
The Effect of Number and Position of P=O/P=S Bridging Units on Cavitand Selectivity toward Methyl Ammonium Salts


Abstract
:1. Introduction
2. Results and Discussion
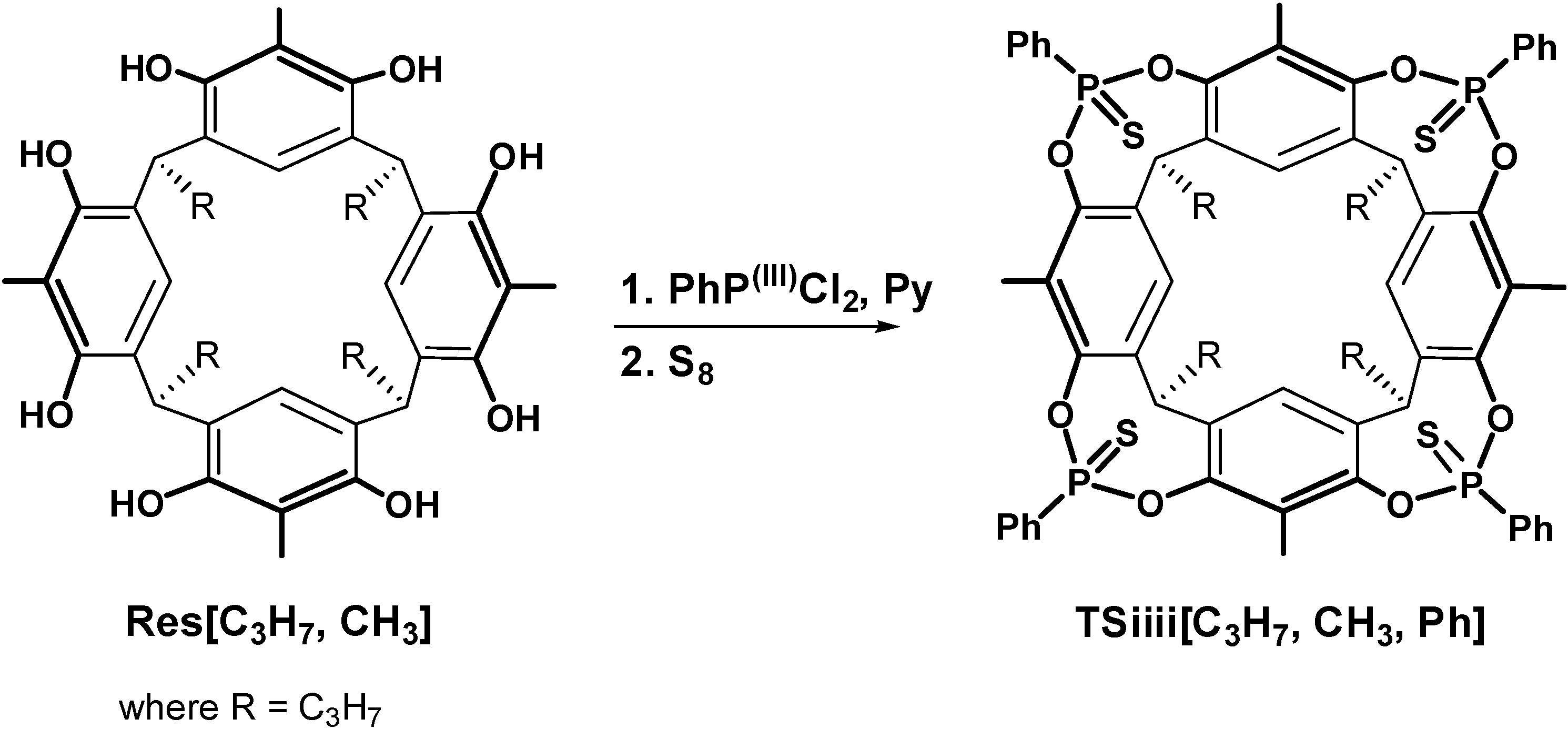
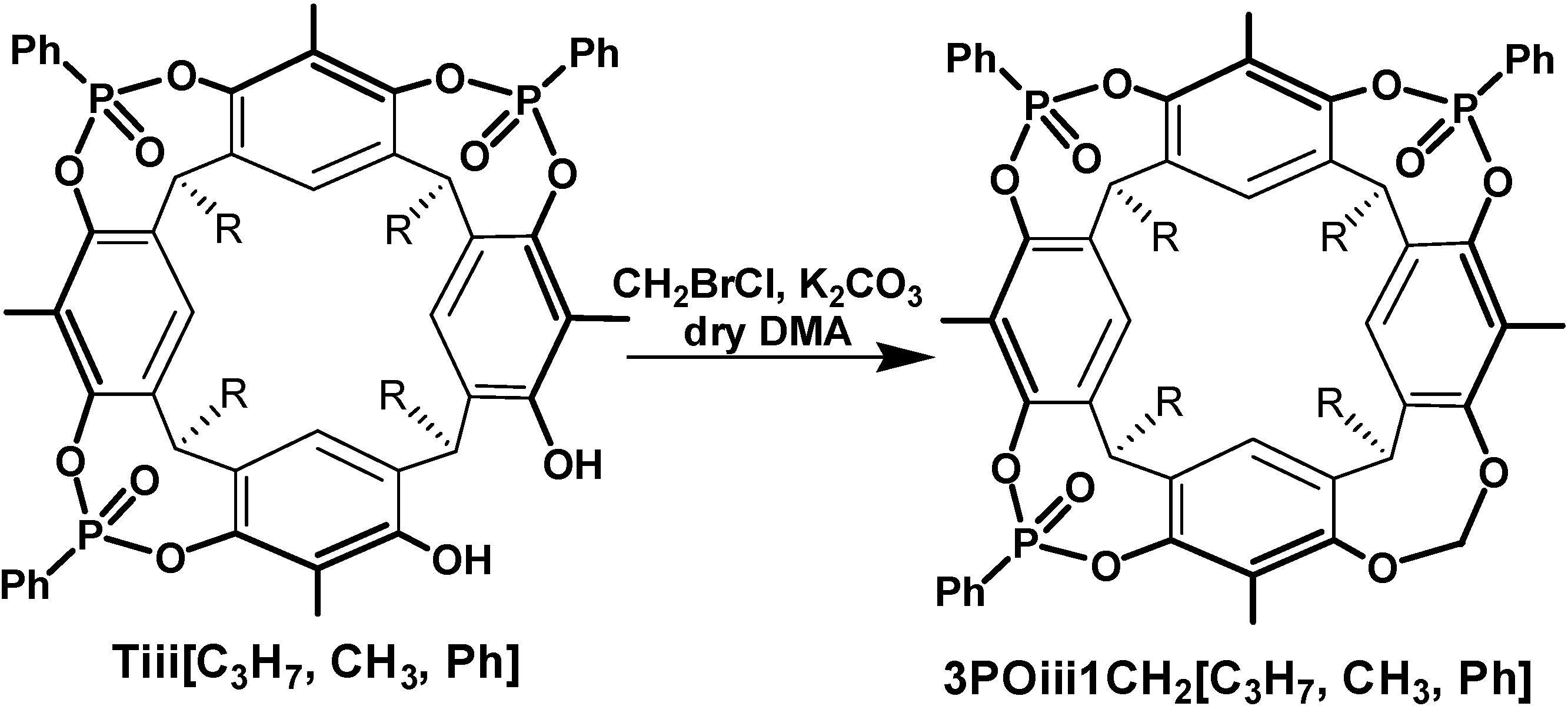
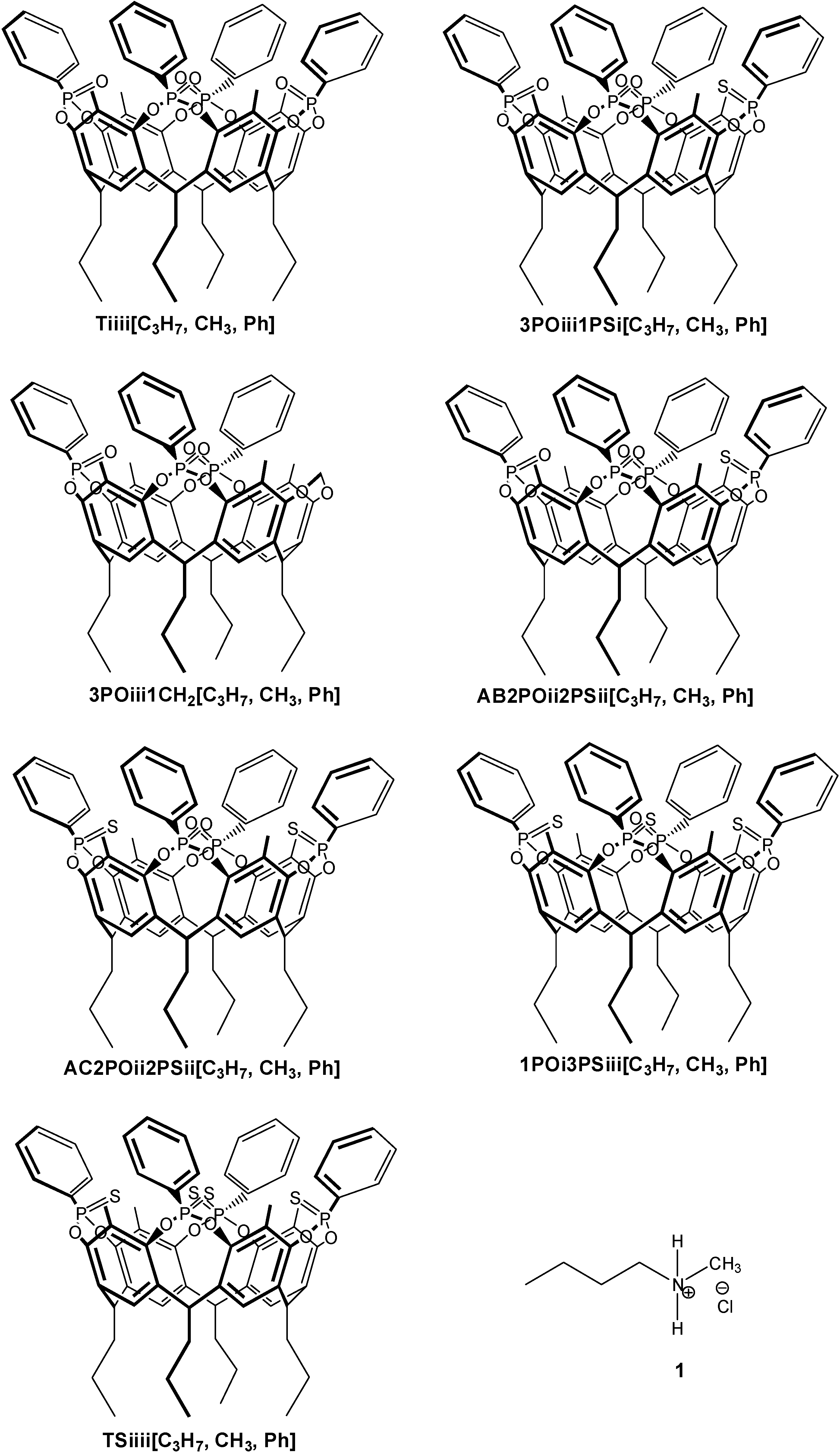
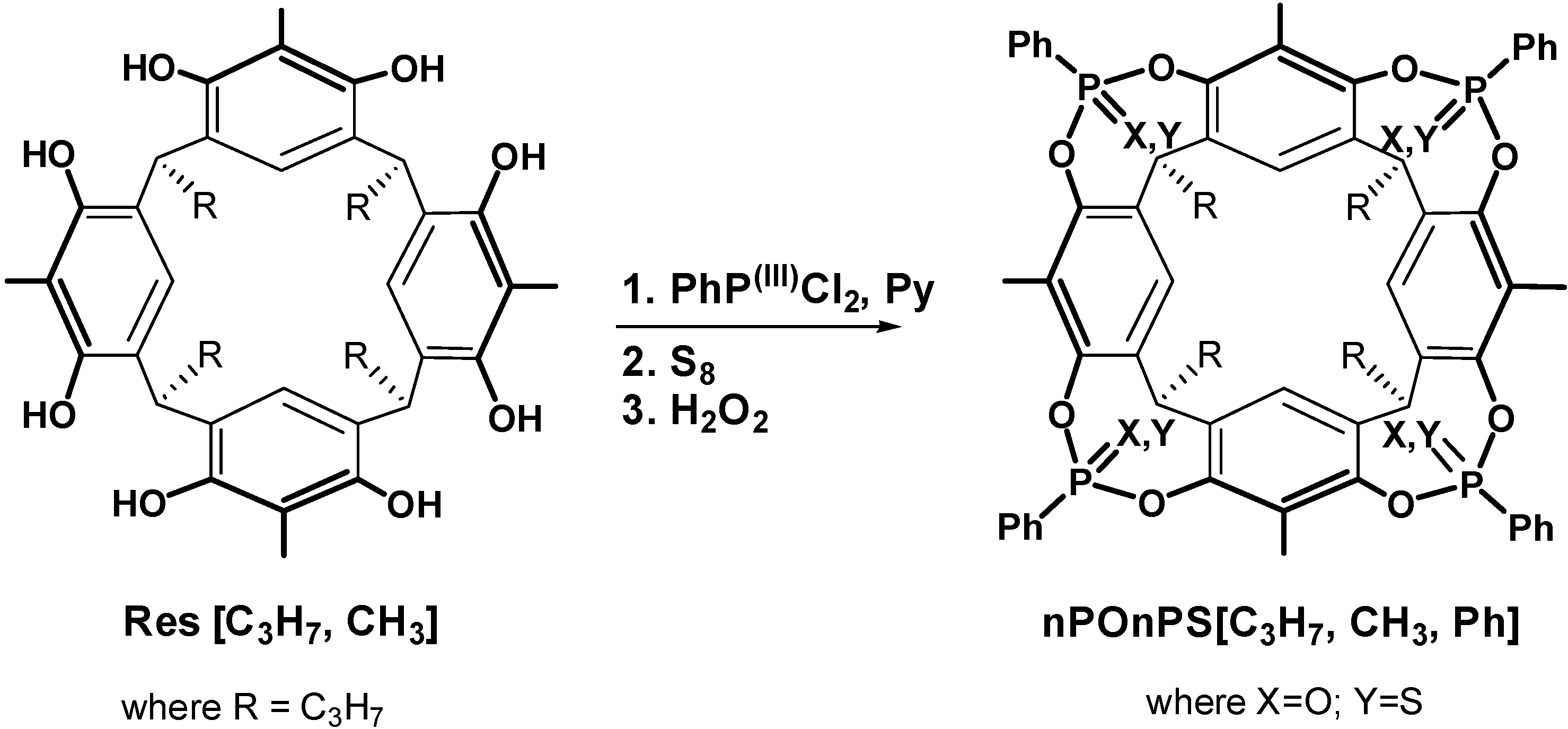
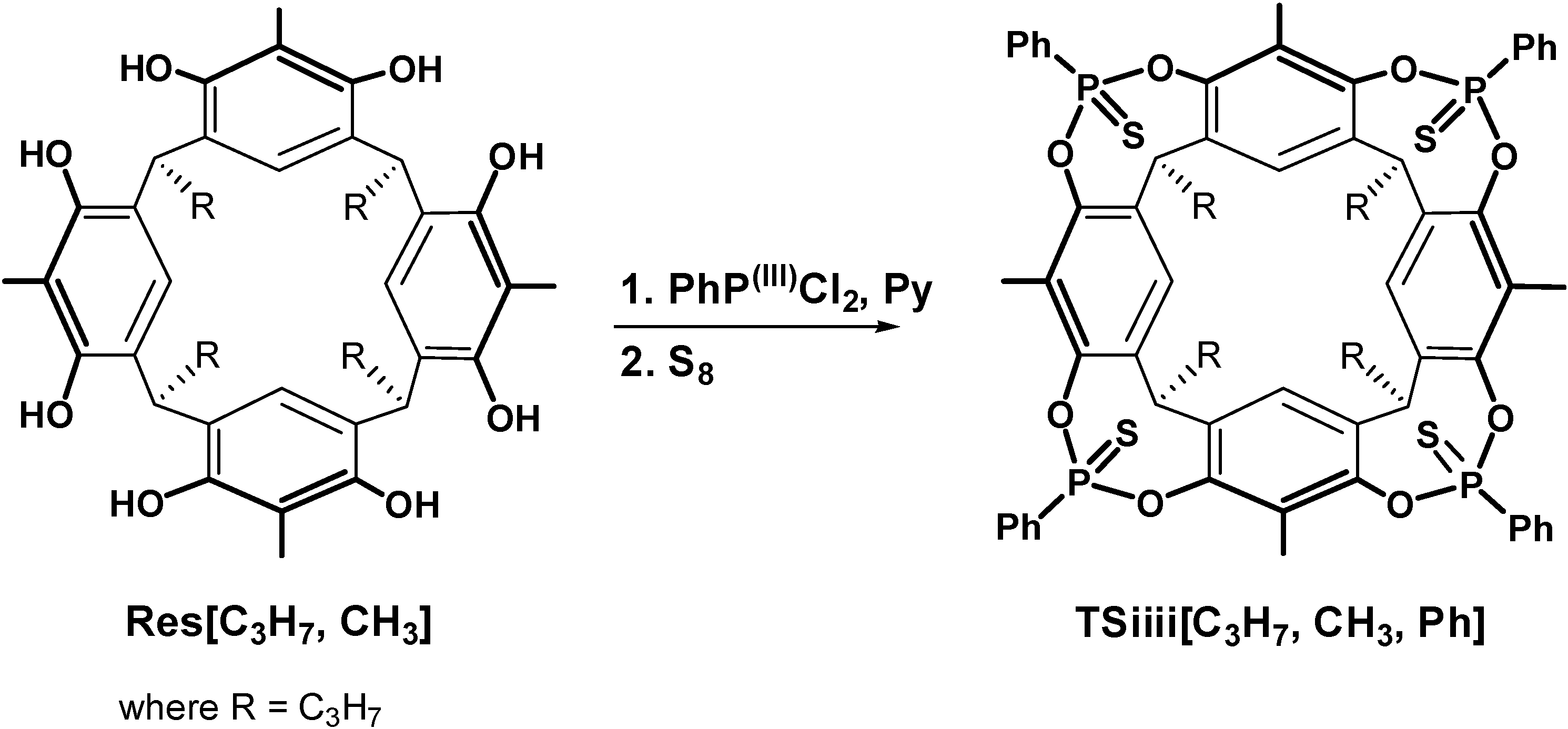
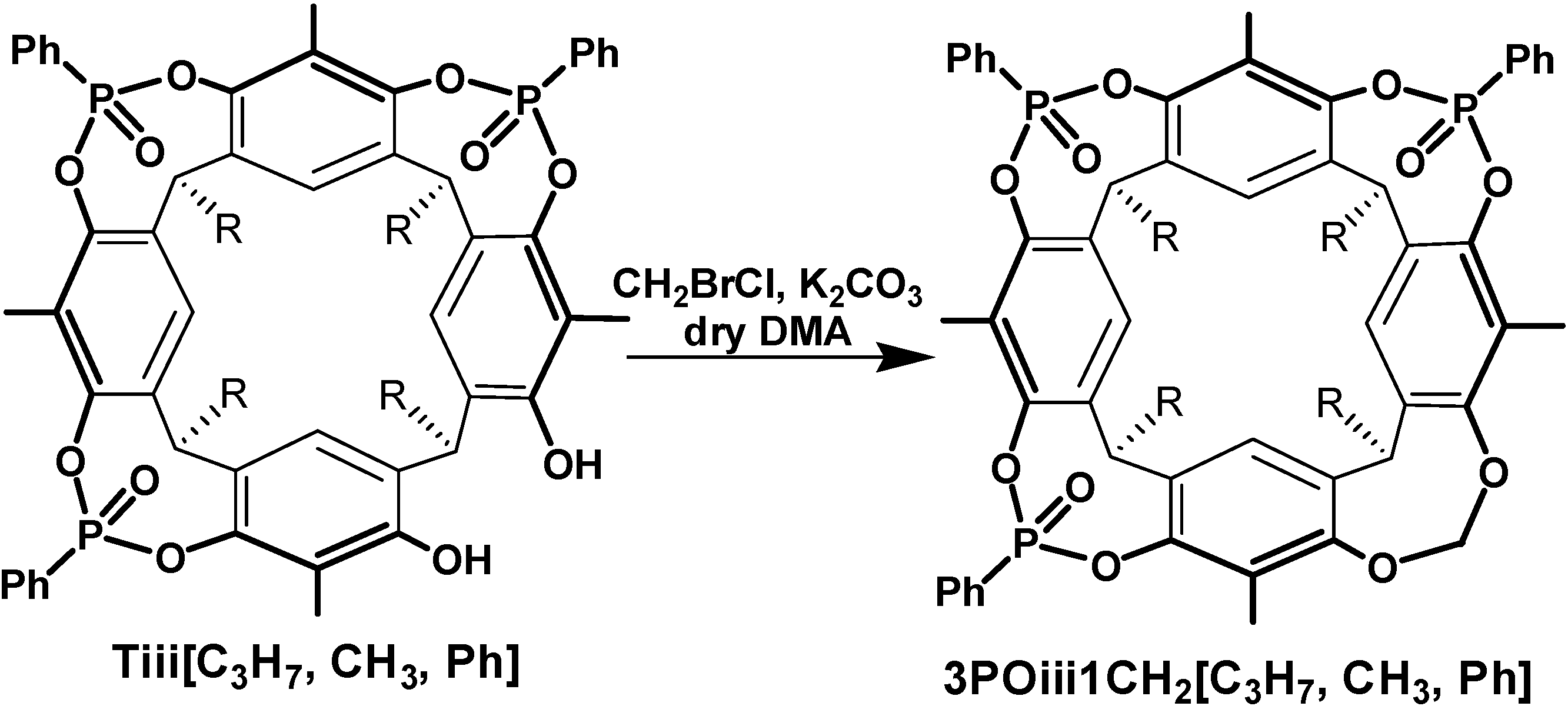
2.1. Synthesis
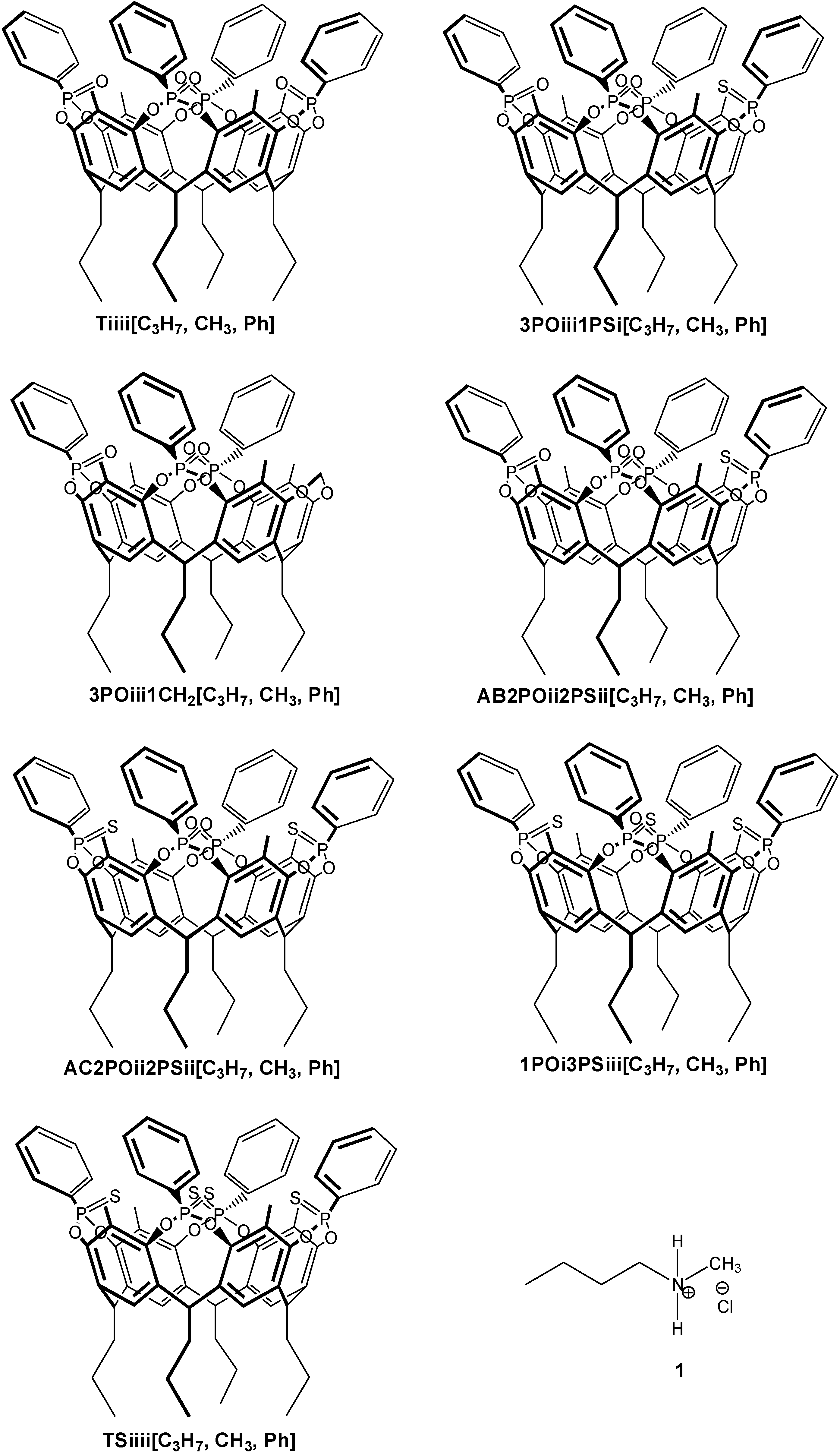
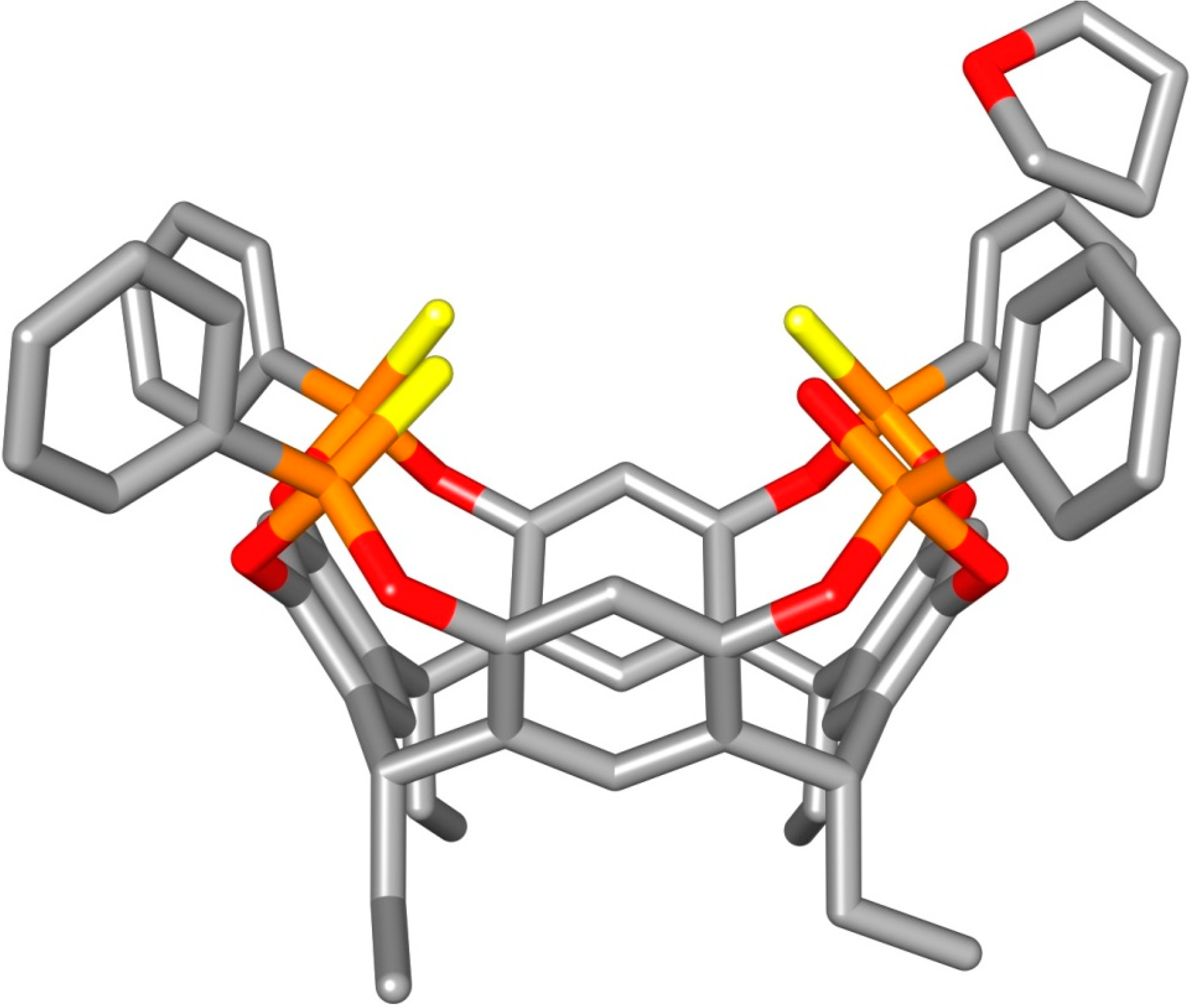
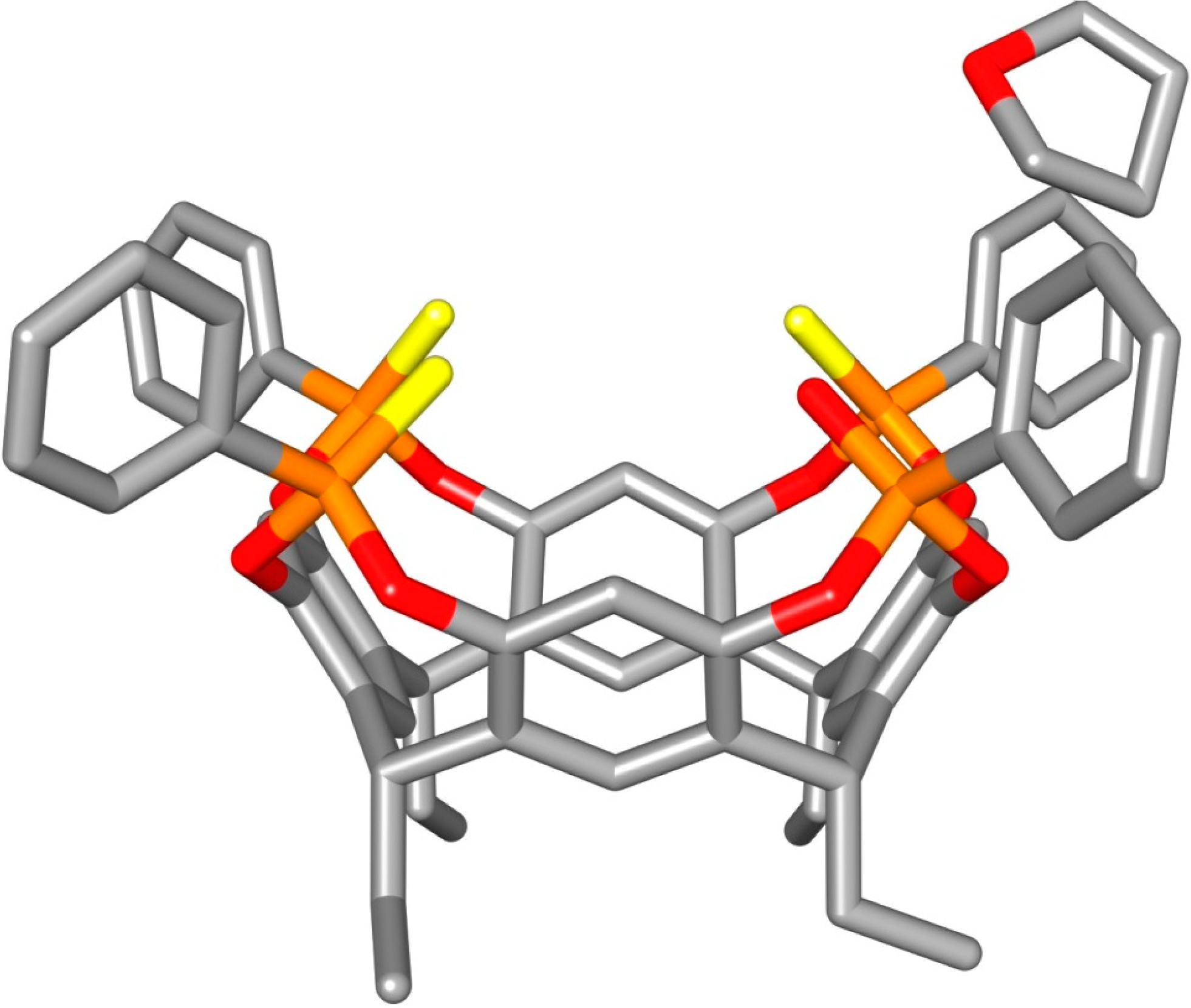
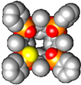
2.2. Crystal Structure of the 1POi3PSiii[C2H5, CH3, Ph] Cavitand
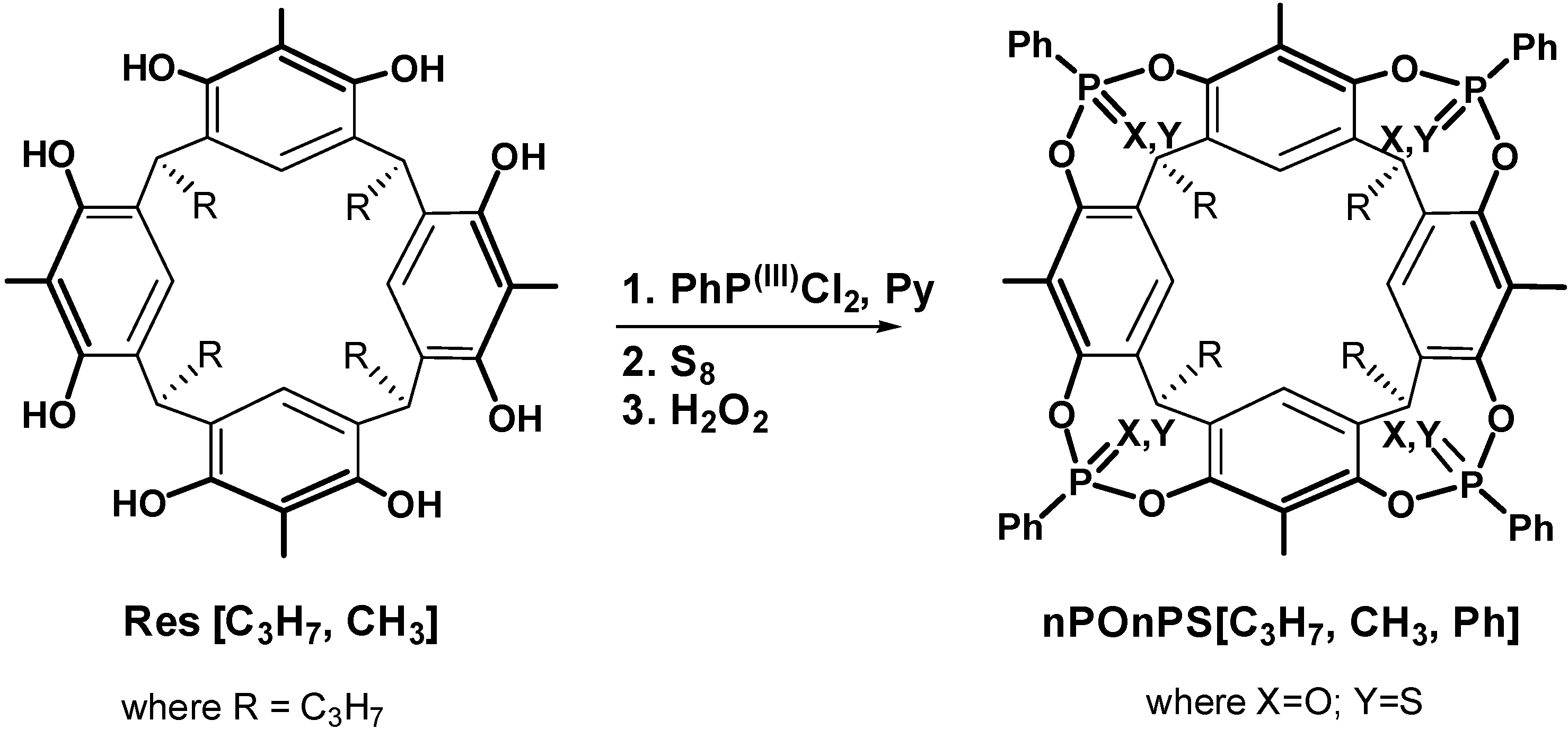
2.3. ITC Measurements

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guest | Host | Solvent | Kass ± δKass (M−1) | ΔH ± δH (KJ·mol−1) | ΔG± δG (KJ·mol−1) | TΔS ± TδS (KJ·mol−1) | |
|---|---|---|---|---|---|---|---|
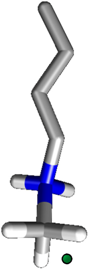 N-methylbutyl ammonium chloride 1 N-methylbutyl ammonium chloride 1 |  | Tiiii[C3H7, CH3, Ph] | DCE | (2.04 ± 0.2) × 105 | −19.04 ± 0.2 | −30.80 ± 1.8 | 11.76 ± 3.4 |
 | 3POiii1PSi[C3H7, CH3, Ph] | DCE | (4.95 ± 0.4) × 105 | −24.2 ± 0.2 | −33.03 ± 0.1 | 8.75 ± 0.1 | |
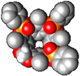 | 3POiii1CH2[C3H7, CH3, Ph] | DCE | (2.05 ± 0.1) × 104 | −24.86 ± 0.1 | −25.01 ± 0.7 | 0.15 ± 1.2 | |
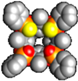 | ABPOii2PSii[C3H7, CH3, Ph] | DCE | (1.63 ± 0.1) × 105 | −28.21 ± 0.2 | −30.24 ± 0.6 | 2.02 ± 0.8 | |
 | ACPOii2PSii[C3H7, CH3, Ph] | DCE | (2.78 ± 0.1) × 103 | −11.22 ± 0.1 | −19.98 ± 1.8 | 8.76 ± 3.0 | |
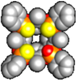 | 1POi3PSiii[C3H7, CH3, Ph] | DCE | Interaction too low to be measured | ||||
 | TSiii[C3H7, CH3, Ph] | DCE | Interaction not detectable | ||||
3. Experimental Section
3.1. General Methods
3.2. Synthesis of Cavitand Hosts
3.2.1. Cavitand 3POiii1PSi[C3H7, CH3, Ph]
3.2.2. Cavitand AB2POii2PSii[C3H7, CH3, Ph] and AC2POii2PSii[C3H7, CH3, Ph]
3.2.3. Cavitand 1POiii3PSi[C3H7, CH3, Ph]
3.2.4. Cavitand TSiiii[C3H7, CH3, Ph]
3.2.5. Cavitand 3POiii1CH2[C3H7, CH3, Ph]
3.3. Crystal Structure of 1POi3PSiii[C2H5, H, Ph]
| Empirical Formula | C72H76O12P4S3 |
|---|---|
| Formula weight (g·mol−1) | 1353.39 |
| Temperature (K) | 190(2) |
| Crystal system | Triclinic |
| Space group | P–1 |
| a (Å) | 11.961(2) |
| b (Å) | 16.442(2) |
| c (Å) | 19.828(3) |
| α (°) | 109.717(3) |
| β (°) | 101.308(3) |
| γ (°) | 95.779(3) |
| V (Å3) | 3540.3(9) |
| Z | 2 |
| ρcalcd | 1.270 |
| μ (mm−1) | 0.254 |
| F(000) | 1424 |
| θ for data collection (°) | 1.13–26.55 |
| Reflections collected/unique | 14587/14587 [R(int) = 0.0] |
| Observed reflections [Fo > 4σ(Fo)] | 8806 |
| Completeness to theta | 98.8 |
| Data/restraints/parameters | 14587/0/720 |
| Goodness-of-fit on F2 a | 1.056 |
| Final R indices [Fo > 4σ(Fo)] b | R1 = 0.0658, wR2 = 0.1991 |
| R indices (all data) | R1 = 0.1000, wR2 = 0.2193 |
| largest diff. peak and hole (e Å3) | 1.027 and −0.568 |
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Biavardi, E.; Battistini, G.; Montalti, M.; Yebeutchou, R.M.; Prodi, L.; Dalcanale, E. Fully reversible guest exchange in tetraphosphonate cavitand complexes probed by fluorescence spectroscopy. Chem. Commun. 2008, 1638–1640. [Google Scholar] [CrossRef]
- Kalenius, E.; Moiani, D.; Dalcanale, E.; Vainiotalo, P. Measuring H-bonding in supramolecular complexes by gas phase ion-molecule reactions. Chem. Commun. 2007, 3865–3867. [Google Scholar] [CrossRef]
- Zhang, Z.; Luo, Y.; Chen, J.; Dong, S.; Yu, Y.; Ma, Z.; Huang, F. Formation of Linear Supramolecular Polymers That Is Driven by CH···π Interactions in Solution and in the Solid State. Angew. Chem. Int. Ed. 2011, 50, 1397–1401. [Google Scholar] [CrossRef]
- Biavardi, E.; Tudisco, C.; Maffei, F.; Motta, A.; Massera, C.; Condorelli, G.G.; Dalcanale, E. Exclusive recognition of sarcosine in water and urine by a cavitand-functionalized silicon surface. Proc. Natl. Acad. Sci. USA 2012, 109, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Biavardi, E.; Federici, S.; Tudisco, C.; Menozzi, D.; Massera, C.; Sottini, A.; Condorelli, G.G.; Bergese, P.; Dalcanale, E. Cavitand-grafted silicon microcantilevers as universal probe for illicit and designer drugs in water. Angew. Chem. Int. Ed. 2014, 53, 9183–9188. [Google Scholar] [CrossRef]
- Biavardi, E.; Favazza, M.; Motta, A.; Fragalà, I.L.; Massera, C.; Prodi, L.; Montalti, M.; Melegari, M.; Condorelli, G.G.; Dalcanale, E. Molecular recognition on a cavitand-functionalized silicon surface. J. Am. Chem. Soc. 2009, 131, 7447–7455. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, M.; Oliviero, G.; Menozzi, D.; Federici, S.; Yebeutchou, R.M.; Schmidtchen, F.P.; Dalcanale, E.; Bergese, P. Nanomechanical recognition of N-methylammonium salts. J. Am. Chem. Soc. 2012, 134, 2392–2398. [Google Scholar] [CrossRef] [PubMed]
- Melegari, M.; Massera, C.; Pinalli, R.; Yebeutchou, R.M.; Dalcanale, E. Supramolecular sensing of short chain alcohols with mixed-bridged thio-phosphonate cavitands. Sens. Actuators B 2013, 179, 74–80. [Google Scholar] [CrossRef]
- Reyntjens-Van Damme, D.; Zeegers-Huyskens, T. Infrared spectrometric study of hydrogen bonding of phenols with triethylthiophosphate. J. Phys. Chem. 1980, 84, 282–285. [Google Scholar] [CrossRef]
- Melegari, M.; Suman, M.; Pirondini, L.; Moiani, D.; Massera, C.; Ugozzoli, F.; Kalenius, E.; Vainiotalo, P.; Mulatier, J.-C.; Dutasta, J.-P.; et al. Supramolecular sensing with phosphonate cavitands. Chem. Eur. J. 2008, 14, 5772–5779. [Google Scholar] [CrossRef] [PubMed]
- Schmidtchen, F.P. Isothermal titration calorimetry in supramolecular chemistry. In Analytical Methods in Supramolecular Chemistry; Schalley, C.A., Ed.; Wiley-VCH: Weinheim, Germany, 2007; pp. 55–78. [Google Scholar]
- Cantadori, B.; Betti, P.; Boccini, F.; Massera, C.; Dalcanale, E. Synthesis of partially bridged phosphonate and thiophosphonate resorcinarenes. Supramol. Chem. 2008, 20, 29–34. [Google Scholar] [CrossRef]
- TWINABS. Twinned Split Crystals Cell; Bruker AXS: Madison, WI, USA, 2004. [Google Scholar]
- SAINT. Software Users Guide; Version 6.0; Bruker Analytical X-ray Systems: Madison, WI, USA, 1999. [Google Scholar]
- Sheldrick, G.M. SADABS v2.03: Area-Detector Absorption Correction; University of Göttingen: Göttingen, Germany, 1999. [Google Scholar]
- Altomare, A.; Burla, M.C.; Camalli, M.; Cascarano, G.L.; Giacovazzo, C.; Guagliardi, A.; Moliterni, A.G.G.; Polidori, G.; Spagna, R. SIR97: A new tool for crystal structure determination and refinement. J. Appl. Cryst. 1999, 32, 115–119. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXL-97.Program for Crystal Structure Refinement; University of Göttingen: Gȍttingen, Germany, 1997. [Google Scholar]
- Farrugia, L.J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. [Google Scholar] [CrossRef]
- Sluis, P.; Spek, A.L. BYPASS: An effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. Sect. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menozzi, D.; Pinalli, R.; Massera, C.; Maffei, F.; Dalcanale, E. The Effect of Number and Position of P=O/P=S Bridging Units on Cavitand Selectivity toward Methyl Ammonium Salts. Molecules 2015, 20, 4460-4472. https://doi.org/10.3390/molecules20034460
Menozzi D, Pinalli R, Massera C, Maffei F, Dalcanale E. The Effect of Number and Position of P=O/P=S Bridging Units on Cavitand Selectivity toward Methyl Ammonium Salts. Molecules. 2015; 20(3):4460-4472. https://doi.org/10.3390/molecules20034460
Chicago/Turabian StyleMenozzi, Daniela, Roberta Pinalli, Chiara Massera, Francesca Maffei, and Enrico Dalcanale. 2015. "The Effect of Number and Position of P=O/P=S Bridging Units on Cavitand Selectivity toward Methyl Ammonium Salts" Molecules 20, no. 3: 4460-4472. https://doi.org/10.3390/molecules20034460
APA StyleMenozzi, D., Pinalli, R., Massera, C., Maffei, F., & Dalcanale, E. (2015). The Effect of Number and Position of P=O/P=S Bridging Units on Cavitand Selectivity toward Methyl Ammonium Salts. Molecules, 20(3), 4460-4472. https://doi.org/10.3390/molecules20034460