
When Phosphosugars Meet Gold: Synthesis and Catalytic Activities of Phostones and Polyhydroxylated Phosphonite Au(I) Complexes


Abstract
:
1. Introduction
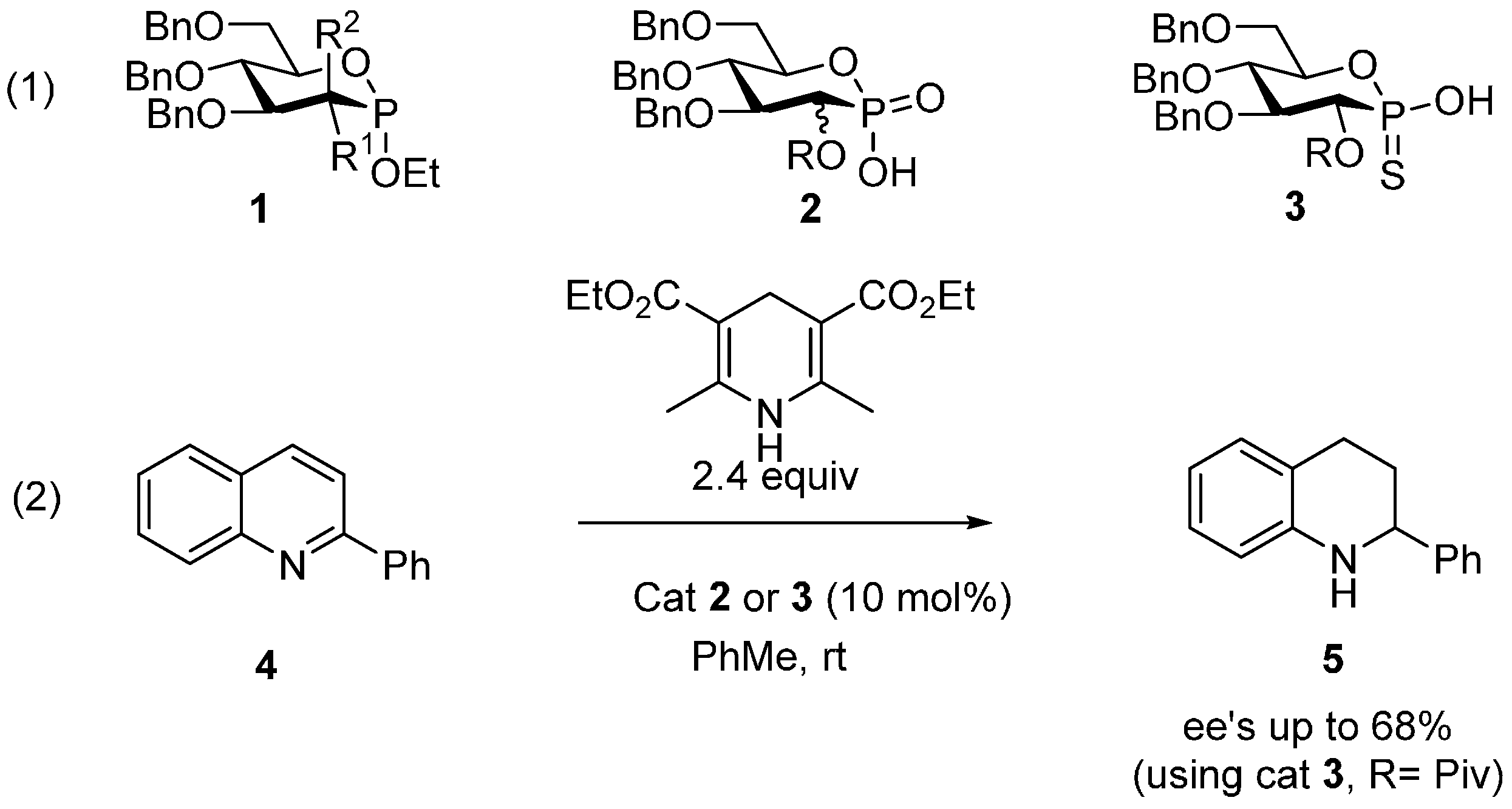
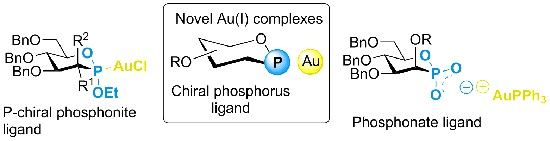
2. Results and Discussion
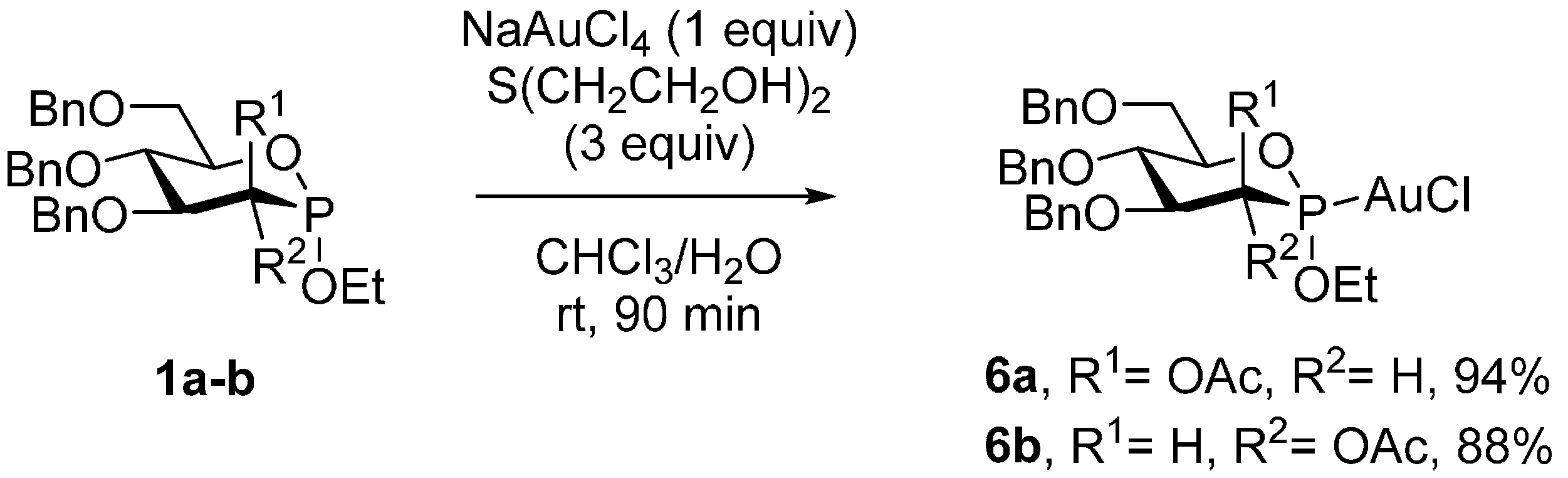

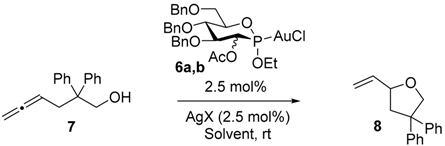
| Entry | Cat. | Solvent | AgX | Yield (%) 2 | ee (%) |
|---|---|---|---|---|---|
| 1 | 6a | PhMe | AgOTs | 90 | 9 |
| 2 | 6a | PhMe | AgSbF6 | 62 | 7 |
| 3 | 6a | PhMe | AgBF4 | 94 | 2 |
| 4 | 6a | PhMe | AgClO4 | 68 | 4 |
| 5 | 6a | PhMe | AgOBz | 94 | 5 |
| 6 | 6b | PhMe | AgOTs | 86 | 6 |
| 7 | 6b | PhMe | AgSbF6 | 64 | 5 |
| 8 | 6b | PhMe | AgBF4 | 70 | 9 |
| 9 | 6b | PhMe | AgClO4 | 66 | 14 |
| 10 | 6b | PhMe | AgOBz | 0 | - |
| 11 | 6a | EtOAc | AgOTs | 98 | 2 |
| 12 | 6a | THF | AgOTs | 98 | 0 |
| 13 | 6a | Acetone | AgOTs | 94 | 0 |
| 14 | 6b | EtOAc | AgOTs | 94 | 7 |
| 15 | 6b | THF | AgOTs | 90 | 4 |
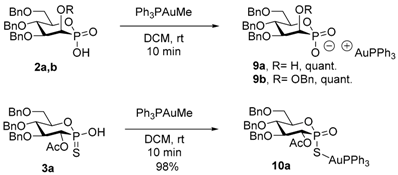
| Entry | Compound | 31P-NMR Chemical Shift |
|---|---|---|
| 1 | 2a | 20.2 |
| 2 | 9a | 27.7 and 19.2 (PPh3) |
| 3 | 2b | 20.7 |
| 4 | 9b | 27.7 and 18.6 (PPh3) |
| 5 | 3a | 79.9 |
| 6 | 10a | 51.4 and 37.4 (PPh3) |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
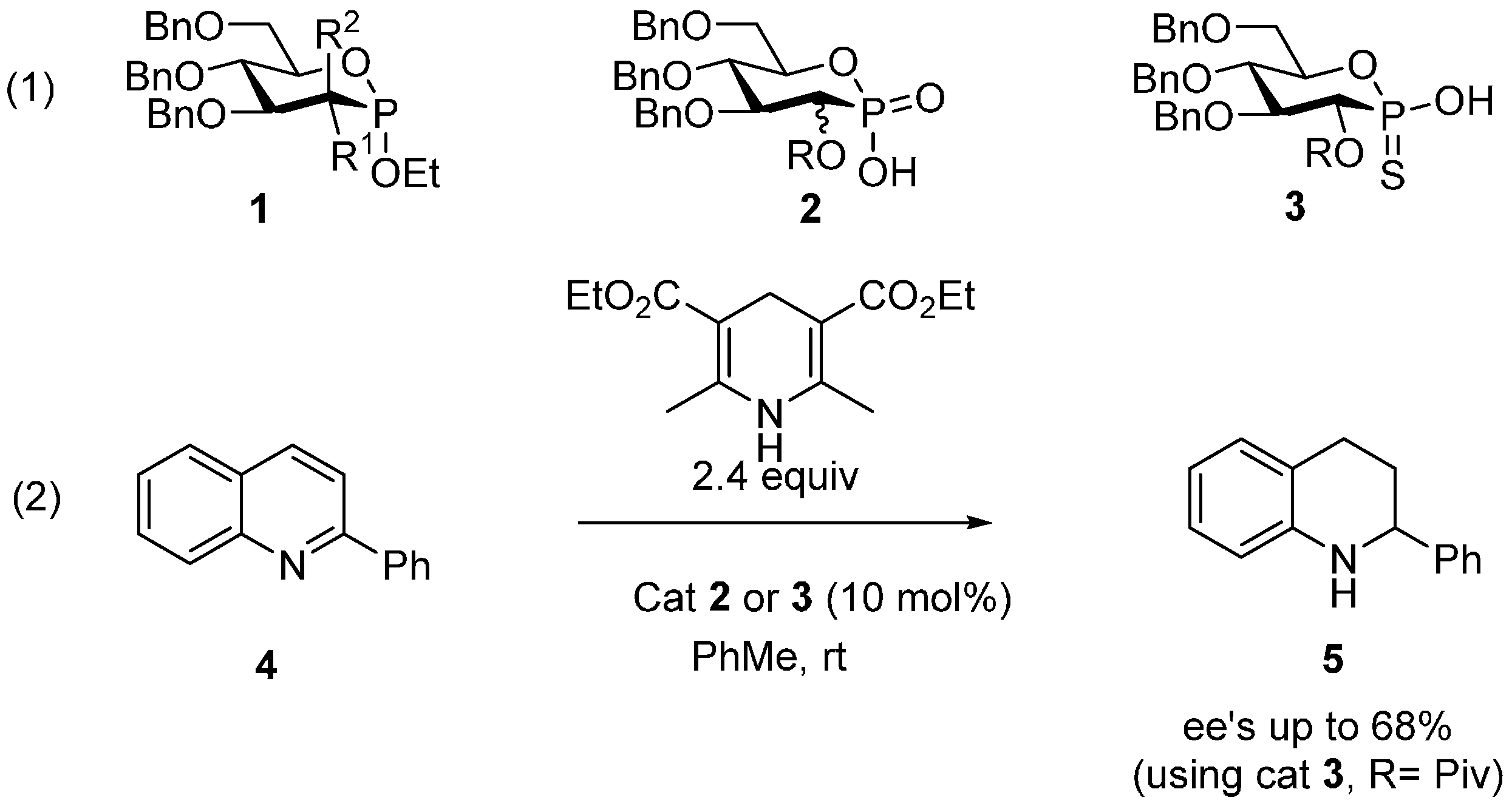
| Entry | Cat. | T (°C) | Yield (%) 2 | ee (%) |
|---|---|---|---|---|
| 1 | 2b | rt | 99 | 7 |
| 2 | 2c | rt | 99 | 14 |
| 3 | 2d | rt | 80 | 4 |
| 4 | 2e | rt | 93 | 15 |
| 5 | 3a or 3b | rt | 0 | - |
| 6 | 3a or 3b | 60 | traces | - |
| 7 3 | 3a | 60 | 25% | 6 |
| 8 4 | 3a | 60 | 27% | 0 |
| 9 4 | 3b | 60 | 59% | 0 |
 | ||||
3. Experimental Section
3.1. General Information
3.2. Synthesis of Chloro[(2S,3R,4S,5S,6R)-4,5-bis(benzyloxy)-6-((benzyloxy)methyl)-2-ethoxy-1,2-oxaphosphinan-3-yl acetate] gold (6a)
3.3 Synthesis of Chloro[(2S,3S,4S,5S,6R)-4,5-bis(benzyloxy)-6-((benzyloxy)methyl)-2-ethoxy-1,2-oxaphosphinan-3-yl acetate] gold (6b)
3.4. Representative Procedure for Gold Catalyzed-Cyclization of Allenol 7
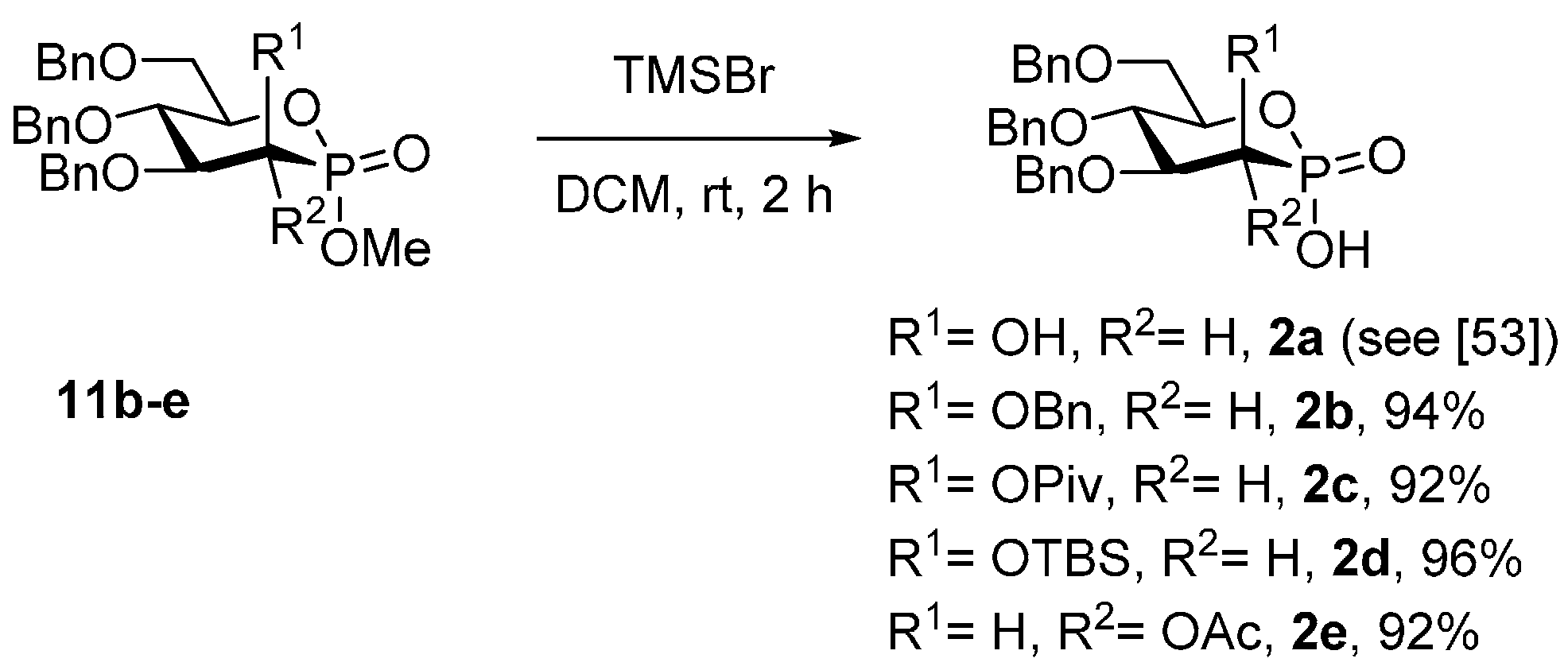
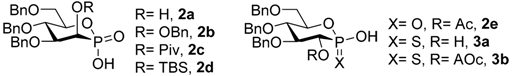
3.5. Synthesis of Phosphonic Acids 2
3.6. Formation of Gold Complexes 9 and 10
3.7. Representative Procedure for Gold/Acid Catalyzed-Cyclization of 12 in 5
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hashmi, A.S.K. Homogeneous gold catalysis beyond assumptions and proposals—Characterized intermediates. Angew. Chem. Int. Ed. 2010, 49, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Bongers, N.; Krause, N. Golden opportunities in stereoselective catalysis. Angew. Chem. Int. Ed. 2008, 47, 2178–2181. [Google Scholar] [CrossRef] [PubMed]
- Fuerstner, A. Gold and platinum catalysis—A convenient tool for generating molecular complexity. Chem. Soc. Rev. 2009, 38, 3208–3221. [Google Scholar] [CrossRef] [PubMed]
- Arcadi, A. Alternative synthetic methods through new developments in catalysis by gold. Chem. Rev. 2008, 108, 3266–3325. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Zhou, Y.; Liu, H. Recent advances in the gold-catalyzed additions to C–C multiple bonds. Beilstein J. Org. Chem. 2011, 7, 897–936. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.; Hashmi, A.S.K. Gold catalysis in total synthesis-an update. Chem. Soc. Rev. 2012, 41, 2448–2462. [Google Scholar] [CrossRef] [PubMed]
- Hashmi, A.S.K.; Rudolph, M. Gold catalysis in total synthesis. Chem. Soc. Rev. 2008, 37, 1766–1775. [Google Scholar] [CrossRef] [PubMed]
- Widenhoefer, R.A.; Han, X. Gold-catalyzed hydroamination of C–C multiple bonds. Eur. J. Org. Chem. 2006, 2006, 4555–4563. [Google Scholar] [CrossRef]
- Dorel, R.; Echavarren, A.M. Gold(I)-catalyzed activation of alkynes for the construction of molecular complexity. Chem. Rev. 2015, 115, 9028–9072. [Google Scholar] [CrossRef] [PubMed]
- Pradal, A.; Toullec, P.Y.; Michelet, V. Recent developments in asymmetric catalysis in the presence of chiral gold complexes. Synthesis 2011, 1501–1514. [Google Scholar]
- Teller, H.; Flügge, S.; Goddard, R.; Fürstner, A. Enantioselective gold catalysis: Opportunities provided by monodentate phosphoramidite ligands with an acyclic taddol backbone. Angew. Chem. Int. Ed. 2010, 49, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Shi, X. Recent advances in asymmetric gold catalysis. ChemCatChem 2010, 2, 609–619. [Google Scholar] [CrossRef]
- Widenhoefer, R.A. Recent developments in enantioselective gold(I) catalysis. Chem. Eur. J. 2008, 14, 5382–5391. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-M.; Lackner, A.D.; Toste, F.D. Development of catalysts and ligands for enantioselective gold catalysis. Acc. Chem. Res. 2014, 47, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-W.; Xu, Q.; Shi, M. Synthesis of axially chiral gold complexes and their applications in asymmetric catalyses. Beilstein J. Org. Chem. 2013, 9, 2224–2232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-M.; Chen, P.; Li, W.; Niu, Y.; Zhao, X.-L.; Zhang, J. A new type of chiral sulfinamide monophosphine ligands: Stereodivergent synthesis and application in enantioselective gold(I)-catalyzed cycloaddition reactions. Angew. Chem. Int. Ed. 2014, 53, 4350–4354. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.Z.; Benitez, D.; Tkatchouk, E.; Goddard, W.A., III; Toste, F.D. Phosphoramidite gold(I)-catalyzed diastereo- and enantioselective synthesis of 3,4-substituted pyrrolidines. J. Am. Chem. Soc. 2011, 133, 5500–5507. [Google Scholar] [CrossRef] [PubMed]
- Raubenheimer, H.G. Monomeric linear diaminocarbene complexes of gold(I) show merit in enantioselective catalysis. Angew. Chem. Int. Ed. 2012, 51, 5042–5044. [Google Scholar] [CrossRef] [PubMed]
- Handa, S.; Slaughter, L.M. Enantioselective alkynylbenzaldehyde cyclizations catalyzed by chiral gold(I) acyclic diaminocarbene complexes containing weak au-arene interactions. Angew. Chem. Int. Ed. 2012, 51, 2912–2915. [Google Scholar] [CrossRef] [PubMed]
- Barbazanges, M.; Fensterbank, L. Chiral acyclic diaminocarbene complexes: A new opportunity for gold asymmetric catalysis. ChemCatChem 2012, 4, 1065–1066. [Google Scholar] [CrossRef]
- Cera, G.; Bandini, M. Enantioselective gold(I) catalysis with chiral monodentate ligands. Isr. J. Chem. 2013, 53, 848–855. [Google Scholar] [CrossRef]
- Francos, J.; Grande-Carmona, F.; Faustino, H.; Iglesias-Sigueenza, J.; Diez, E.; Alonso, I.; Fernandez, R.; Lassaletta, J.M.; Lopez, F.; Mascarenas, J.L. Axially chiral triazoloisoquinolin-3-ylidene ligands in gold(I)-catalyzed asymmetric intermolecular (4 + 2) cycloadditions of allenamides and dienes. J. Am. Chem. Soc. 2012, 134, 14322–14325. [Google Scholar] [CrossRef] [PubMed]
- Gu, P.; Xu, Q.; Shi, M. Development and outlook of chiral carbene-gold(I) complexes catalyzed asymmetric reactions. Tetrahedron Lett. 2014, 55, 577–584. [Google Scholar] [CrossRef]
- Yavari, K.; Aillard, P.; Zhang, Y.; Nuter, F.; Retailleau, P.; Voituriez, A.; Marinetti, A. Helicenes with embedded phosphole units in enantioselective gold catalysis. Angew. Chem. Int. Ed. 2014, 53, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Aillard, P.; Retailleau, P.; Voituriez, A.; Marinetti, A. Synthesis of new phosphahelicene scaffolds and development of gold(I)-catalyzed enantioselective allenene cyclizations. Chem. Eur. J. 2015, 21, 11989–11993. [Google Scholar] [CrossRef] [PubMed]
- Aillard, P.; Voituriez, A.; Dova, D.; Cauteruccio, S.; Licandro, E.; Marinetti, A. Phosphathiahelicenes: Synthesis and uses in enantioselective gold catalysis. Chem. Eur. J. 2014, 20, 12373–12376. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, G.L.; Kang, E.J.; Mba, M.; Toste, F.D. A powerful chiral counterion strategy for asymmetric transition metal catalysis. Science 2007, 317, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Phipps, R.J.; Hamilton, G.L.; Toste, F.D. The progression of chiral anions from concepts to applications in asymmetric catalysis. Nat. Chem. 2012, 4, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Gyurcsik, B.; Nagy, L. Carbohydrates as ligands: Coordination equilibria and structure of the metal complexes. Coord. Chem. Rev. 2000, 203, 81–149. [Google Scholar] [CrossRef]
- Alexeev, Y.E.; Vasilchenko, I.S.; Kharisov, B.I.; Blanco, L.M.; Garnovskii, A.D.; Zhdanov, Y.A. Review: Synthetically modified carbohydrates as ligands. J. Coord. Chem. 2004, 57, 1447–1517. [Google Scholar] [CrossRef]
- Bauer, T.; Majdecki, M. Carbohydrates as ligands for enantioselective synthesis. Curr. Org. Chem. 2014, 18, 1749–1767. [Google Scholar] [CrossRef]
- Boysen, M.M.K. Hydroformylations, hydrovinylations, and hydrocyanations. In Carbohydrates—Tools for Stereoselective Synthesis; Wiley-VCH: Weinheim, Germany, 2013; pp. 183–216. [Google Scholar]
- Boysen, M.M.K. Cyclopropanation. In Carbohydrates—Tools for Stereoselective Synthesi; Wiley-VCH: Weinheim, Germany, 2013; pp. 313–318. [Google Scholar]
- Claver, C.; Castillón, S.; Diéguez, M.; Pàmies, O. Hydrogenation reactions. In Carbohydrates—Tools for Stereoselective Synthesis; Wiley-VCH: Weinheim, Germany, 2013; pp. 155–182. [Google Scholar]
- Díaz, Y.; Matheu, M.I.; Benito, D.; Castillón, S. 1,4-addition of nucleophiles to α,β-unsaturated carbonyl compounds. In Carbohydrates—Tools for Stereoselective Synthesis; Wiley-VCH: Weinheim, Germany, 2013; pp. 253–291. [Google Scholar]
- Diéguez, M.; Pàmies, O. Carbohydrate-derived ligands in asymmetric tsuji–trost reactions. In Carbohydrates—Tools for Stereoselective Synthesis; Wiley-VCH: Weinheim, Germany, 2013; pp. 217–244. [Google Scholar]
- Diéguez, M.; Pàmies, O. Carbohydrate-derived ligands in asymmetric heck reactions. In Carbohydrates—Tools for Stereoselective Synthesis; Wiley-VCH: Weinheim, Germany, 2013; pp. 245–251. [Google Scholar]
- Matheu, M.I.; Díaz, Y.; Marcé, P.; Castillón, S. 1,2-Addition of nucleophiles to carbonyl compounds. In Carbohydrates—Tools for Stereoselective Synthesis; Wiley-VCH: Weinheim, Germany, 2013; pp. 293–312. [Google Scholar]
- Tri-Substituted Phosphinegold(I) 1-thio-beta-d-Glucopyranosides. U.S. Pantent 4096250, 20 June 1978.
- Ferry, A.; Guinchard, X.; Retailleau, P.; Crich, D. Synthesis, characterization, and coupling reactions of six-membered cyclic P-chiral ammonium phosphonite–boranes; reactive H-phosphinate equivalents for the stereoselective synthesis of glycomimetics. J. Am. Chem. Soc. 2012, 134, 12289–12301. [Google Scholar] [CrossRef] [PubMed]
- Ferry, A.; Malik, G.; Retailleau, P.; Guinchard, X.; Crich, D. Alternative synthesis of P-chiral phosphonite-borane complexes: Application to the synthesis of phostone-phostone dimers. J. Org. Chem. 2013, 78, 6858–6867. [Google Scholar] [CrossRef] [PubMed]
- Malik, G.; Ferry, A.; Guinchard, X.; Cresteil, T.; Crich, D. N–O bond as a glycosidic-bond surrogate: Synthetic studies toward polyhydroxylated n-alkoxypiperidines. Chem. Eur. J. 2012, 19, 2168–2179. [Google Scholar] [CrossRef] [PubMed]
- Malik, G.; Ferry, A.; Guinchard, X.; Crich, D. Synthesis of β-hydroxy O-alkyl hydroxylamines from epoxides using a convenient and versatile two-step procedure. Synthesis 2013, 45, 65–74. [Google Scholar] [CrossRef]
- Malik, G.; Guinchard, X.; Crich, D. Asymmetric synthesis of polyhydroxylated N-alkoxypiperidines by ring-closing double reductive amination: Facile preparation of isofagomine and analogues. Org. Lett. 2012, 14, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Ferry, A.; Stemper, J.; Marinetti, A.; Voituriez, A.; Guinchard, X. Thiophostone-derived brønsted acids in the organocatalyzed transfer hydrogenation of quinolines: Influence of the P-stereogenicity. Eur. J. Org. Chem. 2014, 2014, 188–193. [Google Scholar] [CrossRef]
- Rueping, M.; Antonchick, A.P.; Theissmann, T. A highly enantioselective brønsted acid catalyzed cascade reaction: Organocatalytic transfer hydrogenation of quinolines and their application in the synthesis of alkaloids. Angew. Chem. Int. Ed. 2006, 45, 3683–3686. [Google Scholar] [CrossRef] [PubMed]
- Phosphinegold(I) Salts Having Antiarthritic Activity. U.S. Pantent 4171360, 16 October 1979.
- Bruce, M.I.; Horn, E.; Matisons, J.G.; Snow, M.R. Chemistry of the group 1b metals. Xvii. Preparation of some gold(I) acetylide complexes containing group 5 donor ligands: Crystal and molecular structures of Au(C2C6F5)(PPh3). Aust. J. Chem. 1984, 37, 1163–1170. [Google Scholar] [CrossRef]
- Govindaraju, S.; Ananthnag, G.S.; Naik, S.; Mobin, S.M.; Balakrishna, M.S. Allyl functionalized phosphinite and phosphonite ligands: Synthesis, transition metal chemistry and orthopalladation reactions. J. Chem. Sci. 2012, 124, 773–779. [Google Scholar] [CrossRef]
- Payne, N.C.; Ramachandran, R.; Treurnicht, I.; Puddephat, R.J. Easy double metalation of a diphosphinomethane ligand: structure of the remarkable octagold cage complex [Au8Cl2{.mu.3-(MeO)2PCHP(OMe)2}2{.mu.4-(MeO)2PCP(OMe)2}2].cntdot.CHCl3. Organometallics 1990, 9, 880–882. [Google Scholar] [CrossRef]
- Rao, S.; Mague, J.T.; Balakrishna, M.S. Synthesis, transition metal chemistry and catalytic reactions of ferrocenylbis(phosphonite), [Fe{C5H4P(OC6H3(OMe-o)(C3H5-p))2}2]. Dalton Trans. 2013, 42, 11695–11708. [Google Scholar] [CrossRef] [PubMed]
- Raducan, M.; Moreno, M.; Bour, C.; Echavarren, A.M. Phosphate ligands in the gold(I)-catalysed activation of enynes. Chem. Commun. 2012, 48, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Darrow, J.W.; Drueckhammer, D.G. Cyclic phosphonate analogs of hexopyranoses. J. Org. Chem. 1994, 59, 2976–2985. [Google Scholar] [CrossRef]
- Salomon, C.J.; Breuer, E. Efficient and selective dealkylation of phosphonate dilsopropyl esters using me3sibr. Tetrahedron Lett. 1995, 36, 6759–6760. [Google Scholar] [CrossRef]
- Hanessian, S.; Rogel, O. Synthesis of glycophostones: Cyclic phosphonate analogues of biologically relevant sugars. J. Org. Chem. 2000, 65, 2667–2674. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, E.; Hayashi, T.; Tanaka, M. Au(I)-catalyzed highly efficient intermolecular hydroamination of alkynes. Org. Lett. 2003, 5, 3349–3352. [Google Scholar] [CrossRef] [PubMed]
- Vassiliou, S. Cbz-aminomethylphosphonic acid and its structural variations: Synthesis from a common precursor and a stability study. ARKIVOC 2012, 7–14. [Google Scholar] [CrossRef]
- Han, Z.-Y.; Xiao, H.; Chen, X.-H.; Gong, L.-Z. Consecutive intramolecular hydroamination/asymmetric transfer hydrogenation under relay catalysis of an achiral gold complex/chiral brønsted acid binary system. J. Am. Chem. Soc. 2009, 131, 9182–9183. [Google Scholar] [CrossRef] [PubMed]
- Hakkinen, H. The gold-sulfur interface at the nanoscale. Nature Chem. 2012, 4, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Widenhoefer, R.A. Gold(I)-catalyzed intramolecular enantioselective hydroalkoxylation of allenes. Angew. Chem. Int. Ed. 2007, 46, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, G.; Ferry, A.; Guinchard, X. When Phosphosugars Meet Gold: Synthesis and Catalytic Activities of Phostones and Polyhydroxylated Phosphonite Au(I) Complexes. Molecules 2015, 20, 21082-21093. https://doi.org/10.3390/molecules201219755
Malik G, Ferry A, Guinchard X. When Phosphosugars Meet Gold: Synthesis and Catalytic Activities of Phostones and Polyhydroxylated Phosphonite Au(I) Complexes. Molecules. 2015; 20(12):21082-21093. https://doi.org/10.3390/molecules201219755
Chicago/Turabian StyleMalik, Gaëlle, Angélique Ferry, and Xavier Guinchard. 2015. "When Phosphosugars Meet Gold: Synthesis and Catalytic Activities of Phostones and Polyhydroxylated Phosphonite Au(I) Complexes" Molecules 20, no. 12: 21082-21093. https://doi.org/10.3390/molecules201219755
APA StyleMalik, G., Ferry, A., & Guinchard, X. (2015). When Phosphosugars Meet Gold: Synthesis and Catalytic Activities of Phostones and Polyhydroxylated Phosphonite Au(I) Complexes. Molecules, 20(12), 21082-21093. https://doi.org/10.3390/molecules201219755