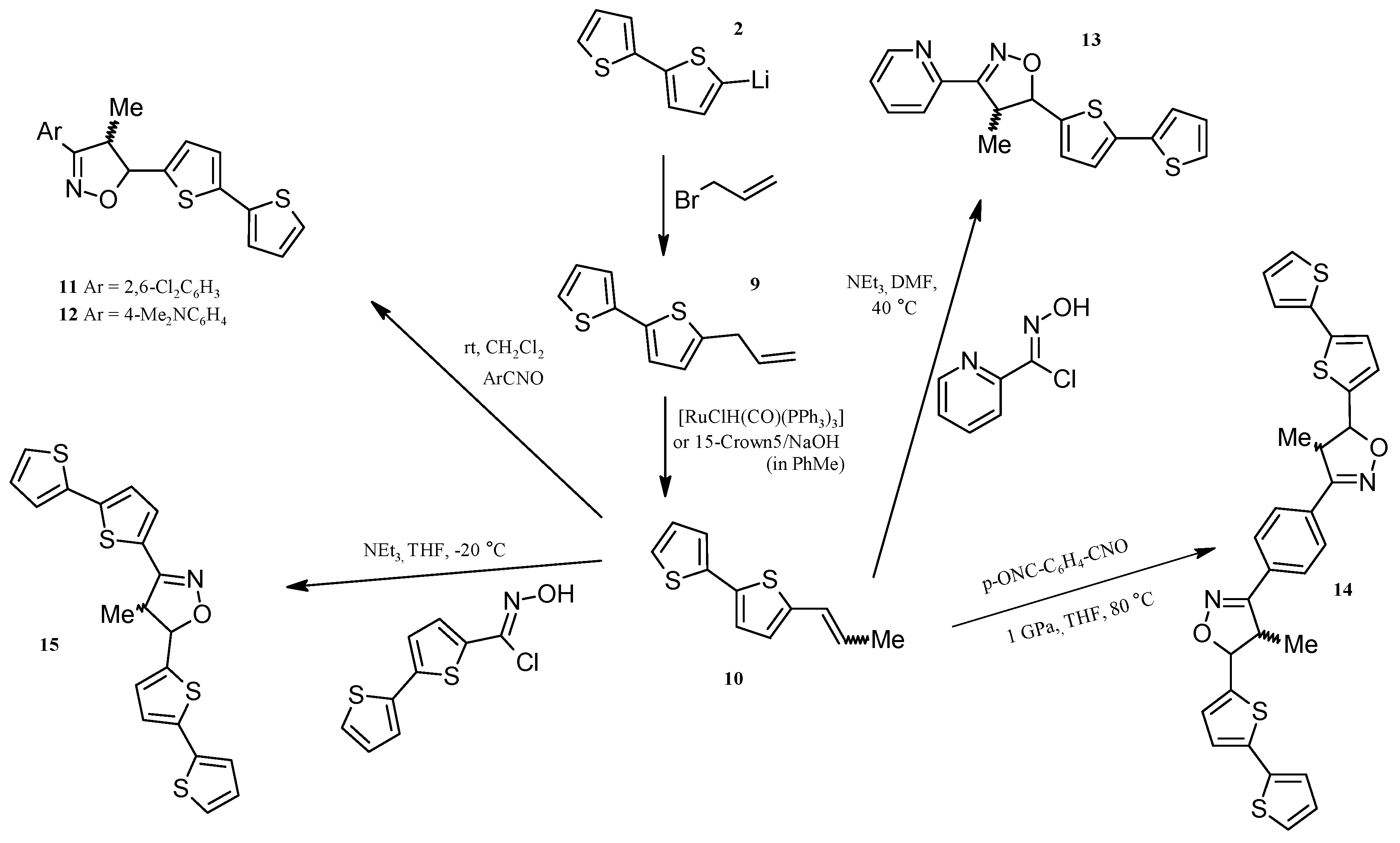
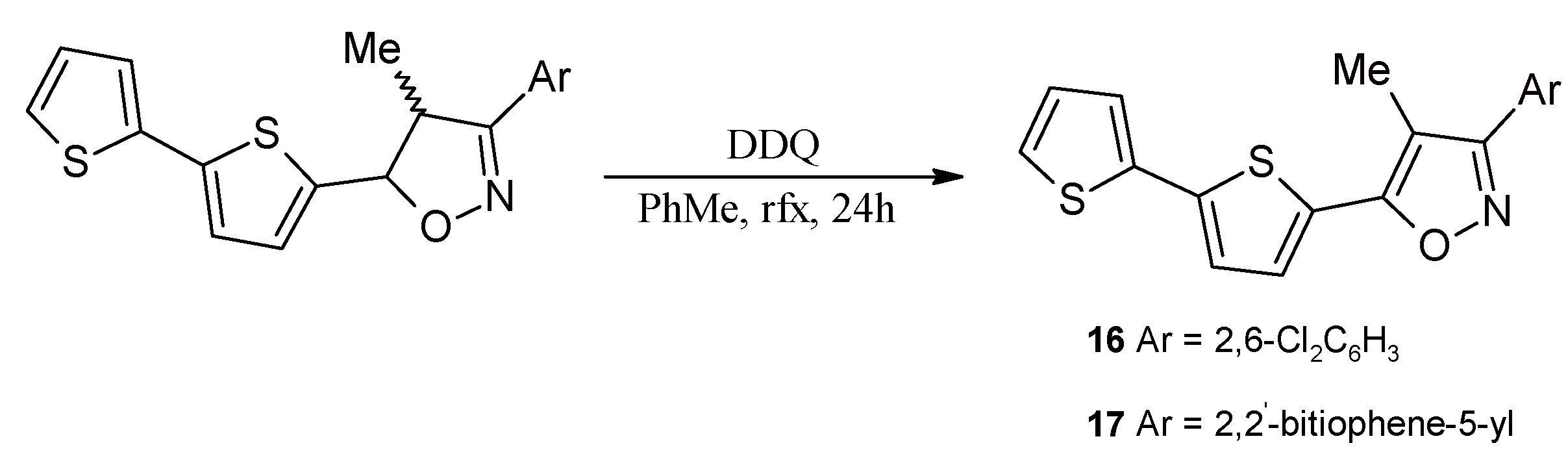
2.3. Isoxazolines With bt Motifs
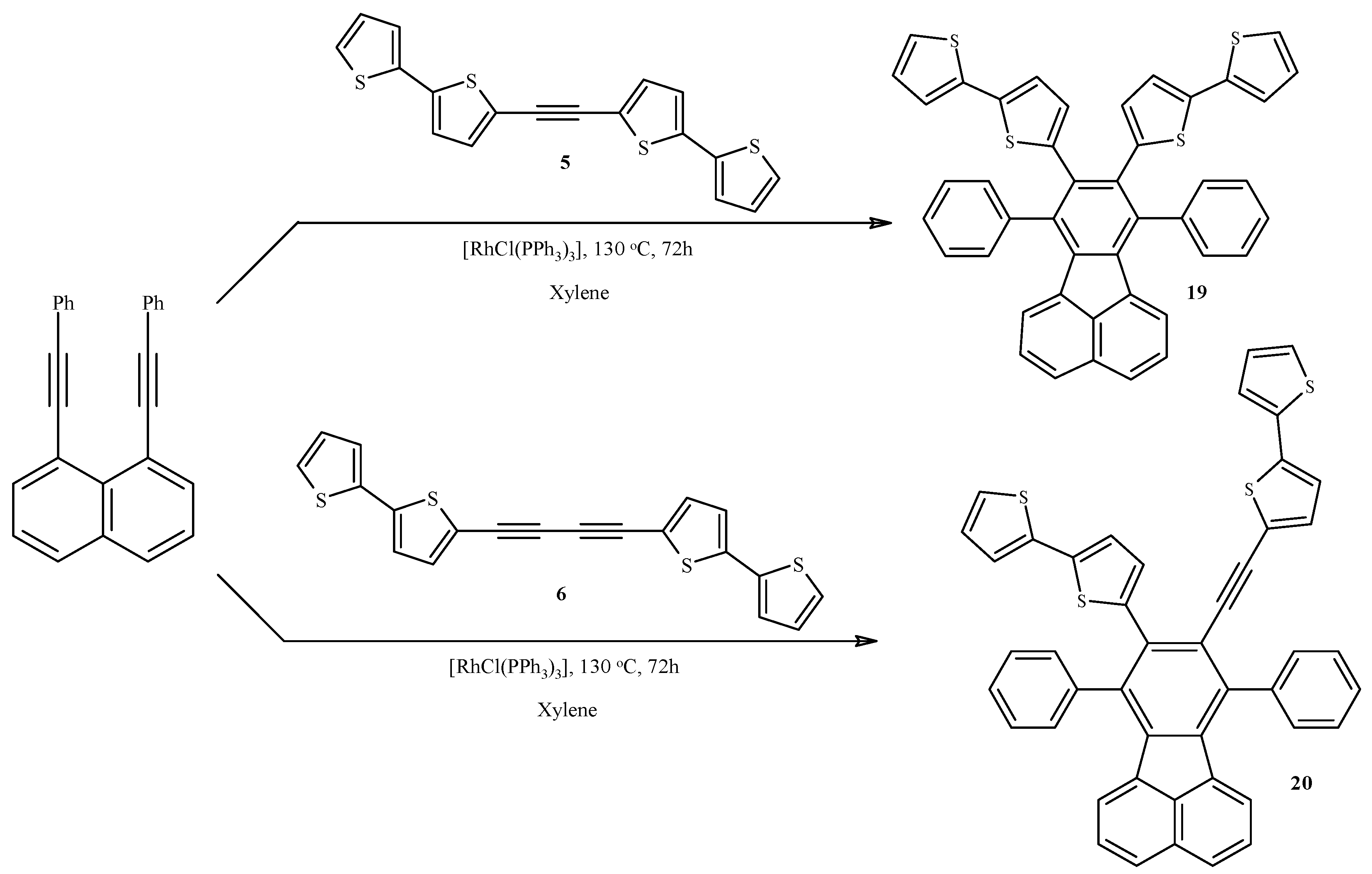
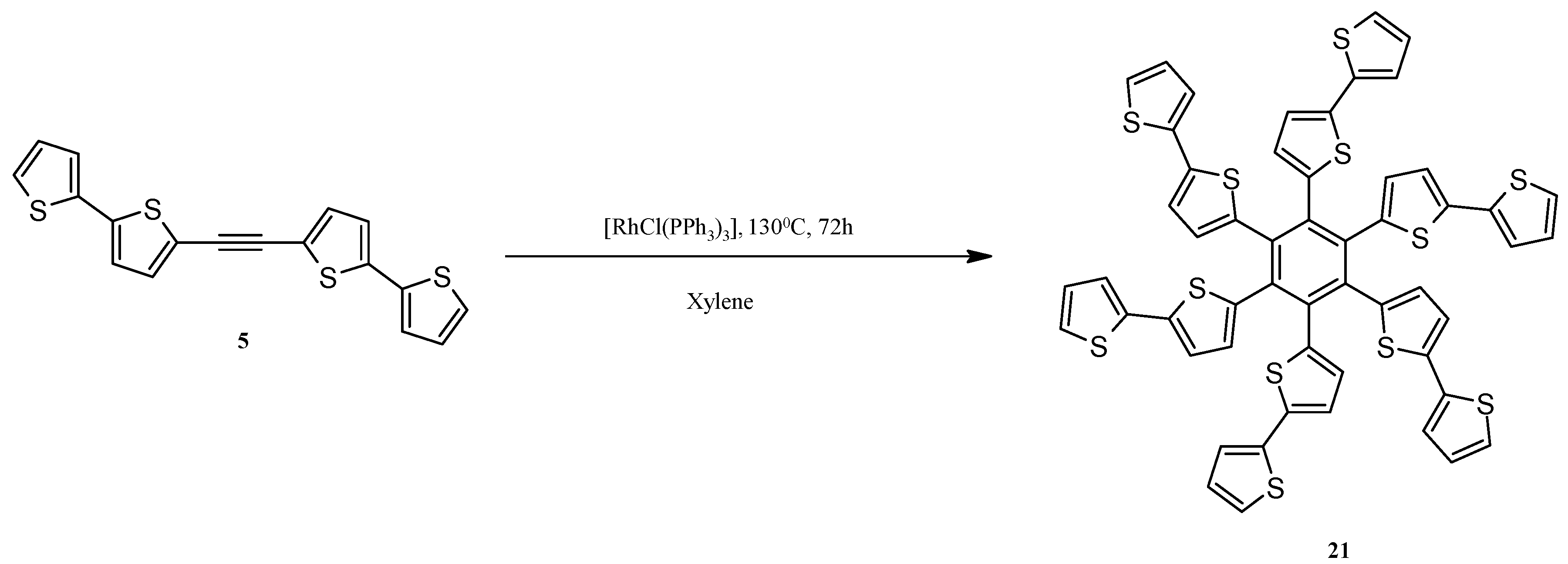
2.4. Rh-Mediated [2 + 2 + 2] Cycloaddition for Synthesis of Fluoranthene and Benzene Derivatives with a bt Motif
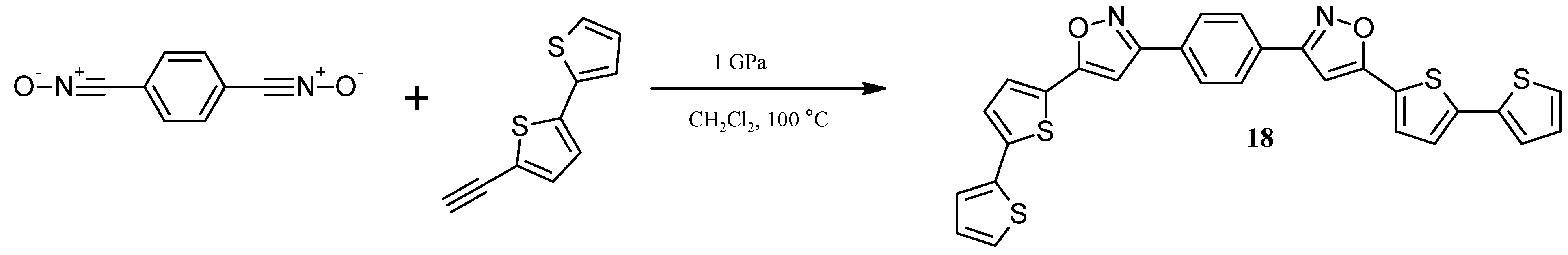
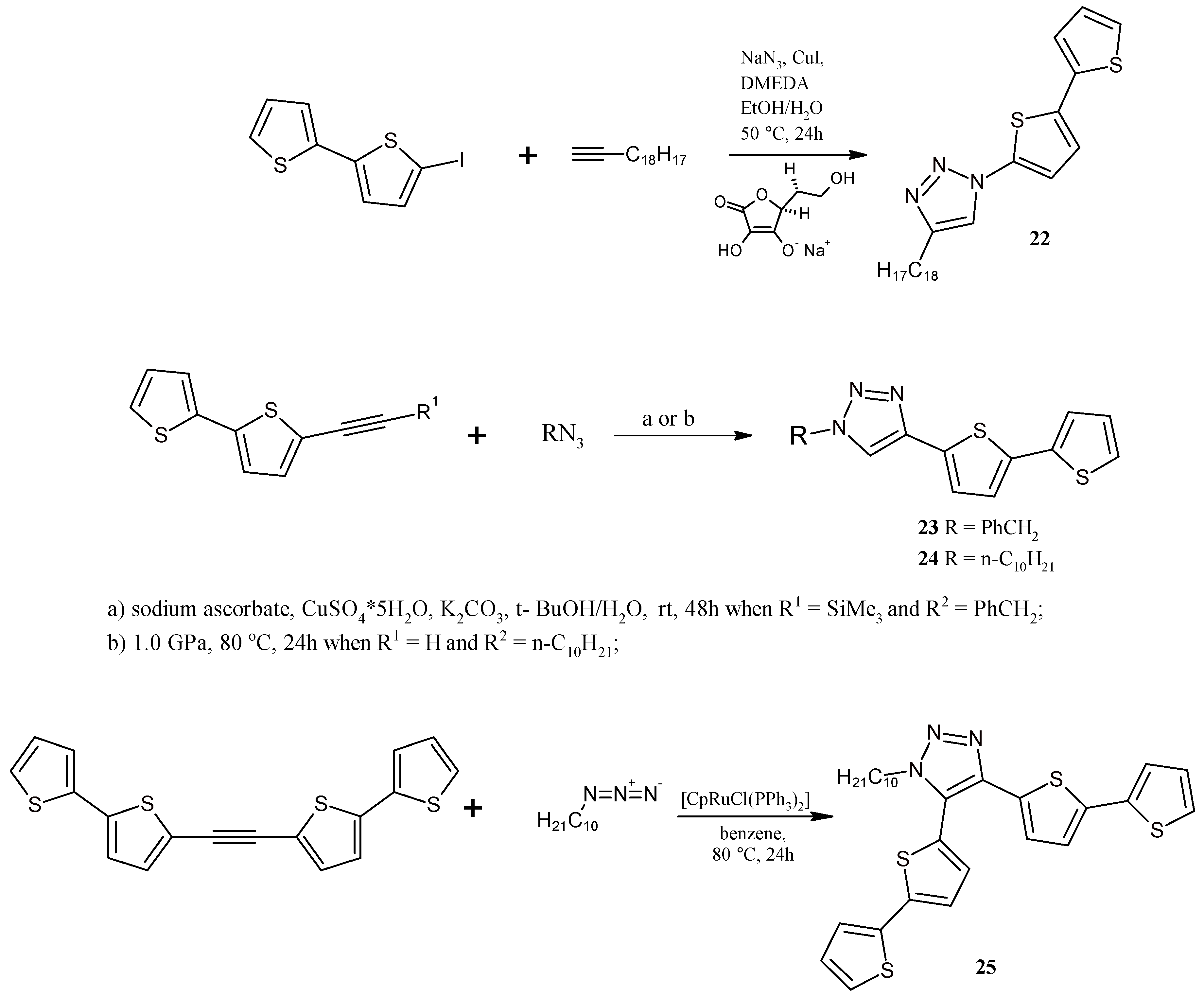
2.5. Catalytically or High Pressure Activated 1,3-DC of Azides to Triple Bond for Synthesis of Triazoles with bt Moiety
2.6. Synthesis of Pyrroles with two bt Moieties via CuCl-Mediated Dihydroamination of 1,4-Bis(2,2'-bithiophene-5-yl)buta-1,3-diyne
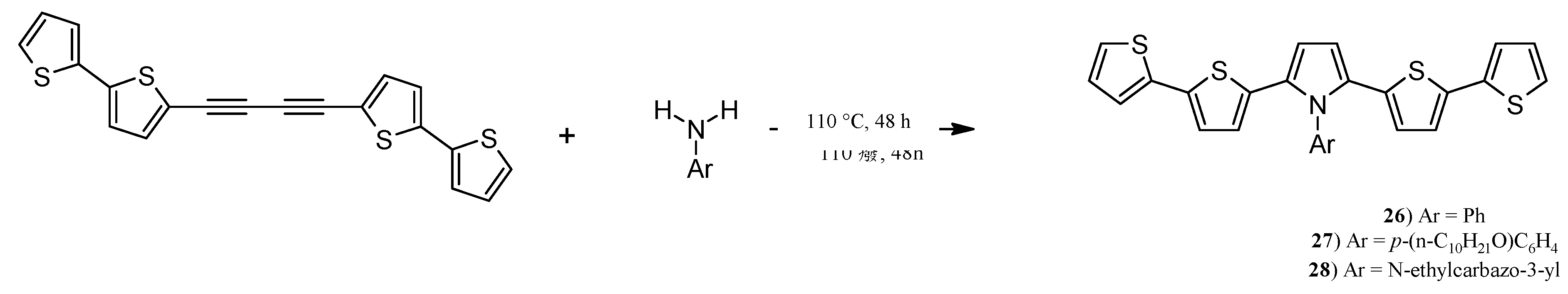
3. Experimental Section
3.1. General Methods
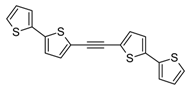
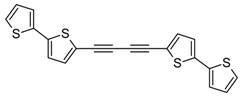
3.2. Synthesis of 1,2-Bis(2,2'-bithiophene-5-yl)acetylene (5) and 1,4-Bis(2,2'-bithiophene-5-yl)buta-1,3-diyne (6)
3.3. Synthesis of 1,4-Bis(2,2'-bithiophene-5-yl)-3-methyl-2-ethoxycarbonylbenzene (7) and 1,4-Bis(2,2'-bithiophene-5-yl)-3-phenyl-2-ethoxycarbonylbenzene (8)
3.4. Synthesis of 5-Allyl-2,2'-bithiophene (9)
Isomerization of 5-Allyl-2,2'-bithiophene (9), N-allylimidazole and Allyl-phenyl sulfide to 1-propenyl derivatives catalysed by 15-crown-5/NaOH
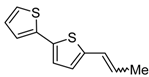
3.5. Synthesis of (E) + (Z)-5-(1-Propenyl)-2,2'-bithiophene (10)
3.6. General Procedure for 1,3-Dipolar Cycloaddition: Synthesis of 11–13
3.7. Synthesis of 1,4-Bis(5-(2,2'-bithiophen-5-ylo)-4-methylisoxazoline-3-yl)benzene (14) under High Pressure
3.8. Synthesis of 3,5-Bis(2,2'-bithiophen-5-yl)-4-methylisoxazoline (15)
3.9. Synthesis of 5-(2,2'-Bithiophen-5-yl)-3-(2,6-dichlorophenyl)-4-methylisoxazole (16) and 3,5-Bis(2,2'-bithiophene-5-yl)-4-methylisoxazole (17)
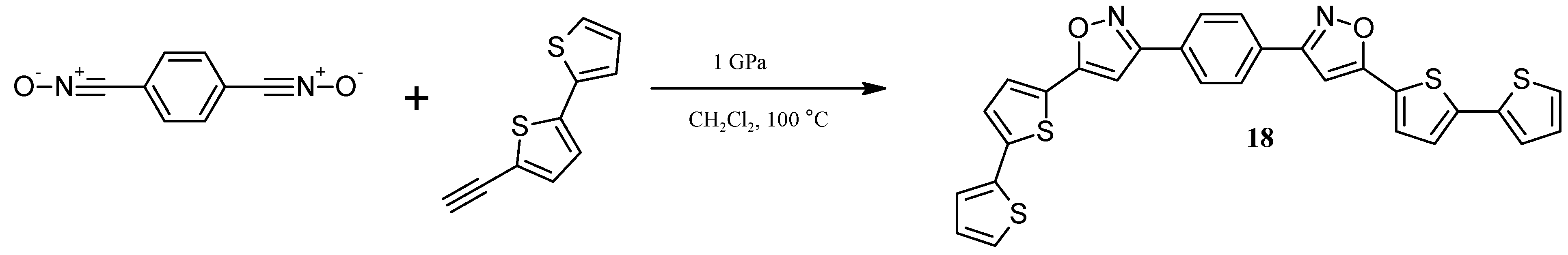
3.10. Synthesis of1,4-Bis[5-(2,2'-bithiophen-5-yl)isoxazol-3-yl]benzene (18) under high pressure
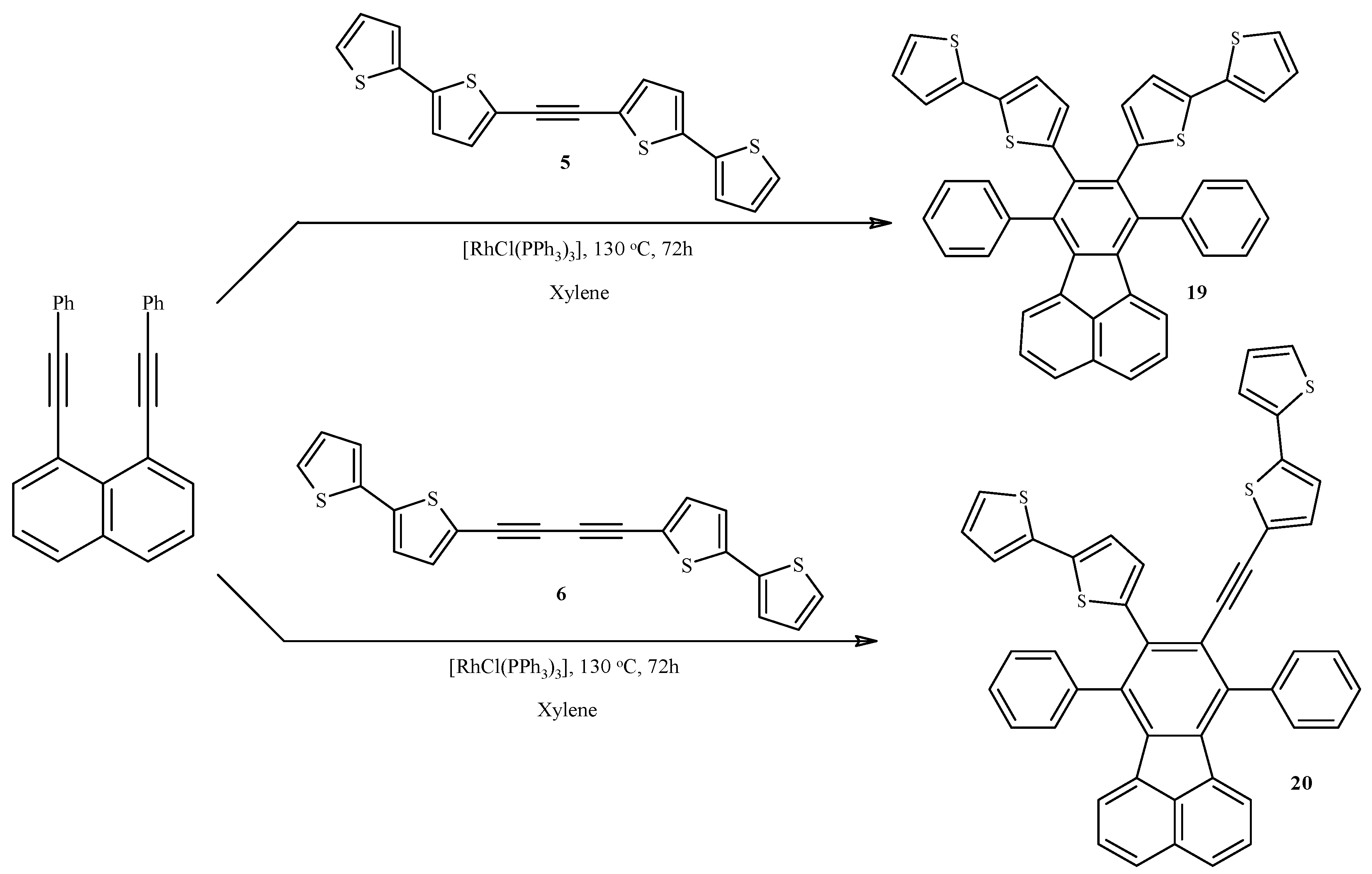
3.11. Synthesis of 8,9-Bis(2,2'-bithiophen-5-yl)-7,10-(diphenyl)fluoranthene (19) and 9-(2,2'-Bithiophene-5-yl)-8-(5-ethynyl-2,2'-bithiophen-5-yl)-7,10-(diphenyl)fluoranthene (20)
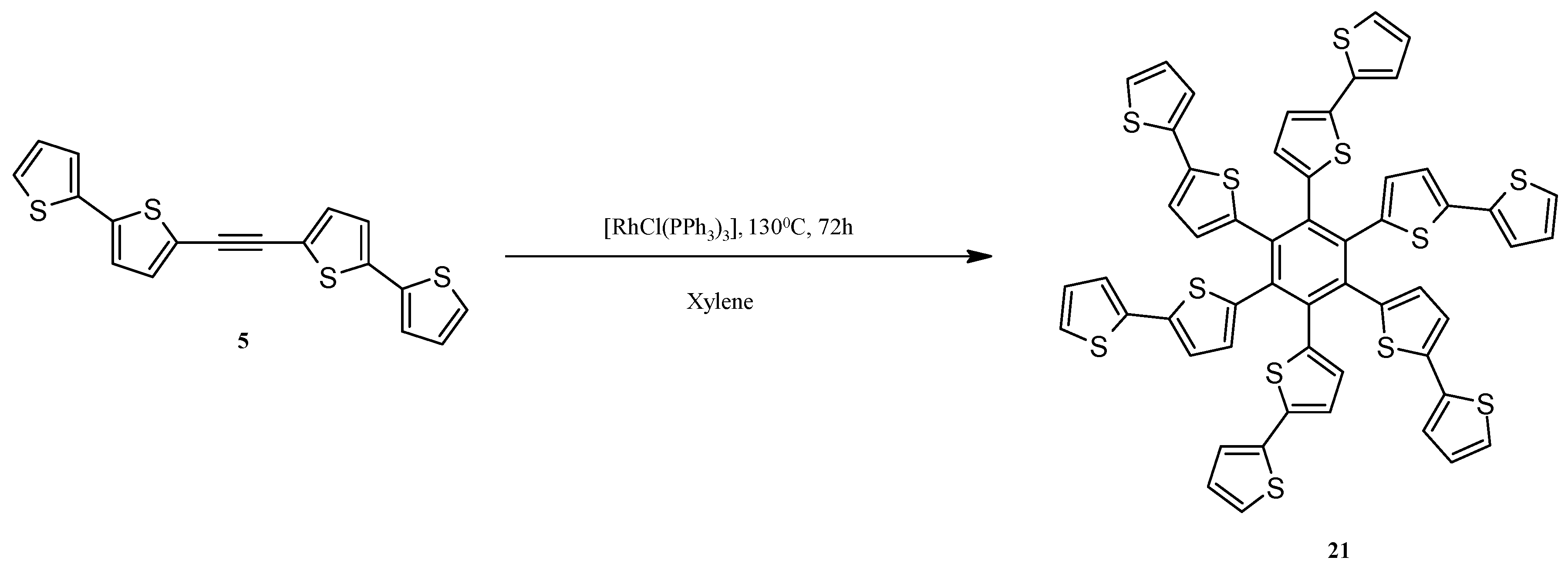
3.12. Synthesis of Hexa(2,2'-bithiophen-5-yl)benzene (21)
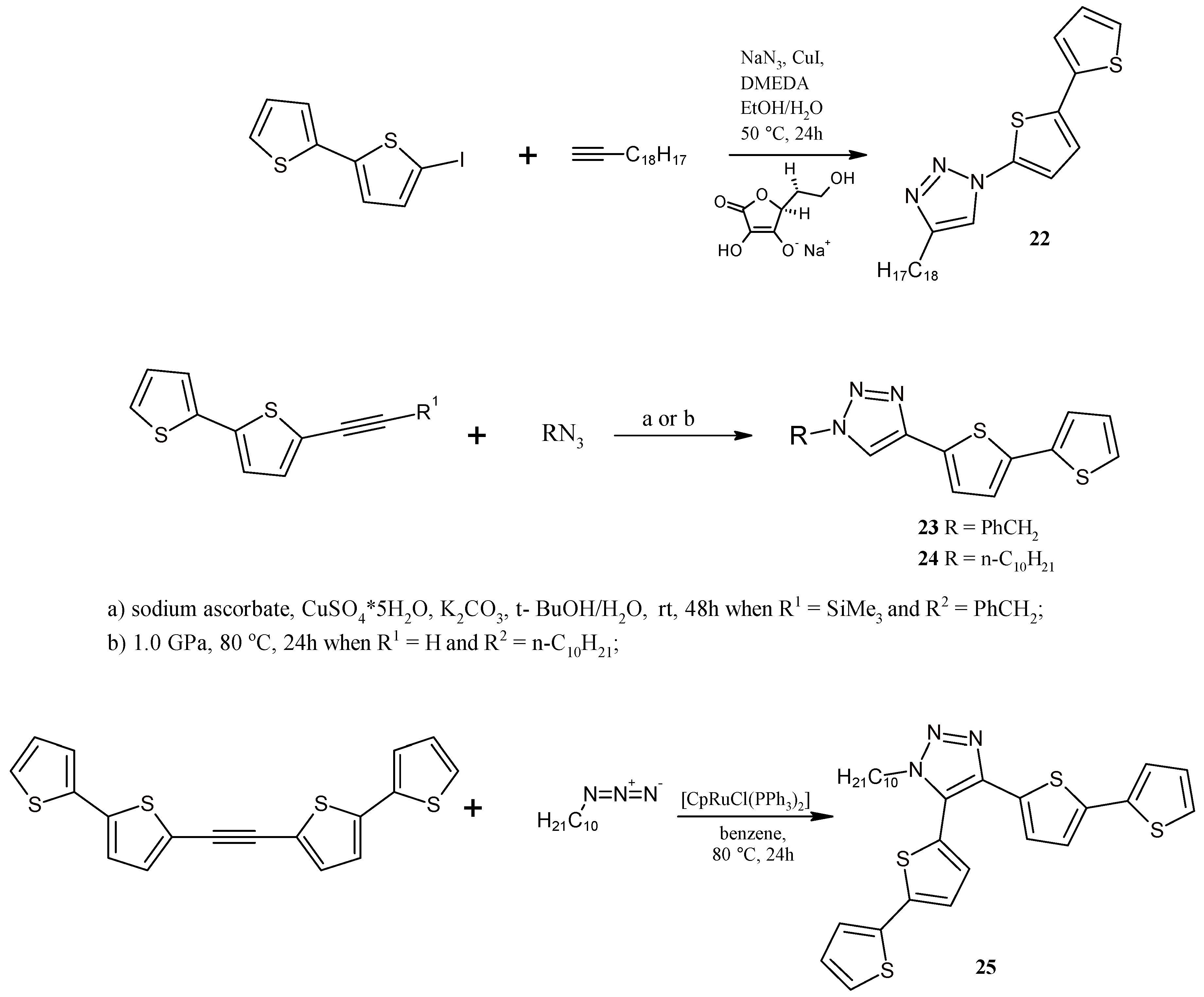
3.13. Synthesis of 1-(2,2'-Bithiophen-5-yl)-4-octyl-1,2,3-triazole (22)
3.14. Synthesis of 1-Benzyl-4-(2,2'-bithiophen-5-yl)-1,2,3-triazole (23)
3.15. Synthesis of 1-Decyl-4-(2,2'-Bithiophen-5-yl)-1,2,3-triazole (24) under High Pressure
3.16. Synthesis of 1-Decyl-4,5-bis(2,2'-bithiophen-5-yl)-1,2,3-triazole (25)
3.17. Synthesis of N-phenyl-2,5-bis(2,2'-bithiophen-5-yl)pyrrole (26), N-(p-decyloxy)phenyl-2,5-bis(2,2'-bithiophen-5-yl)pyrrole (27) and N-(2-carbazolylethyl)-2,5-bis(2,2'-bithiophen-5-yl)pyrrole (28)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mishra, A.; Ma Ch, Q.; Bäuerle, P. Functional Oligothiophenes: Molecular Design for Multidimensional Nanoarchitectures and Their Applications. Chem. Rev. 2009, 109, 1141–1276. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.; Nayak, P.K.; Ali, F.; Patankar, M.P.; Narasimhan, K.L. Tuning of HOMO levels of carbazole derivatives: New molecules for blue OLED. Synth. Met. 2011, 161, 466–473. [Google Scholar] [CrossRef]
- Hung, W.Y.; Chi, L.C.; Chen, W.J.; Chen, Y.M.; Chou, S.H.; Wong, K.T. A new benzimidazole/carbazole hybrid bipolar material for highly efficient deep-blue electrofluorescence, yellow–green electrophosphorescence, and two-color-based white OLEDs. J. Mater. Chem. 2010, 20, 10113–10119. [Google Scholar] [CrossRef]
- Lin, Y.; Li, Y.; Zhan, X. Small molecule semiconductors for high-efficiency organic photovoltaics. Chem. Soc. Rev. 2012, 41, 4245–4272. [Google Scholar] [CrossRef] [PubMed]
- Letizia, J.A.; Rivnay, J.; Facchetti, A.; Ratner, M.A.; Marks, T.J. Variable Temperature Mobility Analysis of n-Channel, p-Channel, and Ambipolar Organic Field-Effect Transistors. Adv. Funct. Mater. 2010, 20, 50–58. [Google Scholar] [CrossRef]
- Hung, W.I.; Liao, Y.Y.; Hsu, C.Y.; Chou, H.H.; Lee, T.H.; Kao, W.S.; Lin, J.T. High-Performance Dye-Sensitized Solar Cells Based on Phenothiazine Dyes Containing Double Anchors and Thiophene Spacers. Chem. Asian J. 2014, 9, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, M.; Pschirer, N.G.; Baumgarten, M.; Mullen, K. Polyphenylene-Based Materials for Organic Photovoltaics. Chem. Rev. 2010, 110, 6817–6855. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.C.; Li, Z.J.; Tang, R.K.; Liu, B.L. Synthesis and evaluation of novel benzothiazole derivatives based on the bithiophene structure as potential radiotracers for β-amyloid plaques in Alzheimer’s disease. Bioorg. Med. Chem. 2010, 18, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.I.; Lee, S.S.; Chou, S.T.; Jan, C.M.; Chang, Y.T.; Lee, A.S.Y.; Lee, C.K. A sensitive bithiophene-based biosensor for interferon-gamma characterization and analysis. Thin Solid Films 2014, in press. [Google Scholar]
- Cui, M.; Li, Z.; Tang, R.; Jia, H.; Liu, B. Novel (E)-5-styryl-2,2'-bithiophene derivatives as ligands for β-amyloid plaques. Eur. J. Med. Chem. 2011, 46, 2908–2916. [Google Scholar] [CrossRef] [PubMed]
- Martínez, M.M.; Peña-López, M.; Sestelo, J.P.; Sarandeses, L.A. Synthesis of functionalized thiophenes and oligothiophenes by selective and iterative cross-coupling reactions using indium organometallics. Org. Biomol. Chem. 2012, 10, 3892–3898. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Shimizu, S.; Taguchi, H.; Kobayashi, A.; Tobita, S.; Nakamura, Y. Synthesis and Electronic, Photophysical, and Electrochemical Properties of a Series of Thienylcarbazoles. J. Org. Chem. 2012, 77, 3222–3232. [Google Scholar] [CrossRef] [PubMed]
- Potratz, S.; Mishra, A.; Bäuerle, P. Thiophene-based donor-acceptor co-oligomers by copper-catalyzed 1,3-dipolar cycloaddition. Beilstein J. Org. Chem. 2012, 8, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Krompiec, S.; Krompiec, M.; Filapek, M.; Flak, T.; Grudzka, I.; Zemlak, K.; Jezierski, Z. Method for the synthesis of 5-alkynyl-2,2'-bithiophenes by using coupling of the preparate containing 5-iodo-2,2'-bitiophene with terminal acetylenes. Appl. No.: P.399817. 5 July 2012. [Google Scholar]
- Krompiec, S.; Grudzka, I.; Zych, D. Method for the synthesis of 1,4-bis(2,2'-bithiophene-5-yl)-1,3-butadiyne. Appl. No.: P.404279. 10 June 2013. [Google Scholar]
- Liu, W.; Pink, M.D.; Lee, D. Conjugated Polymer Sensors Built on π-Extended Borasiloxane Cages. J. Am. Chem. Soc. 2009, 131, 8703–8707. [Google Scholar] [CrossRef] [PubMed]
- Yam, V.W.; Cheung, K.L.; Yip, S.K.; Cheung, K.K. Synthesis, characterization, structure and luminescence studies of mono-, di- and trinuclear gold(I) phosphine alkynyl complexes. J. Organomet. Chem. 2003, 681, 196–209. [Google Scholar] [CrossRef]
- Abbotto, A.; Bradamante, S.; Facchetti, A.; Pagani, G.A. Regioselective Synthesis of Highly Solvatochromic Thiophene-Spaced N-Alkylpyridinium Dicyanomethanides for Second-Harmonic Generation. J. Org. Chem. 1997, 62, 5755–5765. [Google Scholar] [CrossRef]
- Krompiec, S.; Kuźnik, N.; Krompiec, M.; Penczek, R.; Mrzigod, J.; Tórz, A. The role of the functional group in double bond migration in allylic systems catalysed by ruthenium hydride complexes. J. Mol. Catal. A Chem. 2006, 253, 132–146. [Google Scholar] [CrossRef]
- Brorson, M.; King, J.D.; Kiriakidou, K.; Prestopino, F.; Nordlander, E. Metal Cluster in Chemistry; Wiley-VCH: Weinheim, Germany, 1999; pp. 741–781. [Google Scholar]
- Landman, M.; Waldbach, T.; Gorls, H.; Lotz, S. Titanium complexes of π-coordinated thiophene derivatives. J. Organomet. Chem. 2003, 678, 5–14. [Google Scholar] [CrossRef]
- Lockemeyer, J.R.; Rauchfuss, T.B.; Rheingold, A.L.; Wilson, S.R. (Tetramethylthiophene)ruthenium Dichloride Dimer: A Versatile Synthetic Intermediate in Thiophene Coordination Chemistry. J. Am. Chem. Soc. 1989, 111, 8828–8834. [Google Scholar] [CrossRef]
- Uddin, M.N.; Mottalib, M.A.; Begum, N.; Ghosh, S.; Raha, A.K.; Haworth, D.T.; Lindeman, S.V.; Siddiquee, T.A.; Bennett, D.W.; Hogarth, G.; et al. Carbon-Phosphorus Bond Activation of Tri(2-thienyl)phosphine at Dirhenium and Dimanganese Centers. Organometallics 2009, 28, 1514–1523. [Google Scholar] [CrossRef]
- Sanger, M.J.; Angelici, R.J. Dynamic NMR Studies of the Restricted Rotation of Thiophenes (Th) and Selenophenes (Seln) in the Cr(CO)3(η5-Th) and Cr(CO)3 η5-Seln) Complexes. Organometallics 1994, 13, 1821–1831. [Google Scholar] [CrossRef]
- Ganja, E.A.; Rauchfuss, T.M.; Stern, C.L. The chemistry of (ring)Ru2+ (ring = tetramethylthiophene, p-cymene). Organometallics 1991, 10, 270–275. [Google Scholar] [CrossRef]
- Krompiec, M.; Krompiec, S.; Ignasiak, H.; Łapkowski, M.; Kuś, P.; Stanek, Ł.; Penczek, R.; Lis, S.; Staniński, K.; Sajewicz, M.; et al. Synthesis and electropolymerization of 3,5-dithienylpyridines, their complexes and N-methylpyridinium cations. Synth. Met. 2008, 158, 831–838. [Google Scholar] [CrossRef]
- Krompiec, M.; Krompiec, S.; Grudzka, I.; Filapek, M.; Skórka, Ł.; Flak, T.; Łapkowski, M. A cross-linked conjugated metallopolymer comprised of bisaxially coordinated ruthenium tetra-t-butyl phthalocyanine connected by quaterthiophene linkers. Electrochim. Acta 2011, 56, 6824–6830. [Google Scholar] [CrossRef]
- Data, P.; Zassowski, P.; Lapkowski, M.; Domagała, W.; Krompiec, S.; Flak, T.; Penkala, M.; Swist, A.; Soloducho, J.; Danikiewicz, W. Electrochemical and spectroelectrochemical comparison of alternated monomers and their copolymers based on carbazole and thiophene derivatives. Electrochim. Acta 2014, 122, 118–129. [Google Scholar] [CrossRef]
- Krompiec, S.; Filapek, M.; Grudzka, I.; Kula, S.; Słodek, A.; Skórka, Ł.; Danikiewicz, W.; Ledwon, P.; Lapkowski, M. An ambipolar behavior of novel ethynyl-bridged polythiophenes—A comprehensive study. Synth. Met. 2013, 165, 7–16. [Google Scholar] [CrossRef]
- Krompiec, S.; Krompiec, M.; Filapek, M.; Flak, T.; Grudzka, I.; Zemlak, K.; Jezierski, Z. Method for the synthesis of the preparate containing 5-lithio-2,2'-bithiophene. Appl. No.: P.399815. 5 July 2012. [Google Scholar]
- Krompiec, S.; Krompiec, M.; Filapek, M.; Flak, T.; Grudzka, I.; Zemlak, K.; Jezierski, Z. Method for the synthesis of the preparate containing 5-iodo-2,2'-bithiophene. Appl. No.: P.399816. 5 July 2012. [Google Scholar]
- Chuentragool, P.; Vongnam, K.; Rashatasakhon, P.; Sukwattanasinitt, M.; Wacharasindhu, S. Calcium carbide as a cost-effective starting material for symmetrical diarylethynes via Pd-catalyzed coupling reaction. Tetrahedron 2011, 67, 8177–8182. [Google Scholar] [CrossRef]
- Bertrand, G.H.V.; Tortech, L.; Gandon, V.; Aubert, C.; Fichou, D. Synthesis and photovoltaic performances in solution-processed BHJs of oligothiophene-substituted organocobalt complexes [(η4-C4(nT)4)Co(η5-C5H5)]. Chem. Commun. 2014, 50, 8663–8866. [Google Scholar] [CrossRef]
- Kagan, J.; Arora, S.K. Synthesis of alpha-thiophene oligomers via 1,3-butadiynes. J. Org. Chem. 1983, 48, 4317–4320. [Google Scholar] [CrossRef]
- Merkul, E.; Urselmann, D.; Müller, T.J.J. Consecutive One-Pot Sonogashira-Glaser Coupling Sequence—Direct Preparation of Symmetrical Diynes by Sequential Pd/Cu Catalysis. Eur. J. Org. Chem. 2011, 238–242. [Google Scholar] [CrossRef]
- Trotus, I.T.; Zimmermann, T.; Schüth, F. Catalytic Reactions of Acetylene: A Feedstock for the Chemical Industry Revisited. Chem. Rev. 2014, 114, 1761–1782. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.B.; Sporikou, C.N. Trimethylsilylacetylene. Org. Synth. 1987, 65, 61–67. [Google Scholar] [CrossRef]
- McCormick, T.M.; Jahnke, A.A.; Lough, A.J.; Seferos, D.S. Tellurophenes with Delocalized π-Systems and Their Extended Valence Adducts. J. Am. Chem. Soc. 2012, 134, 3542–3548. [Google Scholar] [CrossRef] [PubMed]
- Yoshikai, N.; Zhang, S.L.; Yamagata, K.; Tsuji, H.; Nakamura, E. Mechanistic Study of the Manganese-Catalyzed [2 + 2 + 2] Annulation of 1,3-Dicarbonyl Compounds and Terminal Alkynes. J. Am. Chem. Soc. 2009, 131, 4099–4109. [Google Scholar] [CrossRef] [PubMed]
- Kuninobu, Y.; Nishi, M.; Kawata, A.; Takata, H.; Hanatani, Y.; Yudha, S.S.; Iwai, A.; Takai, K. Rhenium- and Manganese-Catalyzed Synthesis of Aromatic Compounds from 1,3-Dicarbonyl Compounds and Alkynes. J. Org. Chem. 2010, 75, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Broere, D.L.J.; Ruijter, E. Recent Advances in Transition-Metal-Catalyzed [2 + 2 + 2]-Cyclo(co)trimerization Reactions. Synthesis 2012, 44, 2639–2672. [Google Scholar] [CrossRef]
- Savage, G.P. Spiro Isoxazolines via Nitrile Oxide 1,3-Dipolar Cycloaddition Reactions. Curr. Org. Chem. 2010, 14, 1478–1499. [Google Scholar] [CrossRef]
- Minakata, S.; Okumura, S.; Nagamachi, T.; Takeda, Y. Generation of Nitrile Oxides from Oximes Using t-BuOI and Their Cycloaddition. Org. Lett. 2011, 13, 2966–2969. [Google Scholar] [CrossRef] [PubMed]
- Easton, C.J.; Hughes, C.M.M.; Savage, G.P.; Simpson, G.W. Cycloaddition Reactions of Nitrile Oxides with Alkenes; Academic Press: San Diego, CA, USA, 1994; Volume 60, pp. 261–327. [Google Scholar]
- Jager, V.; Colinas, P.A. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products; Wiley & Sons: New York, NY, USA, 2002; pp. 361–472. [Google Scholar]
- Bujak, P.; Krompiec, S.; Malarz, J.; Krompiec, M.; Filapek, M.; Danikiewicz, W.; Kania, M.; Gębarowska, K.; Grudzka, I. Synthesis of 5-aminoisoxazolines from N-allyl compounds and nitrile oxides via tandem isomerization-1,3-dipolar cycloaddition. Tetrahedron 2010, 66, 5972–5981. [Google Scholar] [CrossRef]
- Krompiec, S.; Bujak, P.; Szczepankiewicz, W. Convenient synthesis of isoxazolines via tandem isomerization of allyl compounds to vinylic derivatives and 1,3-dipolar cycloaddition of nitrile oxides to the vinylic compounds. Tetrahedron Lett. 2008, 49, 6071–6074. [Google Scholar] [CrossRef]
- Krompiec, S.; Bujak, P.; Malarz, J.; Krompiec, M.; Skórka, Ł.; Pluta, T.; Danikiewicz, W.; Kania, M.; Kusz, J. An isomerization-1,3-dipolar cycloaddition tandem reaction towards the synthesis of 3-aryl-4-methyl-5-O-substituted isoxazolines from O-allyl compounds. Tetrahedron 2012, 68, 6018–6031. [Google Scholar] [CrossRef]
- Iwakura, Y.; Shiraishi, S.; Akiyama, M.; Yuyama, M. Polymerizations by 1,3-Dipolar Cycloaddition Reactions. V. The 1,3-Dipolar Polycycloadditions of Dinitrile N-Oxides with Diolefins. Bull. Chem. Soc. Jpn. 1968, 41, 1648–1653. [Google Scholar] [CrossRef]
- Imgartinger, H.; Weber, A. Twofold cycloaddition of [60]fullerene to a bifunctional nitrile oxide. Tetrahedron Lett. 1996, 37, 4137–4140. [Google Scholar] [CrossRef]
- Dubrovskiy, A.V.; Jain, P.; Shi, F.; Lushington, G.H.; Santini, C.; Porubsky, P.; Larock, R.C. Solution—Phase Synthesis of a Diverse Library of Benzisoxazoles Utilizing the [3 + 2] Cycloaddition of in Situ-Generated Nitrile Oxides and Arynes. ACS Comb. Sci. 2013, 15, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, C.; Rothberg, L.; Ng, M.K. Surface-initiated growth ofconjugated polymersfor functionalization of electronically active nanoporous networks: synthesis, structure and optical properties. J. Mater. Chem. 2006, 16, 3721–3725. [Google Scholar] [CrossRef]
- Kreyes, A.; Amirkhani, M.; Lieberwirth, I.; Mauer, R.; Laquai, F.; Landfester, K.; Ziener, U. The Longest β-Unsubstituted Oligothiophenes and Their Self - Assembly in Solution. Chem. Mater. 2010, 22, 6453–6458. [Google Scholar] [CrossRef]
- Bumagin, N.A.; Kasatkin, A.N.; Beletskaya, I.P. Reactions of orgamnometallic compounds catalyzed by complexes of transition—Metals. 5. Organomagnesium, organozinc, organocadmium, and organoaluminum coumpounds in allyldemetallation reactions catalyzed by palladium complexes. B Acad. Sci. Ussr CH+ 1984, 33, 1696–1703. [Google Scholar] [CrossRef]
- Urbala, M.; Krompiec, S.; Penkala, M.; Danikiewicz, W. Solvent—Free Ru-catalyzed isomerization of allyloxyalcohols: Methods for highly selective synthesis of 1-propenyloxyalcohols. Appl. Catal. A Gen. 2013, 451, 101–111. [Google Scholar] [CrossRef]
- Kuźnik, N.; Krompiec, S. Transition metal complexes as catalysts of double—Bond migration in O-allyl systems. Coord. Chem. Rev. 2007, 251, 222–233. [Google Scholar] [CrossRef]
- Chen, S.F.; Ho, E.; Mariano, P.S. A protodesilylation route for 2-Aza-1,3-diene synthesis. Tetrahedron 1988, 44, 7013–7026. [Google Scholar] [CrossRef]
- Govindan, C.K.; Taylor, G. Thermal electrocyclic reactions of 2-aza-1,3-butadiene derivatives. A new N-heterocyclic annelation. J. Org. Chem. 1983, 48, 5348–5354. [Google Scholar] [CrossRef]
- Grundmann, C.; Dean, J.M. Stable Aromatic Nitrile Oxides. J. Org. Chem. 1965, 30, 2809–2812. [Google Scholar] [CrossRef]
- Liu, K.C.; Shelton, B.R.; Howe, R.K. A particularly convenient preparation of benzohydroximinoyl chlorides (nitrile oxide precursors). J. Org. Chem. 1980, 45, 3916–3918. [Google Scholar] [CrossRef]
- Benito-Lopez, F.; Egberink, R.J.M.; Reinhoudt, D.N.; Verboom, W. High pressure in organic chemistry on the way to miniaturization. Tetrahedron 2008, 64, 10023–10040. [Google Scholar] [CrossRef]
- Klärner, F.G.; Wurche, F. The Effect of Pressure on Organic Reactions. J. Prakt. Chem. 2000, 342, 609–636. [Google Scholar] [CrossRef]
- Fan, J.C.; Liang, J.; Wang, Y.; Shang, Z.C. Theoretical study of the mechanism of high—Pressure induced 1,3-dipolar cycloadditions of azides with electron-rich olefins. J. Mol. Struct. Theochem. 2007, 821, 145–152. [Google Scholar] [CrossRef]
- Gobis, K.; Foks, H.; Kędzia, A.; Wierzbowska, M.; Zwolska, Z. Synthesis and antibacterial activity of novel pyridine and pyrazine derivatives obtained from amidoximes. J. Heterocycl. Chem. 2009, 46, 1271–1279. [Google Scholar] [CrossRef]
- Van der Peet, P.L.; Connell, T.U.; Gunawan, C.; White, J.M.; Donnelly, P.S.; Williams, S.J. A Click Chemistry Approach to 5,5'-Disubstituted-3,3'-Bisisoxazoles from Dichloroglyoxime and Alkynes: Luminescent Organometallic Iridium and Rhenium Bisisoxazole Complexes. J. Org. Chem. 2013, 78, 7298–7304. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Dell’Erba, C.; Gasarrini, F.; Novi, M.; Patrillo, G.; Sancassan, F.; Tavani, C. Access to 5,5'-diaryl substituted 4,5,4',5'-tetrahydro[3,3']bi-isoxazolyl 2,2'-dioxides, 4,5,4',5'-tetrahydro[3,3'] bi-isoxazolyls and [3,3']bi-isoxazolyls via an initial ring-opening of 3,4-dinitrothiophene. ARKIVOC 2002, 11, 124–129. [Google Scholar]
- Chopade, P.R.; Louie, J. [2+2+2] Cycloaddition Reactions Catalyzed by Transition Metal Complexes. Adv. Synth. Catal. 2006, 348, 2307–2327. [Google Scholar] [CrossRef]
- Wu, Y.T.; Hayama, T.; Baldridge, K.K.; Linden, A.; Siegel, J.S. Synthesis of Fluoranthenes and Indenocorannulenes: Elucidation of Chiral Stereoisomers on the Basis of Static Molecular Bowls. J. Am. Chem. Soc. 2006, 128, 6870–6884. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Linden, A.; Siegel, J.S. Formal [(2+2)+2] and [(2+2)+(2+2)] Nonconjugated Dienediyne Cascade Cycloadditions. Org. Lett. 2005, 7, 4353–4355. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, P.; Venkateswararan, P.T.A.; Thomas, K.R.J. Solution Processable Indoloquinoxaline Derivatives Containing Bulky Polyaromatic Hydrocarbons: Synthesis, Optical Spectra, and Electroluminescence. J. Org. Chem. 2011, 76, 4571–4581. [Google Scholar] [CrossRef] [PubMed]
- House, H.O.; Koepsell, D.G.; Campbell, W.J. Synthesis of some diphenyl and triphenyl derivatives of anthracene and naphthalene. J. Org. Chem. 1972, 37, 1003–1011. [Google Scholar] [CrossRef]
- Huang, X.; Zeng, L.; Zeng, Z.; Wu, J. Intramolecular Domino Electrophilic and Thermal Cyclization of peri-Ethynylene Naphthalene Oligomers. Chem. Eur. J. 2011, 17, 14907–14915. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Fechtenkötter, A.; Müllen, K. Star-like substituted hexaarylbenzenes: Synthesis and mesomorphic properties. J. Mater. Chem. 2001, 11, 1634–1641. [Google Scholar] [CrossRef]
- Bossenbroek, B.; Sanders, D.C.; Curry, H.M.; Shechter, H. Interactions and chemistry of 1,8-bis(phenylethynyl)naphthalene and 1,8-bis(1-alkynyl)naphthalenes. J. Am. Chem. Soc. 1969, 91, 371–379. [Google Scholar] [CrossRef]
- Wang, D.; Denux, D.; Riuz, J.; Astruc, D. The Clicked Pyridyl—Triazole Ligand: From Homogeneous to Robust, Recyclable Heterogeneous Mono- and Polymetallic Palladium Catalysts for Efficient Suzuki–Miyaura, Sonogashira, and Heck Reactions. Adv. Synth. Catal. 2013, 355, 129–142. [Google Scholar] [CrossRef]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef]
- Hein, J.E.; Fokin, V.V. Copper-catalyzedazide–alkynecycloaddition(CuAAC) and beyond: New reactivity of copper(I) acetylides. Chem. Soc. Rev. 2010, 39, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, X.; Xue, P.; Sun, H.H.Y.; Williams, I.D.; Sharpless, K.B.; Fokin, V.V.; Jia, G. Ruthenium-Catalyzed Cycloaddition of Alkynes and Organic Azides. J. Am. Chem. Soc. 2005, 127, 15998–15999. [Google Scholar] [CrossRef] [PubMed]
- Boren, B.C.; Narayan, S.; Rasmussen, L.K.; Zhang, L.; Zhao, H.; Lin, Z.; Jia, G.; Fokin, V.V. Ruthenium-Catalyzed Azide−Alkyne Cycloaddition: Scope and Mechanism. J. Am. Chem. Soc. 2008, 130, 8923–8930. [Google Scholar] [CrossRef] [PubMed]
- Ni, B.B.; Wang, K.; Yan, Q.; Chen, H.; Ma, Y.; Zou, B. Pressure accelerated 1,3-dipolar cycloaddition of azide and alkyne groups in crystals. Chem. Commun. 2013, 49, 10130–10132. [Google Scholar] [CrossRef]
- Aufort, M.; Herscovici, J.; Bouhours, P.; Moreau, N.; Girard, C. Synthesis and antibiotic activity of a small molecules library of 1,2,3-triazole derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 1195–1198. [Google Scholar] [CrossRef] [PubMed]
- Kramer, S.; Madsen, J.L.H.; Rottländer, M.; Skrydstrup, T. Access to 2,5-Diamidopyrroles and 2,5-Diamidofurans by Au(I)-Catalyzed Double Hydroamination or Hydration of 1,3-Diynes. Org. Lett. 2010, 12, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Nun, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S.P. Gold(I)-catalyzed synthesis offuransandpyrrolesviaalkynehydration. Catal. Sci. Technol. 2011, 1, 58–61. [Google Scholar] [CrossRef]
- Duan, H.; Sengupta, S.; Petersen, J.L.; Akhmedov, N.G.; Shi, X. Triazole−Au(I) Complexes: A New Class of Catalysts with Improved Thermal Stability and Reactivity for Intermolecular Alkyne Hydroamination. J. Am. Chem. Soc. 2009, 131, 12100–12102. [Google Scholar] [CrossRef] [PubMed]
- Schulte, K.A.; Reisch, J.; Walker, H. Eine neue Pyrrolsynthese aus Butadiin-Derivaten. Chem. Ber. 1965, 98, 98–103. [Google Scholar] [CrossRef]
- Zheng, Q.; Hua, R. CuCl-catalyzed cycloaddition of 1,3-butadiynes with primary amines: An atom-economic process for synthesis of 1,2,5-trisubsituted pyrroles. Tetrahedron Lett. 2010, 51, 4512–4514. [Google Scholar] [CrossRef]
- Yang, C. Pyrrole-cored push-pull single chromophore. Tetrahedron Lett. 2010, 51, 2007–2009. [Google Scholar] [CrossRef]
- Kowada, T.; Kuwabara, T.; Ohe, K. Synthesis, Structures, and Optical Properties of Heteroarene-Fused Dispiro Compounds. J. Org. Chem. 2010, 75, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, F.B.; Sefer, E.; Koyuncu, S.; Ozdemir, E. The New Branched Multielectrochromic Materials: Enhancing the Electrochromic Performance via Longer Side Alkyl Chain. Macromolecules 2011, 44, 8407–8414. [Google Scholar] [CrossRef]
- Severin, R.; Dove, S. The catalytic hydroamination of alkynes. Chem. Soc. Rev. 2007, 36, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Paluch, M.; Sekuła, M.; Pawlus, S.; Rzoska, S.J.; Roland, C.M.; Zioło, J. Test of the Einstein-Debye Relation in Supercooled Dibutylphthalate at Pressures up to 1.4 GPa. Phys. Rev. Lett. 2003, 90, 175702-1–175702-4. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis RED; Version 1.171.36.24; Oxford Diffraction Ltd. Agilent: Santa Clara, CA, USA, 2012.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H.J. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Block, E.; Tries, F.; He, C.; Guo, C.; Thiruvazhi, M.; Toscano, P.J. Perthio- and Perseleno-1,3-butadienes, -but-1-ene-3-ynes, and -[3]-cumulenes: One-Step Syntheses from 1,4-Dilithio-1,3-butadiyne. Org. Lett. 2003, 5, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds (5)–(28) (except of compound 13a) are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krompiec, S.; Filapek, M.; Grudzka-Flak, I.; Slodek, A.; Kula, S.; Malecki, J.G.; Malarz, J.; Szafraniec-Gorol, G.; Penkala, M.; Schab-Balcerzak, E.; et al. Multifaceted Strategy for the Synthesis of Diverse 2,2'-Bithiophene Derivatives. Molecules 2015, 20, 4565-4593. https://doi.org/10.3390/molecules20034565
Krompiec S, Filapek M, Grudzka-Flak I, Slodek A, Kula S, Malecki JG, Malarz J, Szafraniec-Gorol G, Penkala M, Schab-Balcerzak E, et al. Multifaceted Strategy for the Synthesis of Diverse 2,2'-Bithiophene Derivatives. Molecules. 2015; 20(3):4565-4593. https://doi.org/10.3390/molecules20034565
Chicago/Turabian StyleKrompiec, Stanisław, Michał Filapek, Iwona Grudzka-Flak, Aneta Slodek, Sławomir Kula, Jan Grzegorz Malecki, Joanna Malarz, Grażyna Szafraniec-Gorol, Mateusz Penkala, Ewa Schab-Balcerzak, and et al. 2015. "Multifaceted Strategy for the Synthesis of Diverse 2,2'-Bithiophene Derivatives" Molecules 20, no. 3: 4565-4593. https://doi.org/10.3390/molecules20034565
APA StyleKrompiec, S., Filapek, M., Grudzka-Flak, I., Slodek, A., Kula, S., Malecki, J. G., Malarz, J., Szafraniec-Gorol, G., Penkala, M., Schab-Balcerzak, E., Paluch, M., Mierzwa, M., Matussek, M., Szlapa, A., Pajak, M., Blach, D., Marcol, B., Danikiewicz, W., Boharewicz, B., & Iwan, A. (2015). Multifaceted Strategy for the Synthesis of Diverse 2,2'-Bithiophene Derivatives. Molecules, 20(3), 4565-4593. https://doi.org/10.3390/molecules20034565