
Cross-Coupling Synthesis of Methylallyl Alkenes: Scope Extension and Mechanistic Study




Abstract
:1. Introduction
2. Results and Discussion
2.1. Reaction Conditions Optimization


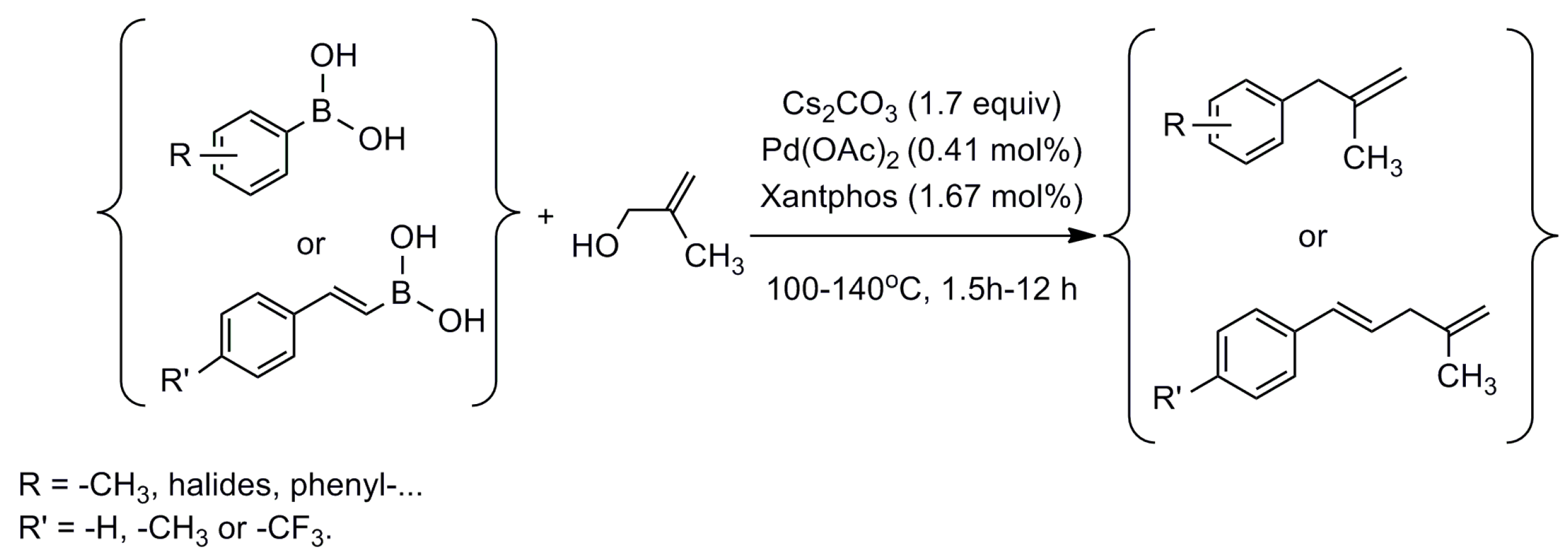{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Alkenes | Side Product | Yield (%) 1,2 | |
|---|---|---|---|---|
| 1 | 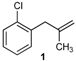 | 1 | 51 | |
| 2 | 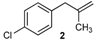 | 2 | 95 | |
| 3 | 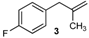 | 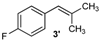 | 3/3′ | 52/38 |
| 4 | 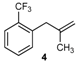 | 4 | 55 | |
| 5 |  | 5 | 77 | |
| 6 | 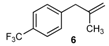 | 6 | 78 | |
| 7 | 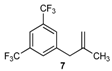 | 7 | 80 | |
| 8 |  | 8 | 57 | |
| 9 | 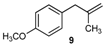 | 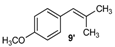 | 9/9′ | 45/30 |
| 10 | 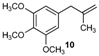 | 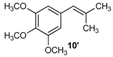 | 10/10′ | 41/42 |
| 11 | 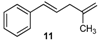 | 11 | 77 | |
| 12 | 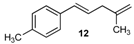 | 12 | 50 | |
| 13 |  | 13 | 46 | |
| Entry | Alkenes | Yield (%) 1 |
|---|---|---|
| 14 | 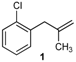 | 61 2 |
| 15 | 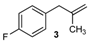 | 74 3 |
| 16 | 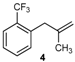 | 97 3 |
| 17 | 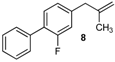 | 78 3 |
| 18 | 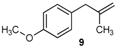 | 97 3 |
| 19 |  | 99 2 |
| 20 | 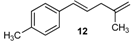 | 92 3 |
| 21 | 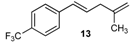 | 88 3 |
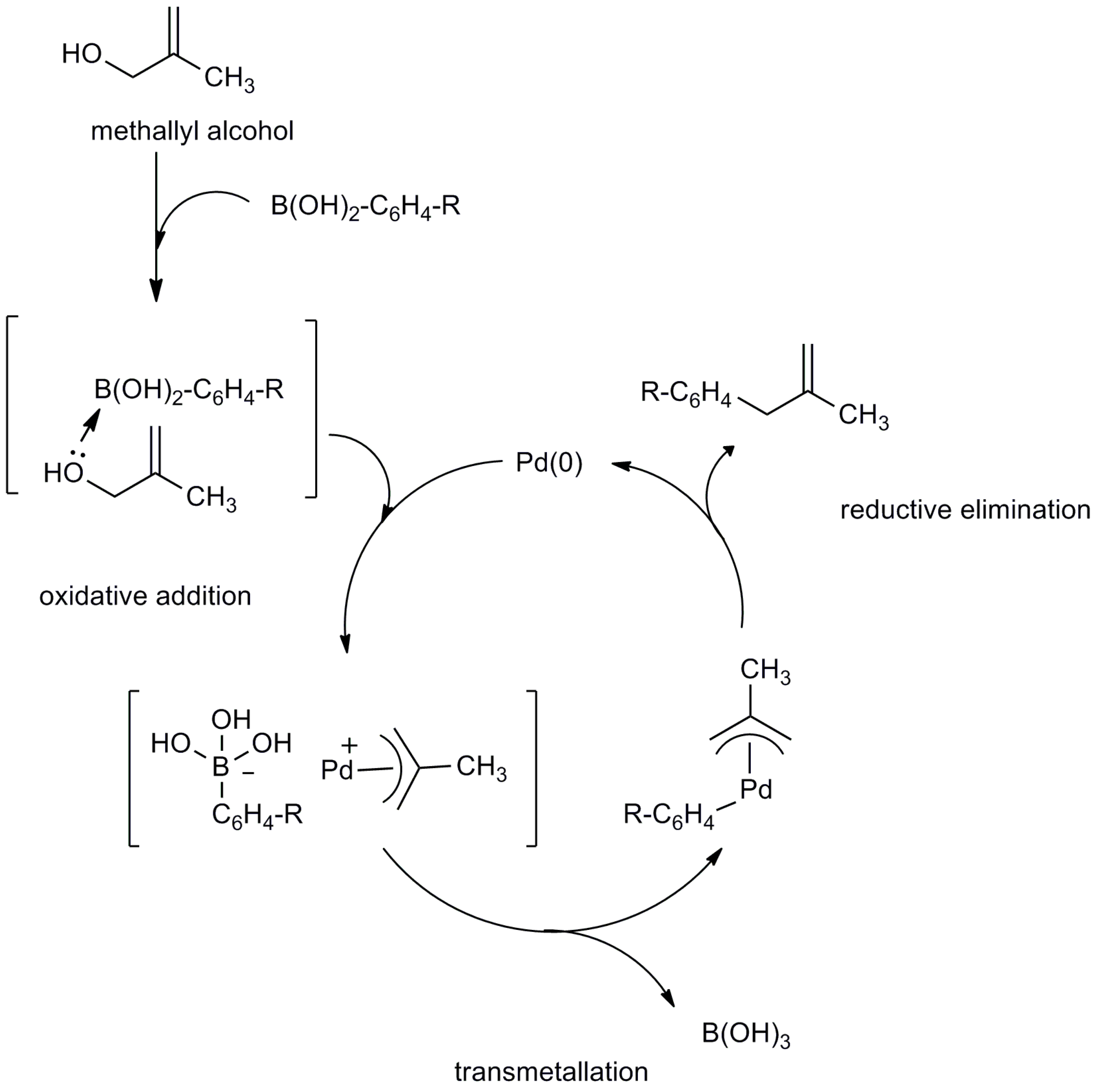
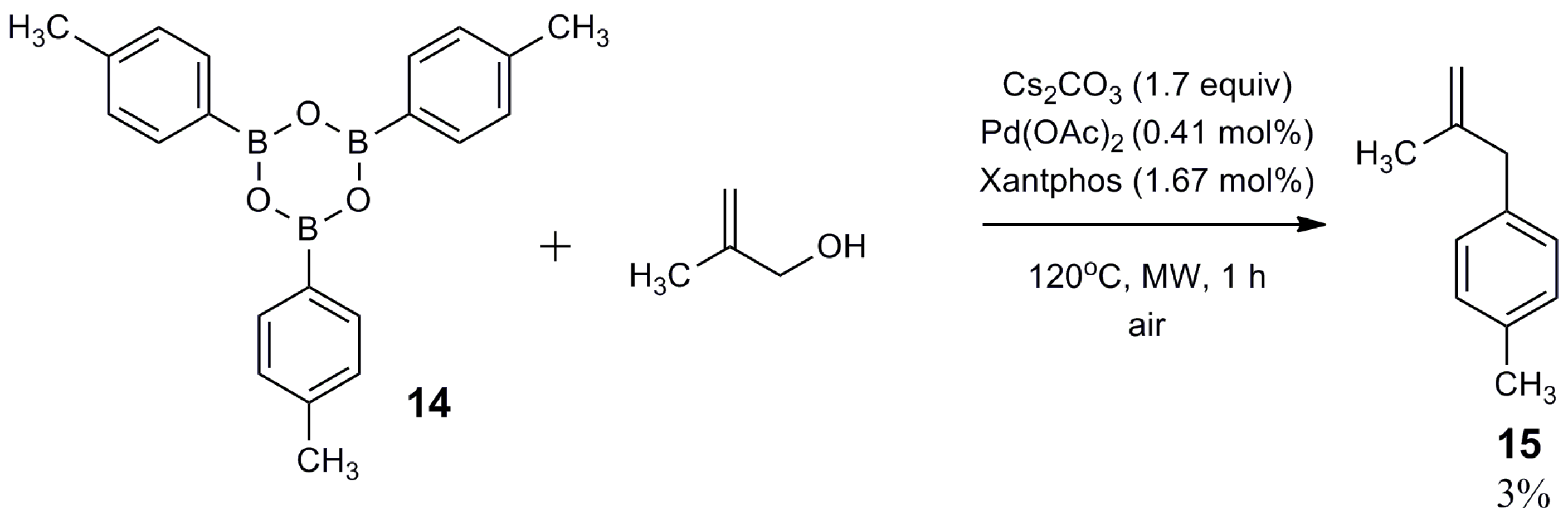
2.2. Mechanistic Study
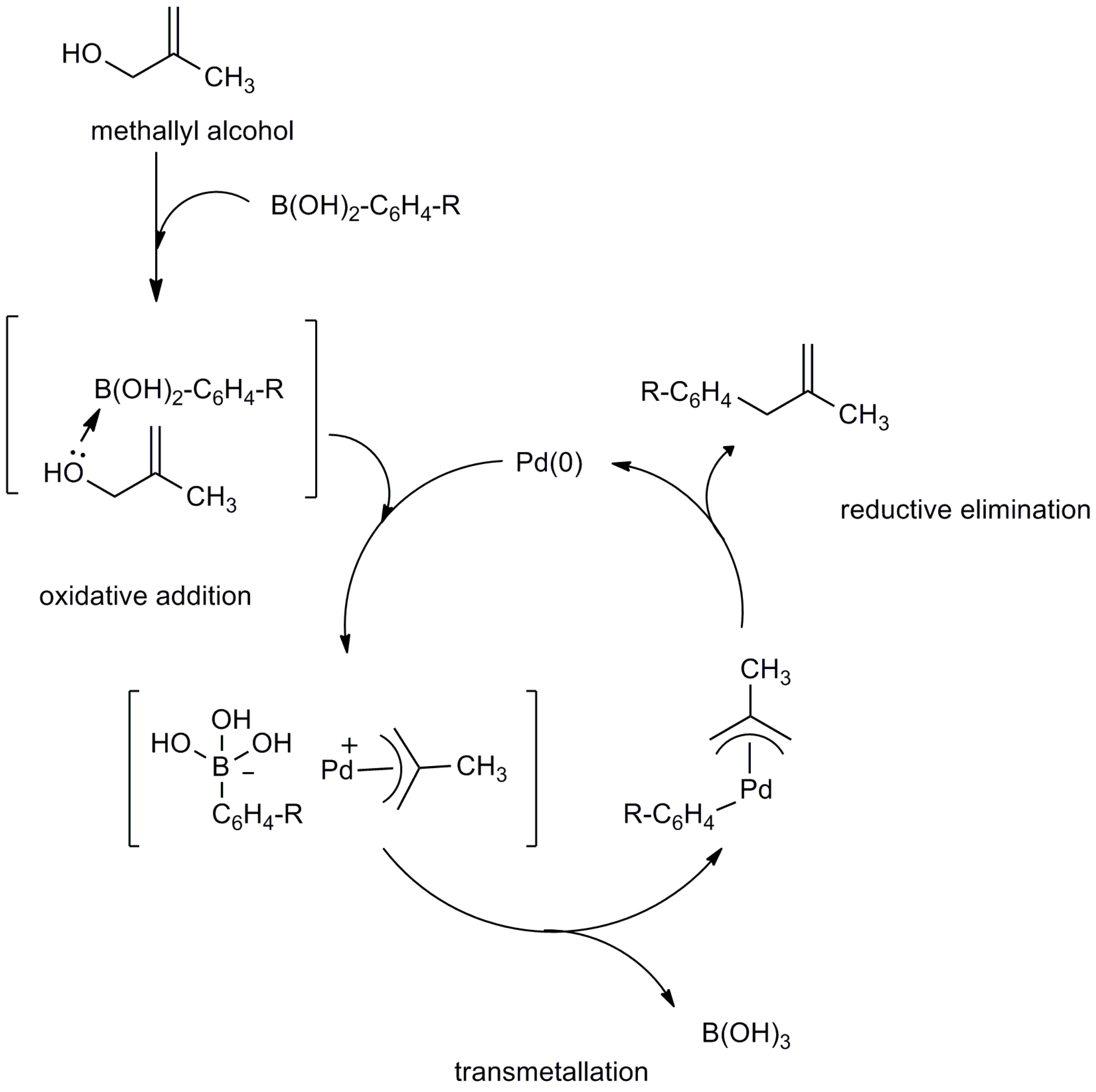
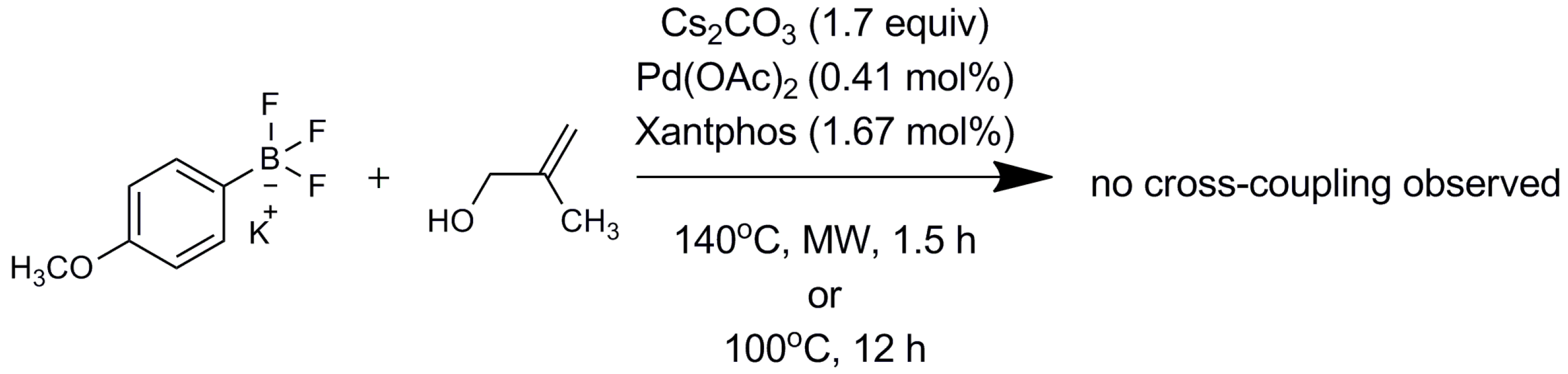
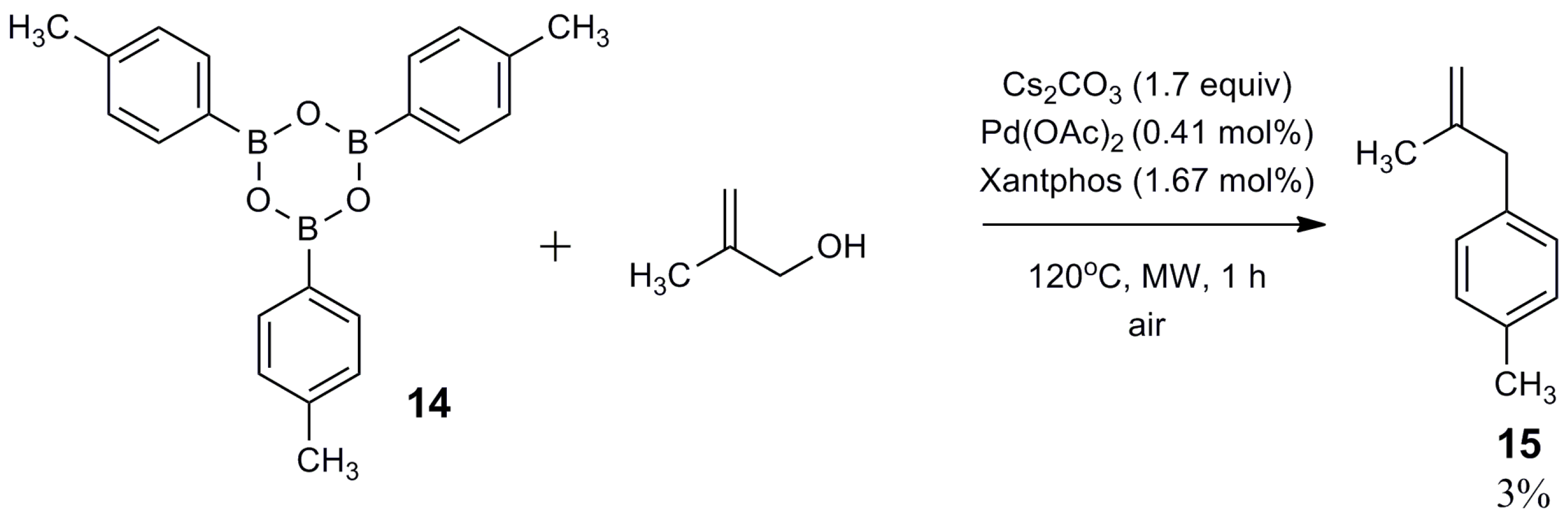
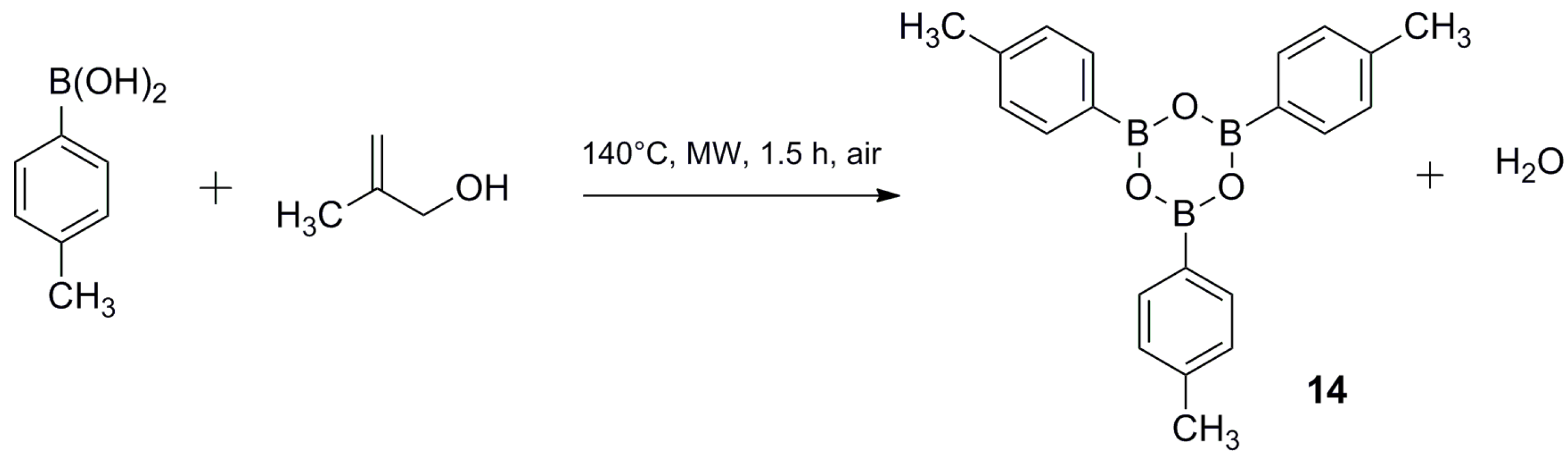
3. Materials and Methods
3.1. General Information
3.2. Synthesis of Methylallyl Alkenes
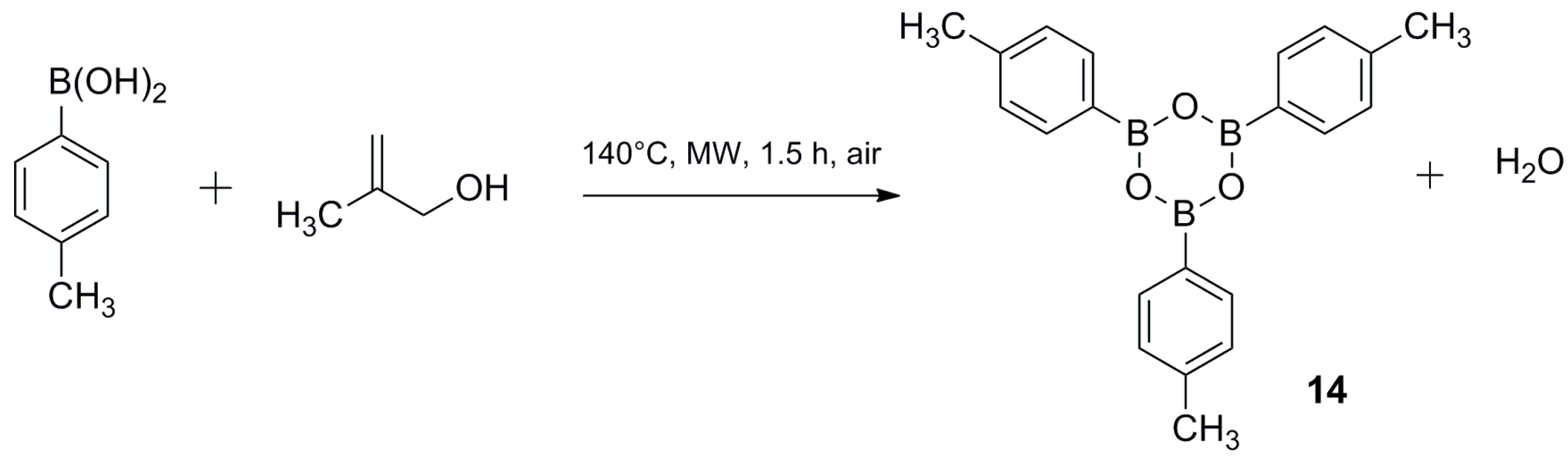
3.3. Synthesis of 2,4,6-tri-p-Tolyl-1,3,5,2,4,6-trioxatriborinane
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tabélé, C.; Curti, C.; Primas, N.; Kabri, Y.; Remusat, V.; Vanelle, P. An efficient method for the synthesis of 2-methylallyl alkenes by cross-coupling reactions. Synthesis 2015, 47, 3339–3346. [Google Scholar]
- Tabélé, C.; Cohen, A.; Curti, C.; Bouhlel, A.; Hutter, S.; Rémusat, V.; Primas, N.; Terme, T.; Azas, N.; Vanelle, P. New series of monoamidoxime derivatives displaying versatile antiparasitic activity. Eur. J. Med. Chem. 2014, 87, 440–453. [Google Scholar] [CrossRef] [PubMed]
- Bouhlel, A.; Curti, C.; Vanelle, P. New methodology for the synthesis of thiobarbiturates mediated by manganese(III) acetate. Molecules 2012, 17, 4313–4325. [Google Scholar] [CrossRef] [PubMed]
- Kuivila, H.; Reuwer, J.; Mangravite, J. Electrophilic displacement reactions: XV. Kinetics and mechanism of the base-catalyzed protodeboronation of areneboronic acids Can. J. Chem. 1963, 41, 3081–3090. [Google Scholar] [CrossRef]
- Kuivila, H.; Reuwer, J.; Mangravite, J. Electrophilic displacement reactions. XVI. Metal ion catalysis in the protodeboronation of areneboronic acids. J. Am. Chem. Soc. 1964, 86, 2666–2670. [Google Scholar] [CrossRef]
- Kinzel, T.; Zhang, Y.; Buchwald, S. A New Palladium Precatalyst Allows for the fast Suzuki—Miyaura coupling reactions of unstable polyfluorophenyl and 2-heteroaryl boronic acids. J. Am. Chem. Soc. 2010, 132, 14073–14075. [Google Scholar] [CrossRef] [PubMed]
- Primas, N.; Mahatsekake, C.; Bouillon, A.; Lancelot, J.C.; Oliveira Santos, J.S.; Lohier, J.F.; Rault, S. A new boronic-acid based strategy to synthesize 4(5)-(het)aryl-1H-imidazoles. Tetrahedron 2008, 64, 4596–4601. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Sato, M.; Kondo, Y. Palladium(0)-catalyzed direct cross-coupling reaction of allyl alcohols with aryl- and vinyl-boronic acids. Chem. Commun. 2004, 10, 1200–1201. [Google Scholar] [CrossRef]
- Baxter, J.; Steinhuebel, D.; Palucki, M.; Davies, I. Stereoselective enol tosylation: Preparation of trisubstituted α,β-unsaturated esters. Org. Lett. 2005, 7, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Uchiyama, T.; Suzuki, T.; Kondo, Y. Palladium(0)-catalyzed direct cross-coupling reaction of allylic alcohols with aryl- and alkenylboronic acids. Org. Biomol. Chem. 2008, 6, 3005–3013. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Zhao, J.; Xu, J.; Mao, Y.; Zhang, Y.J. Pd-Catalyzed stereospecific allyl–aryl coupling of allylic alcohols with arylboronic acids. Chem. Commun. 2013, 49, 9761–9763. [Google Scholar] [CrossRef] [PubMed]
- Kayaki, Y.; Koda, T.; Ikariya, T. A highly effective (triphenyl phosphite)palladium catalyst for a cross-coupling reaction of allylic alcohols with organoboronic acids. Eur. J. Org. Chem. 2004, 2004, 4989–4993. [Google Scholar] [CrossRef]
- Manabe, K.; Nakada, K.; Aoyama, N.; Kobayashi, S. Cross-coupling reactions of allylic alcohols in water. Adv. Synth. Catal. 2005, 347, 1499–1503. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, S.C.; Lu, J.M. N-Heterocyclic carbene-palladium(II)-1-methylimidazole complex catalyzed allyl-aryl coupling of allylic alcohols with arylboronic acids in neat water. Tetrahedron 2015, 71, 544–549. [Google Scholar] [CrossRef]
- Kayaki, Y.; Koda, T.; Ikariya, T. Halide-free dehydrative allylation using allylic alcohols promoted by a palladium-triphenyl phosphite catalyst. J. Org. Chem. 2004, 69, 2595–2597. [Google Scholar] [CrossRef] [PubMed]
- Gooßen, L.J.; Paetzold, J. New synthesis of biaryls via Rh-catalyzed decarbonylative suzuki-coupling of carboxylic anhydrides with arylboroxines. Adv. Synth. Catal. 2004, 346, 1665–1668. [Google Scholar] [CrossRef]
- Martin, R.; Buchwald, S.L. Palladium-catalyzed suzuki-miyaura cross-coupling reactions employing dialkylbiaryl phosphine ligands. Acc. Chem. Res. 2008, 41, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Tsubouchi, A.; Muramatsu, D.; Takeda, T. Copper(I)-catalyzed alkylation of aryl- and alkenylsilanes activated by intramolecular coordination of an alkoxide. Angew. Chem. Int. Ed. 2013, 52, 12719–12722. [Google Scholar] [CrossRef] [PubMed]
- Volla, C.M.R.; Markovic, D.; Reddy Dubbaka, S.; Vogel, P. Ligandless iron-catalyzed desulfinylative C–C allylation reactions using grignard reagents and Alk-2-enesulfonyl chlorides. Eur. J. Org. Chem. 2009, 36, 6281–6288. [Google Scholar] [CrossRef]
- Vedejs, E.; Chapman, R.W.; Fields, S.C.; Lin, S.; Schrimpf, M.R. Conversion of arylboronic acids into potassium aryltrifluoroborates: Convenient precursors of arylboron difluoride lewis acids. J. Org. Chem. 1995, 60, 3020–3027. [Google Scholar] [CrossRef]
- Hayashi, S.; Hirano, K.; Yurimitsu, H.; Oshima, K. Palladium-catalyzed stereo- and regiospecific allylation of aryl halides with homoallyl alcohols via Retro-allylation: Selective generation and use of σ-allylpalladium. J. Am. Chem. Soc. 2006, 128, 2210–2211. [Google Scholar] [CrossRef] [PubMed]
- Rappoport, Z.; Kaspi, J. Vinylic cations from solvolysis. XVIII. Unusual solvent effects and external ion return in the solvolysis of several vinylic compounds in aqueous trifluoroethanol. J. Am. Chem. Soc. 1974, 96, 4518–4530. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1–15 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabélé, C.; Curti, C.; Kabri, Y.; Primas, N.; Vanelle, P. Cross-Coupling Synthesis of Methylallyl Alkenes: Scope Extension and Mechanistic Study. Molecules 2015, 20, 22890-22899. https://doi.org/10.3390/molecules201219886
Tabélé C, Curti C, Kabri Y, Primas N, Vanelle P. Cross-Coupling Synthesis of Methylallyl Alkenes: Scope Extension and Mechanistic Study. Molecules. 2015; 20(12):22890-22899. https://doi.org/10.3390/molecules201219886
Chicago/Turabian StyleTabélé, Clémence, Christophe Curti, Youssef Kabri, Nicolas Primas, and Patrice Vanelle. 2015. "Cross-Coupling Synthesis of Methylallyl Alkenes: Scope Extension and Mechanistic Study" Molecules 20, no. 12: 22890-22899. https://doi.org/10.3390/molecules201219886
APA StyleTabélé, C., Curti, C., Kabri, Y., Primas, N., & Vanelle, P. (2015). Cross-Coupling Synthesis of Methylallyl Alkenes: Scope Extension and Mechanistic Study. Molecules, 20(12), 22890-22899. https://doi.org/10.3390/molecules201219886